
Acute Kidney Injury Induces Oxidative Stress and Hepatic Lipid Accumulation through AMPK Signaling Pathway
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Induction of Renal Ischemia-Reperfusion in Rats
2.2. Cell Culture
2.3. Biochemical Analysis
2.4. Histological Staining
2.5. Quantitative Real-Time PCR
2.6. Western Immunoblotting Analysis
2.7. Statistical Analysis
3. Results
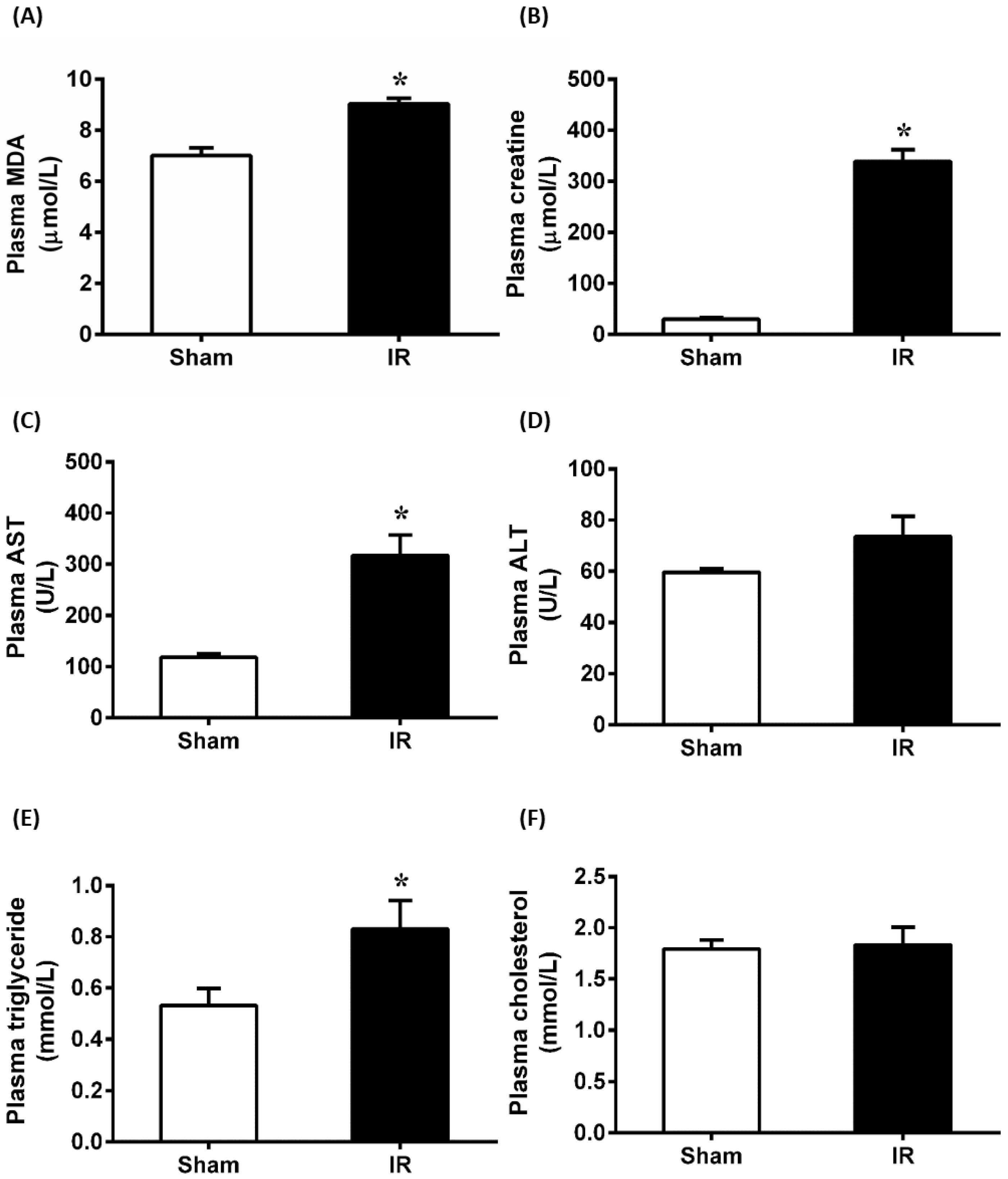
3.1. Renal Ischemia-Reperfusion Impaired Kidney and Liver Function
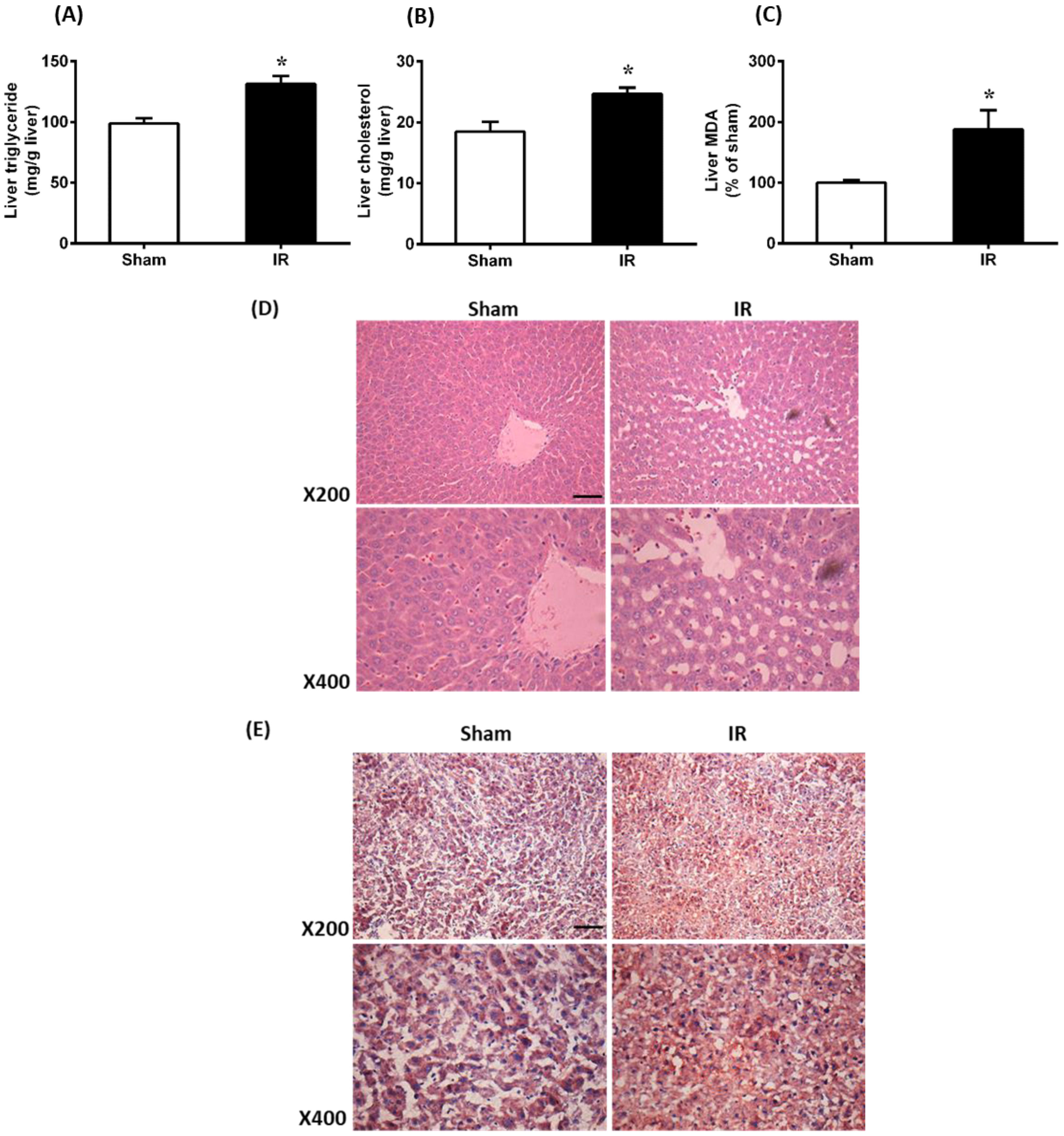
3.2. Renal Ischemia-Reperfusion Caused Oxidative Stress and Increased Lipid Level in the Liver
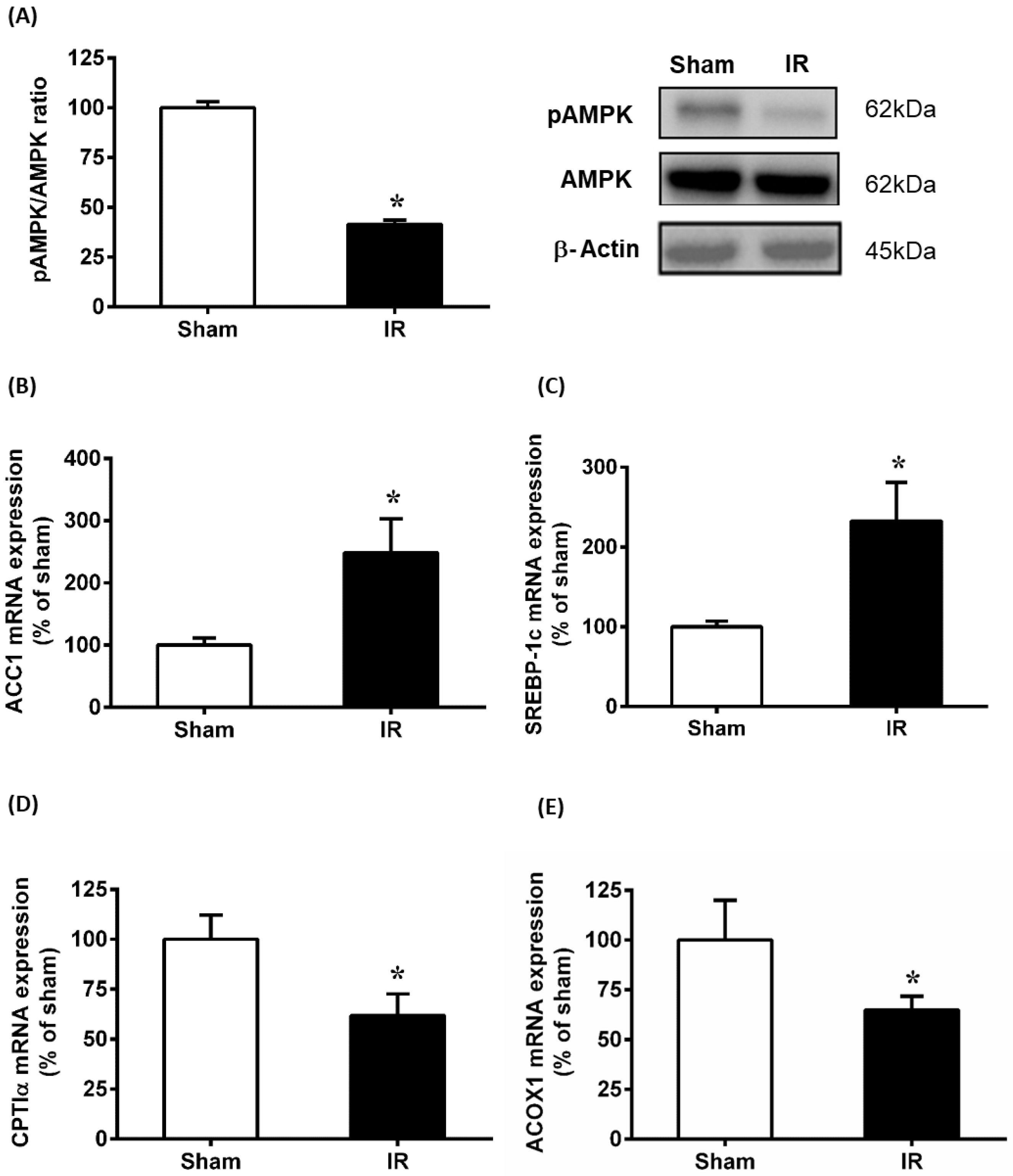
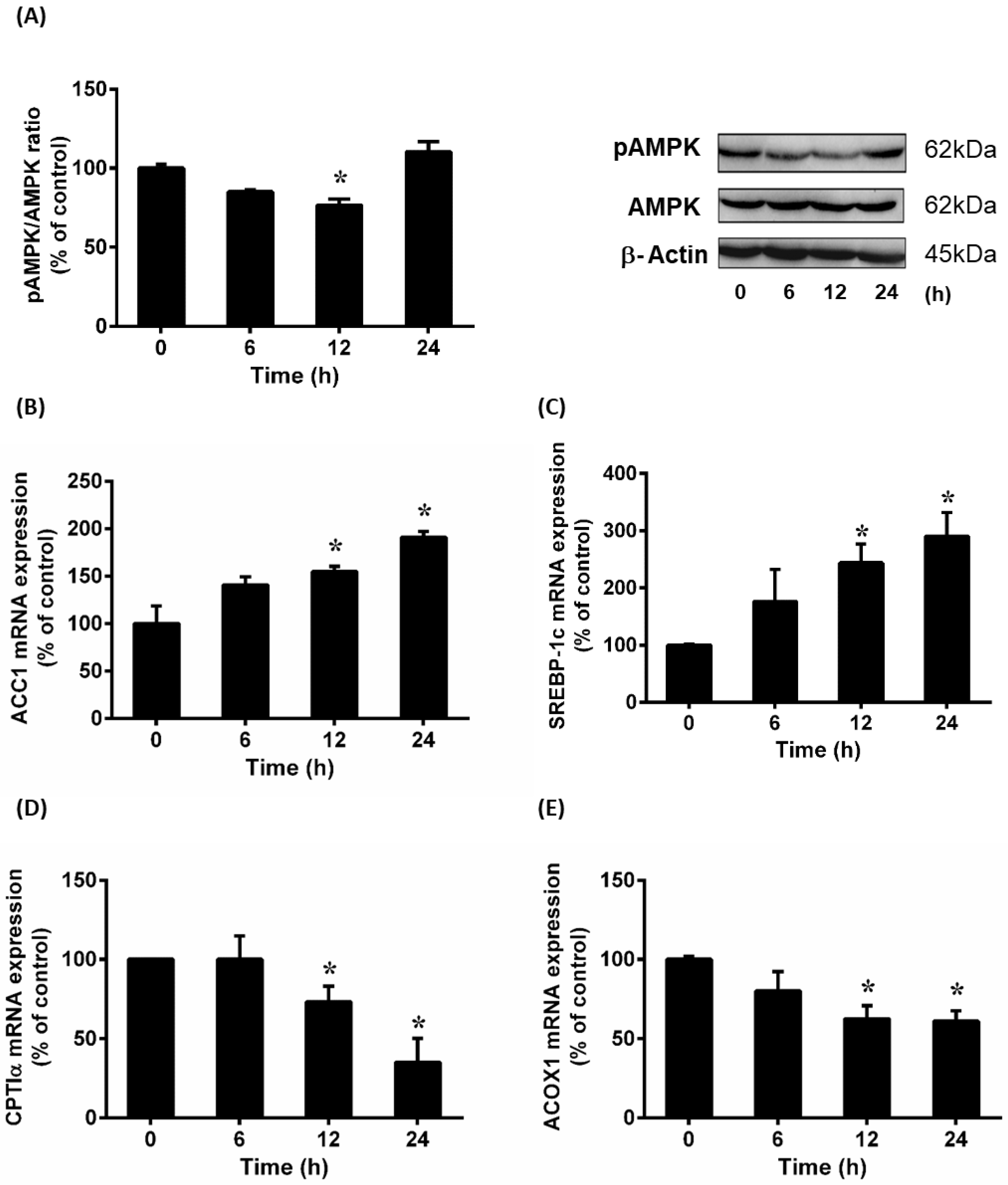
3.3. Renal Ischemia-Reperfusion Downregulated AMPK Pathway and Affected Lipid Metabolism in the Liver
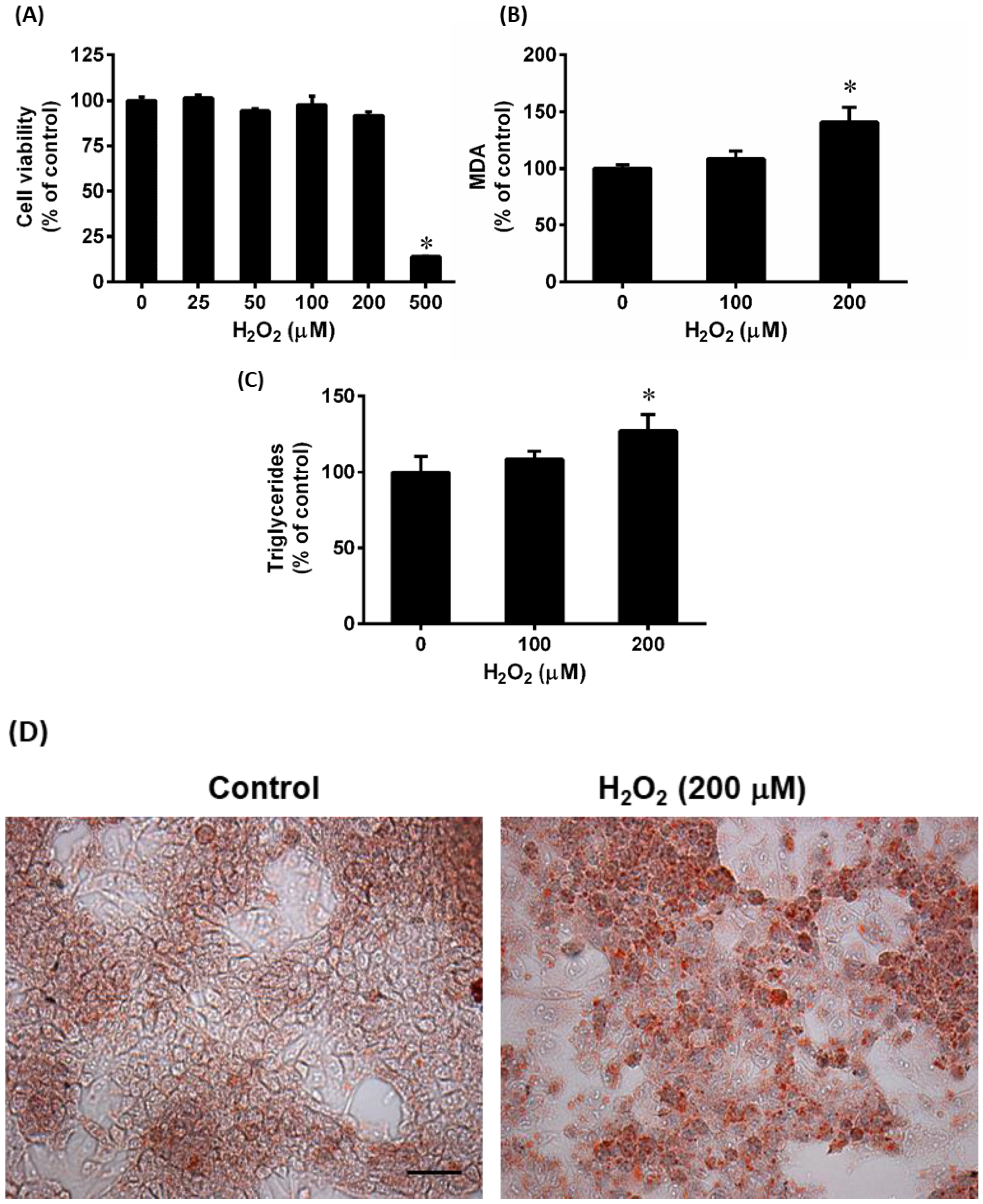
3.4. Effect of Hydrogen Peroxide (H2O2) on Lipid Accumulation in HepG2 Cells
3.5. Effect of Hydrogen Peroxide (H2O2) on AMPK Phosphorylation and Lipid Metabolizing Gene Expression
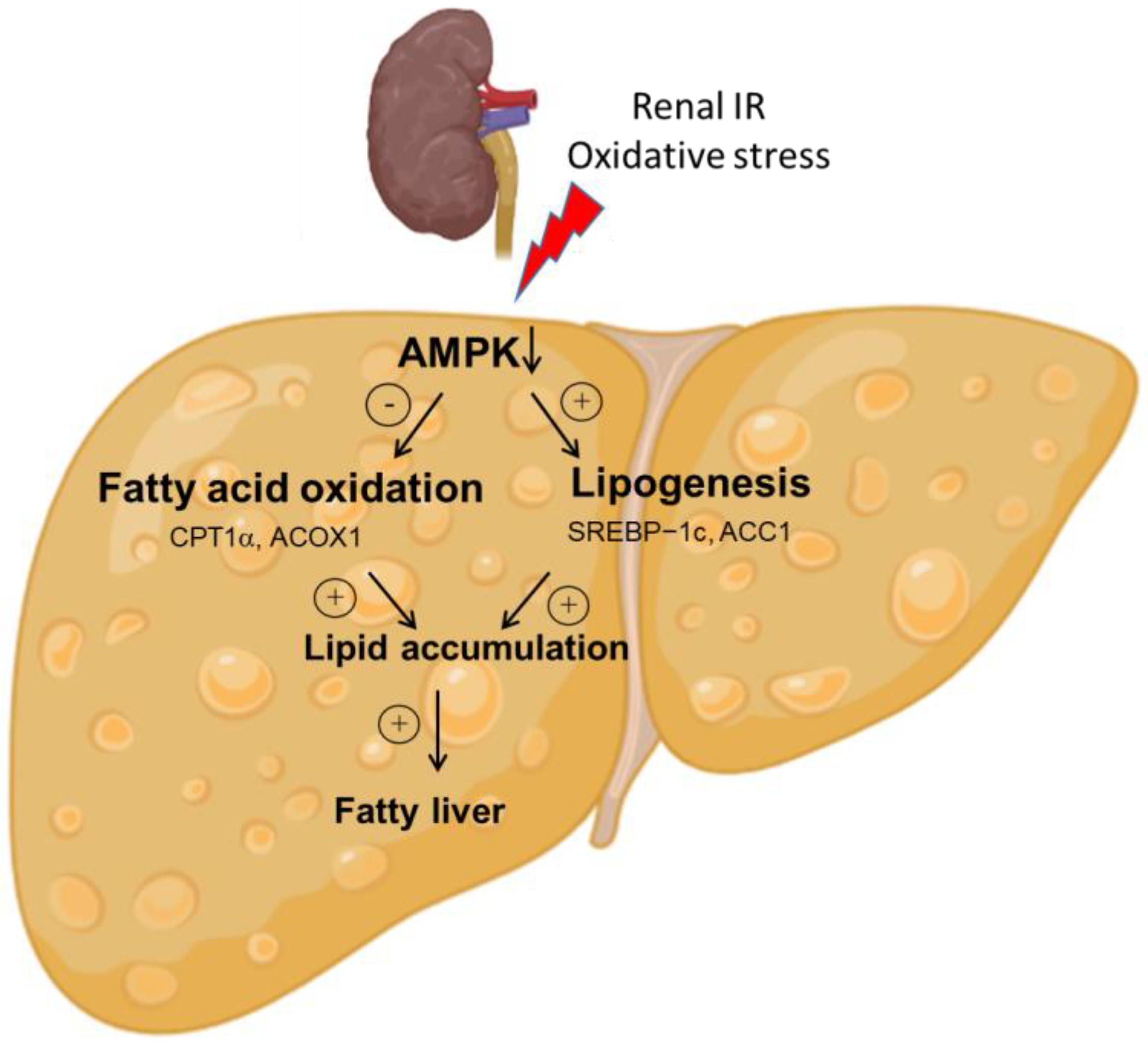
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Makris, K.; Spanou, L. Acute kidney injury: Definition, pathophysiology and clinical phenotypes. Clin. Biochem. Rev. 2016, 37, 85. [Google Scholar]
- Kister, T.S.; Remmler, J.; Schmidt, M.; Federbusch, M.; Eckelt, F.; Isermann, B.; Richter, H.; Wehner, M.; Krause, U.; Halbritter, J.; et al. Acute kidney injury and its progression in hospitalized patients-Results from a retrospective multicentre cohort study with a digital decision support system. PLoS ONE 2021, 16, e0254608. [Google Scholar] [CrossRef] [PubMed]
