
The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis
 , , and
, , and
Abstract
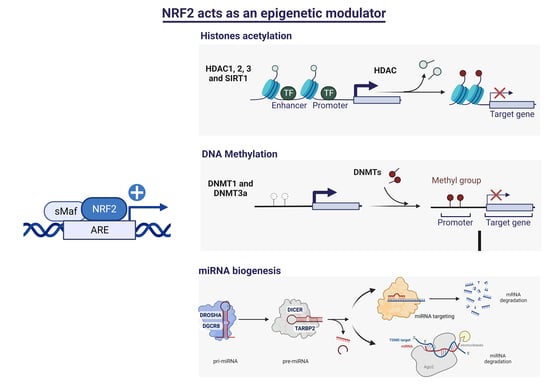
:
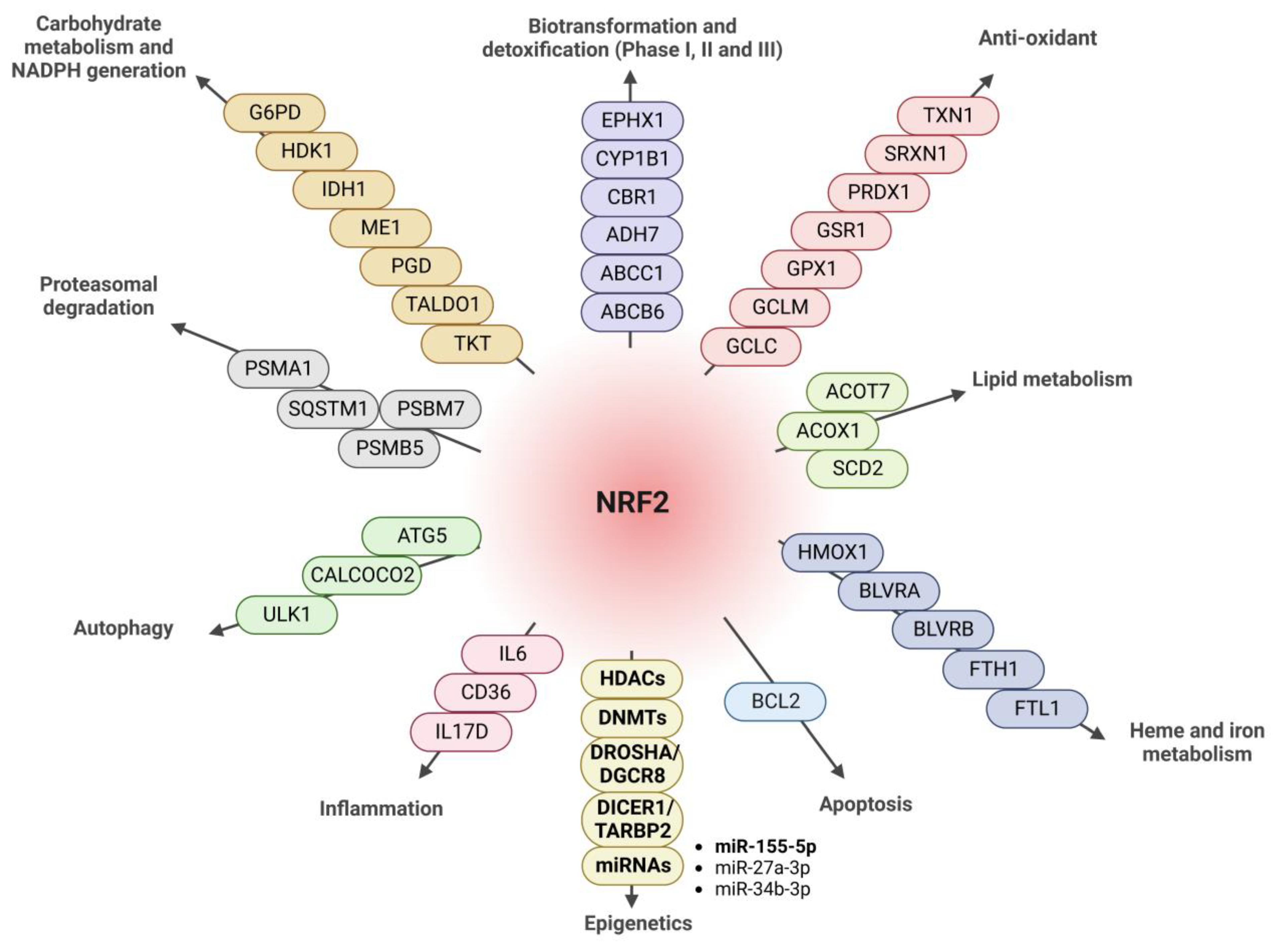
1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis
2.2. Cell Cultures and Treatments
2.3. Analysis of mRNA Levels via Quantitative Real-Time PCR
2.4. Plasmids
2.5. Luciferase Assays
2.6. Immunoblotting
2.7. Antisense Oligonucleotide (ASO) Pull-Down Assay
2.8. Analysis of miRNA Levels via Quantitative Real-Time PCR
2.9. Statistical Analyses
3. Results
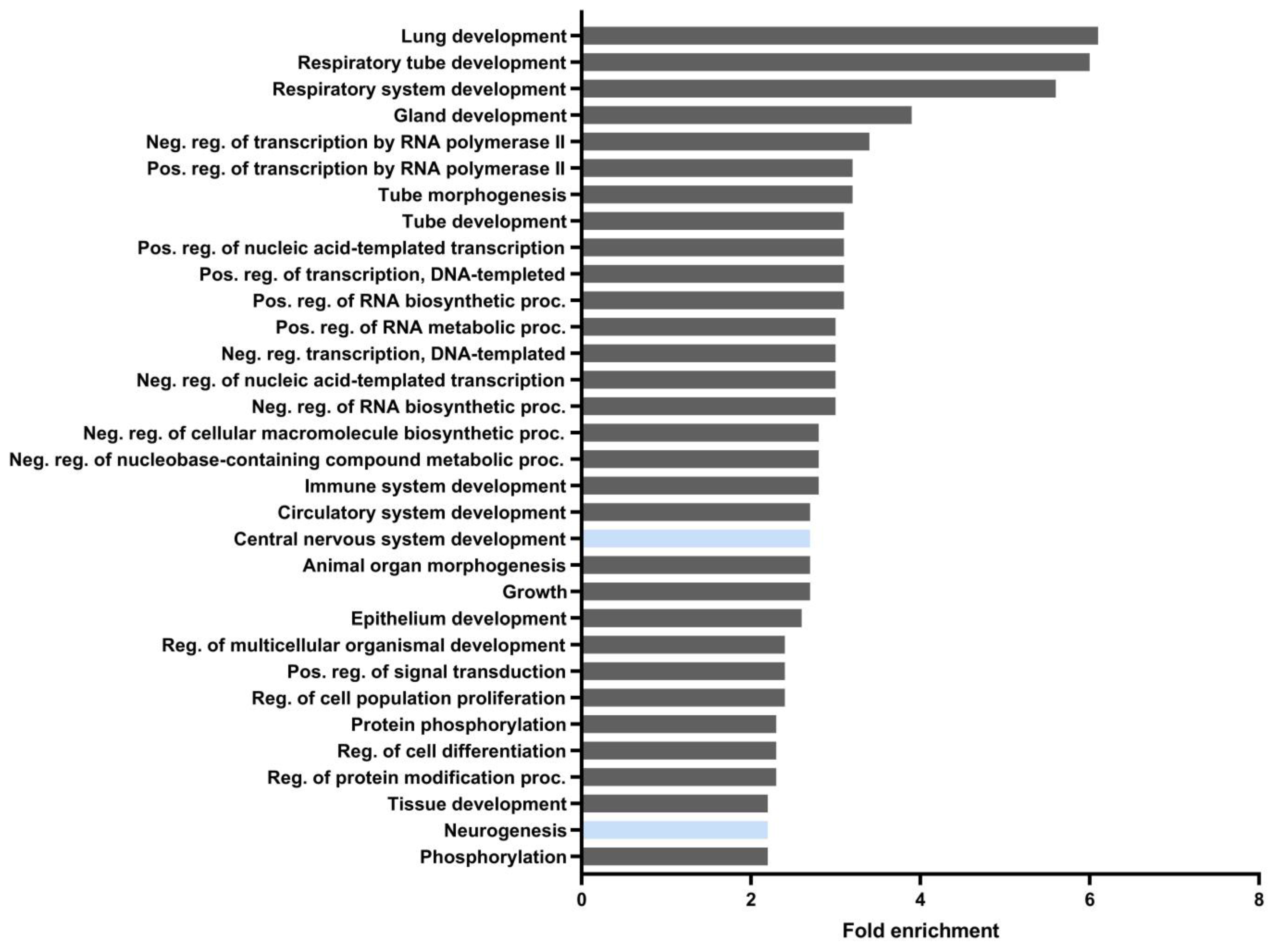
3.1. Identification of Putative ARE Sequences in HDCAs and DNMTs
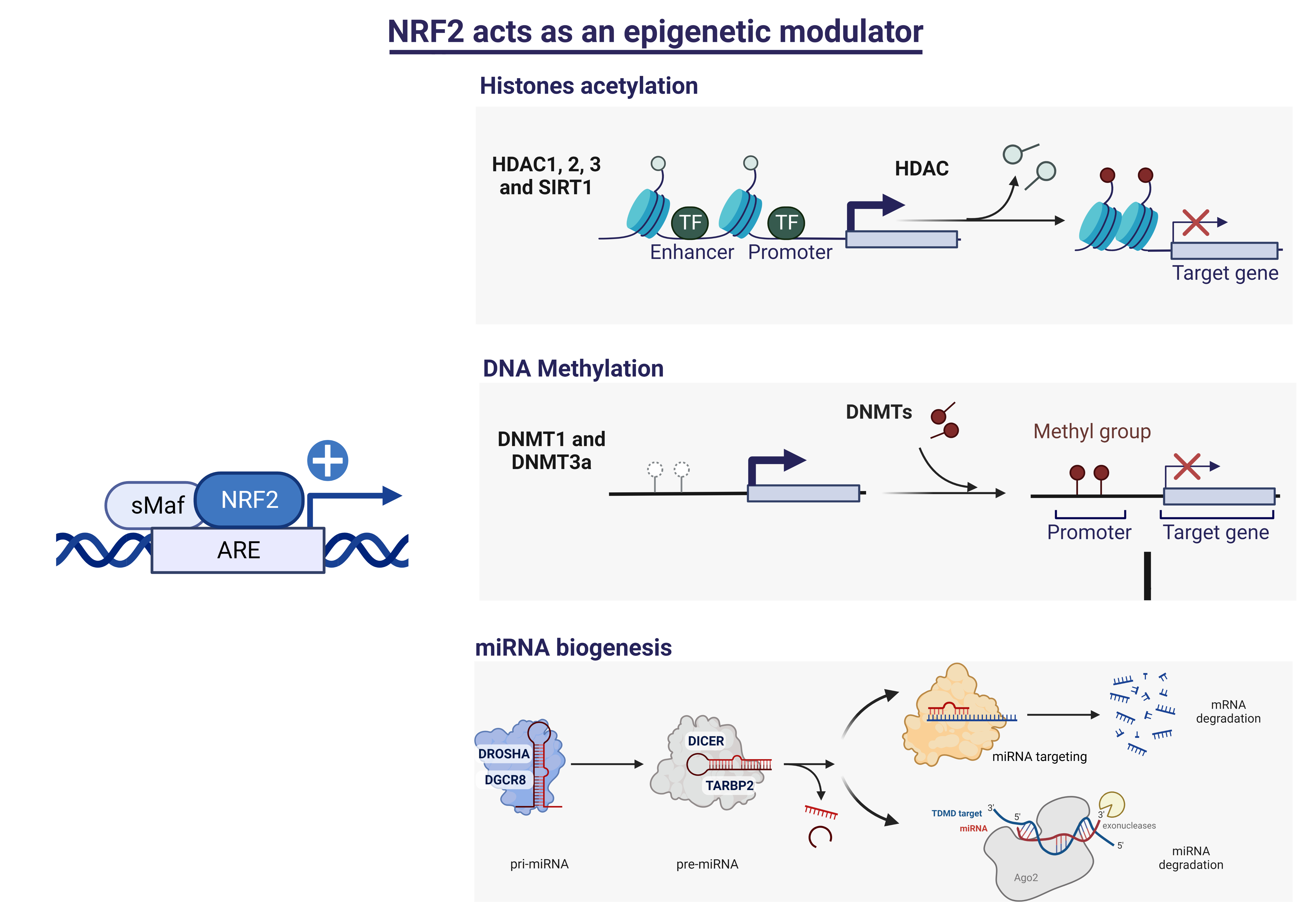
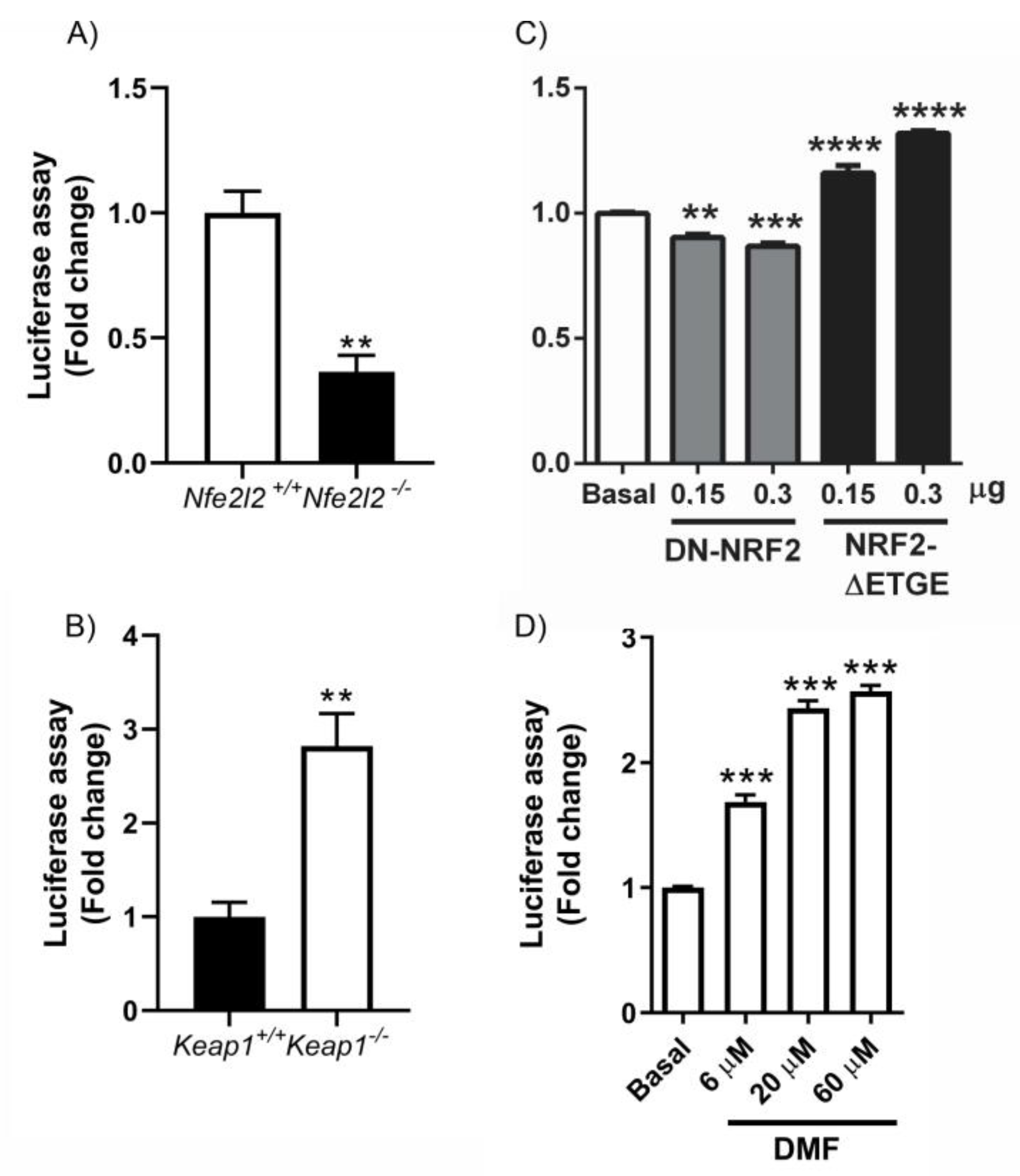
3.2. HDCAs Are NRF2-Dependent Genes
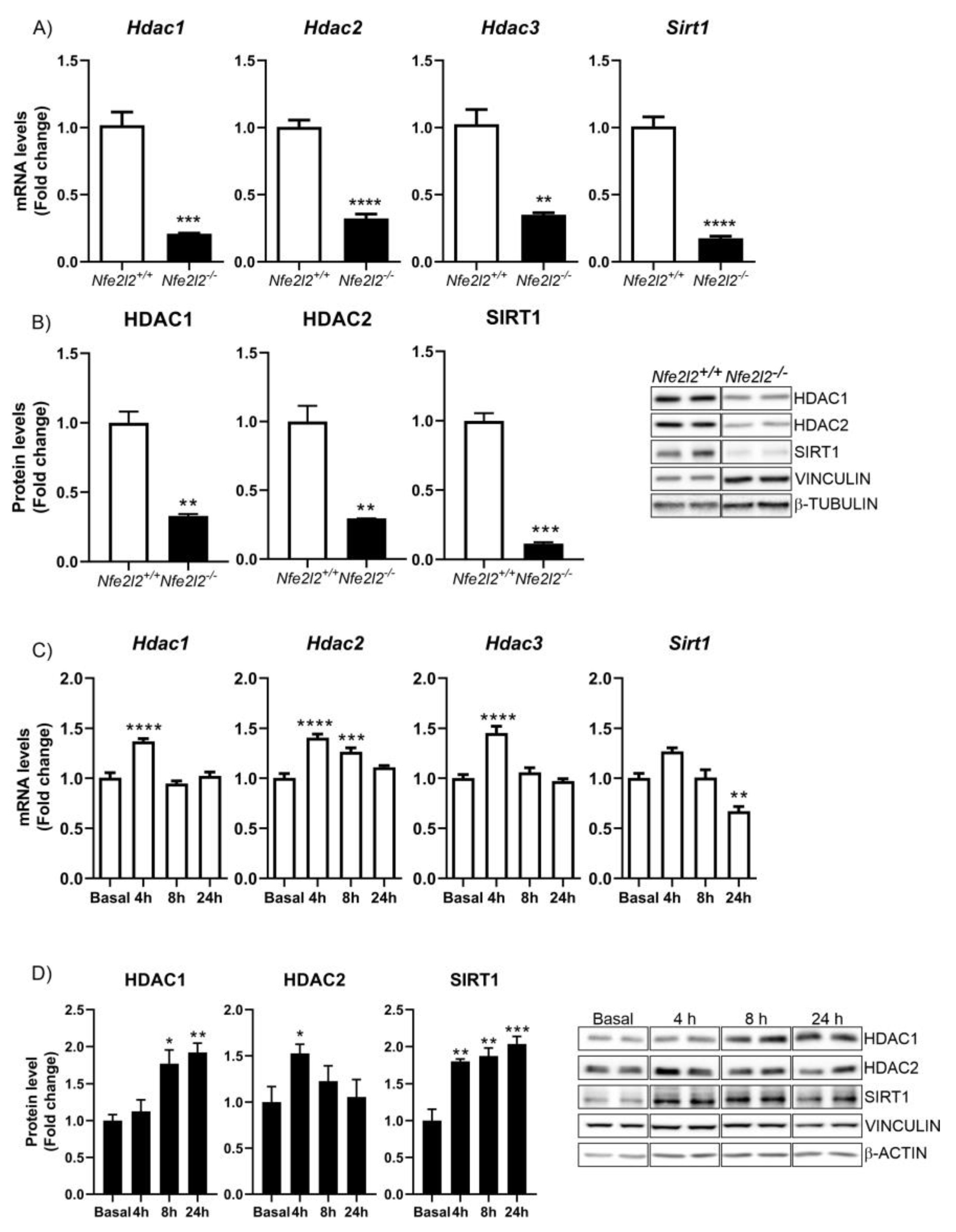
3.3. NRF2 Is a Modulator of DNMTs Expression
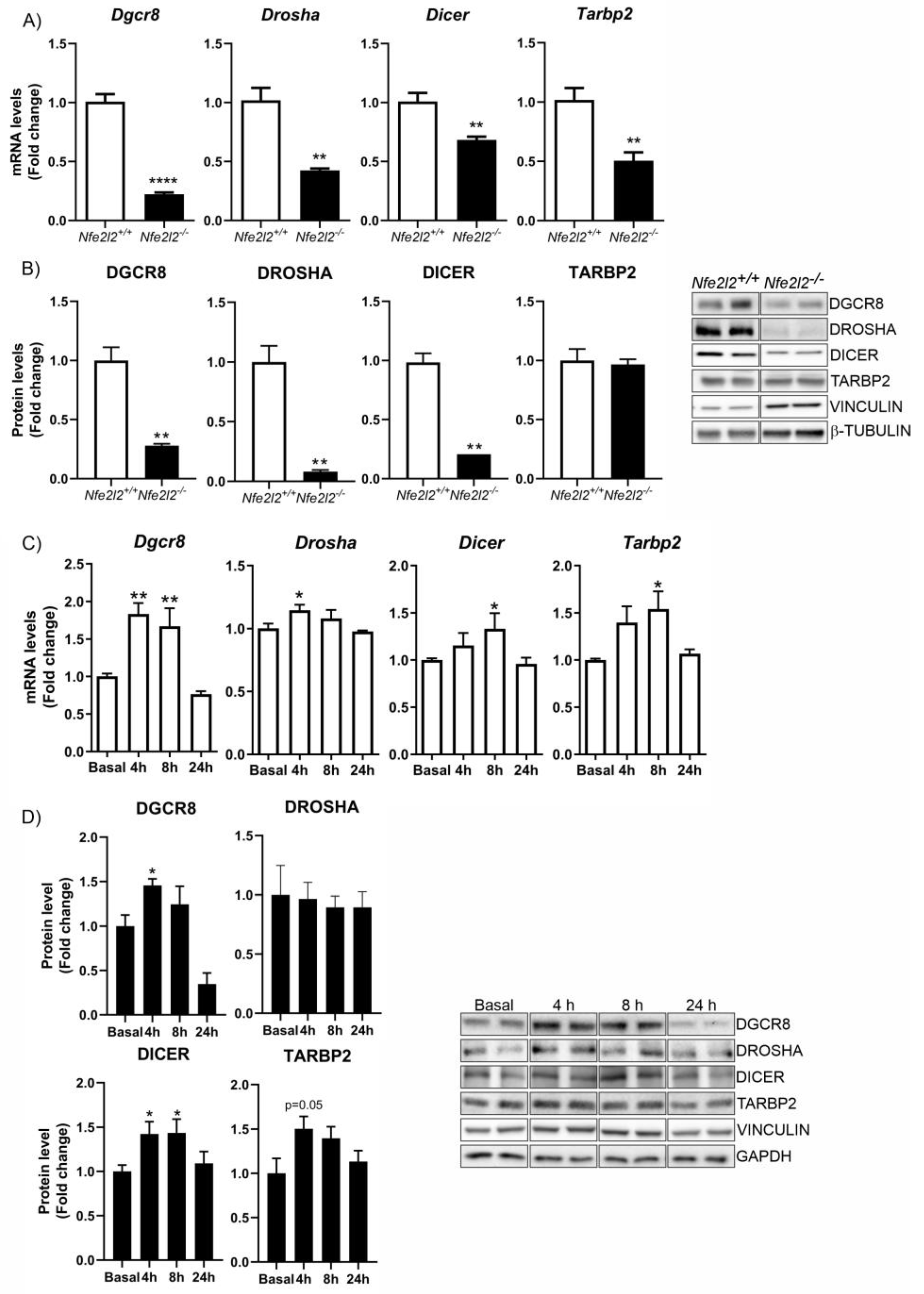
3.4. NRF2 Regulates the Expression of Proteins Implicated in miRNA Biogenesis
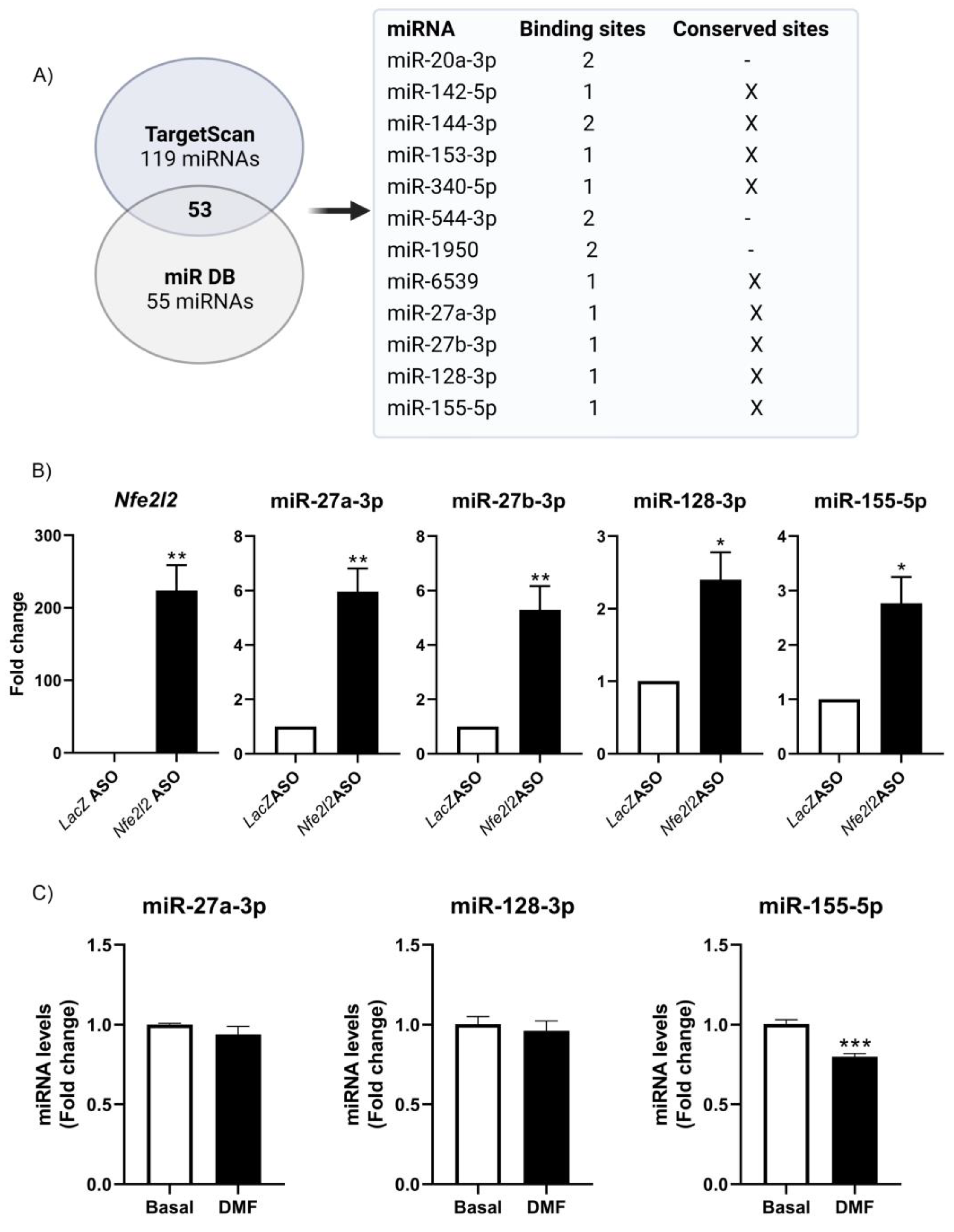
3.5. NRF2 Modulates miRNA Expression
3.6. Implication of NRF2 Expression in Target-Dependent miRNA Degradation (TDMD) of miR-155-5p
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stephens, K.E.; Miaskowski, C.A.; Levine, J.D.; Pullinger, C.R.; Aouizerat, B.E. Epigenetic regulation and measurement of epigenetic changes. Biol. Res. Nurs. 2013, 15, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A. Brain-Protective Mechanisms of Transcription Factor NRF2: Toward a Common Strategy for Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 255–277. [Google Scholar] [CrossRef] [PubMed]
- Bukke, V.N.; Moola, A.; Serviddio, G.; Vendemiale, G.; Bellanti, F. Nuclear factor erythroid 2-related factor 2-mediated signaling and metabolic associated fatty liver disease. World J. Gastroenterol. 2022, 28, 6909–6921. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Khor, T.O.; Cheung, K.L.; Li, W.; Wu, T.Y.; Huang, Y.; Foster, B.A.; Kan, Y.W.; Kong, A.N. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS ONE 2010, 5, e8579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taheri, Z.; Asadzadeh Aghdaei, H.; Irani, S.; Modarressi, M.H.; Zahra, N. Evaluation of the Epigenetic Demethylation of NRF2, a Master Transcription Factor for Antioxidant Enzymes, in Colorectal Cancer. Rep. Biochem. Mol. Biol. 2020, 9, 33–39. [Google Scholar] [CrossRef]
- Shah, N.M.; Rushworth, S.A.; Murray, M.Y.; Bowles, K.M.; MacEwan, D.J. Understanding the role of NRF2-regulated miRNAs in human malignancies. Oncotarget 2013, 4, 1130–1142. [Google Scholar] [CrossRef] [Green Version]
- Quiles, J.M.; Pepin, M.E.; Sunny, S.; Shelar, S.B.; Challa, A.K.; Dalley, B.; Hoidal, J.R.; Pogwizd, S.M.; Wende, A.R.; Rajasekaran, N.S. Identification of Nrf2-responsive microRNA networks as putative mediators of myocardial reductive stress. Sci. Rep. 2021, 11, 11977. [Google Scholar] [CrossRef] [PubMed]
