
Protective Role of Short-Chain Fatty Acids against Ang- II-Induced Mitochondrial Dysfunction in Brain Endothelial Cells: A Potential Role of Heme Oxygenase 2
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Endothelial Cell Culture
2.3. Heme Oxygenase Activity Assay
2.4. Protein Expression
2.5. Measurement of Ca2+ Uptake by Mitochondria
2.6. Measurement of Cytosolic Ca2+
2.7. Measurement of Mitochondrial ROS Production
2.8. Measurement of Cellular Hydrogen Peroxide
2.9. Quantification of Nitric Oxide
2.10. Quantitative Real Time PCR
2.11. Bioenergetics by Seahorse
2.12. Statistical Analysis
3. Results
3.1. SCFAs Reverse Ang-II-Induced Downregulation of HO-2
3.2. SCFAs Improve Ang-II-Induced Endothelial Dysfunction by Regulating HO-2
3.3. SCFAs Reduce Ang-II-Induced Endothelial Inflammation by Regulating HO-2
3.4. The SCFAs/HO-2 Axis Regulates Calcium Homeostasis in Mitochondria from Cerebral ECs
3.5. SCFAs Normalized Mitochondrial Membrane Potential by Mediating HO-2 following Ang-II Treatment
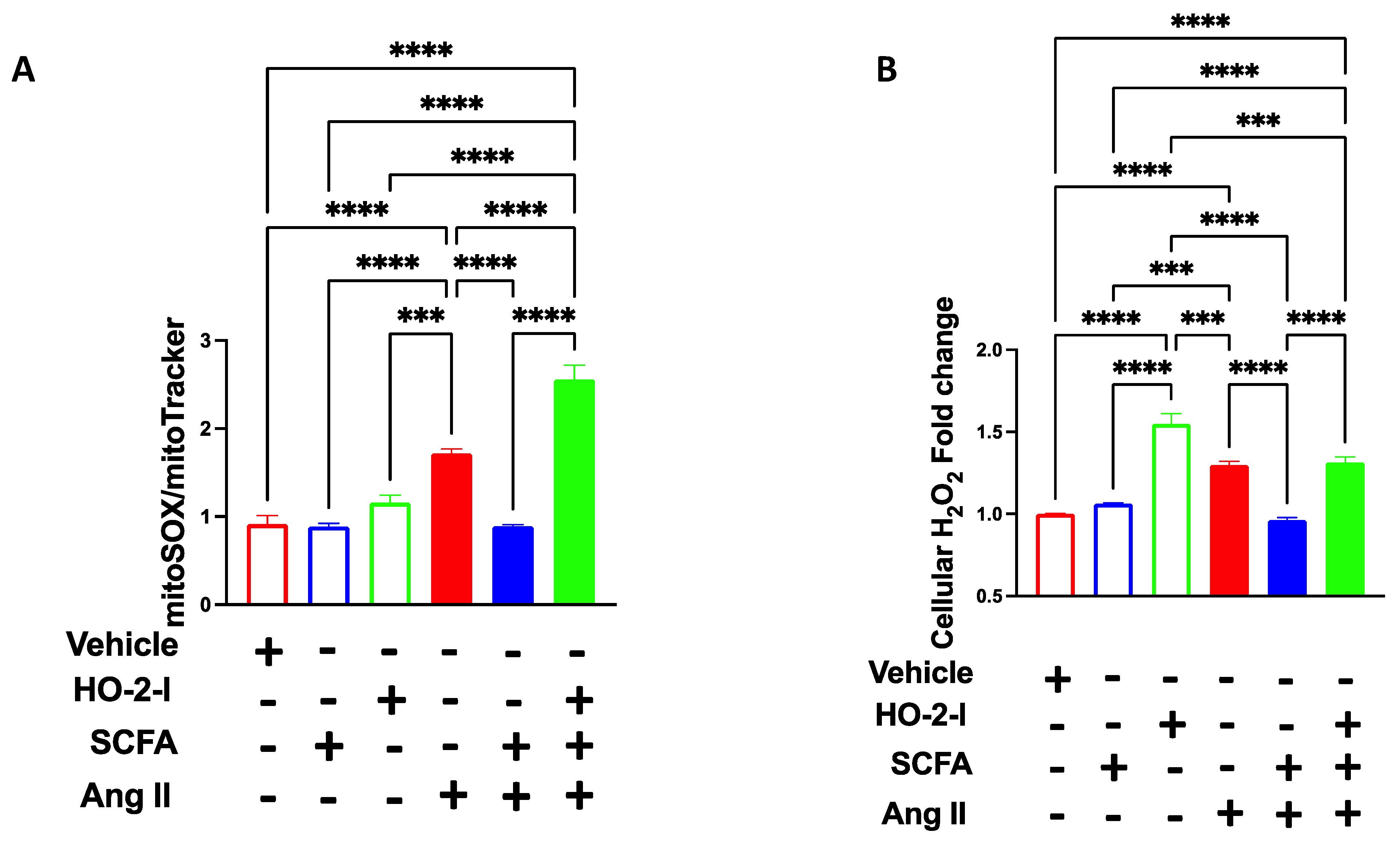
3.6. The SCFAs/HO-2 Axis Regulates Mitochondrial ROS, H2O2 and Mitochondrial Function
3.7. SCFAs Rescued Ang-II-Induced Mitochondrial Respiration Damage by Mediating HO-2
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carvalho, C.; Correia, S.C.; Santos, R.X.; Cardoso, S.; Moreira, P.I.; Clark, T.A.; Zhu, X.; Smith, M.A.; Perry, G. Role of mitochondrial-mediated signaling pathways in Alzheimer disease and hypoxia. J. Bioenerg. Biomembr. 2009, 41, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busija, D.W.; Katakam, P.V. Mitochondrial mechanisms in cerebral vascular control: Shared signaling pathways with preconditioning. J. Vasc. Res. 2014, 51, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Campistrous, A.; Hao, L.; Xiang, W.; Ton, D.; Semchuk, P.; Sander, J.; Ellison, M.J.; Fernandez-Patron, C. Mitochondrial dysfunction in the hypertensive rat brain: Respiratory complexes exhibit assembly defects in hypertension. Hypertension 2008, 51, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letra, L.; Sena, C. Cerebrovascular disease: Consequences of obesity-induced endothelial dysfunction. Obes. Brain Funct. 2017, 19, 163–189. [Google Scholar]
- Hu, S.; Kuwabara, R.; de Haan, B.J.; Smink, A.M.; de Vos, P. Acetate and butyrate improve β-cell metabolism and mitochondrial respiration under oxidative stress. Int. J. Mol. Sci. 2020, 21, 1542. [Google Scholar] [CrossRef] [Green Version]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Besten, G.; Van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhang, P.; Shen, L.; Niu, L.; Tan, Y.; Chen, L.; Zhao, Y.; Bai, L.; Hao, X.; Li, X. Short-chain fatty acids and their association with signalling pathways in inflammation, glucose and lipid metabolism. Int. J. Mol. Sci. 2020, 21, 6356. [Google Scholar] [CrossRef] [PubMed]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyles, L.; Snelling, T.; Umlai, U.-K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome–host systems interactions: Protective effects of propionate upon the blood–brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvigneau, J.C.; Esterbauer, H.; Kozlov, A.V. Role of heme oxygenase as a modulator of heme-mediated pathways. Antioxidants 2019, 8, 475. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Pal, C.; Dey, S.; Goyal, M.; Alam, A.; Iqbal, M.S.; Dutta, S.; Sarkar, S.; Kumar, R.; Maity, P. