
Alzheimer’s Disease: Models and Molecular Mechanisms Informing Disease and Treatments
 and
and
Abstract
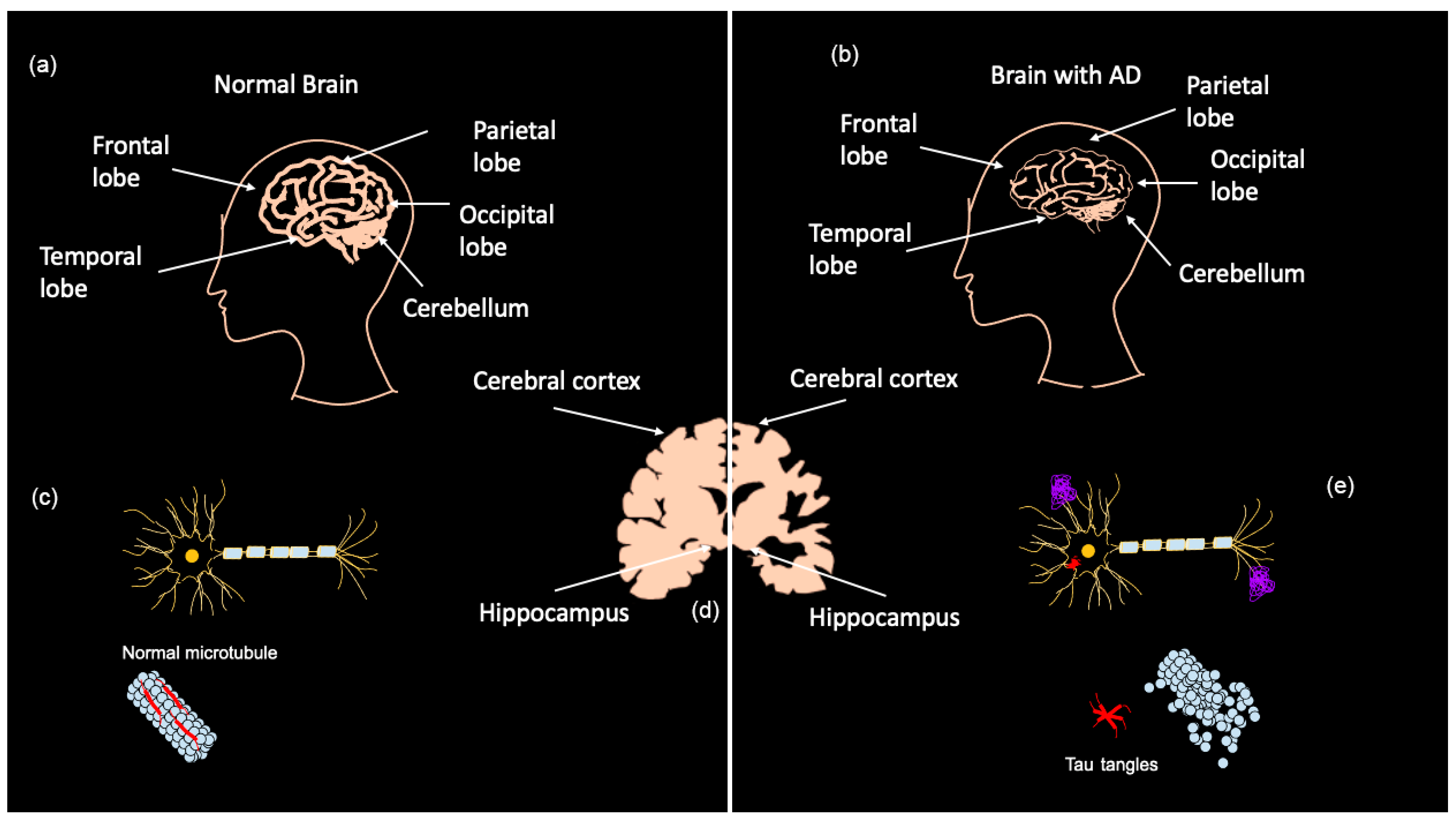
:1. Introduction
Diagnosis

{kind=link}
{kind=link}
{kind=link}
| Serum Levels (pg/mL) | ||||||
|---|---|---|---|---|---|---|
| Biomarker | AD | Depression | Anxiety Disorders | Cardiovascular Disease | Diabetes | Inflammatory Bowel Disease (IBD) |
| TNFα | 1.6 ± 1.4 [37] ↑ | 4.1 ± 0.5 [38] ↑ | 2.4 ± 0.9 [39] ↓ | 3.1 ± 3.4 [40] ↑ | 7.5 ± 2.5 [41] ↑ | 29.4 ± 0.2 [42] |
| IL-1α | 89.2 ± 17.6 [43] ↑ | 3.3 ± 0.4 [38] ↑ | 70.3 ± 3.6 [44] ↑ | - | 0.9 ± 4.8 [45] | 7.6 ± 61.5 [46] ↑ |
| IL-1β | 4.7 ± 2.1 [47] ↑ | 1.2 ± 0.2 [48] ↑ | 5.0 ± 2.3 [49] | 1.7 ± 0.2 [50] ↑ | 3.0 ± 1.0 [45] ↑ | 3.8 ± 43.0 [46] ↑ |
| IL-6 | 4.4 ± 5.1 [37] ↑ | 2.9 ± 0.1 [51] ↑ | 12.6 ± 2.4 [44] ↑ | 4.3 ± 3.5 [50] ↑ | 4.3 ± 2.6 [45] ↑ | 4.5 ± 4.1 [52] ↑ |
| IL-8 | 35.0 ± 4.1 [53] ↓ | 8.9 ± 4.0 [54] ↓ | 44.6 ± 16.2 [44] ↑ | 47.8 ± 71.2 [50] ↑ | 6.5 ± 5.3 [45] ↓ | 5.0 ± 12.1 [55] ↓ |
| sST2 | 27.5 ± 7.1 [23] ↑ | 9000± 3300 [56] ↑ | - | 420.0 ± 49.0 [57] ↑ | 160 ± 60 [58] ↑ | 50.3 ± 52.9 [59] ↑ |
| IL-33 | 5.9 ± 5.5 [60] ↓ | 17.2 ± 5.6 [56] | 635.8 ± 6.7 [61] ↑ | 103.3 ± 19.3 [62] ↓ | 40 ± 7 [63] ↓ | 40 ± 52.5 [64] |
| SDF-1 | 1949.6 ± 427.9 [65] ↓ | 4928.8 ± 589.5 [66] ↑ | 1352 ± 733 [67] ↑ | 1891.8 ± 1044.8 [68] ↑ | 204.2 ± 30.9 [69] ↑ | - |
| progranulin | 45.3 ± 11.8 [70,71] ↑ | - | - | 3.5 × 104 ± 8.2 × 103 [72] | 47.2 ± 4.5 [73] ↑ | - |
| VCAM-1 | 9.5 × 105 ± 1.6 × 105 [74] ↑ | 1.2 × 106 ± 4.5 × 105 [75] ↑ | 6.3 × 105 ± 1.4 × 105 [76] ↑ | 1.7 × 106 ± 3.4 × 105 [77] ↑ | 736.4 ± 267.0 [78] ↑ | 6.0 × 105 ± 1.5 × 105 [79] ↑ |
| ICAM-1 | 3.4 × 105 ± 3.2 × 105 [80] ↑ | 2.7 × 105 ± 8.7 × 104 [75] ↑ | 2.4 × 105 ± 4.3 × 104 [76] ↑ | 1.6 × 106 ± 3.6 × 105 [77] ↑ | 245.4 ± 107.4 [81] | 4.0 × 105 ± 3.4 × 104 [79] ↑ |
| NFL | 19 ± 12 [82] ↑ | 28.8 ± 22.5 [83] ↑ | 72.220 ± 22.8 [84] ↑ | 19.8 ± 12.2 [85] ↑ | 13 ± 4.5 [86] | - |
| neurogranin | 429.2 ± 104.3 [87] ↓ | 100.3 ± 124.3[88] ↑ | - | - | - | - |
| Aß42 | 44.2 ± 10.3 [37] ↑ | 11.4 ± 1.7 [89,90] | 2.06 ± 0.2 [71] | - | - | - |
| Tau | 351.9 ± 50.0 [91] ↑ | 4.3 ± 2.1 [90] ↑ | 0.27 ± 0.6 [71] | - | - | - |
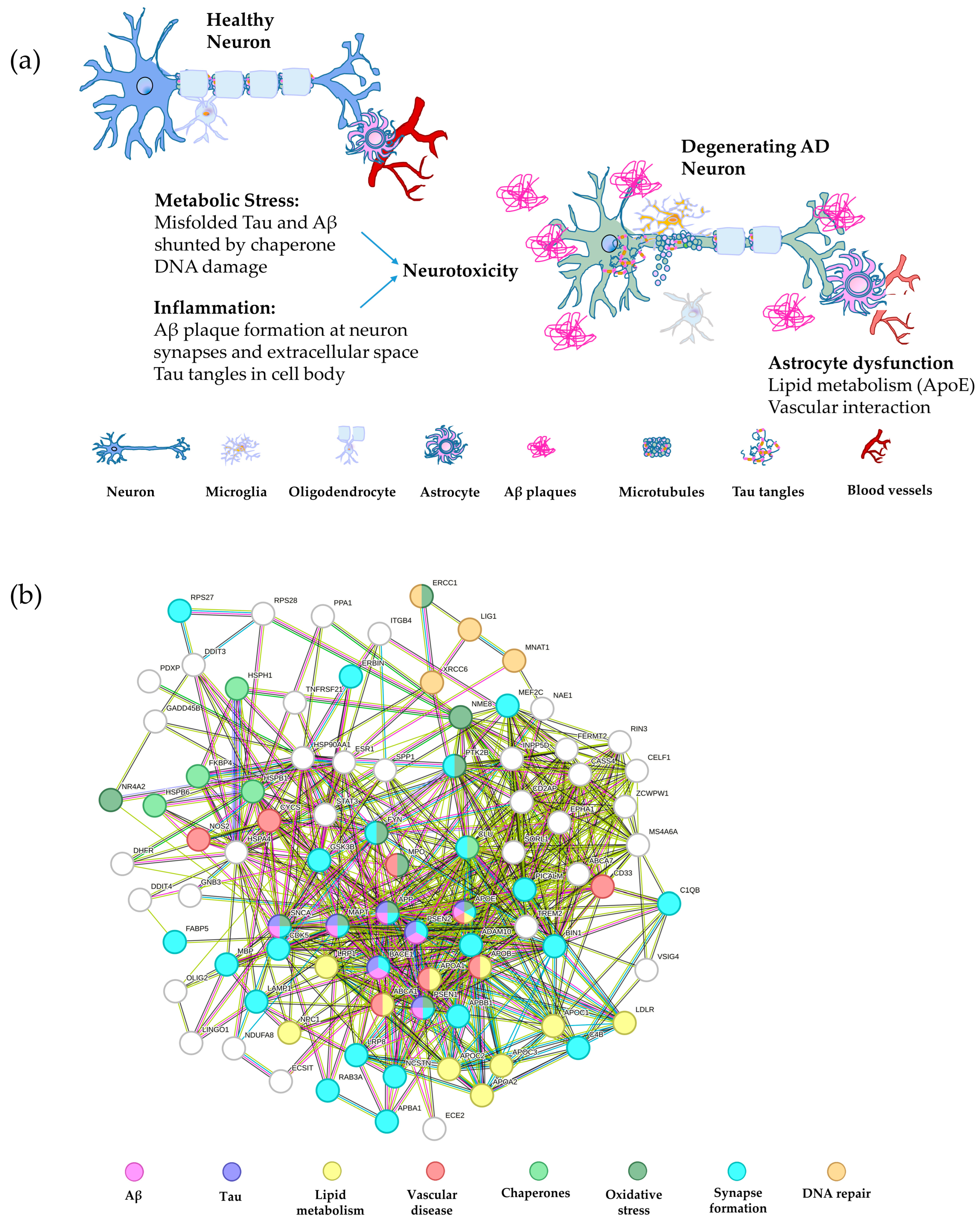
2. Genetics of AD
| Gene and Pathway | Gene Name | Gene Function | AD Relevance from Mutations | AD Variants |
|---|---|---|---|---|
PSEN1     | Presenilin 1 | Encodes PS1 protein, a catalytic subunit of the γ-secretase enzyme that cleaves APP, resulting in Aß production [6,116] | Decreases Aß40 levels increasing Aβ42/Aβ40 ratio [6,109] | EOAD risk: 33 variants (Table S1) |
| PSEN2 | Presenilin 2 | Encodes PS2 protein, a catalytic subunit of the γ-secretase enzyme that cleaves APP, resulting in Aß production [6,116] | Increases Aß42 levels increasing Aβ42/Aβ40 ratio [6,117]. Missense mutations rare cause of EOAD | EOAD risk: 6 variants (Table S1) |
| APP | amyloid precursor protein | Encodes APP protein cleaved to release Aβ [6] | Promotes Aβ production/build-up and increases Aβ42/Aβ40 ratio [109]. Associated with familial EOAD | EOAD risk: 13 variants EOAD protective: 1 variant (Table S1) |
| ECE2 | Endothelin-Converting Enzyme 2 | Endothelin-converting enzyme/breaks down Aß | If ECE2 is not active, then it cannot breakdown Aß, leading to an excess in Aß | LOAD risk: c.556C>T c.2252T>C [118] |
| GNB3 | Guanine Nucleotide-binding protein, Beta-3 | G protein β3 subunit/promotes adrenaline production | Different forms of the code coding for GNB3 can enhance APP expression | AD risk modifier: rs5443 [119] |
| ADRB1 | Beta-1-Adrenergic Receptor | β1-adrenergic receptor/promotes adrenaline production | Different forms of the code coding for ADRB1 can enhance APP expression | AD risk modifier: rs1801253 [119] |
| CR1 | Complement component Receptor 1 | Type-I transmembrane glycoprotein | Involved in eliminating Aβ and tauopathy [120] | AD risk: rs1408077 rs6701713 rs3818361 [121] |
| SLC24A4/RIN3 | Solute Carrier family 24, member 4/Ras and Rab Interactor 3 | Solute carrier | Increases endosomal dysfunction in APP/PSA1 mouse model [122] | Protective: rs10498633 rs12881735 [123,124,125] |
| INPP5D | Inositol Polyphosphate-5-Phosphatase, 145-KD | Inositol polyphosphate-5-phosphatase family | Expression is elevated in microglia and associated with plaque in an AD mouse model [126] | Protective: rs61068452-G [127] LOAD Risk: rs1057258 rs35349669 [124,128,129,130] |
| ECSIT | Evolutionarily Conserved Signaling Intermediate in Toll pathway | Encodes cytoplasmic/signaling adapting protein. Stabilizes mitochondrial respiratory complex [6] | Interacts with PSEN1, PSEN2 and APOE. Molecular link in AD inflammation, oxidative stress, and mitochondrial dysfunction [6,131] | |
| CELF1 | Cugbp- and Elav-Like Family, member 1 | Alternate splicing of pre-mRNA | Affects expression of Aβ42 | AD Risk: rs3740688 rs10838725 [132,133] |
| FERMT2 | Ferm domain-containing kindlin 2 | TGFβ1 receptor binding and actin binding | Involved in metabolism of APP [134] | Risk: rs7160582 rs7143400-T [128,135] Brain Amyloidosis: rs17125944 [136,137] |
| CASS4 | Cas Scaffold Protein Family, member 4 | Tyrosine kinase binding | Possible role via regulation of CASS4 phosphorylation by α2β1 and αVβ1 integrins, which induces Aβ neurotoxicity [138] | Protective: rs7274581 rs6024870 rs6069736 [123] Pathogenic: rs16979934 [128] |
| MAPT | Microtubule-Associated Protein Tau | Encodes Tau protein/stabilizes microtubules | Tau tangles lead to destabilization of microtubules and death of neuron [139] | AD risk modifier: A152T [140] |
| CD2AP | CD2-Associated Protein | Regulation of actin cytoskeleton | Loss causes neuronal toxicity resulting from tau [141] | LOAD risk: rs10948363 [142] rs9349407 [143] |
APOE  | Apolipoprotein E | Metabolizes lipids and cholesterol [6] | APOE ε4 allele increases AD risk while APOE ε2 allele reduces risk. Role in formation of senile plaques from Aβ deposition. Associated with vascular damage and cerebral amyloid angiopathy [6,144,145] | EOAD and LOAD risk: c.127C>T [146] |
| TREM2 | Triggering Receptor Expressed on Myeloid cells 2 | Modifies microglia activity and survival | Increased expression in microglia cells surrounding amyloid | AD risk: rs75932628T [147] |
ABCA1  | ATP-Binding Cassette, subfamily A, member 1 | Regulates cholesterol transport from bloodstream into the brain. Stabilizes APOE lipidation and mediates HDL generation [6] | Increases Aß plaques and eliminates APOE lipidation. Decreases plasma HDL and ApoAI levels, cholesterol accumulation in tissues, and pathogenesis of AD [6] | AD possibly protective: P1059S V399A E1172D [148] |
| ABCA7 | ATP-Binding Cassette, subfamily A, member 7 | ATP-binding cassette transporter | Affects AD pathogenesis through regulation of lipid metabolism and clearing of amyloid [149] | Protective: rs3764650 rs72973581[149] LOAD Risk: rs4147914[128] AD Risk: rs3764650, rs4147929, rs3752246, rs115550680, rs78117248, rs142076058 [149] |
SORL1  | Sortilin-related Receptor | Participates in APP and Aß trafficking | Neurons without SORL1 show downregulation of APOE and CLU [150] | Protective: rs11218343 [123] LOAD Risk: rs2276412 [128] |
| MPO | Myeloperoxidase | Inflammatory enzyme/catalyzes Cl and H2O2 to make HOCl, promotes production of reactive oxygen and nitrogen species | Over production of reactive oxygen species causes oxidative stress, which results in neuroinflammation [151] | EOAD risk: c.2031-2A>C c.1705C>T [146] |
| CD33 | Sialic Acid Binding Ig-Like Lectin 3 | Phosphatase and sialic acid binding activity | Short isoform leads to Aβ1–42 phagocytosis in microglial cells [152] | Risk: rs3865444-C rs12459419-C rs1803254 [128,153,154] LOAD protective: rs3865444-A rs12459419-T [153,155,156,157] |
| CLU | Clusterin | Lipid transport [6] | Promotes/Reduces Aß clearance [6] | LOAD Risk: rs1532278, rs9331947, rs11136000C/T, rs2279590, rs9331888, rs7012010, rs7982, and rs9331949 [128,136,143,158,159] Amyloid Deposition: rs3818361 [137] |
| NME8 | NME-NM23 family, member 8 | Has a catalytically active N-terminal thioredoxin domain and implicated in ciliary function | Certain variants may play a role in reducing neurodegeneration [160] | LOAD Protective: rs2718058 [136,161] |
| ESR | Estrogen Receptor | Binds estrogen | Implicated in neuroinflammation contributing to AD [37] | AD risk: rs6909023 rs2982684 [162] |
| MS4A6A | Membrane-Spanning 4-domains, subfamily A, member 6A | Membrane spanning protein | Over-expression increases neuroinflammation [163] | AD Protective: rs610932-A rs7232-T [164] LOAD Risk: rs12453 [128] Cortical/Hippocampal Atrophy: rs610932 [137] |
| BIN1 | Bridging Integrator 1 | Membrane curvature and endocytosis functions [6] | Participates in Aβ production and modulator of tau and NFT pathology [6,165] | LOAD risk: rs754834233 rs138047593 [166] |
| ADAM10 | A Disintegrin and Metalloproteinase Domain 10 | α-secretase/involved in cutting of APP ectodomain | Certain variants increase Aß levels in vitro and makes APP produce Aß in Tg2576 mice | LOAD risk: Q170H R181G [167] |
| PTK2B | Protein-Tyrosine Kinase 2, Beta | Tyrosine kinase | Plays a role in Aβ-mediated synaptic defects [168] | LOAD Risk: rs4732720 rs28834970 [128,169] |
| MEF2C | Myocyte Enhancer Factor 2C | Member of the MADS box transcription enhancer factor 2 family, plays a role in myogenesis | Knockdown in AD mouse model leads to elevated Aβ levels, downregulation of synaptic proteins and oxidative stress [84] | Protective: rs190982 [170] LOAD Risk: rs9293505 [128] |
| PICALM | Phosphatidylinositol-binding Clathrin Assembly protein | Clathrin assembly | Down-regulated in AD brain correlating with autophagy defect [171] | LOAD Risk: rs7480193 rs510566 rs1237999 rs561655 rs17148741 rs3851179 [128,137,143,172] |
| EPHA1 | Ephrin receptor A1 | Protein tyrosine kinase | Affects neuroinflammation [173] | AD protective: rs11762262 rs11771145 [123,136,174] LOAD Risk: rs11767557 rs11768549 [128,143] |
ZCWPW1  | Zinc finger CW-type domain and PWWP domain-containing protein 1 | Involved in the histone methylation process; possible role in meiosis I | Proposed to play a role via regulation of DNA and via reduction of insulin resistance | LOAD Protective: rs1476679 [132,175] |
| HLA-DRB1/DRB5 | Major histocompatibility complex, class II, DR Beta-1/Beta-5 | Human leukocyte antigen complex proteins | May be involved in AD pathogenesis through its role in the immune system | LOAD Risk: rs6597017 rs9271192 [128,176] |