- Tatum, J.M.; Barmparas, G.; Ko, A.; Dhillon, N.; Smith, E.; Margulies, D.R.; Ley, E.J. Analysis of Survival After Initiation of Continuous Renal Replacement Therapy in a Surgical Intensive Care Unit. JAMA Surg. 2017, 152, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Rabb, H. Impact of acute kidney injury on distant organ function: Recent findings and potential therapeutic targets. Kidney Int. 2016, 89, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Cozzi, M.; Bush, E.L.; Rabb, H. Distant Organ Dysfunction in Acute Kidney Injury: A Review. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2018, 72, 846–856. [Google Scholar] [CrossRef]
- Lara-Prado, J.I.; Pazos-Pérez, F.; Méndez-Landa, C.E.; Grajales-García, D.P.; Feria-Ramírez, J.A.; Salazar-González, J.J.; Cruz-Romero, M.; Treviño-Becerra, A. Acute Kidney Injury and Organ Dysfunction: What Is the Role of Uremic Toxins? Toxins 2021, 13, 551. [Google Scholar] [CrossRef] [PubMed]
- Chancharoenthana, W.; Leelahavanichkul, A. Acute kidney injury spectrum in patients with chronic liver disease: Where do we stand? World J. Gastroenterol. 2019, 25, 3684–3703. [Google Scholar] [CrossRef]
- Cullaro, G.; Kanduri, S.R.; Velez, J.C.Q. Acute Kidney Injury in Patients with Liver Disease. Clin. J. Am. Soc. Nephrol. 2022, 17, 1674–1684. [Google Scholar] [CrossRef]
- Chen, L.; Lv, X.; Kan, M.; Wang, R.; Wang, H.; Zang, H. Critical Overview of Hepatic Factors That Link Non-Alcoholic Fatty Liver Disease and Acute Kidney Injury: Physiology and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 12464. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Siow, Y.L.; Isaak, C.K. Downregulation of glutathione biosynthesis contributes to oxidative stress and liver dysfunction in acute kidney injury. Oxid. Med. Cell Longev. 2016, 2016, 9707292. [Google Scholar] [CrossRef] [Green Version]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical im-plications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Peng, C.; Stewart, A.G.; Woodman, O.L.; Ritchie, R.H.; Qin, C.X. Non-Alcoholic Steatohepatitis: A Review of Its Mechanism, Models and Medical Treatments. Front. Pharmacol. 2020, 11, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a therapeutic target for treating metabolic diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kandula, N.; Kumar, S.; Mandlem, V.K.K.; Siddabathuni, A.; Singh, S.; Kosuru, R. Role of AMPK in Myocardial Ischemia-Reperfusion Injury-Induced Cell Death in the Presence and Absence of Diabetes. Oxidative Med. Cell. Longev. 2022, 2022, 7346699. [Google Scholar] [CrossRef] [PubMed]
- Boudaba, N.; Marion, A.; Huet, C.; Pierre, R.; Viollet, B.; Foretz, M. AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine 2018, 28, 194–209. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [Green Version]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I. Lifestyle, oxidative stress, and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiya, M.; Hiraishi, A.; Touyama, M.; Sakamoto, K. Oxidative stress induced lipid accumulation via SREBP-1c activation in HepG2 cells. Biochem. Biophys. Res. Commun. 