
Oxidative Stress, Inflammation, and Mitochondrial Dysfunction: A Link between Obesity and Atrial Fibrillation

1
Center for Advanced Medical and Pharmaceutical Research, University of Medicine, Pharmacy, Science and Technology “George Emil Palade” of Târgu Mureș, 540142 Târgu Mureș, Romania
2
Physiology Department, University of Medicine, Pharmacy, Science and Technology “George Emil Palade” of Târgu Mureș, 540142 Târgu Mureș, Romania
*
Author to whom correspondence should be addressed.
Antioxidants 2024, 13(1), 117; https://doi.org/10.3390/antiox13010117
Submission received: 18 December 2023
/
Revised: 11 January 2024
/
Accepted: 16 January 2024
/
Published: 17 January 2024
(This article belongs to the Section Health Outcomes of Antioxidants and Oxidative Stress)
Abstract
:The adipose tissue has long been thought to represent a passive source of triglycerides and fatty acids. However, extensive data have demonstrated that the adipose tissue is also a major endocrine organ that directly or indirectly affects the physiological functions of almost all cell types. Obesity is recognized as a risk factor for multiple systemic conditions, including metabolic syndrome, type 2 diabetes mellitus, sleep apnea, cardiovascular disorders, and many others. Obesity-related changes in the adipose tissue induce functional and structural changes in cardiac myocytes, promoting a wide range of cardiovascular disorders, including atrial fibrillation (AF). Due to the wealth of epidemiologic data linking AF to obesity, the mechanisms underlying AF occurrence in obese patients are an area of rich ongoing investigation. However, progress has been somewhat slowed by the complex phenotypes of both obesity and AF. The triad inflammation, oxidative stress, and mitochondrial dysfunction are critical for AF pathogenesis in the setting of obesity via multiple structural and functional proarrhythmic changes at the level of the atria. The aim of this paper is to provide a comprehensive view of the close relationship between obesity-induced oxidative stress, inflammation, and mitochondrial dysfunction and the pathogenesis of AF. The clinical implications of these mechanistic insights are also discussed.
1. Introduction
Obesity represents an important public health issue, with a significant increase in incidence and prevalence over the past 50 years [1]. It is currently thought to affect more than one billion people around the globe, and its prevalence is continuously on the rise. Moreover, the prevalence of overweight or obese children and adolescents has increased more than four-fold in the last 40 years, representing a major societal concern [2]. The deleterious impact of obesity is not exclusively attributable to its presence. Many of the negative effects of obesity result from obesity-related diseases, including cardiovascular diseases, diabetes mellitus, metabolic syndrome, respiratory disturbances, chronic kidney disease, or fatty liver disease [1].
Among cardiovascular diseases, atrial fibrillation (AF) is the most common sustained cardiac rhythm disorder, affecting approximately 2% of the European population, according to recent data. Even though the prevalence of the disease is estimated at less than 1% in individuals under 49 years of age, it increases to more than 15% in those over 80 years of age [3,4]. A series of cardiac (e.g., coronary heart disease, arterial hypertension, heart failure) and non-cardiac (e.g., diabetes, hyperthyroidism, chronic kidney disease) conditions are known to be associated with the occurrence and maintenance of AF [5]. Extensive evidence has associated obesity with AF [6,7,8], with obese individuals having a more than two-fold higher risk of developing AF compared to non-obese individuals [9]. Clinical conditions frequently present in obese people (e.g., hypertension, diabetes, obstructive sleep apnea) are major risk factors for AF [9]. Obesity-related hemodynamic changes and myocardial ischemia further increase the risk of AF. Additionally, the fat tissue itself has major implications in AF [9,10]. Visceral fat promotes metabolic processes such as chronic systemic inflammation, oxidative stress, and increased insulin resistance, which ultimately damage the atria and increase the risk of AF. Meanwhile, the epicardial fat exerts a direct impact on the atria through mechanical and paracrine mechanisms, as well as interaction with the cardiac ganglionated plexi [9,10]. Obesity affects cardiac structure and function and contributes to cardiac disorders pathogenesis, particularly via the triad of inflammation, oxidative stress, and mitochondrial dysfunction [11]. The same triad can also provide an explanation for the complex pathophysiological link between obesity and AF (Figure 1).
Obesity contributes to atrial fibrillation by affecting cardiac structure and function, particularly via the triad of inflammation, oxidative stress, and mitochondrial dysfunction. In turn, atrial fibrillation promotes inflammation, oxidative stress, and mitochondrial dysfunction, thereby leading to a self-perpetuating cycle.
In this paper, we aimed to provide a comprehensive view of the close relationship between obesity-induced oxidative stress, inflammation, and mitochondrial dysfunction and the pathogenesis of AF (Figure 2). The clinical implications of these mechanistic links are also discussed.
2. Inflammation in Obesity and Atrial Fibrillation
Both acute and chronic inflammation play an important role in inducing tissue damage. Intracellular pathways are responsible for the production of various inflammatory mediators involved in the occurrence and progression of numerous chronic disorders, including cardiovascular diseases, non-alcoholic fatty liver disease, acute and chronic kidney disease, and lung diseases [12].
A large body of evidence links obesity with chronic inflammation, with the interrelationship between the two being primarily related to the over-expression of proinflammatory cytokines [13,14]. In parallel with its expansion, in obesity, the adipose tissue becomes infiltrated by immune cells, particularly macrophages [13,14,15,16]. This infiltration is a key contributor to the low-grade inflammation that occurs in the adipose tissue. In addition, the adipose tissue per se is an important source of mediators of inflammation, including proinflammatory cytokines and leptin, which directly contribute to inflammation in obese individuals [16,17,18,19]. Obesity also triggers the activation of immune cells, including T cells and macrophages, leading to the release of additional inflammatory mediators [13,14,15,16]. This immune activation is not limited to the adipose tissue but can also affect other organs, contributing to the typical systemic inflammatory syndrome associated with obesity [13,14,15,16]. Increased levels of proinflammatory cytokines, such as tumor necrosis factor-alpha (TNFα), transforming growth factor-beta (TGF-β), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6) were reported in adult obese patients [15]. All these mediators are produced by the macrophages from the adipose tissue via leptin receptor activation [13]. Leptin was found to positively correlate with IL-6 and TNFα levels, as well as with clinical parameters of obesity (i.e., body mass index (BMI) and abdominal circumference) [16]. Other studies also showed that central obesity is associated with higher levels of leptin, TNFα, and IL-1β [17,18]. In a cohort of 740 patients, IL-6 and C-reactive protein (CRP) levels positively correlated with central obesity [19]. In addition to CRP [20], other acute-phase proteins, such as the proinflammatory adipokine serum amyloid A, could also play a critical role in obesity-associated inflammation [21,22] and in obesity-related complications, including AF. The impact of weight loss on the levels of inflammatory markers is still being discussed. In a study conducted by Greco et al., a modest calorie restriction and weight reduction were associated with a significant decrease in leptin levels [23]. Meanwhile, in the study by Rość et al., a decrease in bodyweight by approximately 9% in morbidly obese patients was not associated with a significant decrease in the levels of inflammatory cytokines (IL-6 and TNFα) [24]. These results suggest that, at least in morbidly obese patients, only substantial decreases in the amount of adipose tissue, probably leading to BMI normalization, may reflect a significant improvement in obesity-related inflammation [24].
Numerous studies, therefore, point to obesity as a proinflammatory disease. In parallel, studies incriminate obesity as a major risk factor for both the occurrence and maintenance of AF [15,25,26], and inflammation appears to represent one of the key elements of the link between obesity and AF. Inflammation increases the vulnerability to AF through both electrical and structural remodeling [27]. The increased prevalence of AF in inflammatory conditions such as myocarditis, pericarditis, endocarditis, or after cardiac surgery strongly supports the contribution of inflammation to AF development [10,28]. In recent studies performed in patients undergoing coronary artery bypass grafting surgery, preoperative CRP and IL-6 serum levels positively correlated with the occurrence of postoperative AF (Table 1) [29,30]. In the study by Li et al., CRP levels also positively correlated with the risk of AF in the general population [31].
The mechanisms through which obesity-induced inflammation contributes to the onset and maintenance of AF are complex and multifactorial (Figure 3). Chronic inflammation leads to atrial myopathy, characterized by substantial changes in the electrical and structural properties of the atrial tissue. Under the effects of TGF-β (via increased α-SMA expression), fibroblasts transdifferentiate into myofibroblasts, specialized profibrotic and proinflammatory cells that modify the structure of the extracellular matrix through increased production of collagen [36]. Collagen then contributes to reducing cardiac compliance, interrupting cell-to-cell connections, and decreasing conduction velocity, thus contributing to the typical pathological remodeling observed in AF [37]. In addition, activin A, a member of the TGF-β family, has been shown to increase the deposition of fibrotic material in cell culture, supporting the direct role of TGF-β in cardiac fibrosis and structural remodeling [38]. In addition, cardiac fibrosis involved in the pathogenesis of AF also appears as a consequence of the upregulation of matrix metalloproteinases and modulation of extracellular matrix degradation via TNFα [39]. The adipose tissue is also an important source of IL-2, IL-6, IL-8, and monocyte chemoattractant protein-1. These cytokines contribute to the occurrence and maintenance of AF through cellular (macrophage and neutrophile) infiltration in the myocardium, as well as through inflammation-induced oxidative stress and subsequent fibrosis [37,38,39,40]. All these structural changes induced by inflammation lead to collagen and fibrous tissue deposition within the atria and contribute to progressive loss of atrial compliance and atrial enlargement and stretching, creating the perfect environment for initiation and maintenance of AF [25,27]. Atrial fibrosis also disrupts electrical communication between the adjacent cells and is associated with altered expression and function of ion channels responsible for the generation and conduction of electrical signals within the atria [25,27]. These changes can then lead to alterations in atrial action potential duration and increased spontaneous activity in the atrial cells, increasing the propensity to atrial ectopic activity and the formation of re-entry circuits, thus favoring AF [25,27].
The increased inflammation associated with obesity also leads to electrical changes within the myocardium, contributing to the typical atrial electrical remodeling observed in AF. TNFα has been shown to alter Ca2+ handling in the cardiomyocytes of the pulmonary veins, probably by decreasing sarcoplasmic reticulum Ca2+ ATPase expression, thereby favoring the occurrence of delayed afterdepolarizations and AF [41,42]. Abnormalities in Ca2+ handling are also mediated by IL-6, with major implications in atrial arrhythmogenesis [43]. An increase in Na+ current density induced by IL-2 via SCN3B overexpression could also contribute to the atrial electrical remodeling observed in AF [44]. In addition, in transgenic mice with cardiac-restricted overexpression of TNFα, connexins 40 and 43 were downregulated and lateralized, respectively, with consequences on atrial conduction and atrial arrhythmias [45].
The NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome, is a complex signaling pathway in the immune system that plays a crucial role in the initiation and regulation of inflammation [46]. The NLRP3 inflammasome is an intracellular multiprotein complex with a role in cleaving pro-IL-β and pro-interleukin-18 (IL-18) via cysteine protease caspase-1 to generate proinflammatory IL-1β and IL-18 [46]. In addition to cytokine maturation, caspase-1 activation triggers pyroptosis, a form of programmed cell death characterized by cell swelling, membrane rupture, and release of cellular contents, which further promotes inflammation [46]. Studies have demonstrated the involvement of the NLRP3 inflammasome pathway in coronary artery disease, acute myocardial infarction, and heart failure, which are major drivers of AF-promoting atrial remodeling [46]. The relationship between NLRP3 activation and AF was demonstrated in an experimental study by Yao and colleagues [47]. In addition, NLRP3 inflammasome activation and regulation seem to have a role in adipose tissue dysfunction and insulin resistance, possibly representing a link between obesity and AF [48].