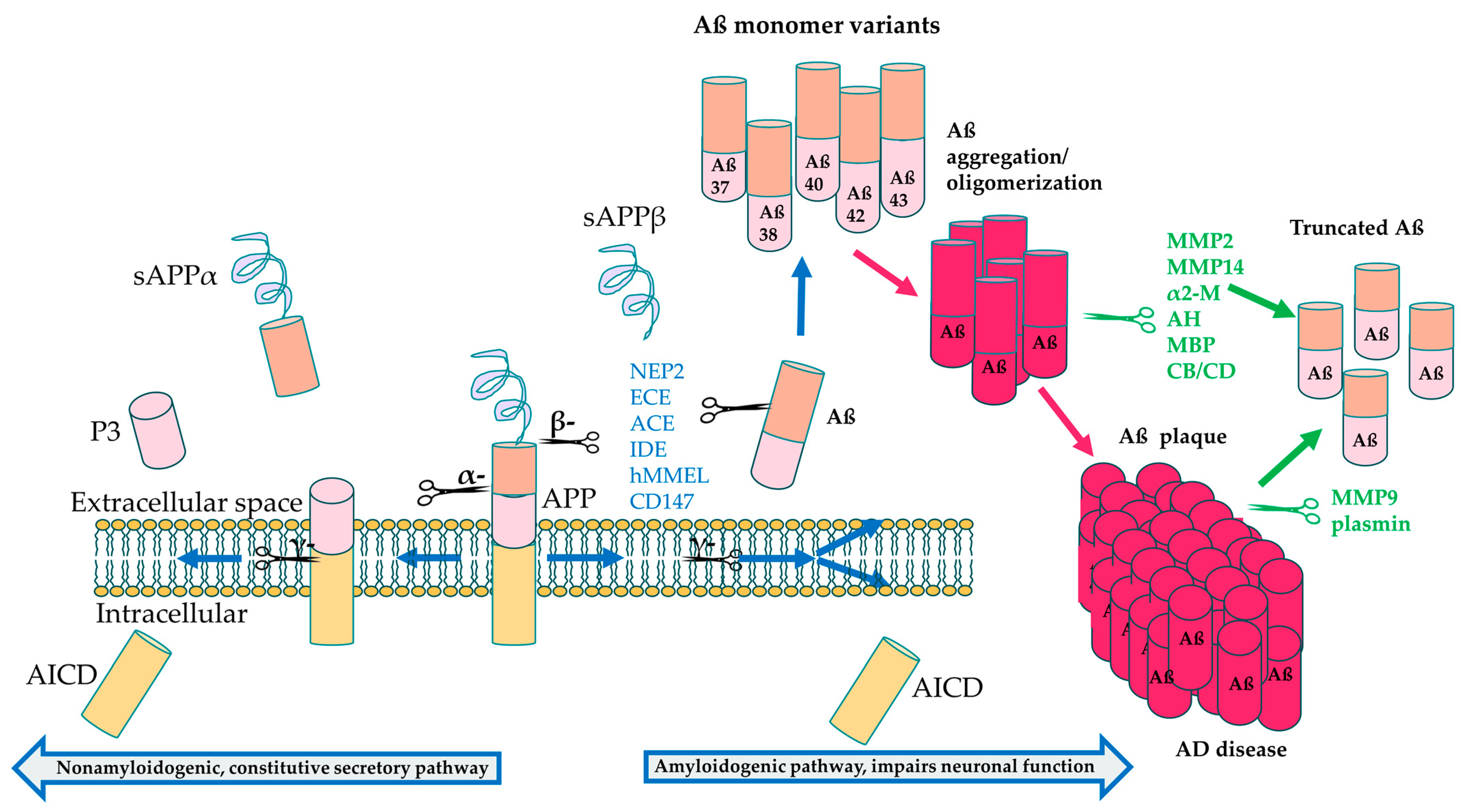
2.1. Amyloid-β Aggregation

| Enzyme Type | Enzyme | Substrate | Significance in AD |
|---|---|---|---|
| Metalloproteinase | MMP2 | Aβ fibrils | Expressed in the healthy state |
| MMP9 | Aβ fibrils and Compact plaques | Expressed in the healthy and pathological states | |
| MMP14 | Aβ fibrils | Expressed in the healthy and pathological states | |
| NEP | Synthetic Aβ oligomers | Expressed in healthy and pathological states | |
| NEP2 | Aβ monomers [188] | Expressed in healthy physiological state | |
| ECE1 | Aβ monomers | Expressed in healthy physiological state | |
| ECE2 | Aβ monomers | Expressed in healthy physiological state | |
| ACE | Aβ monomers | Expressed in healthy physiological state | |
| IDE | Aβ monomers | Expressed in healthy physiological state | |
| hMMEL | Aβ monomers | Expressed in healthy physiological state | |
| CD147 [193] | Aβ monomers [194] | Expressed in healthy physiological state | |
| α2-M [195] | Aβ fibrils | Overexpressed in pathological state | |
| Aminopeptidases | plasmin | Aβ monomers, fibrils and compact aggregates | Expressed in the pathological state |
| AH | Aβ oligomers | Expressed in the pathological state | |
| MBP | Aβ fibrils | Expressed in healthy physiological state | |
| ACT | Aβ monomers | Increases Aβ polymerization | |
| CB | Aβ fibrils | Expressed in healthy physiological state | |
| CD | Aβ fibrils | Expressed in healthy physiological state | |
| BACE1 | APP | Expressed in healthy and pathological physiological states | |
| BACE2 | APP | Expressed in healthy and pathological physiological states | |
| Proteasome | Aβ monomers | Expressed in healthy physiological state |
2.2. Tau Accumulation
2.3. Lipid Metabolism
2.4. Acetylcholine
2.5. Chaperones
3. Environmental Risk Factors
3.1. Air Pollution and Heavy Metals
3.2. Diet and the Gut Microbiome
3.3. Pre-Existing Conditions
4. Models of AD
4.1. AD Modeling in Humans Using Single-Cell Genomics
4.2. Animal Models of AD
4.2.1. Non-Human Primate Models
4.2.2. Mouse/Rat Models
4.2.3. Canines
4.2.4. Zebrafish Models
4.2.5. Invertebrate Models
5. Current Treatments
5.1. Donepezil
5.2. Rivastigmine
5.3. Galantamine
5.4. Memantine
5.5. Aducanumab
5.6. Lecanemab
5.7. Upcoming Treatments
| Pathway | Intervention | Mechanism | Clinical Trials | FDA Approved Treatments |
|---|---|---|---|---|
| Amyloid | BACE1 inhibitors: MK-8931, AZD3293, JNJ-54861911, E2609 and CNP520 | Inhibits β-secretase, an enzyme that cleaves APP at a site that leads to formation of toxic Aβ monomers | NCT01953601 (2013–2018), NCT02245737 (2014–2018), NCT01978548 (2013–2015), NCT02956486 (2016–2020), NCT03131453 (2017–2020) | None |
| γ-secretase inhibitors: LY411575, LY-450139, BMS-708163 | Inhibits γ-secretase, which cleaves APP | NCT00594568 (2008–2011), NCT00890890 (2009–2013) | None | |
| Contraloid Acetate | Disassembly of Aβ oligomers into monomers | NCT04711486, NCT03955380, NCT03944460 | None | |
| Monoclonal antibody: Aducanumab, Lecanemab, LY3372993, Crenezumab | Recognize and bind Aβ or proteins in the Aβ pathway | NCT03720548, NCT03977584 | Aducanumab and Lecanemab | |
| Electromagnetic field (EMF) stimulation | low and high frequency pulsed EMF stimulation, transcranial direct current stimulation (tDCS), transcranial alternate current stimulation (tACS) | Clearance of protein aggregates, chaperone-mediated degradation, improved mitochondrial function | NCT02873546, NCT04045990, NCT05784298, NCT01481961 | None |
| NFT pathway | Inhibitors of tau aggregation: LMTM, ACI3024, Curcumin | Inhibits formation of aggregated tau NFTs | NCT03446001, NCT01383161 | None |
| Tau inhibitors: BIIB080 | Inhibits tau protein production | NCT05399888 | None | |
| Antibodies: RG7345, Gosuranemab, Semorinemab, Zagotenemab, JNJ63733657 | Recognize and binds tau protein | NCT02281786, NCT03068468, NCT03289143, NCT03518073, NCT04619420 | None | |
| Acetylcholine | Cholinesterase inhibitors | Inhibits the enzyme that breaks down acetylcholine | NCT02087865, NCT01951118, NCT02079246, NCT00428389 | Donepezil, Rivastigmine and Galantamine |
5.8. Holistic Treatments
6. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, Y.-T.; Clare, L.; Hindle, J.V.; Nelis, S.M.; Martyr, A.; Matthews, F.E. Dementia Subtype and Living Well: Results from the Improving the Experience of Dementia and Enhancing Active Life (IDEAL) Study. BMC Med. 2018, 16, 140. [Google Scholar] [CrossRef] [PubMed]
- Elahi, F.M.; Miller, B.L. A Clinicopathological Approach to the Diagnosis of Dementia. Nat. Rev. Neurol. 2017, 13, 457–476. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A. Continuum: Lifelong Learning in Neurology—Neurology of Dementia, Volume 28, Issue 3, June 2022. Continuum 2022, 28, 834–851. [Google Scholar] [CrossRef] [PubMed]
- Amjad, H.; Roth, D.L.; Sheehan, O.C.; Lyketsos, C.G.; Wolff, J.L.; Samus, Q.M. Underdiagnosis of Dementia: An Observational Study of Patterns in Diagnosis and Awareness in US Older Adults. J. Gen. Intern. Med. 2018, 33, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Balasa, M.; Gelpi, E.; Antonell, A.; Rey, M.J.; Sánchez-Valle, R.; Molinuevo, J.L.; Lladó, A. Neurological Tissue Bank/University of Barcelona/Hospital Clínic NTB/UB/HC Collaborative Group Clinical Features and APOE Genotype of Pathologically Proven Early-Onset Alzheimer Disease. Neurology 2011, 76, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- 2023 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef]
- Guo, L.; Zhong, M.B.; Zhang, L.; Zhang, B.; Cai, D. Sex Differences in Alzheimer’s Disease: Insights from the Multiomics Landscape. Biol. Psychiatry 2022, 91, 61–71. [Google Scholar] [CrossRef]
- Zhu, D.; Montagne, A.; Zhao, Z. Alzheimer’s Pathogenic Mechanisms and Underlying Sex Difference. Cell Mol. Life Sci. 2021, 78, 4907–4920. [Google Scholar] [CrossRef]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef]
- Reitz, C.; Rogaeva, E.; Beecham, G.W. Late-Onset vs Nonmendelian Early-Onset Alzheimer Disease. Neurol. Genet. 2020, 6, e512. [Google Scholar] [CrossRef] [PubMed]
- Wingo, T.S.; Lah, J.J.; Levey, A.I.; Cutler, D.J. Autosomal Recessive Causes Likely in Early-Onset Alzheimer Disease. Arch. Neurol. 2012, 69, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Barber, I.S.; Braae, A.; Clement, N.; Patel, T.; Guetta-Baranes, T.; Brookes, K.; Medway, C.; Chappell, S.; Guerreiro, R.; Bras, J.; et al. Mutation Analysis of Sporadic Early-Onset Alzheimer’s Disease Using the NeuroX Array. Neurobiol. Aging 2017, 49, 215.e1–215.e8. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, S.; Peetoom, K.; Bakker, C.; van der Flier, W.M.; Papma, J.M.; Koopmans, R.; Verhey, F.R.J.; de Vugt, M.; Köhler, S.; Young-Onset Dementia Epidemiology Study Group; et al. Global Prevalence of Young-Onset Dementia: A Systematic Review and Meta-Analysis. JAMA Neurol. 2021, 78, 1080–1090. [Google Scholar] [CrossRef]
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; McAninch, E.A.; Wilson, R.S.; Evans, D.A. Population Estimate of People with Clinical AD and Mild Cognitive Impairment in the United States (2020–2060). Alzheimers Dement. 2021, 17, 1966–1975. [Google Scholar] [CrossRef]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Klyucherev, T.O.; Olszewski, P.; Shalimova, A.A.; Chubarev, V.N.; Tarasov, V.V.; Attwood, M.M.; Syvänen, S.; Schiöth, H.B. Advances in the Development of New Biomarkers for Alzheimer’s Disease. Transl. Neurodegener. 2022, 11, 25. [Google Scholar] [CrossRef]
- Palmqvist, S.; Zetterberg, H.; Mattsson, N.; Johansson, P.; Minthon, L.; Blennow, K.; Olsson, M.; Hansson, O. Detailed Comparison of Amyloid PET and CSF Biomarkers for Identifying Early Alzheimer Disease. Neurology 2015, 85, 1240–1249. [Google Scholar] [CrossRef]
- Bagad, D.M.; Chowdhury, D.; Khan, Z. Towards Understanding Alzheimer’s Disease: An Overview. Res. J. Pharm. Biol. Chem. Sci. 2013, 4, 286–298. [Google Scholar]
- Dursun, E.; Gezen-Ak, D.; Hanağası, H.; Bilgiç, B.; Lohmann, E.; Ertan, S.; Atasoy, İ.L.; Alaylıoğlu, M.; Araz, Ö.S.; Önal, B.; et al. The Interleukin 1 Alpha, Interleukin 1 Beta, Interleukin 6 and Alpha-2-Macroglobulin Serum Levels in Patients with Early or Late Onset Alzheimer’s Disease, Mild Cognitive Impairment or Parkinson’s Disease. J. Neuroimmunol. 2015, 283, 50–57. [Google Scholar] [CrossRef]
- Zuliani, G.; Cavalieri, M.; Galvani, M.; Passaro, A.; Munari, M.R.; Bosi, C.; Zurlo, A.; Fellin, R. Markers of Endothelial Dysfunction in Older Subjects with Late Onset Alzheimer’s Disease or Vascular Dementia. J. Neurol. Sci. 2008, 272, 164–170. [Google Scholar] [CrossRef]
- Rentzos, M.; Michalopoulou, M.; Nikolaou, C.; Cambouri, C.; Rombos, A.; Dimitrakopoulos, A.; Kapaki, E.; Vassilopoulos, D. Serum Levels of Soluble Intercellular Adhesion Molecule-1 and Soluble Endothelial Leukocyte Adhesion Molecule-1 in Alzheimer’s Disease. J. Geriatr. Psychiatry Neurol. 2004, 17, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Marventano, I.; Piancone, F.; La Rosa, F.; Galimberti, D.; Fenoglio, C.; Scarpini, E.; Clerici, M. IL-33 and Its Decoy sST2 in Patients with Alzheimer’s Disease and Mild Cognitive Impairment. J. Neuroinflamm. 2020, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, R.; Kishi, T.; Iwata, N. Plasma Levels of IL-6 in Patients with Untreated Major Depressive Disorder: Comparison with Catecholamine Metabolites. Neuropsychiatr. Dis. Treat. 2019, 15, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Lyra e Silva, N.M.; Gonçalves, R.A.; Pascoal, T.A.; Lima-Filho, R.A.; Resende, E.D.; Vieira, E.L.M.; Teixeira, A.L.; de Souza, L.C.; Peny, J.A.; Fortuna, J.T.S.; et al. Pro-Inflammatory Interleukin-6 Signaling Links Cognitive Impairments and Peripheral Metabolic Alterations in Alzheimer’s Disease. Transl. Psychiatry 2021, 11, 251. [Google Scholar] [CrossRef] [PubMed]
- Baghai, T.C.; Varallo-Bedarida, G.; Born, C.; Häfner, S.; Schüle, C.; Eser, D.; Zill, P.; Manook, A.; Weigl, J.; Jooyandeh, S.; et al. Classical Risk Factors and Inflammatory Biomarkers: One of the Missing Biological Links between Cardiovascular Disease and Major Depressive Disorder. Int. J. Mol. Sci. 2018, 19, 1740. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.-F.; Wu, W.; Wong, H.Y.; Ouyang, L.; Qiao, Y.; Xu, J.; Lau, J.H.-Y.; Wong, C.; Jiang, Y.; Holtzman, D.M.; et al. The VCAM1–ApoE Pathway Directs Microglial Chemotaxis and Alleviates Alzheimer’s Disease Pathology. Nat. Aging 2023, 3, 1219–1236. [Google Scholar] [CrossRef]