2008, 375, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.; Kamel, M.A.; El-Nabi, S.E.H.; Ismail, A.H.A.; Ullah, S.; Al-Ghamdi, A.; Hathout, H.M.; El-Garawani, I.M. The regulation of HBP1, SIRT1, and SREBP-1c genes and the related microRNAs in non-alcoholic fatty liver rats: The association with the folic acid anti-steatosis. PLoS ONE 2022, 17, e0265455. [Google Scholar] [CrossRef]
- Li, G.; Zhou, F.; Chen, Y.; Zhang, W.; Wang, N. Kukoamine A attenuates insulin resistance and fatty liver through downregulation of Srebp-1c. Biomed. Pharmacother. 2017, 89, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2017, 8, 1. [Google Scholar] [PubMed]
- Esquejo, R.M.; Salatto, C.T.; Delmore, J.; Albuquerque, B.; Reyes, A.; Shi, Y.; Moccia, R.; Cokorinos, E.; Peloquin, M.; Monetti, M. Activation of liver AMPK with PF-06409577 corrects NAFLD and lowers cholesterol in rodent and primate preclinical models. EBioMedicine 2018, 31, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.-P.; O’neill, H.M.; Ford, R.J.; Palanivel, R.; O’brien, M. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; Madduma Hewage, S.; Wijerathne, C.U.; Siow, Y.L.; Isaak, C.K.; O, K. Kidney ischemia-reperfusion elicits acute liver injury and inflammatory response. Front. Med. 2020, 7, 201. [Google Scholar] [CrossRef]
- Madduma Hewage, S.; Prashar, S.; O, K.; Siow, Y.L. Lingonberry Improves Non-Alcoholic Fatty Liver Disease by Reducing Hepatic Lipid Accumulation, Oxidative Stress and Inflammatory Response. Antioxidants 2021, 10, 565. [Google Scholar] [CrossRef]
- Choi, Y.-J.; Zhou, D.; Barbosa, A.C.S.; Niu, Y.; Guan, X.; Xu, M.; Ren, S.; Nolin, T.D.; Liu, Y.; Xie, W. Activation of Constitutive Androstane Receptor Ameliorates Renal Ischemia-Reperfusion–Induced Kidney and Liver Injury. Mol. Pharmacol. 2018, 93, 239. [Google Scholar] [CrossRef] [PubMed]
