
The NADPH Link between the Renin Angiotensin System and the Antioxidant Mechanisms in Dopaminergic Neurons

 , , and
, , and 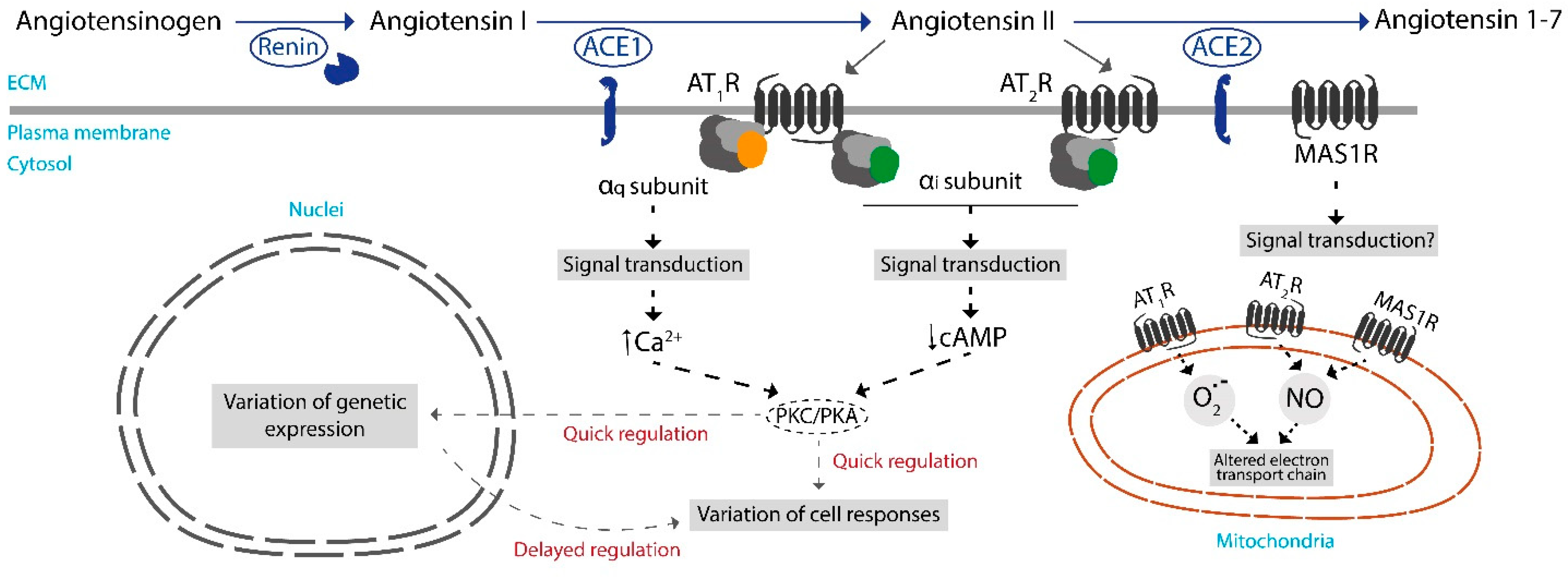{kind=link}
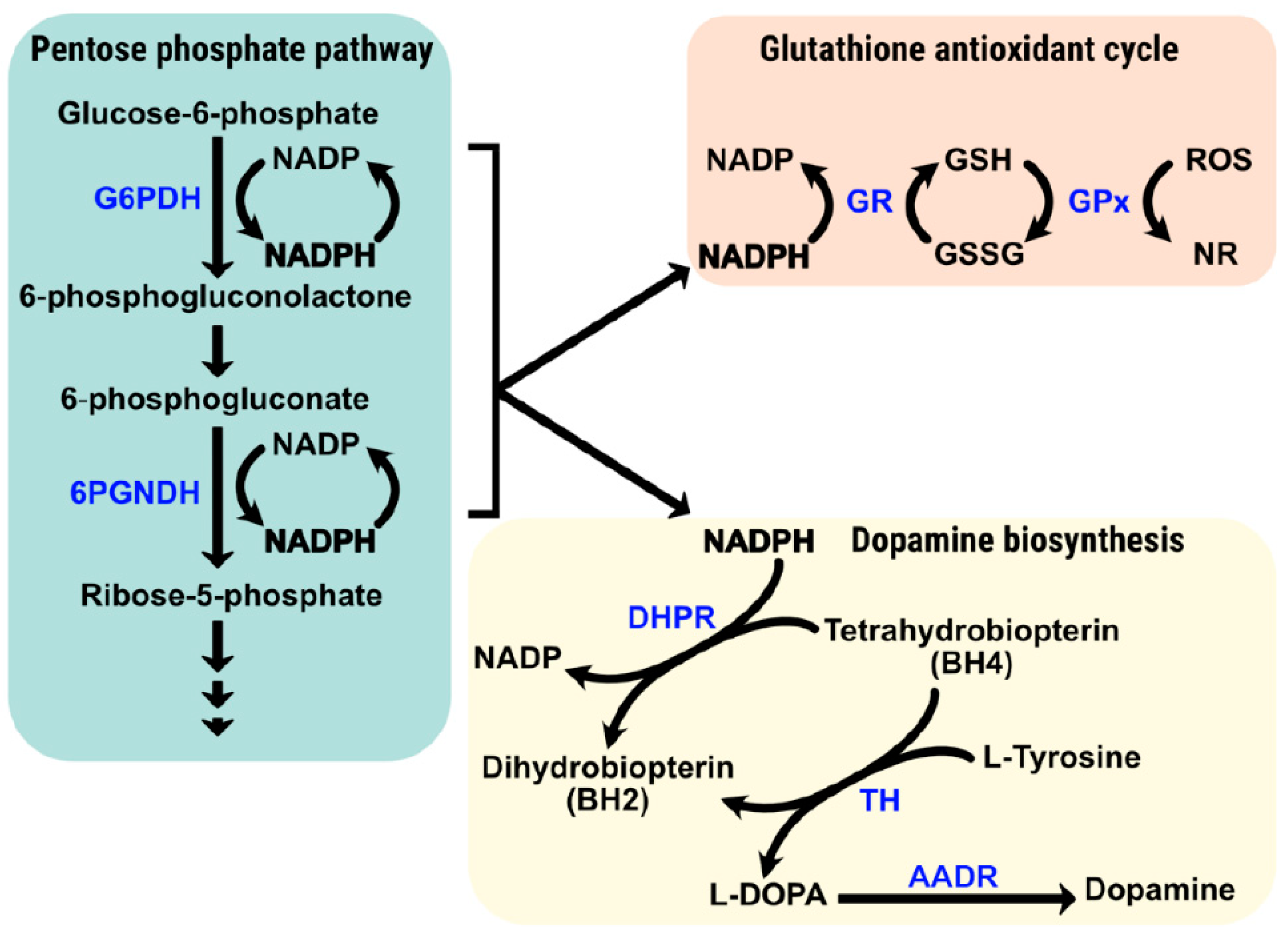{kind=link}
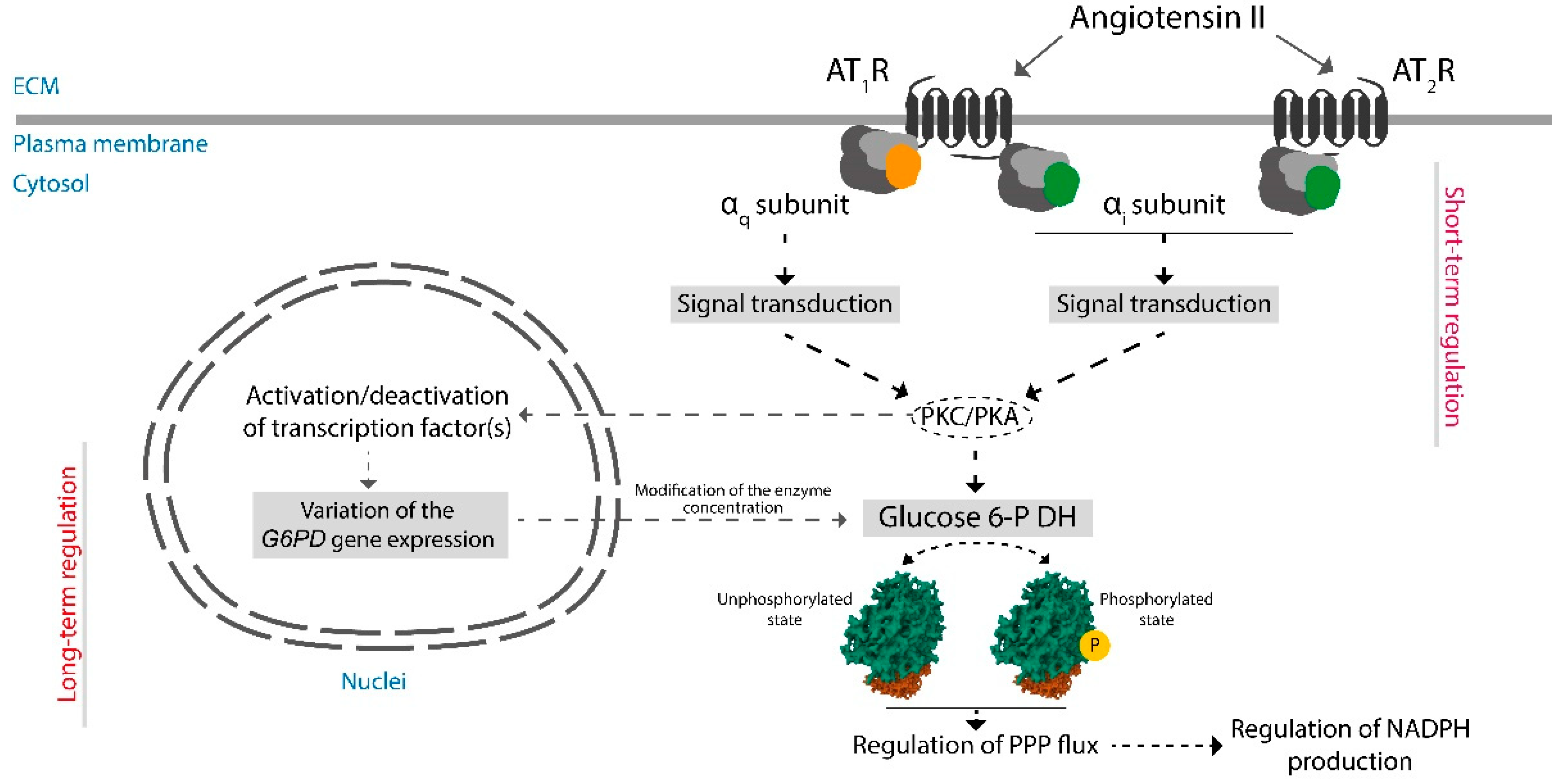{kind=link}
Abstract
:1. Introduction
2. Components of the RAS System
3. RAS in the CNS
4. NADPH Is Needed for Keeping REDOX Homeostasis in the CNS
4.1. Transgenic Expression of the Gene Coding for Glucose-6-Phosphate Dehydrogenase Prevents Nigral Neurodegeneration
4.2. Role of the Pentose Phosphate Pathway in the CNS: Evidence from Patient Samples and from Preclinical Studies in Animal Models of Neurodegeneration
5. Peripheral RAS and Glucose Availability to the Brain
6. Regulation by RAS of the Expression of Genes Coding for Protein Needed in Antioxidant Routes
6.1. Effect of RAS-Targeted Anti-Hypertensive Drugs
6.2. Evidence of Angiotensin II Effects on AT1 and AT2 Receptors Using AT1R KO Mice
6.3. Expression of G6PDH in Cells Isolated from Patients with Preeclampsia
7. Assessing the Potential of RAS to Regulate NADPH Production
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Labandeira-Garcia, J.L.; Valenzuela, R.; Costa-Besada, M.A.; Villar-Cheda, B.; Rodriguez-Perez, A.I. The intracellular renin-angiotensin system: Friend or foe. Some light from the dopaminergic neurons. Prog. Neurobiol. 2021, 199, 101919. [Google Scholar] [CrossRef]
- Valenzuela, R.; Rodriguez-Perez, A.I.; Costa-Besada, M.A.; Rivas-Santisteban, R.; Garrido-Gil, P.; Lopez-Lopez, A.; Navarro, G.; Lanciego, J.L.; Franco, R.; Labandeira-Garcia, J.L. An ACE2/Mas-related receptor MrgE axis in dopaminergic neuron mitochondria. Redox Biol. 2021, 46, 102078. [Google Scholar] [CrossRef]
- Franco, R.; Casanovas, B.; Camps, J.; Navarro, G.; Martínez-Pinilla, E. Antixoxidant supplements versus health benefits of brief/intermittent exposure to potentially toxic physical or chemical agents. Curr. Issues Mol. Biol. 2021, 43, 650–664. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Franco, R.; Serrano-Marín, J. Can chronic therapeutic drug use by the elderly affect Alzheimers disease risk and rate of progression? Explor. Neuroprot. Ther. 2023, 3, 8–23. [Google Scholar] [CrossRef]
- Zamarbide, M.; Gil-Bea, F.J.; Bannenberg, P.; Martínez-Pinilla, E.; Sandoval, J.; Franco, R.; Pérez-Mediavilla, A. Maternal imprinting on cognition markers of wild type and transgenic Alzheimer’s disease model mice. Sci. Rep. 2018, 8, 6434. [Google Scholar] [CrossRef]
- DiDonato, S.; Zeviani, M.; Giovannini, P.; Savarese, N.; Rimoldi, M.; Mariotti, C.; Girotti, F.; Caraceni, T. Respiratory chain and mitochondrial DNA in muscle and brain in Parkinson’s disease patients. Neurology 1993, 43, 2262–2268. [Google Scholar] [CrossRef]
- Zeviani, M.; Viscomi, C. Mitochondrial Neurodegeneration. Cells 2022, 11, 637. [Google Scholar] [CrossRef]
