
Biocatalytic Insights for The Synthesis of New Potential Prodrugs: Design of two Ibuprofen Derivatives

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Computational Analysis
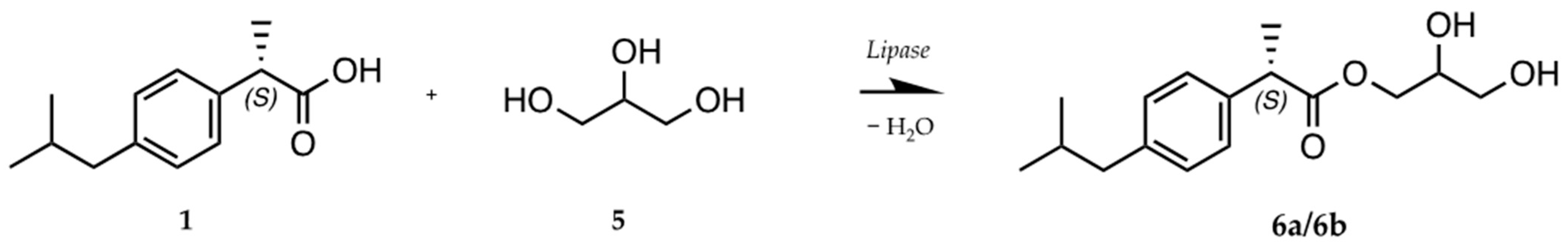
2.3. Biocatalytic Synthesis of the Ibuprofen Esters 3a/3b and 6
2.4. Thin-Layer Chromatography (TLC)
2.5. Purification and Spectroscopic Characterization of the Prodrugs
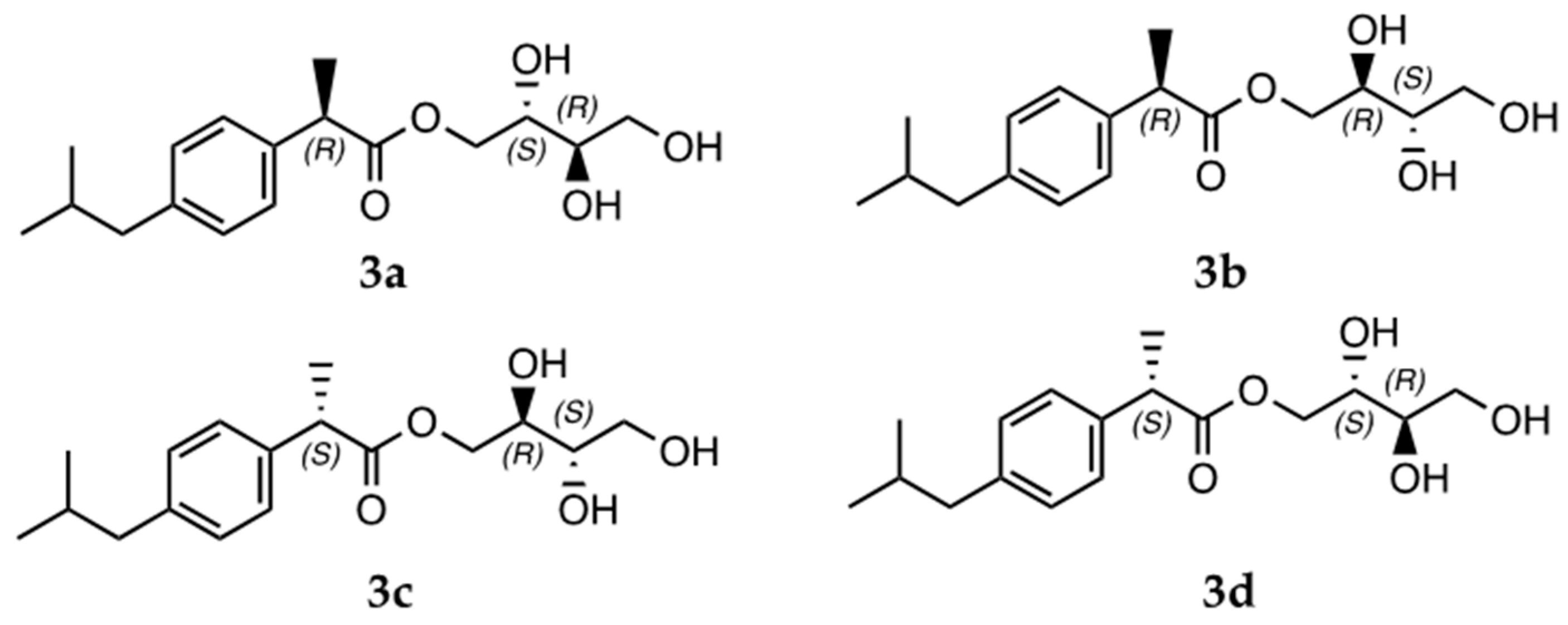
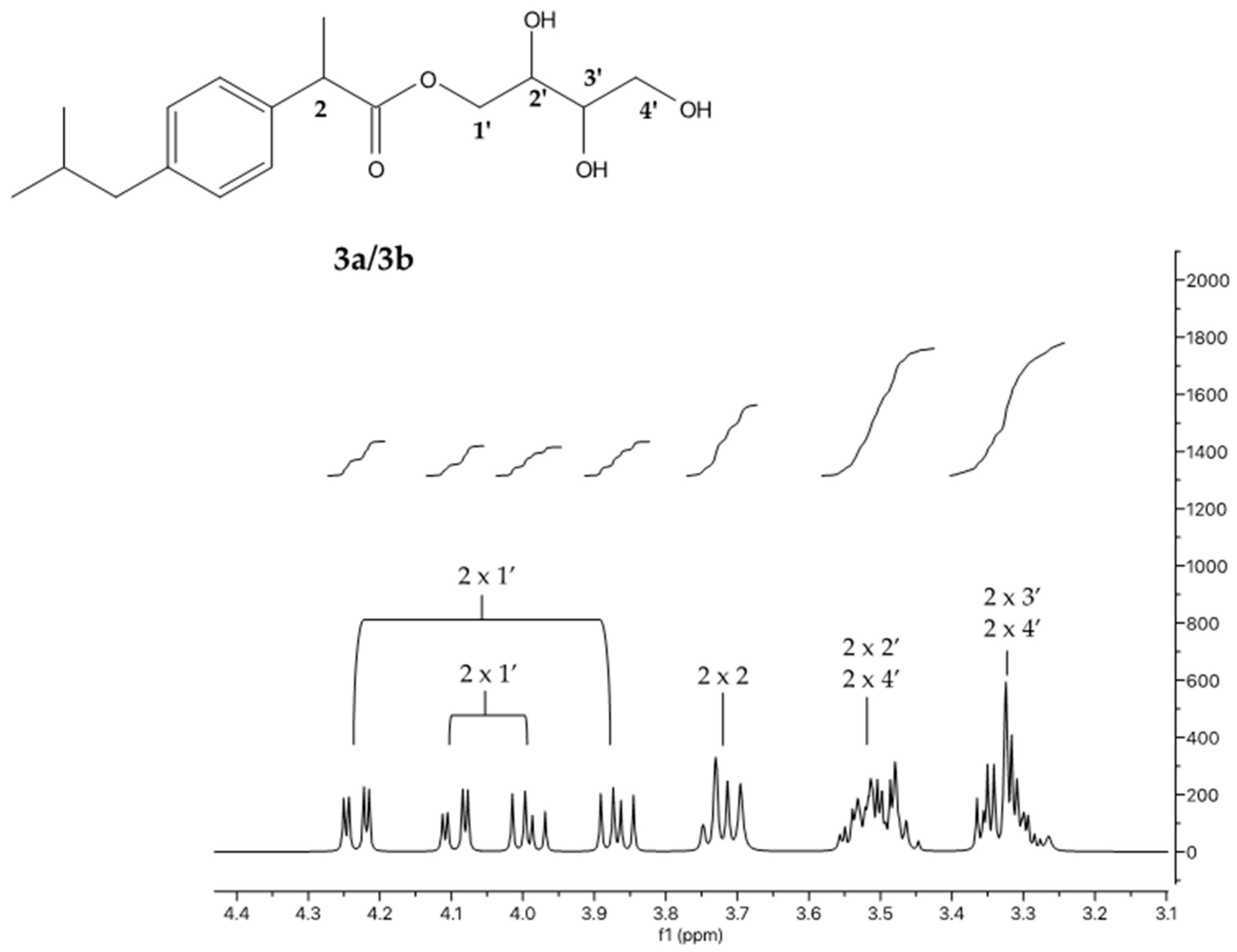
- IBU-erythritol ester (3a/3b; (2S,3R)-2,3,4-trihydroxybutyl (R)-2-(4-isobutylphenyl)propanoate and (2R,3S)-2,3,4-trihydroxybutyl (R)-2-(4-isobutylphenyl)propanoate): 1H NMR (400 MHz, DMSO-d6) δ 7.18 (dd, J = 8.1, 1.9 Hz, 2 H); 7.08 (dd, J = 8.2, 2.0 Hz, 2 H); 4.23 (dd, J = 11.3, 2.9 Hz); 4.09 (dd, J = 11.3, 2.8 Hz); 3.99 (dd, J = 11.3, 7.2 Hz); 3.87 (dd, J = 11.3, 7.0 Hz); 3.71 (q, J = 7.5 Hz, 1 H); 3.51 (m, 2 H); 3.33 (m, 2 H); 2.39 (d, J = 7.1 Hz, 2 H); 1.81 (m, 1 H); 1.36 (dd, J = 7.1, 1.9 Hz, 3 H); 0.83 (d, J = 6.6 Hz, 6 H). 