
Novel Strategies in the Early Detection and Treatment of Endothelial Cell-Specific Mitochondrial Dysfunction in Coronary Artery Disease

Abstract
:1. Introduction
2. Mitochondrial Dysfunction as an Instigator of Endothelial Dysfunction in Coronary Artery Disease
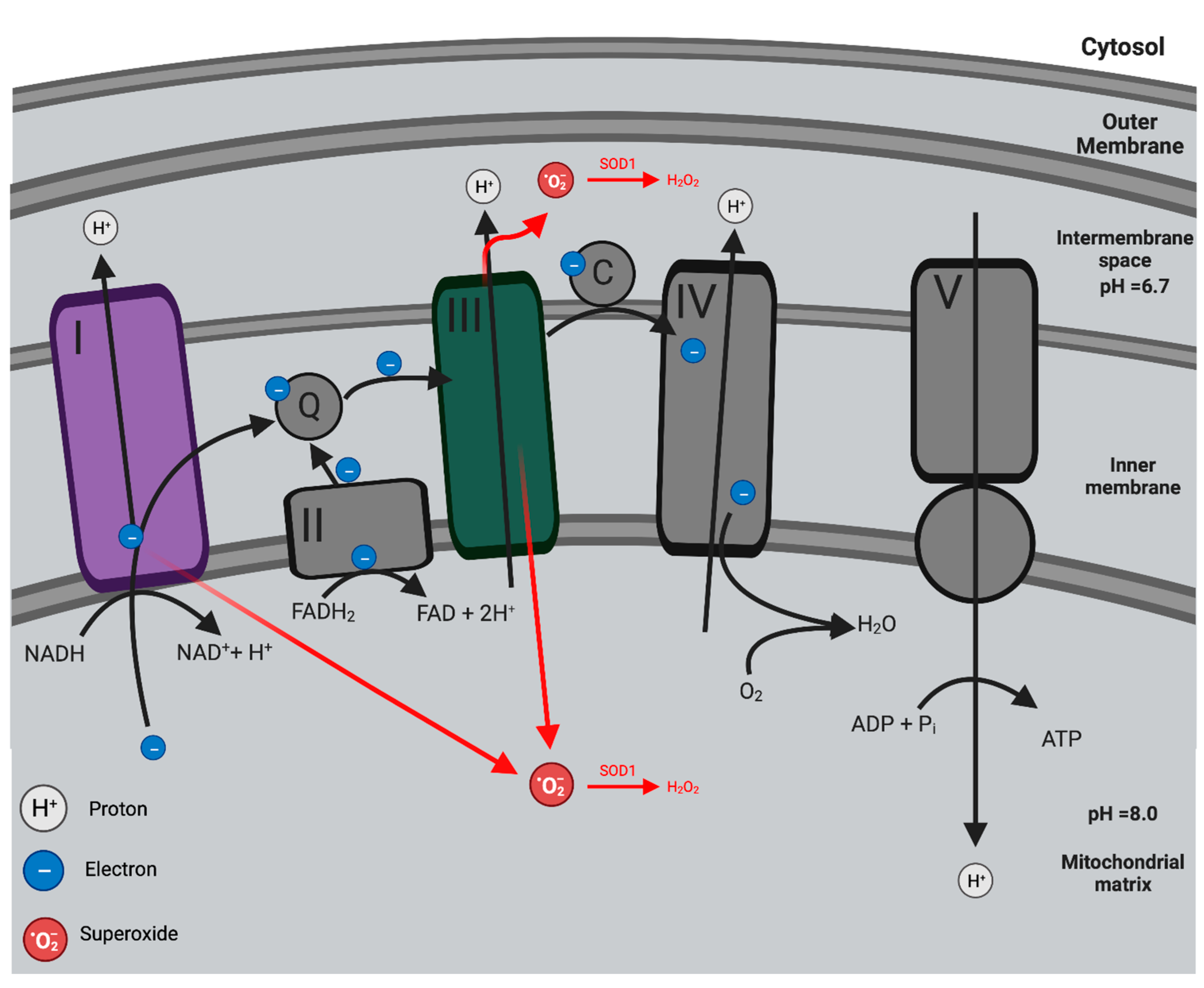
2.1. Mitochondria: Oxidative Phosphorylation and the Production of Mitochondria ROS

2.2. Pathogenesis of Mitochondrial Dysfunction in Endothelial Dysfunction
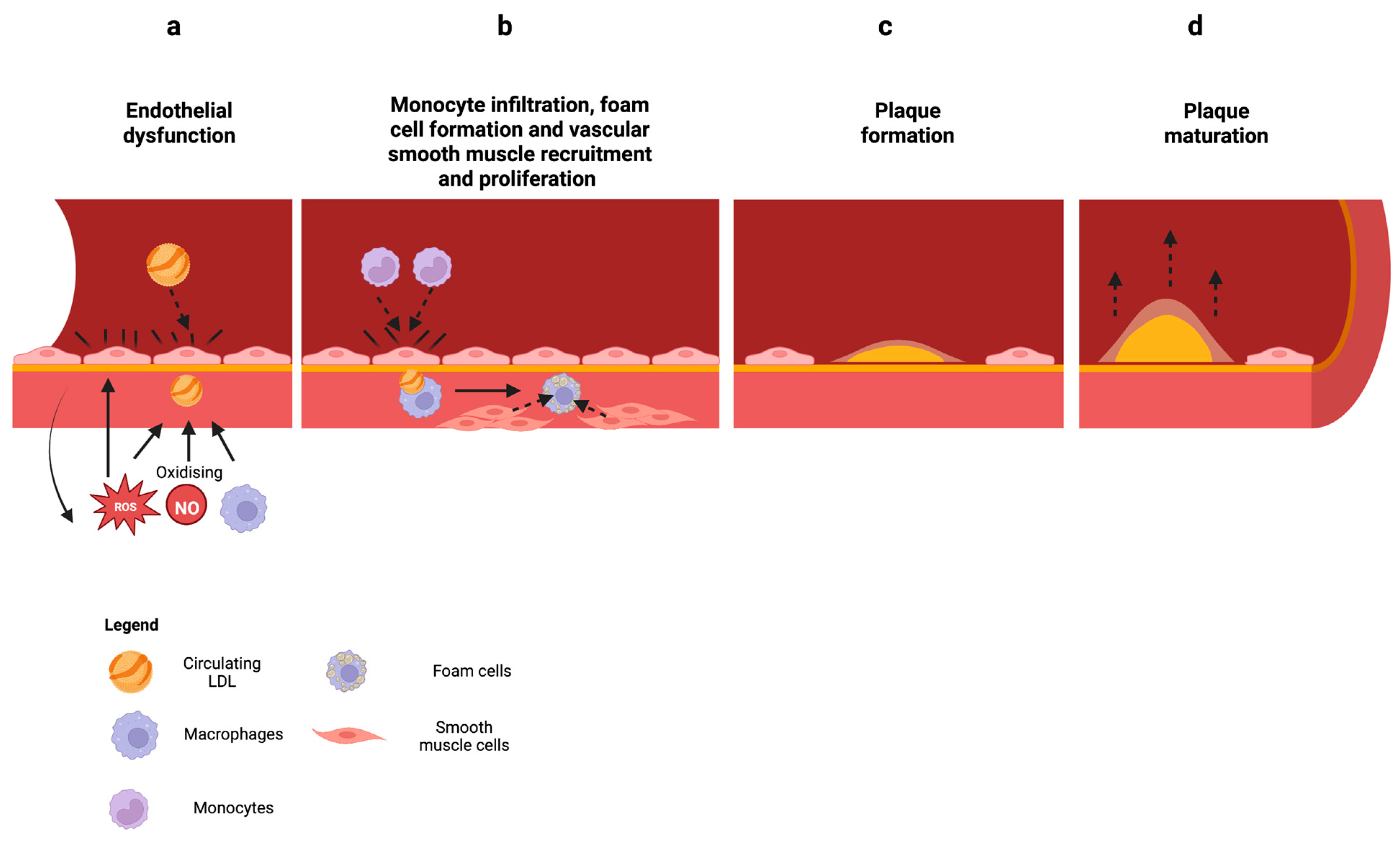
2.3. Mitochondrial Dysfunction in Atherosclerosis

3. Mitochondrial Dysfunction as a Biomarker in the Early Detection of Coronary Artery Disease
3.1. Targeting Mitochondria ROS and Oxidative Stress
3.2. Potential Targets to Reduce Mitochondrial ROS Production and Oxidative Stress
3.2.1. Mitochondria Outer Membrane Proteins
3.2.2. Mitochondria Inner Membrane Proteins
3.3. Mitochondria DNA
3.3.1. P66SHC
3.3.2. P2X7 Receptor
3.4. Myeloperoxidase
4. Potential Therapeutic Strategies to Target Mitochondrial ROS and Oxidative Stress
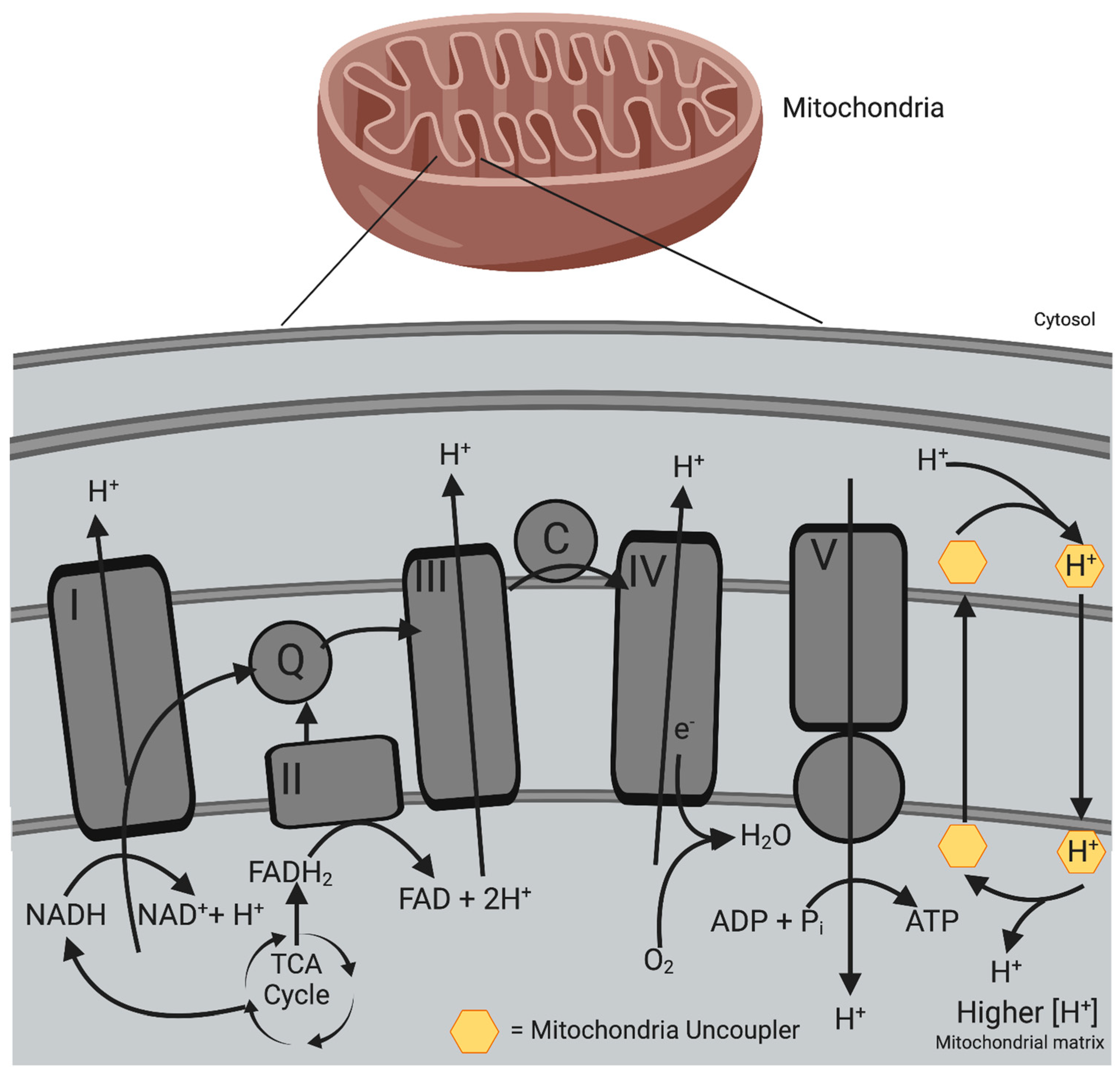
4.1. Mitochondrial Uncouplers
4.2. ROS Scavengers
4.3. P2X7R Antagonists
4.4. Colchicine
4.5. MPO Inhibitors
5. Future Directions
5.1. Endothelial Colony Forming Cells and Importance in Biomarker and Drug Discovery
5.2. Phenotypic High Throughput Screening for Treating Mitochondrial Dysfunction in Endothelial Cells
5.3. Omic Approaches to Drug and Biomarker Discovery with ECFCs
5.4. Animal Models of Atherosclerosis to Study Endothelial Mitochondrial Dysfunction
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dawber, T.R.; Moore, F.E.; Mann, G.V., II. Coronary Heart Disease in the Framingham Study. Am. J. Public Health Nations Health 1957, 47, 4–24. [Google Scholar] [CrossRef]
- Prediction of Coronary Heart Disease Using Risk Factor Categories|Circulation. Available online: https://www.ahajournals.org/doi/10.1161/01.cir.97.18.1837?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed (accessed on 26 August 2022).
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 24 April 2023).
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.-T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on Cardiovascular Disease Prevention in Clinical Practice. Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Lloyd-Jones, D.M.; Bennett, G.; Coady, S.; D’Agostino, R.B.; Gibbons, R.; Greenland, P.; Lackland, D.T.; Levy, D.; O’Donnell, C.J.; et al. 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk. Circulation 2014, 129, S49–S73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernon, S.T.; Coffey, S.; Bhindi, R.; Soo Hoo, S.Y.; Nelson, G.I.; Ward, M.R.; Hansen, P.S.; Asrress, K.N.; Chow, C.K.; Celermajer, D.S.; et al. Increasing Proportion of ST Elevation Myocardial Infarction Patients with Coronary Atherosclerosis Poorly Explained by Standard Modifiable Risk Factors. Eur. J. Prev. Cardiol. 2017, 24, 1824–1830. [Google Scholar] [CrossRef]
- Figtree, G.A.; Vernon, S.T.; Hadziosmanovic, N.; Sundström, J.; Alfredsson, J.; Arnott, C.; Delatour, V.; Leósdóttir, M.; Hagström, E. Mortality in STEMI Patients without Standard Modifiable Risk Factors: A Sex-Disaggregated Analysis of SWEDEHEART Registry Data. Lancet 2021, 397, 1085–1094. [Google Scholar] [CrossRef]
