
Hereditary Optic Neuropathies: A Systematic Review on the Interplay between Biomaterials and Induced Pluripotent Stem Cells


Abstract
:1. Introduction
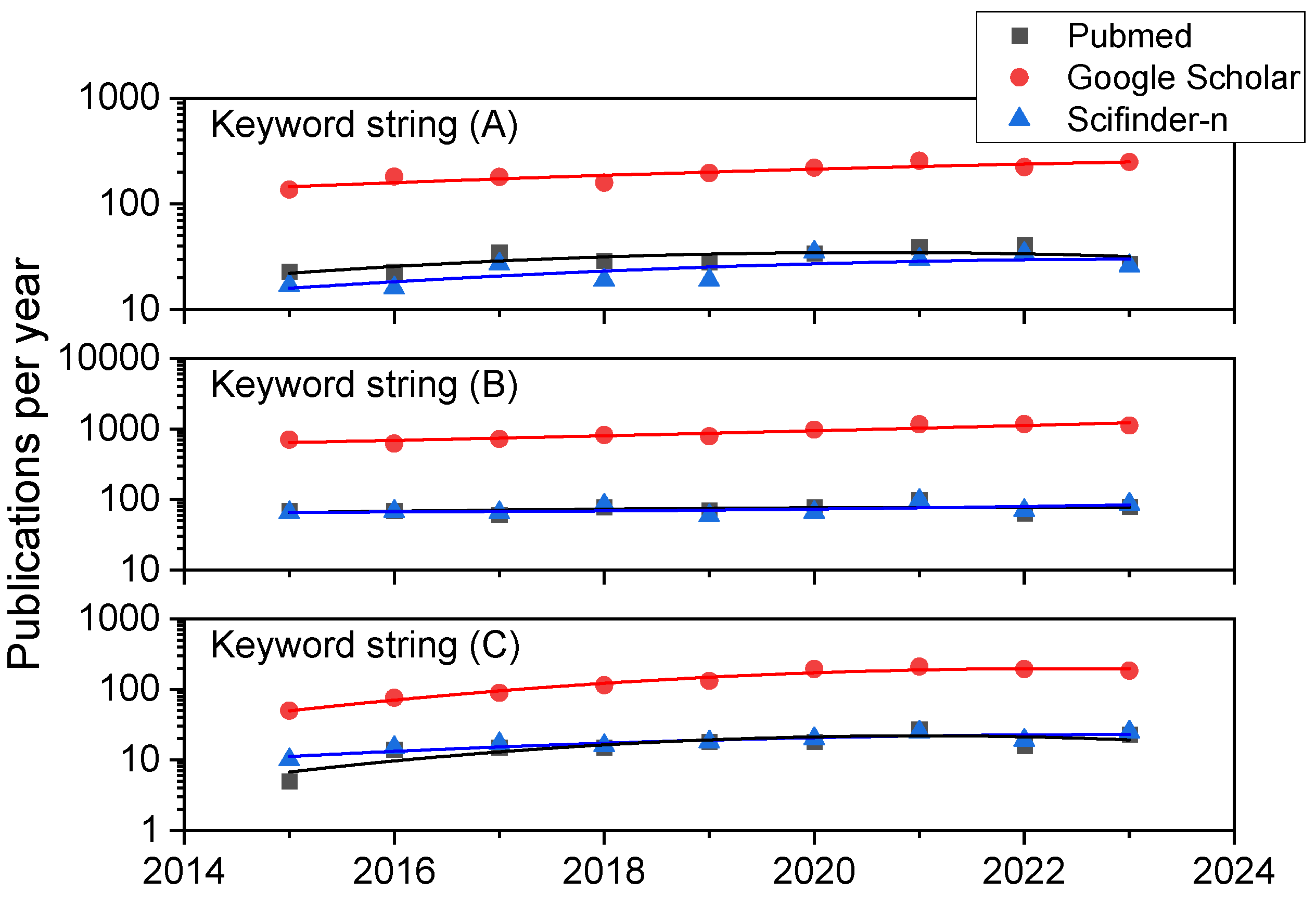
2. Methodology
3. An Approach to Hereditary Optic Neuropathies
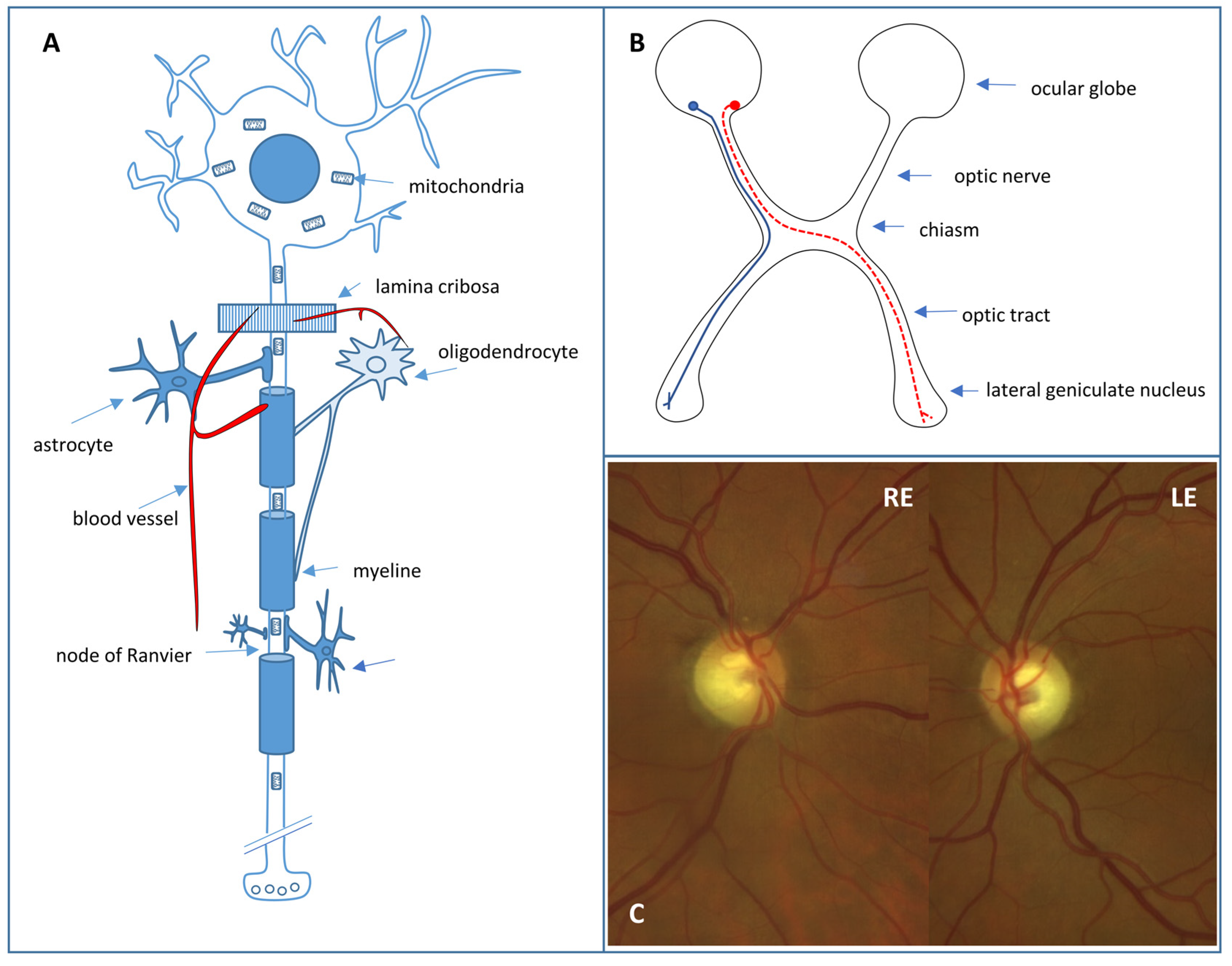
3.1. Retinal Ganglion Cells
3.2. The Hereditary Optic Neuropathies
3.2.1. Leber Hereditary Optic Neuropathy
3.2.2. Autosomal Dominant Optic Atrophy
3.3. Treatment of Hereditary Optic Neuropathies
3.3.1. Gene Therapy
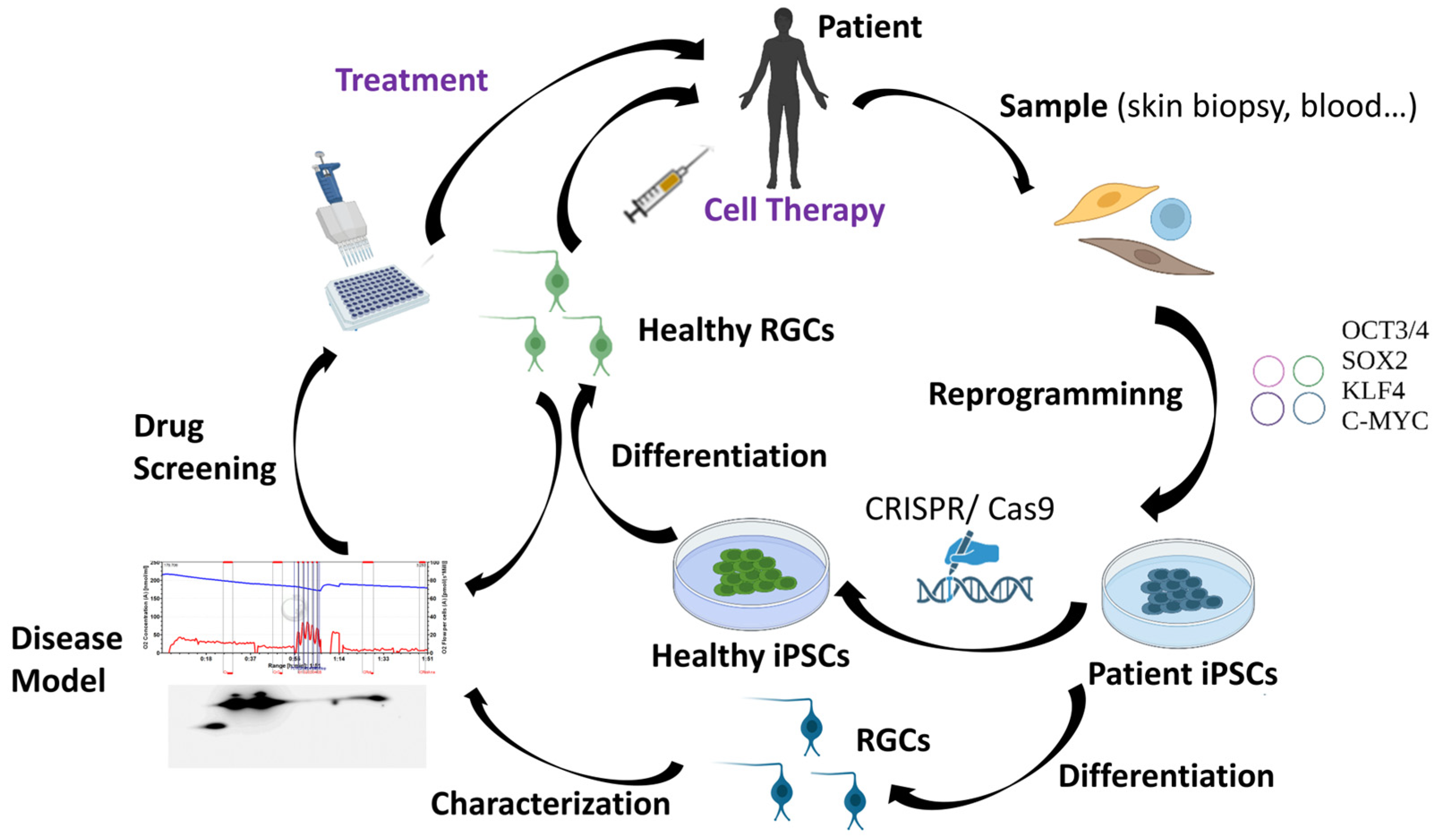
3.3.2. Cell Replacement Therapies: An Alternative Approach

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Locus | Function | Phenotypes | Reference |
|---|---|---|---|---|
| OPA1 | 3q28–q29 | Mitochondrial fusion | DOA (AD) | [40] |
| DOA PLUS (AD) | [41] | |||
| Behr syndrome (AR, AD) | [42] | |||
| OPA3 | 19q13.2–q13.3 | Mitochondrial shape and apoptosis | DOA and cataract (AD) | [43] |
| Costeff syndrome (AR) | [44] | |||
| MFN2 | 1p36.22 | Mitochondrial fusion | Charcot Marie Tooth type 2 A (AD, AR) | [45] |
| Hereditary motor and sensory neuropathy type VI (AD) | [46] | |||
| NDUFS2 | 2q23.3 | Respiratory chain complex I deficiency | LHON fenotype (AR) | [47] |
| NDUFS1 | 2q33.3. | Respiratory chain complex I deficiency | Optic atrophy and multisystem neurological disorder (AR XLD) | [48] |
| [49] | ||||
| WFS1 | 4q16.1 | Endoplasmic reticulum–mitochondria interactions and calcium homeostasis | DOA and hearing loss (AD) | [50] |
| Wolfram syndrome (AR) | [51] | |||
| NR2F1 | 5q15 | Transcriptional regulation | Bosch–Boonstra–Schaaf syndrome (AD) | [52] |
| SSBP1 | 7q34 | mtDNA replication | DOA and retinopathy (AD) | [53] |
| DOA, retinopathy, nephropathy, and deafness (AD) | [54] | |||
| SGP7 | 16q24.3 | Mitochondrial quality control | DOA (AD) | [55] |
| Hereditary spastic paraplegia type 7 (AD/AR) | [56] | |||
| AFG3L2 | 18q11.21 | Mitochondrial quality control | DOA(AR) | [57] |
| Spinocerebellar ataxia type 28 (AD) | [58] | |||
| Spastic ataxia type 5 (AR) | [59] | |||
| TIMM8A | Xq22.1 | Translocase of inner mitochondrial membrane | Mohr–Tranebjaerg syndrome (XLR) | [60] |
| FXN | 9q21.11 | Frataxin (mitochondrial respiratory chain) | Friedrich ataxia (AR) | [61] |
| ACO2 | 22q13.2 | Krebs cycle | Isolated optic atrophy | [62] |
4. iPSC Technology for Studying and Treating Hereditary Optic Neuropathies
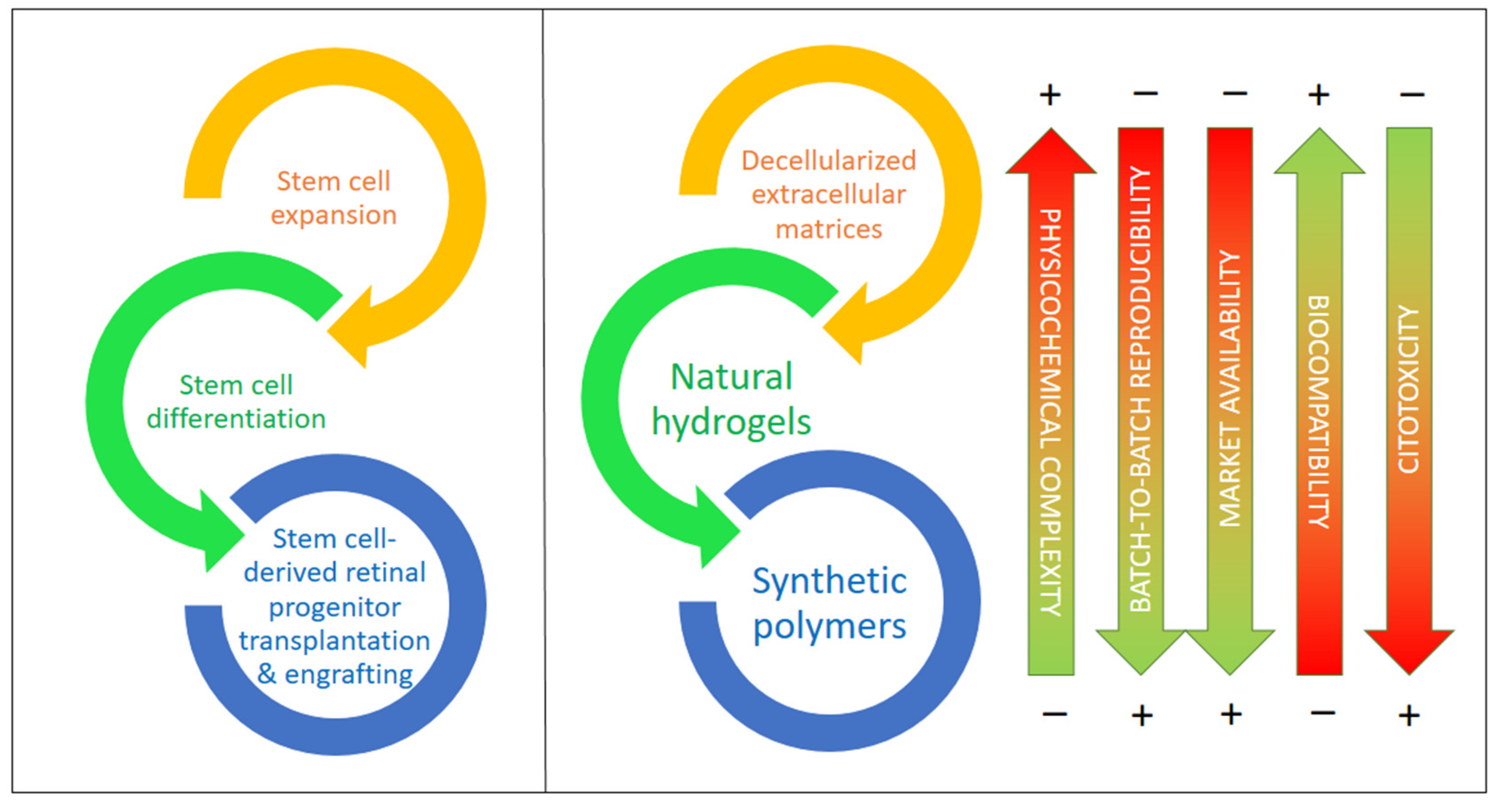
5. The Role of Biomaterials in Optic Neuropathies: Present Developments
5.1. Extracellular Matrix-Based Biomaterials
5.2. Synthetic Polymers and Copolymers
5.3. Natural Hydrogels Based on Polysaccharides and/or Proteins
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Z.; Keel, S.; Liu, C.; He, M. Can artificial intelligence make screening faster, more accurate, and more accessible? Asia-Pacific J. Ophthalmol. 2018, 7, 436–441. [Google Scholar]
- Chen, B.S.; Harvey, J.P.; Gilhooley, M.J.; Jurkute, N.; Yu-Wai-Man, P. Mitochondria and the eye—manifestations of mitochondrial diseases and their management. Eye 2023, 37, 2416–2425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.J.; Jin, K.; Jin, Z.B. Stem cells and genetic engineering empower therapeutic development for blinding eye diseases. Eye 2023, 4, 139–170. [Google Scholar]
- Ortuño-Costela, M.D.C.; Cerrada, V.; García-López, M.; Gallardo, M.E. The challenge of bringing iPSCs to the patient. Int. J. Mol. Sci. 2019, 20, 6305. [Google Scholar] [CrossRef] [PubMed]
- Kharbikar, B.N.; Mohindra, P.; Desai, T.A. Biomaterials to enhance stem cell transplantation. Cell Stem Cell 2022, 29, 692–721. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Min, S.; Choi, Y.S.; Jo, S.H.; Jung, J.H.; Han, K.; Kim, J.; An, S.; Ji, Y.W.; Kim, Y.G.; et al. Tissue extracellular matrix hydrogels as alternatives to Matrigel for culturing gastrointestinal organoids. Nat. Commun. 2022, 13, 1692. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.C.; Chuang, J.H.; Buddhakosai, W.; Wu, W.J.; Lee, C.J.; Chen, W.S.; Yang, Y.P.; Li, M.C.; Peng, C.H.; Chen, S.J. Elongation of axon extension for human ipsc-derived retinal ganglion cells by a nano-imprinted scaffold. Int. J. Mol. Sci. 2017, 18, 2013. [Google Scholar] [CrossRef]
