
Pharmacologic Ascorbate Radiosensitizes Pancreatic Cancer but Radioprotects Normal Tissue: The Role of Oxidative Stress-Induced Lipid Peroxidation
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Sample and Staining
2.2. In Vivo Orthotopic Mouse Experiment
2.3. Cell Culture
2.4. Reagent/Drug and Ionizing Radiation Treatment
2.5. Western Blotting
2.6. Clonogenic Survival Assay
2.7. Measurement of Lipid Peroxidation
2.8. Activity Assays, Catalase and GPx4
2.9. Glutathione Levels
2.10. Statistical Methods
3. Results
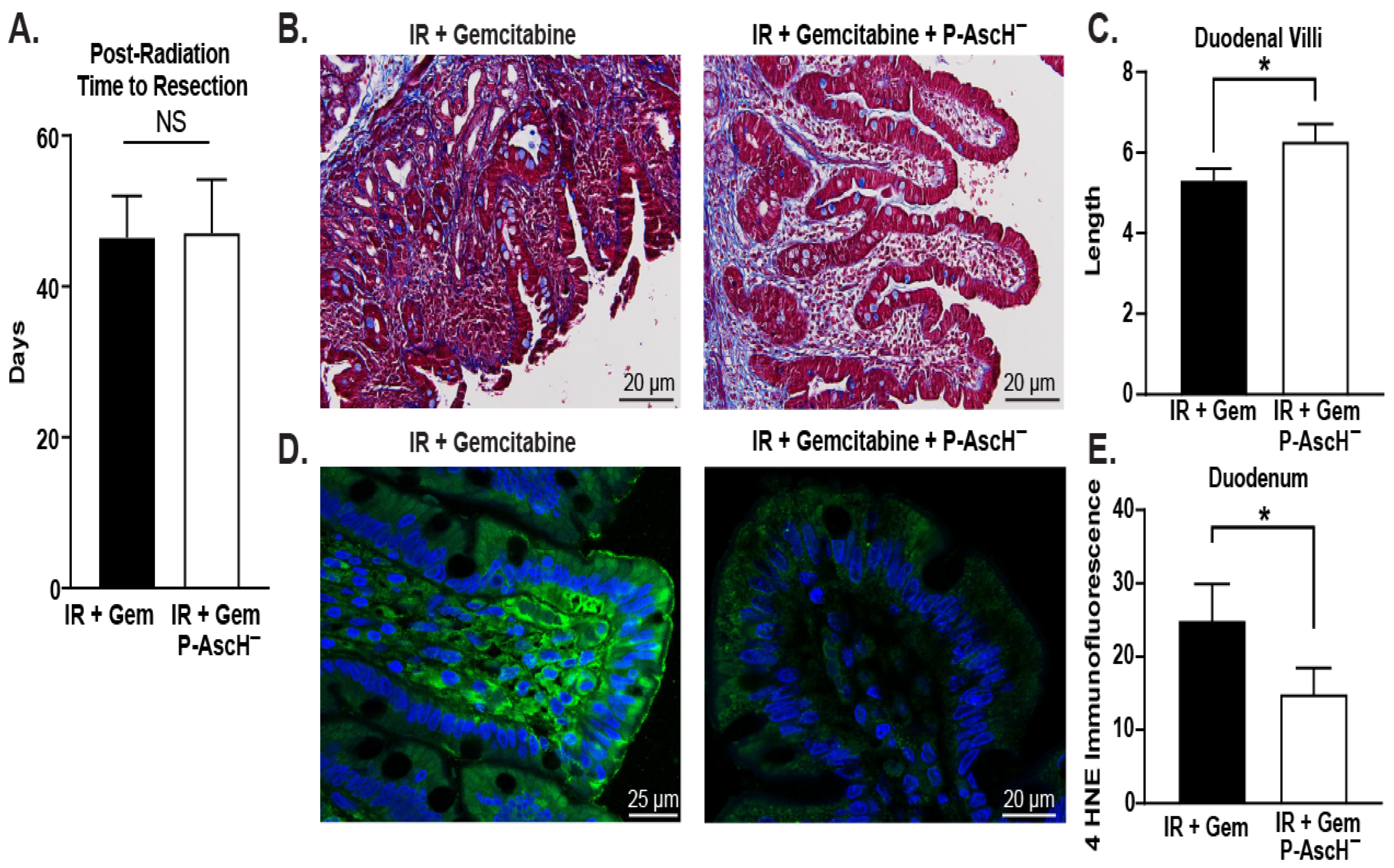
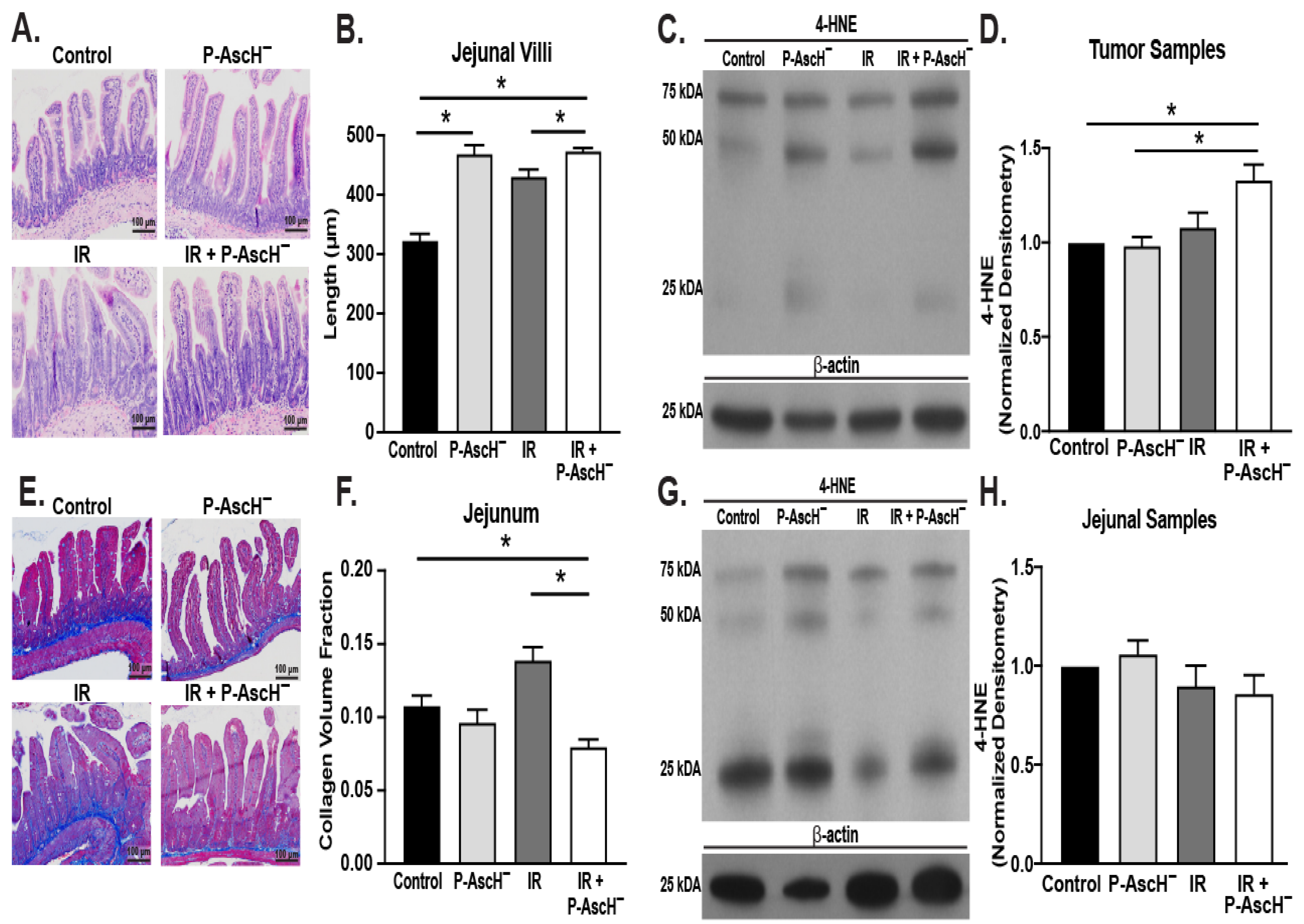
3.1. P-AscH− Ameliorates Radiation-Induced Intestinal Damage
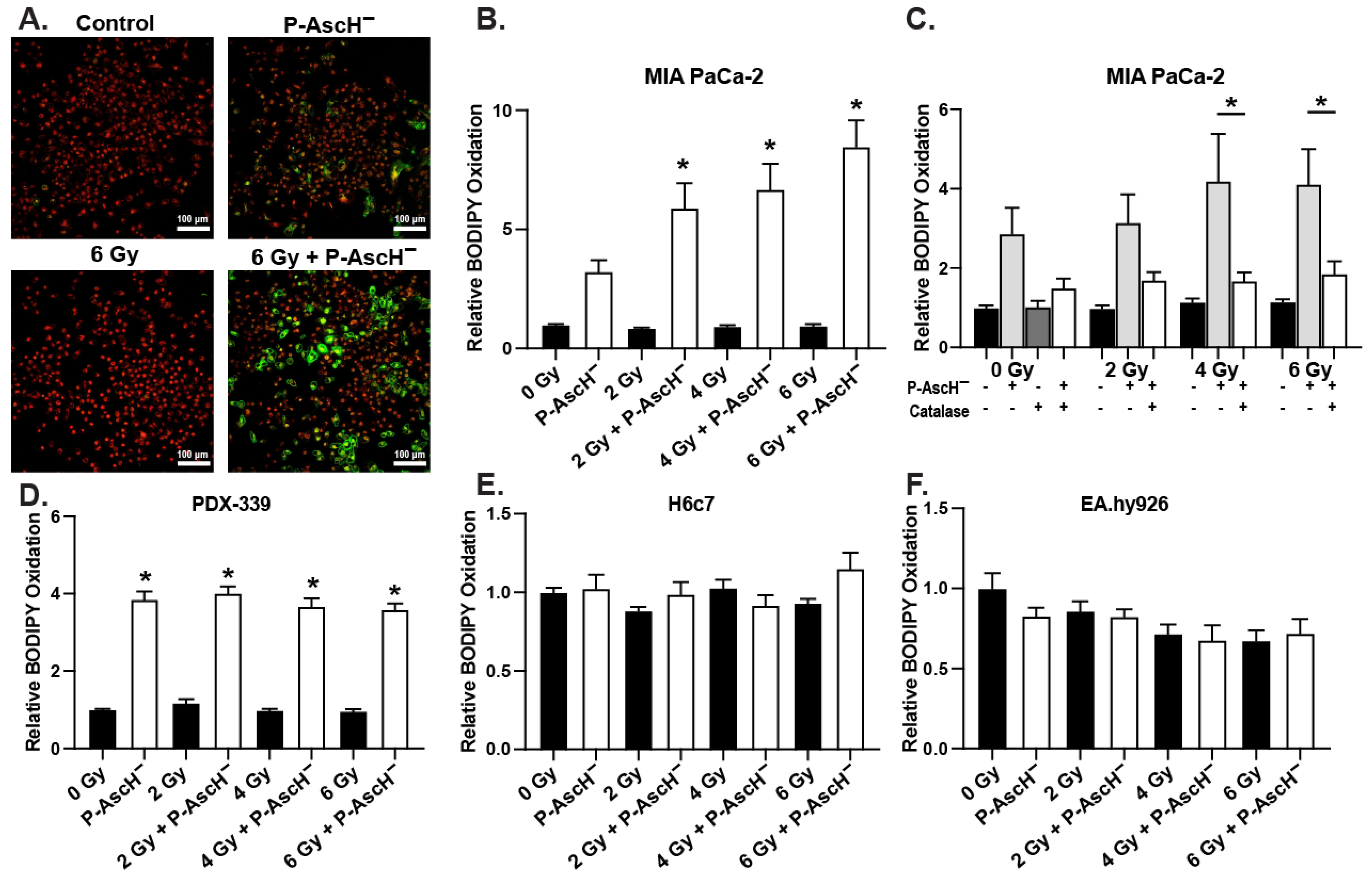
3.2. Lipid Peroxidation Contributes to P-AscH−-Induced Radiosensitivity
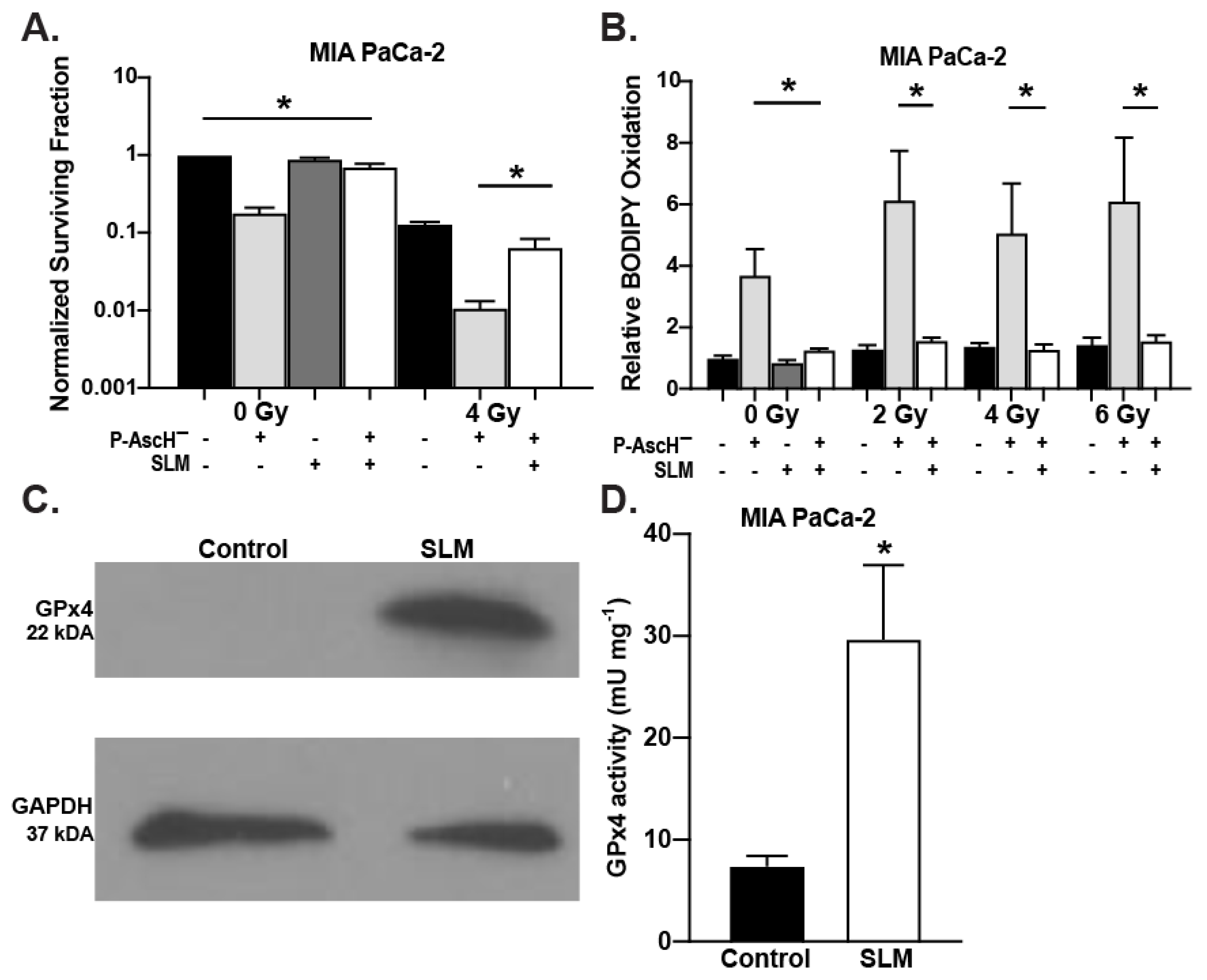
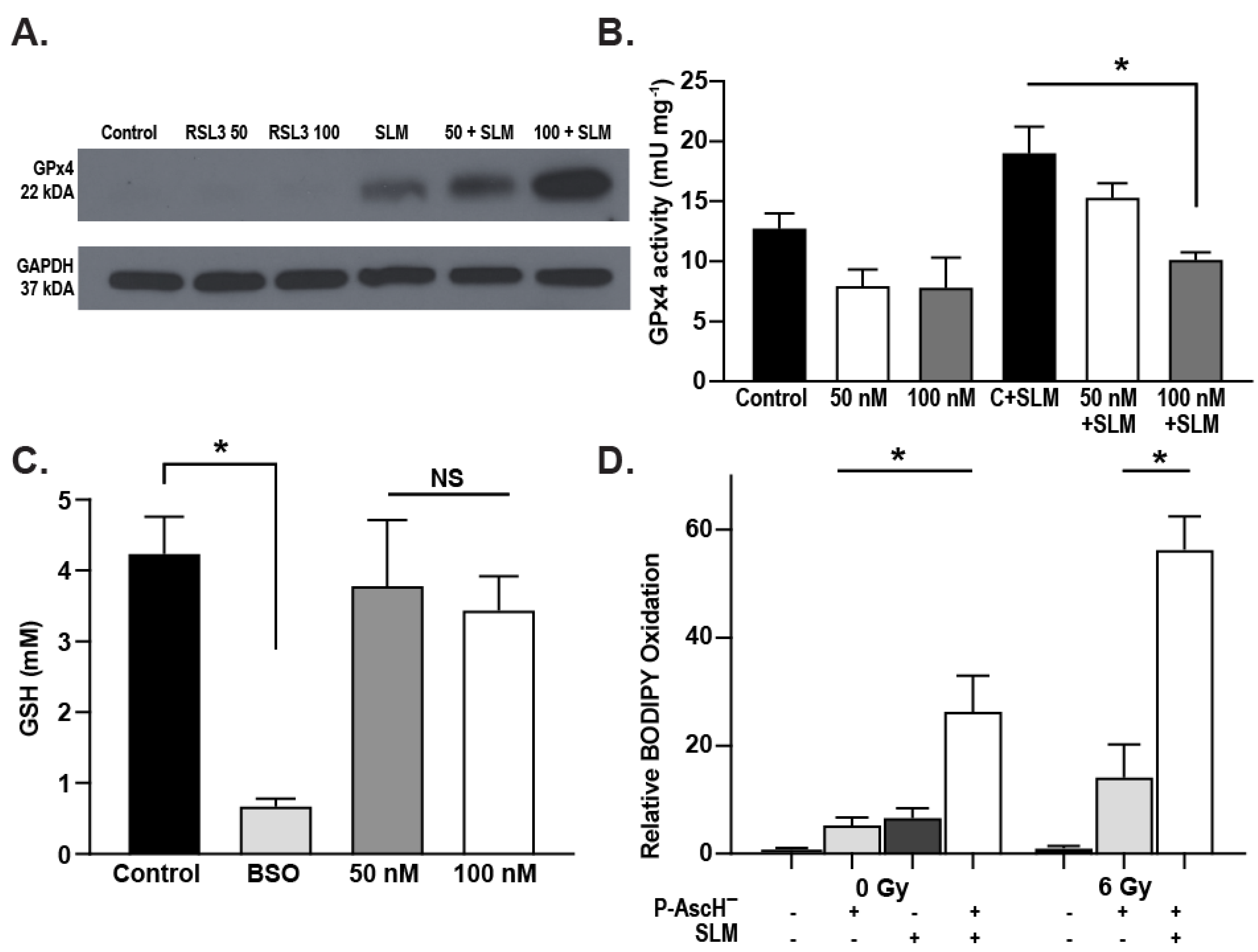
3.3. Selenomethionine Reverses the Effects of P-AscH− on PDAC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute. Cancer Stat Facts: Pancreatic Cancer. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 1 February 2023).
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef]
- Powell, S.; McMillan, T.J. DNA damage and repair following treatment with ionizing radiation. Radiother. Oncol. 1990, 19, 95–108. [Google Scholar] [CrossRef]