- Chen, J.; Dai, A.-X.; Tang, H.-L.; Lu, C.-H.; Liu, H.-X.; Hou, T.; Lu, Z.-J.; Kong, N.; Peng, X.-Y.; Lin, K.-X.; et al. Increase of ALCAM and VCAM-1 in the Plasma Predicts the Alzheimer’s Disease. Front. Immunol. 2023, 13, 1097409. [Google Scholar] [CrossRef]
- Müller, N. The Role of Intercellular Adhesion Molecule-1 in the Pathogenesis of Psychiatric Disorders. Front. Pharmacol. 2019, 10, 1251. [Google Scholar] [CrossRef]
- Drake, J.D.; Chambers, A.B.; Ott, B.R.; Daiello, L.A. Peripheral Markers of Vascular Endothelial Dysfunction Show Independent but Additive Relationships with Brain-Based Biomarkers in Association with Functional Impairment in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 1553–1565. [Google Scholar] [CrossRef]
- Mattsson, N.; Cullen, N.C.; Andreasson, U.; Zetterberg, H.; Blennow, K. Association Between Longitudinal Plasma Neurofilament Light and Neurodegeneration in Patients with Alzheimer Disease. JAMA Neurol. 2019, 76, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Gräber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum Neurofilament Dynamics Predicts Neurodegeneration and Clinical Progression in Presymptomatic Alzheimer’s Disease. Nat. Med. 2019, 25, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Stomrud, E.; Palmqvist, S.; Zetterberg, H.; van Westen, D.; Jeromin, A.; Song, L.; Hanlon, D.; Tan Hehir, C.A.; Baker, D.; et al. Plasma β-Amyloid in Alzheimer’s Disease and Vascular Disease. Sci. Rep. 2016, 6, 26801. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.; Blennow, K.; Alzheimer’s Disease Neuroimaging Initiative; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N.; et al. Prediction of Future Alzheimer’s Disease Dementia Using Plasma Phospho-Tau Combined with Other Accessible Measures. Nat. Med. 2021, 27, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased Synaptic Proteins in Neuronal Exosomes of Frontotemporal Dementia and Alzheimer’s Disease. FASEB J. 2016, 30, 4141–4148. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Nwosu, A.; Ang, T.F.A.; Liu, Y.; Devine, S.; Au, R.; Doraiswamy, P.M. Sex-Specific Biomarkers in Alzheimer’s Disease Progression: Framingham Heart Study. Alzheimer’s Dement. 2022, 14, e12369. [Google Scholar] [CrossRef]
- Gao, W.; Xu, Y.; Liang, J.; Sun, Y.; Zhang, Y.; Shan, F.; Ge, J.; Xia, Q. Comparison of Serum Cytokines Levels in Normal-Weight and Overweight Patients with First-Episode Drug-Naïve Major Depressive Disorder. Front. Endocrinol. 2022, 13, 1048337. [Google Scholar] [CrossRef]
- Chandrashekara, S.; Jayashree, K.; Veeranna, H.B.; Vadiraj, H.S.; Ramesh, M.N.; Shobha, A.; Sarvanan, Y.; Vikram, Y.K. Effects of Anxiety on TNF-α Levels during Psychological Stress. J. Psychosom. Res. 2007, 63, 65–69. [Google Scholar] [CrossRef]
- Dunlay, S.M.; Weston, S.A.; Redfield, M.M.; Killian, J.M.; Roger, V.L. Tumor Necrosis Factor Alpha (TNFα) and Mortality in Heart Failure: A Community Study. Circulation 2008, 118, 625–631. [Google Scholar] [CrossRef]
- Alzamil, H. Elevated Serum TNF-α Is Related to Obesity in Type 2 Diabetes Mellitus and Is Associated with Glycemic Control and Insulin Resistance. J. Obes. 2020, 2020, 5076858. [Google Scholar] [CrossRef]
- Avdagić, N.; Babić, N.; Šeremet, M.; Delić-Šarac, M.; Drače, Z.; Denjalić, A.; Nakaš-Ićindić, E. Tumor Necrosis Factor-Alpha Serum Level in Assessment of Disease Activity in Inflammatory Bowel Diseases. Med. Glas 2013, 10, 211–216. [Google Scholar]
- Cacabelos, R.; Franco-Maside, A.; Alvarez, X.A. Interleukin-1 in Alzheimer’s Disease and Multi-Infarct Dementia: Neuropsychological Correlations. Methods Find Exp. Clin. Pharmacol. 1991, 13, 703–708. [Google Scholar] [PubMed]
- Tang, Z.; Ye, G.; Chen, X.; Pan, M.; Fu, J.; Fu, T.; Liu, Q.; Gao, Z.; Baldwin, D.S.; Hou, R. Peripheral Proinflammatory Cytokines in Chinese Patients with Generalised Anxiety Disorder. J. Affect Disord. 2018, 225, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Klimontov, V.V.; Mavlianova, K.R.; Orlov, N.B.; Semenova, J.F.; Korbut, A.I. Serum Cytokines and Growth Factors in Subjects with Type 1 Diabetes: Associations with Time in Ranges and Glucose Variability. Biomedicines 2023, 11, 2843. [Google Scholar] [CrossRef] [PubMed]
- Vounotrypidis, P.; Kouklakis, G.; Anagnostopoulos, K.; Zezos, P.; Polychronidis, A.; Maltezos, E.; Efremidou, E.; Pitiakoudis, M.; Lyratzopoulos, N. Interleukin-1 Associations in Inflammatory Bowel Disease and the Enteropathic Seronegative Spondylarthritis. Auto Immun. Highlights 2013, 4, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Lee, K.J.; Kim, J.Y.; Kim, H. The Association of Blood-Based Inflammatory Factors IL-1β, TGF-β and CRP with Cognitive Function in Alzheimer’s Disease and Mild Cognitive Impairment. Psychiatry Investig. 2021, 18, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ogłodek, E. Changes in the Serum Levels of Cytokines: IL-1β, IL-4, IL-8 and IL-10 in Depression with and without Posttraumatic Stress Disorder. Brain Sci. 2022, 12, 387. [Google Scholar] [CrossRef]
- Shan, L.-L.; Wang, Y.-L.; Qiao, T.-C.; Bian, Y.-F.; Huo, Y.-J.; Guo, C.; Liu, Q.-Y.; Yang, Z.-D.; Li, Z.-Z.; Liu, M.-Y.; et al. Association of Serum Interleukin-8 and Serum Amyloid A With Anxiety Symptoms in Patients With Cerebral Small Vessel Disease. Front. Neurol. 2022, 13, 938655. [Google Scholar] [CrossRef]
- Liu, S.; Wang, C.; Guo, J.; Yang, Y.; Huang, M.; Li, L.; Wang, Y.; Qin, Y.; Zhang, M. Serum Cytokines Predict the Severity of Coronary Artery Disease Without Acute Myocardial Infarction. Front. Cardiovasc. Med. 2022, 9, 896810. [Google Scholar] [CrossRef]
- Nishuty, N.L.; Khandoker, M.M.H.; Karmoker, J.R.; Ferdous, S.; Shahriar, M.; Qusar, M.M.A.S.; Islam, M.S.; Kadir, M.F.; Islam, M.R. Evaluation of Serum Interleukin-6 and C-Reactive Protein Levels in Drug-Naïve Major Depressive Disorder Patients. Cureus 2019, 11, e3868. [Google Scholar] [CrossRef]
- Mavropoulou, E.; Mechie, N.-C.; Knoop, R.; Petzold, G.; Ellenrieder, V.; Kunsch, S.; Pilavakis, Y.; Amanzada, A. Association of Serum Interleukin-6 and Soluble Interleukin-2-Receptor Levels with Disease Activity Status in Patients with Inflammatory Bowel Disease: A Prospective Observational Study. PLoS ONE 2020, 15, e0233811. [Google Scholar] [CrossRef] [PubMed]
- Hesse, R.; Wahler, A.; Gummert, P.; Kirschmer, S.; Otto, M.; Tumani, H.; Lewerenz, J.; Schnack, C.; von Arnim, C.A.F. Decreased IL-8 Levels in CSF and Serum of AD Patients and Negative Correlation of MMSE and IL-1β. BMC Neurol. 2016, 16, 185. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Feng, R.; Yang, Y. Changes in the Serum Levels of Inflammatory Cytokines in Antidepressant Drug-Naïve Patients with Major Depression. PLoS ONE 2018, 13, e0197267. [Google Scholar] [CrossRef] [PubMed]
- Bertani, L.; Caviglia, G.P.; Antonioli, L.; Pellicano, R.; Fagoonee, S.; Astegiano, M.; Saracco, G.M.; Bugianesi, E.; Blandizzi, C.; Costa, F.; et al. Serum Interleukin-6 and -8 as Predictors of Response to Vedolizumab in Inflammatory Bowel Diseases. J. Clin. Med. 2020, 9, 1323. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wu, G. The Clinical Significance of Serum IL-33 and sST2 Alterations in the Post-Stroke Depression. J. Multidiscip. Health 2021, 14, 2009–2015. [Google Scholar] [CrossRef] [PubMed]
- Fhaid, N.W.; Mostafa, T.M.; Ibrahim, O.M.; Ashmawy, M.M. Correlation Between sST2, IL-34 and Mortality in CHF Egyptian Patients. Int. J. Pharm. Sci. Rev. Res. 2019, 54, 37–44. [Google Scholar]
- Demyanets, S.; Kaun, C.; Kaider, A.; Speidl, W.; Prager, M.; Oravec, S.; Hohensinner, P.; Wojta, J.; Rega-Kaun, G. The Pro-Inflammatory Marker Soluble Suppression of Tumorigenicity-2 (ST2) Is Reduced Especially in Diabetic Morbidly Obese Patients Undergoing Bariatric Surgery. Cardiovasc. Diabetol. 2020, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Jiménez, D.; Núñez, L.E.; Beltrán, C.J.; Candia, E.; Suazo, C.; Alvarez-Lobos, M.; González, M.-J.; Hermoso, M.A.; Quera, R. Soluble ST2: A New and Promising Activity Marker in Ulcerative Colitis. World J. Gastroenterol. 2011, 17, 2181–2190. [Google Scholar] [CrossRef]
- Toppi, E.; Sireno, L.; Lembo, M.; Banaj, N.; Messina, B.; Golesorkhtafti, S.; Spalletta, G.; Bossù, P. IL-33 and IL-10 Serum Levels Increase in MCI Patients Following Homotaurine Treatment. Front. Immunol. 2022, 13, 813951. [Google Scholar] [CrossRef]
- Ogłodek, E.A.; Just, M.J. The Association between Inflammatory Markers (iNOS, HO-1, IL-33, MIP-1β) and Depression with and without Posttraumatic Stress Disorder. Pharmacol. Rep. 2018, 70, 1065–1072. [Google Scholar] [CrossRef]
- Segiet, O.A.; Romuk, E.; Nowalany-Kozielska, E.; Wojciechowska, C.; Piecuch, A.; Wojnicz, R. The Concentration of Interleukin-33 in Heart Failure with Reduced Ejection Fraction. Anatol. J. Cardiol. 2019, 21, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Khadanga, S.; Goel, S.K.; Majumder, S.; Baig, M.S.; Bhatia, V.; Chaudhary, N.; Saluja, R. Evaluation of Interleukin-33 & sST2 Levels in Type-2 Diabetic Mellitus Patients with or without Metabolic Syndrome. Indian J. Med. Res. 2023, 157, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Saadah, O.I.; Al-Harthi, S.E.; Al-Mughales, J.A.; Bin-Taleb, Y.Y.; Baeshen, R.S. Serum Interleukin-33 Level in Saudi Children with Inflammatory Bowel Disease. Int. J. Clin. Exp. Pathol. 2015, 8, 16000–16006. [Google Scholar] [PubMed]
- Laske, C.; Stellos, K.; Eschweiler, G.W.; Leyhe, T.; Gawaz, M. Decreased CXCL12 (SDF-1) Plasma Levels in Early Alzheimer’s Disease: A Contribution to a Deficient Hematopoietic Brain Support? J. Alzheimers Dis. 2008, 15, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Ogłodek, E.A.; Szota, A.; Just, M.J.; Moś, D.; Araszkiewicz, A. Comparison of Chemokines (CCL-5 and SDF-1), Chemokine Receptors (CCR-5 and CXCR-4) and IL-6 Levels in Patients with Different Severities of Depression. Pharmacol. Rep. 2014, 66, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Almuwaqqat, Z.; Martini, A.; Liu, C.; Ko, Y.-A.; Sullivan, S.; Dong, T.; Shah, A.J.; Bremner, J.D.; Pearce, B.D.; et al. Mental Stress-Induced Change in Plasma Stromal Cell-Derived Factor-1 and Adverse Cardiovascular Outcomes: A Cohort Study. CJC Open 2023, 5, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Liu, C.; Aviv, A.; Ho, J.E.; Courchesne, P.; Muntendam, P.; Larson, M.G.; Cheng, S.; Wang, T.J.; Mehta, N.N.; et al. Stromal Cell-Derived Factor 1 as a Biomarker of Heart Failure and Mortality Risk. Arter. Thromb. Vasc. Biol. 2014, 34, 2100–2105. [Google Scholar] [CrossRef]