- Vernon, S.T.; Coffey, S.; D’Souza, M.; Chow, C.K.; Kilian, J.; Hyun, K.; Shaw, J.A.; Adams, M.; Roberts-Thomson, P.; Brieger, D.; et al. ST-Segment–Elevation Myocardial Infarction (STEMI) Patients without Standard Modifiable Cardiovascular Risk Factors—How Common Are They, and What Are Their Outcomes? J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2019, 8, e013296. [Google Scholar] [CrossRef]
- Nelson, A.J.; Ardissino, M.; Psaltis, P.J. Current Approach to the Diagnosis of Atherosclerotic Coronary Artery Disease: More Questions than Answers. Ther. Adv. Chronic Dis. 2019, 10, 2040622319884819. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Arnold, P.K.; Finley, L.W.S. Regulation and Function of the Mammalian Tricarboxylic Acid Cycle. J. Biol. Chem. 2022, 299, 102838. [Google Scholar] [CrossRef]
- Cooper, G.M. The Mechanism of Oxidative Phosphorylation. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- The Oxidative Phosphorylation System in Mammalian Mitochondria|SpringerLink. Available online: https://link.springer.com/chapter/10.1007/978-94-007-2869-1_1 (accessed on 31 August 2022).
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial Proton and Electron Leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X. Targeting Mitochondrial Reactive Oxygen Species as Novel Therapy for Inflammatory Diseases and Cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Canali, R.; Rettori, D.; Kaplowitz, N. Effect of Glutathione Depletion on Sites and Topology of Superoxide and Hydrogen Peroxide Production in Mitochondria. Mol. Pharmacol. 2003, 64, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial Dysfunction in Atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The Role of Mitochondrial Function and Cellular Bioenergetics in Ageing and Disease. Br. J. Dermatol. 2013, 169, 1–8. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in Innate Immune Responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and Endothelial Function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [Green Version]
- Qu, K.; Yan, F.; Qin, X.; Zhang, K.; He, W.; Dong, M.; Wu, G. Mitochondrial Dysfunction in Vascular Endothelial Cells and Its Role in Atherosclerosis. Front. Physiol. 2022, 13, 1084604. [Google Scholar] [CrossRef] [PubMed]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; Yazbi, A.E.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci.-Landmark 2022, 27, 105. [Google Scholar] [CrossRef] [PubMed]
- Widlansky, M.; Gutterman, D. Regulation of Endothelial Function by Mitochondrial Reactive Oxygen Species. Antioxid. Redox Signal. 2011, 15, 1517–1530. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, C.M. Endothelium. Arterioscler. Thromb. Vasc. Biol. 2016, 36, e26–e31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, B.K.; Chan, K.-G.; Goh, B.H.; Yap, W.H. The Role of Natural Products in Targeting Cardiovascular Diseases via Nrf2 Pathway: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial Function and Dysfunction. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative Stress and Reactive Oxygen Species in Endothelial Dysfunction Associated with Cardiovascular and Metabolic Diseases. Vascul. Pharmacol. 2018, 100, 26–33. [Google Scholar] [CrossRef]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Leonart, M.E. Oxidative Stress and Cancer: An Overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Neurodegenerative Diseases and Oxidative Stress|Nature Reviews Drug Discovery. Available online: https://www.nature.com/articles/nrd1330 (accessed on 26 September 2022).
