
SARS-CoV-2 Variants in Lebanon: Evolution and Current Situation

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
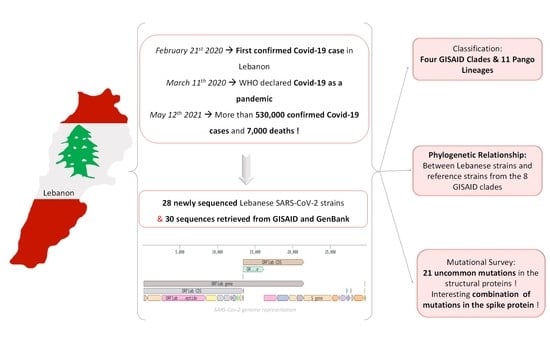
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Processing and SARS-CoV-2 Genome Sequencing
2.3. Bioinformatic Analysis
3. Results
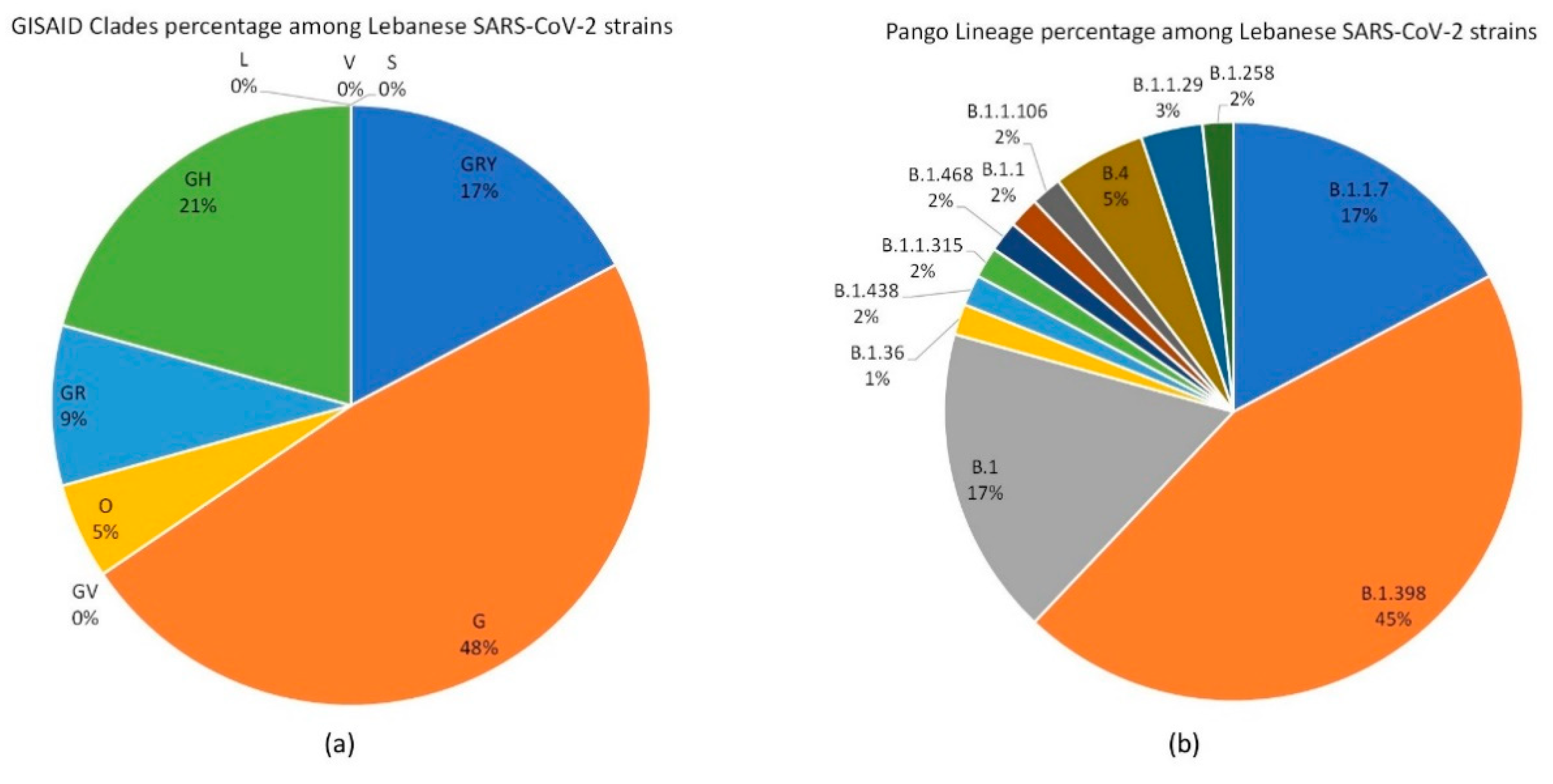
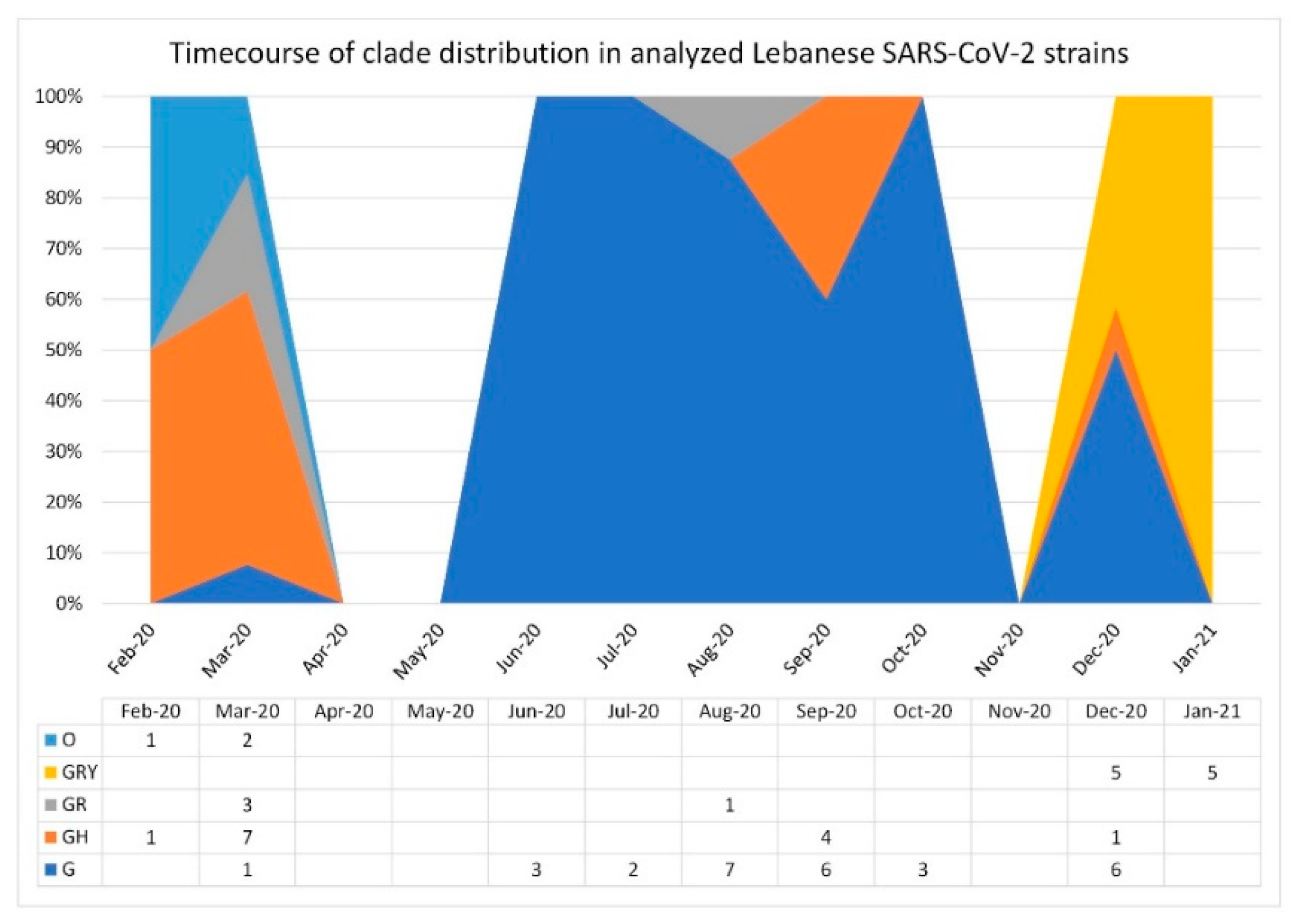
3.1. SARS-CoV-2 Lebanese Strains: Sequences and Classification
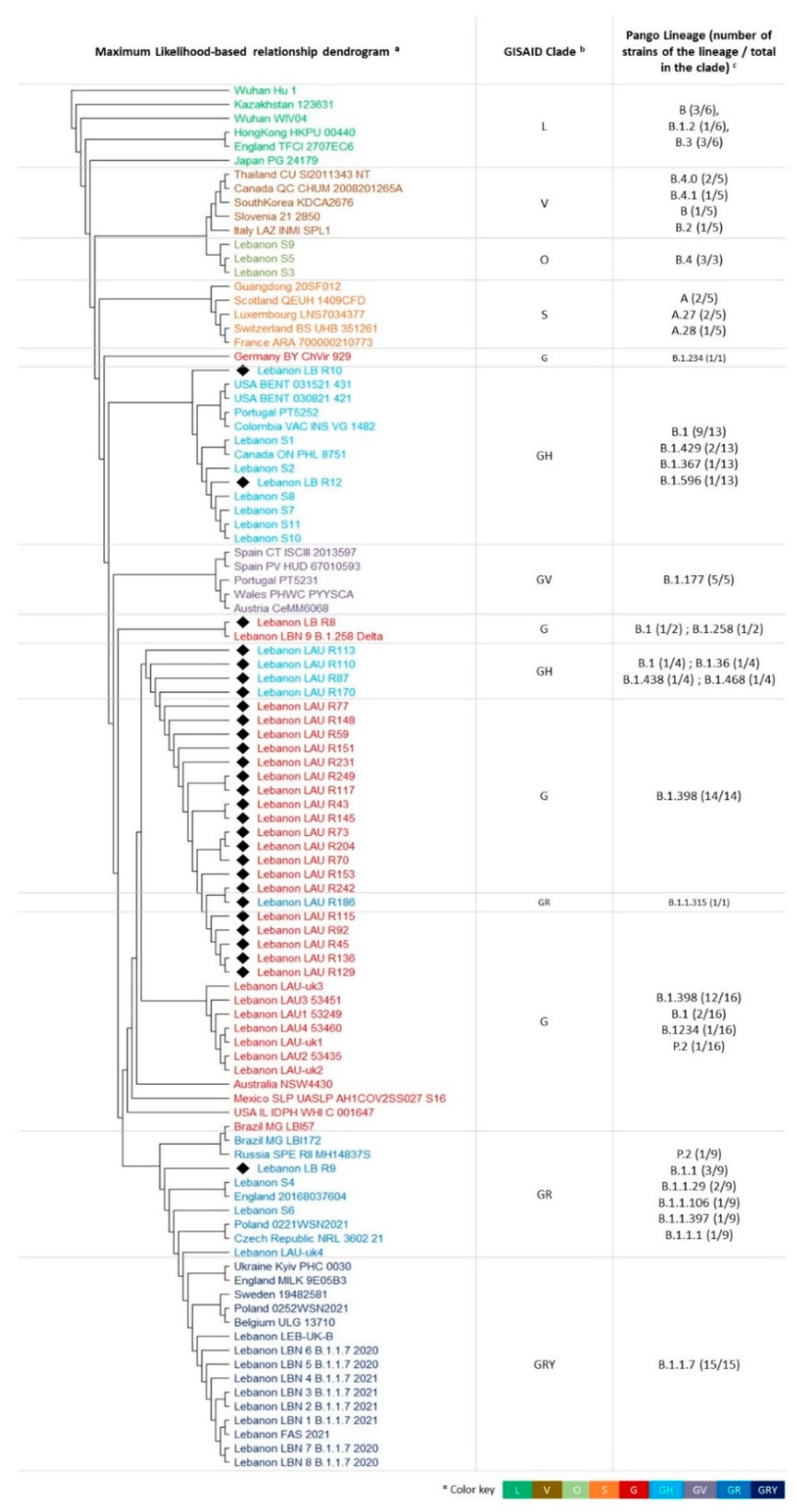
3.2. Phylogenetic Relationships
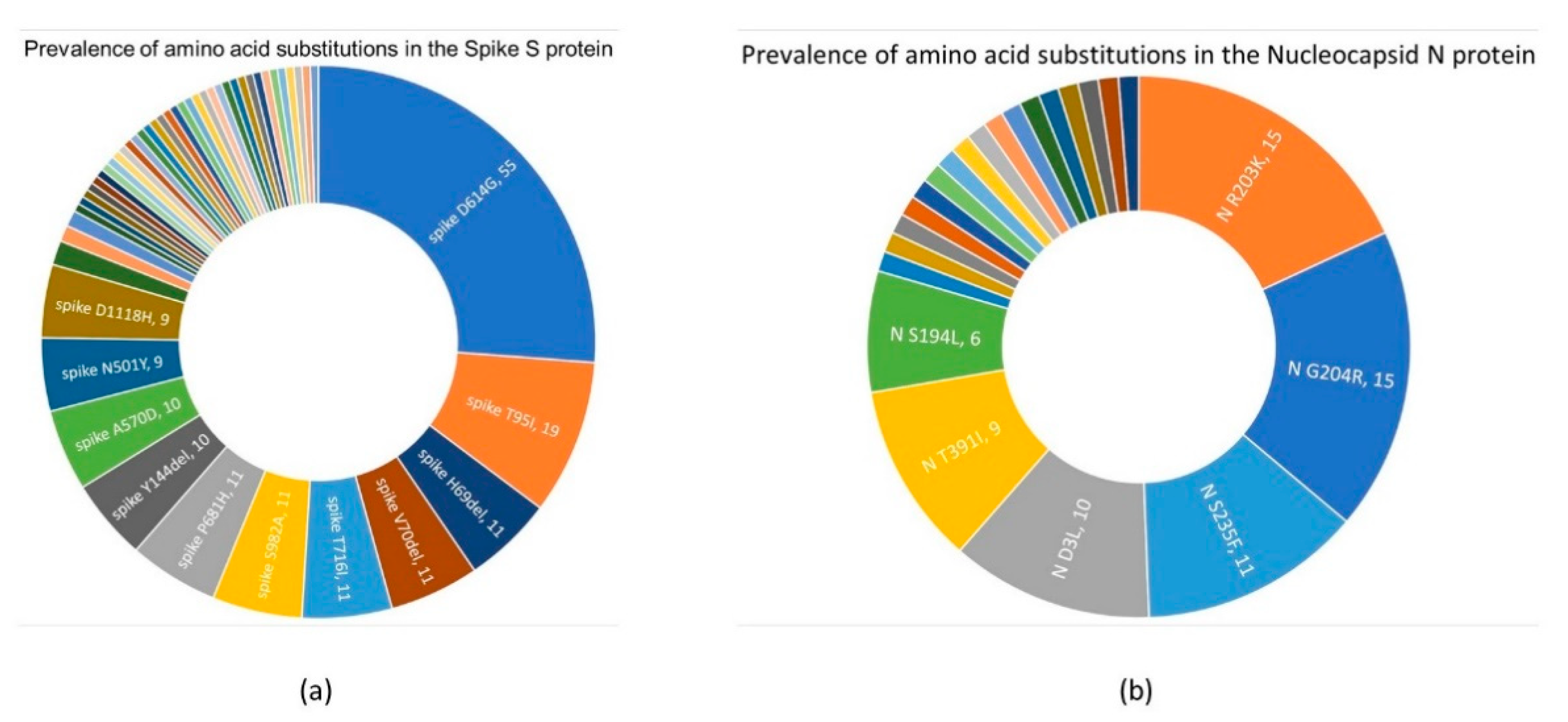
3.3. Mutation Survey: A Focus on Structural Proteins
3.3.1. Spike S Protein
3.3.2. Nucleocapsid N Protein
3.3.3. Membrane M and Envelope E proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Z.; Xiao, K.; Zhang, X.; Roy, A.; Shen, Y. Emergence of SARS-like coronavirus in China: An update. J. Infect. 2020, 80, e28–e29. [Google Scholar] [CrossRef]