- Kaundal, R.K.; Datusalia, A.K.; Sharma, S.S. Posttranscriptional regulation of Nrf2 through miRNAs and their role in Alzheimer’s disease. Pharmacol. Res. 2022, 175, 106018. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, E.; Dobre, M.; Cucos, C.A.; Rojo, A.I.; Jiménez-Villegas, J.; Capetillo-Zarate, E.; Matute, C.; Piñol-Ripoll, G.; Manda, G.; Cuadrado, A. Whole Blood Expression Pattern of Inflammation and Redox Genes in Mild Alzheimer’s Disease. J. Inflamm. Res. 2021, 14, 6085–6102. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Ernst, J.; Wilder, S.P.; Kundaje, A.; Harris, R.S.; Libbrecht, M.; Giardine, B.; Ellenbogen, P.M.; Bilmes, J.A.; Birney, E.; et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013, 41, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; Kaul, R.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Zou, Z.; Ohta, T.; Miura, F.; Oki, S. ChIP-Atlas 2021 update: A data-mining suite for exploring epigenomic landscapes by fully integrating ChIP-seq, ATAC-seq and Bisulfite-seq data. Nucleic Acids Res. 2022, 50, W175–W182. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinform. 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, R.K.; Pedersen, B.S.; Quinlan, A.R. Pybedtools: A flexible Python library for manipulating genomic datasets and annotations. Bioinform. (Oxf. Engl.) 2011, 27, 3423–3424. [Google Scholar] [CrossRef] [Green Version]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Lemma, R.B.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Pérez, N.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef] [PubMed]
- Martin-Hurtado, A.; Martin-Morales, R.; Robledinos-Antón, N.; Blanco, R.; Palacios-Blanco, I.; Lastres-Becker, I.; Cuadrado, A.; Garcia-Gonzalo, F.R. NRF2-dependent gene expression promotes ciliogenesis and Hedgehog signaling. Sci. Rep. 2019, 9, 13896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Ginés, R.; Encinar, J.A.; Hayes, J.D.; Oliva, B.; Rodríguez-Franco, M.I.; Rojo, A.I.; Cuadrado, A. An inhibitor of interaction between the transcription factor NRF2 and the E3 ubiquitin ligase adapter β-TrCP delivers anti-inflammatory responses in mouse liver. Redox Biol. 2022, 55, 102396. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martin-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-kappaB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; de Lago, E.; Martínez, A.; Fernández-Ruiz, J. New Statement about NRF2 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Biomolecules 2022, 12, 1200. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, W.G. Targeting histone deacetylases for cancer therapy: From molecular mechanisms to clinical implications. Int. J. Biol. Sci. 2014, 10, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Alseksek, R.K.; Ramadan, W.S.; Saleh, E.; El-Awady, R. The Role of HDACs in the Response of Cancer Cells to Cellular Stress and the Potential for Therapeutic Intervention. Int. J. Mol. Sci. 2022, 23, 8141. [Google Scholar] [CrossRef]