The relationship between obesity, inflammation, and AF could also be mediated by hypoxia [49]. In AF, an up-regulation of the hypoxia-inducible factor (HIF) pathway and an increased expression of hypoxic and angiogenic markers were observed [49], and the transcription factor HIF-1α is upregulated in the adipose tissue in obesity. In obesity, HIF-1α contributes to chronic inflammation by promoting the expression of proinflammatory cytokines and recruiting M1 macrophages [50]. The increased expression of HIF-1α leads to the activation of a profibrotic transcriptional program, resulting in collagen I, III, IV, and lysyl oxidase synthesis, ultimately causing fibrosis in the adipose tissue [51,52]. HIF-1α has also been implicated in the pathophysiology of AF, particularly through structural remodeling, including fibrosis [53]. Ogi et al. observed increased atrial fibrosis in patients with AF that may be secondary to myocardial hypoxia, implicating HIF-1α as a key mediator in this process [53]. Moreover, inhibition of HIF-1α expression reduced the level of cytokines involved in atrial fibrosis (e.g., TGF-β1, MMP-9) and attenuated atrial structural changes [54]. Although a direct relationship between HIF-1α and electrical remodeling has not yet been demonstrated, cytokines released due to increased HIF-1α activity, such as IL-6 and TNFα, have been implicated in proarrhythmic electrical remodeling of the atria [55,56]. This suggests a potential link between HIF-1α-mediated inflammation and electrical disturbances contributing to AF. Together, these data suggest that the HIF-1α pathway, which is activated in obesity and contributes to chronic inflammation and fibrosis, may also play a role in the pathophysiology of AF. Other transcription factors, including the high-mobility group protein AT-hook 1 (HMGA1), have been identified as participants in the hypoxia-induced inflammatory responses within the adipose tissue [57]. Functioning as an architectural transcription factor, HMGA1 plays a central role in a range of biological processes, including inflammation, tumorigenesis, and metabolism [57,58]. Several studies have shown that HMGA1 physically and/or functionally interacts with nuclear factor-kappa B (NF-kB) and HIF-1, particularly in the context of hypoxia-associated inflammation, leading to the subsequent release of numerous proinflammatory cytokines [58].
In addition to the considerable amount of data regarding the mechanisms by which inflammation contributes to the occurrence of AF, the inflammation induced by AF per se is also non-negligible. The increased levels of TNFα and IL-6 and the activation of the renin–angiotensin–aldosterone and the sympathetic nervous systems commonly associated with AF all contribute to the proinflammatory status observed in the presence of the arrhythmia [59]. Thus, inflammation may also be a result of AF, with a bidirectional relationship between inflammation and AF being present in most patients. This complex relationship may contribute to the development and maintenance of AF and may represent a promising therapeutic target in the treatment of arrhythmia.
The relationship between obesity and AF has been less studied from a genetic point of view. However, variants in genes such as those encoding for cholesteryl transfer protein (CETP), CRP, and G protein-coupled inward rectifier K(+) channel 4 (GIRK4) have been shown to play crucial roles in influencing susceptibility to AF, particularly in the context of obesity. TaqIB of CETP (B2 allele as a protective factor) and CRP 1444 C/T polymorphism may contribute to the susceptibility to AF [60]. In men, these genetic variants were associated with BMI, suggesting a gender-specific genetic influence on the relationship between obesity and AF [60]. Abnormal expression of GIRK4 has also been associated with AF [61]. The connection between GIRK4 expression and AF was previously highlighted and was shown to be closely related to obesity and metabolic syndrome, suggesting that genetic variations in GIRK4 may contribute to the link between AF and obesity [61].
Several microRNAs (miRNAs) have been identified as common regulators in both obesity and cardiac remodeling. These miRNAs may influence shared pathways related to endothelial dysfunction and fibrosis, providing a possible molecular link between obesity and AF. Although a direct link to AF is not explicitly mentioned, Zou et al. demonstrated that miRNA-410-5p is markedly upregulated in the cardiac tissue of obese rats [62]. The TGF-β signaling pathway activation via miRNA-410-5p suggests a potential mechanism for cardiac fibrosis and dysfunction in obesity [62]. As cardiac fibrosis is a common feature in the pathogenesis of AF, miRNA-410-5p could play a role in the relationship between obesity and AF [9]. Altered concentrations of hsa-miR-125a-5p, hsa-miR-342-3p, and hsa-miR-365b-3p in the plasma of obese children with endothelial dysfunction raise the possibility of a connection between these miRNAs and the relationship between AF and obesity [63]. The emerging evidence that endothelial dysfunction is implicated in the promotion and maintenance of atrial arrhythmic substrate and predicts adverse outcomes in AF supports the idea that these miRNAs could play a role in the complex interplay between AF, obesity, and endothelial dysfunction [63,64]. MiR-1-3p and miR-133a-3p were upregulated in extracellular vesicles released from the epicardial adipose tissue [65]. Overexpression of these miRNAs was associated with conduction slowing and reduced KCNJ2 and KCNJ12 expression, suggesting that they may act as mediators of epicardial adipose tissue-induced arrhythmogenicity [65]. As the amount of epicardial adipose tissue is directly proportional to the degree of obesity, these findings suggest a link between miR-1-3p and miR-133a-3p and the obesity-AF relationship [65]. In a mouse model of atrial fibrosis induced by a high-fat diet, upregulated miR-205-5p was associated with decreased atrial fibrosis, suggesting that miR-205-5p could represent a therapeutic target for atrial fibrosis-related arrhythmias [66]. The role of these miRNAs and others in the obesity-AF relationship remains to be investigated in future studies.
Inflammation and oxidative stress are interconnected processes, and a complex inter-relationship exists between the two. On the one hand, inflammation stimulates reactive oxygen species (ROS) production. For example, during the inflammatory reaction, immune cells such as macrophages can release ROS to destroy pathogens [67]. On the other hand, inflammation can induce the activity of antioxidant enzymes and oxidative stress defense factors to counteract the harmful effects of ROS [67]. In its turn, oxidative stress also contributes to inflammation [38]. ROS functions as alarm signals for the immune system, initiating inflammation. For example, ROS activates the NF-κB pathway, a key regulator of the inflammatory response [67]. ROS also directly affects lipid and protein inflammatory molecules by causing chemical and functional changes in these molecules [67].
3. Oxidative Stress in Obesity and Atrial Fibrillation
The relationship between obesity and oxidative stress is complex and involves chronic inflammation, production of free radicals, impairment of mitochondrial function, antioxidant system imbalance, and other metabolic disorders [10,68,69,70]. All these factors contribute to a bidirectional relationship between obesity and oxidative stress, with obesity increasing oxidative stress and increased oxidative stress contributing to the development of metabolic disorders and diseases associated with obesity [69,70].
Oxidative stress designates an imbalance between ROS production and antioxidants [71]. ROS molecules are produced in the majority of body cells as a result of mitochondrial aerobic metabolism, cytoplasmic enzymatic reactions, or from exogenous oxidant sources (e.g., X-rays, pollutants, cigarette smoking) [72,73], and ROS overproduction increases oxidative stress [74]. At a chemical level, ROS are formed as products of physiological oxygen (peroxide, superoxide, hydroxyl, and singlet oxygen) metabolism [75]. Mitochondria are responsible for the production of over 90% of the superoxide anion by transferring electrons to the oxygen molecule through electron transport chain complexes I/III [73]. In the case of hypoxia, the mitochondrial electron transport chain is disrupted, leading to an incomplete reduction in oxygen molecules and increased generation of ROS [68]. In turn, increased oxidative stress can modify proteins, lipids, and DNA, activating various signaling pathways and leading to cell apoptosis [75].
Even though the white adipose tissue is not considerably rich in mitochondria, normal mitochondrial function is essential in this tissue for the production of energy required for adipocyte differentiation and maturation [73]. Prolonged exposure to ROS of the adipose tissue leads to DNA impairment, resulting in mitochondrial dysfunction and, consecutively, adipogenesis and adipocyte hypertrophy [73]. Oxidative stress, expressed as an overproduction of hydrogen peroxide determined by catalase deficiency, was shown to be involved in obesity through lipo- and adipogenesis [68]. Experimental studies have shown that a high-fat (at least 41% fat) diet in aging C57Bl/6 mice is associated with protein oxidation and increased oxidative stress [76]. In parallel, the activity of antioxidant enzymes glutathioneperoxidase, catalase, and superoxide dismutase is decreased in obese patients [77]. Importantly, weight loss using caloric restriction [78] or bariatric surgery [79] appears to be associated with an improvement in oxidative stress parameters. The use of dietary and supplement antioxidants has also been shown to mitigate oxidative stress and improve obesity and obesity-related conditions [69].
A series of features commonly associated with obesity, such as hyperglycemia, raised levels of plasma lipids and leptin, chronic low-grade inflammation, and altered response to muscle activity, has been associated with the presence of oxidative stress in obesity. Overproduction of nicotinamide adenine dinucleotide and dihydroflavine-adenine dinucleotide as a consequence of an increase in intracellular glucose leads to increased proton gradient across the mitochondrial inner membrane and to superoxide production [80]. Elevated free fatty acid levels promote the production of free oxygen radicals at the mitochondrial level via inhibition of adenine nucleotide translocation [81]. The chronic low-grade inflammation typically associated with obesity also contributes to oxidative stress promotion. Increased levels of TNFα and IL-6, commonly encountered in obesity, augment superoxide anion production and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) activity [82,83]. The elevation in plasma leptin further amplifies oxidative stress via NOX activation and the production of reactive intermediates [84]. Increased levels of lipid hydroperoxide, isoprostane, and protein carbonyl were observed in animal models following leptin administration [85]. Furthermore, in obese patients, muscle activity during exercise is associated with an abnormal increase in the rate of cellular respiration and oxygen consumption and, consequently, with abnormally high post-exercise lipid hydroperoxide levels [86,87].
In parallel, clinical and experimental studies have demonstrated that oxidative stress contributes to proarrhythmic atrial electrical and structural remodeling through a series of complex mechanisms, thus increasing the susceptibility to AF. Increased levels of ROS (e.g., superoxide and H2O2) and the ratio of oxidized to reduced glutathione, a marker of oxidative stress, have both been associated with AF [88,89]. Atrial structural remodeling associated with AF seems to be promoted by oxidative damage of myofibrils via hydroxyl and peroxynitrite radicals [90,91]. In addition, mitochondrial DNA damage induced by oxidative stress modulates Ca2+ channels and Ca2+ handling proteins, leading to Ca2+ overload and atrial electrical remodeling [92]. Meanwhile, treatment with antioxidants seems to decrease the risk of postoperative AF. For instance, treatment with vitamin C has been shown to decrease the incidence of AF after cardiac surgery, as well as arrhythmia recurrence after electrical cardioversion of persistent AF [93,94]. Treatment with other antioxidants has also been associated with a lower risk of AF (Table 2). Administration of N-acetyl cysteine was shown to reduce the risk of AF by increasing the density of L-type calcium current [95]. Probucol, xanthine oxidase inhibitors (e.g., allopurinol), selective NOX inhibitors (e.g., apocynin), sodium nitroprusside, and statins, which have potent antioxidant effects, also demonstrated a positive impact on the risk of developing AF [67]. Treatment with antioxidants to reduce ROS and AF risk could also decrease oxidative stress and provide additional benefits in other organs. For instance, n-3 polyunsaturated fatty acids have been shown not only to decrease the incidence of postoperative AF but also to improve musculoskeletal health related to sarcopenia [96]. A healthy lifestyle, with the adoption of a Mediterranean diet, consumption of olive oil, and weight loss, has also been associated with a reduction in oxidative stress and, thus, in the risk of AF [67,97].
Several sources of ROS have been identified in the setting of AF. Among these, NOX, xanthine oxidase, nitric oxide synthase uncoupling, mitochondrial dysfunction, myeloperoxidases, and monoamine oxidases are the most studied sources [67]. The molecular mechanisms underlying the increase in ROS in AF are complex. Although NOX isoform/subunit levels were unchanged, NOX-dependent ROS production and highly upregulated Rac1 expression were observed in a model of pacing-induced AF [112]. Cardiac-specific Rac1 overexpression was also associated with increased prevalence of AF in aged mice, whereas Rac1 downregulation using statins reduced the incidence of angiotensin II-induced AF in endothelial nitric oxide synthase null mice [113,114]. In agreement with animal studies, significant upregulation of Rac1 GTPase and NOX activities has also been observed in patients with AF [114], whereas the use of diphenyleneiodonium and apocynin was shown to inhibit the production of superoxide via the inhibition of flavin-containing oxidases and of p47phox translocation, respectively [115]. Overproduction of NADPH-dependent superoxide was also significantly increased in patients with postoperative AF, whereas atorvastatin treatment attenuated this process, probably by inhibiting Rac1-dependent NOX activation [115,116].