- World Health Organization. WHO Director General’s Opening Remarks at the Media Briefing on COVID-19. 11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 5 May 2021).
- Siddiqui, A.J.; Jahan, S.; Ashraf, S.A.; Alreshidi, M.; Ashraf, M.S.; Patel, M.; Snoussi, M.; Singh, R.; Adnan, M. Current Status and Strategic Possibilities on Potential Use of Combinational Drug Therapy against COVID-19 Caused by SARS-CoV-2. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Ciotti, M.; Angeletti, S.; Minieri, M.; Giovannetti, M.; Benvenuto, D.; Pascarella, S.; Sagnelli, C.; Bianchi, M.; Bernardini, S.; Ciccozzi, M. COVID-19 outbreak: An overview. Chemotherapy 2020, 64, 215–223. [Google Scholar] [CrossRef]
- Ellis, P.; Somogyvári, F.; Virok, D.P.; Noseda, M.; Mclean, G.R. Decoding COVID-19 with the SARS-CoV-2 genome. Curr. Genet. Med. Rep. 2021, 9, 1–12. [Google Scholar] [CrossRef]
- Gültekin, V.; Allmer, J. Novel perspectives for SARS-CoV-2 genome browsing. J. Integr. Bioinform. 2021, 18, 19–26. [Google Scholar] [CrossRef]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.W. Decoding SARS-CoV-2 Transmission and Evolution and Ramifications for COVID-19 Diagnosis, Vaccine, and Medicine. J. Chem. Inf. Model. 2020, 60, 5853–5865. [Google Scholar] [CrossRef]
- Shu, Y.; Mccauley, J. GISAID: Global initiative on sharing all influenza data-from vision to reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Alm, E.; Broberg, E.K.; Connor, T.; Hodcroft, E.B.; Komissarov, A.B.; Maurer-Stroh, S.; Melidou, A.; Neher, R.A.; O’Toole, Á.; Pereyaslov, D.; et al. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020. Euro Surveill. 2020, 25, 1–8. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and genomic distribution of SARS-CoV-2 mutations. Front. Microbiol. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Sengupta, A.; Sarif, S.; Pal, P. Clade GR and clade GH isolates of SARS-CoV-2 in Asia show highest amount of SNPs. Infect. Genet. Evol. 2020, 89, 104724. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Crawford, K.H.D.; Bloom, J.D.; Veesler, D.; Vaughan, T.G.; Comas, I.; Candelas, F.G.; Stadler, T.; et al. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. MedRxiv 2020. [Google Scholar] [CrossRef]
- Young, B.E.; Wei, W.E.; Fong, S.-W.; Mak, T.-M.; Anderson, D.E.; Chan, Y.-H.; Pung, R.; Heng, C.S.; Ang, L.W.; Zheng, A.K.E.; et al. Association of SARS-CoV-2 clades with clinical, inflammatory and virologic outcomes: An observational study. EBioMedicine 2021, 66, 103319. [Google Scholar] [CrossRef] [PubMed]
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, G. The SARS-CoV-2 spike variant D614G favors an open conformational state. Biophys. J. 2021, 120, 298a. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Lebanese Ministry of Public Health. Novel Coronavirus 2019-Lebanon. Available online: https://www.moph.gov.lb/en/Pages/2/24870/novel-coronavirus-2019 (accessed on 6 May 2021).