- Behtaj, S.; Karamali, F.; Najafian, S.; Masaeli, E.; Esfahani, M.H.N.; Rybachuk, M. The role of PGS/PCL scaffolds in promoting differentiation of human embryonic stem cells into retinal ganglion cells. Acta Biomater. 2021, 126, 238–248. [Google Scholar] [CrossRef]
- Behtaj, S.; Karamali, F.; Masaeli, E.; Anissimov, Y.G.; Rybachuk, M. Electrospun PGS/PCL, PLLA/PCL, PLGA/PCL and pure PCL scaffolds for retinal progenitor cell cultivation. Biochem. Eng. J. 2021, 166, 107846. [Google Scholar] [CrossRef]
- Hunt, N.C.; Hallam, D.; Chichagova, V.; Steel, D.H.; Lako, M. The application of biomaterials to tissue engineering neural retina and retinal pigment epithelium. Adv. Healthc. Mater. 2018, 7, 1800226. [Google Scholar] [CrossRef]
- Glaser, T.; Bueno, V.B.; Cornejo, D.R.; Petri, D.F.S.; Ulrich, H. Neuronal adhesion, proliferation and differentiation of embryonic stem cells on hybrid scaffolds made of xanthan and magnetite nanoparticles. Biomed. Mater. 2015, 10, 045002. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-Hernández, D.D.; Canales-Aguirre, A.A.; Matias-Guiu, J.; Gomez-Pinedo, U.; Mateos-Díaz, J.C. Potential of chitosan and its derivatives for biomedical applications in the central nervous system. Front. Bioeng. Biotechnol. 2020, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Mohammadinejad, R.; Kumar, A.; Ranjbar-Mohammadi, M.; Ashrafizadeh, M.; Han, S.S.; Khang, G.; Roveimiab, Z. Recent advances in natural gum-based biomaterials for tissue engineering and regenerative medicine: A review. Polymers (Basel) 2020, 12, 176. [Google Scholar] [CrossRef] [PubMed]
- Habibi, H.; Khosravi-Darani, K. Effective variables on production and structure of xanthan gum and its food applications: A review. Biocatal. Agric. Biotechnol. 2017, 10, 130–140. [Google Scholar] [CrossRef]
- Smith, C.A.; Vianna, J.R.; Chauhan, B.C. Assessing retinal ganglion cell damage. Eye 2017, 31, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.S.; Mahroo, O.A.; Mollon, J.D.; Yu-Wai-Man, P. Retinal Ganglion Cells—Diversity of Cell Types and Clinical Relevance. Front. Neurol. 2021, 12, 661938. [Google Scholar] [CrossRef]
- Newman, N.J.; Yu-Wai-Man, P.; Biousse, V.; Carelli, V. Understanding the molecular basis and pathogenesis of hereditary optic neuropathies: Towards improved diagnosis and management. Lancet Neurol. 2023, 22, 172–188. [Google Scholar] [CrossRef]
- Carelli, V.; La Morgia, C.; Yu-Wai-Man, P. Mitochondrial optic neuropathies. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2023; Volume 194, pp. 23–42. [Google Scholar]
- Kamel, K.; Farrell, M.; O’Brien, C. Mitochondrial dysfunction in ocular disease: Focus on glaucoma. Mitochondrion 2017, 35, 44–53. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Newman, N.J. Inherited eye-related disorders due to mitochondrial dysfunction. Hum. Mol. Genet. 2017, 26, R12–R20. [Google Scholar] [CrossRef]
- Fraser, J.A.; Biousse, V.; Newman, N.J. The neuro-ophthalmology of mitochondrial disease. Surv. Ophthalmol. 2010, 55, 299–334. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Mancuso, M.; Pareyson, D.; Burgunder, J.M.; Klopstock, T. Mitochondrial disorders of the retinal ganglion cells and the optic nerve. Mitochondrion 2018, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Taylor, R.W. Mitochondrial DNA abnormalities in ophthalmological disease. Saudi J. Ophthalmol. 2011, 25, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Schrier, S.A.; Falk, M.J. Mitochondrial disorders and the eye. Curr. Opin. Ophthalmol. 2011, 22, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.S.; Yu-Wai-Man, P.; Newman, N.J. Developments in the Treatment of Leber Hereditary Optic Neuropathy. Curr. Neurol. Neurosci. Rep. 2022, 22, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Carelli, V.; Taiel, M.; Yu-Wai-Man, P. Visual Outcomes in Leber Hereditary Optic Neuropathy Patients with the m.11778G>A (MTND4) Mitochondrial DNA Mutation. J. Neuro-Ophthalmol. 2020, 40, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, N.J.; Chakrabarti, R.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial disorders and the eye. Eye Brain 2011, 3, 29–47. [Google Scholar]