- Valenzuela, R.; Costa-Besada, M.A.M.A.; Iglesias-Gonzalez, J.; Perez-Costas, E.; Villar-Cheda, B.; Garrido-Gil, P.; Melendez-Ferro, M.; Soto-Otero, R.; Lanciego, J.L.J.L.; Henrion, D.; et al. Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging-related vulnerability to neurodegeneration. Cell Death Dis. 2016, 7, e2427. [Google Scholar] [CrossRef]
- Franco, R.; Serrano-Marín, J. The unbroken Krebs cycle. Hormonal-like regulation and mitochondrial signaling to control mitophagy and prevent cell death. BioEssays 2022, 45, 2200194. [Google Scholar] [CrossRef]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The concise guide to pharmacology 2021/22: G protein-coupled receptors. Br. J. Pharmacol. 2021, 178, S27–S156. [Google Scholar] [CrossRef]
- Pringle, K.G.; Tadros, M.A.; Callister, R.J.; Lumbers, E.R. The expression and localization of the human placental prorenin/renin-angiotensin system throughout pregnancy: Roles in trophoblast invasion and angiogenesis? Placenta 2011, 32, 956–962. [Google Scholar] [CrossRef]
- Gregório, J.F.; Rodrigues-Machado, M.d.G.; Santos, R.A.S.; Carvalho-Ribeiro, I.A.; Nunes, O.M.; Oliveira, I.F.A.; Vasconcellos, A.V.d.O.; Campagnole-Santos, M.J.; Magalhães, G.S. Asthma: Role of the angiotensin-(1-7)/Mas (MAS1) pathway in pathophysiology and therapy. Br. J. Pharmacol. 2021, 178, 4428–4439. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.R.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef]
- Kuriakose, J.; Montezano, A.C.; Touyz, R.M. ACE2/Ang-(1-7)/Mas1 axis and the vascular system: Vasoprotection to COVID-19-associated vascular disease. Clin. Sci. 2021, 135, 387–407. [Google Scholar] [CrossRef]
- Karnik, S.S.; Singh, K.D.; Tirupula, K.; Unal, H. Significance of angiotensin 1-7 coupling with MAS1 receptor and other GPCRs to the renin-angiotensin system: IUPHAR Review 22. Br. J. Pharmacol. 2017, 174, 737–753. [Google Scholar] [CrossRef]
- Young, D.; Waitches, G.; Birchmeier, C.; Fasano, O.; Wigler, M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 1986, 45, 711–719. [Google Scholar] [CrossRef]
- Jackson, T.R.; Blair, L.A.C.; Marshall, J.; Goedert, M.; Hanley, M.R. The mas oncogene encodes an angiotensin receptor. Nature 1988, 335, 437–440. [Google Scholar] [CrossRef]
- Gaidarov, I.; Adams, J.; Frazer, J.; Anthony, T.; Chen, X.; Gatlin, J.; Semple, G.; Unett, D.J. Angiotensin (1-7) does not interact directly with MAS1, but can potently antagonize signaling from the AT1 receptor. Cell. Signal. 2018, 50, 9–24. [Google Scholar] [CrossRef]
- Musnier, A.; Blanchot, B.; Reiter, E.; Crépieux, P. GPCR signalling to the translation machinery. Cell. Signal. 2010, 22, 707–716. [Google Scholar] [CrossRef]
- Gobeil, F.; Fortier, A.; Zhu, T.; Bossolasco, M.; Leduc, M.; Grandbois, M.; Heveker, N.; Bkaily, G.; Chemtob, S.; Barbaz, D. G-protein-coupled receptors signalling at the cell nucleus: An emerging paradigm. Can. J. Physiol. Pharmacol. 2006, 84, 287–297. [Google Scholar] [CrossRef]