- Bizri, A.R.; Khachfe, H.H.; Fares, M.Y.; Musharrafieh, U. COVID-19 pandemic: An insult over injury for Lebanon. J. Community Health 2020. [Google Scholar] [CrossRef]
- El Deeb, O.; Jalloul, M. The dynamics of COVID-19 spread: Evidence from Lebanon. Math. Biosci. Eng. 2020, 17, 5618–5632. [Google Scholar] [CrossRef]
- Younis, N.K.; Rahm, M.; Bitar, F.; Arabi, M. COVID-19 in the MENA Region: Facts and findings. J. Infect. Dev. Ctries. 2021, 15, 342–349. [Google Scholar] [CrossRef]
- Lebanese Ministry of Public Health. Epidemiological Surveillance Program of COVID-19 in Lebanon. Available online: https://moph.gov.lb/en/Media/view/43750/monitoring-of-covid-19 (accessed on 7 May 2021).
- Abou-Hamdan, M.; Hamze, K.; Abdel, A.; Akl, H.; El-Zein, N.; Dandache, I.; Abdel-Sater, F. Variant analysis of the first Lebanese SARS-CoV-2 isolates. Genomics 2020, 113, 892–895. [Google Scholar] [CrossRef]
- Feghali, R.; Merhi, G.; Kwasiborski, A.; Hourdel, V.; Ghosn, N.; Tokajian, S. Genomic characterization and phylogenetic analysis of the first SARS-CoV-2 variants introduced in Lebanon. PeerJ 2021, 9, 1–15. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- GISAID. hCoV-19 Latest Updates by GISAID; GISID: Munich, Germany, 2021. [Google Scholar]
- Astuti, I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Kumari, S.; Pandey, B.; Mistry, H.; Bihani, S.C.; Das, A.; Prashar, V.; Gupta, G.D.; Panicker, L.; Kumar, M. Structural insights into SARS-CoV-2 proteins. J. Mol. Biol. 2021, 433. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Rapid Increase of a SARS-CoV-2 Variant with Multiple Spike Protein Mutations Observed in the United Kingdom. Available online: http://covid19-country-overviews.ecdc.europa.eu/#34_United_Kingdom (accessed on 6 May 2021).
- Zhou, H.; Chen, Y.; Zhang, S.; Niu, P.; Qin, K.; Jia, W.; Huang, B.; Zhang, S.; Lan, J.; Zhang, L.; et al. Structural definition of a neutralization epitope on the N-terminal domain of MERS-CoV spike glycoprotein. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Nelde, A.; Bilich, T.; Heitmann, J.S.; Maringer, Y.; Salih, H.R.; Roerden, M.; Lübke, M.; Bauer, J.; Rieth, J.; Wacker, M.; et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat. Immunol. 2021, 22, 74–85. [Google Scholar] [CrossRef]
- The World Bank. Population, Total-Lebanon. Available online: https://data.worldbank.org/indicator/SP.POP.TOTL?locations=LB (accessed on 7 May 2021).
- Center for Disease Control and Prevention. Genomic Surveillance for SARS-CoV-2 Variants. Available online: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance.html#print (accessed on 7 May 2021).
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, 3055. [Google Scholar] [CrossRef]
- Firth, A.E.; Brierley, I. Non-canonical translation in RNA viruses. J. Gen. Virol. 2012, 93, 1385–1409. [Google Scholar] [CrossRef] [PubMed]
- Namy, O.; Rousset, J.-P. Specification of Standard Amino Acids by Stop Codons. In Recording: Expansion of Decoding Rules Enriches Gene Expression; Springer: New York, NY, USA, 2010; pp. 79–100. ISBN 9780387893822. [Google Scholar]
- Lebanese SARS-CoV-2 sequences on GISAID. Available online: https://www.epicov.org/epi3/frontend#30f5b0 (accessed on 5 May 2021).