- Derakhshan, R.; Arababadi, M.K.; Ahmadi, Z.; Karimabad, M.N.; Salehabadi, V.A.; Abedinzadeh, M.; Khorramdelazad, H.; Balaei, P.; Kennedy, D.; Hassanshahi, G. Increased Circulating Levels of SDF-1 (CXCL12) in Type 2 Diabetic Patients Are Correlated to Disease State but Are Unrelated to Polymorphism of the SDF-1β Gene in the Iranian Population. Inflammation 2012, 35, 900–904. [Google Scholar] [CrossRef]
- Almeida, M.R.; Baldeiras, I.; Ribeiro, M.H.; Santiago, B.; Machado, C.; Massano, J.; Guimarães, J.; Resende Oliveira, C.; Santana, I. Progranulin Peripheral Levels as a Screening Tool for the Identification of Subjects with Progranulin Mutations in a Portuguese Cohort. Neurodegener. Dis. 2014, 13, 214–223. [Google Scholar] [CrossRef]
- Pierce, M.E.; Hayes, J.; Huber, B.R.; Jeromin, A.; Fortier, C.B.; Fonda, J.R.; Lasseter, H.; Chaby, L.; McGlinchey, R.; Milberg, W. Plasma Biomarkers Associated with Deployment Trauma and Its Consequences in Post-9/11 Era Veterans: Initial Findings from the TRACTS Longitudinal Cohort. Transl. Psychiatry 2022, 12, 80. [Google Scholar] [CrossRef]
- Nádró, B.; Lőrincz, H.; Juhász, L.; Szentpéteri, A.; Sztanek, F.; Varga, É.; Páll, D.; Paragh, G.; Harangi, M. Determination of Serum Progranulin in Patients with Untreated Familial Hypercholesterolemia. Biomedicines 2022, 10, 771. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, B.; Li, H.; Liu, J.; Du, J.; Zang, W.; Wu, S.; Sun, H. Serum Levels of Progranulin Are Closely Associated with Microvascular Complication in Type 2 Diabetes. Dis. Markers 2015, 2015, 357279. [Google Scholar] [CrossRef] [PubMed]
- Hosny, S.S.; Bahaaeldin, A.M.; Khater, M.S.; Bekhet, M.M.; Hebah, H.A.; Hasanin, G.A. Role of Inflammatory Markers in Elderly Type 2 Diabetic Patients with Mild Cognitive Impairment. Curr. Diabetes Rev. 2019, 15, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Tchalla, A.E.; Wellenius, G.A.; Sorond, F.A.; Travison, T.G.; Dantoine, T.; Lipsitz, L.A. Elevated Circulating Vascular Cell Adhesion Molecule-1 (sVCAM-1) Is Associated with Concurrent Depressive Symptoms and Cerebral White Matter Hyperintensities in Older Adults. BMC Geriatr. 2015, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Sumner, J.A.; Chen, Q.; Roberts, A.L.; Winning, A.; Rimm, E.B.; Gilsanz, P.; Glymour, M.M.; Tworoger, S.S.; Koenen, K.C.; Kubzansky, L.D. Posttraumatic Stress Disorder Onset and Inflammatory and Endothelial Function Biomarkers in Women. Brain Behav. Immun. 2018, 69, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Lino, D.O.C.; Freitas, I.A.; Meneses, G.C.; Martins, A.M.C.; Daher, E.F.; Rocha, J.H.C.; Silva, G.B. Interleukin-6 and Adhesion Molecules VCAM-1 and ICAM-1 as Biomarkers of Post-Acute Myocardial Infarction Heart Failure. Braz. J. Med. Biol. Res. 2019, 52, e8658. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, R.; Iantorno, M.; Quon, M.J. An Integrated View of Insulin Resistance and Endothelial Dysfunction. Endocrinol. Metab. Clin. N. Am. 2008, 37, 685–711. [Google Scholar] [CrossRef]
- Battat, R.; Dulai, P.S.; Vande Casteele, N.; Evans, E.; Hester, K.D.; Webster, E.; Jain, A.; Proudfoot, J.A.; Mairalles, A.; Neill, J.; et al. Biomarkers Are Associated With Clinical and Endoscopic Outcomes With Vedolizumab Treatment in Ulcerative Colitis. Inflamm. Bowel Dis. 2019, 25, 410–420. [Google Scholar] [CrossRef]
- Rentzos, M.; Michalopoulou, M.; Nikolaou, C.; Cambouri, C.; Rombos, A.; Dimitrakopoulos, A.; Vassilopoulos, D. The Role of Soluble Intercellular Adhesion Molecules in Neurodegenerative Disorders. J. Neurol. Sci. 2005, 228, 129–135. [Google Scholar] [CrossRef]
- Bagg, W.; Ferri, C.; Desideri, G.; Gamble, G.; Ockelford, P.; Braatvedt, G.D. The Influences of Obesity and Glycemic Control on Endothelial Activation in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 5491–5497. [Google Scholar] [CrossRef]
- Vermunt, L.; Otte, M.; Verberk, I.M.W.; Killestein, J.; Lemstra, A.W.; van der Flier, W.M.; Pijnenburg, Y.A.L.; Vijverberg, E.G.B.; Bouwman, F.H.; Gravesteijn, G.; et al. Age- and Disease-Specific Reference Values for Neurofilament Light Presented in an Online Interactive Support Interface. Ann. Clin. Transl. Neurol. 2022, 9, 1832–1837. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-H.; Liu, Y.-L.; Kuo, H.-W.; Tsai, S.-J.; Hsu, J.-W.; Huang, K.-L.; Tu, P.-C.; Bai, Y.-M. Neurofilament Light Chain Is a Novel Biomarker for Major Depression and Related Executive Dysfunction. Int. J. Neuropsychopharmacol. 2022, 25, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.-Y.; Liu, P.-P.; Li, J.; Li, Y.-Q.; Zhang, L.-J.; Chen, G.-H.; Dong, F.-Y.; Hu, D.; Zhang, M. Changes in Telomere Length and Serum Neurofilament Light Chain Levels in Female Patients with Chronic Insomnia Disorder. J. Clin. Sleep Med. 2022, 18, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Korley, F.K.; Goldstick, J.; Mastali, M.; Van Eyk, J.E.; Barsan, W.; Meurer, W.J.; Sussman, J.; Falk, H.; Levine, D. Serum Neurofilament Light Chain Levels and Incident Stroke In Adults with Diabetes. Stroke 2019, 50, 1669–1675. [Google Scholar] [CrossRef]
- Thota, R.N.; Chatterjee, P.; Pedrini, S.; Hone, E.; Ferguson, J.J.A.; Garg, M.L.; Martins, R.N. Association of Plasma Neurofilament Light Chain With Glycaemic Control and Insulin Resistance in Middle-Aged Adults. Front. Endocrinol. 2022, 13, 915449. [Google Scholar] [CrossRef] [PubMed]
- Canturk, I.B.; Kalkan, A.; Es, A.K.; Bozan, O.; Unver, S.S.; Senturk, M.; Ferhatlar, M.E.; Tayfun, B.D. Serum Neurogranin Measurement as a Biomarker of Central Nervous System Infections: A Preliminary Study. Keio J. Med 2022, 71, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Bruno, D.; Reichert Plaska, C.; Clark, D.P.A.; Zetterberg, H.; Blennow, K.; Verbeek, M.M.; Pomara, N. CSF α-Synuclein Correlates with CSF Neurogranin in Late-Life Depression. Int. J. Neurosci. 2021, 131, 357–361. [Google Scholar] [CrossRef]
- Nakamura, T.; Kawarabayashi, T.; Nakahata, N.; Itoh, K.; Ihara, K.; Nakaji, S.; Ikeda, Y.; Takatama, M.; Shoji, M. Annual Stability of the Plasma Aß40/42 Ratio and Associated Factors. Ann. Clin. Transl. Neurol. 2023, 10, 879–891. [Google Scholar] [CrossRef]
- Liu, F.-R.; Yang, L.-Y.; Zheng, H.-F.; Zhou, Y.; Chen, B.-B.; Xu, H.; Zhang, Y.-W.; Shen, D.-Y. Plasma Levels of Interleukin 18 but Not Amyloid-β or Tau Are Elevated in Female Depressive Patients. Compr. Psychiatry 2020, 97, 152159. [Google Scholar] [CrossRef]
- Nam, E.; Lee, Y.-B.; Moon, C.; Chang, K.-A. Serum Tau Proteins as Potential Biomarkers for the Assessment of Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2020, 21, 5007. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tonk, S.; Kumar, S.; Vijayan, M.; Kandimalla, R.; Kuruva, C.S.; Reddy, A.P. A Critical Evaluation of Neuroprotective and Neurodegenerative MicroRNAs in Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 2017, 483, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, S.; Bramanti, P.; Mazzon, E. Role of miRNAs in Alzheimer’s Disease and Possible Fields of Application. Int. J. Mol. Sci. 2019, 20, 3979. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Fan, M.; Zheng, Q.; Hao, S.; Yang, L.; Xia, Q.; Qi, C.; Ge, J. MicroRNAs in Alzheimer’s Disease: Potential Diagnostic Markers and Therapeutic Targets. Biomed. Pharmacother. 2022, 148, 112681. [Google Scholar] [CrossRef]
- Tan, X.; Luo, Y.; Pi, D.; Xia, L.; Li, Z.; Tu, Q. MiR-340 Reduces the Accumulation of Amyloid-β Through Targeting BACE1 (β-Site Amyloid Precursor Protein Cleaving Enzyme 1) in Alzheimer’s Disease. Curr. Neurovasc. Res. 2020, 17, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Wang, X.; Cai, J.; Du, X.; Sun, H.; Zhang, N. MiR-22-3p Regulates Amyloid β Deposit in Mice Model of Alzheimer’s Disease by Targeting Mitogen-Activated Protein Kinase 14. Curr. Neurovasc. Res. 2019, 16, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Liu, Z.; Sun, G. Diagnostic Value of miR-193a-3p in Alzheimer’s Disease and miR-193a-3p Attenuates Amyloid-β Induced Neurotoxicity by Targeting PTEN. Exp. Gerontol. 2020, 130, 110814. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, W.; Ge, H.; Li, K. Aberrant Expression of miR-148a-3p in Alzheimer’s Disease and Its Protective Role against Amyloid-β Induced Neurotoxicity. Neurosci. Lett. 2021, 756, 135953. [Google Scholar] [CrossRef]
- Fu, Y.; Hu, X.; Zheng, C.; Sun, G.; Xu, J.; Luo, S.; Cao, P. Intrahippocampal miR-342-3p Inhibition Reduces β-Amyloid Plaques and Ameliorates Learning and Memory in Alzheimer’s Disease. Metab. Brain Dis. 2019, 34, 1355–1363. [Google Scholar] [CrossRef]
- Koronyo, Y.; Rentsendorj, A.; Mirzaei, N.; Regis, G.C.; Sheyn, J.; Shi, H.; Barron, E.; Cook-Wiens, G.; Rodriguez, A.R.; Medeiros, R.; et al. Retinal Pathological Features and Proteome Signatures of Alzheimer’s Disease. Acta Neuropathol. 2023, 145, 409–438. [Google Scholar] [CrossRef]
- den Haan, J.; Verbraak, F.D.; Visser, P.J.; Bouwman, F.H. Retinal Thickness in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Alzheimers Dement. 2017, 6, 162–170. [Google Scholar] [CrossRef]
- Querques, G.; Borrelli, E.; Sacconi, R.; De Vitis, L.; Leocani, L.; Santangelo, R.; Magnani, G.; Comi, G.; Bandello, F. Functional and Morphological Changes of the Retinal Vessels in Alzheimer’s Disease and Mild Cognitive Impairment. Sci. Rep. 2019, 9, 63. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Mok, V.; Foster, P.J.; Trucco, E.; Chen, C.; Wong, T.Y. Retinal Imaging in Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 2021, 92, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Elahi, F.M.; Ashimatey, S.B.; Bennett, D.J.; Walters, S.M.; La Joie, R.; Jiang, X.; Wolf, A.; Cobigo, Y.; Staffaroni, A.M.; Rosen, H.J.; et al. Retinal Imaging Demonstrates Reduced Capillary Density in Clinically Unimpaired APOE Ε4 Gene Carriers. Alzheimers Dement. 2021, 13, e12181. [Google Scholar] [CrossRef] [PubMed]
- Mavilio, A.; Sisto, D.; Prete, F.; Guadalupi, V.; Dammacco, R.; Alessio, G. RE-PERG in Early-Onset Alzheimer’s Disease: A Double-Blind, Electrophysiological Pilot Study. PLoS ONE 2020, 15, e0236568. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, X.; Xiao, Y.; Zhao, Q. Retinal Examination Modalities in the Early Detection of Alzheimer’s Disease: Seeing Brain Through the Eye. J. Transl. Int. Med. 2022, 10, 185–187. [Google Scholar] [CrossRef]
- How Is Alzheimer’s Disease Diagnosed? Available online: https://www.nia.nih.gov/health/how-alzheimers-disease-diagnosed (accessed on 11 October 2023).