13C NMR (101 MHz, DMSO-d6) δ 174.53; 140.13; 138.41; 129.45 (2 C); 127.57 (2 C); 72.75; 69.83; 67.03; 63.42; 44.71; 44.66; 30.03; 22.64 (2 C); 19.20.
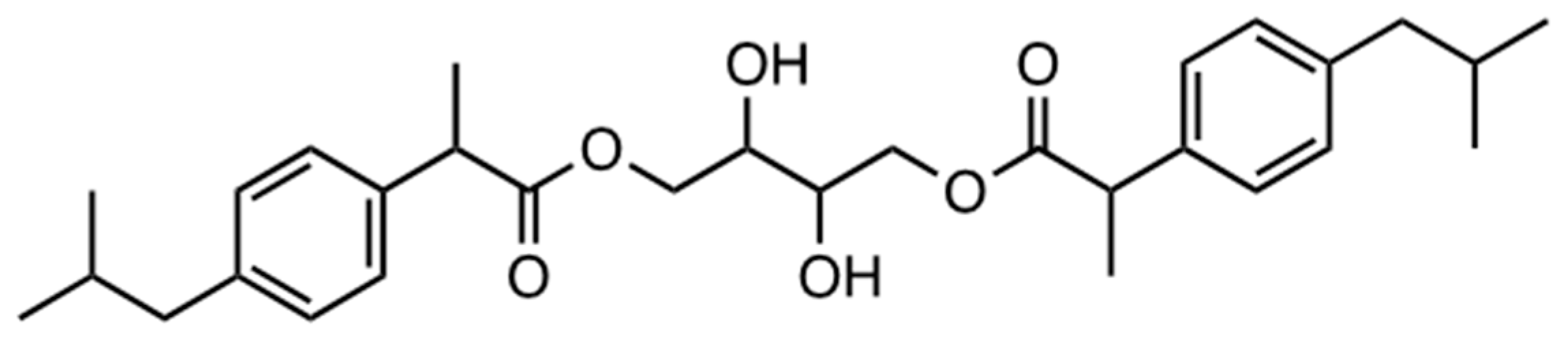
- 1,3-diacylated IBU-erythritol ester (4; three possible configurations: (2R,3S)-2,3-dihydroxybutane-1,4-diyl (2R,2′R)-bis(2-(4-isobutylphenyl)propanoate), (2R,3S)-2,3-dihydroxybutane-1,4-diyl (2S,2′S)-bis(2-(4-isobutylphenyl)propanoate), and (2R,3S)-2,3-dihydroxy-4-(((R)-2-(4-isobutylphenyl)propanoyl)oxy)butyl (S)-2-(4-isobutylphenyl)propanoate): 1H NMR (400 MHz, DMSO-d6) δ 7.16 (dd, J = 8.0, 2.0 4 H); 7.06 (dd, J = 8.1, 2.1 Hz, 4 H); 4.18 (dd, J = 11.2, 1.2 Hz, 1 H); 4.05 (dd, J = 11.3, 2.7 Hz, 1 H); 3.97 (m, 1 H); 3.86 (m, 1 H); 3.70 (m, 2 H); 3.51 (m, 1 H); 2.37 (d, J = 7.2 Hz, 5 H); 1.76 (m, 2 H); 1.34 (m, 6 H); 0.82 (d, J = 6.7 Hz, 12 H). 13C NMR (101 MHz, DMSO-d6) δ 174.47; 140.14; 138.37; 129.46; 127.57; 69.66; 66.61; 66.49; 53,75; 44.66; 30.03; 22.62; 19.15.