- Abu-Amero, K.K.; Kondkar, A.A.; Chalam, K.V. Mitochondrial aberrations and ophthalmic diseases. J. Transl. Sci. 2016, 3, 1–11. [Google Scholar] [CrossRef]
- Bagli, E.; Zikou, A.K.; Agnantis, N.; Kitsos, G. Mitochondrial membrane dynamics and inherited optic neuropathies. In Vivo 2017, 31, 511–525. [Google Scholar]
- Lenaers, G.; Neutzner, A.; Le Dantec, Y.; Jüschke, C.; Xiao, T.; Decembrini, S.; Swirski, S.; Kieninger, S.; Agca, C.; Kim, U.S.; et al. Dominant optic atrophy: Culprit mitochondria in the optic nerve. Prog. Retin. Eye Res. 2021, 83, 100935. [Google Scholar] [CrossRef]
- Davila-Siliezar, P.; Carter, M.; Milea, D.; Lee, A.G. Leber hereditary optic neuropathy: New and emerging therapies. Curr. Opin. Ophthalmol. 2022, 33, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Chun, B.Y.; Rizzo, J.F. Dominant Optic Atrophy and Leber’s Hereditary Optic Neuropathy: Update on Clinical Features and Current Therapeutic Approaches. Semin. Pediatr. Neurol. 2017, 24, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, C.; Van Stavern, G.; McClelland, C. Leber hereditary optic neuropathy: Current perspectives. Clin. Ophthalmol. 2015, 9, 1165–1176. [Google Scholar] [PubMed]
- DeBusk, A.; Moster, M.L. Gene therapy in optic nerve disease. Curr. Opin. Ophthalmol. 2018, 29, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Feuer, W.J.; Schiffman, J.C.; Davis, J.L.; Porciatti, V.; Gonzalez, P.; Koilkonda, R.D.; Yuan, H.; Lalwani, A.; Lam, B.L.; Guy, J. Gene therapy for leber hereditary optic neuropathy initial results. Ophthalmology 2016, 123, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Luis, J.; Eastlake, K.; Lamb, W.D.B.; Limb, G.A.; Jayaram, H.; Khaw, P.T. Cell-Based Therapies for Glaucoma. Transl. Vis. Sci. Technol. 2023, 12, 23. [Google Scholar] [CrossRef]
- Rabesandratana, O.; Chaffiol, A.; Mialot, A.; Slembrouck-Brec, A.; Joffrois, C.; Nanteau, C.; Rodrigues, A.; Gagliardi, G.; Reichman, S.; Sahel, J.A.; et al. Generation of a Transplantable Population of Human iPSC-Derived Retinal Ganglion Cells. Front. Cell Dev. Biol. 2020, 8, 1–20. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.A.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Pelloquin, L.; Belenguer, P.; Hamel, C.P. OPA1 (Kjer type) dominant optic atrophy: A novel mitochondrial disease. Mol. Genet. Metab. 2002, 75, 97–107. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Reynier, P.; Amati-Bonneau, P.; Verny, C.; Olichon, A.; Simard, G.; Guichet, A.; Bonnemains, C.; Malecaze, F.; Malinge, M.C.; Pelletier, J.B.; et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J. Med. Genet. 2004, 41, 110. [Google Scholar] [CrossRef] [PubMed]
- Anikster, Y.; Kleta, R.; Shaag, A.; Gahl, W.A.; Elpeleg, O. Type III 3-methylglutaconic aciduria (optic atrophy plus syndrome, or costeff optic atrophy syndrome): Identification of the OPA3 gene and its founder mutation in Iraqi Jews. Am. J. Hum. Genet. 2001, 69, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Barbullushi, K.; Abati, E.; Rizzo, F.; Bresolin, N.; Comi, G.P.; Corti, S. Disease Modeling and Therapeutic Strategies in CMT2A: State of the Art. Mol. Neurobiol. 2019, 56, 6460–6471. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; De Jonghe, P.; Jordanova, A.; Claeys, K.G.; Guergueltcheva, V.; Cherninkova, S.; Hamilton, S.R.; Van Stavern, G.; Krajewski, K.M.; Stajich, J.; et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 2006, 59, 276–281. [Google Scholar] [CrossRef]
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, M.S.; Paris, A.; Alban, J.; Quiles, M.; Delettre, C.; et al. Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain 2017, 140, 2586–2596. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Chen, A.T.; Brady, L.; Bulman, D.E.; Sundaram, A.N.E.; Rodriguez, A.R.; Margolin, E.; Waye, J.S.; Tarnopolsky, M.A. An evaluation of genetic causes and environmental risks for bilateral optic atrophy. PLoS ONE 2019, 14, e0225656. [Google Scholar] [CrossRef]