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The Genetic Landscape of Alzheimer Disease: Clinical Implications and Perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef]
- KHANAHMADI, M.; FARHUD, D.D.; MALMIR, M. Genetic of Alzheimer’s Disease: A Narrative Review Article. Iran J. Public Health 2015, 44, 892–901. [Google Scholar]
- Gharbi-Meliani, A.; Dugravot, A.; Sabia, S.; Regy, M.; Fayosse, A.; Schnitzler, A.; Kivimäki, M.; Singh-Manoux, A.; Dumurgier, J. The Association of APOE Ε4 with Cognitive Function over the Adult Life Course and Incidence of Dementia: 20 Years Follow-up of the Whitehall II Study. Alzheimers Res. Ther. 2021, 13, 5. [Google Scholar] [CrossRef]
- Hu, M.L.; Quinn, J.; Xue, K. Interactions between Apolipoprotein E Metabolism and Retinal Inflammation in Age-Related Macular Degeneration. Life 2021, 11, 635. [Google Scholar] [CrossRef]
- Simmons, K.T.; Mazzilli, J.L.; Mueller-Ortiz, S.L.; Domozhirov, A.Y.; Garcia, C.A.; Zsigmond, E.M.; Wetsel, R.A. Complement Receptor 1 (CR1/CD35)-Expressing Retinal Pigment Epithelial Cells as a Potential Therapy for Age-Related Macular Degeneration. Mol. Immunol. 2020, 118, 91–98. [Google Scholar] [CrossRef]
- Ho, W.-L.; Leung, Y.; Tsang, A.W.-T.; So, K.-F.; Chiu, K.; Chang, R.C.-C. Review: Tauopathy in the Retina and Optic Nerve: Does It Shadow Pathological Changes in the Brain? Mol. Vis. 2012, 18, 2700–2710. [Google Scholar] [PubMed]
- Yoneda, S.; Hara, H.; Hirata, A.; Fukushima, M.; Inomata, Y.; Tanihara, H. Vitreous Fluid Levels of Beta-Amyloid((1-42)) and Tau in Patients with Retinal Diseases. Jpn J. Ophthalmol. 2005, 49, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein-Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Bergmans, B.A.; De Strooper, B. Gamma-Secretases: From Cell Biology to Therapeutic Strategies. Lancet Neurol. 2010, 9, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; An, S.S.A.; Kim, S. Mutations in Presenilin 2 and Its Implications in Alzheimer’s Disease and Other Dementia-Associated Disorders. Clin. Interv. Aging 2015, 10, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Cai, F.; Sun, Z.; Zhang, Y.; Wang, J.; Jiao, B.; Guo, J.; Li, J.; Liu, X.; Guo, L.; et al. Identification of Alzheimer’s Disease-Associated Rare Coding Variants in the ECE2 Gene. JCI Insight 2020, 5, e135119. [Google Scholar] [CrossRef]
- Bullido, M.J.; Ramos, M.C.; Ruiz-Gómez, A.; Tutor, A.S.; Sastre, I.; Frank, A.; Coria, F.; Gil, P.; Mayor, F.; Valdivieso, F. Polymorphism in Genes Involved in Adrenergic Signaling Associated with Alzheimer’s. Neurobiol. Aging 2004, 25, 853–859. [Google Scholar] [CrossRef]
- Zhu, X.-C.; Yu, J.-T.; Jiang, T.; Wang, P.; Cao, L.; Tan, L. CR1 in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 753–765. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.; Singh Pahwa, J.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-Wide Association Study Identifies Variants at CLU and PICALM Associated with Alzheimer’s Disease, and Shows Evidence for Additional Susceptibility Genes. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Shen, R.; Murphy, C.J.; Xu, X.; Hu, M.; Ding, J.; Wu, C. Ras and Rab Interactor 3: From Cellular Mechanisms to Human Diseases. Front. Cell Dev. Biol. 2022, 10, 824961. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Protective Variants in Alzheimer’s Disease. Curr. Genet. Med. Rep. 2019, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.P.; Lin, P.B.-C.; Dong, C.; Moutinho, M.; Casali, B.T.; Liu, Y.; Lamb, B.T.; Landreth, G.E.; Oblak, A.L.; Nho, K. INPP5D Expression Is Associated with Risk for Alzheimer’s Disease and Induced by Plaque-Associated Microglia. Neurobiol. Dis. 2021, 153, 105303. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Risacher, S.L.; Nho, K.; Saykin, A.J.; Wang, Z.; Shen, L. Targeted Genetic Analysis of Cerebral Blood Flow Imaging Phenotypes Implicates the INPP5D Gene. Neurobiol. Aging 2019, 81, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lopez, O.L.; Sweet, R.A.; Becker, J.T.; DeKosky, S.T.; Barmada, M.M.; Demirci, F.Y.; Kamboh, M.I. Genetic Determinants of Disease Progression in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 43, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Zhu, J.-X.; Wang, H.-F.; Zhang, W.; Zheng, Z.-J.; Kong, L.-L.; Tan, C.-C.; Wang, Z.-X.; Tan, L.; Tan, L. INPP5D Rs35349669 Polymorphism with Late-Onset Alzheimer’s Disease: A Replication Study and Meta-Analysis. Oncotarget 2016, 7, 69225–69230. [Google Scholar] [CrossRef]
- Samuels, J.D.; Moore, K.A.; Ennerfelt, H.E.; Johnson, A.M.; Walsh, A.E.; Price, R.J.; Lukens, J.R. The Alzheimer’s Disease Risk Factor INPP5D Restricts Neuroprotective Microglial Responses in Amyloid Beta-Mediated Pathology. Alzheimer’s Dement. 2023, 19, 4908–4921. [Google Scholar] [CrossRef]
- Soler-López, M.; Badiola, N.; Zanzoni, A.; Aloy, P. Towards Alzheimer’s Root Cause: ECSIT as an Integrating Hub between Oxidative Stress, Inflammation and Mitochondrial Dysfunction. Hypothetical Role of the Adapter Protein ECSIT in Familial and Sporadic Alzheimer’s Disease Pathogenesis. Bioessays 2012, 34, 532–541. [Google Scholar] [CrossRef]
- Rosenthal, S.L.; Kamboh, M.I. Late-Onset Alzheimer’s Disease Genes and the Potentially Implicated Pathways. Curr. Genet. Med. Rep. 2014, 2, 85–101. [Google Scholar] [CrossRef]
- Tan, M.-S.; Yang, Y.-X.; Xu, W.; Wang, H.-F.; Tan, L.; Zuo, C.-T.; Dong, Q.; Tan, L.; Suckling, J.; Yu, J.-T. Associations of Alzheimer’s Disease Risk Variants with Gene Expression, Amyloidosis, Tauopathy, and Neurodegeneration. Alzheimers Res. Ther. 2021, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Gao, Z.; Jiang, X.; Jiang, L.; Qin, Y.; Zhang, D.; Tian, P.; Wang, W.; Zhang, Q.; Zhang, R.; et al. Alzheimer’s Risk Factor FERMT2 Promotes the Progression of Colorectal Carcinoma via Wnt/β-Catenin Signaling Pathway and Contributes to the Negative Correlation between Alzheimer and Cancer. PLoS ONE 2022, 17, e0278774. [Google Scholar] [CrossRef] [PubMed]
- Eysert, F.; Coulon, A.; Boscher, E.; Vreulx, A.-C.; Flaig, A.; Mendes, T.; Hughes, S.; Grenier-Boley, B.; Hanoulle, X.; Demiautte, F.; et al. Alzheimer’s Genetic Risk Factor FERMT2 (Kindlin-2) Controls Axonal Growth and Synaptic Plasticity in an APP-Dependent Manner. Mol. Psychiatry 2021, 26, 5592–5607. [Google Scholar] [CrossRef] [PubMed]
- Stage, E.; Risacher, S.L.; Lane, K.A.; Gao, S.; Nho, K.; Saykin, A.J.; Apostolova, L.G. Association of the Top 20 Alzheimer’s Disease Risk Genes with [18F]Flortaucipir PET. Alzheimers Dement. 2022, 14, e12308. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, L.G.; Risacher, S.L.; Duran, T.; Stage, E.C.; Goukasian, N.; West, J.D.; Do, T.M.; Grotts, J.; Wilhalme, H.; Nho, K.; et al. Associations of the Top 20 Alzheimer Disease Risk Variants with Brain Amyloidosis. JAMA Neurol. 2018, 75, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.N.; Nicolas, E.; Kopp, M.C.; Golemis, E.A. Adaptors for Disorders of the Brain? The Cancer Signaling Proteins NEDD9, CASS4, and PTK2B in Alzheimer’s Disease. Oncoscience 2014, 1, 486–503. [Google Scholar] [CrossRef]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT Mutations, Tauopathy, and Mechanisms of Neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Coppola, G.; Chinnathambi, S.; Lee, J.J.; Dombroski, B.A.; Baker, M.C.; Soto-Ortolaza, A.I.; Lee, S.E.; Klein, E.; Huang, A.Y.; Sears, R.; et al. Evidence for a Role of the Rare p.A152T Variant in MAPT in Increasing the Risk for FTD-Spectrum and Alzheimer’s Diseases. Hum. Mol. Genet. 2012, 21, 3500–3512. [Google Scholar] [CrossRef]
- Tao, Q.-Q.; Chen, Y.-C.; Wu, Z.-Y. The Role of CD2AP in the Pathogenesis of Alzheimer’s Disease. Aging Dis. 2019, 10, 901–907. [Google Scholar] [CrossRef]
- Beecham, G.W.; Hamilton, K.; Naj, A.C.; Martin, E.R.; Huentelman, M.; Myers, A.J.; Corneveaux, J.J.; Hardy, J.; Vonsattel, J.-P.; Younkin, S.G.; et al. Genome-Wide Association Meta-Analysis of Neuropathologic Features of Alzheimer’s Disease and Related Dementias. PLoS Genet. 2014, 10, e1004606. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common Variants in MS4A4/MS4A6E, CD2uAP, CD33, and EPHA1 Are Associated with Late-Onset Alzheimer’s Disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms, and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. Role of Apolipoprotein E in Neurodegenerative Diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Hooli, B.; Mullin, K.; Jin, S.C.; Cella, M.; Ulland, T.K.; Wang, Y.; Tanzi, R.; Colonna, M. Alzheimer’s Disease-Associated TREM2 Variants Exhibit Either Decreased or Increased Ligand-Dependent Activation. Alzheimers Dement. 2017, 13, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Lupton, M.K.; Proitsi, P.; Lin, K.; Hamilton, G.; Daniilidou, M.; Tsolaki, M.; Powell, J.F. The Role of ABCA1 Gene Sequence Variants on Risk of Alzheimer’s Disease. J. Alzheimers Dis. 2014, 38, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Dib, S.; Pahnke, J.; Gosselet, F. Role of ABCA7 in Human Health and in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4603. [Google Scholar] [CrossRef]
- Lee, H.; Aylward, A.J.; Pearse, R.V.; Lish, A.M.; Hsieh, Y.-C.; Augur, Z.M.; Benoit, C.R.; Chou, V.; Knupp, A.; Pan, C.; et al. Cell-Type-Specific Regulation of APOE and CLU Levels in Human Neurons by the Alzheimer’s Disease Risk Gene SORL1. Cell Rep. 2023, 42, 112994. [Google Scholar] [CrossRef]
- Tzikas, S.; Schlak, D.; Sopova, K.; Gatsiou, A.; Stakos, D.; Stamatelopoulos, K.; Stellos, K.; Laske, C. Increased Myeloperoxidase Plasma Levels in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2014, 39, 557–564. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; St. Laurent, C.D.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 Short Isoform Is a Gain-of-Function Variant That Enhances Aβ1–42 Phagocytosis in Microglia. Mol. Neurodegener. 2021, 16, 19. [Google Scholar] [CrossRef]
- Zhao, L. CD33 in Alzheimer’s Disease—Biology, Pathogenesis, and Therapeutics: A Mini-Review. Gerontology 2018, 65, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Raj, T.; Ryan, K.J.; Replogle, J.M.; Chibnik, L.B.; Rosenkrantz, L.; Tang, A.; Rothamel, K.; Stranger, B.E.; Bennett, D.A.; Evans, D.A.; et al. CD33: Increased Inclusion of Exon 2 Implicates the Ig V-Set Domain in Alzheimer’s Disease Susceptibility. Hum. Mol. Genet. 2014, 23, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common Variants in ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP Are Associated with Alzheimer’s Disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, L.R.; Pimassoni, L.H.S.; Sena, G.G.S.; Camporez, D.; Belcavello, L.; Trancozo, M.; Morelato, R.L.; Errera, F.I.V.; Bueno, M.R.P.; de Paula, F. Validating GWAS Variants from Microglial Genes Implicated in Alzheimer’s Disease. J. Mol. Neurosci. 2017, 62, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Simpson, J.F.; Parikh, I.; Wilfred, B.R.; Fardo, D.W.; Nelson, P.T.; Estus, S. CD33 Alzheimer’s Risk-Altering Polymorphism, CD33 Expression, and Exon 2 Splicing. J. Neurosci. 2013, 33, 13320–13325. [Google Scholar] [CrossRef]
- Braskie, M.N.; Jahanshad, N.; Stein, J.L.; Barysheva, M.; McMahon, K.L.; de Zubicaray, G.I.; Martin, N.G.; Wright, M.J.; Ringman, J.M.; Toga, A.W.; et al. Common Alzheimer’s Disease Risk Variant within the CLU Gene Affects White Matter Microstructure in Young Adults. J. Neurosci. 2011, 31, 6764–6770. [Google Scholar] [CrossRef]
- Tan, L.; Wang, H.-F.; Tan, M.-S.; Tan, C.-C.; Zhu, X.-C.; Miao, D.; Yu, W.-J.; Jiang, T.; Tan, L.; Yu, J.-T. Effect of CLU Genetic Variants on Cerebrospinal Fluid and Neuroimaging Markers in Healthy, Mild Cognitive Impairment and Alzheimer’s Disease Cohorts. Sci. Rep. 2016, 6, 26027. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, J.-T.; Wang, H.-F.; Hao, X.-K.; Yang, Y.-F.; Jiang, T.; Zhu, X.-C.; Cao, L.; Zhang, D.-Q.; Tan, L. Association between NME8 Locus Polymorphism and Cognitive Decline, Cerebrospinal Fluid and Neuroimaging Biomarkers in Alzheimer’s Disease. PLoS ONE 2014, 9, e114777. [Google Scholar] [CrossRef]
- Liu, S.-L.; Wang, X.-C.; Tan, M.-S.; Wang, H.-F.; Zhang, W.; Wang, Z.-X.; Yu, J.-T.; Tan, L. NME8 Rs2718058 Polymorphism with Alzheimer’s Disease Risk: A Replication and Meta-Analysis. Oncotarget 2016, 7, 36014–36020. [Google Scholar] [CrossRef]
- Janicki, S.C.; Park, N.; Cheng, R.; Clark, L.N.; Lee, J.H.; Schupf, N. Estrogen Receptor α Variants Affect Age at Onset of Alzheimer’s Disease in a Multiethnic Female Cohort. Dement. Geriatr. Cogn. Disord. 2014, 38, 200–213. [Google Scholar] [CrossRef]
- Lacher, S.E.; Alazizi, A.; Wang, X.; Bell, D.A.; Pique-Regi, R.; Luca, F.; Slattery, M. A Hypermorphic Antioxidant Response Element Is Associated with Increased MS4A6A Expression and Alzheimer’s Disease. Redox Biol. 2017, 14, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, W.; Tan, L.; Wang, H.-F.; Wan, Y.; Sun, F.-R.; Tan, C.-C.; Yu, J.-T.; Tan, L. MS4A6A Genotypes Are Associated with the Atrophy Rates of Alzheimer’s Disease Related Brain Structures. Oncotarget 2016, 7, 58779–58788. [Google Scholar] [CrossRef] [PubMed]
- Holler, C.J.; Davis, P.R.; Beckett, T.L.; Platt, T.L.; Webb, R.L.; Head, E.; Murphy, M.P. Bridging Integrator 1 (BIN1) Protein Expression Increases in the Alzheimer’s Disease Brain and Correlates with Neurofibrillary Tangle Pathology. J. Alzheimers Dis. 2014, 42, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Perdigão, C.; Barata, M.A.; Burrinha, T.; Guimas Almeida, C. Alzheimer’s Disease BIN1 Coding Variants Increase Intracellular Aβ Levels by Interfering with BACE1 Recycling. J. Biol. Chem. 2021, 297, 101056. [Google Scholar] [CrossRef]
- Kim, M.; Suh, J.; Romano, D.; Truong, M.H.; Mullin, K.; Hooli, B.; Norton, D.; Tesco, G.; Elliott, K.; Wagner, S.L.; et al. Potential Late-Onset Alzheimer’s Disease-Associated Mutations in the ADAM10 Gene Attenuate α-Secretase Activity. Hum. Mol. Genet. 2009, 18, 3987–3996. [Google Scholar] [CrossRef]
- Brody, A.H.; Nies, S.H.; Guan, F.; Smith, L.M.; Mukherjee, B.; Salazar, S.A.; Lee, S.; Lam, T.K.T.; Strittmatter, S.M. Alzheimer Risk Gene Product Pyk2 Suppresses Tau Phosphorylation and Phenotypic Effects of Tauopathy. Mol. Neurodegener. 2022, 17, 32. [Google Scholar] [CrossRef]
- Li, Y.-Q.; Tan, M.-S.; Wang, H.-F.; Tan, C.-C.; Zhang, W.; Zheng, Z.-J.; Kong, L.-L.; Wang, Z.-X.; Tan, L.; Jiang, T.; et al. Common Variant in PTK2B Is Associated with Late-Onset Alzheimer’s Disease: A Replication Study and Meta-Analyses. Neurosci. Lett. 2016, 621, 83–87. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, Y. Progress on the Roles of MEF2C in Neuropsychiatric Diseases. Mol. Brain 2022, 15, 8. [Google Scholar] [CrossRef]
- Fyfe, I. Alzheimer Disease-Associated Gene Increases Tau Pathology. Nat. Rev. Neurol. 2020, 16, 128. [Google Scholar] [CrossRef]
- Xu, W.; Tan, C.-C.; Cao, X.-P.; Tan, L. Association of Alzheimer’s Disease Risk Variants on the PICALM Gene with PICALM Expression, Core Biomarkers, and Feature Neurodegeneration. Aging 2020, 12, 21202–21219. [Google Scholar] [CrossRef]
- Ma, J.; Wang, Z.; Chen, S.; Sun, W.; Gu, Q.; Li, D.; Zheng, J.; Yang, H.; Li, X. EphA1 Activation Induces Neuropathological Changes in a Mouse Model of Parkinson’s Disease Through the CXCL12/CXCR4 Signaling Pathway. Mol. Neurobiol. 2021, 58, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-F.; Tan, L.; Hao, X.-K.; Jiang, T.; Tan, M.-S.; Liu, Y.; Zhang, D.-Q.; Yu, J.-T. Alzheimer’s Disease Neuroimaging Initiative Effect of EPHA1 Genetic Variation on Cerebrospinal Fluid and Neuroimaging Biomarkers in Healthy, Mild Cognitive Impairment and Alzheimer’s Disease Cohorts. J. Alzheimers Dis. 2015, 44, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tan, M.-S.; Wang, H.-F.; Zhang, W.; Wang, Z.-X.; Jiang, T.; Yu, J.-T.; Tan, L. ZCWPW1 Is Associated with Late-Onset Alzheimer’s Disease in Han Chinese: A Replication Study and Meta-Analyses. Oncotarget 2016, 7, 20305–20311. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-C.; Yang, W.; Tan, L.; Sun, F.-R.; Tan, M.-S.; Zhang, W.; Wang, H.-F.; Tan, L. Association of HLA-DRB1 Polymorphism with Alzheimer’s Disease: A Replication and Meta-Analysis. Oncotarget 2017, 8, 93219–93226. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Peng, Z.; Boix, C.A.; Victor, M.B.; Leary, N.; Babu, S.; Abdelhady, G.; Jiang, X.; Ng, A.P.; Ghafari, K.; et al. Single-Cell Atlas Reveals Correlates of High Cognitive Function, Dementia, and Resilience to Alzheimer’s Disease Pathology. Cell 2023, 186, 4365–4385. [Google Scholar] [CrossRef] [PubMed]