- Kobayashi, J.; Iwabuchi, K.; Miyagawa, K.; Sonoda, E.; Suzuki, K.; Takata, M.; Tauchi, H. Current topics in DNA double-strand break repair. J. Radiat. Res. 2008, 49, 93–103. [Google Scholar] [CrossRef]
- Spiliopoulos, S.; Zurlo, M.T.; Casella, A.; Laera, L.; Surico, G.; Surgo, A.; Fiorentino, A.; de’Angelis, N.; Calbi, R.; Memeo, R.; et al. Current status of non-surgical treatment of locally advanced pancreatic cancer. World J. Gastrointest. Oncol. 2021, 13, 2064–2075. [Google Scholar] [CrossRef]
- Landau, E.; Kalnicki, S. The Evolving Role of Radiation in Pancreatic Cancer. Surg. Clin. N. Am. 2018, 98, 113–125. [Google Scholar] [CrossRef]
- Pavic, M.; Niyazi, M.; Wilke, L.; Corradini, S.; Vornhulz, M.; Mansmann, U.; Al Tawil, A.; Fritsch, R.; Horner-Rieber, J.; Debus, J.; et al. MR-guided adaptive stereotactic body radiotherapy (SBRT) of primary tumor for pain control in metastatic pancreatic ductal adenocarcinoma (mPDAC): An open randomized, multicentric, parallel group clinical trial (MASPAC). Radiat. Oncol. 2022, 17, 18. [Google Scholar] [CrossRef]
- Abi Jaoude, J.; Thunshelle, C.P.; Kouzy, R.; Nguyen, N.D.; Lin, D.; Prakash, L.; Bumanlag, I.M.; Noticewala, S.S.; Niedzielski, J.S.; Beddar, S.; et al. Stereotactic Versus Conventional Radiation Therapy for Patients With Pancreatic Cancer in the Modern Era. Adv. Radiat. Oncol. 2021, 6, 100763. [Google Scholar] [CrossRef]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J., 2nd; et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef]