- The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Campos, A.C.; Molognoni, F.; Melo, F.H.; Galdieri, L.C.; Carneiro, C.R.; D’Almeida, V.; Correa, M.; Jasiulionis, M.G. Oxidative stress modulates DNA methylation during melanocyte anchorage blockade associated with malignant transformation. Neoplasia 2007, 9, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- Hedman, Å.K.; Zilmer, M.; Sundström, J.; Lind, L.; Ingelsson, E. DNA methylation patterns associated with oxidative stress in an ageing population. BMC Med. Genom. 2016, 9, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathelier, A.; Zhao, X.; Zhang, A.W.; Parcy, F.; Worsley-Hunt, R.; Arenillas, D.J.; Buchman, S.; Chen, C.-y.; Chou, A.; Ienasescu, H.; et al. JASPAR 2014: An extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2013, 42, D142–D147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.C.; Engström, P.G.; Lithwick, S.; Arenillas, D.; Eriksson, P.; Lenhard, B.; Wasserman, W.W.; Odeberg, J. In silico detection of sequence variations modifying transcriptional regulation. PLoS Comput. Biol. 2008, 4, e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, A.T.; Arenillas, D.J.; Worsley Hunt, R.; Wasserman, W.W. oPOSSUM-3: Advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 (Bethesda Md.) 2012, 2, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Kugler, S.; Lastres-Becker, I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef]
- Carbonell, T.; Gomes, A.V. MicroRNAs in the regulation of cellular redox status and its implications in myocardial ischemia-reperfusion injury. Redox Biol. 2020, 36, 101607. [Google Scholar] [CrossRef]
- Mayya, V.K.; Duchaine, T.F. Ciphers and Executioners: How 3’-Untranslated Regions Determine the Fate of Messenger RNAs. Front. Genet. 2019, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Ghini, F.; Rubolino, C.; Climent, M.; Simeone, I.; Marzi, M.J.; Nicassio, F. Endogenous transcripts control miRNA levels and activity in mammalian cells by target-directed miRNA degradation. Nat. Commun. 2018, 9, 3119. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Dinger, M.E. Endogenous microRNA sponges: Evidence and controversy. Nat. Rev. Genet. 2016, 17, 272–283. [Google Scholar] [CrossRef]
- Rüegger, S.; Großhans, H. MicroRNA turnover: When, how, and why. Trends Biochem. Sci. 2012, 37, 436–446. [Google Scholar] [CrossRef]
- de la Mata, M.; Gaidatzis, D.; Vitanescu, M.; Stadler, M.B.; Wentzel, C.; Scheiffele, P.; Filipowicz, W.; Großhans, H. Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep. 2015, 16, 500–511. [Google Scholar] [CrossRef] [Green Version]