- Lebanese SARS-CoV-2 sequences on NCBI’s GenBank. Available online: https://www.ncbi.nlm.nih.gov/nuccore/?term=lebanon%20covid-19 (accessed on 5 May 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Name | GISAID Accession Number (Genbank if Available) | Collection Date | GISAID Clade | Pango Lineage |
|---|---|---|---|---|
| FAS/2021 | EPI_ISL_1159375 | 4-January-2021 | GRY | B.1.1.7 |
| LAU1-53249 | EPI_ISL_637110 | 13-August-2020 | G | B.1.398 |
| LAU2-53435 | EPI_ISL_637111 | August-2020 | G | B.1.398 |
| LAU3-53451 | EPI_ISL_637112 | 14-August-2020 | G | B.1.398 |
| LAU4-53460 | EPI_ISL_637113 | 14-August-2020 | G | B.1.398 |
| LAU-R110 1 | EPI_ISL_1009085 | 1-September-2020 | GH | B.1 |
| LAU-R113 1 | EPI_ISL_1009123 | 1-September-2020 | GH | B.1.36 |
| LAU-R115 1 | EPI_ISL_1009127 | 1-September-2020 | G | B.1.398 |
| LAU-R117 1 | EPI_ISL_1009128 | 1-September-2020 | G | B.1.398 |
| LAU-R129 1 | EPI_ISL_1009159 | 1-September-2020 | G | B.1.398 |
| LAU-R136 1 | EPI_ISL_1009160 | 1-October-2020 | G | B.1.398 |
| LAU-R145 1 | EPI_ISL_1009213 | 1-October-2020 | G | B.1.398 |
| LAU-R148 1 | EPI_ISL_1009214 | 1-October-2020 | G | B.1.398 |
| LAU-R151 1 | EPI_ISL_1009617 | 1-September-2020 | G | B.1.398 |
| LAU-R153 1 | EPI_ISL_1009626 | 1-September-2020 | G | B.1.398 |
| LAU-R170 1 | EPI_ISL_1009628 | 1-September-2020 | GH | B.1.438 |
| LAU-R186 1 | EPI_ISL_1009629 | 1-August-2020 | GR | B.1.1.315 |
| LAU-R204 1 | EPI_ISL_1009630 | 1-July 2020 | G | B.1.398 |
| LAU-R231 1 | EPI_ISL_1009631 | 1-August-2020 | G | B.1.398 |
| LAU-R242 1 | EPI_ISL_1009656 | 1-December-2020 | G | B.1.398 |
| LAU-R249 1 | EPI_ISL_1009658 | 1-December-2020 | G | B.1.398 |
| LAU-R43 1 | EPI_ISL_982298 | 1-June-2020 | G | B.1.398 |
| LAU-R45 1 | EPI_ISL_1009011 | 1-June-2020 | G | B.1.398 |
| LAU-R59 1 | EPI_ISL_1009012 | 1-June-2020 | G | B.1.398 |
| LAU-R70 1 | EPI_ISL_1009013 | 1-July 2020 | G | B.1.398 |
| LAU-R73 1 | EPI_ISL_1009014 | 1-August-2020 | G | B.1.398 |
| LAU-R77 1 | EPI_ISL_1009015 | 1-August-2020 | G | B.1.398 |
| LAU-R87 1 | EPI_ISL_1009016 | 1-September-2020 | GH | B.1.468 |
| LAU-R92 1 | EPI_ISL_1009017 | 1-September-2020 | G | B.1.398 |
| LAU-uk1 | EPI_ISL_768751 | 21-December-2020 | G | B.1.398 |
| LAU-uk2 | EPI_ISL_768798 | 21-December-2020 | G | B.1.398 |
| LAU-uk3 | EPI_ISL_768799 | 21-December-2020 | G | B.1.398 |
| LAU-uk4 | EPI_ISL_768743 | 22-December-2020 | GR | B.1.1 |
| LB-R8 1 | EPI_ISL_498551 (MT801000) | 19-March-2020 | G | B.1 |
| LB-R9 1 | EPI_ISL_498552 (MT801001) | 18-March-2020 | GR | B.1.1.106 |
| LB-R10 1 | EPI_ISL_498554 (MT801002) | 20-March-2020 | GH | B.1 |
| LB-R12 1 | EPI_ISL_498556 (MT801003) | 20-March-2020 | GH | B.1 |
| LEB-UK-B | EPI_ISL_1072985 | 30-Decmember-2020 | GRY | B.1.1.7 |
| S1_758 | EPI_ISL_450508 | 4-March-2020 | GH | B.1 |
| S2_759 | EPI_ISL_450509 | 15-March-2020 | GH | B.1 |
| S3_760 | EPI_ISL_450510 | 4-March-2020 | O | B.4 |
| S4_761 | EPI_ISL_450511 | 4-March-2020 | GR | B.1.1.29 |
| S5_762 | EPI_ISL_450512 | 27-February-2020 | O | B.4 |
| S6_766 | EPI_ISL_450513 | 9-March-2020 | GR | B.1.1.29 |
| S7_763 | EPI_ISL_454420 | 21-February-2020 | GH | B.1 |