- Du, J.; Martin, S.M.; Levine, M.; Wagner, B.A.; Buettner, G.R.; Wang, S.H.; Taghiyev, A.F.; Du, C.; Knudson, C.M.; Cullen, J.J. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 509–520. [Google Scholar] [CrossRef]
- Cieslak, J.A.; Strother, R.K.; Rawal, M.; Du, J.; Doskey, C.M.; Schroeder, S.R.; Button, A.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Manganoporphyrins and ascorbate enhance gemcitabine cytotoxicity in pancreatic cancer. Free. Radic. Biol. Med. 2015, 83, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Rawal, M.; Schroeder, S.R.; Wagner, B.A.; Cushing, C.M.; Welsh, J.L.; Button, A.M.; Du, J.; Sibenaller, Z.A.; Buettner, G.R.; Cullen, J.J. Manganoporphyrins increase ascorbate-induced cytotoxicity by enhancing H2O2 generation. Cancer Res. 2013, 73, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Espey, M.G.; Chen, P.; Chalmers, B.; Drisko, J.; Sun, A.Y.; Levine, M.; Chen, Q. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free. Radic. Biol. Med. 2011, 50, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.R.; Ruppenkamp, E.K.; Steers, G.J.; Du, J.; Carroll, R.S.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Pharmacological Ascorbate Enhances Chemotherapies in Pancreatic Ductal Adenocarcinoma. Pancreas 2022, 51, 684–693. [Google Scholar] [CrossRef]