- Song, M.Y.; Lee, D.Y.; Chun, K.S.; Kim, E.H. The Role of NRF2/KEAP1 Signaling Pathway in Cancer Metabolism. Int. J. Mol. Sci. 2021, 22, 4376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.W.; Wang, M.; Sun, N.X.; Qing, Y.; Yin, T.F.; Li, C.; Wu, D. Sulforaphane-induced epigenetic regulation of Nrf2 expression by DNA methyltransferase in human Caco-2 cells. Oncol. Lett. 2019, 18, 2639–2647. [Google Scholar] [CrossRef] [Green Version]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltby, V.E.; Lea, R.A.; Ribbons, K.A.; Sanders, K.A.; Kennedy, D.; Min, M.; Scott, R.J.; Lechner-Scott, J. DNA methylation changes in CD4(+) T cells isolated from multiple sclerosis patients on dimethyl fumarate. Mult. Scler. J.-Exp. Transl. Clin. 2018, 4, 2055217318787826. [Google Scholar] [CrossRef] [Green Version]
- Carlström, K.E.; Ewing, E.; Granqvist, M.; Gyllenberg, A.; Aeinehband, S.; Enoksson, S.L.; Checa, A.; Badam, T.V.S.; Huang, J.; Gomez-Cabrero, D.; et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ROS pathway in monocytes. Nat. Commun. 2019, 10, 3081. [Google Scholar] [CrossRef] [Green Version]
- Pouremamali, F.; Pouremamali, A.; Dadashpour, M.; Soozangar, N.; Jeddi, F. An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Commun. Signal. 2022, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Castro-Muñoz, L.J.; Ulloa, E.V.; Sahlgren, C.; Lizano, M.; De La Cruz-Hernández, E.; Contreras-Paredes, A. Modulating epigenetic modifications for cancer therapy (Review). Oncol. Rep. 2023, 49, 1–23. [Google Scholar] [CrossRef]
- Wang, N.; Ma, T.; Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct. Target. Ther. 2023, 8, 69. [Google Scholar] [CrossRef]
- Potaczek, D.P.; Alashkar Alhamwe, B.; Miethe, S.; Garn, H. Epigenetic Mechanisms in Allergy Development and Prevention. Handb. Exp. Pharmacol. 2022, 268, 331–357. [Google Scholar] [CrossRef]
- Sundaramoorthy, T.H.; Castanho, I. The Neuroepigenetic Landscape of Vertebrate and Invertebrate Models of Neurodegenerative Diseases. Epigenetics Insights 2022, 15, 25168657221135848. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, C.B.; Zimmer-Bensch, G. Role of DNMTs in the Brain. Adv. Exp. Med. Biol. 2022, 1389, 363–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Duan, S.; Xie, Z.; Bao, W.; Xu, B.; Yang, W.; Zhou, L. Epigenetic Therapeutics Targeting NRF2/KEAP1 Signaling in Cancer Oxidative Stress. Front. Pharmacol. 2022, 13, 924817. [Google Scholar] [CrossRef]
- Camiña, N.; Penning, T.M. Genetic and epigenetic regulation of the NRF2-KEAP1 pathway in human lung cancer. Br. J. Cancer 2022, 126, 1244–1252. [Google Scholar] [CrossRef]