- Murdock, M.H.; Tsai, L.-H. Insights into Alzheimer’s Disease from Single-Cell Genomic Approaches. Nat. Neurosci. 2023, 26, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Lodato, M.A.; Rodin, R.E.; Bohrson, C.L.; Coulter, M.E.; Barton, A.R.; Kwon, M.; Sherman, M.A.; Vitzthum, C.M.; Luquette, L.J.; Yandava, C.N.; et al. Aging and Neurodegeneration Are Associated with Increased Mutations in Single Human Neurons. Science 2018, 359, 555–559. [Google Scholar] [CrossRef]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.; Yamakawa, H.; Pao, P.-C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef]
- Cárdenas-Aguayo, M.D.; Silva-Lucero, M.D.C.; Cortes-Ortiz, M.; Jiménez-Ramos, B.; Gómez-Virgilio, L.; Ramírez-Rodríguez, G.; Vera- Arroyo, E.; Fiorentino-Pérez, R.; García, U.; Luna-Muñoz, J.; et al. Physiological Role of Amyloid Beta in Neural Cells: The Cellular Trophic Activity. In Neurochemistry; IntechOpen: London, UK, 2014; ISBN 978-953-51-1237-2. [Google Scholar]
- Steiner, H.; Fukumori, A.; Tagami, S.; Okochi, M. Making the Final Cut: Pathogenic Amyloid-β Peptide Generation by γ-Secretase. Cell Stress 2018, 2, 292–310. [Google Scholar] [CrossRef]
- Delabio, R.; Rasmussen, L.; Mizumoto, I.; Viani, G.-A.; Chen, E.; Villares, J.; Costa, I.-B.; Turecki, G.; Linde, S.A.; Smith, M.C.; et al. PSEN1 and PSEN2 Gene Expression in Alzheimer’s Disease Brain: A New Approach. J. Alzheimers Dis. 2014, 42, 757–760. [Google Scholar] [CrossRef]
- Leissring, M.A. Aβ-Degrading Proteases:Therapeutic Potential in Alzheimer Disease. CNS Drugs 2016, 30, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Hu, X.; Song, H.; Yin, K.; Bateman, R.J.; Cirrito, J.R.; Xiao, Q.; Hsu, F.F.; Turk, J.W.; Xu, J.; et al. Matrix Metalloproteinase-9 Degrades Amyloid-Beta Fibrils in Vitro and Compact Plaques in Situ. J. Biol. Chem. 2006, 281, 24566–24574. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Kweon, H.-S.; Cho, E.; Lee, J.-Y.; Byun, H.-R.; Kim, D.H.; Kim, Y.-H.; Han, P.-L.; Koh, J.-Y. Upregulation of tPA/Plasminogen Proteolytic System in the Periphery of Amyloid Deposits in the Tg2576 Mouse Model of Alzheimer’s Disease. Neurosci. Lett. 2007, 423, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Dresser, L.; Hunter, P.; Yendybayeva, F.; Hargreaves, A.L.; Howard, J.A.L.; Evans, G.J.O.; Leake, M.C.; Quinn, S.D. Amyloid-β Oligomerization Monitored by Single-Molecule Stepwise Photobleaching. Methods 2021, 193, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiological Brain Activity Causes DNA Double Strand Breaks in Neurons—Exacerbation by Amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Blacker, D.; Gómez-lsla, T.; Betensky, R.A.; Growdon, J.H.; Johnson, K.; Frosch, M.P.; Sperling, R.A.; et al. Mild to Moderate Alzheimer Dementia with Insufficient Neuropathological Changes. Ann. Neurol. 2014, 75, 597–601. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Saido, T.; Leissring, M.A. Proteolytic Degradation of Amyloid β-Protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef]
- Kanyenda, L.J.; Verdile, G.; Boulos, S.; Krishnaswamy, S.; Taddei, K.; Meloni, B.P.; Mastaglia, F.L.; Martins, R.N. The Dynamics of CD147 in Alzheimer’s Disease Development and Pathology. J. Alzheimers Dis. 2011, 26, 593–605. [Google Scholar] [CrossRef]
- Zhou, S.; Zhou, H.; Walian, P.J.; Jap, B.K. CD147 Is a Regulatory Subunit of the Gamma-Secretase Complex in Alzheimer’s Disease Amyloid Beta-Peptide Production. Proc. Natl. Acad. Sci. USA 2005, 102, 7499–7504. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.-W.; Murray, E.J.B.; Murray, S.S. Fibroblastic Synoviocytes Secrete Plasma Proteins via A2 -Macroglobulins Serving as Intracellular and Extracellular Chaperones. J. Cell Biochem. 2015, 116, 2563–2576. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Pîrşcoveanu, D.F.V.; Pirici, I.; Tudorică, V.; Bălşeanu, T.A.; Albu, V.C.; Bondari, S.; Bumbea, A.M.; Pîrşcoveanu, M. Tau Protein in Neurodegenerative Diseases—A Review. Rom. J. Morphol. Embryol. 2017, 58, 1141–1150. [Google Scholar] [PubMed]
- Bu, G. Apolipoprotein E and Its Receptors in Alzheimer’s Disease: Pathways, Pathogenesis and Therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 Mutations in Early-Onset Alzheimer Disease: A Genetic Screening Study of Familial and Sporadic Cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.-C. ApoE in Alzheimer’s Disease: Pathophysiology and Therapeutic Strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef]
- Michikawa, M.; Fan, Q.W.; Isobe, I.; Yanagisawa, K. Apolipoprotein E Exhibits Isoform-Specific Promotion of Lipid Efflux from Astrocytes and Neurons in Culture. J. Neurochem. 2000, 74, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid Droplet Accumulating Microglia Represent a Dysfunctional and Pro-Inflammatory State in the Aging Brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Garai, K.; Verghese, P.B.; Baban, B.; Holtzman, D.M.; Frieden, C. The Binding of Apolipoprotein E to Oligomers and Fibrils of Amyloid-β Alters the Kinetics of Amyloid Aggregation. Biochemistry 2014, 53, 6323–6331. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.-W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.; Apostolova, L.G.; Atri, A.; Salloway, S.; Weiner, M. Aducanumab: Appropriate Use Recommendations. J. Prev. Alzheimers Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Gibb, G.M.; Pearce, J.; Betts, J.C.; Lovestone, S.; Hoffmann, M.M.; Maerz, W.; Blackstock, W.P.; Anderton, B.H. Differential Effects of Apolipoprotein E Isoforms on Phosphorylation at Specific Sites on Tau by Glycogen Synthase Kinase-3 Beta Identified by Nano-Electrospray Mass Spectrometry. FEBS Lett. 2000, 485, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Goedert, M.; Weisgraber, K.H.; Dong, L.M.; Jakes, R.; Huang, D.Y.; Pericak-Vance, M.; Schmechel, D.; Roses, A.D. Isoform-Specific Interactions of Apolipoprotein E with Microtubule-Associated Protein Tau: Implications for Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1994, 91, 11183–11186. [Google Scholar] [CrossRef]
- Therriault, J.; Benedet, A.L.; Pascoal, T.A.; Mathotaarachchi, S.; Chamoun, M.; Savard, M.; Thomas, E.; Kang, M.S.; Lussier, F.; Tissot, C.; et al. Association of Apolipoprotein E Ε4 With Medial Temporal Tau Independent of Amyloid-β. JAMA Neurol. 2020, 77, 470–479. [Google Scholar] [CrossRef]
- A Armstrong, R. Risk Factors for Alzheimer’s Disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef]
- Armstrong, R.A. What Causes Alzheimer’s Disease? Folia Neuropathol. 2013, 51, 169–188. [Google Scholar] [CrossRef]
- Tittelmeier, J.; Nachman, E.; Nussbaum-Krammer, C. Molecular Chaperones: A Double-Edged Sword in Neurodegenerative Diseases. Front. Aging Neurosci. 2020, 12, 581374. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular Chaperones in Protein Folding and Proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Wankhede, N.L.; Kale, M.B.; Upaganlawar, A.B.; Taksande, B.G.; Umekar, M.J.; Behl, T.; Abdellatif, A.A.H.; Bhaskaran, P.M.; Dachani, S.R.; Sehgal, A.; et al. Involvement of Molecular Chaperone in Protein-Misfolding Brain Diseases. Biomed. Pharmacother. 2022, 147, 112647. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, M.M.M.; Boelens, W.C.; Otte-Höller, I.; Kamps, B.; de Waal, R.M.W.; Verbeek, M.M. Small Heat Shock Proteins Inhibit Amyloid-Beta Protein Aggregation and Cerebrovascular Amyloid-Beta Protein Toxicity. Brain Res. 2006, 1089, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Mulder, S.D.; Nielsen, H.M.; Blankenstein, M.A.; Eikelenboom, P.; Veerhuis, R. Apolipoproteins E and J Interfere with Amyloid-Beta Uptake by Primary Human Astrocytes and Microglia in Vitro. Glia 2014, 62, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.-A.; Condello, C.; Freilich, R.; Gillies, A.; Arhar, T.; Oroz, J.; Kadavath, H.; Julien, O.; Assimon, V.A.; Rauch, J.N.; et al. Mapping Interactions with the Chaperone Network Reveals Factors That Protect against Tau Aggregation. Nat. Struct. Mol. Biol. 2018, 25, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.R.; Ward, S.M.; Combs, B.; Voss, K.; Kanaan, N.M.; Morfini, G.; Brady, S.T.; Gamblin, T.C.; Binder, L.I. Heat Shock Protein 70 Prevents Both Tau Aggregation and the Inhibitory Effects of Preexisting Tau Aggregates on Fast Axonal Transport. Biochemistry 2011, 50, 10300–10310. [Google Scholar] [CrossRef]
- Eroglu, B.; Moskophidis, D.; Mivechi, N.F. Loss of Hsp110 Leads to Age-Dependent Tau Hyperphosphorylation and Early Accumulation of Insoluble Amyloid Beta. Mol. Cell Biol. 2010, 30, 4626–4643. [Google Scholar] [CrossRef]
- Nachman, E.; Wentink, A.S.; Madiona, K.; Bousset, L.; Katsinelos, T.; Allinson, K.; Kampinga, H.; McEwan, W.A.; Jahn, T.R.; Melki, R.; et al. Disassembly of Tau Fibrils by the Human Hsp70 Disaggregation Machinery Generates Small Seeding-Competent Species. J. Biol. Chem. 2020, 295, 9676–9690. [Google Scholar] [CrossRef]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C.; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated Neurodegeneration through Chaperone-Mediated Oligomerization of Tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [PubMed]
- Campanella, C.; Pace, A.; Caruso Bavisotto, C.; Marzullo, P.; Marino Gammazza, A.; Buscemi, S.; Palumbo Piccionello, A. Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial Defects and Oxidative Stress in Alzheimer Disease and Parkinson Disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Marino Gammazza, A.; Bavisotto, C.C.; Barone, R.; de Macario, E.C.; Macario, A.J.L. Alzheimer’s Disease and Molecular Chaperones: Current Knowledge and the Future of Chaperonotherapy. Curr. Pharm. Des. 2016, 22, 4040–4049. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, T.; Ma, D.; Li, H.; Hua, L.; He, Q.; Deng, X. The Impact of Air Pollution on Neurodegenerative Diseases. Ther. Drug Monit. 2021, 43, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.R.; Wang, M.-X. APP Expression, Distribution and Accumulation Are Altered by Aluminum in a Rodent Model for Alzheimer’s Disease. J. Inorg. Biochem. 2009, 103, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Sanders, T.; Liu, Y.; Buchner, V.; Tchounwou, P.B. Neurotoxic Effects and Biomarkers of Lead Exposure: A Review. Rev. Env. Health 2009, 24, 15–45. [Google Scholar] [CrossRef]
- Zhou, C.-C.; Gao, Z.-Y.; Wang, J.; Wu, M.-Q.; Hu, S.; Chen, F.; Liu, J.-X.; Pan, H.; Yan, C.-H. Lead Exposure Induces Alzheimers’s Disease (AD)-like Pathology and Disturbes Cholesterol Metabolism in the Young Rat Brain. Toxicol. Lett. 2018, 296, 173–183. [Google Scholar] [CrossRef]
- Notarachille, G.; Arnesano, F.; Calò, V.; Meleleo, D. Heavy Metals Toxicity: Effect of Cadmium Ions on Amyloid Beta Protein 1-42. Possible Implications for Alzheimer’s Disease. Biometals 2014, 27, 371–388. [Google Scholar] [CrossRef]
- Cristofori, F.; Dargenio, V.N.; Dargenio, C.; Miniello, V.L.; Barone, M.; Francavilla, R. Anti-Inflammatory and Immunomodulatory Effects of Probiotics in Gut Inflammation: A Door to the Body. Front. Immunol. 2021, 12, 578386. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Lukiw, W.J. Alzheimer’s Disease and the Microbiome. Front. Cell Neurosci. 2013, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Mitew, S.; Kirkcaldie, M.T.K.; Dickson, T.C.; Vickers, J.C. Altered Synapses and Gliotransmission in Alzheimer’s Disease and AD Model Mice. Neurobiol. Aging 2013, 34, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.-Q.; Shen, L.-L.; Li, W.-W.; Fu, X.; Zeng, F.; Gui, L.; Lü, Y.; Cai, M.; Zhu, C.; Tan, Y.-L.; et al. Gut Microbiome Is Altered in Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 1–10. [Google Scholar] [CrossRef]
- Liu, P.; Wu, L.; Peng, G.; Han, Y.; Tang, R.; Ge, J.; Zhang, L.; Jia, L.; Yue, S.; Zhou, K.; et al. Altered Microbiomes Distinguish Alzheimer’s Disease from Amnestic Mild Cognitive Impairment and Health in a Chinese Cohort. Brain Behav. Immun. 2019, 80, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, J.; Chen, Y. Regulation of Neurotransmitters by the Gut Microbiota and Effects on Cognition in Neurological Disorders. Nutrients 2021, 13, 2099. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.-H.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, A.L.; Choi, J.; Ryou, J.; Newcomer, E.P.; Thompson, R.; Bollinger, R.M.; Hall-Moore, C.; Ndao, I.M.; Sax, L.; Benzinger, T.L.S.; et al. Gut Microbiome Composition May Be an Indicator of Preclinical Alzheimer’s Disease. Sci. Transl. Med. 2023, 15, eabo2984. [Google Scholar] [CrossRef]
- Brenner, S.R. Blue-Green Algae or Cyanobacteria in the Intestinal Micro-Flora May Produce Neurotoxins Such as Beta-N-Methylamino-l-Alanine (BMAA) Which May Be Related to Development of Amyotrophic Lateral Sclerosis, Alzheimer’s Disease and Parkinson-Dementia-Complex in Humans and Equine Motor Neuron Disease in Horses. Med. Hypotheses 2013, 80, 103. [Google Scholar] [CrossRef]
- Tran, L.; Greenwood-Van Meerveld, B. Age-Associated Remodeling of the Intestinal Epithelial Barrier. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1045–1056. [Google Scholar] [CrossRef]
- Komatsu, A.; Iida, I.; Nasu, Y.; Ito, G.; Harada, F.; Kishikawa, S.; Moss, S.J.; Maeda, T.; Terunuma, M. Ammonia Induces Amyloidogenesis in Astrocytes by Promoting Amyloid Precursor Protein Translocation into the Endoplasmic Reticulum. J. Biol. Chem. 2022, 298, 101933. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut Microbiome Alterations in Alzheimer’s Disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Sisodia, S.S.; Vassar, R.J. The Gut Microbiome in Alzheimer’s Disease: What We Know and What Remains to Be Explored. Mol. Neurodegener. 2023, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.D.; Montenigro, P.H.; Alvarez, V.E.; Xia, W.; Crary, J.F.; Tripodis, Y.; Daneshvar, D.H.; Mez, J.; Solomon, T.; Meng, G.; et al. Beta-Amyloid Deposition in Chronic Traumatic Encephalopathy. Acta Neuropathol. 2015, 130, 21. [Google Scholar] [CrossRef] [PubMed]
- Fortea, J.; Zaman, S.H.; Hartley, S.; Rafii, M.S.; Head, E.; Carmona-Iragui, M. Alzheimer’s Disease Associated with Down Syndrome: A Genetic Form of Dementia. Lancet Neurol. 2021, 20, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Doran, E.; Keator, D.; Head, E.; Phelan, M.J.; Kim, R.; Totoiu, M.; Barrio, J.R.; Small, G.W.; Potkin, S.G.; Lott, I.T. Down Syndrome, Partial Trisomy 21, and Absence of Alzheimer’s Disease: The Role of APP. J. Alzheimers Dis. 2017, 56, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Siletti, K.; Hodge, R.; Mossi Albiach, A.; Lee, K.W.; Ding, S.-L.; Hu, L.; Lönnerberg, P.; Bakken, T.; Casper, T.; Clark, M.; et al. Transcriptomic Diversity of Cell Types across the Adult Human Brain. Science 2023, 382, eadd7046. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; van Velthoven, C.T.J.; Kunst, M.; Zhang, M.; McMillen, D.; Lee, C.; Jung, W.; Goldy, J.; Abdelhak, A.; Baker, P.; et al. A High-Resolution Transcriptomic and Spatial Atlas of Cell Types in the Whole Mouse Brain. bioRxiv 2023, arXiv:2023.03.06.531121. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-Cell Chromatin Accessibility Reveals Principles of Regulatory Variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, B.; LaFave, L.M.; Earl, A.S.; Chiang, Z.; Hu, Y.; Ding, J.; Brack, A.; Kartha, V.K.; Tay, T.; et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 2020, 183, 1103–1116. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An Introduction to Spatial Transcriptomics for Biomedical Research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- Lau, S.-F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-Nucleus Transcriptome Analysis Reveals Dysregulation of Angiogenic Endothelial Cells and Neuroprotective Glia in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N.; et al. A Human Brain Vascular Atlas Reveals Diverse Mediators of Alzheimer’s Risk. Nature 2022, 603, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Gazestani, V.; Kamath, T.; Nadaf, N.M.; Dougalis, A.; Burris, S.J.; Rooney, B.; Junkkari, A.; Vanderburg, C.; Pelkonen, A.; Gomez-Budia, M.; et al. Early Alzheimer’s Disease Pathology in Human Cortex Involves Transient Cell States. Cell 2023, 186, 4438–4453. [Google Scholar] [CrossRef] [PubMed]