- Eiberg, H.; Kjer, B.; Kjer, P.; Rosenberg, T. Dominant optic atrophy (OPA1) mapped to chromosome 3q region. I. Linkage analysis. Hum. Mol. Genet. 1994, 3, 977–980. [Google Scholar] [CrossRef]
- Inoue, M.; Himori, N.; Kunikata, H.; Takeshita, T.; Aizawa, N.; Shiga, Y.; Omodaka, K.; Nishiguchi, K.M.; Takahashi, H.; Nakazawa, T. The reduction of temporal optic nerve head microcirculation in autosomal dominant optic atrophy. Acta Ophthalmol. 2016, 94, e580–e585. [Google Scholar] [CrossRef]
- Bosch, D.G.M.; Boonstra, F.N.; Gonzaga-Jauregui, C.; Xu, M.; De Ligt, J.; Jhangiani, S.; Wiszniewski, W.; Muzny, D.M.; Yntema, H.G.; Pfundt, R.; et al. NR2F1 mutations cause optic atrophy with intellectual disability. Am. J. Hum. Genet. 2014, 94, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Jurkute, N.; Leu, C.; Pogoda, H.M.; Arno, G.; Robson, A.G.; Nürnberg, G.; Altmüller, J.; Thiele, H.; Motameny, S.; Toliat, M.R.; et al. SSBP1 mutations in dominant optic atrophy with variable retinal degeneration. Ann. Neurol. 2019, 86, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Del Dotto, V.; Ullah, F.; Di Meo, I.; Magini, P.; Gusic, M.; Maresca, A.; Caporali, L.; Palombo, F.; Tagliavini, F.; Baugh, E.H. SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder. Am. Soc. Clin. Investig. 2020, 130, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Klebe, S.; Depienne, C.; Gerber, S.; Challe, G.; Anheim, M.; Charles, P.; Fedirko, E.; Lejeune, E.; Cottineau, J.; Brusco, A.; et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012, 135, 2980–2993. [Google Scholar] [CrossRef] [PubMed]
- Casari, G.; De Fusco, M.; Ciarmatori, S.; Zeviani, M.; Mora, M.; Fernandez, P.; De Michele, G.; Filla, A.; Cocozza, S.; Marconi, R.; et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998, 93, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Charif, M.; Roubertie, A.; Salime, S.; Mamouni, S.; Goizet, C.; Hamel, C.P.; Lenaers, G. A novel mutation of AFG3L2 might cause dominant optic atrophy in patients with mild intellectual disability. Front. Genet. 2015, 6, 311. [Google Scholar] [CrossRef]
- Di Bella, D.; Lazzaro, F.; Brusco, A.; Plumari, M.; Battaglia, G.; Pastore, A.; Finardi, A.; Cagnoli, C.; Tempia, F.; Frontali, M. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 2010, 42, 313–321. [Google Scholar] [CrossRef]
- Pierson, T.M.; Adams, D.; Bonn, F.; Martinelli, P.; Cherukuri, P.F.; Teer, J.K.; Hansen, N.F.; Cruz, P.; Mullikin, J.C.; Blakesley, R.W.; et al. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011, 7, e1002325. [Google Scholar] [CrossRef]
- Jin, H.; May, M.; Tranebjaerg, L.; Kendall, E.; Fontan, G.; Jackson, J.; Subramony, S.H.; Arena, F.; Lubs, H.; Smith, S.; et al. A novel X-linked gene, DDP, shows mutations in families with deafness (DFN-1), dystonia, mental deficiency and blindness. Nat. Genet. 1996, 14, 177–180. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Charif, M.; Gueguen, N.; Ferré, M.; Elkarhat, Z.; Khiati, S.; Lemao, M.; Chevrollier, A.; Desquiret-Dumas, V.; Goudenège, D.; Bris, C.; et al. Dominant ACO2 mutations are a frequent cause of isolated optic atrophy. Brain Commun. 2021, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Liu, D.T.L.; Chiang, S.W.Y.; Choy, K.W.; Pang, C.P.; Lam, D.S.C.; Yam, G.H.F. Immunopanning purification and long-term culture of human retinal ganglion cells. Mol. Vis. 2010, 16, 2867–2872. [Google Scholar] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Moradi, S.; Mahdizadeh, H.; Šarić, T.; Kim, J.; Harati, J.; Shahsavarani, H.; Greber, B.; Moore, J.B. Research and therapy with induced pluripotent stem cells (iPSCs): Social, legal, and ethical considerations. Stem Cell Res. Ther. 2019, 10, 341. [Google Scholar] [CrossRef] [PubMed]