- Tang, X.; Luo, Y.-X.; Chen, H.-Z.; Liu, D.-P. Mitochondria, Endothelial Cell Function, and Vascular Diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 Deficiency Alters Mitochondrial Function and Exacerbates the Development of Atherosclerosis. Antioxid. Redox Signal. 2011, 14, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing Mitochondrial Superoxide Production Blocks Three Pathways of Hyperglycaemic Damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, X.; Bi, H.; Zhu, Q.; Wu, J.; Xia, X.; Qiushi, R.; Ho, P.C.P. Expression Modification of Uncoupling Proteins and MnSOD in Retinal Endothelial Cells and Pericytes Induced by High Glucose: The Role of Reactive Oxygen Species in Diabetic Retinopathy. Exp. Eye Res. 2006, 83, 807–816. [Google Scholar] [CrossRef]
- Lee, K.-U.; Lee, I.K.; Han, J.; Song, D.-K.; Kim, Y.M.; Song, H.S.; Kim, H.S.; Lee, W.J.; Koh, E.H.; Song, K.-H.; et al. Effects of Recombinant Adenovirus-Mediated Uncoupling Protein 2 Overexpression on Endothelial Function and Apoptosis. Circ. Res. 2005, 96, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Ando, S.; Dajani, H.R.; Floras, J.S. Frequency Domain Characteristics of Muscle Sympathetic Nerve Activity in Heart Failure and Healthy Humans. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1997, 273, R205–R212. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting MiR-155 Restores Abnormal Microglia and Attenuates Disease in SOD1 Mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dromparis, P.; Michelakis, E.D. Mitochondria in Vascular Health and Disease. Annu. Rev. Physiol. 2013, 75, 95–126. [Google Scholar] [CrossRef] [PubMed]
- Quintero, M.; Colombo, S.L.; Godfrey, A.; Moncada, S. Mitochondria as Signaling Organelles in the Vascular Endothelium. Proc. Natl. Acad. Sci. USA 2006, 103, 5379–5384. [Google Scholar] [CrossRef] [Green Version]
- Dranka, B.P.; Hill, B.G.; Darley-Usmar, V.M. Mitochondrial Reserve Capacity in Endothelial Cells: The Impact of Nitric Oxide and Reactive Oxygen Species. Free Radic. Biol. Med. 2010, 48, 905–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial Dysfunction: A Comprehensive Appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial Dysfunction: The Early Predictor of Atherosclerosis. Cardiovasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef]
- Barthelmes, J.; Nägele, M.P.; Ludovici, V.; Ruschitzka, F.; Sudano, I.; Flammer, A.J. Endothelial Dysfunction in Cardiovascular Disease and Flammer Syndrome—Similarities and Differences. EPMA J. 2017, 8, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Widder, J.D.; Fraccarollo, D.; Galuppo, P.; Hansen, J.M.; Jones, D.P.; Ertl, G.; Bauersachs, J. Attenuation of Angiotensin II-Induced Vascular Dysfunction and Hypertension by Overexpression of Thioredoxin-2. Hypertension 2009, 54, 338–344. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Malakar, A.K.; Choudhury, D.; Halder, B.; Paul, P.; Uddin, A.; Chakraborty, S. A Review on Coronary Artery Disease, Its Risk Factors, and Therapeutics. J. Cell. Physiol. 2019, 234, 16812–16823. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, B.; Vilahur, G.; Badimon, J.J. Plaque Progression and Regression in Atherothrombosis. J. Thromb. Haemost. 2007, 5, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Padró, T.; Vilahur, G. Atherosclerosis, Platelets and Thrombosis in Acute Ischaemic Heart Disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianazza, E.; Brioschi, M.; Martinez Fernandez, A.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid Peroxidation in Atherosclerotic Cardiovascular Diseases. Antioxid. Redox Signal. 2021, 34, 49–98. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Y.; Lei, Y.; Tzvetkov, N.T.; Liu, X.; Yeung, A.W.K.; Xu, S.; Atanasov, A.G. Targeting Foam Cell Formation in Atherosclerosis: Therapeutic Potential of Natural Products. Pharmacol. Rev. 2019, 71, 596–670. [Google Scholar] [CrossRef]
- Yu, X.-H.; Fu, Y.-C.; Zhang, D.-W.; Yin, K.; Tang, C.-K. Foam Cells in Atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.-J.; Libby, P. Progression of Atheroma. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1370–1380. [Google Scholar] [CrossRef] [Green Version]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial Pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; Smith, R.A.J. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Szeto, H.H.; Schiller, P.W. Novel Therapies Targeting Inner Mitochondrial Membrane—From Discovery to Clinical Development. Pharm. Res. 2011, 28, 2669–2679. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Hartley, R.C.; Murphy, M.P. Mitochondria-Targeted Small Molecule Therapeutics and Probes. Antioxid. Redox Signal. 2011, 15, 3021–3038. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Harrison, D.G. Methods for Detection of Mitochondrial and Cellular Reactive Oxygen Species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, R.; Hilhorst, M.; Zhang, H.; Zeisbrich, M.; Berry, G.J.; Wallis, B.B.; Harrison, D.G.; Giacomini, J.C.; Goronzy, J.J.; Weyand, C.M. Glucose Metabolism Controls Disease-Specific Signatures of Macrophage Effector Functions. JCI Insight 2018, 3, e123047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owada, T.; Yamauchi, H.; Saitoh, S.; Miura, S.; Machii, H.; Takeishi, Y. Resolution of Mitochondrial Oxidant Stress Improves Aged-Cardiovascular Performance. Coron. Artery Dis. 2017, 28, 33. [Google Scholar] [CrossRef] [Green Version]
- Shchepinova, M.M.; Cairns, A.G.; Prime, T.A.; Logan, A.; James, A.M.; Hall, A.R.; Vidoni, S.; Arndt, S.; Caldwell, S.T.; Prag, H.A.; et al. MitoNeoD: A Mitochondria-Targeted Superoxide Probe. Cell Chem. Biol. 2017, 24, 1285–1298.e12. [Google Scholar] [CrossRef] [Green Version]
- Robb, E.L.; Gawel, J.M.; Aksentijević, D.; Cochemé, H.M.; Stewart, T.S.; Shchepinova, M.M.; Qiang, H.; Prime, T.A.; Bright, T.P.; James, A.M.; et al. Selective Superoxide Generation within Mitochondria by the Targeted Redox Cycler MitoParaquat. Free Radic. Biol. Med. 2015, 89, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Guarini, G.; Kiyooka, T.; Ohanyan, V.; Pung, Y.F.; Marzilli, M.; Chen, Y.R.; Chen, C.L.; Kang, P.T.; Hardwick, J.P.; Kolz, C.L.; et al. Impaired Coronary Metabolic Dilation in the Metabolic Syndrome Is Linked to Mitochondrial Dysfunction and Mitochondrial DNA Damage. Basic Res. Cardiol. 2016, 111, 29. [Google Scholar] [CrossRef] [Green Version]
- Duicu, O.M.; Lighezan, R.; Sturza, A.; Balica, R.; Vaduva, A.; Feier, H.; Gaspar, M.; Ionac, A.; Noveanu, L.; Borza, C.; et al. Assessment of Mitochondrial Dysfunction and Monoamine Oxidase Contribution to Oxidative Stress in Human Diabetic Hearts. Oxid. Med. Cell. Longev. 2016, 2016, 8470394. [Google Scholar] [CrossRef] [Green Version]
- Avram, V.F.; Merce, A.P.; Hâncu, I.M.; Bătrân, A.D.; Kennedy, G.; Rosca, M.G.; Muntean, D.M. Impairment of Mitochondrial Respiration in Metabolic Diseases: An Overview. Int. J. Mol. Sci. 2022, 23, 8852. [Google Scholar] [CrossRef] [PubMed]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W.; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Metabolomic Profiling Identifies Novel Circulating Biomarkers of Mitochondrial Dysfunction Differentially Elevated in Heart Failure with Preserved Versus Reduced Ejection Fraction: Evidence for Shared Metabolic Impairments in Clinical Heart Failure. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2016, 5, e003190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a Multi-Functional Mitochondrial Protein Regulating Cell Life and Death. Mol. Aspects Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. VDAC Regulation by the Bcl-2 Family of Proteins. Cell Death Differ. 2000, 7, 1174–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahimi-Horn, M.C.; Giuliano, S.; Saland, E.; Lacas-Gervais, S.; Sheiko, T.; Pelletier, J.; Bourget, I.; Bost, F.; Féral, C.; Boulter, E.; et al. Knockout of Vdac1 Activates Hypoxia-Inducible Factor through Reactive Oxygen Species Generation and Induces Tumor Growth by Promoting Metabolic Reprogramming and Inflammation. Cancer Metab. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.; Linck, V.; Zhai, Y.-J.; Galarza-Paez, L.; Li, L.; Yue, Q.; Al-Khalili, O.; Bao, H.-F.; Ma, H.-P.; Thai, T.L.; et al. Knockout of Mitochondrial Voltage-Dependent Anion Channel Type 3 Increases Reactive Oxygen Species (ROS) Levels and Alters Renal Sodium Transport. J. Biol. Chem. 2018, 293, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Shuvo, S.R.; Wiens, L.M.; Subramaniam, S.; Treberg, J.R.; Court, D.A. Increased Reactive Oxygen Species Production and Maintenance of Membrane Potential in VDAC-Less Neurospora Crassa Mitochondria. J. Bioenerg. Biomembr. 2019, 51, 341–354. [Google Scholar] [CrossRef]