- Bovilla, V.R.; Kuruburu, M.G.; Bettada, V.G.; Krishnamurthy, J.; Sukocheva, O.A.; Thimmulappa, R.K.; Shivananju, N.S.; Balakrishna, J.P.; Madhunapantula, S.V. Targeted Inhibition of Anti-Inflammatory Regulator Nrf2 Results in Breast Cancer Retardation In Vitro and In Vivo. Biomedicines 2021, 9, 1119. [Google Scholar] [CrossRef]
- Ballout, F.; Lu, H.; Chen, Z.; Hu, T.; Chen, L.; Washington, M.K.; El-Rifai, W.; Peng, D. Targeting NRF2 Sensitizes Esophageal Adenocarcinoma Cells to Cisplatin through Induction of Ferroptosis and Apoptosis. Antioxidants 2022, 11, 1859. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Somasekharan, S.P.; Wang, Y.; Cheng, H.; Negri, G.L.; Pan, M.; Wang, X.Q.; Delaidelli, A.; Rafn, B.; Cran, J.; et al. Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep. 2019, 20, e48375. [Google Scholar] [CrossRef]
- McMahon, M.; Campbell, K.H.; MacLeod, A.K.; McLaughlin, L.A.; Henderson, C.J.; Wolf, C.R. HDAC inhibitors increase NRF2-signaling in tumour cells and blunt the efficacy of co-adminstered cytotoxic agents. PLoS ONE 2014, 9, e114055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, F.; Huang, J.W.; Ding, P.Y.; Zang, H.G.; Kou, Z.J.; Li, T.; Fan, J.; Peng, Z.W.; Yan, W.J. Nrf2/antioxidant defense pathway is involved in the neuroprotective effects of Sirt1 against focal cerebral ischemia in rats after hyperbaric oxygen preconditioning. Behav. Brain Res. 2016, 309, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pan, W.; Zhang, Y.; Tan, M.; Yin, Y.; Li, Y.; Zhang, L.; Han, L.; Bai, J.; Jiang, T.; et al. Comprehensive overview of Nrf2-related epigenetic regulations involved in ischemia-reperfusion injury. Theranostics 2022, 12, 6626–6645. [Google Scholar] [CrossRef]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, S.; Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Crow, J.P.; Beal, M.F. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 22, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-H.; Peng, W.-N.; Deng, Y.; Li, J.-J.; Tian, X.-R. Action of trichostatin A on Alzheimer’s disease-like pathological changes in SH-SY5Y neuroblastoma cells. Neural Regen. Res. 2020, 15, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rabano, A.; Kugler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Wang, L.; Chen, B.; Zheng, P.; He, Y.; Ding, Y.; Deng, Y.; Lu, X.; Guo, X.; Zhang, Y.; et al. DNA Demethylation Upregulated Nrf2 Expression in Alzheimer’s Disease Cellular Model. Front. Aging Neurosci. 2015, 7, 244. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Chen, F.; Zhang, L.; Wei, A.; Wang, Y.; Wu, Z.; Cao, W. Inhibition of DNA methyltransferase aberrations reinstates antioxidant aging suppressors and ameliorates renal aging. Aging Cell 2022, 21, e13526. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, H.Q.; Liu, F. DNA Methyltransferase Inhibitors and their Therapeutic Potential. Curr. Top. Med. Chem. 2018, 18, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Kurinna, S.; Werner, S. NRF2 and microRNAs: New but awaited relations. Biochem. Soc. Trans. 2015, 43, 595–601. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, X.; Luo, Q.; Zhang, X. MicroRNAs Involved in Oxidative Stress Processes Regulating Physiological and Pathological Responses. MicroRNA 2021, 10, 164–180. [Google Scholar] [CrossRef]