- Galera-Monge, T.; Zurita-Díaz, F.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernández, A.F.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with an optic atrophy “plus” phenotype due to a mutation in the OPA1 gene. Stem Cell Res. 2016, 16, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Galera, T.; Zurita, F.; González-Páramos, C.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernández, A.F.; Garesse, R.; Gallardo, M.E. Generation of a human control iPSC line with a European mitochondrial haplogroup U background. Stem Cell Res. 2016, 16, 88–91. [Google Scholar] [CrossRef]
- Zurita-Díaz, F.; Galera-Monge, T.; Moreno-Izquierdo, A.; Corton, M.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Establishment of a human DOA “plus” iPSC line, IISHDOi003-A, with the mutation in the OPA1 gene: c.1635C > A; p.Ser545Arg. Stem Cell Res. 2017, 24, 81–84. [Google Scholar] [CrossRef]
- Ortuño-Costela, M.D.C.; Rodríguez-Mancera, N.; García-López, M.; Zurita-Díaz, F.; Moreno-Izquierdo, A.; Lucía, A.; Martín, M.Á.; Garesse, R.; Gallardo, M.E. Establishment of a human iPSC line (IISHDOi001-A) from a patient with McArdle disease. Stem Cell Res. 2017, 23, 188–192. [Google Scholar] [CrossRef]
- Ortuño-Costela, M.D.C.; Moreno-Izquierdo, A.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line, IISHDOi002-A, with a 46, XY/47, XYY mosaicism and belonging to an African mitochondrial haplogroup. Stem Cell Res. 2018, 28, 131–135. [Google Scholar] [CrossRef]
- Pareja-galeano, H.; Sanchis-gomar, F.; Pérez, L.M.; Emanuele, E.; Lucia, A.; Gálvez, B.G.; Esther, M. iPSCs-based anti-aging therapies: Recent discoveries and future challenges. Ageing Res. Rev. 2021, 27, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.S.; Mooney, D.J. Regenerative medicine: Current therapies and future directions. Proc. Natl. Acad. Sci. USA 2015, 112, 14452–14459. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.C.B.; Lim, S.Y.; Hung, S.S.C.; Jackson, S.; Khan, S.; Van Bergen, N.J.; De Smit, E.; Liang, H.H.; Kearns, L.S.; Clarke, L.; et al. Mitochondrial replacement in an iPSC model of Leber’s hereditary optic neuropathy. Aging 2017, 9, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.R.; Wang, A.G.; Chen, Y.T.; Yarmishyn, A.A.; Buddhakosai, W.; Yang, T.C.; Hwang, D.K.; Yang, Y.P.; Shen, C.N.; Lee, H.C.; et al. Bioactivity and gene expression profiles of hiPSC-generated retinal ganglion cells in MT-ND4 mutated Leber’s hereditary optic neuropathy. Exp. Cell Res. 2018, 363, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.C.; Yarmishyn, A.A.; Yang, Y.P.; Lu, P.C.; Chou, S.J.; Wang, M.L.; Lin, T.C.; Hwang, D.K.; Chou, Y.B.; Chen, S.J.; et al. Mitochondrial transport mediates survival of retinal ganglion cells in affected LHON patients. Hum. Mol. Genet. 2020, 29, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Riazifar, H.; Guan, M.X.; Huang, T. Modeling autosomal dominant optic atrophy using induced pluripotent stem cells and identifying potential therapeutic targets. Stem Cell Res. Ther. 2016, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Zurita-Díaz, F.; Ortuño-Costela, M.D.C.; Moreno-Izquierdo, A.; Galbis, L.; Millán, J.M.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Establishment of a human iPSC line, IISHDOi004-A, from a patient with Usher syndrome associated with the mutation c.2276G>T; p.Cys759Phe in the USH2A gene. Stem Cell Res. 2018, 31, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Su, X.; Hu, C.; Zhang, Z.; Wang, M.; Zou, W.; Shen, L.; Liu, Y.; Liang, C.; Du, Y.; et al. Generation of an induced pluripotent stem cell line from a patient with leber’s hereditary optic neuropathy carrying a homoplasmic m.3635G > A mutation in the mitochondrial ND1 gene. Stem Cell Res 2022, 63, 102858. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, S.; Jin, Z.B.; Wang, N. Stem cell-based regeneration and restoration for retinal ganglion cell: Recent advancements and current challenges. Biomolecules 2021, 11, 987. [Google Scholar] [CrossRef]
- García-López, M.; Arenas, J.; Gallardo, M.E. Hereditary optic neuropathies: Induced pluripotent stem cell-based 2D/3D approaches. Genes (Basel) 2021, 12, 112. [Google Scholar] [CrossRef]