Although the increase in oxidative stress seems to precede the appearance of AF, it could also be a consequence of the arrhythmia. In experimental studies, pacing-induced AF led to NOX-dependent ROS production and upregulation of Rac1 expression, demonstrating that oxidative stress is not only a cause but also a consequence of AF. Moreover, the increase in ROS is also incriminated in the occurrence of consequences of AF, such as thrombosis, inflammation, and even the “AF begets AF” phenomenon [112].
Emerging research has highlighted the pivotal role played by mitochondrial-derived ROS in the intricate landscape of AF [102,117]. Beyond their conventional implications in oxidative stress, mitochondrial ROS are now recognized as crucial components in AF genesis and maintenance. While the focal point often revolves around ROS generated through electron leakage within the mitochondrial respiratory chain, an important role is also played by ROS accumulation resulting from mitochondrial calcium overload.
4. Mitochondrial Dysfunction in Obesity and Atrial Fibrillation
The mitochondria’s main function is the production of adenosine triphosphate (ATP) from food substrates, thus playing a central role in energy metabolism [118]. The role of mitochondria extends, however, far beyond the production of energy [118]. During the reactions responsible for ATP production, ROS is also produced at the level of the mitochondria, which represent the main seat of ROS production [118,119]. An exact definition of mitochondrial dysfunction is difficult to formulate. Although it is classically defined as the inability of the mitochondria to generate and sustain sufficient levels of ATP for the cell, metabolic disorders of substrate, Ca2+ buffering, mitochondrial DNA mutations, changes in mitochondrial size and morphology, and/or ROS production are also commonly present and can be seen as part of the definition of mitochondrial dysfunction [118,119].
Since excessive nutrient consumption affects mitochondrial function, it is not surprising that obesity is a strong contributor to mitochondrial dysfunction [118]. An increase in free fatty acid concentrations, hyperglycemia, and ROS production, as a consequence of excessive nutrient intake, compromises mitochondrial function at the level of the adipocytes [118]. Further, mitochondrial dysfunction decreases the rate of β-oxidation, compromising adipogenesis, fatty acid esterification, lipolysis, and adiponectin production [118]. The relationship between mitochondrial dysfunction and obesity does not seem to be limited, however, to the adipose tissue. A reduction in mitochondrial function and size and in mitochondrial fission, with consequent alteration of mitochondrial dynamics (balance between mitochondrial fusion and fission), were also observed in skeletal muscles in mice with both genetic and diet-induced obesity [120]. As a consequence, reduction in fatty acid oxidation and inhibition of glucose transport occurs in muscle tissues in the presence of obesity [121]. Alterations of mitochondrial function associated with obesity have also been shown to occur in the liver. In rats fed for 14 weeks with a high-fat diet, pathological changes included significant fat deposition, liver steatosis, and a disruption in the hepatic mitochondrial quality control processes, evidenced by increased mitochondrial ROS production, mitochondrial DNA damage, impaired mitochondrial biogenesis, and disrupted mitochondrial fusion [122]. Hepatic mitochondrial fission processes are increased, with consequences on mitochondrial respiratory capacity and protein expression, including peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) [123]. Under normal conditions, only a small amount of the oxygen uptaken by the mitochondria is released in the form of ROS [124]. In the presence of mitochondrial dysfunction, there is a significant increase in the production of ROS, with consequences on mitochondrial and nuclear nucleic acids, membrane lipids and proteins, and the enzymes of the mitochondrial respiratory chain [124]. This entire process is significantly amplified by the aging of the adipose organ, which leads to alterations in adipogenesis, insulin resistance, abnormal adipokine secretion, inflammation, mitochondrial dysfunction, and cellular and tissue senescence [125,126].
At the level of the heart, more than two-thirds of the energy required for sustained contraction and relaxation comes from the oxidation of fatty acids in the mitochondria. The price of this process is the concomitant production of a certain amount of ROS [127]. However, in physiological conditions, this amount is negligible, as it is rapidly removed by antioxidants [127]. In patients with insulin resistance, such as those with obesity, fatty acid oxidation becomes exceedingly important for energy production, the cardiomyocytes shifting away from glucose utilization [127,128]. Consequently, there is an increase in electron leakage and ROS production, ultimately leading to mitochondrial dysfunction [128,129].
Both experimental and clinical studies have intensively studied the issue of mitochondrial dysfunction in AF. In patients with AF, oxidative stress is increased, and damage to the mitochondrial DNA occurs, altering the bioenergetic function of the mitochondria [92,130]. In parallel, oxidative stress and mitochondrial dysfunction have been shown to contribute to the onset and progression of AF. Together, these data indicate the existence of a bidirectional relationship between mitochondrial dysfunction and AF [92,117]. Data suggest, however, that changes in the function of the mitochondria are already present before AF onset and that, once installed, AF accelerates further changes in mitochondrial function [131]. The exact mechanisms through which mitochondrial dysfunction contributes to AF are incompletely understood. Mitochondrial alterations have been shown to contribute to the electro-pathology of the arrhythmia [132]. Mitochondrial functional and structural remodeling appears to be involved in the pathogenesis of AF by increasing energy deficit and metabolic dysregulation in the human and mouse atria [132]. Low ATP levels affect the intracellular ion balance, decrease the efficiency of all energy-dependent enzymatic reactions, and alter myocardial contraction and relaxation, all of which have been involved in the pathogenesis of AF [131]. Increased ROS (especially superoxide anion) generation and apoptotic cascade activation associated with mitochondrial dysfunction also contribute to AF occurrence [132]. Increased O2- oxidizes numerous intracellular targets, including the ryanodine receptor 2 of the sarcoplasmic reticulum and the sarcolemmal inward Na+ channels, altering cardiomyocyte excitability and intercellular coupling, and thus contributing to maintaining re-entry circuits [133].
Considering the association between mitochondrial dysfunction and AF, it is not surprising that restorers of mitochondrial function have a positive impact on the pathogenesis of AF. Studies have pointed to SS31, a ROS scavenger that improves mitochondrial function by normalizing ATP levels, mitochondrial membrane potential, and mitochondrial morphology, as a promising therapeutic compound in AF [131]. Dipeptidyl peptidase-4 inhibitors, a class of oral antidiabetics, have been shown to improve mitochondrial membrane potential and mitochondrial biogenesis via activation of the PGC-1α/NRF1/Tfam signaling pathway and to decrease the duration of pacing-induced AF in a rabbit model of heart failure [134,135]. Sodium-glucose co-transporter 2 inhibitors reduced tachypacing-induced AF susceptibility by approximately 50% in rats with high-fat diet/streptozotocin-induced diabetes mellitus [136]. The mechanisms potentially involved in this effect are suppression of mitochondrial ROS production, preservation of the barrier function of cardiac microvascular endothelial cells via adenosine monophosphate protein kinase activation-induced mitochondrial fission inhibition, as well as restoration of mitochondrial membrane potential and mitochondrial respiratory rate [136,137]. Other therapeutic strategies that could have a positive impact on the pathogenesis of AF by modifying mitochondrial function are presented in Table 3.
5. Clinical Implications
Accumulating evidence supports a close relationship between the presence of obesity and AF pathogenesis. Given the continuous rise in the prevalence of obesity in the general population, the burden of obesity-related AF is expected to become increasingly important in the near future. Further research will have to provide new therapeutic options with an impact on the genesis and persistence of AF in obese subjects. Several studies have evaluated the effects of proinflammatory activity, oxidative stress, and mitochondrial dysfunction modulation through weight loss, physical activity, and various drugs in obese patients. Although most of these therapeutic interventions managed to efficiently reduce the inflammatory status, ROS, and mitochondrial dysfunction, more aggressive interventions may be needed to modulate the critical processes involved in AF pathogenesis.
Inflammatory mechanisms are implicated in obesity and are critical contributors to AF occurrence and persistence. Different studies have suggested weight loss as a prevention strategy for AF [8]. Reduction in inflammation in obese patients using direct or indirect/pleiotropic anti-inflammatory agents has not managed to demonstrate a clear benefit in the studies carried out so far, suggesting that multitarget pharmacological interventions along with weight loss may be required and that such strategies may need to be applied at an early stage of obesity. The most appropriate therapeutic strategy aimed at preventing atrial remodeling and AF occurrence in obese people remains to be established. The relative impact of inflammation in different stages of atrial remodeling also remains to be elucidated.
Although the prognostic role of inflammatory markers in AF has been well established, the identification of biomarkers that can be used for the diagnosis and prediction of AF in obese patients would be of interest. To date, a series of such biomarkers have been proposed, but no cut-off points have been established, and, more importantly, none of them is specific to AF. Whether such inflammatory markers have additive value in obese people beyond conventional clinical and echocardiographic risk factors needs further confirmation. In recent years, extensive studies have demonstrated the role of miRNAs in inflammation associated with both AF and obesity [65,66]. However, the full impact of miRNAs in obese patients with AF is still unknown. Further studies should evaluate if there are specific miRNAs present both in obesity and AF and, if there are, evaluate whether these miRNAs can be used for diagnosis and/or prediction or as therapeutic targets in obese patients with AF.
Increased oxidative stress observed in obese patients activates a series of processes (including atrial inflammation, fibrosis, and electrical remodeling) that are, in turn, involved in the occurrence of AF [153,154]. But, these processes, including oxidative stress, are also a consequence of AF. As a result, ROS inhibition in obese subjects may serve as a new therapeutic strategy for breaking the oxidative stress-AF vicious circle. A series of agents with antioxidant properties (e.g., vitamin C alone or in combination with vitamin E or n-acetylcysteine, statins) have been extensively studied in AF, but the results of clinical trials have been rather unconvincing. The answer could come from more specific strategies that specifically inhibit ROS-generating pathways or from siRNAs that target different NOX isoforms.
Considering that the increase in ROS in obese people precedes the appearance of AF, a series of markers of oxidative stress could emerge as biomarkers for the prediction of AF in obese people. The strong correlation that appears to exist between AF initiation and oxidative stress could also be useful for identifying novel biomarkers for AF diagnosis in obese patients. Changes in ROS have also been shown to occur alongside the progression of AF. For example, increased expression of p22phox and NOX2 was observed in patients with postoperative AF. This was not the case in patients with permanent AF [155], suggesting that oxidative stress is a dynamic process and that certain biomarkers could be used to determine different evolutionary stages of AF. Determination of these stages using various biomarkers could have implications in the decision to convert patients to sinus rhythm. However, considering the high degree of variability of oxidative stress, finding a biomarker from this spectrum is a real challenge, at least at this moment.
The contribution of mitochondrial dysfunction to AF and obesity and the molecular mechanisms potentially involved in the obesity-AF relationship have been intensively studied. However, pharmaceutical and nutraceutical compounds directed at mitochondrial dysfunction as a treatment strategy in AF have not been investigated in detail yet. Although many drugs with an impact on mitochondrial dysfunction have been studied both experimentally and clinically, there are still gaps in the knowledge regarding their true impact on the prevention of AF and its clinical outcomes. Further randomized clinical trials could contribute to expanding the indications of certain drugs that are already used in cardiovascular diseases or diabetes for AF prevention in obese people or for prevention of AF-related clinical consequences.
The role of the microtubule network in mitochondrial and cardiac function is well known. Recent studies have shown a key role of microtubule disruption in AF. In cardiomyocytes, overexpression of histone deacetylase 6 (HDAC6), a compound that mediates α-tubulin acetylation, responsible for the traffic of metabolites through the cell, results in deacetylation and degradation of microtubules [156]. Experimental and clinical studies have shown that both HDAC6-induced deacetylation and degradation of the microtubule network may underlie mitochondrial dysfunction in AF [156]. However, data regarding the manipulation of the microtubule network in obese patients remain scarce. Elucidating the role of the microtubule network in mitochondrial function may provide further insights into mitochondrial dynamics in obese people and reveal new therapeutic strategies in AF, as suggested by some experimental studies [157]. Biomarkers of mitochondrial dysfunction, such as 8-hydroxy2′-deoxyguanosine, circulating cell-free mitochondrial DNA, and heat shock proteins, could also emerge as promising clinical tools for the prediction and diagnosis of AF in obese people.