- Reina, S.; Nibali, S.C.; Tomasello, M.F.; Magrì, A.; Messina, A.; De Pinto, V. Voltage Dependent Anion Channel 3 (VDAC3) Protects Mitochondria from Oxidative Stress. Redox Biol. 2022, 51, 102264. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Armstrong, J.S. Mitochondrial Medicine: Pharmacological Targeting of Mitochondria in Disease. Br. J. Pharmacol. 2007, 151, 1154–1165. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial Complex III Is Required for Hypoxia-Induced ROS Production and Cellular Oxygen Sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.; Crofts, A.R.; Kramer, D.M. Multiple Q-Cycle Bypass Reactions at the Qo Site of the Cytochrome Bc1 Complex. Biochemistry 2002, 41, 7866–7874. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Valuparampil Varghese, M.; Vasilyev, M.; Langlais, P.R.; Tofovic, S.P.; Rafikova, O.; Rafikov, R. Complex III Inhibition-Induced Pulmonary Hypertension Affects the Mitochondrial Proteomic Landscape. Int. J. Mol. Sci. 2020, 21, 5683. [Google Scholar] [CrossRef] [PubMed]
- Hirschenson, J.; Melgar-Bermudez, E.; Mailloux, R.J. The Uncoupling Proteins: A Systematic Review on the Mechanism Used in the Prevention of Oxidative Stress. Antioxidants 2022, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Ma, S.; Tse, G.; Wong, W.T.; Huang, Y. Uncoupling Protein 2 in Cardiovascular Health and Disease. Front. Physiol. 2018, 9, 1060. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.-W.; Hong, K.H.; Maeng, J.H.; Kim, J.-B.; Ko, J.; Park, J.Y.; Lee, K.-U.; Hong, M.K.; Park, S.W.; Kim, Y.H.; et al. Overexpression of Uncoupling Protein 2 in THP1 Monocytes Inhibits Β2 Integrin-Mediated Firm Adhesion and Transendothelial Migration. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 864–870. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Le, P.K.; Tse, S.; Wallace, D.C.; Huang, T. Heterozygous Mutation of Opa1 in Drosophila Shortens Lifespan Mediated through Increased Reactive Oxygen Species Production. PLoS ONE 2009, 4, e4492. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 Mutation and Late-Onset Cardiomyopathy: Mitochondrial Dysfunction and MtDNA Instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef] [Green Version]
- Robert, P.; Nguyen, P.M.C.; Richard, A.; Grenier, C.; Chevrollier, A.; Munier, M.; Grimaud, L.; Proux, C.; Champin, T.; Lelièvre, E.; et al. Protective Role of the Mitochondrial Fusion Protein OPA1 in Hypertension. FASEB J. 2021, 35, e21678. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Nan, J.; Nan, C.; Ye, J.; Qian, L.; Geng, Y.; Xing, D.; Rahman, M.S.U.; Huang, M. EGCG Protects Cardiomyocytes against Hypoxia-Reperfusion Injury through Inhibition of OMA1 Activation. J. Cell Sci. 2019, 132, jcs220871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shemiakova, T.; Ivanova, E.; Grechko, A.V.; Gerasimova, E.V.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Baker, L.; Harrison, J.; Figg, N.; Mercer, J.; Calvert, P.; Vidal-Puig, A.; Murphy, M.; Bennett, M. Mitochondrial DNA Damage Promotes Atherosclerosis and Is Associated with Vulnerable Plaque. Lancet 2013, 381, S117. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Stowe, D.F.; Camara, A.K.S. Mitochondrial Reactive Oxygen Species Production in Excitable Cells: Modulators of Mitochondrial and Cell Function. Antioxid. Redox Signal. 2009, 11, 1373–1414. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA Damage Is More Extensive and Persists Longer than Nuclear DNA Damage in Human Cells Following Oxidative Stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA Oxidative Damage and Repair in Aging and Alzheimer’s Disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef]
- Pitkanen, S.; Robinson, B.H. Mitochondrial Complex I Deficiency Leads to Increased Production of Superoxide Radicals and Induction of Superoxide Dismutase. J. Clin. Investig. 1996, 98, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Sinyov, V.; Sazonova, M.; Ryzhkova, A.; Galitsyna, E.; Melnichenko, A.; Anton, P.; Orekhov, A.; Grechko, A.; Sobenin, I. Potential Use of Buccal Epithelium for Genetic Diagnosis of Atherosclerosis Using MtDNA Mutations. Vessel Plus 2017, 1, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Yoshinaga, N.; Numata, K. Rational Designs at the Forefront of Mitochondria-Targeted Gene Delivery: Recent Progress and Future Perspectives. ACS Biomater. Sci. Eng. 2022, 8, 348–359. [Google Scholar] [CrossRef]
- Taylor, R.W.; Chinnery, P.F.; Turnbull, D.M.; Lightowlers, R.N. Selective Inhibition of Mutant Human Mitochondrial DNA Replication in Vitro by Peptide Nucleic Acids. Nat. Genet. 1997, 15, 212–215. [Google Scholar] [CrossRef]
- Lone, A.; Harris, R.A.; Singh, O.; Betts, D.H.; Cumming, R.C. P66Shc Activation Promotes Increased Oxidative Phosphorylation and Renders CNS Cells More Vulnerable to Amyloid Beta Toxicity. Sci. Rep. 2018, 8, 17081. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron Transfer between Cytochrome c and P66Shc Generates Reactive Oxygen Species That Trigger Mitochondrial Apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Martin-Padura, I.; de Nigris, F.; Giorgio, M.; Mansueto, G.; Somma, P.; Condorelli, M.; Sica, G.; De Rosa, G.; Pelicci, P. Deletion of the P66Shc Longevity Gene Reduces Systemic and Tissue Oxidative Stress, Vascular Cell Apoptosis, and Early Atherogenesis in Mice Fed a High-Fat Diet. Proc. Natl. Acad. Sci. USA 2003, 100, 2112–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camici, G.G.; Schiavoni, M.; Francia, P.; Bachschmid, M.; Martin-Padura, I.; Hersberger, M.; Tanner, F.C.; Pelicci, P.; Volpe, M.; Anversa, P.; et al. Genetic Deletion of P66Shc Adaptor Protein Prevents Hyperglycemia-Induced Endothelial Dysfunction and Oxidative Stress. Proc. Natl. Acad. Sci. USA 2007, 104, 5217–5222. [Google Scholar] [CrossRef] [Green Version]
- Laviola, L.; Orlando, M.R.; Incalza, M.A.; Caccioppoli, C.; Melchiorre, M.; Leonardini, A.; Cignarelli, A.; Tortosa, F.; Labarbuta, R.; Martemucci, S.; et al. TNFα Signals via P66Shc to Induce E-Selectin, Promote Leukocyte Transmigration and Enhance Permeability in Human Endothelial Cells. PLoS ONE 2013, 8, e81930. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, Z.; Liu, X.; Yin, H.-Y.; Tang, Y.; Cao, X. P2X7 Receptor–Mediated Inflammation in Cardiovascular Disease. Front. Pharmacol. 2021, 12, 654425. [Google Scholar] [CrossRef] [PubMed]
- North, R.A. Molecular Physiology of P2X Receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef] [Green Version]
- Franco, M.; Bautista-Pérez, R.; Pérez-Méndez, O. Purinergic Receptors in Tubulointerstitial Inflammatory Cells: A Pathophysiological Mechanism of Salt-Sensitive Hypertension. Acta Physiol. 2015, 214, 75–87. [Google Scholar] [CrossRef]
- Shokoples, B.G.; Paradis, P.; Schiffrin, E.L. P2X7 Receptors. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 186–199. [Google Scholar] [CrossRef]
- Hung, S.-C.; Choi, C.H.; Said-Sadier, N.; Johnson, L.; Atanasova, K.R.; Sellami, H.; Yilmaz, Ö.; Ojcius, D.M. P2X4 Assembles with P2X7 and Pannexin-1 in Gingival Epithelial Cells and Modulates ATP-Induced Reactive Oxygen Species Production and Inflammasome Activation. PLoS ONE 2013, 8, e70210. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, R.; Yerbury, J.J.; Sluyter, R. P2X7 Receptor Activation Induces Reactive Oxygen Species Formation and Cell Death in Murine EOC13 Microglia. Mediat. Inflamm. 2013, 2013, 271813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Dubyak, G.R. P2X7 Receptors Regulate Multiple Types of Membrane Trafficking Responses and Non-Classical Secretion Pathways. Purinergic Signal. 2009, 5, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apolloni, S.; Parisi, C.; Pesaresi, M.G.; Rossi, S.; Carrì, M.T.; Cozzolino, M.; Volonté, C.; D’Ambrosi, N. The NADPH Oxidase Pathway Is Dysregulated by the P2X7 Receptor in the SOD1-G93A Microglia Model of Amyotrophic Lateral Sclerosis. J. Immunol. 2013, 190, 5187–5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.; Zhang, Y.; Li, G.-G.; Yu, H.-H.; Bai, S.; Guo, G.-Y.; Guo, W.-L.; Ma, Y.; Wang, J.-H.; Liu, N.; et al. P2X7 Receptor Activation Aggravates NADPH Oxidase 2-Induced Oxidative Stress after Intracerebral Hemorrhage. Neural Regen. Res. 2021, 16, 1582–1591. [Google Scholar] [CrossRef]