| S8_767 | EPI_ISL_450514 | 13-March-2020 | GH | B.1 |
| S9_764 | EPI_ISL_450515 | 11-March-2020 | O | B.4 |
| S10_768 | EPI_ISL_450516 | 15-March-2020 | GH | B.1 |
| S11_765 | EPI_ISL_450517 | 9-March-2020 | GH | B.1 |
| LBN_1-B.1.1.7_2021 | (MW686007) | 4-January-2021 | GRY | B.1.1.7 |
| LBN_2-B.1.1.7_2021 | (MW692113) | 4-January-2021 | GRY | B.1.1.7 |
| LBN_3-B.1.1.7_2021 | (MW692114) | 8-January-2021 | GRY | B.1.1.7 |
| LBN_4-B.1.1.7_2021 | (MW692115) | 8-January-2021 | GRY | B.1.1.7 |
| LBN_5-B.1.1.7_2020 | (MW692116) | 28-December-2020 | GRY | B.1.1.7 |
| LBN_6-B.1.1.7_2020 | (MW692117) | 28-December-2020 | GRY | B.1.1.7 |
| LBN_7-B.1.1.7_2020 | (MW692118) | 24-December-2020 | GRY | B.1.1.7 |
| LBN_8-B.1.1.7_2020 | (MW692119) | 24-December-2020 | GRY | B.1.1.7 |
| LBN_9-B.1.258Delta | (MW720771) | 12-December-2020 | G | B.1.258 |
| Virus Name 1 | GISAID Accession Number (Genbank if Available) | Collection Date | GISAID Clade | Pango Lineage |
|---|---|---|---|---|
| England MLK-9E05B3 | EPI_ISL_601443 | 20-September-2020 | GRY | B.1.1.7 |
| Ukraine Kyiv-PHC-0030 | EPI_ISL_1495138 | 18-March-2021 | GRY | B.1.1.7 |
| Poland 0252WSN2021_wsseol | EPI_ISL_1493349 | 25-March-2021 | GRY | B.1.1.7 |
| Belgium ULG-13710 | EPI_ISL_1493104 | 19-March-2021 | GRY | B.1.1.7 |
| Sweden 19482581 | EPI_ISL_1492593 | 15-February-2021 | GRY | B.1.1.7 |
| England 20168037604 | EPI_ISL_466615 | 16-February-2020 | GR | B.1.1.1 |
| Brazil MG-LBI172 | EPI_ISL_1494976 | 22-February-2021 | GR | P.2 |
| Poland 0221WSN2021_wsseol | EPI_ISL_1493318 | 22-March-2021 | GR | B.1.1 |
| Czech Republic NRL-3602-21 | EPI_ISL_1492191 | 10-February-2021 | GR | B.1.1 |
| Russia SPE-RII-MH14837S | EPI_ISL_1491732 | 9-February-2021 | GR | B.1.1.397 |
| Spain CT-ISCIII-2013597 | EPI_ISL_539548 | 26-June-2020 | GV | B.1.177 |
| Spain PV-HUD-67010593 | EPI_ISL_1495978 | 1-February-2021 | GV | B.1.177 |
| Austria CeMM6068 | EPI_ISL_1495837 | 6-November-2020 | GV | B.1.177 |
| Portugal PT5231 | EPI_ISL_1494894 | 9-February-2021 | GV | B.1.177 |
| Wales PHWC-PYYSCA | EPI_ISL_1476485 | 14-March-2021 | GV | B.1.177 |
| Canada ON-PHL-8751 | EPI_ISL_418345 | 1-February-2020 | GH | B.1.* |
| USA OR-TRACE-BENT-031521-431 | EPI_ISL_1494938 | 14-March-2021 | GH | B.1.429 |
| Portugal PT5252 | EPI_ISL_1494914 | 9-February-2021 | GH | B.1.367 |
| Colombia VAC-INS-VG-1482 | EPI_ISL_1494952 | 5-March-2021 | GH | B.1.596 |
| USA OR-TRACE-BENT-030821-421 | EPI_ISL_1494930 | 8-March-2021 | GH | B.1.429 |
| Wuhan WIV04 2019 | EPI_ISL_402124 | 30-December-2019 | L | B |
| England TFCI-2707EC6 | EPI_ISL_1476828 | 3-April-2020 | L | B.3 |
| Japan PG-24179 | EPI_ISL_1429761 | 19-February-2020 | L | B.12 |
| Kazakhstan 123631 | EPI_ISL_1341150 | 1-March-2020 | L | B |
| Hong Kong HKPU-00440 | EPI_ISL_1289439 | 26-March-2020 | L | B.3 |
| Wuhan-Hu-1 | (NC_045512.2) | 1-December-2019 | L | B |
| Italy LAZ-INMI-SPL1 | EPI_ISL_412974 | 29-January-2020 | V | B.2 |
| South Korea KDCA2676 | EPI_ISL_1490167 | 18-February-2020 | V | B.41 |
| Canada QC-CHUM-2008201265A | EPI_ISL_1378973 | 22-March-2020 | V | B.40 |