- Ge, J.; Barnes, N.M. Alterations in angiotensin AT1 and AT2 receptor subtype levels in brain regions from patients with neurodegenerative disorders. Eur. J. Pharmacol. 1996, 297, 299–306. [Google Scholar] [CrossRef]
- De Dios, L.; Collazo, C.; Inostroza-Nieves, Y. Renin-angiotensin-system increases phosphorylated tau and Reactive Oxygen Species in human cortical neuron cell line. Biochem. Biophys. Rep. 2022, 32, 101355. [Google Scholar] [CrossRef]
- Takuwa, H.; Masamoto, K.; Yamazaki, K.; Kawaguchi, H.; Ikoma, Y.; Tajima, Y.; Obata, T.; Tomita, Y.; Suzuki, N.; Kanno, I.; et al. Long-term adaptation of cerebral hemodynamic response to somatosensory stimulation during chronic hypoxia in awake mice. J. Cereb. Blood Flow. Metab. 2013, 33, 774–779. [Google Scholar] [CrossRef]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 34. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Perier, C. Neurodegeneration: The Size Takes It All. Curr. Biol. 2015, 25, R797–R800. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Fedorow, H.; Tribl, F.; Halliday, G.; Gerlach, M.; Riederer, P.; Double, K.L. Neuromelanin in human dopamine neurons: Comparison with peripheral melanins and relevance to Parkinson’s disease. Prog. Neurobiol. 2005, 75, 109–124. [Google Scholar] [CrossRef]
- Marsden, C.D. Pigmentation in the nucleus substantiae nigrae of mammals. J. Anat. 1961, 95, 256. [Google Scholar]
- Nakaoka, H.; Mogi, M.; Kan-No, H.; Tsukuda, K.; Ohshima, K.; Wang, X.L.; Chisaka, T.; Bai, H.Y.; Shan, B.S.; Kukida, M.; et al. Angiotensin II type 2 receptor signaling affects dopamine levels in the brain and prevents binge eating disorder. J. Renin. Angiotensin. Aldosterone. Syst. 2015, 16, 749–757. [Google Scholar] [CrossRef]
- Jackson, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef]
- Kobiec, T.; Otero-Losada, M.; Chevalier, G.; Udovin, L.; Bordet, S.; Menéndez-Maissonave, C.; Capani, F.; Pérez-Lloret, S. The Renin-Angiotensin System Modulates Dopaminergic Neurotransmission: A New Player on the Scene. Front. Synaptic Neurosci. 2021, 13, 638519. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Rodriguez-Pallares, J.; Dominguez-Meijide, A.; Valenzuela, R.; Villar-Cheda, B.; Rodríguez-Perez, A.I. Dopamine-angiotensin interactions in the basal ganglia and their relevance for Parkinson’s disease. Mov. Disord. 2013, 28, 1337–1342. [Google Scholar] [CrossRef]
- Costa-Besada, M.A.; Valenzuela, R.; Garrido-Gil, P.; Villar-Cheda, B.; Parga, J.A.; Lanciego, J.L.; Labandeira-Garcia, J.L. Paracrine and Intracrine Angiotensin 1-7/Mas Receptor Axis in the Substantia Nigra of Rodents, Monkeys, and Humans. Mol. Neurobiol. 2018, 55, 5847–5867. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.; Rodríguez-Perez, A.; Garrido-Gil, P.; Rodriguez-Pallares, J.; Lanciego, J.; Guerra, M. Brain renin-angiotensin system and microglial polarization: Implications for aging and neurodegeneration. Front. Aging Neurosci. 2017, 9, 129. [Google Scholar] [CrossRef]