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J. Biol. Chem. 2011, 286, 39387–39402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Schragenheim, J.; Cao, J.; Falck, J.R.; Abraham, N.G.; Bellner, L. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat. 2016, 125, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. A review on hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxidative Med. Cell. Longev. 2014, 2014, 604981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waltz, P.K.; Kautza, B.; Luciano, J.; Dyer, M.; Stolz, D.B.; Loughran, P.; Neal, M.D.; Sperry, J.L.; Rosengart, M.R.; Zuckerbraun, B.S. Heme oxygenase-2 localizes to mitochondria and regulates hypoxic responses in hepatocytes. Oxidative Med. Cell. Longev. 2018, 2018, 2021645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait-Aissa, K.; Kadlec, A.O.; Hockenberry, J.; Gutterman, D.D.; Beyer, A.M. Telomerase reverse transcriptase protects against angiotensin II-induced microvascular endothelial dysfunction. Am. J. Physiol.-Heart Circ. Physiol. 2018, 314, H1053–H1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.J.; Koval, O.M.; Sebag, S.C.; Ait-Aissa, K.; Allen, B.G.; Spitz, D.R.; Grumbach, I.M. Inhibition of CaMKII in mitochondria preserves endothelial barrier function after irradiation. Free Radic. Biol. Med. 2020, 146, 287–298. [Google Scholar] [CrossRef]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koval, O.M.; Nguyen, E.K.; Santhana, V.; Fidler, T.P.; Sebag, S.C.; Rasmussen, T.P.; Mittauer, D.J.; Strack, S.; Goswami, P.C.; Abel, E.D. Loss of MCU prevents mitochondrial fusion in G1-S phase and blocks cell cycle progression and proliferation. Sci. Signal. 2019, 12, eaav1439. [Google Scholar] [CrossRef]
- Raut, G.K.; Chakrabarti, M.; Pamarthy, D.; Bhadra, M.P. Glucose starvation-induced oxidative stress causes mitochondrial dysfunction and apoptosis via Prohibitin 1 upregulation in human breast cancer cells. Free Radic. Biol. Med. 2019, 145, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Pulliam, D.A.; Liu, Y.; Hamilton, R.T.; Jernigan, A.L.; Bhattacharya, A.; Sloane, L.B.; Qi, W.; Chaudhuri, A.; Buffenstein, R. Reduced mitochondrial ROS, enhanced antioxidant defense, and distinct age-related changes in oxidative damage in muscles of long-lived Peromyscus leucopus. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2013, 304, R343–R355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkels, R.; Dachs, C.; Roesen, R.; Klaus, W. Simultaneous measurement of intracellular Ca2+ and nitric oxide: A new method. Cell Calcium 2000, 27, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, R.; Potunuru, U.R.; Nastasijević, B.; Joksić, G.; Dixit, M. Inhibition of vascular smooth muscle cell proliferation by Gentiana lutea root extracts. PLoS ONE 2013, 8, e61393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pober, J.S.; Sessa, W.C. Inflammation and the blood microvascular system. Cold Spring Harb. Perspect. Biol. 2015, 7, a016345. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Boehning, D.; Sedaghat, L.; Sedlak, T.W.; Snyder, S.H. Heme oxygenase-2 is activated by calcium-calmodulin. J. Biol. Chem. 2004, 279, 30927–30930. [Google Scholar] [CrossRef] [Green Version]
- Regan, C.W.L.; Parfenova, H. HO-2 provides endogenous protection against oxidative. Am. J. Physiol. Cell Physiol. 2006, 291, C897–C908. [Google Scholar]