- Zhang, J.S.; Johnston, R. Retinal ganglion cell generation and loss in human retinal organoids. Invest. Ophthalmol. Vis. Sci. 2023, 64, 3179. [Google Scholar]
- Fischer, D.; Harvey, A.R.; Pernet, V.; Lemmon, V.P.; Park, K.K. Optic nerve regeneration in mammals: Regenerated or spared axons? Exp. Neurol. 2017, 296, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Vrathasha, V.; Nikonov, S.; Bell, B.A.; He, J.; Bungatavula, Y.; Uyhazi, K.E.; Murthy Chavali, V.R. Transplanted human induced pluripotent stem cells- derived retinal ganglion cells embed within mouse retinas and are electrophysiologically functional. iScience 2022, 25, 105308. [Google Scholar] [CrossRef] [PubMed]
- Dorgau, B.; Felemban, M.; Hilgen, G.; Kiening, M.; Zerti, D.; Hunt, N.C.; Doherty, M.; Whitfield, P.; Hallam, D.; White, K.; et al. Decellularised extracellular matrix-derived peptides from neural retina and retinal pigment epithelium enhance the expression of synaptic markers and light responsiveness of human pluripotent stem cell derived retinal organoids. Biomaterials 2019, 199, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Behtaj, S.; Öchsner, A.; Anissimov, Y.G.; Rybachuk, M. Retinal tissue bioengineering, materials and methods for the treatment of glaucoma. Tissue Eng. Regen. Med. 2020, 17, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, X.; Hong, H.; Hu, R.; Liu, J.; Liu, C. Decellularized extracellular matrix scaffolds: Recent trends and emerging strategies in tissue engineering. Bioact. Mater. 2022, 10, 15–31. [Google Scholar] [CrossRef]
- Ren, T.; van der Merwe, Y.; Steketee, M.B. Developing extracellular matrix technology to treat retinal or optic nerve injury. eNeuro 2015, 2, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Topuz, B.; Aydin, H.M. Preparation of decellularized optic nerve grafts. Artif. Organs 2022, 46, 618–632. [Google Scholar] [CrossRef]
- Maqueda, M.; Mosquera, J.L.; García-Arumí, J.; Veiga, A.; Duarri, A. Repopulation of decellularized retinas with hiPSC-derived retinal pigment epithelial and ocular progenitor cells shows cell engraftment, organization and differentiation. Biomaterials 2021, 276, 121049. [Google Scholar] [CrossRef]
- Reinhard, J.; Roll, L.; Faissner, A. Tenascins in retinal and optic nerve neurodegeneration. Front. Integr. Neurosci. 2017, 11, 30. [Google Scholar] [CrossRef]
- Zhang, R.; Li, B.; Li, H. Extracellular-matrix mechanics regulate the ocular physiological and pathological activities. J. Ophthalmol. 2023, 2023, 7626920. [Google Scholar] [CrossRef] [PubMed]
- McGrady, N.R.; Pasini, S.; Baratta, R.O.; Del Buono, B.J.; Schlumpf, E.; Calkins, D.J. Restoring the extracellular matrix: A neuroprotective role for collagen mimetic peptides in experimental glaucoma. Front. Pharmacol. 2021, 12, 764709. [Google Scholar] [CrossRef] [PubMed]
- Soucy, J.R.; Aguzzi, E.A.; Cho, J.; Gilhooley, M.J.; Keuthan, C.; Luo, Z.; Monavarfeshani, A.; Saleem, M.A.; Wang, X.-W.; Wohlschlegel, J.; et al. Retinal ganglion cell repopulation for vision restoration in optic neuropathy: A roadmap from the RReSTORe Consortium. Mol. Neurodegener. 2023, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.; Magli, S.; Rabbachin, L.; Sampaolesi, S.; Nicotra, F.; Russo, L. 3D extracellular matrix mimics: Fundamental concepts and role of materials chemistry to influence stem cell fate. Biomacromolecules 2020, 21, 1968–1994. [Google Scholar] [CrossRef]
- Han, I.C.; Bohrer, L.R.; Gibson-Corley, K.N.; Wiley, L.A.; Shrestha, A.; Harman, B.E.; Jiao, C.; Sohn, E.H.; Wendland, R.; Allen, B.N.; et al. Biocompatibility of human induced pluripotent stem cell–derived retinal progenitor cell grafts in immunocompromised rats. Cell Transplant. 2022, 31, 09636897221104451. [Google Scholar] [CrossRef]
- Xue, J.; Wu, T.; Qiu, J.; Xia, Y. Accelerating cell migration along radially aligned nanofibers through the addition of electrosprayed nanoparticles in a radial density gradient. Part. Part. Syst. Charact. 2022, 39, 2100280. [Google Scholar] [CrossRef]