- Morabito, S.; Miyoshi, E.; Michael, N.; Shahin, S.; Martini, A.C.; Head, E.; Silva, J.; Leavy, K.; Perez-Rosendahl, M.; Swarup, V. Single-Nucleus Chromatin Accessibility and Transcriptomic Characterization of Alzheimer’s Disease. Nat. Genet. 2021, 53, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Gamache, J.; Gingerich, D.; Shwab, E.K.; Barrera, J.; Garrett, M.E.; Hume, C.; Crawford, G.E.; Ashley-Koch, A.E.; Chiba-Falek, O. Integrative Single-Nucleus Multi-Omics Analysis Prioritizes Candidate Cis and Trans Regulatory Networks and Their Target Genes in Alzheimer’s Disease Brains. Cell Biosci. 2023, 13, 185. [Google Scholar] [CrossRef] [PubMed]
- Stonebarger, G.A.; Bimonte-Nelson, H.A.; Urbanski, H.F. The Rhesus Macaque as a Translational Model for Neurodegeneration and Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 734173. [Google Scholar] [CrossRef]
- Uno, H.; Walker, L.C. The Age of Biosenescence and the Incidence of Cerebral Beta-Amyloidosis in Aged Captive Rhesus Monkeys. Ann. N. Y. Acad. Sci. 1993, 695, 232–235. [Google Scholar] [CrossRef]
- Voytko, M.L. Impairments in Acquisition and Reversals of Two-Choice Discriminations by Aged Rhesus Monkeys. Neurobiol. Aging 1999, 20, 617–627. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Zhang, Y. Animal Models of Alzheimer’s Disease: Applications, Evaluation, and Perspectives. Zool Res. 2022, 43, 1026–1040. [Google Scholar] [CrossRef]
- Li, Z.-H.; He, X.-P.; Li, H.; He, R.-Q.; Hu, X.-T. Age-Associated Changes in Amyloid-β and Formaldehyde Concentrations in Cerebrospinal Fluid of Rhesus Monkeys. Zool Res. 2020, 41, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Sani, S.; Traul, D.; Klink, A.; Niaraki, N.; Gonzalo-Ruiz, A.; Wu, C.-K.; Geula, C. Distribution, Progression and Chemical Composition of Cortical Amyloid-β Deposits in Aged Rhesus Monkeys: Similarities to the Human. Acta Neuropathol. 2003, 105, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.B.; et al. Alzheimer’s Disease-Like Pathology Induced by Amyloid-β Oligomers in Nonhuman Primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, Y.; Min, F.; Li, Z.; Huang, J.; Huang, R. A Nonhuman Primate Model of Alzheimer’s Disease Generated by Intracranial Injection of Amyloid-Β42 and Thiorphan. Metab. Brain Dis. 2010, 25, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Li, W.; Luo, Y.; Wang, D.; Zhu, C.; Huang, Z.-X.; Tan, X. Exploring the Differences between Mouse mAβ1–42 and Human hAβ1–42 for Alzheimer’s Disease Related Properties and Neuronal Cytotoxicity. Chem. Commun. 2013, 49, 5865–5867. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F. Alzheimer-Type Neuropathology in Transgenic Mice Overexpressing V717F Beta-Amyloid Precursor Protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Immunization with Amyloid-β Attenuates Alzheimer-Disease-Like Pathology in the PDAPP Mouse|Nature. Available online: https://www.nature.com/articles/22124 (accessed on 26 October 2023).
- Chen, G.; Chen, K.S.; Knox, J.; Inglis, J.; Bernard, A.; Martin, S.J.; Justice, A.; McConlogue, L.; Games, D.; Freedman, S.B.; et al. A Learning Deficit Related to Age and β-Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Nature 2000, 408, 975–979. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Abeta Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two Amyloid Precursor Protein Transgenic Mouse Models with Alzheimer Disease-like Pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.-Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-Level Neuronal Expression of Aβ1–42 in Wild-Type Human Amyloid Protein Precursor Transgenic Mice: Synaptotoxicity without Plaque Formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef]
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramár, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C.; et al. Systematic Phenotyping and Characterization of the 5xFAD Mouse Model of Alzheimer’s Disease. Sci. Data 2021, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Tsuchiya, A.; Nishizaki, T. Hyperphosphorylation of Tau at Ser396 Occurs in the Much Earlier Stage than Appearance of Learning and Memory Disorders in 5XFAD Mice. Behav. Brain Res. 2014, 274, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Frontiers|Differences Between Human and Murine Tau at the N-Terminal End. Available online: https://www.frontiersin.org/articles/10.3389/fnagi.2020.00011/full (accessed on 26 October 2023).
- Umeda, T.; Maekawa, S.; Kimura, T.; Takashima, A.; Tomiyama, T.; Mori, H. Neurofibrillary Tangle Formation by Introducing Wild-Type Human Tau into APP Transgenic Mice. Acta Neuropathol. 2014, 127, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary Tangles, Amyotrophy and Progressive Motor Disturbance in Mice Expressing Mutant (P301L) Tau Protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Frauen, H.D.; Zhang, B.; Nordhues, B.A.; Bijan, S.; Lin, Y.-C.; Zamudio, F.; Hernandez, L.D.; Sabbagh, J.J.; Selenica, M.-L.B.; et al. Tau Depletion Prevents Progressive Blood-Brain Barrier Damage in a Mouse Model of Tauopathy. Acta Neuropathol. Commun. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau Suppression in a Neurodegenerative Mouse Model Improves Memory Function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Javonillo, D.I.; Tran, K.M.; Phan, J.; Hingco, E.; Kramár, E.A.; da Cunha, C.; Forner, S.; Kawauchi, S.; Milinkeviciute, G.; Gomez-Arboledas, A.; et al. Systematic Phenotyping and Characterization of the 3xTg-AD Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2021, 15, 785276. [Google Scholar] [CrossRef]
- Zeng, H.; Huang, J.; Zhou, H.; Meilandt, W.J.; Dejanovic, B.; Zhou, Y.; Bohlen, C.J.; Lee, S.-H.; Ren, J.; Liu, A.; et al. Integrative in Situ Mapping of Single-Cell Transcriptional States and Tissue Histopathology in a Mouse Model of Alzheimer’s Disease. Nat. Neurosci. 2023, 26, 430–446. [Google Scholar] [CrossRef]
- Steffen, J.; Krohn, M.; Paarmann, K.; Schwitlick, C.; Brüning, T.; Marreiros, R.; Müller-Schiffmann, A.; Korth, C.; Braun, K.; Pahnke, J. Revisiting Rodent Models: Octodon Degus as Alzheimer’s Disease Model? Acta Neuropathol. Commun. 2016, 4, 91. [Google Scholar] [CrossRef]
- Flood, D.G.; Lin, Y.-G.; Lang, D.M.; Trusko, S.P.; Hirsch, J.D.; Savage, M.J.; Scott, R.W.; Howland, D.S. A Transgenic Rat Model of Alzheimer’s Disease with Extracellular Abeta Deposition. Neurobiol. Aging 2009, 30, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Orozco, I.J.; Planel, E.; Wen, Y.; Bretteville, A.; Krishnamurthy, P.; Wang, L.; Herman, M.; Figueroa, H.; Yu, W.H.; et al. A Transgenic Rat That Develops Alzheimer’s Disease-like Amyloid Pathology, Deficits in Synaptic Plasticity and Cognitive Impairment. Neurobiol. Dis. 2008, 31, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Leon, W.C.; Canneva, F.; Partridge, V.; Allard, S.; Ferretti, M.T.; DeWilde, A.; Vercauteren, F.; Atifeh, R.; Ducatenzeiler, A.; Klein, W.; et al. A Novel Transgenic Rat Model with a Full Alzheimer’s-like Amyloid Pathology Displays Pre-Plaque Intracellular Amyloid-Beta-Associated Cognitive Impairment. J. Alzheimers Dis. 2010, 20, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A Transgenic Alzheimer Rat with Plaques, Tau Pathology, Behavioral Impairment, Oligomeric Aβ, and Frank Neuronal Loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef] [PubMed]
- Neilson, J.C.; Hart, B.L.; Cliff, K.D.; Ruehl, W.W. Prevalence of Behavioral Changes Associated with Age-Related Cognitive Impairment in Dogs. J. Am. Vet. Med. Assoc. 2001, 218, 1787–1791. [Google Scholar] [CrossRef] [PubMed]
- Siwak-Tapp, C.T.; Head, E.; Muggenburg, B.A.; Milgram, N.W.; Cotman, C.W. Region Specific Neuron Loss in the Aged Canine Hippocampus Is Reduced by Enrichment. Neurobiol. Aging 2008, 29, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.J.; Hart, B.L.; Cliff, K.D.; Ruehl, W.W. Predicting Behavioral Changes Associated with Age-Related Cognitive Impairment in Dogs. J. Am. Vet. Med. Assoc. 2001, 218, 1792–1795. [Google Scholar] [CrossRef]
- Shen, J.; Bronson, R.T.; Chen, D.F.; Xia, W.; Selkoe, D.J.; Tonegawa, S. Skeletal and CNS Defects in Presenilin-1-Deficient Mice. Cell 1997, 89, 629–639. [Google Scholar] [CrossRef]
- Wong, P.C.; Zheng, H.; Chen, H.; Becher, M.W.; Sirinathsinghji, D.J.; Trumbauer, M.E.; Chen, H.Y.; Price, D.L.; Van der Ploeg, L.H.; Sisodia, S.S. Presenilin 1 Is Required for Notch1 and DII1 Expression in the Paraxial Mesoderm. Nature 1997, 387, 288–292. [Google Scholar] [CrossRef]
- Herreman, A.; Hartmann, D.; Annaert, W.; Saftig, P.; Craessaerts, K.; Serneels, L.; Umans, L.; Schrijvers, V.; Checler, F.; Vanderstichele, H.; et al. Presenilin 2 Deficiency Causes a Mild Pulmonary Phenotype and No Changes in Amyloid Precursor Protein Processing but Enhances the Embryonic Lethal Phenotype of Presenilin 1 Deficiency. Proc. Natl. Acad. Sci. USA 1999, 96, 11872–11877. [Google Scholar] [CrossRef]
- Nornes, S.; Groth, C.; Camp, E.; Ey, P.; Lardelli, M. Developmental Control of Presenilin1 Expression, Endoproteolysis, and Interaction in Zebrafish Embryos. Exp. Cell Res. 2003, 289, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.A.; Yang, H.; Zetterberg, H.; Baulac, S.; Sears, J.A.; Liu, T.; Wong, S.T.C.; Zhong, T.P.; Xia, W. Zebrafish Lacking Alzheimer Presenilin Enhancer 2 (Pen-2) Demonstrate Excessive P53-Dependent Apoptosis and Neuronal Loss. J. Neurochem. 2006, 96, 1423–1440. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Pimplikar, S.W. Knockdown of Amyloid Precursor Protein in Zebrafish Causes Defects in Motor Axon Outgrowth. PLoS ONE 2012, 7, e34209. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.; Ebrahimie, E.; Lardelli, M. Using the Zebrafish Model for Alzheimer’s Disease Research. Front. Genet. 2014, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an Emerging Model for Studying Complex Brain Disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef]
- Best, J.D.; Berghmans, S.; Hunt, J.J.F.G.; Clarke, S.C.; Fleming, A.; Goldsmith, P.; Roach, A.G. Non-Associative Learning in Larval Zebrafish. Neuropsychopharmacology 2008, 33, 1206–1215. [Google Scholar] [CrossRef]
- Link, C.D. Invertebrate Models of Alzheimer’s Disease. Genes Brain Behav. 2005, 4, 147–156. [Google Scholar] [CrossRef]
- Griffin, E.F.; Scopel, S.E.; Stephen, C.A.; Holzhauer, A.C.; Vaji, M.A.; Tuckey, R.A.; Berkowitz, L.A.; Caldwell, K.A.; Caldwell, G.A. ApoE-Associated Modulation of Neuroprotection from Aβ-Mediated Neurodegeneration in Transgenic Caenorhabditis Elegans. Dis. Model Mech. 2019, 12, dmm037218. [Google Scholar] [CrossRef]
- Knight, D.; Iliadi, K.; Charlton, M.P.; Atwood, H.L.; Boulianne, G.L. Presynaptic Plasticity and Associative Learning Are Impaired in a Drosophila Presenilin Null Mutant. Dev. Neurobiol. 2007, 67, 1598–1613. [Google Scholar] [CrossRef]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.-K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human Wild-Type Tau Interacts with Wingless Pathway Components and Produces Neurofibrillary Pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Greeve, I.; Kretzschmar, D.; Tschäpe, J.-A.; Beyn, A.; Brellinger, C.; Schweizer, M.; Nitsch, R.M.; Reifegerste, R. Age-Dependent Neurodegeneration and Alzheimer-Amyloid Plaque Formation in Transgenic Drosophila. J. Neurosci. 2004, 24, 3899–3906. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J. Donepezil in Alzheimer’s Disease: An Evidence-Based Review of Its Impact on Clinical and Economic Outcomes. Core Evid. 2006, 1, 195–219. [Google Scholar] [PubMed]
- Tariot, P.N.; Cummings, J.L.; Katz, I.R.; Mintzer, J.; Perdomo, C.A.; Schwam, E.M.; Whalen, E. A Randomized, Double-Blind, Placebo-Controlled Study of the Efficacy and Safety of Donepezil in Patients with Alzheimer’s Disease in the Nursing Home Setting. J. Am. Geriatr. Soc. 2001, 49, 1590–1599. [Google Scholar] [PubMed]
- Seltzer, B.; Zolnouni, P.; Nunez, M.; Goldman, R.; Kumar, D.; Ieni, J.; Richardson, S. Donepezil “402” Study Group Efficacy of Donepezil in Early-Stage Alzheimer Disease: A Randomized Placebo-Controlled Trial. Arch. Neurol. 2004, 61, 1852–1856. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.R.R.; Charles, H.C.; Doraiswamy, P.M.; Mintzer, J.; Weisler, R.; Yu, X.; Perdomo, C.; Ieni, J.R.; Rogers, S. Randomized, Placebo-Controlled Trial of the Effects of Donepezil on Neuronal Markers and Hippocampal Volumes in Alzheimer’s Disease. Am. J. Psychiatry 2003, 160, 2003–2011. [Google Scholar] [CrossRef]
- Rösler, M.; Anand, R.; Cicin-Sain, A.; Gauthier, S.; Agid, Y.; Dal-Bianco, P.; Stähelin, H.B.; Hartman, R.; Gharabawi, M. Efficacy and Safety of Rivastigmine in Patients with Alzheimer’s Disease: International Randomised Controlled Trial. BMJ 1999, 318, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Forette, F.; Anand, R.; Gharabawi, G. A Phase II Study in Patients with Alzheimer’s Disease to Assess the Preliminary Efficacy and Maximum Tolerated Dose of Rivastigmine (Exelon). Eur. J. Neurol. 1999, 6, 423–429. [Google Scholar] [CrossRef]
- Wilkinson, D.; Murray, J.; Galantamine Research Group. Galantamine: A Randomized, Double-Blind, Dose Comparison in Patients with Alzheimer’s Disease. Int. J. Geriatr. Psychiatry 2001, 16, 852–857. [Google Scholar] [CrossRef]
- Rockwood, K.; Mintzer, J.; Truyen, L.; Wessel, T.; Wilkinson, D. Effects of a Flexible Galantamine Dose in Alzheimer’s Disease: A Randomised, Controlled Trial. J. Neurol. Neurosurg. Psychiatry 2001, 71, 589–595. [Google Scholar] [CrossRef]
- Wilcock, G.K.; Lilienfeld, S.; Gaens, E. Efficacy and Safety of Galantamine in Patients with Mild to Moderate Alzheimer’s Disease: Multicentre Randomised Controlled Trial. Galantamine International-1 Study Group. BMJ 2000, 321, 1445–1449. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine Study Group Memantine in Moderate-to-Severe Alzheimer’s Disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Kishi, T.; Iwata, N. Memantine Monotherapy for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0123289. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli-Ardakani, M.; Abbaspour, H.; Farhadi Nasab, A.; Mazaheri Meibodi, A.; Kheradmand, A. Study of the Effect of Memantine on Negative Sign in Patients with Schizophrenia and Schizoaffective Disorders. Iran J. Pharm. Res. 2018, 17, 122–129. [Google Scholar] [PubMed]
- Di Iorio, G.; Baroni, G.; Lorusso, M.; Montemitro, C.; Spano, M.C.; di Giannantonio, M. Efficacy of Memantine in Schizophrenic Patients: A Systematic Review. J. Amino Acids 2017, 2017, 7021071. [Google Scholar] [CrossRef] [PubMed]
- Padda, I.S.; Parmar, M. Aducanumab. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Commissioner, O. Of the FDA Grants Accelerated Approval for Alzheimer’s Drug. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 25 October 2023).