- Prathapasinghe, G.A.; Siow, Y.L.; O, K. Detrimental role of homocysteine in renal ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 2007, 292, F1354–F1363. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Lees, M.; Sloanestanley, G. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar]
- Madduma Hewage, S.; Au-Yeung, K.K.; Prashar, S.; Wijerathne, C.U.; O, K.; Siow, Y.L. Lingonberry Improves Hepatic Lipid Metabolism by Targeting Notch1 Signaling. Antioxidants 2022, 11, 472. [Google Scholar] [CrossRef]
- Ding, R.; Wu, W.; Sun, Z.; Li, Z. AMP-activated protein kinase: An attractive therapeutic target for ischemia-reperfusion injury. Eur. J. Pharmacol. 2020, 888, 173484. [Google Scholar] [CrossRef]
- Ransy, C.; Vaz, C.; Lombès, A.; Bouillaud, F. Use of H2O2 to Cause Oxidative Stress, the Catalase Issue. Int. J. Mol. Sci. 2020, 21, 9149. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-C.; Hardie, D.G. AMPK: Sensing glucose as well as cellular energy status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Guo, X.; Cui, S.; Wu, Y.; Zhang, Y.; Shen, X.; Xie, C.; Li, J. Dephosphorylation of AMP-activated protein kinase exacerbates ischemia/reperfusion-induced acute kidney injury via mitochondrial dysfunction. Kidney Int. 2022, 101, 315–330. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, Y.J.; et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Liang, K.; Zhao, S.; Jia, W.; Liu, Y.; Wu, H.; Lv, J.; Cao, C.; Chen, T.; Zhuang, S. Chemoproteomics reveals baicalin activates hepatic CPT1 to ameliorate diet-induced obesity