- Alexander, M.S.; Wilkes, J.G.; Schroeder, S.R.; Buettner, G.R.; Wagner, B.A.; Du, J.; Gibson-Corley, K.; O’Leary, B.R.; Spitz, D.R.; Buatti, J.M.; et al. Pharmacologic Ascorbate Reduces Radiation-Induced Normal Tissue Toxicity and Enhances Tumor Radiosensitization in Pancreatic Cancer. Cancer Res. 2018, 78, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cieslak, J.A., 3rd; Welsh, J.L.; Sibenaller, Z.A.; Allen, B.G.; Wagner, B.A.; Kalen, A.L.; Doskey, C.M.; Strother, R.K.; Button, A.M.; et al. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer Res. 2015, 75, 3314–3326. [Google Scholar] [CrossRef] [PubMed]
- Loehrer, P.J., Sr.; Feng, Y.; Cardenes, H.; Wagner, L.; Brell, J.M.; Cella, D.; Flynn, P.; Ramanathan, R.K.; Crane, C.H.; Alberts, S.R.; et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: An Eastern Cooperative Oncology Group trial. J. Clin. Oncol. 2011, 29, 4105–4112. [Google Scholar] [CrossRef]
- Roy, I.; Zimmerman, N.P.; Mackinnon, A.C.; Tsai, S.; Evans, D.B.; Dwinell, M.B. CXCL12 chemokine expression suppresses human pancreatic cancer growth and metastasis. PLoS ONE 2014, 9, e90400. [Google Scholar] [CrossRef]
- Kim, M.P.; Evans, D.B.; Wang, H.; Abbruzzese, J.L.; Fleming, J.B.; Gallick, G.E. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc. 2009, 4, 1670–1680. [Google Scholar] [CrossRef]