- Welcome, M.O.; Mastorakis, N.E. Emerging Concepts in Brain Glucose Metabolic Functions: From Glucose Sensing to How the Sweet Taste of Glucose Regulates Its Own Metabolism in Astrocytes and Neurons. Neuromolecular Med. 2018, 20, 281–300. [Google Scholar] [CrossRef]
- Tu, D.; Gao, Y.; Yang, R.; Guan, T.; Hong, J.S.; Gao, H.M. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J. Neuroinflammation 2019, 16, 255. [Google Scholar] [CrossRef]
- Gruetter, R. Glycogen: The forgotten cerebral energy store. J. Neurosci. Res. 2003, 74, 179–183. [Google Scholar] [CrossRef]
- Ben-Yoseph, O.; Boxer, P.A.; Ross, B.D. Assessment of the role of the glutathione and pentose phosphate pathways in the protection of primary cerebrocortical cultures from oxidative stress. J. Neurochem. 1996, 66, 2329–2337. [Google Scholar] [CrossRef]
- Herken, H.; Lange, K.; Kolbe, H. Brain disorders induced by pharmacological blockade of the pentose phosphate pathway. Biochem. Biophys. Res. Commun. 1969, 36, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Attilio, P.J.; Snapper, D.M.; Rusnak, M.; Isaac, A.; Soltis, A.R.; Wilkerson, M.D.; Dalgard, C.L.; Symes, A.J. Transcriptomic Analysis of Mouse Brain after Traumatic Brain Injury Reveals That the Angiotensin Receptor Blocker Candesartan Acts Through Novel Pathways. Front. Neurosci. 2021, 15, 636259. [Google Scholar] [CrossRef] [PubMed]
- Mejías, R.; Villadiego, J.; Pintado, C.O.; Vime, P.J.; Gao, L.; Toledo-Aral, J.J.; Echevarría, M.; López-Barneo, J. Neuroprotection by transgenic expression of glucose-6-phosphate dehydrogenase in dopaminergic nigrostriatal neurons of mice. J. Neurosci. 2006, 26, 4500–4508. [Google Scholar] [CrossRef]
- Romero-Ruiz, A.; Mejías, R.; Díaz-Martín, J.; López-Barneo, J.; Gao, L. Mesencephalic and striatal protein profiles in mice over-expressing glucose-6-phosphate dehydrogenase in dopaminergic neurons. J. Proteom. 2010, 73, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, S.; Almeida, A.; Bolaños, J.P. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 2012, 443, 3–12. [Google Scholar] [CrossRef]
- Silver, I.; Erecińska, M. Oxygen and ion concentrations in normoxic and hypoxic brain cells. Adv. Exp. Med. Biol. 1998, 454, 7–16. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Goyal, M.S.; Hawrylycz, M.; Miller, J.A.; Snyder, A.Z.; Raichle, M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014, 19, 49–57. [Google Scholar] [CrossRef]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- García-Nogales, P.; Almeida, A.; Bolaños, J.P. Peroxynitrite protects neurons against nitric oxide-mediated apoptosis. A key role for glucose-6-phosphate dehydrogenase activity in neuroprotection. J. Biol. Chem. 2003, 278, 864–874. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Gutterer, J.M.; Kussmaul, L.; Hamprecht, B.; Dringen, R. Microglial cells in culture express a prominent glutathione system for the defense against reactive oxygen species. Dev. Neurosci. 2000, 22, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C. Why are nigral catecholaminergic neurons more vulnerable than other cells in Parkinson’s disease? Ann. Neurol. 1992, 32 (Suppl. S1), S88–S93. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Han, L.L.; Du, C.M.; Yu, Z.Y. Redox potentials of dopamine and its supramolecular complex with aspartic acid. Russ. J. Phys. Chem. A 2014, 88, 1085–1090. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. On the Role of Aminochrome in Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Parkinson’s Disease. Front. Neurosci. 2019, 13, 437984. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Jang, Y.J.; Kim, H.J.; Hwang, O. Tetrahydrobiopterin is released from and causes preferential death of catecholaminergic cells by oxidative stress. Mol. Pharmacol. 2000, 58, 633–640. [Google Scholar] [CrossRef]
- Perry, T.L.; Godin, D.V.; Hansen, S. Parkinson’s disease: A disorder due to nigral glutathione deficiency? Neurosci. Lett. 1982, 33, 305–310. [Google Scholar] [CrossRef]
- Bellinger, F.P.; Bellinger, M.T.; Seale, L.A.; Takemoto, A.S.; Raman, A.V.; Miki, T.; Manning-Boǧ, A.B.; Berry, M.J.; White, L.R.; Ross, G.W. Glutathione Peroxidase 4 is associated with Neuromelanin in Substantia Nigra and Dystrophic Axons in Putamen of Parkinson’s brain. Mol. Neurodegener. 2011, 6, 8. [Google Scholar] [CrossRef]
- Kish, S.J.; Morito, C.; Hornykiewicz, O. Glutathione peroxidase activity in Parkinson’s disease brain. Neurosci. Lett. 1985, 58, 343–346. [Google Scholar] [CrossRef]
- Ridet, J.L.; Bensadoun, J.C.; Déglon, N.; Aebischer, P.; Zurn, A.D. Lentivirus-mediated expression of glutathione peroxidase: Neuroprotection in murine models of Parkinson’s disease. Neurobiol. Dis. 2006, 21, 29–34. [Google Scholar] [CrossRef]
- Bensadoun, J.C.; Mirochnitchenko, O.; Inouye, M.; Aebischer, P.; Zurn, A.D. Attenuation of 6-OHDA-induced neurotoxicity in glutathione peroxidase transgenic mice. Eur. J. Neurosci. 1998, 10, 3231–3236. [Google Scholar] [CrossRef] [PubMed]
- Mnafgui, K.; Ghazouani, L.; Hajji, R.; Tlili, A.; Derbali, F.; da Silva, F.I.; Araújo, J.L.; de Oliveira Schinoff, B.; Bachega, J.F.R.; da Silva Santos, A.L.; et al. Oleuropein Protects Against Cerebral Ischemia Injury in Rats: Molecular Docking, Biochemical and Histological Findings. Neurochem. Res. 2021, 46, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.X.; Li, J.Y.; Tang, Z.Z.; Jiang, S.; Liu, Y.H.; Deng, J.G.; Gao, C.H. Marinoid J, a phenylglycoside from Avicennia marina fruit, ameliorates cognitive impairment in rat vascular dementia: A quantitative iTRAQ proteomic study. Pharm. Biol. 2020, 58, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.A.; Pincus, M.R. Henry’s Clinical Diagnosis and Management by Laboratory Methods; Saunders: Philadelphia, PA, USA, 2011; ISBN 9781455726844. [Google Scholar]