and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5896–E5905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.S.; Cho, S.Y.; Lee, E.H.; Lee, S.J.; Chang, I.S.; Lee, T.R. Positive regulation of hepatic carnitine palmitoyl transferase 1A (CPT1A) activities by soy isoflavones and L–carnitine. Eur. J. Nutr. 2006, 45, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Hashimoto, T. Peroxisomal β-oxidation and peroxisome proliferator–activated receptor α: An adaptive metabolic system. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly-Y, M.; Carling, D. Liver-specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsky, V.W.; Chan, A.Y.; Robillard Frayne, I.; Light, P.E.; Des Rosiers, C.; Dyck, J.R. Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation 2009, 119, 1643–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saberi, B.; Shinohara, M.; Ybanez, M.D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N.; Han, D. Regulation of H2O2-induced necrosis by PKC and AMP-activated kinase signaling in primary cultured hepatocytes. Am. J. Physiol. Cell Physiol. 2008, 295, C50–C63. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Oka, S.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, Y.; Hong, K.; Luo, F.; Gu, Q.; Lu, N.; Bai, A. AMPK inhibition blocks ROS-NFκB signaling and attenuates endotoxemia-induced liver injury. PLoS ONE 2014, 9, e86881. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [Green Version]
- Auciello, F.R.; Ross, F.A.; Ikematsu, N.; Hardie, D.G. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett. 2014, 588, 3361–3366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef]