- Larrouyet-Sarto, M.L.; Tamura, A.S.; Alves, V.S.; Santana, P.T.; Ciarlini-Magalhães, R.; Rangel, T.P.; Siebert, C.; Hartwig, J.R.; dos Santos, T.M.; Wyse, A.T.S.; et al. P2X7 Receptor Deletion Attenuates Oxidative Stress and Liver Damage in Sepsis. Purinergic Signal. 2020, 16, 561–572. [Google Scholar] [CrossRef]
- Green, J.P.; Souilhol, C.; Xanthis, I.; Martinez-Campesino, L.; Bowden, N.P.; Evans, P.C.; Wilson, H.L. Atheroprone Flow Activates Inflammation via Endothelial ATP-Dependent P2X7-P38 Signalling. Cardiovasc. Res. 2018, 114, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Milner, P.; Bodin, P.; Loesch, A.; Burnstock, G. Rapid Release of Endothelin and ATP from Isolated Aortic Endothelial Cells Exposed to Increased Flow. Biochem. Biophys. Res. Commun. 1990, 170, 649–656. [Google Scholar] [CrossRef]
- Zhou, R.; Dang, X.; Sprague, R.S.; Mustafa, S.J.; Zhou, Z. Alteration of Purinergic Signaling in Diabetes: Focus on Vascular Function. J. Mol. Cell. Cardiol. 2020, 140, 1–9. [Google Scholar] [CrossRef]
- Cheng, D.; Talib, J.; Stanley, C.P.; Rashid, I.; Michaëlsson, E.; Lindstedt, E.-L.; Croft, K.D.; Kettle, A.J.; Maghzal, G.J.; Stocker, R. Inhibition of MPO (Myeloperoxidase) Attenuates Endothelial Dysfunction in Mouse Models of Vascular Inflammation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1448–1457. [Google Scholar] [CrossRef]
- Sugiyama, S.; Kugiyama, K.; Aikawa, M.; Nakamura, S.; Ogawa, H.; Libby, P. Hypochlorous Acid, a Macrophage Product, Induces Endothelial Apoptosis and Tissue Factor Expression. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1309–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteman, M.; Rose, P.; Siau, J.L.; Cheung, N.S.; Tan, G.S.; Halliwell, B.; Armstrong, J.S. Hypochlorous Acid-Mediated Mitochondrial Dysfunction and Apoptosis in Human Hepatoma HepG2 and Human Fetal Liver Cells: Role of Mitochondrial Permeability Transition. Free Radic. Biol. Med. 2005, 38, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, G.; Di Stefano, A.; Eleuteri, E.; Anzalone, R.; Magno, F.; Corrao, S.; Loria, T.; Martorana, A.; Di Gangi, C.; Colombo, M.; et al. Oxidative Stress Induces Myeloperoxidase Expression in Endocardial Endothelial Cells from Patients with Chronic Heart Failure. Basic Res. Cardiol. 2009, 104, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Alsahli, M.A.; Rahmani, A.H. Myeloperoxidase as an Active Disease Biomarker: Recent Biochemical and Pathological Perspectives. Med. Sci. 2018, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Baldus, S.; Heeschen, C.; Meinertz, T.; Zeiher, A.M.; Eiserich, J.P.; Münzel, T.; Simoons, M.L.; Hamm, C.W. Myeloperoxidase Serum Levels Predict Risk in Patients with Acute Coronary Syndromes. Circulation 2003, 108, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Dorighello, G.G.; Rovani, J.C.; Paim, B.A.; Rentz, T.; Assis, L.H.P.; Vercesi, A.E.; Oliveira, H.C.F. Mild Mitochondrial Uncoupling Decreases Experimental Atherosclerosis, A Proof of Concept. J. Atheroscler. Thromb. 2022, 29, 825–838. [Google Scholar] [CrossRef]
- Ma, M.-H.; Li, F.-F.; Li, W.-F.; Zhao, H.; Jiang, M.; Yu, Y.-Y.; Dong, Y.-C.; Zhang, Y.-X.; Li, P.; Bu, W.-J.; et al. Repurposing Nitazoxanide as a Novel Anti-Atherosclerotic Drug Based on Mitochondrial Uncoupling Mechanisms. Br. J. Pharmacol. 2023, 180, 62–79. [Google Scholar] [CrossRef]
- Kenwood, B.M.; Weaver, J.L.; Bajwa, A.; Poon, I.K.; Byrne, F.L.; Murrow, B.A.; Calderone, J.A.; Huang, L.; Divakaruni, A.S.; Tomsig, J.L.; et al. Identification of a Novel Mitochondrial Uncoupler That Does Not Depolarize the Plasma Membrane. Mol. Metab. 2013, 3, 114–123. [Google Scholar] [CrossRef]
- Dhanasekaran, A.; Kotamraju, S.; Kalivendi, S.V.; Matsunaga, T.; Shang, T.; Keszler, A.; Joseph, J.; Kalyanaraman, B. Supplementation of Endothelial Cells with Mitochondria-Targeted Antioxidants Inhibit Peroxide-Induced Mitochondrial Iron Uptake, Oxidative Damage, and Apoptosis. J. Biol. Chem. 2004, 279, 37575–37587. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M. Endothelial Mitochondria and Heart Disease. Cardiovasc. Res. 2010, 88, 58–66. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Zhang, H.; Chen, R.; Lv, F.; Li, Z.; Jiang, T.; Lin, D.; Zhang, H.; Yang, L.; et al. The Antioxidant MitoQ Protects Against CSE-Induced Endothelial Barrier Injury and Inflammation by Inhibiting ROS and Autophagy in Human Umbilical Vein Endothelial Cells. Int. J. Biol. Sci. 2019, 15, 1440–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-Targeted Antioxidant (MitoQ) Ameliorates Age-Related Arterial Endothelial Dysfunction in Mice. J. Physiol. 2014, 592, 2549–2561. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; O’Connell, K.A.; Hecker, P.A.; Murphy, M.P. MitoQ Improves Mitochondrial Dysfunction in Heart Failure Induced by Pressure Overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Ivanovski, O.; Szumilak, D.; Nguyen-Khoa, T.; Ruellan, N.; Phan, O.; Lacour, B.; Descamps-Latscha, B.; Dreeke, T.B.; Massy, Z.A. The Antioxidant N-Acetylcysteine Prevents Accelerated Atherosclerosis in Uremic Apolipoprotein E Knockout Mice. Kidney Int. 2005, 67, 2288–2294. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Chen, X.; Wang, Y.; Xiao, H.; Peng, Y.; Lin, L.; Xia, W.; Long, M.; Tao, J.; Shuai, X. Aortic Plaque-Targeted Andrographolide Delivery with Oxidation-Sensitive Micelle Effectively Treats Atherosclerosis via Simultaneous ROS Capture and Anti-Inflammation. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2215–2226. [Google Scholar] [CrossRef]
- Hansen, T.; Bubb, K.; Kassiou, M.; Figtree, G. Abstract 17368: The Novel P2X7 Receptor Antagonist SMW139 Inhibits Inflammasome Activation in STEMI Monocytes. Circulation 2018, 138, A17368. [Google Scholar] [CrossRef]
- Hansen, T.; Karimi Galougahi, K.; Besnier, M.; Genetzakis, E.; Tsang, M.; Finemore, M.; O’Brien-Brown, J.; Di Bartolo, B.A.; Kassiou, M.; Bubb, K.J.; et al. The Novel P2X7 Receptor Antagonist PKT100 Improves Cardiac Function and Survival in Pulmonary Hypertension by Direct Targeting of the Right Ventricle. Am. J. Physiol.-Heart Circ. Physiol. 2020, 319, H183–H191. [Google Scholar] [CrossRef]
- Territo, P.R.; Zarrinmayeh, H. P2X7 Receptors in Neurodegeneration: Potential Therapeutic Applications from Basic to Clinical Approaches. Front. Cell. Neurosci. 2021, 15, 617036. [Google Scholar] [CrossRef]
- Keystone, E.C.; Wang, M.M.; Layton, M.; Hollis, S.; McInnes, I.B.; on behalf of the D1520C00001 Study Team. Clinical Evaluation of the Efficacy of the P2X7 Purinergic Receptor Antagonist AZD9056 on the Signs and Symptoms of Rheumatoid Arthritis in Patients with Active Disease despite Treatment with Methotrexate or Sulphasalazine. Ann. Rheum. Dis. 2012, 71, 1630–1635. [Google Scholar] [CrossRef]