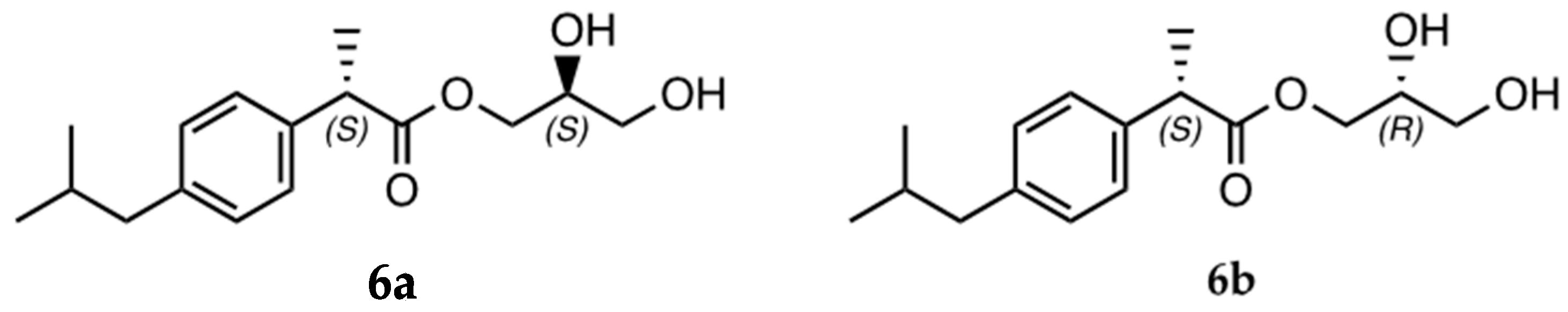
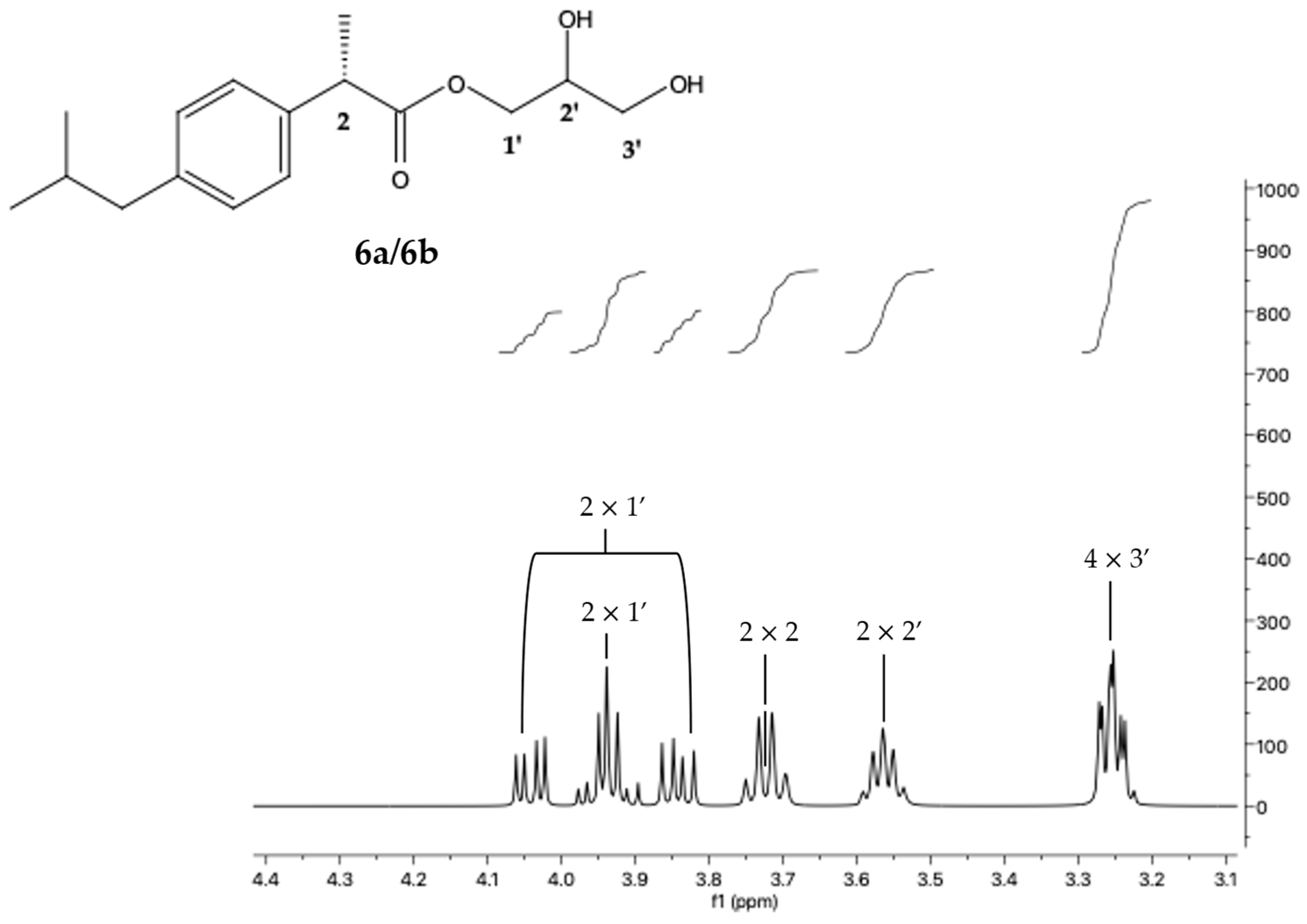
- IBU-glycerol ester (6a/6b; (S)-2,3-dihydroxypropyl (S)-2-(4-isobutylphenyl)propanoate and (R)-2,3-dihydroxypropyl (S)-2-(4-isobutylphenyl)propanoate): 1H NMR (400 MHz, DMSO-d6) δ 7.17 (dd, J = 8.0, 1.8 Hz, 2 H); 7.08 (dd, J = 8.1, 1.9 Hz, 2 H); 4.04 (dd, J = 11.1, 4.5 Hz); 3.94 (m, 1 H); 3.84 (dd, J = 11.1, 6.2 Hz); 3.72 (q, J = 7.2 Hz, 1 H); 3.56 (m, 1 H); 3.25 (m, 2 H); 2.39 (d, J = 7.2 Hz, 2 H); 1.78 (hept, J = 6.8 Hz, 1 H); 1.35 (d, J = 7.2 Hz, 3 H); 0.83 (d, J = 6.6 Hz, 5 H). 13C NMR (101 MHz, DMSO- d6) δ 174.39; 140.17; 138.36; 129.48 (2 C); 127.53 (2 C); 69.68; 66.40; 62.98; 44.64; 44.42; 30.03; 22.63 (2 C); 19.10.