- Kašparová, S.; Brezová, V.; Valko, M.; Horecký, J.; Mlynárik, V.; Liptaj, T.; Vančová, O.G.; Uličná, O.G.; Dobrota, D. Study of the oxidative stress in a rat model of chronic brain hypoperfusion. Neurochem. Int. 2005, 46, 601–611. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, S.; Sun, J.; Wang, X.; Chen, N.; Zhou, Y.; Tian, Y.; Ye, D. Berberine protects against ischemia-reperfusion injury: A review of evidence from animal models and clinical studies. Pharmacol. Res. 2019, 148, 104385. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, L.; Huang, K.; Wang, C.; Chiang, C.; Liu, S. Quercetin attenuates renal ischemia/reperfusion injury via an activation of AMP-activated protein kinase-regulated autophagy pathway. J. Nutr. Biochem. 2014, 25, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Tujios, S.R.; Hynan, L.S.; Vazquez, M.A.; Larson, A.M.; Seremba, E.; Sanders, C.M.; Lee, W.M.; Group, A.L.F.S. Risk factors and outcomes of acute kidney injury in patients with acute liver failure. Clin. Gastroenterol. Hepatol. 2015, 13, 352–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Scherer, P.E. Immunologic and endocrine functions of adipose tissue: Implications for kidney disease. Nat. Rev. Nephrol. 2018, 14, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.M.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208.e196. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; Wong, K.I.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer (5′−3′) | Reverse Primer (5′−3′) | Size (bp) | Accession Number |
|---|---|---|---|---|
| Rat | ||||
| SREBP-1c | GGCCCTGTGTGTACTGGTCT | AGCATCAGAGGGAGTGAGGA | 88 | NM_001276708.1 |