- Chen, T.C.; She, P.Y.; Chen, D.F.; Lu, J.H.; Yang, C.H.; Huang, D.S.; Chen, P.Y.; Lu, C.Y.; Cho, K.S.; Chen, H.F.; et al. Polybenzyl glutamate biocompatible scaffold promotes the efficiency of retinal differentiation toward retinal ganglion cell lineage from human-induced pluripotent stem cells. Int. J. Mol. Sci. 2019, 20, 178. [Google Scholar] [CrossRef]
- Sluch, V.M.; Davis, C.H.O.; Ranganathan, V.; Kerr, J.M.; Krick, K.; Martin, R.; Berlinicke, C.A.; Marsh-Armstrong, N.; Diamond, J.S.; Mao, H.Q.; et al. Differentiation of human ESCs to retinal ganglion cells using a CRISPR engineered reporter cell line. Sci. Rep. 2015, 5, 16595. [Google Scholar] [CrossRef]
- Laughter, M.R.; Ammar, D.A.; Bardill, J.R.; Pena, B.; Kahook, M.Y.; Lee, D.J.; Park, D. A self-assembling injectable biomimetic microenvironment encourages retinal ganglion cell axon extension in vitro. ACS Appl. Mater. Interfaces 2016, 8, 20540–20548. [Google Scholar] [CrossRef]
- Kador, K.E.; Grogan, S.P.; Dorthé, E.W.; Venugopalan, P.; Malek, M.F.; Goldberg, J.L.; D’Lima, D.D. Control of retinal ganglion cell positioning and neurite growth: Combining 3D printing with radial electrospun scaffolds. Tissue Eng. Part A 2016, 22, 286–294. [Google Scholar] [CrossRef]
- Kador, K.E.; Alsehli, H.S.; Zindell, A.N.; Lau, L.W.; Andreopoulos, F.M.; Watson, B.D.; Goldberg, J.L. Retinal ganglion cell polarization using immobilized guidance cues on a tissue-engineered scaffold. Acta Biomater. 2014, 10, 4939–4946. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhao, B.; Liu, X.; Li, X.; Zeng, C.; Shi, H.; Xu, X.; Lin, T.; Dai, L.; Liu, Y. Aligned nanofibers from polypyrrole/graphene as electrodes for regeneration of optic nerve via electrical stimulation. ACS Appl. Mater. Interfaces 2016, 8, 6834–6840. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Lin, Y.Y.; Yang, T.C.; Yarmishyn, A.A.; Lin, T.W.; Chang, Y.L.; Hwang, D.K.; Wang, C.Y.; Liu, Y.Y.; Lo, W.L.; et al. P3HT:Bebq2-based photovoltaic device enhances differentiation of hiPSC-derived retinal ganglion cells. Int. J. Mol. Sci. 2019, 20, 2661. [Google Scholar] [CrossRef] [PubMed]
- Davari, N.; Bakhtiary, N.; Khajehmohammadi, M.; Sarkari, S.; Tolabi, H.; Ghorbani, F.; Ghalandari, B. Protein-based hydrogels: Promising materials for tissue engineering. Polymers 2022, 14, 986. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Peng, J.; Xiao, H.; Xu, X.; Qian, Z. Polysaccharide hydrogels: Functionalization, construction and served as scaffold for tissue engineering. Carbohydr. Polym. 2022, 278, 118952. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.C.; Hallam, D.; Karimi, A.; Mellough, C.B.; Chen, J.; Steel, D.H.W.; Lako, M. 3D culture of human pluripotent stem cells in RGD-alginate hydrogel improves retinal tissue development. Acta Biomater. 2017, 49, 329–343. [Google Scholar] [CrossRef]
- Roozafzoon, R.; Lashay, A.; Vasei, M.; Ai, J.; Khoshzaban, A.; Keshel, S.H.; Barabadi, Z.; Bahrami, H. Dental pulp stem cells differentiation into retinal ganglion-like cells in a three dimensional network. Biochem. Biophys. Res. Commun. 2015, 457, 154–160. [Google Scholar] [CrossRef]
- Zhu, D.; Gao, J.; Tang, C.; Xu, Z.; Sun, T. Evaluation of the potential effects of retinol and alginate/gelatin-based scaffolds on differentiation capacity of mouse mesenchymal stem cells (MSCs) into retinal cells. Int. J. Stem Cells 2021, 15, 183–194. [Google Scholar]
- Wang, J.J.; Wang, T.Z.; Guan, B.; Liu, X.X.; Gong, Z.; Li, Y.; Li, L.L.; Ke, L.N.; Nan, K.H. Implantable patches assembled with mesenchymal stem cells and gelatin/silk fibroin composite microspheres for the treatment of traumatic optic neuropathy. Appl. Mater. Today 2022, 26, 101278. [Google Scholar] [CrossRef]
- Park, J.; Baranov, P.; Aydin, A.; Abdelgawad, H.; Singh, D.; Niu, W.; Kurisawa, M.; Spector, M.; Young, M.J. In situ cross-linking hydrogel as a vehicle for retinal progenitor cell transplantation. Cell Transplant. 2019, 28, 596–606. [Google Scholar] [CrossRef]