3. Results and Discussion
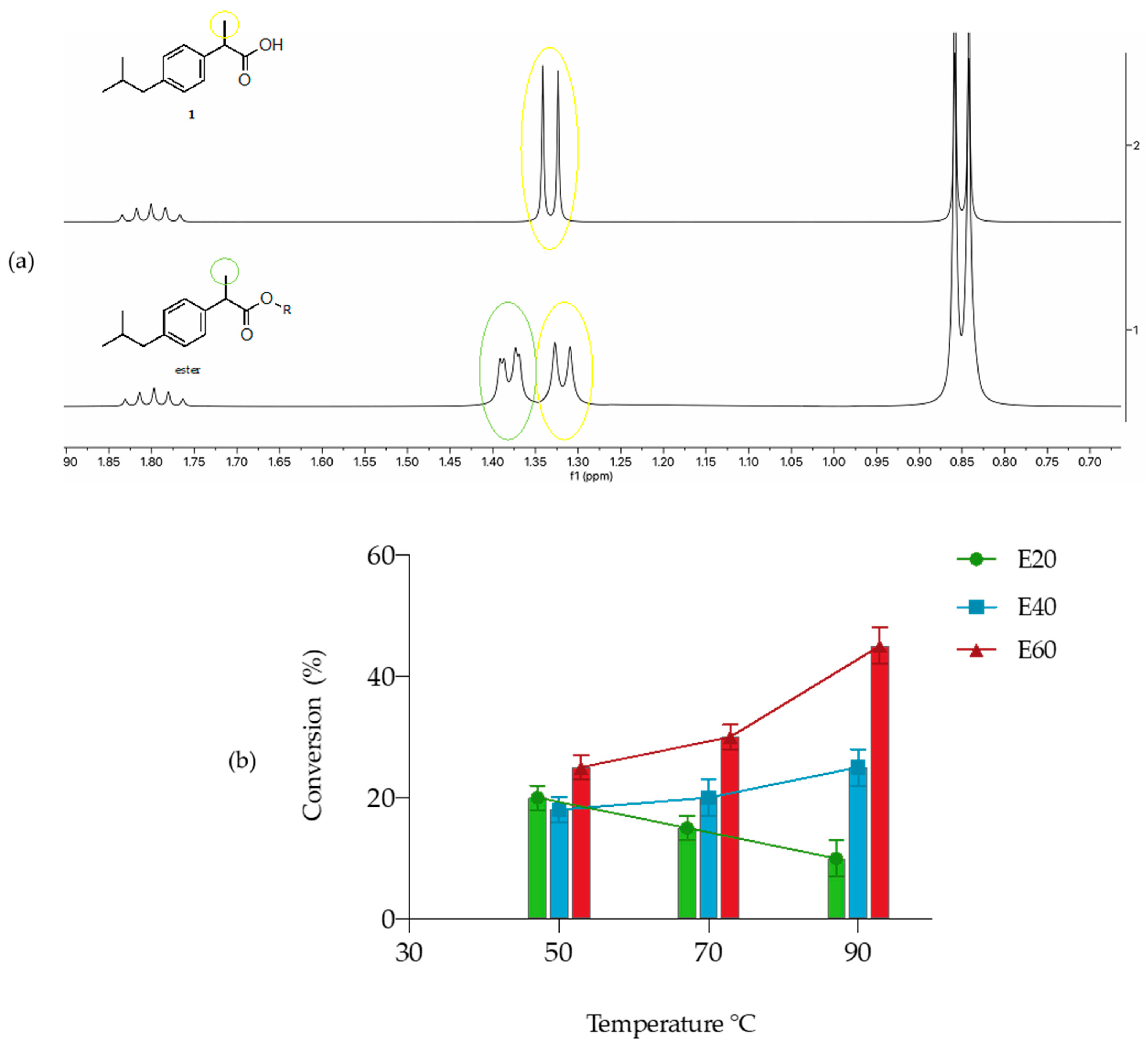
3.1. Screening of Free and Immobilized Lipases for the Synthesis of Ibuprofen (IBU)–Erythritol Ester
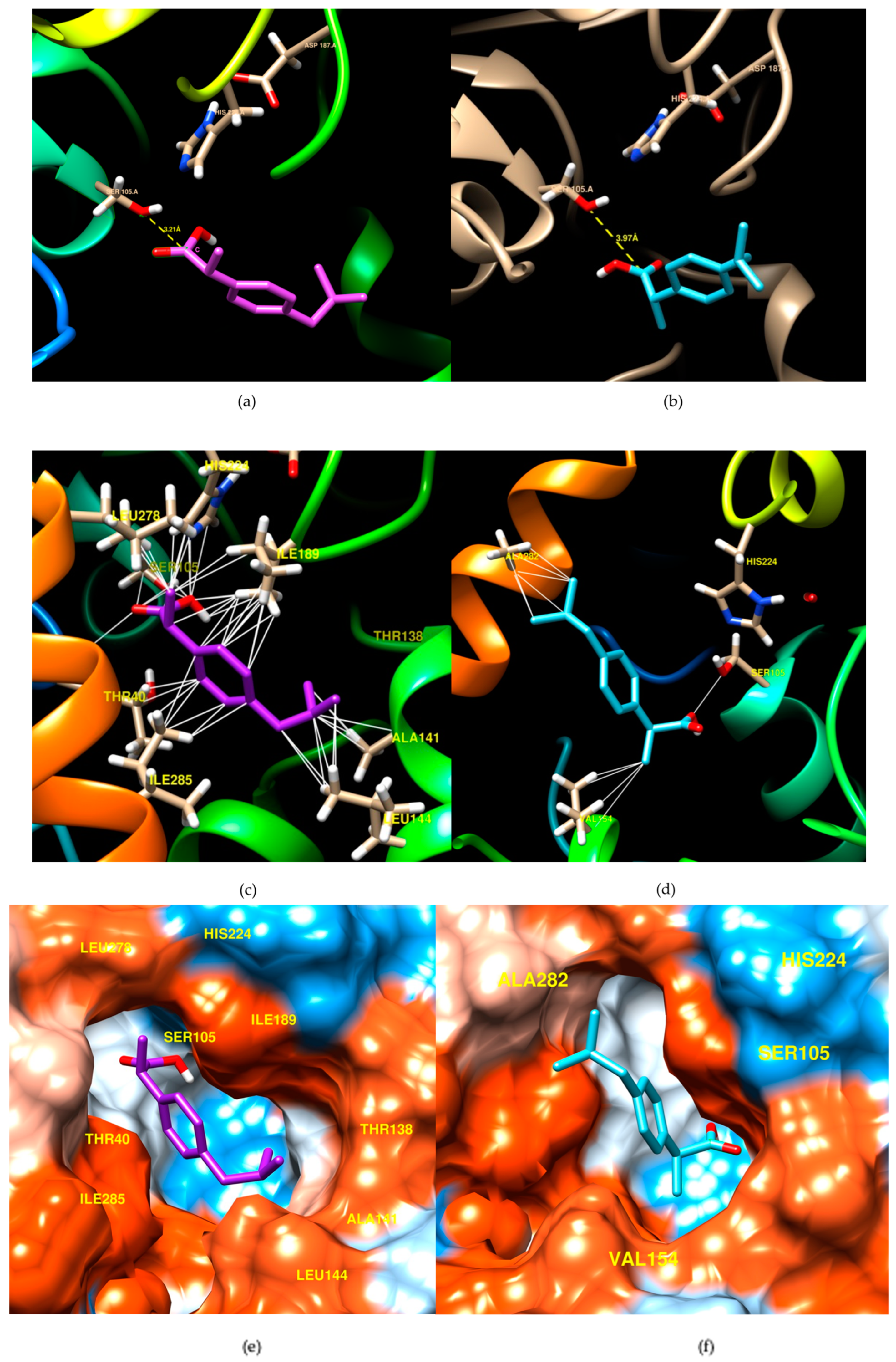
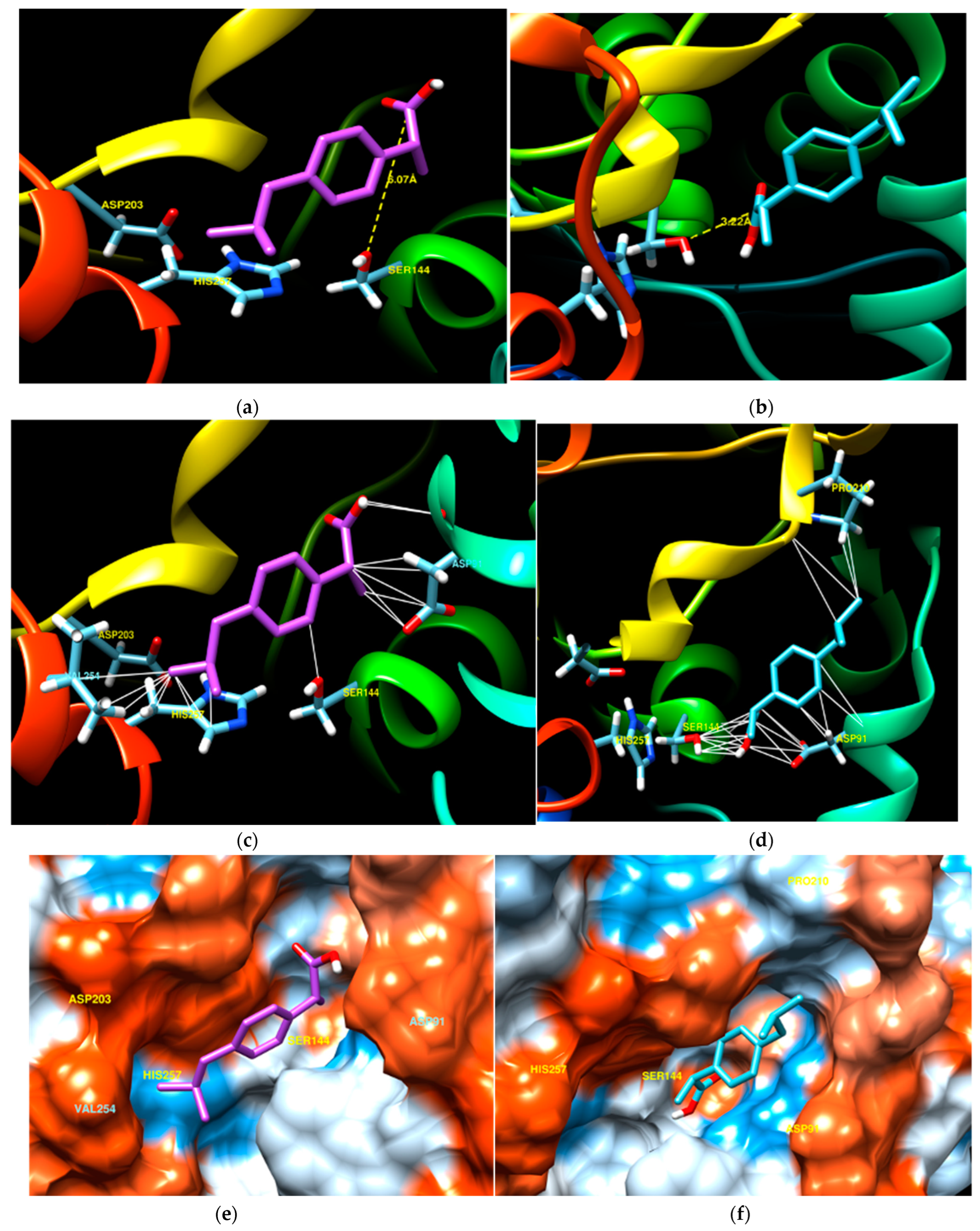
3.2. Computational Analysis for the Evaluation of the CalB Stereoselectivity
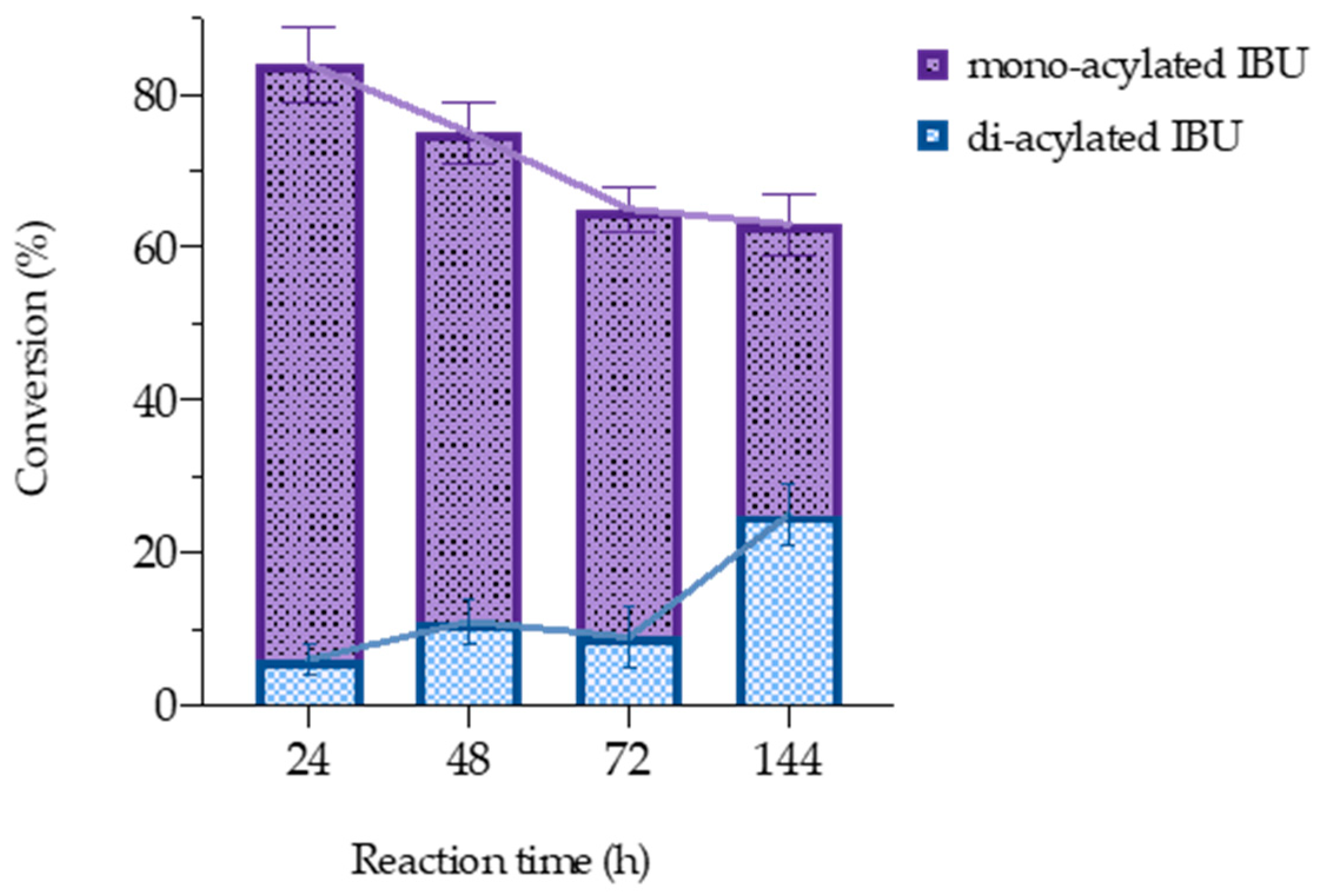
3.3. Experimental Validation of the Computational Study
3.4. Lipozyme RM IM-Catalyzed Synthesis of (S)-IBU–Glycerol Ester
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Konstan, M.W.; Hoppel, C.L.; Chai, B.L.; Davis, P.B. Ibuprofen in Children with Cystic Fibrosis: Pharmacokinetics and Adverse Effects. J. Pediatr. 1991, 118, 956–964. [Google Scholar] [CrossRef]
- Zappaterra, F.; Tupini, C.; Summa, D.; Cristofori, V.; Costa, S.; Trapella, C.; Lampronti, I.; Tamburini, E. Xylitol as a Hydrophilization Moiety for a Biocatalytically Synthesized Ibuprofen Prodrug. Int. J. Mol. Sci. 2022, 23, 2026. [Google Scholar] [CrossRef]
- Yalkowsky, S.; Dannenfelser, R. The AQUASOL DATAbASE of Aqueous Solubility; CRC Press: Boca Raton, FL, USA, 1992. [Google Scholar]
- Stoyanova, K.; Vinarov, Z.; Tcholakova, S. Improving Ibuprofen Solubility by Surfactant-Facilitated Self-Assembly into Mixed Micelles. J. Drug Deliv. Sci. Technol. 2016, 36, 208–215. [Google Scholar] [CrossRef]