| Thailand CU-SI2011343-NT | EPI_ISL_1296450 | 2-April-2020 | V | B.40 |
| Slovenia 21-2850 | EPI_ISL_1289874 | 30-March-2020 | V | B |
| Guangdong 20SF012 | EPI_ISL_403932 | 14-January-2021 | S | A |
| France ARA-700000210773 | EPI_ISL_1490236 | 11-March-2021 | S | A.27 |
| Scotland QEUH-1409CFD | EPI_ISL_1389947 | 18-March-2021 | S | A |
| Switzerland BS-UHB-351261 | EPI_ISL_1388124 | 25-January-2021 | S | A.27 |
| Luxembourg LNS7034377 | EPI_ISL_1383855 | 1-February-2021 | S | A.28 |
| Germany BY-ChVir-929 | EPI_ISL_406862 | 28-January-2020 | G | B.1 |
| Brazil MG-LBI57 | EPI_ISL_1494965 | 27-January-2021 | G | P.2 |
| USA IL-IDPH-WHI-C-001647 | EPI_ISL_1494503 | 14-July 2020 | G | B.1.234 |
| Australia NSW4430 | EPI_ISL_1494721 | 3-April-2021 | G | B.1 |
| Mexico SLP-UASLP-AH1COV2SS027_S16 | EPI_ISL_1494725 | 22-May-2020 | G | B.1 |
| SARS-CoV-2 Strain | AA Substitution | Prevalence in Lebanese Strains | Prevalence between the Strains Reported on GISAID 1 | First/Only Report According to GISAID’s CoVsurver | Number of Countries | Strain Collection Date |
|---|---|---|---|---|---|---|
| Spike protein | ||||||
| LAU-R186 | spike F59Y | 1 | 106 | First | 4 | 01-Aug-20 |
| spike G35S | 1 | 6 | First | 2 | ||
| spike N17S | 1 | 797 | First | 8 | ||
| spike R44S | 1 | 2 | First | 2 | ||
| spike Y38stop | 1 | 1 | Only | 1 | ||
| LAU-R45 | spike P39T | 1 | 8 | First | 6 | 01-Jun-20 |
| spike S31Y | 1 | 1 | Only | 1 | ||
| spike V11D | 1 | 1 | Only | 1 | ||
| LAU-R204 | spike L10R | 1 | 1 | Only | 1 | 01-July 20 |
| spike L5P | 1 | 2 | First | 2 | ||
| LAU-R87 | spike D40V | 1 | 1 | Only | 1 | 01-Sep-20 |
| LAU-R129 | spike L611R | 1 | 5 | First | 2 | 01-September-20 |
| LAU-R151 | spike P82T | 1 | 3 | First | 2 | 01-September-20 |
| LAU-R43 | spike Y91stop | 1 | 1 | Only | 1 | 01-Jun-20 |
| spike F92L | 1 | 2 | Last | 2 | ||
| Nucleocapsid protein | ||||||
| LAU-R145 | N E367stop | 1 | 3 | First | 3 | 01-Oct-20 |
| LAU-R231 | N L352stop | 1 | 1 | Only | 1 | 01-Aug-20 |
| LAU-R43 | N L407M | 1 | 1 | Only | 1 | 01-Jun-20 |
| LAU-R153 | N M411K | 1 | 1 | Only | 1 | 01-September-20 |
| LAU-R92 | N S318W | 1 | 1 | Only | 1 | 01-September-20 |
| Envelope protein | ||||||
| LAU-R110 | E C44S | 1 | 6 | First | 3 | 01-September-20 |
| S6 | E V62D | 1 | 1 | Only | 1 | 09-Mar-20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fayad, N.; Abi Habib, W.; Kandeil, A.; El-Shesheny, R.; Kamel, M.N.; Mourad, Y.; Mokhbat, J.; Kayali, G.; Goldstein, J.; Abdallah, J. SARS-CoV-2 Variants in Lebanon: Evolution and Current Situation. Biology 2021, 10, 531. https://doi.org/10.3390/biology10060531
Fayad N, Abi Habib W, Kandeil A, El-Shesheny R, Kamel MN, Mourad Y, Mokhbat J, Kayali G, Goldstein J, Abdallah J. SARS-CoV-2 Variants in Lebanon: Evolution and Current Situation. Biology. 2021; 10(6):531. https://doi.org/10.3390/biology10060531
Chicago/Turabian StyleFayad, Nancy, Walid Abi Habib, Ahmed Kandeil, Rabeh El-Shesheny, Mina Nabil Kamel, Youmna Mourad, Jacques Mokhbat, Ghazi Kayali, Jimi Goldstein, and Jad Abdallah. 2021. "SARS-CoV-2 Variants in Lebanon: Evolution and Current Situation" Biology 10, no. 6: 531. https://doi.org/10.3390/biology10060531