- Tesauro, M.; Mauriello, A.; Rovella, V.; Annicchiarico-Petruzzelli, M.; Cardillo, C.; Melino, G.; Di Daniele, N. Arterial ageing: From endothelial dysfunction to vascular calcification. J. Intern. Med. 2017, 281, 471–482. [Google Scholar] [CrossRef]
- Sare, G.M.; Gray, L.J.; Bath, P.M.W. Effect of antihypertensive agents on cerebral blood flow and flow velocity in acute ischaemic stroke: Systematic review of controlled studies. J. Hypertens. 2008, 26, 1058–1064. [Google Scholar] [CrossRef]
- Alosco, M.L.; Gunstad, J.; Xu, X.; Clark, U.S.; Labbe, D.R.; Riskin-Jones, H.H.; Terrero, G.; Schwarz, N.F.; Walsh, E.G.; Poppas, A.; et al. The impact of hypertension on cerebral perfusion and cortical thickness in older adults. J. Am. Soc. Hypertens. 2014, 8, 561–570. [Google Scholar] [CrossRef]
- Conlin, P.R. Angiotensin II Antagonists in the Treatment of Hypertension: More Similarities Than Differences. J. Clin. Hypertens. 2000, 2, 253–257. [Google Scholar]
- Sever, P.S. Clinical profile of the novel angiotensin II type I blocker candesartan cilexetil. J. Hypertens. Suppl. 1997, 15, 9–12. [Google Scholar] [CrossRef]
- Pouleur, H.G. Clinical overview of irbesartan: A new angiotensin II receptor antagonist. Am. J. Hypertens. 1997, 10, 318S–324S. [Google Scholar] [CrossRef]
- Gillis, J.C.; Markham, A. Irbesartan. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in the management of hypertension. Drugs 1997, 54, 885–902. [Google Scholar] [CrossRef]
- Ozdemir, S.; Tandogan, B.; Ulusu, N.; Turan, B. Angiotensin II receptor blockage prevents diabetes-induced oxidative damage in rat heart. Folia Biol. 2009, 55, 11–16. [Google Scholar]
- Matsui, R.; Xu, S.; Maitland, K.A.; Hayes, A.; Leopold, J.A.; Handy, D.E.; Loscalzo, J.; Cohen, R.A. Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin II. Circulation 2005, 112, 257–263. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira-Junior, E.B.; Bustamante, J.; Newburger, P.E.; Condino-Neto, A. The Human NADPH Oxidase: Primary and Secondary Defects Impairing the Respiratory Burst Function and the Microbicidal Ability of Phagocytes. Scand. J. Immunol. 2011, 73, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Giardino, G.; Cicalese, M.P.; Delmonte, O.; Migliavacca, M.; Palterer, B.; Loffredo, L.; Cirillo, E.; Gallo, V.; Violi, F.; Pignata, C. NADPH Oxidase Deficiency: A Multisystem Approach. Oxid. Med. Cell. Longev. 2017, 2017, 4590127. [Google Scholar] [CrossRef]