- Levis, K.A.; Lane, M.E.; Corrigan, O.I. Effect of Buffer Media Composition on the Solubility and Effective Permeability Coefficient of Ibuprofen. Int. J. Pharm. 2003, 253, 49–59. [Google Scholar] [CrossRef]
- Shanbhag, V.R.; Crider, A.M.; Gokhale, R.; Harpalani, A.; Dick, R.M. Ester and Amide Prodrugs of Ibuprofen and Naproxen: Synthesis, Anti-inflammatory Activity, and Gastrointestinal Toxicity. J. Pharm. Sci. 1992, 81, 149–154. [Google Scholar] [CrossRef]
- Choudhary, D.; Goykar, H.; Kalyane, D.; Tekade, R.K.; Sreeharsha, N. Prodrug Design for Improving the Biopharmaceutical Properties of Therapeutic Drugs; INC: New York, NY, USA, 2020. [Google Scholar] [CrossRef]
- Toledo, M.V.; Briand, L.E.; Ferreira, M.L. A Simple Molecular Model to Study the Substrate Diffusion into the Active Site of a Lipase-Catalyzed Esterification of Ibuprofen and Ketoprofen with Glycerol. Top. Catal. 2022, 65, 944–956. [Google Scholar] [CrossRef]
- Toledo, M.V.; José, C.; Suster, C.R.L.; Collins, S.E.; Portela, R.; Bañares, M.A.; Briand, L.E. Catalytic and Molecular Insights of the Esterification of Ibuprofen and Ketoprofen with Glycerol. Mol. Catal. 2021, 513, 111811. [Google Scholar] [CrossRef]
- Ravelo, M.; Fuente, E.; Blanco, Á.; Ladero, M.; García-Ochoa, F. Esterification of Glycerol and Ibuprofen in Solventless Media Catalyzed by Free CALB: Kinetic Modelling. Biochem. Eng. J. 2015, 101, 228–236. [Google Scholar] [CrossRef]
- Ravelo, M.; Gallardo, M.E.; Ladero, M.; Garcia-Ochoa, F. Synthesis of Ibuprofen Monoglyceride Using Novozym ® 435: Biocatalyst Activation and Stabilization in Multiphasic Systems. Catalysts 2022, 12, 1531. [Google Scholar] [CrossRef]