6. Conclusions
Atrial fibrillation is the most common cardiac arrhythmia and is associated with increased morbidity and mortality. Studies suggest that up to 20% of AF cases can be attributed to being overweight or obese. Although a plethora of obesity-related diseases, including obstructive sleep apnea, diabetes, metabolic syndrome, and dyslipidemia, contribute to the onset and maintenance of AF, obesity per se has a major contribution to AF occurrence, mainly via inflammation, mitochondrial dysfunction, and oxidative stress. These three processes are interdependent, creating a self-perpetuating cycle that contributes to atrial proarrhythmic structural and functional changes, eventually leading to the appearance and maintenance of AF. It is, therefore, not surprising that weight loss is a broad treatment that addresses several central AF mediators, as it is associated in several studies with a decrease in AF load. Meanwhile, a series of anti-inflammatory and/or antioxidant strategies act at the molecular level, counteracting the harmful effects of inflammation, oxidative stress, and mitochondrial dysfunction, and may thus emerge as possible therapeutic agents with a role in AF prophylaxis. Further studies will have to clarify if and when these therapeutic strategies should be used to prevent the occurrence of AF and/or decrease the arrhythmic load.
Author Contributions
A.I.B. and V.B.H. prepared the figures and drafted the manuscript. A.S. edited and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the University of Medicine, Pharmacy, Science and Technology “George Emil Palade” of Târgu Mureș, grant number 164/6/2023.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lin, X.; Li, H. Obesity: Epidemiology, Pathophysiology, and Therapeutics. Front. Endocrinol. 2021, 12, 706978. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://www.who.int/health-topics/obesity#tab=tab_1 (accessed on 23 June 2023).
- Wilke, T.; Growth, A.; Mueller, S.; Pfannkuche, M.; Verheyen, F.; Linder, R.; Maywald, U.; Bauersachs, R.; Breithardt, G. Incidence and prevalence of atrial fibrillation: An analysis based on 8.3 million patients. Europace 2013, 15, 486–493. [Google Scholar] [CrossRef]
- Zoni-Berisso, M.; Lercari, F.; Carazza, T.; Domenicucci, S. Epidemiology of atrial fibrillation: European perspective. Clin. Epidemiol. 2014, 6, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.; Smit, M.D.; Nguyen, B.O.; Rienstra, M.; Van Gelder, I.C. Risk Factor Management in Atrial Fibrillation. Arrhythm. Electrophysiol. Rev. 2018, 7, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.A.; Giulianini, F.; Geelhoed, B.; Lunetta, K.L.; Misialek, J.R.; Niemeijer, M.N.; Rienstra, M.; Rose, L.M.; Smith, A.V.; Arking, D.E.; et al. Genetic Obesity and the Risk of Atrial Fibrillation: Causal Estimates from Mendelian Randomization. Circulation 2017, 135, 741–754. [Google Scholar] [CrossRef]
- Nalliah, C.J.; Sanders, P.; Kottkamp, H.; Kalman, J.M. The role of obesity in atrial fibrillation. Eur. Heart J. 2016, 37, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Parise, H.; Levy, D.; D’Agostino, R.B.; Wolf, P.A.; Vasan, R.S.; Benjamin, E.J. Obesity and the risk of new-onset atrial fibrillation. JAMA 2004, 292, 2471–2477. [Google Scholar] [CrossRef]
- Goudis, C.A.; Korantzopoulos, P.; Ntalas, I.V.; Kallergis, E.M.; Ketikoglou, D.G. Obesity and atrial fibrillation: A comprehensive review of the pathophysiological mechanisms and links. J. Cardiol. 2015, 66, 361–369. [Google Scholar] [CrossRef]
- Scridon, A.; Dobreanu, D.; Chevalier, P.; Şerban, R.C. Inflammation, a link between obesity and atrial fibrillation. Inflamm. Res. 2015, 64, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Homan, E.A.; Reyes, M.V.; Hickey, K.T.; Morrow, J.P. Clinical Overview of Obesity and Diabetes Mellitus as Risk Factors for Atrial Fibrillation and Sudden Cardiac Death. Front. Physiol. 2019, 9, 1847. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Tuomisto, K.; Jousilahti, P.; Havulinna, A.S.; Borodulin, K.; Männistö, S.; Salomaa, V. Role of inflammation markers in the prediction of weight gain and development of obesity in adults—A prospective study. Metabol. Open 2019, 3, 100016. [Google Scholar] [CrossRef] [PubMed]
- Battineni, G.; Sagaro, G.G.; Chintalapudi, N.; Amenta, F.; Tomassoni, D.; Tayebati, S.K. Impact of Obesity-Induced Inflammation on Cardiovascular Diseases (CVD). Int. J. Mol. Sci. 2021, 22, 4798. [Google Scholar] [CrossRef]
- Borges, M.D.; Franca, E.L.; Fujimori, M.; Silva, S.M.C.; de Marchi, P.G.F.; Deluque, A.L.; Honorio-Franca, A.C.; de Abreu, L.C. Relationship between Proinflammatory Cytokines/Chemokines and Adipokines in Serum of Young Adults with Obesity. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 260–267. [Google Scholar] [CrossRef] [PubMed]
- El-Wakkad, A.; Hassan, N.-M.; Sibaii, H.; El-Zayat, S.R. Proinflammatory, anti-inflammatory cytokines and adipokines in students with central obesity. Cytokine 2013, 61, 682–687. [Google Scholar] [CrossRef]
- Arcidiacono, B.; Chiefari, E.; Foryst-Ludwig, A.; Currò, G.; Navarra, G.; Brunetti, F.S.; Mirabelli, M.; Corigliano, D.M.; Kintscher, U.; Britti, D.; et al. Obesity-related hypoxia via miR-128 decreases insulin-receptor expression in human and mouse adipose tissue promoting systemic insulin resistance. EBioMedicine 2020, 59, 102912. [Google Scholar] [CrossRef]
- Dahlén, E.M.; Tengblad, A.; Länne, T.; Clinchy, B.; Ernerudh, J.; Nystrom, F.H.; Östgren, C.J. Abdominal obesity and low-grade systemic inflammation as markers of subclinical organ damage in type 2 diabetes. Diabetes Metab. 2014, 40, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, M.; Silambanan, S.; Chandran, A.S.; Prabhakaran, A.A.; Ramakrishnan, R. C-Reactive Protein (CRP) and Leptin Receptor in Obesity: Binding of Monomeric CRP to Leptin Receptor. Front. Immunol. 2018, 9, 1167. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhao, H.; Yin, C.; Lan, X.; Wu, L.; Du, X.; Griffiths, H.R.; Gao, D. Adipokines, Hepatokines and Myokines: Focus on Their Role and Molecular Mechanisms in Adipose Tissue Inflammation. Front. Endocrinol. 2022, 13, 873699. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pollin, T.I.; Ryan, A.S.; Nicklas, B.J.; Snitker, S.; Horenstein, R.B.; Hull, K.; Goldberg, N.H.; et al. Acute-phase serum amyloid A: An inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006, 3, e287. [Google Scholar] [CrossRef]
- Greco, M.; Chiefari, E.; Montalcini, T.; Accattato, F.; Costanzo, F.S.; Pujia, A.; Foti, D.; Brunetti, A.; Gulletta, E. Early effects of a hypocaloric, Mediterranean diet on laboratory parameters in obese individuals. Mediat. Inflamm. 2014, 2014, 750860. [Google Scholar] [CrossRef] [PubMed]
- Rość, D.; Adamczyk, P.; Boinska, J.; Szafkowski, R.; Ponikowska, I.; Stankowska, K.; Góralczyk, B.; Ruszkowska-Ciastek, B. CRP, but not TNF-α or IL-6, decreases after weight loss in patients with morbid obesity exposed to intensive weight reduction and balneological treatment. J. Zhejiang Univ. Sci. B 2015, 16, 404–411. [Google Scholar] [CrossRef]
- Korantzopoulos, P.; Letsas, K.P.; Tse, G.; Fragakis, N.; Goudis, C.A.; Liu, T. Inflammation and atrial fibrillation: A comprehensive review. J. Arrhythm. 2018, 34, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Al-Kaisey, A.M.; Kalman, J.M. Obesity and Atrial Fibrillation: Epidemiology, Pathogenesis and Effect of Weight Loss. Arrhythm. Electrophysiol. Rev. 2021, 10, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Dudley, S.C., Jr. Evidence for Inflammation as a Driver of Atrial Fibrillation. Front. Cardiovasc. Med. 2020, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Nso, N.; Bookani, K.R.; Metzl, M.; Radparvar, F. Role of inflammation in atrial fibrillation: A comprehensive review of current knowledge. J. Arrhythm. 2020, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Olesen, O.J.; Vinding, N.E.; Østergaard, L.; Butt, J.H.; Gislason, G.H.; Torp-Pedersen, C.; Køber, L.; Fosbøl, E.L. C-reactive protein after coronary artery bypass graft surgery and its relationship with postoperative atrial fibrillation. Europace 2020, 22, 1182–1188. [Google Scholar] [CrossRef]
- Weymann, A.; Popov, A.F.; Sabashnikov, A.; Ali-Hasan-Al-Saegh, S.; Ryazanov, M.; Tse, G.; Mirhosseini, S.J.; Liu, T.; Lotfaliani, M.; Sedaghat, M.; et al. Baseline and postoperative levels of C-reactive protein and interleukins as inflammatory predictors of atrial fibrillation following cardiac surgery: A systematic review and meta-analysis. Kardiol. Pol. 2018, 76, 440–451. [Google Scholar] [CrossRef]
- Li, X.; Peng, S.; Wu, X.; Guan, B.; Tse, G.; Chen, S.; Zhou, G.; Wei, Y.; Gong, C.; Lu, X.; et al. C-reactive protein and atrial fibrillation: Insights from epidemiological and Mendelian randomization studies. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Ionin, V.; Baraschkova, E.; Zaslavskaya, E.; Nifontov, S.; Bazhenova, E.; Belyaeva, O.; Baranova, E. Biomarkers of inflammation, parameters characterizing obesity and cardiac remodeling in patients with atrial fibrillation and metabolic syndrome. Russ. J. Cardiol. 2021, 26, 4343. [Google Scholar] [CrossRef]
- Kondo, H.; Abe, I.; Gotoh, K.; Fukui, A.; Takanari, H.; Ishii, Y.; Ikebe, Y.; Kira, S.; Oniki, T.; Saito, S.; et al. Interleukin 10 treatment ameliorates high-fat diet-induced inflammatory atrial remodeling and fibrillation. Circ. Arrhythm. Electrophysiol. 2018, 11, e006040. [Google Scholar] [CrossRef] [PubMed]
- Podzolkov, V.I.; Tarzimanova, A.I.; Bragina, A.E.; Gataulin, R.G.; Oganesyan, K.A.; Pokrovskaya, A.E.; Osadchy, K.K. The importance of matrix metalloproteinases in the development of atrial fibrillation in obesity. Ter. Arkhiv 2021, 93, 1451–1456. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R.; Lau, D.H.; Brooks, A.G.; Shipp, N.J.; Wood, J.P.M.; Manavis, J.; Samuel, C.S.; Patel, K.P.; Finnie, J.W.; Alasady, M.; et al. Atrial fibrillation and obesity: Reverse remodeling of atrial substrate with weight reduction. JACC Clin. Electrophysiol. 2021, 7, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Lee, D.Y.; White, E.S.; Cui, Z.; Larios, J.M.; Chacon, R.; Horowitz, J.C.; Day, R.M.; Thomas, P.E. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 2003, 278, 12384–12389. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Yndestad, A.; Ueland, T.; Øie, E.; Florholmen, G.; Halvorsen, B.; Attramadal, H.; Simonsen, S.; Frøland, S.S.; Gullestad, L.; Christensen, G.; et al. Elevated levels of activin A in heart failure: Potential role in myocardial remodeling. Circulation 2004, 109, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Liew, R.; Khairunnisa, K.; Gu, Y.; Tee, N.; Yin, N.O.; Naylynn, T.M.; Moe, K.T. Role of tumor necrosis factor-α in the pathogenesis of atrial fibrosis and development of an arrhythmogenic substrate. Circ. J. 2013, 77, 1171–1179. [Google Scholar] [CrossRef]