- Szanto, I.; Pusztaszeri, M.; Mavromati, M. H2O2 Metabolism in Normal Thyroid Cells and in Thyroid Tumorigenesis: Focus on NADPH Oxidases. Antioxidants 2019, 8, 126. [Google Scholar] [CrossRef]
- De Deken, X.; Wang, D.; Dumont, J.E.; Miot, F. Characterization of ThOX proteins as components of the thyroid H2O2-generating system. Exp. Cell Res. 2002, 273, 187–196. [Google Scholar] [CrossRef]
- Dupuy, C.; Virion, A.; Ohayon, R.; Kaniewski, J.; Dème, D.; Pommier, J. Mechanism of hydrogen peroxide formation catalyzed by NADPH oxidase in thyroid plasma membrane. J. Biol. Chem. 1991, 266, 3739–3743. [Google Scholar] [CrossRef]
- Pessôa, B.S.; Peixoto, E.B.; Papadimitriou, A.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Spironolactone improves nephropathy by enhancing glucose-6-phosphate dehydrogenase activity and reducing oxidative stress in diabetic hypertensive rat. J. Renin-Angiotensin-Aldosterone Syst. 2012, 13, 56–66. [Google Scholar] [CrossRef]
- Moriyama, T.; Tanaka, S.; Nakayama, Y.; Fukumoto, M.; Tsujimura, K.; Yamada, K.; Bamba, T.; Yoneda, Y.; Fukusaki, E.; Oka, M. Two isoforms of TALDO1 generated by alternative translational initiation show differential nucleocytoplasmic distribution to regulate the global metabolic network. Sci. Rep. 2016, 6, 34648. [Google Scholar] [CrossRef]
- Eyaid, W.; Al Harbi, T.; Anazi, S.; Wamelink, M.M.C.; Jakobs, C.; Al Salammah, M.; Al Balwi, M.; Alfadhel, M.; Alkuraya, F.S. Transaldolase deficiency: Report of 12 new cases and further delineation of the phenotype. J. Inherit. Metab. Dis. 2013, 36, 997–1004. [Google Scholar] [CrossRef]
- Hanczko, R.; Fernandez, D.R.; Doherty, E.; Qian, Y.; Vas, G.; Niland, B.; Telarico, T.; Garba, A.; Banerjee, S.; Middleton, F.A.; et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J. Clin. Investig. 2009, 119, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Makhanova, N.A.; Crowley, S.D.; Griffiths, R.C.; Coffman, T.M. Gene expression profiles linked to AT1 angiotensin receptors in the kidney. Physiol. Genom. 2010, 42, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Irani, R.A.; Xia, Y. The Functional Role of the Renin–Angiotensin System in Pregnancy and Preeclampsia. Placenta 2008, 29, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.M. Role of the renin-angiotensin system in the pathogenesis of preeclampsia. Am. J. Physiol. Ren. Physiol. 2005, 288, F614–F625. [Google Scholar] [CrossRef] [PubMed]
- Afzal-Ahmed, I.; Mann, G.E.; Shennan, A.H.; Poston, L.; Naftalin, R.J. Preeclampsia inactivates glucose-6-phosphate dehydrogenase and impairs the redox status of erythrocytes and fetal endothelial cells. Free Radic. Biol. Med. 2007, 42, 1781–1790. [Google Scholar] [CrossRef]
- Mommsen, T.P.; Moon, T.W. Metabolic actions of glucagon-family hormones in liver. Fish Physiol. Biochem. 1989, 7, 279–288. [Google Scholar] [CrossRef]
- Exton, J.H. Mechanisms of hormonal regulation of hepatic glucose metabolism. Diabetes. Metab. Rev. 1987, 3, 163–183. [Google Scholar] [CrossRef]