- Ravelo, M.; Wojtusik, M.; Ladero, M.; García-Ochoa, F. Synthesis of Ibuprofen Monoglyceride in Solventless Medium with Novozym®435: Kinetic Analysis. Catalysts 2020, 10, 76. [Google Scholar] [CrossRef]
- Ravelo, M.; Esteban, J.; Ladero, M.; García-Ochoa, F. Enzymatic Synthesis of Ibuprofen Monoglycerides Catalyzed by Free: Candida Antarctica Lipase B in a Toluene-Glycerol Biphasic Medium. RSC Adv. 2016, 6, 69658–69669. [Google Scholar] [CrossRef]
- Zappaterra, F.; Rodriguez, M.E.M.; Summa, D.; Semeraro, B.; Costa, S.; Tamburini, E. Biocatalytic Approach for Direct Esterification of Ibuprofen with Sorbitol in Biphasic Media. Int. J. Mol. Sci. 2021, 22, 3066. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, S.A.B.E.; Van Den Berg, D.J.; Tromp, M.N.J.L.; Griffioen, D.H.; Van Bennekom, W.P.; Van Der Vijgh, W.J.F.; Bast, A. Structural Aspects of Antioxidant Activity of Flavonoids. Free Radic. Biol. Med. 1996, 20, 331–342. [Google Scholar] [CrossRef]
- Munro, I.C.; Bernt, W.O.; Borzelleca, J.F.; Flamm, G.; Lynch, B.S.; Kennepohl, E.; Bär, E.A.; Modderman, J. Erythritol: An Interpretive Summary of Biochemical, Metabolic, Toxicological and Clinical Data. Food Chem. Toxicol. 1998, 36, 1139–1174. [Google Scholar] [CrossRef]