- Frustaci, A.; Chimenti, C.; Bellocci, F.; Morgante, E.; Russo, M.A.; Maseri, A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation 1997, 96, 1180–1184. [Google Scholar] [CrossRef]
- Lee, S.H.; Chen, Y.C.; Chen, Y.J.; Chang, S.L.; Tai, C.T.; Wongcharoen, W.; Yeh, H.I.; Lin, C.I.; Chen, S.A. Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci. 2007, 80, 1806–1815. [Google Scholar] [CrossRef]
- Kao, Y.H.; Chen, Y.C.; Cheng, C.C.; Lee, T.I.; Chen, Y.J.; Chen, S.A. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit. Care Med. 2010, 38, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Zhang, S.; Yang, S.; Lu, Y.; Lu, K.; Wu, Y.; Wu, Q.; Zhao, N.; Dong, Q.; Chen, L.; et al. Interleukin-6-Mediated-Ca2+ Handling Abnormalities Contributes to Atrial Fibrillation in Sterile Pericarditis Rats. Front. Immunol. 2021, 12, 758157. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, Q.; Zeng, Z.; Li, Q.; Zhou, S.; Zhou, M.; Xue, Y.; Cheng, X.; Xia, Y.; Wang, Q.; et al. Regulation of SCN3B/scn3b by Interleukin 2 (IL-2): IL-2 modulates SCN3B/scn3b transcript expression and increases sodium current in myocardial cells. BMC Cardiovasc. Disord. 2016, 16, 1. [Google Scholar] [CrossRef]
- Sawaya, S.E.; Rajawat, Y.S.; Rami, T.G.; Szalai, G.; Price, R.L.; Sivasubramanian, N.; Mann, D.L.; Khoury, D.S. Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1561–H1567. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Heijman, J.; Hiram, R.; Li, N.; Nattel, S. Inflammatory signalling in atrial cardiomyocytes: A novel unifying principle in atrial fibrillation pathophysiology. Nat. Rev. Cardiol. 2023, 20, 145–167. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Wani, K.; AlHarthi, H.; Alghamdi, A.; Sabico, S.; Al-Daghri, N.M. Role of NLRP3 Inflammasome Activation in Obesity-Mediated Metabolic Disorders. Int. J. Environ. Res. Public Health 2021, 18, 511. [Google Scholar] [CrossRef]
- Gramley, F.; Lorenzen, J.; Jedamzik, B.; Gatter, K.; Koellensperger, E.; Munzel, T.; Pezzella, F. Atrial fibrillation is associated with cardiac hypoxia. Cardiovasc. Pathol. 2010, 19, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Yuan, P.; Du, X.; Jin, H.; Du, J.; Huang, Y. Hypoxia inducible factor-1α is an important regulator of macrophage biology. Heliyon 2023, 9, e17167. [Google Scholar] [CrossRef] [PubMed]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Halberg, N.; Khan, M.; Magalang, U.J.; Scherer, P.E. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol. Cell. Biol. 2013, 33, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Ogi, H.; Nakano, Y.; Niida, S.; Dote, K.; Hirai, Y.; Suenari, K.; Tonouchi, Y.; Oda, N.; Makita, Y.; Ueda, S.; et al. Is structural remodeling of fibrillated atria the consequence of tissue hypoxia? Circ. J. 2010, 74, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Zhang, W.; Chen, Y.; Ma, L.; Zhang, H.; Wang, F. Significance of hypoxia-inducible factor-1α expression with atrial fibrosis in rats induced with isoproterenol. Exp. Ther. Med. 2014, 8, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Chung, H.S.; Lee, B.R.; Kim, S.J.; Yoo, S.J.; Hong, S.H.; Kim, H.M. Expression of proinflammatory cytokines via HIF-1alpha and NF-kappaB activation on desferrioxamine-stimulated HMC-1 cells. Biochem. Biophys. Res. Commun. 2003, 306, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Ihara, K.; Sasano, T. Role of Inflammation in the Pathogenesis of Atrial Fibrillation. Front. Physiol. 2022, 13, 862164. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Liu, C.; Cai, Z.; Xie, Q.; Hu, T.; Duan, M.; Wu, H.; Yuan, Y.; Tang, Q. High-mobility group AT-hook 1 promotes cardiac dysfunction in diabetic cardiomyopathy via autophagy inhibition. Cell Death Dis. 2020, 11, 160. [Google Scholar] [CrossRef]
- Su, Q.; Lv, X.; Sun, Y.; Yang, H.; Ye, Z.; Li, L. Role of high mobility group A1/nuclear factor-kappa B signaling in coronary microembolization-induced myocardial injury. Biomed. Pharmacother. 2018, 105, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Boos, C.J.; Lip, G.Y. Inflammation and atrial fibrillation: Cause or effect? Heart 2008, 94, 133–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.Y.; Xu, L.X.; Zhang, H.Q. Association of CETP and CRP gene polymorphisms with non-valvular atrial fibrillation in Chinese Han population. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2008, 25, 225–229. [Google Scholar] [PubMed]
- Kang, Y.A.; Hu, Y.R.; Li, N.F. Advances in research on G protein-coupled inward rectifier K(+) channel gene. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2012, 34, 426–430. [Google Scholar]
- Zou, T.; Zhu, M.; Ma, Y.C.; Xiao, F.; Yu, X.; Xu, L.; Ma, L.Q.; Yang, J.; Dong, J.Z. MicroRNA-410-5p exacerbates high-fat diet-induced cardiac remodeling in mice in an endocrine fashion. Sci. Rep. 2018, 8, 8780. [Google Scholar] [CrossRef]
- Khalyfa, A.; Kheirandish-Gozal, L.; Bhattacharjee, R.; Khalyfa, A.A.; Gozal, D. Circulating microRNAs as Potential Biomarkers of Endothelial Dysfunction in Obese Children. Chest 2016, 149, 786–800. [Google Scholar] [CrossRef]
- Black, N.; Mohammad, F.; Saraf, K.; Morris, G. Endothelial function and atrial fibrillation: A missing piece of the puzzle? J. Cardiovasc. Electrophysiol. 2022, 33, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Ernault, A.C.; de Winter, R.; Fabrizi, B.; Bracht, J.W.P.; Hau, C.; van Amersfoorth, S.C.M.; Meulendijks, E.R.; Tijsen, A.J.; Ortega, L.C.; van der Made, I.; et al. MicroRNAs in extracellular vesicles released from epicardial adipose tissue promote arrhythmogenic conduction slowing. Heart Rhythm O2 2023, 4, 805–814. [Google Scholar] [CrossRef]
- Xiao, Z.; Xie, Y.; Huang, F.; Yang, J.; Liu, X.; Lin, X.; Zhu, P.; Zheng, S. MicroRNA-205-5p plays a suppressive role in the high-fat diet-induced atrial fibrosis through regulation of the EHMT2/IGFBP3 axis. Genes Nutr. 2022, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Korantzopoulos, P.; Letsas, K.; Fragakis, N.; Tse, G.; Liu, T. Oxidative stress and atrial fibrillation: An update. Free Radic. Res. 2018, 52, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.K.; Cho, H.W.; Song, S.E.; Im, S.S.; Bae, J.H.; Song, D.K. Oxidative stress resulting from the removal of endogenous catalase induces obesity by promoting hyperplasia and hypertrophy of white adipocytes. Redox Biol. 2020, 37, 101749. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Castrejón-Téllez, V.; Soto, M.E.; Rubio-Ruiz, M.E.; Manzano-Pech, L.; Guarner-Lans, V. Oxidative Stress, Plant Natural Antioxidants, and Obesity. Int. J. Mol. Sci. 2021, 22, 1786. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef]
- Azzi, A. Oxidative Stress: What Is It? Can It Be Measured? Where Is It Located? Can It Be Good or Bad? Can It Be Prevented? Can It Be Cured? Antioxidants 2022, 11, 1431. [Google Scholar] [CrossRef]
- Dikalov, S.; Itani, H.; Richmond, B.; Vergeade, A.; Rahman, S.M.J.; Boutaud, O.; Blackwell, T.; Massion, P.P.; Harrison, D.G.; Dikalova, A. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H639–H646. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, C.; Friederich-Persson, M.; Palacios-Ramirez, R.; Nguyen Dinh Cat, A. Mitochondrial oxidative stress in obesity: Role of the mineralocorticoid receptor. J. Endocrinol. 2018, 238, R143–R159. [Google Scholar] [CrossRef]
- Panda, P.; Verma, H.K.; Lakkakula, S.; Merchant, N.; Kadir, F.; Rahman, S.; Jeffree, M.S.; Lakkakula, B.V.K.S.; Rao, P.V. Biomarkers of Oxidative Stress Tethered to Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2022, 2022, 9154295. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kang, P.M. Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases. Antioxidants 2020, 9, 1292. [Google Scholar] [CrossRef]
- Morrison, C.D.; Pistell, P.J.; Ingram, D.K.; Johnson, W.D.; Liu, Y.; Fernandez-Kim, S.O.; White, C.L.; Purpera, M.N.; Uranga, R.M.; Bruce-Keller, A.J.; et al. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: Implications for decreased Nrf2 signaling. J. Neurochem. 2010, 114, 1581–1589. [Google Scholar] [CrossRef]
- Olusi, S.O. Obesity is an independent risk factor for plasma lipid peroxidation and depletion of erythrocyte cytoprotectic enzymes in humans. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1159–1164. [Google Scholar] [CrossRef]
- Kanikowska, D.; Kanikowska, A.; Swora-Cwynar, E.; Grzymisławski, M.; Sato, M.; Bręborowicz, A.; Witowski, J.; Korybalska, K. Moderate Caloric Restriction Partially Improved Oxidative Stress Markers in Obese Humans. Antioxidants 2021, 10, 1018. [Google Scholar] [CrossRef] [PubMed]
- Metere, A.; Graves, C.E.; Pietraforte, D.; Casella, G. The Effect of Sleeve Gastrectomy on Oxidative Stress in Obesity. Biomedicines 2020, 8, 168. [Google Scholar] [CrossRef]
- Fan, L.; Cacicedo, J.M.; Ido, Y. Impaired nicotinamide adenine dinucleotide (NAD+) metabolism in diabetes and diabetic tissues: Implications for nicotinamide-related compound treatment. J. Diabetes Investig. 2020, 11, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.J.; IJzerman, R.G.; Teerlink, T.; Westerhoff, H.V.; Gans, R.O.; Heine, R.J. Cytosolic triglycerides and oxidative stress in central obesity: The missing link between excessive atherosclerosis, endothelial dysfunction, and beta-cell failure? Atherosclerosis 2000, 148, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Pervaiz, S. TNF receptor superfamily-induced cell death: Redox-dependent execution. FASEB J. 2006, 20, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Fortuño, A.; Bidegain, J.; Baltanás, A.; Moreno, M.U.; Montero, L.; Landecho, M.F.; Beloqui, O.; Díez, J.; Zalba, G. Is leptin involved in phagocytic NADPH oxidase overactivity in obesity? Potential clinical implications. J. Hypertens. 2010, 28, 1944–1950. [Google Scholar] [CrossRef]