- Ishizaka, N.; Griendling, K.K. Heme oxygenase-1 is regulated by angiotensin II in rat vascular smooth muscle cells. Hypertension 1997, 29, 790–795. [Google Scholar] [CrossRef]
- Yang, L.; Quan, S.; Nasjletti, A.; Laniado-Schwartzman, M.; Abraham, N.G. Heme oxygenase-1 gene expression modulates angiotensin II–induced increase in blood pressure. Hypertension 2004, 43, 1221–1226. [Google Scholar] [CrossRef] [Green Version]
- Calderón-Pérez, L.; Gosalbes, M.J.; Yuste, S.; Valls, R.M.; Pedret, A.; Llauradó, E.; Jimenez-Hernandez, N.; Artacho, A.; Pla-Pagà, L.; Companys, J. Gut metagenomic and short chain fatty acids signature in hypertension: A cross-sectional study. Sci. Rep. 2020, 10, 6436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Xu, H.; Tu, X.; Gao, Z. The Role of Short-Chain Fatty Acids of Gut Microbiota Origin in Hypertension. Front. Microbiol. 2021, 12, 730809. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.N.; Zerin, F.; Pandey, A.K.; Rahman, T.; Hasan, R. Gut Microbiota-Derived Short Chain Fatty Acids Stimulate Mesenteric Artery Vasodilation. 2021. Available online: https://ursa.mercer.edu/handle/10898/12572 (accessed on 12 May 2022).
- Pluznick, J.L. Microbial short-chain fatty acids and blood pressure regulation. Curr. Hypertens. Rep. 2017, 19, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Bosch, C.; Boorman, E.; Zunszain, P.A.; Mann, G.E. Short-chain fatty acids as modulators of redox signaling in health and disease. Redox Biol. 2021, 47, 102165. [Google Scholar] [CrossRef] [PubMed]
- Robles-Vera, I.; Toral, M.; de la Visitación, N.; Aguilera-Sánchez, N.; Redondo, J.M.; Duarte, J. Protective effects of short-chain fatty acids on endothelial dysfunction induced by angiotensin II. Front. Physiol. 2020, 11, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; van Esch, B.C.; Wagenaar, G.T.; Garssen, J.; Folkerts, G.; Henricks, P.A. Pro-and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur. J. Pharmacol. 2018, 831, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; van Esch, B.C.; Henricks, P.A.; Folkerts, G.; Garssen, J. The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide-or tumor necrosis factor α-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front. Pharmacol. 2018, 9, 533. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.J.; Zaloga, G.P.; Hoggatt, A.; Labarrere, C.; Faulk, W.P. Short-chain fatty acids modulate gene expression for vascular endothelial cell adhesion molecules. Nutrition 2005, 21, 740–748. [Google Scholar] [CrossRef]
- Clark, A.; Mach, N. The crosstalk between the gut microbiota and mitochondria during exercise. Front. Physiol. 2017, 8, 319. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl. Psychiatry 2018, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, P.; Wojtczak, A.B.; Geelen, M.J.; Kunz, W.; Wojtczak, L. On the mechanism of the so-called uncoupling effect of medium-and short-chain fatty acids. Biochim. Biophys. Acta (BBA)-Bioenerg. 1988, 936, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Kyrou, I.; Tan, B.K.; Dimitriadis, G.K.; Ramanjaneya, M.; Tripathi, G.; Patel, V.; James, S.; Kawan, M.; Chen, J. Short-chain fatty acid acetate stimulates adipogenesis and mitochondrial biogenesis via GPR43 in brown adipocytes. Endocrinology 2016, 157, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 2015, 64, 1744–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Chen, H.; Gao, Y.; An, N.; Li, X.; Pan, X.; Yang, X.; Tian, L.; Sun, J.; Xiong, X. Gut microbiota-derived short-chain fatty acids and hypertension: Mechanism and treatment. Biomed. Pharmacother. 2020, 130, 110503. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benakis, C.; Brea, D.; Caballero, S.; Faraco, G.; Moore, J.; Murphy, M.; Sita, G.; Racchumi, G.; Ling, L.; Pamer, E.G.; et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med. 2016, 22, 516–523. [Google Scholar] [CrossRef]