- Yokozawa, T.; Kim, H.Y.; Cho, E.J. Erythritol Attenuates the Diabetic Oxidative Stress through Modulating Glucose Metabolism and Lipid Peroxidation in Streptozotocin-Induced Diabetic Rats. J. Agric. Food Chem. 2002, 50, 5485–5489. [Google Scholar] [CrossRef]
- Zappaterra, F.; Costa, S.; Summa, D.; Bertolasi, V.; Semeraro, B.; Pedrini, P.; Buzzi, R.; Vertuani, S. Biotransformation of Cortisone with Rhodococcus Rhodnii: Synthesis of New Steroids. Molecules 2021, 26, 1352. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Summa, D.; Zappaterra, F.; Blo, R.; Tamburini, E. Aspergillus oryzae Grown on Rice Hulls Used as an Additive for Pretreatment of Starch-Containing Wastewater from the Pulp and Paper Industry. Fermentation 2021, 7, 317. [Google Scholar] [CrossRef]
- Máximo, F.; Asensi, M.; Serrano-Arnaldos, M.; Ortega-Requena, S.; Montiel, C.; Bastida, J. Biocatalytic Intensified Process for the Synthesis of Neopentyl Glycol Dicaprylate/Dicaprate. Sustain. Chem. Pharm. 2022, 30, 100882. [Google Scholar] [CrossRef]
- Rădoi, I.-I.; Bedolla, D.E.; Vaccari, L.; Todea, A.; Zappaterra, F.; Volkov, A.; Gardossi, L. FTIR Microscopy for Direct Observation of Conformational Changes on Immobilized ω-Transaminase: Effect of Water Activity and Organic Solvent on Biocatalyst Performance. Catal. Sci. Technol. 2023, 13, 4955–4967. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Delano, W.L. The PyMOL Molecular Graphics System; Schrödinger, LLC: New York, NY, USA, 2002; Available online: https://pymol.org/2/ (accessed on 15 January 2023).