- Commissioner, O. Of the FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 25 October 2023).
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Bastrup, J.; Hansen, K.H.; Poulsen, T.B.G.; Kastaniegaard, K.; Asuni, A.A.; Christensen, S.; Belling, D.; Helboe, L.; Stensballe, A.; Volbracht, C. Anti-Aβ Antibody Aducanumab Regulates the Proteome of Senile Plaques and Closely Surrounding Tissue in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2021, 79, 249–265. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A Randomized, Double-Blind, Phase 2b Proof-of-Concept Clinical Trial in Early Alzheimer’s Disease with Lecanemab, an Anti-Aβ Protofibril Antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Shi, M.; Chu, F.; Zhu, F.; Zhu, J. Impact of Anti-Amyloid-β Monoclonal Antibodies on the Pathology and Clinical Profile of Alzheimer’s Disease: A Focus on Aducanumab and Lecanemab. Front. Aging Neurosci. 2022, 14, 870517. [Google Scholar] [CrossRef]
- Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, Q.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.-J.J.; et al. Chronic Treatment with the Gamma-Secretase Inhibitor LY-411,575 Inhibits Beta-Amyloid Peptide Production and Alters Lymphopoiesis and Intestinal Cell Differentiation. J. Biol. Chem. 2004, 279, 12876–12882. [Google Scholar] [CrossRef] [PubMed]
- Mitani, Y.; Yarimizu, J.; Saita, K.; Uchino, H.; Akashiba, H.; Shitaka, Y.; Ni, K.; Matsuoka, N. Differential Effects between γ-Secretase Inhibitors and Modulators on Cognitive Function in Amyloid Precursor Protein-Transgenic and Nontransgenic Mice. J. Neurosci. 2012, 32, 2037–2050. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and Tolerability of the γ-Secretase Inhibitor Avagacestat in a Phase 2 Study of Mild to Moderate Alzheimer Disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Vassar, R.; Golde, T. The Secretases: Enzymes with Therapeutic Potential in Alzheimer Disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Cebers, G.; Alexander, R.C.; Haeberlein, S.B.; Han, D.; Goldwater, R.; Ereshefsky, L.; Olsson, T.; Ye, N.; Rosen, L.; Russell, M.; et al. AZD3293: Pharmacokinetic and Pharmacodynamic Effects in Healthy Subjects and Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 1039–1053. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Maloney, B.; Long, J.M.; Greig, N.H. Lessons from a BACE1 Inhibitor Trial: Off-Site but Not off Base. Alzheimers Dement. 2014, 10, S411–S419. [Google Scholar] [CrossRef]
- Bazzari, F.H.; Bazzari, A.H. BACE1 Inhibitors for Alzheimer’s Disease: The Past, Present and Any Future? Molecules 2022, 27, 8823. [Google Scholar] [CrossRef]
- Mullard, A. Alzheimer Prevention Failure Rattles Field, Anew. Nat. Rev. Drug Discov. 2019, 18, 656. [Google Scholar] [CrossRef]
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.H.M.A.; Fakhoury, M.; Lawand, N. Electromagnetic Field in Alzheimer’s Disease: A Literature Review of Recent Preclinical and Clinical Studies. Curr. Alzheimer Res. 2020, 17, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Dhaynaut, M.; Sprugnoli, G.; Cappon, D.; Macone, J.; Sanchez, J.S.; Normandin, M.D.; Guehl, N.J.; Koch, G.; Paciorek, R.; Connor, A.; et al. Impact of 40 Hz Transcranial Alternating Current Stimulation on Cerebral Tau Burden in Patients with Alzheimer’s Disease: A Case Series. J. Alzheimers Dis. 2022, 85, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; Kang, G.-Y.; Kwon, J.H.; Choi, H.-D.; Pack, J.-K.; Kim, N.; Lee, Y.-S.; Lee, H.-J. 1950 MHz Electromagnetic Fields Ameliorate Aβ Pathology in Alzheimer’s Disease Mice. Curr. Alzheimer Res. 2015, 12, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Mori, T.; Dorsey, M.; Gonzalez, R.; Tajiri, N.; Borlongan, C. Electromagnetic Treatment to Old Alzheimer’s Mice Reverses β-Amyloid Deposition, Modifies Cerebral Blood Flow, and Provides Selected Cognitive Benefit. PLoS ONE 2012, 7, e35751. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Bradshaw, P.C.; Mamcarz, M.; Lin, X.; Wang, L.; Cao, C.; Arendash, G.W. Long-Term Electromagnetic Field Treatment Enhances Brain Mitochondrial Function of Both Alzheimer’s Transgenic Mice and Normal Mice: A Mechanism for Electromagnetic Field-Induced Cognitive Benefit? Neuroscience 2011, 185, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, F.; Botarelli, E.; Mele, G.; Polo, L.; Zoncu, D.; Renati, P.; Sgarlata, C.; Rollone, M.; Ricevuti, G.; Maurizi, N.; et al. An Innovative Intervention for the Treatment of Cognitive Impairment-Emisymmetric Bilateral Stimulation Improves Cognitive Functions in Alzheimer’s Disease and Mild Cognitive Impairment: An Open-Label Study. Neuropsychiatr Dis. Treat. 2015, 11, 2391–2404. [Google Scholar] [CrossRef]
- Andel, R.; Crowe, M.; Feychting, M.; Pedersen, N.L.; Fratiglioni, L.; Johansson, B.; Gatz, M. Work-Related Exposure to Extremely Low-Frequency Magnetic Fields and Dementia: Results from the Population-Based Study of Dementia in Swedish Twins. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65A, 1220–1227. [Google Scholar] [CrossRef]
- Sandyk, R. Alzheimer’s Disease: Improvement of Visual Memory and Visuoconstructive Performance by Treatment with Picotesla Range Magnetic Fields. Int. J. Neurosci. 1994, 76, 185–225. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Barten, D.M.; Meredith, J.E.; Zaczek, R.; Houston, J.G.; Albright, C.F. Gamma-Secretase Inhibitors for Alzheimer’s Disease: Balancing Efficacy and Toxicity. Drugs R D 2006, 7, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Widyastuti, Y.; Febrisiantosa, A.; Tidona, F. Health-Promoting Properties of Lactobacilli in Fermented Dairy Products. Front. Microbiol. 2021, 12, 673890. [Google Scholar] [CrossRef] [PubMed]
- Śliwińska, S.; Jeziorek, M. The Role of Nutrition in Alzheimer’s Disease. Rocz. Panstw. Zakl. Hig. 2021, 72, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Seethaler, B.; Nguyen, N.K.; Basrai, M.; Kiechle, M.; Walter, J.; Delzenne, N.M.; Bischoff, S.C. Short-Chain Fatty Acids Are Key Mediators of the Favorable Effects of the Mediterranean Diet on Intestinal Barrier Integrity: Data from the Randomized Controlled LIBRE Trial. Am. J. Clin. Nutr. 2022, 116, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Solch, R.J.; Aigbogun, J.O.; Voyiadjis, A.G.; Talkington, G.M.; Darensbourg, R.M.; O’Connell, S.; Pickett, K.M.; Perez, S.R.; Maraganore, D.M. Mediterranean Diet Adherence, Gut Microbiota, and Alzheimer’s or Parkinson’s Disease Risk: A Systematic Review. J. Neurol. Sci. 2022, 434, 120166. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Chan, P.; Wang, T.; Hong, Z.; Wang, S.; Kuang, W.; He, J.; Pan, X.; Zhou, Y.; Ji, Y.; et al. A 36-Week Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group, Phase 3 Clinical Trial of Sodium Oligomannate for Mild-to-Moderate Alzheimer’s Dementia. Alzheimers Res. Ther. 2021, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Tom, S.E.; Hubbard, R.A.; Crane, P.K.; Haneuse, S.J.; Bowen, J.; McCormick, W.C.; McCurry, S.; Larson, E.B. Characterization of Dementia and Alzheimer’s Disease in an Older Population: Updated Incidence and Life Expectancy with and without Dementia. Am. J. Public Health 2015, 105, 408–413. [Google Scholar] [CrossRef]
- Ganguli, M.; Dodge, H.H.; Shen, C.; Pandav, R.S.; DeKosky, S.T. Alzheimer Disease and Mortality: A 15-Year Epidemiological Study. Arch. Neurol. 2005, 62, 779–784. [Google Scholar] [CrossRef]
- Waring, S.C.; Doody, R.S.; Pavlik, V.N.; Massman, P.J.; Chan, W. Survival among Patients with Dementia from a Large Multi-Ethnic Population. Alzheimer Dis. Assoc. Disord. 2005, 19, 178–183. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Corrada, M.M.; Curriero, F.C.; Kawas, C. Survival Following a Diagnosis of Alzheimer Disease. Arch. Neurol. 2002, 59, 1764–1767. [Google Scholar] [CrossRef]
- Larson, E.B.; Shadlen, M.-F.; Wang, L.; McCormick, W.C.; Bowen, J.D.; Teri, L.; Kukull, W.A. Survival after Initial Diagnosis of Alzheimer Disease. Ann. Intern. Med. 2004, 140, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Helzner, E.P.; Scarmeas, N.; Cosentino, S.; Tang, M.X.; Schupf, N.; Stern, Y. Survival in Alzheimer Disease: A Multiethnic, Population-Based Study of Incident Cases. Neurology 2008, 71, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Brayne, C.; Matthews, F.E. Medical Research Council Cognitive Function and Ageing Study collaborators Survival Times in People with Dementia: Analysis from Population Based Cohort Study with 14 Year Follow-Up. BMJ 2008, 336, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Brodaty, H.; Seeher, K.; Gibson, L. Dementia Time to Death: A Systematic Literature Review on Survival Time and Years of Life Lost in People with Dementia. Int. Psychogeriatr. 2012, 24, 1034–1045. [Google Scholar] [CrossRef]
- Todd, S.; Barr, S.; Roberts, M.; Passmore, A.P. Survival in Dementia and Predictors of Mortality: A Review. Int. J. Geriatr. Psychiatry 2013, 28, 1109–1124. [Google Scholar] [CrossRef]

| Changes in AD Patients | miRNA Biomarkers |
|---|---|
| Down-regulated | miR-15b-5p, miR-19b-3p, miR-23a, miR-26a, miR-26b, miR-26b-5p, miR-19c-3, miR-31, miR-34a-5p, miR-103, miR-125b, miR-146a, miR-181c, miR-191-5p, miR-193b, miR-222, and Let-7d-5p [17] |
| Up-regulated | miR-34c, miR-132, miR-181c, miR-206, miR-411, and miR-502-3p [17] |
| Model | Aß Build-Up | Tau or Tau-like Tangles | Neurodegeneration or Synaptic Deficits |
|---|---|---|---|
| Mice | |||
| 5xFAD | X | X | |
| PDAPP | X | X | |
| Tg2576 | X | ||
| APP23 | X | X | |
| J20 | X | X | |
| TgCRND8 | X | ||
| PS2APP | X | ||
| APPswe/PSEN1dE9 | X | ||
| Tg-ArcSwe | X | ||
| A7 | X | ||
| NL-G-F | X | ||
| rTg4510 | X | X | |
| PS19 | X | X | |
| 3xTg | X | X | X |
| pR5-183 | X | X | |
| Rat | |||
| Tg478/Tg1116 | X | ||
| PSAPP | X | ||
| McGill-R-Thy1-APP | X | ||
| TgF344 | X | X | |
| NHP | |||
| Aging rhesus macaque | X | X | |
| Aging stump-tail macaque | X | X | |
| Aβ oligomer injection in rhesus macaque | X | X | |
| Aβ and thiorphan injection in rhesus macaques | X | X | |
| Canine | |||
| canine cognitive dysfunction (CCD) model | X | X | |
| Zebra fish | |||
| psen1 mutant | |||
| psen2 mutant | |||
| app-a and app-b mutants | X | X | |
| Fruit flies | |||
| psn mutant | X | ||
| tau mutant | X | X | |
| Aβ mutant | X | X | |
| Aβ mutation in the eye | |||
| C. elegans | |||
| Aβ42 mutation in the muscle | X | X | |
| Aβ mutant + human APOE ε4 transgene | X | X |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nystuen, K.L.; McNamee, S.M.; Akula, M.; Holton, K.M.; DeAngelis, M.M.; Haider, N.B. Alzheimer’s Disease: Models and Molecular Mechanisms Informing Disease and Treatments. Bioengineering 2024, 11, 45. https://doi.org/10.3390/bioengineering11010045
Nystuen KL, McNamee SM, Akula M, Holton KM, DeAngelis MM, Haider NB. Alzheimer’s Disease: Models and Molecular Mechanisms Informing Disease and Treatments. Bioengineering. 2024; 11(1):45. https://doi.org/10.3390/bioengineering11010045
Chicago/Turabian StyleNystuen, Kaden L., Shannon M. McNamee, Monica Akula, Kristina M. Holton, Margaret M. DeAngelis, and Neena B. Haider. 2024. "Alzheimer’s Disease: Models and Molecular Mechanisms Informing Disease and Treatments" Bioengineering 11, no. 1: 45. https://doi.org/10.3390/bioengineering11010045