- Eser, A.; Colombel, J.-F.; Rutgeerts, P.; Vermeire, S.; Vogelsang, H.; Braddock, M.; Persson, T.; Reinisch, W. Safety and Efficacy of an Oral Inhibitor of the Purinergic Receptor P2X7 in Adult Patients with Moderately to Severely Active Crohn’s Disease: A Randomized Placebo-Controlled, Double-Blind, Phase IIa Study. Inflamm. Bowel Dis. 2015, 21, 2247–2253. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Nidorf, S.M.; Fiolet, A.T.L.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; Bax, W.A.; Budgeon, C.A.; Tijssen, J.G.P.; Mosterd, A.; Cornel, J.H.; et al. The Effect of Low-Dose Colchicine in Patients with Stable Coronary Artery Disease: The LoDoCo2 Trial Rationale, Design, and Baseline Characteristics. Am. Heart J. 2019, 218, 46–56. [Google Scholar] [CrossRef]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Brindle, K.M.; Buckingham, J.A.; Harper, J.A.; Rolfe, D.F.; Stuart, J.A. The Significance and Mechanism of Mitochondrial Proton Conductance. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1999, 23 (Suppl. 6), S4–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Nanayakkara, G.; Shao, Y.; Cueto, R.; Wang, L.; Yang, W.Y.; Tian, Y.; Wang, H.; Yang, X. Mitochondrial Proton Leak Plays a Critical Role in Pathogenesis of Cardiovascular Diseases. Adv. Exp. Med. Biol. 2017, 982, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. Mitochondrial Uncoupling, ROS Generation and Cardioprotection. Biochim. Biophys. Acta BBA—Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Papa, S.; Skulachev, V.P. Reactive Oxygen Species, Mitochondria, Apoptosis and Aging. Mol. Cell. Biochem. 1997, 174, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Figarola, J.L.; Singhal, J.; Tompkins, J.D.; Rogers, G.W.; Warden, C.; Horne, D.; Riggs, A.D.; Awasthi, S.; Singhal, S.S. SR4 Uncouples Mitochondrial Oxidative Phosphorylation, Modulates AMP-Dependent Kinase (AMPK)-Mammalian Target of Rapamycin (MTOR) Signaling, and Inhibits Proliferation of HepG2 Hepatocarcinoma Cells. J. Biol. Chem. 2015, 290, 30321–30341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, J.G. Targeting Energy Expenditure via Fuel Switching and Beyond. Diabetologia 2011, 54, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Grundlingh, J.; Dargan, P.I.; El-Zanfaly, M.; Wood, D.M. 2,4-Dinitrophenol (DNP): A Weight Loss Agent with Significant Acute Toxicity and Risk of Death. J. Med. Toxicol. 2011, 7, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.H.; Law, C.Y.; Chen, M.L.; Lam, Y.H.; Chan, A.Y.W.; Mak, T.W.L. 2,4-Dinitrophenol: A Threat to Chinese Body-Conscious Groups. J. Chin. Med. Assoc. 2014, 77, 443–445. [Google Scholar] [CrossRef] [Green Version]
- Colman, E. Dinitrophenol and Obesity: An Early Twentieth-Century Regulatory Dilemma. Regul. Toxicol. Pharmacol. 2007, 48, 115–117. [Google Scholar] [CrossRef]
- Bleasdale, E.E.; Thrower, S.N.; Petróczi, A. Would You Use It with a Seal of Approval? Important Attributes of 2,4-Dinitrophenol (2,4-DNP) as a Hypothetical Pharmaceutical Product. Front. Psychiatry 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Caldeira da Silva, C.C.; Cerqueira, F.M.; Barbosa, L.F.; Medeiros, M.H.G.; Kowaltowski, A.J. Mild Mitochondrial Uncoupling in Mice Affects Energy Metabolism, Redox Balance and Longevity. Aging Cell 2008, 7, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.; Caldwell, S.T.; Quin, C.; Prime, T.A.; James, A.M.; Cairns, A.G.; Murphy, M.P.; McCarron, J.G.; Hartley, R.C. Selective Uncoupling of Individual Mitochondria within a Cell Using a Mitochondria-Targeted Photoactivated Protonophore. J. Am. Chem. Soc. 2012, 134, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, S.J.; Chen, S.-Y.; Brandon, A.E.; Salamoun, J.M.; Byrne, F.L.; Garcia, C.J.; Beretta, M.; Olzomer, E.M.; Shah, D.P.; Philp, A.M.; et al. Mitochondrial Uncoupler BAM15 Reverses Diet-Induced Obesity and Insulin Resistance in Mice. Nat. Commun. 2020, 11, 2397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fu, Y.; Yang, P.; Liu, X.; Li, Y.; Gu, Z. ROS Scavenging Biopolymers for Anti-Inflammatory Diseases: Classification and Formulation. Adv. Mater. Interfaces 2020, 7, 2000632. [Google Scholar] [CrossRef]
- Chiumiento, L.; Bruschi, F. Enzymatic Antioxidant Systems in Helminth Parasites. Parasitol. Res. 2009, 105, 593. [Google Scholar] [CrossRef]
- Dobrakowski, M.; Zalejska-Fiolka, J.; Wielkoszyński, T.; Świętochowska, E.; Kasperczyk, S. The Effect of Occupational Exposure to Lead on the Non-Enzymatic Antioxidant System. Med. Pr. 2014, 65, 443–451. [Google Scholar] [CrossRef]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the Balance between ROS and Antioxidants: When to Use the Synthetic Antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.J.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective Targeting of an Antioxidant to Mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Villalba, J.M.; Parrado, C.; Santos-Gonzalez, M.; Alcain, F.J. Therapeutic Use of Coenzyme Q10 and Coenzyme Q10-Related Compounds and Formulations. Expert Opin. Investig. Drugs 2010, 19, 535–554. [Google Scholar] [CrossRef]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and Functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef]
- Sifuentes-Franco, S.; Sánchez-Macías, D.C.; Carrillo-Ibarra, S.; Rivera-Valdés, J.J.; Zuñiga, L.Y.; Sánchez-López, V.A. Antioxidant and Anti-Inflammatory Effects of Coenzyme Q10 Supplementation on Infectious Diseases. Healthcare 2022, 10, 487. [Google Scholar] [CrossRef]
- Crane, F.L. Biochemical Functions of Coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.V.e.; Gallia, M.C.; Luz, J.R.D.d.; Rezende, A.A.d.; Bongiovanni, G.A.; Araujo-Silva, G.; Almeida, M.d.G. Antioxidant Effect of Coenzyme Q10 in the Prevention of Oxidative Stress in Arsenic-Treated CHO-K1 Cells and Possible Participation of Zinc as a Pro-Oxidant Agent. Nutrients 2022, 14, 3265. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.; Davison, G. Targeted Antioxidants in Exercise-Induced Mitochondrial Oxidative Stress: Emphasis on DNA Damage. Antioxidants 2020, 9, 1142. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Cochemé, H.M.; Smith, R.A.J.; Murphy, M.P. Interactions of Mitochondria-Targeted and Untargeted Ubiquinones with the Mitochondrial Respiratory Chain and Reactive Oxygen Species: IMPLICATIONS FOR THE USE OF EXOGENOUS UBIQUINONES AS THERAPIES AND EXPERIMENTAL TOOLS. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, B.D.; Herlein, J.A.; Yorek, M.A.; Fenner, A.M.; Kerns, R.J.; Sivitz, W.I. Bioenergetic Effects of Mitochondrial-Targeted Coenzyme Q Analogs in Endothelial Cells. J. Pharmacol. Exp. Ther. 2012, 342, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Dos Tenório, M.C.S.; Graciliano, N.G.; Moura, F.A.; de Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Bavarsad Shahripour, R.; Harrigan, M.R.; Alexandrov, A.V. N-Acetylcysteine (NAC) in Neurological Disorders: Mechanisms of Action and Therapeutic Opportunities. Brain Behav. 2014, 4, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Liu, H.; Yang, Y.; Yang, Y.; Jiao, Y.; Tay, F.R.; Chen, J. Biological Activities and Potential Oral Applications of N-Acetylcysteine: Progress and Prospects. Oxid. Med. Cell. Longev. 2018, 2018, 2835787. [Google Scholar] [CrossRef]
- Hosseini, E.; Ghasemzadeh, M.; Atashibarg, M.; Haghshenas, M. ROS Scavenger, N-Acetyl-l-Cysteine and NOX Specific Inhibitor, VAS2870 Reduce Platelets Apoptosis While Enhancing Their Viability during Storage. Transfusion 2019, 59, 1333–1343. [Google Scholar] [CrossRef]
- Paul, M.; Thushara, R.M.; Jagadish, S.; Zakai, U.I.; West, R.; Kemparaju, K.; Girish, K.S. Novel Sila-Amide Derivatives of N-Acetylcysteine Protects Platelets from Oxidative Stress-Induced Apoptosis. J. Thromb. Thrombolysis 2017, 43, 209–216. [Google Scholar] [CrossRef]
- Kerksick, C.; Willoughby, D. The Antioxidant Role of Glutathione and N-Acetyl-Cysteine Supplements and Exercise-Induced Oxidative Stress. J. Int. Soc. Sports Nutr. 2005, 2, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Tabassum, H.; Alam, M.M.; Parvez, S. N-Acetyl-L-Cysteine Ameliorates Mitochondrial Dysfunction in Ischemia/Reperfusion Injury via Attenuating Drp-1 Mediated Mitochondrial Autophagy. Life Sci. 2022, 293, 120338. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Terluk, M.R.; Orchard, P.J.; Cloyd, J.C.; Kartha, R.V. N-Acetylcysteine Reverses the Mitochondrial Dysfunction Induced by Very Long-Chain Fatty Acids in Murine Oligodendrocyte Model of Adrenoleukodystrophy. Biomedicines 2021, 9, 1826. [Google Scholar] [CrossRef]
- Shen, H.-M.; Yang, C.-F.; Ding, W.-X.; Liu, J.; Ong, C.-N. Superoxide Radical–Initiated Apoptotic Signalling Pathway in Selenite-Treated HepG2 Cells: Mitochondria Serve as the Main Target. Free Radic. Biol. Med. 2001, 30, 9–21. [Google Scholar] [CrossRef]
- Baroja-Mazo, A.; Pelegrín, P. Modulating P2X7 Receptor Signaling during Rheumatoid Arthritis: New Therapeutic Approaches for Bisphosphonates. J. Osteoporos. 2012, 2012, 408242. [Google Scholar] [CrossRef] [Green Version]