- Alexander, M.S.; O’Leary, B.R.; Wilkes, J.G.; Gibson, A.R.; Wagner, B.A.; Du, J.; Sarsour, E.; Hwang, R.F.; Buettner, G.R.; Cullen, J.J. Enhanced Pharmacological Ascorbate Oxidation Radiosensitizes Pancreatic Cancer. Radiat. Res. 2019, 191, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.A.; Buettner, G.R. Stability of aqueous solutions of ascorbate for basic research and for intravenous administration. Adv. Redox Res. 2023, 9, 100077. [Google Scholar] [CrossRef] [PubMed]
- Doskey, C.M.; van ‘t Erve, T.J.; Wagner, B.A.; Buettner, G.R. Moles of a Substance per Cell Is a Highly Informative Dosing Metric in Cell Culture. PLoS ONE 2015, 10, e0132572. [Google Scholar] [CrossRef]
- Allen, B.G.; Bhatia, S.K.; Buatti, J.M.; Brandt, K.E.; Lindholm, K.E.; Button, A.M.; Szweda, L.I.; Smith, B.J.; Spitz, D.R.; Fath, M.A. Ketogenic diets enhance oxidative stress and radio-chemo-therapy responses in lung cancer xenografts. Clin. Cancer Res. 2013, 19, 3905–3913. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Beers, R.F., Jr.; Sizer, I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef]
- Stolwijk, J.M.; Falls-Hubert, K.C.; Searby, C.C.; Wagner, B.A.; Buettner, G.R. Simultaneous detection of the enzyme activities of GPx1 and GPx4 guide optimization of selenium in cell biological experiments. Redox Biol. 2020, 32, 101518. [Google