| ACOX1 | CTGATGAAATACGCCCAGGT | GGTCCCATACGTCAGCTTGT | 75 | NM_001414015.1 |
| ACC1 | TGAGGAGGACCGCATTTATC | AAGCTTCCTTCGTGACCAGA | 221 | NM_022193.2 |
| CPTIα | CAGCTCGCACATTACAAGGA | TGCACAAAGTTGCAGGACTC | 128 | XM_039102321.1 |
| β-actin | ACAACCTTCTTGCAGCTCCTC | GACCCATACCCACCATCACA | 198 | NM_031144.3 |
| Human | ||||
| SREBP-1c | ACACAGCAACCAGAAACTCAAG | AGTGTGTCCTCCACCTCAGTCT | 153 | NM_001005291.3 |
| ACOX1 | GGCGCATACATGAAGGAGACCT | AGGTGAAAGCCTTCAGTCCAGC | 112 | NM_001185039.2 |
| ACC1 | TTCACTCCACCTTGTCAGCGGA | GTCAGAGAAGCAGCCCATCACT | 99 | XM_047435894.1 |
| CPTIα | CGATGTTACGACAGGTGGTTTGACA | AGTGCCCATCCTCCGCATAG | 172 | NM_001876.4 |
| β-actin | AGATCAAGATCATTGCTCCTCCT | GATCCACATCTGCTGGAAGG | 95 | NM_001101.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Au-Yeung, K.K.W.; Shang, Y.; Wijerathne, C.U.B.; Madduma Hewage, S.; Siow, Y.L.; O, K. Acute Kidney Injury Induces Oxidative Stress and Hepatic Lipid Accumulation through AMPK Signaling Pathway. Antioxidants 2023, 12, 883. https://doi.org/10.3390/antiox12040883
Au-Yeung KKW, Shang Y, Wijerathne CUB, Madduma Hewage S, Siow YL, O K. Acute Kidney Injury Induces Oxidative Stress and Hepatic Lipid Accumulation through AMPK Signaling Pathway. Antioxidants. 2023; 12(4):883. https://doi.org/10.3390/antiox12040883
Chicago/Turabian StyleAu-Yeung, Kathy K. W., Yue Shang, Charith U. B. Wijerathne, Susara Madduma Hewage, Yaw L. Siow, and Karmin O. 2023. "Acute Kidney Injury Induces Oxidative Stress and Hepatic Lipid Accumulation through AMPK Signaling Pathway" Antioxidants 12, no. 4: 883. https://doi.org/10.3390/antiox12040883