- Bełtowski, J.; Wójcicka, G.; Jamroz, A. Leptin decreases plasma paraoxonase 1 (PON1) activity and induces oxidative stress: The possible novel mechanism for proatherogenic effect of chronic hyperleptinemia. Atherosclerosis 2003, 170, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Vincent, H.K.; Morgan, J.W.; Vincent, K.R. Obesity exacerbates oxidative stress levels after acute exercise. Med. Sci. Sports Exerc. 2004, 36, 772–779. [Google Scholar] [CrossRef]
- Vincent, H.K.; Vincent, K.R.; Bourguignon, C.; Braith, R.W. Obesity and postexercise oxidative stress in older women. Med. Sci. Sports. Exerc. 2005, 37, 213–219. [Google Scholar] [CrossRef]
- Chang, J.P.; Chen, M.C.; Liu, W.H.; Yang, C.H.; Chen, C.J.; Chen, Y.L.; Pan, K.L.; Tsai, T.H.; Chang, H.W. Atrial myocardial nox2 containing NADPH oxidase activity contribution to oxidative stress in mitral regurgitation: Potential mechanism for atrial remodeling. Cardiovasc. Pathol. 2011, 20, 99–106. [Google Scholar] [CrossRef]
- Neuman, R.B.; Bloom, H.L.; Shukrullah, I.; Darrow, L.A.; Kleinbaum, D.; Jones, D.P.; Dudley, S.C., Jr. Oxidative stress markers are associated with persistent atrial fibrillation. Clin. Chem. 2007, 53, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Babusíková, E.; Kaplán, P.; Lehotský, J.; Jesenák, M.; Dobrota, D. Oxidative modification of rat cardiac mitochondrial membranes and myofibrils by hydroxyl radicals. Gen. Physiol. Biophys. 2004, 23, 327–335. [Google Scholar] [PubMed]
- Mihm, M.J.; Yu, F.; Carnes, C.A.; Reiser, P.J.; McCarthy, P.M.; Van Wagoner, D.R.; Bauer, J.A. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 2001, 104, 174–180. [Google Scholar] [CrossRef]
- Bukowska, A.; Schild, L.; Keilhoff, G.; Hirte, D.; Neumann, M.; Gardemann, A.; Neumann, K.H.; Röhl, F.W.; Huth, C.; Goette, A.; et al. Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp. Biol. Med. 2008, 233, 558–574. [Google Scholar] [CrossRef] [PubMed]
- Carnes, C.A.; Chung, M.K.; Nakayama, T.; Nakayama, H.; Baliga, R.S.; Piao, S.; Kanderian, A.; Pavia, S.; Hamlin, R.L.; McCarthy, P.M.; et al. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ. Res. 2001, 89, E32–E38. [Google Scholar] [CrossRef] [PubMed]
- Korantzopoulos, P.; Kolettis, T.M.; Kountouris, E.; Dimitroula, V.; Karanikis, P.; Pappa, E.; Siogas, K.; Goudevenos, J.A. Oral vitamin C administration reduces early recurrence rates after electrical cardioversion of persistent atrial fibrillation and attenuates associated inflammation. Int. J. Cardiol. 2005, 102, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Carnes, C.A.; Janssen, P.M.; Ruehr, M.L.; Nakayama, H.; Nakayama, T.; Haase, H.; Bauer, J.A.; Chung, M.K.; Fearon, I.M.; Gillinov, A.M.; et al. Atrial glutathione content, calcium current, and contractility. J. Biol. Chem. 2007, 282, 28063–28073. [Google Scholar] [CrossRef] [PubMed]
- Therdyothin, A.; Phiphopthatsanee, N.; Isanejad, M. The Effect of Omega-3 Fatty Acids on Sarcopenia: Mechanism of Action and Potential Efficacy. Mar. Drugs 2023, 21, 399. [Google Scholar] [CrossRef]
- Biccirè, F.G.; Bucci, T.; Menichelli, D.; Cammisotto, V.; Pignatelli, P.; Carnevale, R.; Pastori, D. Mediterranean Diet: A Tool to Break the Relationship of Atrial Fibrillation with the Metabolic Syndrome and Non-Alcoholic Fatty Liver Disease. Nutrients 2022, 14, 1260. [Google Scholar] [CrossRef] [PubMed]
- Papoulidis, P.; Ananiadou, O.; Chalvatzoulis, E.; Ampatzidou, F.; Koutsogiannidis, C.; Karaiskos, T.; Madesis, A.; Drossos, G. The role of ascorbic acid in the prevention of atrial fibrillation after elective on-pump myocardial revascularization surgery: A single-center experience—A pilot study. Interact. Cardiovasc. Thorac. Surg. 2011, 12, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guo, M.; Chen, T.; Cheng, H.; Yang, Q.; Zhao, Z.; She, R.; Yang, X.; Xiao, W.; Yang, X.; et al. Walking and taking vitamin C alleviates oxidative stress and inflammation in overweight students, even in the short-term. Front. Public Health 2022, 10, 1024864. [Google Scholar] [CrossRef]
- Sisto, T.; Paajanen, H.; Metsä-Ketelä, T.; Harmoinen, A.; Nordback, I.; Tarkka, M. Pretreatment with antioxidants and allopurinol diminishes cardiac onset events in coronary artery bypass grafting. Ann. Thorac. Surg. 1995, 59, 1519–1523. [Google Scholar] [CrossRef]
- Alcalá, M.; Sánchez-Vera, I.; Sevillano, J.; Herrero, L.; Serra, D.; Ramos, M.P.; Viana, M. Vitamin E reduces adipose tissue fibrosis, inflammation, and oxidative stress and improves metabolic profile in obesity. Obesity 2015, 23, 1598–1606. [Google Scholar] [CrossRef]
- Antoniades, C.; Demosthenous, M.; Reilly, S.; Margaritis, M.; Zhang, M.H.; Antonopoulos, A.; Marinou, K.; Nahar, K.; Jayaram, R.; Tousoulis, D.; et al. Myocardial redox state predicts in-hospital clinical outcome after cardiac surgery effects of short-term pre-operative statin treatment. J. Am. Coll. Cardiol. 2012, 59, 60–70. [Google Scholar] [CrossRef]
- Pengrattanachot, N.; Cherngwelling, R.; Jaikumkao, K.; Pongchaidecha, A.; Thongnak, L.; Swe, M.T.; Chatsudthipong, V.; Lungkaphin, A. Atorvastatin attenuates obese-induced kidney injury and impaired renal organic anion transporter 3 function through inhibition of oxidative stress and inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165741. [Google Scholar] [CrossRef] [PubMed]
- Heidt, M.C.; Vician, M.; Stracke, S.K.; Stadlbauer, T.; Grebe, M.T.; Boening, A.; Vogt, P.R.; Erdogan, A. Beneficial effects of intravenously administered N-3 fatty acids for the prevention of atrial fibrillation after coronary artery bypass surgery: A prospective randomized study. Thorac. Cardiovasc. Surg. 2009, 57, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Awada, M.; Meynier, A.; Soulage, C.O.; Hadji, L.; Géloën, A.; Viau, M.; Ribourg, L.; Benoit, B.; Debard, C.; Guichardant, M.; et al. n-3 PUFA added to high-fat diets affect differently adiposity and inflammation when carried by phospholipids or triacylglycerols in mice. Nutr. Metab. 2013, 10, 23. [Google Scholar] [CrossRef]
- Ozaydin, M.; Peker, O.; Erdogan, D.; Kapan, S.; Turker, Y.; Varol, E.; Ozguner, F.; Dogan, A.; Ibrisim, E. N-acetylcysteine for the prevention of postoperative atrial fibrillation: A prospective, randomized, placebo-controlled pilot study. Eur. Heart J. 2008, 29, 625–631. [Google Scholar] [CrossRef]
- Tada, F.; Abe, M.; Kawasaki, K.; Miyake, T.; Shiyi, C.; Hiasa, Y.; Matsuura, B.; Onji, M. B cell activating factor in obesity is regulated by oxidative stress in adipocytes. J. Clin. Biochem. Nutr. 2013, 52, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Shimano, M.; Tsuji, Y.; Inden, Y.; Kitamura, K.; Uchikawa, T.; Harata, S.; Nattel, S.; Murohara, T. Pioglitazone, a peroxisome proliferator-activated receptor-gamma activator, attenuates atrial fibrosis and atrial fibrillation promotion in rabbits with congestive heart failure. Heart Rhythm 2008, 5, 451–459. [Google Scholar] [CrossRef]
- Ohtomo, S.; Izuhara, Y.; Takizawa, S.; Yamada, N.; Kakuta, T.; van Ypersele de Strihou, C.; Miyata, T. Thiazolidinediones provide better renoprotection than insulin in an obese, hypertensive type II diabetic rat model. Kidney Int. 2007, 72, 1512–1519. [Google Scholar] [CrossRef]
- Gong, Y.T.; Li, W.M.; Li, Y.; Yang, S.S.; Sheng, L.; Yang, N.; Shan, H.B.; Xue, H.J.; Liu, W.; Yang, B.F.; et al. Probucol attenuates atrial autonomic remodeling in a canine model of atrial fibrillation produced by prolonged atrial pacing. Chin. Med. J. 2009, 122, 74–82. [Google Scholar]
- Wu, H.M.; Huang, N.J.; Yang, Y.V.; Fan, L.P.; Tang, T.Y.; Liu, L.; Xu, Y.; Liu, D.T.; Cai, Z.X.; Ren, X.Y.; et al. Probucol mitigates high-fat diet-induced cognitive and social impairments through disruption of redox-inflammation association. bioRxiv 2023. [Google Scholar] [CrossRef]
- Dudley, S.C., Jr.; Hoch, N.E.; McCann, L.A.; Honeycutt, C.; Diamandopoulos, L.; Fukai, T.; Harrison, D.G.; Dikalov, S.I.; Langberg, J. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005, 112, 1266–1273. [Google Scholar]
- Yagi, S.; Akaike, M.; Aihara, K.; Ishikawa, K.; Iwase, T.; Ikeda, Y.; Soeki, T.; Yoshida, S.; Sumitomo-Ueda, Y.; Matsumoto, T.; et al. Endothelial nitric oxide synthase-independent protective action of statin against angiotensin II-induced atrial remodeling via reduced oxidant injury. Hypertension 2010, 55, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Adam, O.; Frost, G.; Custodis, F.; Sussman, M.A.; Schäfers, H.J.; Böhm, M.; Laufs, U. Role of Rac1 GTPase activation in atrial fibrillation. J. Am. Coll. Cardiol. 2007, 50, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. Association of atrial nicotinamide adenine dinucleotide phosphate oxidase activity with the development of atrial fibrillation after cardiac surgery. J. Am. Coll. Cardiol. 2008, 51, 68–74. [Google Scholar] [CrossRef]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443. [Google Scholar] [CrossRef]
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef]
- Hernández-Aguilera, A.; Rull, A.; Rodríguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menéndez, J.A.; Joven, J. Mitochondrial dysfunction: A basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediat. Inflamm. 2013, 2013, 135698. [Google Scholar] [CrossRef]
- Cioffi, F.; Giacco, A.; Petito, G.; de Matteis, R.; Senese, R.; Lombardi, A.; de Lange, P.; Moreno, M.; Goglia, F.; Lanni, A.; et al. Altered Mitochondrial Quality Control in Rats with Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD) Induced by High-Fat Feeding. Genes 2022, 13, 315. [Google Scholar] [CrossRef]
- Holmström, M.H.; Iglesias-Gutierrez, E.; Zierath, J.R.; Garcia-Roves, P.M. Tissue-specific control of mitochondrial respiration in obesity-related insulin resistance and diabetes. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E731–E739. [Google Scholar] [CrossRef] [PubMed]
- Rogge, M.M. The role of impaired mitochondrial lipid oxidation in obesity. Biol. Res. Nurs. 2009, 10, 356–373. [Google Scholar] [CrossRef]