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S.; Derewenda, U.; Dodson, G.G. The Crystal and Molecular Structure of the Rhizomucor Miehei Triacylglyceride Lipase at 1.9 Å Resolution. J. Mol. Biol. 1992, 227, 818–839. [Google Scholar] [CrossRef] [PubMed]
- Muralidhar, R.V.; Chirumamilla, R.R.; Marchant, R.; Ramachandran, V.N.; Ward, O.P.; Nigam, P. Understanding Lipase Stereoselectivity. World J. Microbiol. Biotechnol. 2002, 18, 81–97. [Google Scholar] [CrossRef]
- Zappaterra, F.; Costa, S.; Summa, D.; Semeraro, B.; Cristofori, V.; Trapella, C.; Tamburini, E. Glyceric Prodrug of Ursodeoxycholic Acid (UDCA): Novozym 435-Catalyzed Synthesis of UDCA-Monoglyceride. Molecules 2021, 25, 5966. [Google Scholar] [CrossRef]
- Evans, A.M. Clinical Rheumatology Comparative Pharmacology of S(+)-Ibuprofen and (RS)-Ibuprofen. Clin. Rheumatol. 2001, 20, 9–14. [Google Scholar] [CrossRef]
- José, C.; Toledo, M.V.; Briand, L.E. Enzymatic Kinetic Resolution of Racemic Ibuprofen: Past, Present and Future. Crit. Rev. Biotechnol. 2016, 36, 891–903. [Google Scholar] [CrossRef]
- Todea, A.; Fortuna, S.; Ebert, C.; Asaro, F.; Tomada, S.; Cespugli, M.; Hollan, F.; Gardossi, L. Rational Guidelines for the Two-Step Scalability of Enzymatic Polycondensation: Experimental and Computational Optimization of the Enzymatic Synthesis of Poly(glycerolazelate). ChemSusChem 2022, 15, e202102657. [Google Scholar] [CrossRef]
- Guarneri, A.; Cutifani, V.; Cespugli, M.; Pellis, A.; Vassallo, R.; Asaro, F.; Ebert, C.; Gardossi, L. Functionalization of Enzymatically Synthesized Rigid Poly(Itaconate)s via Post-Polymerization Aza-Michael Addition of Primary Amines. Adv. Synth. Catal. 2019, 361, 2559–2573. [Google Scholar] [CrossRef]
- Chellan, P.; Land, K.M.; Shokar, A.; Au, A.; An, S.H.; Taylor, D.; Smith, P.J.; Riedel, T.; Dyson, P.J.; Chibale, K.; et al. Synthesis and Evaluation of New Polynuclear Organometallic Ru(Ii), Rh(Iii) and Ir(Iii) Pyridyl Ester Complexes as in Vitro Antiparasitic and Antitumor Agents. Dalt. Trans. 2014, 43, 513–526. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Zappaterra, F.; Summa, D.; Semeraro, B.; Buzzi, R.; Trapella, C.; Ladero, M.; Costa, S.; Tamburini, E. Enzymatic Esterification as Potential Strategy to Enhance the Sorbic Acid Behavior as Food and Beverage Preservative. Fermentation 2020, 6, 96. [Google Scholar] [CrossRef]
- Mendes, A.A.; Oliveira, P.C.; De Castro, H.F. Properties and Biotechnological Applications of Porcine Pancreatic Lipase. J. Mol. Catal. B Enzym. 2012, 78, 119–134. [Google Scholar] [CrossRef]
- Chen, J.P. Production of Ethyl Butyrate Using Gel-Entrapped Candida Cylindracea Lipase. J. Ferment. Bioeng. 1996, 82, 404–407. [Google Scholar] [CrossRef]
- López-Belmonte, M.T.; Alcántara, A.R.; Sinisterra, J.V. Enantioselective Esterification of 2-Arylpropionic Acids Catalyzed by Immobilized Rhizomucor Miehei Lipase. J. Org. Chem. 1997, 62, 1831–1840. [Google Scholar] [CrossRef]
- Chen, J.C.; Tsai, S.W. Enantioselective Synthesis of (s)-Ibuprofen Ester Prodrug in Cyclohexane by Candida Rugosa Lipase Immobilized on Accurel MP1000. Biotechnol. Prog. 2000, 16, 986–992. [Google Scholar] [CrossRef]
- Saktaweewong, S.; Phinyocheep, P.; Ulmer, C.; Marie, E.; Durand, A.; Inprakhon, P. Lipase Activity in Biphasic Media: Why Interfacial Area Is a Significant Parameter? J. Mol. Catal. B Enzym. 2011, 70, 8–16. [Google Scholar] [CrossRef]
- Zappaterra, F.; Renzi, M.; Piccardo, M.; Spennato, M.; Asaro, F.; Serio, M.D.; Vitiello, R.; Turco, R.; Todea, A.; Gardossi, L. Understanding Marine Biodegradation of Bio-Based Oligoesters and Plasticizers. Polymers 2023, 15, 1536. [Google Scholar] [CrossRef]
- Vagenende, V.; Yap, M.G.S.; Trout, B.L. Mechanisms of Protein Stabilization and Prevention of Protein Aggregation by Glycerol. Biochemistry 2009, 48, 11084–11096. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
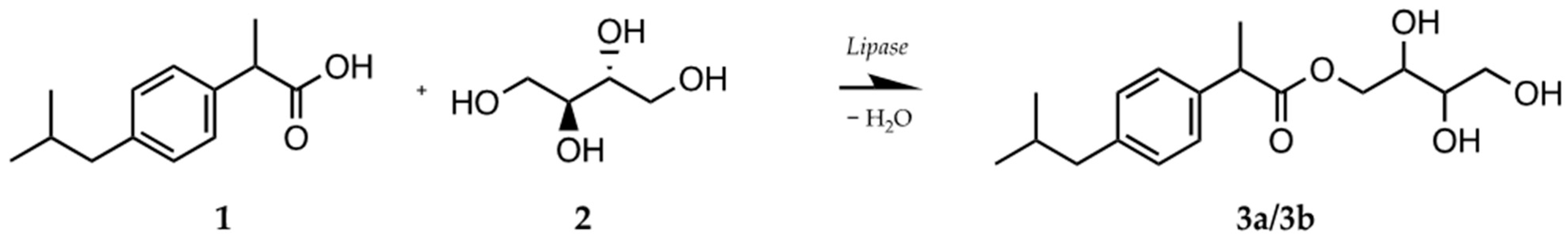{kind=link}
{kind=link}
| Lipase | Ligand | E (kcal/mol) | d (Å) | ϑ (°) |
|---|---|---|---|---|
| CalB | (R)-ibuprofen | −6.7 | 3.21 | 67.7 |
| (S)-ibuprofen | −6.2 | 3.97 | 101.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zappaterra, F.; Presini, F.; Venturi, V.; Lerin, L.A.; Giovannini, P.P.; Costa, S. Biocatalytic Insights for The Synthesis of New Potential Prodrugs: Design of two Ibuprofen Derivatives. Appl. Sci. 2023, 13, 9852. https://doi.org/10.3390/app13179852
Zappaterra F, Presini F, Venturi V, Lerin LA, Giovannini PP, Costa S. Biocatalytic Insights for The Synthesis of New Potential Prodrugs: Design of two Ibuprofen Derivatives. Applied Sciences. 2023; 13(17):9852. https://doi.org/10.3390/app13179852
Chicago/Turabian StyleZappaterra, Federico, Francesco Presini, Valentina Venturi, Lindomar Alberto Lerin, Pier Paolo Giovannini, and Stefania Costa. 2023. "Biocatalytic Insights for The Synthesis of New Potential Prodrugs: Design of two Ibuprofen Derivatives" Applied Sciences 13, no. 17: 9852. https://doi.org/10.3390/app13179852