- Cheng, X.; Ku, C.H.; Siow, R.C. Regulation of the Nrf2 antioxidant pathway by microRNAs: New players in micromanaging redox homeostasis. Free Radic. Biol. Med. 2013, 64, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Lettieri-Barbato, D.; Aquilano, K.; Punziano, C.; Minopoli, G.; Faraonio, R. MicroRNAs, Long Non-Coding RNAs, and Circular RNAs in the Redox Control of Cell Senescence. Antioxidants 2022, 11, 480. [Google Scholar] [CrossRef]
- Bu, H.; Wedel, S.; Cavinato, M.; Jansen-Dürr, P. MicroRNA Regulation of Oxidative Stress-Induced Cellular Senescence. Oxidative Med. Cell. Longev. 2017, 2017, 2398696. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Li, F.; Liu, Z.; Xu, Z.; Sun, B.; Cao, J.; Liu, Y. MicroRNA-27b inhibition promotes Nrf2/ARE pathway activation and alleviates intracerebral hemorrhage-induced brain injury. Oncotarget 2017, 8, 70669–70684. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.R.; Zhang, Z.; Gao, M.; Li, L.; Sun, P.Y.; Xu, L.N.; Qi, Y.; Yin, L.H.; Peng, J.Y. MicroRNA-27a-3p aggravates renal ischemia/reperfusion injury by promoting oxidative stress via targeting growth factor receptor-bound protein 2. Pharmacol. Res. 2020, 155, 104718. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dong, D.; Reece, E.A.; Wang, A.R.; Yang, P. Oxidative stress-induced miR-27a targets the redox gene nuclear factor erythroid 2-related factor 2 in diabetic embryopathy. Am. J. Obstet. Gynecol. 2018, 218, 136.e110–136.e131. [Google Scholar] [CrossRef]
- Yang, H.; Li, T.W.; Zhou, Y.; Peng, H.; Liu, T.; Zandi, E.; Martínez-Chantar, M.L.; Mato, J.M.; Lu, S.C. Activation of a novel c-Myc-miR27-prohibitin 1 circuitry in cholestatic liver injury inhibits glutathione synthesis in mice. Antioxid. Redox Signal. 2015, 22, 259–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodbury, M.E.; Freilich, R.W.; Cheng, C.J.; Asai, H.; Ikezu, S.; Boucher, J.D.; Slack, F.; Ikezu, T. miR-155 Is Essential for Inflammation-Induced Hippocampal Neurogenic Dysfunction. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 9764–9781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, Y.; Song, Z.; Ding, Z.; Wang, J.; Wang, R.; Hou, X. Upregulation of Spinal miR-155-5p Contributes to Mechanical Hyperalgesia by Promoting Inflammatory Activation of Microglia in Bone Cancer Pain Rats. Life 2022, 12, 1349. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Mandrekar-Colucci, S.; Hall, J.C.; Sweet, D.R.; Schmitt, P.J.; Xu, X.; Guan, Z.; Mo, X.; Guerau-de-Arellano, M.; Popovich, P.G. miR-155 Deletion in Mice Overcomes Neuron-Intrinsic and Neuron-Extrinsic Barriers to Spinal Cord Repair. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 8516–8532. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Coordinates (hg19 Genome) | Motif | Relative Score | Strand | Regulatory Element * | TFs |
|---|---|---|---|---|---|---|
| HDAC1 | chr1:32757324-32757335 | GTCACTCAGCC | 0.835 | - | TSS | MAFF |
| DNMT1 | chr19:10266742-10266753 | ATGACTTGGCC | 0.855 | + | T | MAFK |
| DNMT1 | chr19:10266727-10266738 | ATGACTGAGGA | 0.836 | - | T | MAFK |
| DNMT1 | chr19:10284277-10284288 | CTGACTCAGCC | 0.883 | + | T | NFE2L2 |
| DNMT1 | chr19:10288124-10288135 | ATTACTAAGCT | 0.827 | + | T | MAFK |
| DNMT1 | chr19:10288156-10288167 | AGGACTAAGCA | 0.874 | + | T | MAFK |
| DNMT3A | chr2:25473227-25473238 | CTGACTCAACA | 0.836 | + | TSS, R, PF | MAFK |
| DNMT3A | chr2:25524509-25524520 | CTGACTCAGCT | 0.869 | + | R, E | MAFK, NFE2L2, BACH1 |
| DNMT3A | chr2:25524530-25524541 | ATGACTAATCC | 0.816 | + | R, E | MAFK, NFE2L2, BACH1 |
| DNMT3A | chr2:25524581-25524592 | CTGACCCTGCA | 0.833 | + | R, E | MAFK, NFE2L2, BACH1 |
| DNMT3A | chr2:25565466-25565477 | CTGCCTCAGCA | 0.836 | + | TSS, CTCF | MAFK, MAFF |
| Gene | Coordinates (mm10 Genome) | Motif | Relative Score | Strand | Regulatory Element * | TFs |
|---|---|---|---|---|---|---|
| Hdac2 | chr10:37001063-37001074 | ATGAGTCAGCA | 0.92 | - | dELS | Mafk |
| Hdac2 | chr10:37000993-37001004 | ATGATTGGGCA | 0.82 | - | dELS | Mafk |
| Hdac3 | chr18:37949598-37949609 | ATGACTCAGCT | 0.93 | + | None | Mafk |
| Dnmt1 | chr9:20943516-20943527 | CTGCCACAGCA | 0.82 | + | None | Maff |
| Dnmt1 | chr9:20946724-20946735 | ATGACTCAGCA | 1.00 | + | None | Mafk, Mafg |
| Dnmt1 | chr9:20952657-20952668 | ATGCCTCGGCA | 0.83 | + | PLS | Mafk, Bach1 |
| Dnmt1 | chr9:20953035-20953046 | GTGGCTCGGCA | 0.83 | + | PLS | Nfe2l2, Mafk |
| Dnmt1 | chr9:20961672-20961683 | GTGACTCAGTC | 0.83 | - | dELS | Maff, Mafk, Bach1 |
| Dnmt3a | chr12:3811310-3811321 | TTGACTCAGCG | 0.86 | + | None | Maff |
| Dnmt3a | chr12:3846390-3846401 | TTGACTCAGCA | 0.92 | - | None | Mafk |
| Dnmt3a | chr12:3846370-3846381 | ATGACTAACCA | 0.87 | - | None | Mafk |
| Dnmt3a | chr12:3846273-3846284 | ATGGCTTTGCA | 0.81 | - | None | Mafk |
| Dnmt3a | chr12:3846068-3846079 | GTGACCAATCA | 0.82 | - | None | Mafk |
| Dnmt3a | chr12:3854616-3854627 | ATGAGATTGCA | 0.82 | - | dELS | Bach1 |
| Dnmt3a | chr12:3856263-3856274 | ATAACCCAGCA | 0.85 | + | dELS | Mafk, Bach1 |
| Dnmt3a | chr12:3890847-3890858 | CTGTCTCAGCA | 0.84 | - | None | Makf |
| Dnmt3b | chr2:153671696-153671707 | ATGAACCAGCA | 0.85 | + | None | Mafk |