- de Lange, P.; Lombardi, A.; Silvestri, E.; Cioffi, F.; Giacco, A.; Iervolino, S.; Petito, G.; Senese, R.; Lanni, A.; Moreno, M. Physiological Approaches Targeting Cellular and Mitochondrial Pathways Underlying Adipose Organ Senescence. Int. J. Mol. Sci. 2023, 24, 11676. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Kosiol, M.; Mayr, M.; Schulz, R.; Rohrbach, S. Mitochondria and ageing: Role in heart, skeletal muscle and adipose tissue. J. Cachexia Sarcopenia Muscle 2017, 8, 349–369. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef]
- Lin, P.H.; Lee, S.H.; Su, C.P.; Wei, Y.H. Oxidative damage to mitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic. Biol. Med. 2003, 35, 1310–1318. [Google Scholar] [CrossRef]
- Wiersma, M.; van Marion, D.M.S.; Wüst, R.C.I.; Houtkooper, R.H.; Zhang, D.; Groot, N.M.S.; Henning, R.H.; Brundel, B.J.J.M. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, C.; Li, Z.; Kim, G.; Jeevanandam, V.; Uriel, N. Molecular Mechanism of the Association Between Atrial Fibrillation and Heart Failure Includes Energy Metabolic Dysregulation Due to Mitochondrial Dysfunction. J. Card. Fail. 2019, 25, 911–920. [Google Scholar] [CrossRef]
- Liu, M.; Liu, H.; Dudley, S.C., Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 2010, 107, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Yang, Y.; Suo, Y.; Liu, R.; Qiu, J.; Zhao, Y.; Jiang, N.; Liu, C.; Tse, G.; et al. Alogliptin prevents diastolic dysfunction and preserves left ventricular mitochondrial function in diabetic rabbits. Cardiovasc. Diabetol. 2018, 17, 160. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shimano, M.; Inden, Y.; Takefuji, M.; Yanagisawa, S.; Yoshida, N.; Tsuji, Y.; Hirai, M.; Murohara, T. Alogliptin, a dipeptidyl peptidase-4 inhibitor, regulates the atrial arrhythmogenic substrate in rabbits. Heart Rhythm 2015, 12, 1362–1369. [Google Scholar] [CrossRef]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Testai, L.; Colletti, A.; Cicero, A.F.G. Coenzyme Q10: Clinical Applications in Cardiovascular Diseases. Antioxidants 2020, 9, 341. [Google Scholar] [CrossRef]
- Mortensen, A.L.; Rosenfeldt, F.; Filipiak, K.J. Effect of coenzyme Q10 in Europeans with chronic heart failure: A sub-group analysis of the Q-SYMBIO randomized double-blind trial. Cardiol. J. 2019, 26, 147–156. [Google Scholar] [CrossRef]
- Kunitomo, M.; Yamaguchi, Y.; Kagota, S.; Otsubo, K. Beneficial effect of coenzyme Q10 on increased oxidative and nitrative stress and inflammation and individual metabolic components developing in a rat model of metabolic syndrome. J. Pharmacol. Sci. 2008, 107, 128–137. [Google Scholar] [CrossRef]
- Sun, D.; Yang, F. Metformin improves cardiac function in mice with heart failure after myocardial infarction by regulating mitochondrial energy metabolism. Biochem. Biophys. Res. Commun. 2017, 486, 329–335. [Google Scholar] [CrossRef]
- Liu, Y.; Bai, F.; Liu, N.; Zhang, B.; Qin, F.; Tu, T.; Li, B.; Li, J.; Ma, Y.; Ouyang, F.; et al. Metformin improves lipid metabolism and reverses the Warburg effect in a canine model of chronic atrial fibrillation. BMC Cardiovasc. Disord. 2020, 20, 50. [Google Scholar] [CrossRef]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e5. [Google Scholar] [CrossRef] [PubMed]
- Augustyniak, J.; Lenart, J.; Gaj, P.; Kolanowska, M.; Jazdzewski, K.; Stepien, P.P.; Buzanska, L. Bezafibrate Upregulates Mitochondrial Biogenesis and Influence Neural Differentiation of Human-Induced Pluripotent Stem Cells. Mol. Neurobiol. 2019, 56, 4346–4363. [Google Scholar] [CrossRef] [PubMed]
- Hanna, I.R.; Heeke, B.; Bush, H.; Brosius, L.; King-Hageman, D.; Dudley, S.C., Jr.; Beshai, J.F.; Langberg, J.J. Lipid-lowering drug use is associated with reduced prevalence of atrial fibrillation in patients with left ventricular systolic dysfunction. Heart Rhythm 2006, 3, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Lee, M.; Lee, D.; Jang, J.; Shin, S.S.; Yoon, M. Fenofibrate Regulates Visceral Obesity and Nonalcoholic Steatohepatitis in Obese Female Ovariectomized C57BL/6J Mice. Int. J. Mol. Sci. 2021, 22, 3675. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Shangguan, W.; Zhang, Y.; Li, C.; Li, G. Effects of trimetazidine on mitochondrial respiratory function, biosynthesis, and fission/fusion in rats with acute myocardial ischemia. Anatol. J. Cardiol. 2017, 18, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chaolan, L.; Chengcheng, W.; Xi, H.; Yingying, W.; Jiaqiu, L.; Wei, H. GW28-e0789 Trimetazidine decreases inducibility and duration of atrial fibrillation in a dog model of congestive heart failure. J. Am. Coll. Cardiol. 2017, 70, C29. [Google Scholar] [CrossRef]
- Zhang, W.; You, B.; Qi, D.; Qiu, L.; Ripley-Gonzalez, J.W.; Zheng, F.; Fu, S.; Li, C.; Dun, Y.; Liu, S. Trimetazidine and exercise provide comparable improvements to high fat diet-induced muscle dysfunction through enhancement of mitochondrial quality control. Sci. Rep. 2021, 11, 19116. [Google Scholar] [CrossRef]
- Zou, D.; Geng, N.; Chen, Y.; Ren, L.; Liu, X.; Wan, J.; Guo, S.; Wang, S. Ranolazine improves oxidative stress and mitochondrial function in the atrium of acetylcholine-CaCl2 induced atrial fibrillation rats. Life Sci. 2016, 156, 7–14. [Google Scholar] [CrossRef]
- Guerra, F.; Romandini, A.; Barbarossa, A.; Belardinelli, L.; Capucci, A. Ranolazine for rhythm control in atrial fibrillation: A systematic review and meta-analysis. Int. J. Cardiol. 2017, 227, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Al Batran, R.; Gopal, K.; Aburasayn, H.; Eshreif, A.; Almutairi, M.; Greenwell, A.A.; Campbell, S.A.; Saleme, B.; Court, E.A.; Eaton, F.; et al. The antianginal ranolazine mitigates obesity-induced nonalcoholic fatty liver disease and increases hepatic pyruvate dehydrogenase activity. JCI Insight 2019, 4, e124643. [Google Scholar] [CrossRef]
- Serban, R.C.; Balan, A.I.; Perian, M.; Pintilie, I.; Somkereki, C.; Huţanu, A.; Scridon, A. Atrial electrical remodeling induced by chronic ischemia and inflammation in patients with stable coronary artery disease. Chin. J. Physiol. 2019, 62, 11–16. [Google Scholar] [PubMed]
- Scridon, A.; Şerban, R.C.; Chevalier, P. Atrial fibrillation: Neurogenic or myogenic? Arch. Cardiovasc. Dis. 2018, 111, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.N.; Jayaram, R.; Nahar, K.; Antoniades, C.; Verheule, S.; Channon, K.M.; Alp, N.J.; Schotten, U.; Casadei, B. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: Implications for the antiarrhythmic effect of statins. Circulation 2011, 124, 1107–1117. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; van Marion, D.M.S.; Zhang, D.; Brundel, B.J.J.M. Heat shock protein inducer GGA*-59 reverses contractile and structural remodeling via restoration of the microtubule network in experimental Atrial Fibrillation. J. Mol. Cell. Cardiol. 2019, 134, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, H.; Liang, D.; Liu, Y.; Liu, Y.; Zhao, H.; Li, J.; Peng, L.; Chen, Y.H. Taxol, a microtubule stabilizer, prevents atrial fibrillation in in vitro atrial fibrillation models using rabbit hearts. Med. Sci. Monit. 2010, 16, BR353–BR360. [Google Scholar]
Figure 1.
The close relationship between the pathogenesis of atrial fibrillation and obesity-induced oxidative stress, inflammation, and mitochondrial dysfunction.
Figure 1.
The close relationship between the pathogenesis of atrial fibrillation and obesity-induced oxidative stress, inflammation, and mitochondrial dysfunction.

Figure 2.
Systemic and local obesity-related factors involved in the pathogenesis of atrial fibrillation. The adipose tissue promotes the occurrence and maintenance of atrial fibrillation both through local effects (Ca2+ handling abnormalities, structural, connexin, and electrical remodeling) and systemic effects (metabolic, neurohormonal, and proinflammatory factors). AERP—atrial effective refractory period; AF—atrial fibrillation; ANS—autonomic nervous system; CM—cardiomyocyte; DAD—delayed afterdepolarization; ICa-L—L-type Ca2+ current; IK1—inward rectifier K+ current; IKr—rapid component of the delayed rectifier K+ current; IKur—ultra-rapid component of the delayed rectifier K+ current; INa—Na+ current; Ito—transient outward K+ current; SERCA2a—sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2.
Figure 2.
Systemic and local obesity-related factors involved in the pathogenesis of atrial fibrillation. The adipose tissue promotes the occurrence and maintenance of atrial fibrillation both through local effects (Ca2+ handling abnormalities, structural, connexin, and electrical remodeling) and systemic effects (metabolic, neurohormonal, and proinflammatory factors). AERP—atrial effective refractory period; AF—atrial fibrillation; ANS—autonomic nervous system; CM—cardiomyocyte; DAD—delayed afterdepolarization; ICa-L—L-type Ca2+ current; IK1—inward rectifier K+ current; IKr—rapid component of the delayed rectifier K+ current; IKur—ultra-rapid component of the delayed rectifier K+ current; INa—Na+ current; Ito—transient outward K+ current; SERCA2a—sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2.

Figure 3.
The arrhythmogenic substrate of atrial fibrillation induced by obesity-associated inflammation. A variety of proinflammatory factors originating from the adipose tissue promote the development of proarrhythmic atrial cardiomyopathy involving Ca2+ handling abnormalities and structural, connexin, and electrical remodeling. AF—atrial fibrillation; IL-6—interleukin 6; MCP-1—monocyte chemoattractant protein-1; MMPs—matrix metalloproteinases; SERCA2a—sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2; TGF-β—transforming growth factor-beta; TNFα—tumor necrosis factor-alpha.
Figure 3.
The arrhythmogenic substrate of atrial fibrillation induced by obesity-associated inflammation. A variety of proinflammatory factors originating from the adipose tissue promote the development of proarrhythmic atrial cardiomyopathy involving Ca2+ handling abnormalities and structural, connexin, and electrical remodeling. AF—atrial fibrillation; IL-6—interleukin 6; MCP-1—monocyte chemoattractant protein-1; MMPs—matrix metalloproteinases; SERCA2a—sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2; TGF-β—transforming growth factor-beta; TNFα—tumor necrosis factor-alpha.