Scholar] [CrossRef]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, S.; He, C.; Wang, C.; Wang, L.; Piao, M.; Chi, G.; Luo, Y.; Ge, P. RSL3 induced autophagic death in glioma cells via causing glycolysis dysfunction. Biochem. Biophys. Res. Commun. 2019, 518, 590–597. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Alexander, M.S.; Waldron, T.J.; Sibenaller, Z.A.; Spitz, D.R.; Buettner, G.R.; Allen, B.G.; Cullen, J.J. Pharmacological Ascorbate as a Means of Sensitizing Cancer Cells to Radio-Chemotherapy While Protecting Normal Tissue. Semin. Radiat. Oncol. 2019, 29, 25–32. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.R.; Alexander, M.S.; Du, J.; Moose, D.L.; Henry, M.D.; Cullen, J.J. Pharmacological ascorbate inhibits pancreatic cancer metastases via a peroxide-mediated mechanism. Sci. Rep. 2020, 10, 17649. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Carroll, R.S.; Steers, G.J.; Wagner, B.A.; O’Leary, B.R.; Jensen, C.S.; Buettner, G.R.; Cullen, J.J. Catalase Modulates the Radio-Sensitization of Pancreatic Cancer Cells by Pharmacological Ascorbate. Antioxidants 2021, 10, 614. [Google Scholar] [CrossRef]
- Mao, C.; Lei, G.; Horbath, A.; Gan, B. Assessment of lipid peroxidation in irradiated cells. Methods Cell Biol. 2022, 172, 37–50. [Google Scholar] [CrossRef]