| Gene | Coordinates (hg19 Genome) | Motif | Relative Score | Strand | Regulatory. Element | TFs |
|---|---|---|---|---|---|---|
| DGCR8 | chr22:20078164-20078175 | ATGACTCAGTG | 0.837 | + | T | NFE2L2 |
| DGCR8 | chr22:20078089-20078100 | CTGAAAAAGCA | 0.801 | - | T | NFE2L2 |
| DICER1 | chr14:95568484-95568495 | CTTACTCTGCA | 0.801 | - | R, T | MAFF, MAFK |
| DICER1 | chr14:95574241-95574252 | CTGATTCAGCA | 0.879 | - | R, T | MAFK, MAFF |
| DICER1 | chr14:95599297-95599308 | ATGACTAAACT | 0.802 | + | R, T | MAFK |
| DICER1 | chr14:95606901-95606912 | GTCATTAAGCA | 0.81 | + | R, T, E | MAFK |
| DICER1 | chr14:95606922-95606933 | GTAATTTAGCA | 0.804 | + | R, T, E | MAFK |
| DICER1 | chr14:95606976-95606987 | GTGATTCATCA | 0.829 | - | R, T, E | MAFK |
| DROSHA | chr5:31470983-31470994 | CTGACTCAGCA | 0.941 | + | T, E, WE | MAFF, BACH1, MAFK |
| DROSHA | chr5:31531143-31531154 | ATGACTCAGTG | 0.837 | - | TSS | MAFK, MAFF |
| DROSHA | chr5:31532503-31532514 | GTGACTCCGCG | 0.841 | + | TSS | MAFK |
| DROSHA | chr5:31537176-31537187 | AAGACTCAGCA | 0.895 | + | T | MAFK |
| DROSHA | chr5:31537251-31537262 | GTGAGTAGGC | 0.801 | + | T | MAFK |
| DROSHA | chr5:31470983-31470994 | CTGACTCAGCA | 0.941 | + | T, E, WE | MAFF, BACH1, MAFK |
| TARBP2 | chr12:53892564-53892575 | ATGCCACAGCT | 0.804 | + | TSS | BACH1 |
| TARBP2 | chr12:53892583-53892594 | ATGCCACAGCT | 0.804 | + | TSS, E | BACH1 |
| Gene | Coordinates (mm10 Genome) | Motif | Relative Score | Strand | Regulatory Element | TFs |
|---|---|---|---|---|---|---|
| Dicer1 | chr12:104699200-104699211 | CTGAGTCAGCA | 0.87 | + | None | Maff, Mafk |
| Dicer1 | chr12:104709040-104709051 | CTGGCTCAGCA | 0.836 | + | None | Maff, Mafk |
| Dicer1 | chr12:104709107-104709118 | ATGAGTCACCA | 0.82 | - | None | Mafk |
| Dicer1 | chr12:104728875-104728886 | CTGGCTCAGCA | 0.84 | + | None | Maff, Mafk |
| Drosha | chr15:12838880-12838891 | GTGACTCTGCA | 0.94 | + | None | Nfe2l2 |
| Drosha | chr15:12848397-12848408 | GTGACTCAGGA | 0.89 | - | dELS | Mafk |
| Drosha | chr15:12858197-12858208 | CTTACTCTGCA | 0.80 | - | None | Mafk |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-Llanes, I.; Shin, C.H.; Jiménez-Villegas, J.; Gorospe, M.; Lastres-Becker, I. The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis. Antioxidants 2023, 12, 641. https://doi.org/10.3390/antiox12030641
Silva-Llanes I, Shin CH, Jiménez-Villegas J, Gorospe M, Lastres-Becker I. The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis. Antioxidants. 2023; 12(3):641. https://doi.org/10.3390/antiox12030641
Chicago/Turabian StyleSilva-Llanes, Ignacio, Chang Hoon Shin, José Jiménez-Villegas, Myriam Gorospe, and Isabel Lastres-Becker. 2023. "The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis" Antioxidants 12, no. 3: 641. https://doi.org/10.3390/antiox12030641