- Colombe Dromel, P.; Singh, D.; Alexander-Katz, A.; Kurisawa, M.; Spector, M.; Young, M. Injectable gelatin hydroxyphenyl propionic acid hydrogel protects human retinal progenitor cells (hRPCs) from shear stress applied during small-bore needle injection. Appl. Mater. Today 2020, 19, 100602. [Google Scholar] [CrossRef]




| Material Type | Components | Pros | Cons and Needs |
|---|---|---|---|
| Decellularized extracellular matrix (dECM) | collagen, elastin, fibronectin, laminin, tenascins, heparan, growth factors | Adequate 3D structure Presence of growth factors and chemical cues Presence of cell adhesion No ligands or ligands with low presence of immunogenic cellular components Experience at clinical level for some tissues (skin: AlloDerm®, Oasis®; tendon and ligaments: GraftJacket®, heart: CardioCel®…) | Presence of residual toxic chemicals used for decellularization (or other toxins) Low–medium batch-to-batch reproducibility due to origin variability Finely tuned organ/tissue selection to avoid risks of disease transmission Adequate material shape for transport and surgery (application) Full functional integration (need for vascularization, innervation) Non-optimal decellularization leading to tissue architecture degradation |
| Synthetic polymers and mixtures | poly(ε-caprolactone) poly(glycerol sebacate) polylactic-co-glycolic acid poly-(d,l)-lactide polybenzyl glutamate poly(ethylene-co-vinyl acetate) polypyrrole/graphene poly-3-hexylthiophene poly(lactic-co-glycolic acid) poly(serinol hexamethylene urea) | Simple chemical nature (but it can be more complex if additives or natural polymers are mixed with synthetic polymers (netrin-1, laminin, fibronectin, cell adhesion peptides, etc.)) Tunable physical properties (viscoelasticity, stiffness, permeability, etc.) Tunable volume shaping and surface topography (femtosecond laser, two photon lithography, 3D printing, electrospinning) Batch-to-batch reproducibility | Low biocompatibility (these materials lack the chemical signals typical in dECMs; thus they are usually decorated with laminin, fibronectin, or adhesion peptides (RGD, IKVAV, YIGSR, GVMGFO) typical in integrins, laminins, collagen…, glycans, growth factors, etc.) Higher toxicity due to synthesis additives and monomers |
| Natural polymers and hydrogels | polysaccharides (hyaluronic acid, alginate, heparin, carrageenan, fucoidan, dextran, chitosan, cellulose, pullulan, cyclodextrins, etc.) proteins (collagen, gelatin, albumin, elastin, ketatin, resilin, silk) | Simple chemical nature (depending on the nature of the polymer and polymer mixtures) Higher inherent biodegradability, biocompatibility, and bio-responsive functions compared to synthetic polymers Batch-to-batch reproducibility (but lower than in synthetic polymers) | Need for intramolecular interaction modulation (chemical modifications to reach adequate physical properties and structure) Low stability at physiological conditions (for example, due to temperature) Not enough biocompatibility (still requires polymer mixing (i.e., polysaccharides with proteins) or copolymerization (i.e., with synthetic monomers such as hydroxyphenyl propionic acid or natural monomers such as tyramine) and cell adhesion motifs are still needed) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ladero, M.; Reche-Sainz, J.A.; Gallardo, M.E. Hereditary Optic Neuropathies: A Systematic Review on the Interplay between Biomaterials and Induced Pluripotent Stem Cells. Bioengineering 2024, 11, 52. https://doi.org/10.3390/bioengineering11010052
Ladero M, Reche-Sainz JA, Gallardo ME. Hereditary Optic Neuropathies: A Systematic Review on the Interplay between Biomaterials and Induced Pluripotent Stem Cells. Bioengineering. 2024; 11(1):52. https://doi.org/10.3390/bioengineering11010052
Chicago/Turabian StyleLadero, Miguel, Jose Alberto Reche-Sainz, and M. Esther Gallardo. 2024. "Hereditary Optic Neuropathies: A Systematic Review on the Interplay between Biomaterials and Induced Pluripotent Stem Cells" Bioengineering 11, no. 1: 52. https://doi.org/10.3390/bioengineering11010052