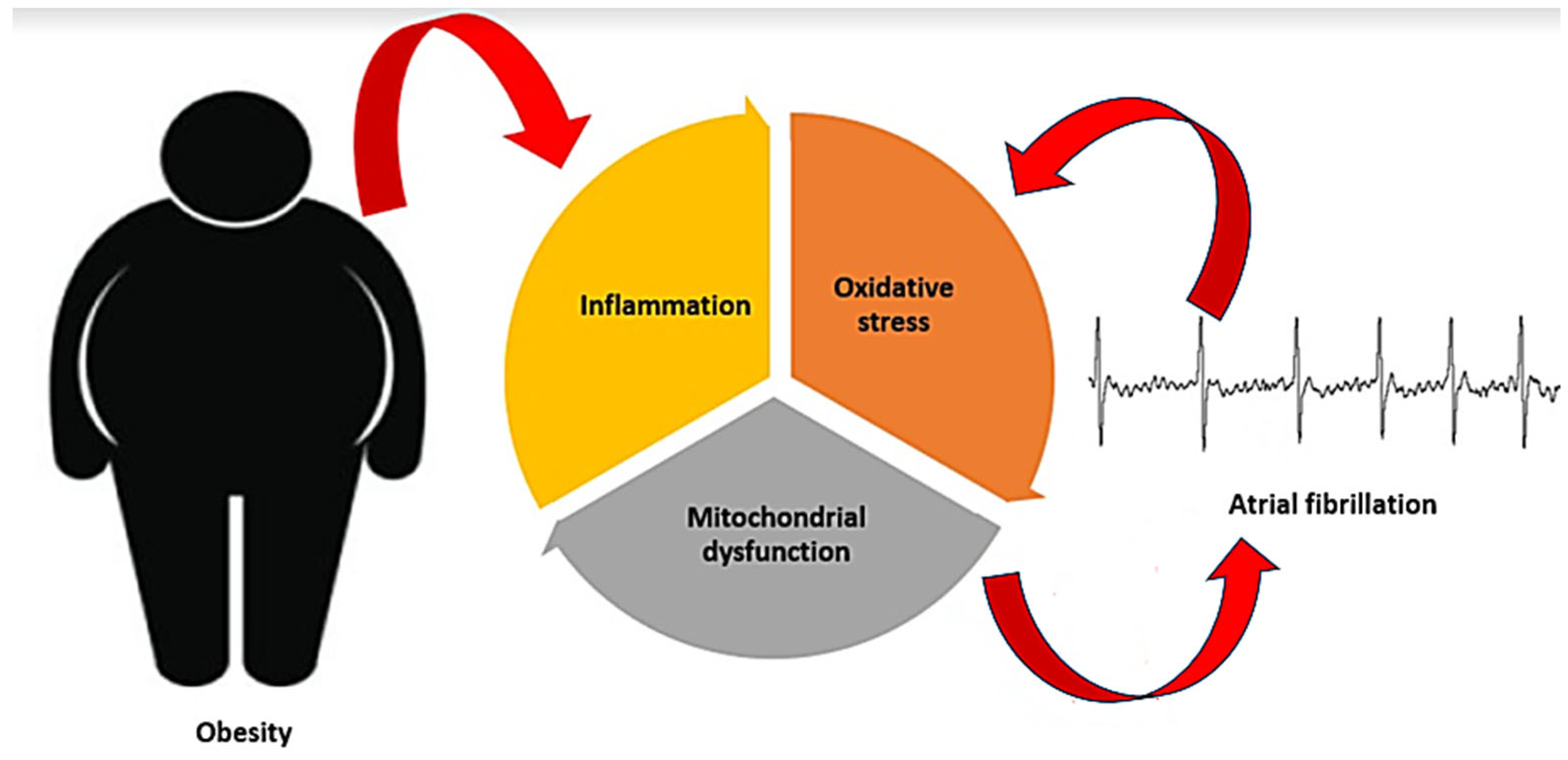{kind=link}
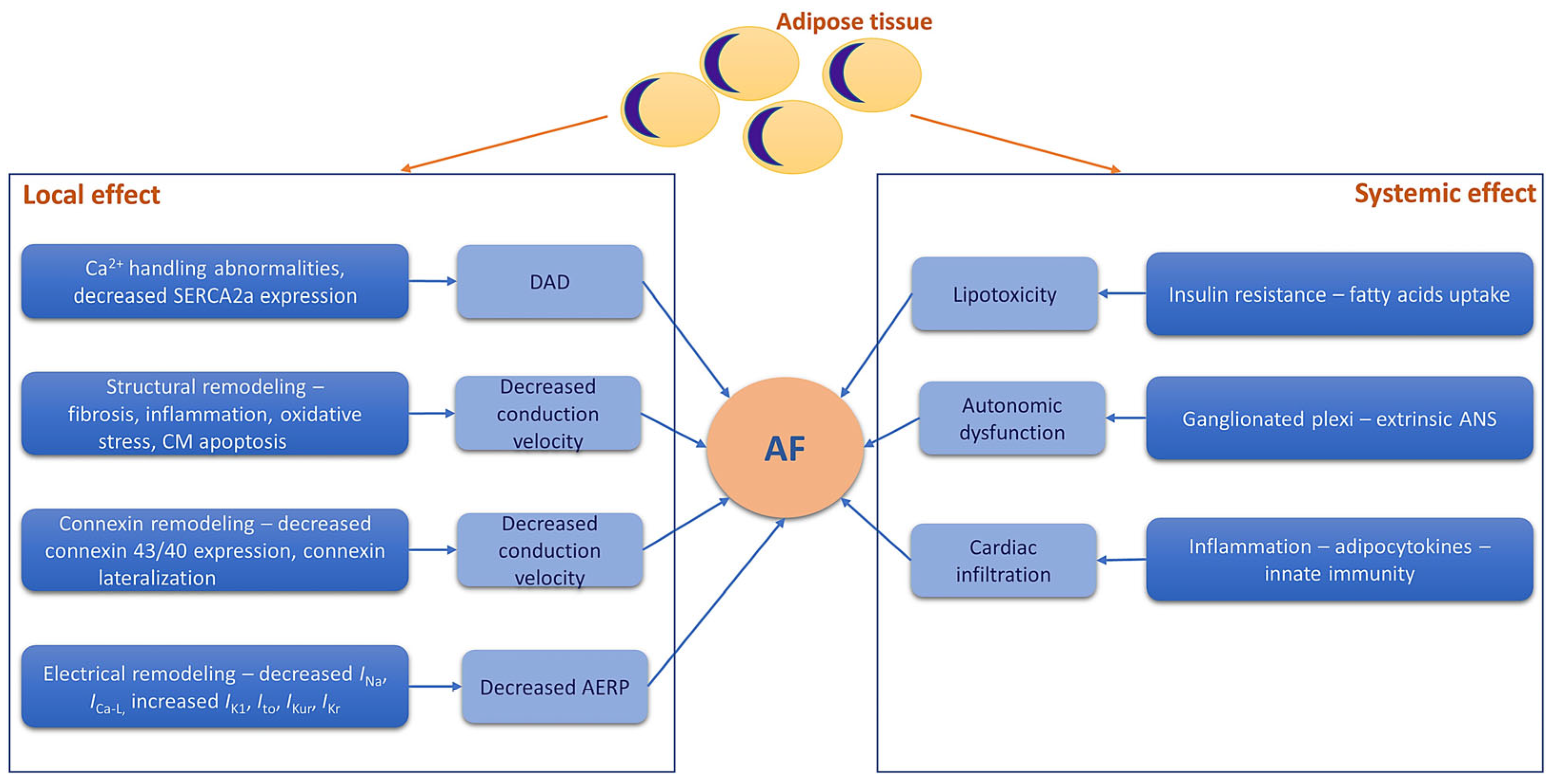{kind=link}
{kind=link}
Table 1.
Inflammatory markers associated with atrial fibrillation and obesity.
| Marker | Study Population | Results | References |
|---|---|---|---|
| IL-6 | 407 patients with metabolic syndrome, of which 128 patients with AF | Increased levels in patients with AF and metabolic syndrome compared with patients with AF but without metabolic syndrome; positively correlated with left and right atrial volumes and epicardial fat thickness. | [32] |
| CRP | 407 patients with metabolic syndrome, of which 128 with AF | Increased levels in patients with AF and metabolic syndrome vs. patients with AF but without metabolic syndrome; positively correlated with epicardial fat thickness. | [32] |
| IL-10 | CL57/B6 mice divided into high-fat and normal-fat diet groups | Reduced serum levels of IL-10 in high-fat diet-induced obesity. | [33] |
| MMP-9 | 105 patients with BMI > 30 kg/m2 | Significantly higher in patients with obesity and paroxysmal AF vs. patients with obesity without AF; significantly correlated with left atrial volume. | [34] |
| TGF-β | Sheep with and without calorie-dense diet | Increased atrial TGF-β, atrial fibrosis, epicardial fat infiltration, and duration of induced AF. | [35] |
| TNFα | 407 patients with metabolic syndrome, of which 128 with AF | Increased levels in patients with AF and metabolic syndrome compared with patients with AF, but without metabolic syndrome; positively correlated with epicardial fat thickness. | [32] |
AF—atrial fibrillation; CRP—C-reactive protein; IL-6—interleukin-6; MMP-9—matrix metalloproteinase-9; TGF-β—transforming growth factor-β; TNFα—tumor necrosis factor-alpha.
Table 2.
Therapeutic strategies with positive impact on oxidative stress and atrial fibrillation and obesity pathogenesis.
Table 2.
Therapeutic strategies with positive impact on oxidative stress and atrial fibrillation and obesity pathogenesis.
| Antioxidant | Disease | Population/ Model | Mechanism | Results | References |
|---|---|---|---|---|---|
| Vitamin C | AF | Patients with elective coronary artery bypass grafting | Downregulation of nicotinamide adenine dinucleotide phosphate oxidase | ↓ incidence of post-coronary artery bypass grafting AF | [98] |
| Obesity | Overweight students | ROS reduction | Attenuation of urinary 8-hydroxy-2′ -deoxyguanosine levels | [99] | |
| Vitamin E | AF | Patients with elective coronary artery bypass grafting | ROS reduction | ↓ incidence of post-coronary artery bypass grafting AF | [100] |
| Obesity | C57BL/6J mice fed a high-fat diet | Decreases oxidative stress | Increased levels of lipid peroxidation and advanced oxidation protein products | [101] | |
| Statins | AF | Patients undergoing cardiac surgery | Reduces myocardial O2 and ONOO− | ↓ incidence of post-coronary artery bypass grafting AF | [102] |
| Obesity | Rats fed a high-fat diet | Reduces renal oxidative stress | Decreased malondialdehyde and glutathione levels; decreased membrane expression of Nox4 and p67phox | [103] | |
| n-3 polyunsaturated fatty acids | AF | Patients with coronary artery bypass grafting | Increase electrical stability | ↓ incidence of postoperative AF | [104] |
| Obesity | C57BL/6 mice fed a high-fat diet | Oxidative stress reduction | Reduced 4-hydroxy-2-nonenal | [105] | |
| N-acetylcysteine | AF | Patients undergoing coronary artery bypass and/or valve surgery | ROS reduction | ↓ incidence of post-coronary artery bypass grafting AF | [106] |
| Obesity | 3T3-L1 and C3H/10 T 1/2-clone 8 (C3H) adipocytes | ROS reduction | Inhibited hydrogen peroxide-induced oxidative stress | [107] | |
| Thiazolidinediones | AF | Rabbits with congestive heart failure | Decrease nicotinamide adenine dinucleotide phosphate; induce antioxidant enzymes such as Cu/Zn superoxide dismutase | Attenuated atrial structural remodeling and inhibited AF promotion | [108] |
| Obesity | Obese, hypertensive, type II diabetes rat model | Reduced renal oxidative stress | Reduced nicotinamide adenine dinucleotide phosphate oxidase in kidney tissues | [109] | |
| Probucol | AF | Right atrial pacing AF model | Reduces atrial oxidative stress and increases total antioxidant capacity | Reduces AF promotion and maintenance | [110] |
| Obesity | Mice fed a high-fat diet | Reduces oxidative stress | Reduced blood levels of oxidized low-density lipoprotein and malondialdehyde | [111] |
↓—decreased; AF—atrial fibrillation; ROS—reactive oxygen species.
Table 3.
Therapeutic strategies with positive impact on mitochondrial function and atrial fibrillation and obesity pathogenesis.
Table 3.
Therapeutic strategies with positive impact on mitochondrial function and atrial fibrillation and obesity pathogenesis.
| Medication | Mechanism at the Mitochondrial Level | Effect on AF | Effect on Obesity |
|---|---|---|---|
| Ubiquinone (coenzyme Q10) | Improves mitochondrial function Antioxidant involved in the electron transport from complex I to complex II and from complex II to complex III of the respiratory chain, membrane stabilizer, and cofactor of mitochondrial uncoupling proteins [138] | AF prevention [139] | Reduction in obesity, oxidative stress, and inflammation in metabolic syndrome [140] |
| Metformin | Preserves mitochondrial function (through AMPK activation), mitochondrial respiration, and mitochondrial biogenesis (probably via upregulation of PGC-1α) [141] | Decreased the incidence of AF by 19% [142] | Activated AMPK and improved mitochondrial respiration in obesity [143] |
| Fibrates | Improve mitochondrial biogenesis via increased PPARGC1A, GFAP, S100B, DCX NRF1, and TFAM genes expression [144] | Decreased AF prevalence [145] | Regulates visceral obesity and inflammation via PPARα activation in obese females [146] |
| Trimetazidine | Improves ATP synthesis via inhibition of beta-oxidation; improves mitochondrial fusion/fission dynamics via normalization of the expression of factors that regulate mitochondrial biogenesis [147] | Prevented tachycardia-induced atrial ultrastructural remodeling, decreased AF inducibility, and shortened AF duration [148] | Successfully mimics exercise to enhance mitochondrial quality control [149] |
| Ranolazine | Improves mitochondrial function, attenuates oxidative stress, suppresses apoptosis [150] | Attenuated AF [151] | Attenuated obesity-induced non-alcoholic fatty liver disease and increased hepatic pyruvate dehydrogenase activity [152] |
AF—atrial fibrillation; AMPK—adenosine monophosphate protein kinase; ATP—adenosine triphosphate; PGC-1α—peroxisome proliferator-activated receptor-γ coactivator-1α.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Balan, A.I.; Halațiu, V.B.; Scridon, A. Oxidative Stress, Inflammation, and Mitochondrial Dysfunction: A Link between Obesity and Atrial Fibrillation. Antioxidants 2024, 13, 117. https://doi.org/10.3390/antiox13010117
AMA Style
Balan AI, Halațiu VB, Scridon A. Oxidative Stress, Inflammation, and Mitochondrial Dysfunction: A Link between Obesity and Atrial Fibrillation. Antioxidants. 2024; 13(1):117. https://doi.org/10.3390/antiox13010117
Chicago/Turabian StyleBalan, Alkora Ioana, Vasile Bogdan Halațiu, and Alina Scridon. 2024. "Oxidative Stress, Inflammation, and Mitochondrial Dysfunction: A Link between Obesity and Atrial Fibrillation" Antioxidants 13, no. 1: 117. https://doi.org/10.3390/antiox13010117
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.