- Cherrington, A.D.; Exton, J.H. Studies on the role of cAMP-dependent protein kinase in the actions of glucagon and catecholamines on liver glycogen metabolism. Metabolism. 1976, 25, 1351–1354. [Google Scholar] [CrossRef]
- Jakob, A.; Diem, S. Metabolic responses of perfused rat livers to alpha- and beta-adrenergic agonists, glucagon and cyclic AMP. Biochim. Biophys. Acta 1975, 404, 57–66. [Google Scholar] [CrossRef]
- Sherline, P.; Lynch, A.; Glinsmann, W.H. Cyclic AMP and adrenergic receptor control of rat liver glycogen metabolism. Endocrinology 1972, 91, 680–690. [Google Scholar] [CrossRef]
- Exton, J.H.; Assimacopoulos-Jeannet, F.D.; Blackmore, P.F.; Cherrington, A.D.; Chan, T.M. Metabolic responses of perfused rat livers to alpha- and beta-adrenergic agonists, glucagon and cyclic AMP. Adv. Cycl. Nucleotide Res. 1978, 9, 441–452. [Google Scholar]
- Amiri, F.; Garcia, R. Regulation of angiotensin II receptors and PKC isoforms by glucose in rat mesangial cells. Am. J. Physiol.-Ren. Physiol. 1999, 276, F691–F699. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.; Apse, K.; Pang, J.; Stanton, R.C. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J. Biol. Chem. 2000, 275, 40042–40047. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Osborne, B.W.; Stanton, R.C. Diabetes causes inhibition of glucose-6-phosphate dehydrogenase via activation of PKA, which contributes to oxidative stress in rat kidney cortex. Am. J. Physiol.-Ren. Physiol. 2005, 289, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Dieni, C.A.; Storey, K.B. Regulation of glucose-6-phosphate dehydrogenase by reversible phosphorylation in liver of a freeze tolerant frog. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 2010, 180, 1133–1142. [Google Scholar] [CrossRef]
- Gupte, R.S.; Ata, H.; Rawat, D.; Abe, M.; Taylor, M.S.; Ochi, R.; Gupte, S.A. Glucose-6-Phosphate Dehydrogenase Is a Regulator of Vascular Smooth Muscle Contraction. Antioxid. Redox Signal. 2011, 14, 543–558. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, R.; Serrano-Marín, J.; Navarro, G.; Rivas-Santisteban, R. The NADPH Link between the Renin Angiotensin System and the Antioxidant Mechanisms in Dopaminergic Neurons. Antioxidants 2023, 12, 1869. https://doi.org/10.3390/antiox12101869
Franco R, Serrano-Marín J, Navarro G, Rivas-Santisteban R. The NADPH Link between the Renin Angiotensin System and the Antioxidant Mechanisms in Dopaminergic Neurons. Antioxidants. 2023; 12(10):1869. https://doi.org/10.3390/antiox12101869
Chicago/Turabian StyleFranco, Rafael, Joan Serrano-Marín, Gemma Navarro, and Rafael Rivas-Santisteban. 2023. "The NADPH Link between the Renin Angiotensin System and the Antioxidant Mechanisms in Dopaminergic Neurons" Antioxidants 12, no. 10: 1869. https://doi.org/10.3390/antiox12101869