- Cheng, N.; Zhang, L.; Liu, L. Understanding the Role of Purinergic P2X7 Receptors in the Gastrointestinal System: A Systematic Review. Front. Pharmacol. 2021, 12, 3518. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Sobey, C.G.; Latz, E.; Mansell, A.; Drummond, G.R. IL-1β and IL-18: Inflammatory Markers or Mediators of Hypertension? Br. J. Pharmacol. 2014, 171, 5589–5602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigi, R.D.; Kertesy, S.B.; Aquilina, G.; Dubyak, G.R. Oxidized ATP (OATP) Attenuates Proinflammatory Signaling via P2 Receptor-Independent Mechanisms. Br. J. Pharmacol. 2003, 140, 507–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F. Novel Data Point to a Broader Mechanism of Action of Oxidized ATP: The P2X7 Receptor Is Not the Only Target. Br. J. Pharmacol. 2003, 140, 441–443. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, E.; Orioli, E.; Dal Ben, D.; Adinolfi, E. Chapter Two—P2X7 Receptor as a Therapeutic Target. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Ion Channels as Therapeutic Targets, Part B; Academic Press: Cambridge, MA, USA, 2016; Volume 104, pp. 39–79. [Google Scholar]
- Angelidis, C.; Kotsialou, Z.; Kossyvakis, C.; Vrettou, A.-R.; Zacharoulis, A.; Kolokathis, F.; Kekeris, V.; Giannopoulos, G. Colchicine Pharmacokinetics and Mechanism of Action. Curr. Pharm. Des. 2018, 24, 659–663. [Google Scholar] [CrossRef]
- Malik, J.; Javed, N.; Ishaq, U.; Khan, U.; Laique, T. Is There a Role for Colchicine in Acute Coronary Syndromes? A Literature Review. Cureus 2020, 12, e8166. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lv, H.; Liu, Q.; Zhang, L.; Zhang, R.; Huang, X.; Wang, X.; Han, B.; Hou, S.; Liu, D.; et al. Colchicine Alleviates Cholesterol Crystal-Induced Endothelial Cell Pyroptosis through Activating AMPK/SIRT1 Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 9173530. [Google Scholar] [CrossRef] [PubMed]
- Marques-da-Silva, C.; Chaves, M.; Castro, N.; Coutinho-Silva, R.; Guimaraes, M. Colchicine Inhibits Cationic Dye Uptake Induced by ATP in P2X2 and P2X7 Receptor-Expressing Cells: Implications for Its Therapeutic Action. Br. J. Pharmacol. 2011, 163, 912–926. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Galijasevic, S. The Development of Myeloperoxidase Inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 1–7. [Google Scholar] [CrossRef]
- Figtree, G.A.; Kovacic, J.C.; McGuire, H.M. Human Susceptibility to Coronary Artery Disease: Lessons from Chimpanzee Resilience. Nat. Rev. Cardiol. 2022, 19, 497–498. [Google Scholar] [CrossRef]
- Varki, A.; Altheide, T.K. Comparing the Human and Chimpanzee Genomes: Searching for Needles in a Haystack. Genome Res. 2005, 15, 1746–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, S.; Hoffman, K.; Bartel, L.; Schwandt, M.; Morris, J.; Fritz, J. Normal Hematologic and Serum Clinical Chemistry Values for Captive Chimpanzees (Pan troglodytes). Comp. Med. 2003, 53, 413–423. [Google Scholar]
- Herndon, J.G.; Tigges, J. Hematologic and Blood Biochemical Variables of Captive Chimpanzees: Cross-Sectional and Longitudinal Analyses. Comp. Med. 2001, 51, 60–69. [Google Scholar]
- Hainsey, B.M.; Hubbard, G.B.; Leland, M.M.; Brasky, K.M. Clinical Parameters of the Normal Baboons (Papio Species) and Chimpanzees (Pan troglodytes). Lab. Anim. Sci. 1993, 43, 236–243. [Google Scholar] [PubMed]
- Varki, N.; Anderson, D.; Herndon, J.G.; Pham, T.; Gregg, C.J.; Cheriyan, M.; Murphy, J.; Strobert, E.; Fritz, J.; Else, J.G.; et al. ORIGINAL ARTICLE: Heart Disease Is Common in Humans and Chimpanzees, but Is Caused by Different Pathological Processes. Evol. Appl. 2009, 2, 101–112. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Animal Models of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1104–1115. [Google Scholar] [CrossRef] [Green Version]
- Bentzon, J.F.; Falk, E. Atherosclerotic Lesions in Mouse and Man: Is It the Same Disease? Curr. Opin. Lipidol. 2010, 21, 434. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Polinsky, P.; Sadoun, E.; Rosenfeld, M.E.; Schwartz, S.M. Atherosclerotic Lesions in the Common Coronary Arteries of ApoE Knockout Mice. Cardiovasc. Pathol. 2005, 14, 120–125. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of Putative Progenitor Endothelial Cells for Angiogenesis. Science 1997, 275, 964–966. [Google Scholar] [CrossRef]
- Lin, Y.; Weisdorf, D.J.; Solovey, A.; Hebbel, R.P. Origins of Circulating Endothelial Cells and Endothelial Outgrowth from Blood. J. Clin. Investig. 2000, 105, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Paschalaki, K.E.; Randi, A.M. Recent Advances in Endothelial Colony Forming Cells Toward Their Use in Clinical Translation. Front. Med. 2018, 5, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prater, D.N.; Case, J.; Ingram, D.A.; Yoder, M.C. Working Hypothesis to Redefine Endothelial Progenitor Cells. Leukemia 2007, 21, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Egorova, A.D.; DeRuiter, M.C.; de Boer, H.C.; van de Pas, S.; Gittenberger-de Groot, A.C.; van Zonneveld, A.J.; Poelmann, R.E.; Hierck, B.P. Endothelial Colony-Forming Cells Show a Mature Transcriptional Response to Shear Stress. Vitro Cell. Dev. Biol.—Anim. 2012, 48, 21–29. [Google Scholar] [CrossRef]
- Fujisawa, T.; Tura-Ceide, O.; Hunter, A.; Mitchell, A.; Vesey, A.; Medine, C.; Gallogly, S.; Hadoke, P.W.F.; Keith, C.; Sproul, A.; et al. Endothelial Progenitor Cells Do Not Originate from the Bone Marrow. Circulation 2019, 140, 1524–1526. [Google Scholar] [CrossRef] [PubMed]
- Tura, O.; Skinner, E.M.; Barclay, G.R.; Samuel, K.; Gallagher, R.C.J.; Brittan, M.; Hadoke, P.W.F.; Newby, D.E.; Turner, M.L.; Mills, N.L. Late Outgrowth Endothelial Cells Resemble Mature Endothelial Cells and Are Not Derived from Bone Marrow. Stem Cells 2013, 31, 338–348. [Google Scholar] [CrossRef]
- Besnier, M.; Finemore, M.; Yu, C.; Kott, K.A.; Vernon, S.T.; Seebacher, N.A.; Genetzakis, E.; Furman, A.; Tang, O.; Davis, R.L.; et al. Patient Endothelial Colony-Forming Cells to Model Coronary Artery Disease Susceptibility and Unravel the Role of Dysregulated Mitochondrial Redox Signalling. Antioxidants 2021, 10, 1547. [Google Scholar] [CrossRef]
- de Boer, S.; Bowman, M.; Notley, C.; Mo, A.; Lima, P.; de Jong, A.; Dirven, R.; Weijers, E.; Lillicrap, D.; James, P.; et al. Endothelial Characteristics in Healthy Endothelial Colony Forming Cells; Generating a Robust and Valid Ex Vivo Model for Vascular Disease. J. Thromb. Haemost. 2020, 18, 2721–2731. [Google Scholar] [CrossRef] [PubMed]
- Avery, V.M.; Camp, D.; Carroll, A.R.; Jenkins, I.D.; Quinn, R.J. 3.07—The Identification of Bioactive Natural Products by High Throughput Screening (HTS). In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 177–203. ISBN 978-0-08-045382-8. [Google Scholar]
- Liu, Z.; Chen, H.; Wold, E.A.; Zhou, J. 2.13—Small-Molecule Inhibitors of Protein–Protein Interactions. In Comprehensive Medicinal Chemistry III; Chackalamannil, S., Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; pp. 329–353. ISBN 978-0-12-803201-5. [Google Scholar]
- Aherne, W.; Garrett, M.; McDonald, T.; Workman, P. Chapter 14—Mechanism-based high throughput screening for novel drug discovery. In Anticancer Drug Development; Baguley, B.C., Kerr, D.J., Eds.; Academic Press: San Diego, CA, USA, 2002; pp. 249–267. ISBN 978-0-12-072651-6. [Google Scholar]
- Etzion, Y.; Muslin, A.J. The Application of Phenotypic High-Throughput Screening Techniques to Cardiovascular Research. Trends Cardiovasc. Med. 2009, 19, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Muslin, A.J. Phenotypic High-Throughput Screening in Atherosclerosis Research: Focus on Macrophages. J. Cardiovasc. Transl. Res. 2010, 3, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Nieland, T.J.F.; Penman, M.; Dori, L.; Krieger, M.; Kirchhausen, T. Discovery of Chemical Inhibitors of the Selective Transfer of Lipids Mediated by the HDL Receptor SR-BI. Proc. Natl. Acad. Sci. USA 2002, 99, 15422–15427. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Xu, Y.; Yang, Y.; Yang, Y.; Zheng, Z.; Jiang, W.; Hong, B.; Yan, X.; Si, S. Identification of Upregulators of Human ATP-Binding Cassette Transporter A1 via High-Throughput Screening of a Synthetic and Natural Compound Library. J. Biomol. Screen. 2008, 13, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Etzion, Y.; Hackett, A.; Proctor, B.M.; Ren, J.; Nolan, B.; Ellenberger, T.; Muslin, A.J. An Unbiased Chemical Biology Screen Identifies Agents That Modulate Uptake of Oxidized LDL by Macrophages. Circ. Res. 2009, 105, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and Their Role in Atherosclerosis: Pathophysiology and Transcriptome Analysis. BioMed Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [Green Version]