- Flohe, L.; Gunzler, W.A.; Schock, H.H. Glutathione peroxidase: A selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Gregolin, C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim. Biophys. Acta 1985, 839, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Stolwijk, J.M.; Garje, R.; Sieren, J.C.; Buettner, G.R.; Zakharia, Y. Understanding the Redox Biology of Selenium in the Search of Targeted Cancer Therapies. Antioxidants 2020, 9, 420. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, G.Y.; O’Leary, B.R.; Du, J.; Carroll, R.S.; Steers, G.J.; Buettner, G.R.; Cullen, J.J. Pharmacologic Ascorbate Radiosensitizes Pancreatic Cancer but Radioprotects Normal Tissue: The Role of Oxidative Stress-Induced Lipid Peroxidation. Antioxidants 2024, 13, 361. https://doi.org/10.3390/antiox13030361
Chen GY, O’Leary BR, Du J, Carroll RS, Steers GJ, Buettner GR, Cullen JJ. Pharmacologic Ascorbate Radiosensitizes Pancreatic Cancer but Radioprotects Normal Tissue: The Role of Oxidative Stress-Induced Lipid Peroxidation. Antioxidants. 2024; 13(3):361. https://doi.org/10.3390/antiox13030361
Chicago/Turabian StyleChen, Gloria Y., Brianne R. O’Leary, Juan Du, Rory S. Carroll, Garett J. Steers, Garry R. Buettner, and Joseph J. Cullen. 2024. "Pharmacologic Ascorbate Radiosensitizes Pancreatic Cancer but Radioprotects Normal Tissue: The Role of Oxidative Stress-Induced Lipid Peroxidation" Antioxidants 13, no. 3: 361. https://doi.org/10.3390/antiox13030361