- Nikpay, M.; Soubeyrand, S.; Tahmasbi, R.; McPherson, R. Multiomics Screening Identifies Molecular Biomarkers Causally Associated with the Risk of Coronary Artery Disease. Circ. Genom. Precis. Med. 2020, 13, e002876. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R. Mouse Models of Atherosclerosis: Explaining Critical Roles of Lipid Metabolism and Inflammation. J. Appl. Genet. 2013, 54, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Lin, H.Y.; Chan, Y.W.F.; Li, K.H.C.; To, O.T.L.; Yan, B.P.; Liu, T.; Li, G.; Wong, W.T.; Keung, W.; et al. Mouse Models of Atherosclerosis: A Historical Perspective and Recent Advances. Lipids Health Dis. 2017, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setälä, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe Hypercholesterolemia and Atherosclerosis in Apolipoprotein E-Deficient Mice Created by Homologous Recombination in ES Cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Plump, A.S.; Breslow, J.L. Apolipoprotein E and the Apolipoprotein E-Deficient Mouse. Annu. Rev. Nutr. 1995, 15, 495–518. [Google Scholar] [CrossRef]
- Plump, A.S.; Scott, C.J.; Breslow, J.L. Human Apolipoprotein A-I Gene Expression Increases High Density Lipoprotein and Suppresses Atherosclerosis in the Apolipoprotein E-Deficient Mouse. Proc. Natl. Acad. Sci. USA 1994, 91, 9607–9611. [Google Scholar] [CrossRef] [Green Version]
- Pászty, C.; Maeda, N.; Verstuyft, J.; Rubin, E.M. Apolipoprotein AI Transgene Corrects Apolipoprotein E Deficiency-Induced Atherosclerosis in Mice. J. Clin. Investig. 1994, 94, 899–903. [Google Scholar] [CrossRef] [Green Version]
- Getz, G.S.; Reardon, C.A. Diet and Murine Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 242–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiselli, G.; Schaefer, E.J.; Gascon, P.; Breser, H.B. Type III Hyperlipoproteinemia Associated with Apolipoprotein E Deficiency. Science 1981, 214, 1239–1241. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Plump, A.S.; Raines, E.W.; Breslow, J.L.; Ross, R. ApoE-Deficient Mice Develop Lesions of All Phases of Atherosclerosis throughout the Arterial Tree. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Reddick, R.L.; Zhang, S.H.; Maeda, N. Atherosclerosis in Mice Lacking Apo E. Evaluation of Lesional Development and Progression. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Pendse, A.A.; Arbones-Mainar, J.M.; Johnson, L.A.; Altenburg, M.K.; Maeda, N. Apolipoprotein E Knock-out and Knock-in Mice: Atherosclerosis, Metabolic Syndrome, and Beyond. J. Lipid Res. 2009, 50, S178–S182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knouff, C.; Hinsdale, M.E.; Mezdour, H.; Altenburg, M.K.; Watanabe, M.; Quarfordt, S.H.; Sullivan, P.M.; Maeda, N. Apo E Structure Determines VLDL Clearance and Atherosclerosis Risk in Mice. J. Clin. Investig. 1999, 103, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a Lipid Transport and Signaling Protein in the Blood, Liver, and Artery Wall. J. Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef] [Green Version]
- VanderLaan, P.A.; Reardon, C.A.; Thisted, R.A.; Getz, G.S. VLDL Best Predicts Aortic Root Atherosclerosis in LDL Receptor Deficient Mice. J. Lipid Res. 2009, 50, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, H.H.; Russell, D.W.; Brown, M.S.; Goldstein, J.L. The LDL Receptor Locus in Familial Hypercholesterolemia: Mutational Analysis of a Membrane Protein. Annu. Rev. Genet. 1990, 24, 133–170. [Google Scholar] [CrossRef]
- Herijgers, N.; Van Eck, M.; Groot, P.H.E.; Hoogerbrugge, P.M.; Van Berkel, T.J.C. Effect of Bone Marrow Transplantation on Lipoprotein Metabolism and Atherosclerosis in LDL Receptor–Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1995–2003. [Google Scholar] [CrossRef]
- Merat, S.; Fruebis, J.; Sutphin, M.; Silvestre, M.; Reaven, P.D. Effect of Aging on Aortic Expression of the Vascular Cell Adhesion Molecule-1 and Atherosclerosis in Murine Models of Atherosclerosis. J. Gerontol. A Biol. Sci. Med. Sci. 2000, 55, B85–B94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenji Ardestani, S.; Eftedal, I.; Pedersen, M.; Jeppesen, P.B.; Nørregaard, R.; Matchkov, V.V. Endothelial Dysfunction in Small Arteries and Early Signs of Atherosclerosis in ApoE Knockout Rats. Sci. Rep. 2020, 10, 15296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Luo, Y.; Zhang, W.; He, Y.; Dai, S.; Zhang, R.; Huang, Y.; Bernatchez, P.; Giordano, F.J.; Shadel, G.; et al. Endothelial-Specific Expression of Mitochondrial Thioredoxin Improves Endothelial Cell Function and Reduces Atherosclerotic Lesions. Am. J. Pathol. 2007, 170, 1108–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Compound | Mechanism of Action | Mitochondria Specificity? | Used in Cardiovascular Setting? | Clinical Trials? | References |
|---|---|---|---|---|---|
| 2,4 dinitrophenol (2,4 DNP) | Mitochondrial uncoupling | No | No | No | [121] |
| Nitazoxanide | Mitochondrial uncoupling | No | Yes. Shown to reduce formation of atherosclerotic plaques in atherosclerosis-model mice fed western diet. | No | [122] |
| BAM15 | Mitochondrial uncoupling | Yes | No | No | [123] |
| MitoVit-E | ROS scavenging | Yes | No | No | [124,125] |
| Mitoquinone (MitoQ) | ROS scavenging | Yes | Yes. Shown to reduce hydrogen peroxide production, improve mitochondrial respiration and mitochondrial permeability transition pore (mPTP) opening. | Yes. None for CAD. | [126,127,128] |
| N-acetylcysteine (NAC) | ROS scavenging | No | Yes. Shown to reduce atheroma progression in uremic-enhanced apolipoprotein E knockout mice. | Yes. None for CAD. | [129] |
| ROS-scavenging biopolymers | ROS scavenging | No | Yes. Micelles combined with loading andrographolide have been shown to act as a responsive and stimulating drug carrier to release encapsulated compounds and demonstrate the ability to consume ROS at pathological sites. | No | [130] |
| PKT100 | P2X7R antagonists | No | Yes. Human peripheral blood mononuclear cells (PBMCs) derived from STEMI patients have inhibited inflammasome. Mice with pulmonary fibrosis and hypertension have improved cardiac function and survival. Bleomycin-treated mice have reduced right-ventricular (RV) dysfunction, improved right ventricular systolic excursion velocity, survival and reduced IL-1β. | No | [131,132] |
| AZD9056 | P2X7R antagonists | No | No | Yes. None for CAD. | [133,134,135] |
| Colchicine | Anti-inflammatory, non-specific | No | Yes. Low dose colchicine (0.5 mg/day) significantly decreased risk of cardiovascular adverse events. | Yes, LoDoCo (pilot study) and LoDoCo2 (randomized follow-up trial). However, incidence of death from non-cardiovascular disease was higher in the colchicine group | [136,137] |
| AZM198 | MPO Inhibitor | No | Yes. Shown to reduce endothelial dysfunction in mouse models of vascular inflammation and atherosclerosis. | No | [115] |
| Genetic Model | Advantages | Disadvantages |
|---|---|---|
| ApoE−/− |
| |
| LDLR−/− |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, W.E.; Genetzakis, E.; Figtree, G.A. Novel Strategies in the Early Detection and Treatment of Endothelial Cell-Specific Mitochondrial Dysfunction in Coronary Artery Disease. Antioxidants 2023, 12, 1359. https://doi.org/10.3390/antiox12071359
Lee WE, Genetzakis E, Figtree GA. Novel Strategies in the Early Detection and Treatment of Endothelial Cell-Specific Mitochondrial Dysfunction in Coronary Artery Disease. Antioxidants. 2023; 12(7):1359. https://doi.org/10.3390/antiox12071359
Chicago/Turabian StyleLee, Weiqian E., Elijah Genetzakis, and Gemma A. Figtree. 2023. "Novel Strategies in the Early Detection and Treatment of Endothelial Cell-Specific Mitochondrial Dysfunction in Coronary Artery Disease" Antioxidants 12, no. 7: 1359. https://doi.org/10.3390/antiox12071359