- Xiong, Z.; Peng, K.; Song, S.; Zhu, Y.; Gu, J.; Huang, C.; Li, X. Cerebral Intraparenchymal Hemorrhage Changes Patients’ Gut Bacteria Composition and Function. Front. Cell. Infect. Microbiol. 2022, 16, 829491. [Google Scholar] [CrossRef] [PubMed]
- Saji, N.; Murotani, K.; Hisada, T.; Tsuduki, T.; Sugimoto, T.; Kimura, A.; Niida, S.; Toba, K.; Sakurai, T. The Association between Cerebral Small Vessel Disease and the Gut Microbiome: A Cross-Sectional Analysis. J. Stroke Cerebrovasc. Dis. 2021, 30, 105568. [Google Scholar] [CrossRef]
- Shikata, F.; Shimada, K.; Sato, H.; Ikedo, T.; Kuwabara, A.; Furukawa, H.; Korai, M.; Kotoda, M.; Yokosuka, K.; Makino, H.; et al. Potential Influences of Gut Microbiota on the Formation of Intracranial Aneurysm. Hypertension 2019, 73, 491–496. [Google Scholar] [CrossRef]
- Tran, S.M.; Mohajeri, M.H. The Role of Gut Bacterial Metabolites in Brain Development, Aging and Disease. Nutrients 2021, 13, 732. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Meng, L.; Shen, L. Multiple roles of short-chain fatty acids in Alzheimer disease. Nutrition 2022, 93, 111499. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pan, J.; Chen, H.; Li, Y.; Amakye, W.K.; Liang, J.; Ma, B.; Chu, X.; Mao, L.; Zhang, Z. Fecal Short-Chain Fatty Acids Levels Were Not Associated With Autism Spectrum Disorders in Chinese Children: A Case-Control Study. Front. Neurosci. 2019, 13, 1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzdorf, J.; Tönges, L. Short-chain fatty acids in the context of Parkinson’s disease. Neural Regen. Res. 2021, 16, 2015–2016. [Google Scholar] [PubMed]
- Sadler, R.; Cramer, J.V.; Heindl, S.; Kostidis, S.; Betz, D.; Zuurbier, K.R.; Northoff, B.H.; Heijink, M.; Goldberg, M.P.; Plautz, E.J.; et al. Short-Chain Fatty Acids Improve Poststroke Recovery via Immunological Mechanisms. J. Neurosci. 2020, 40, 1162–1173. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kassan, M.; Kwon, Y.; Munkhsaikhan, U.; Sahyoun, A.M.; Ishrat, T.; Galán, M.; Gonzalez, A.A.; Abidi, A.H.; Kassan, A.; Ait-Aissa, K. Protective Role of Short-Chain Fatty Acids against Ang- II-Induced Mitochondrial Dysfunction in Brain Endothelial Cells: A Potential Role of Heme Oxygenase 2. Antioxidants 2023, 12, 160. https://doi.org/10.3390/antiox12010160
Kassan M, Kwon Y, Munkhsaikhan U, Sahyoun AM, Ishrat T, Galán M, Gonzalez AA, Abidi AH, Kassan A, Ait-Aissa K. Protective Role of Short-Chain Fatty Acids against Ang- II-Induced Mitochondrial Dysfunction in Brain Endothelial Cells: A Potential Role of Heme Oxygenase 2. Antioxidants. 2023; 12(1):160. https://doi.org/10.3390/antiox12010160
Chicago/Turabian StyleKassan, Modar, Youngin Kwon, Undral Munkhsaikhan, Amal M. Sahyoun, Tauheed Ishrat, María Galán, Alexis A. Gonzalez, Ammaar H. Abidi, Adam Kassan, and Karima Ait-Aissa. 2023. "Protective Role of Short-Chain Fatty Acids against Ang- II-Induced Mitochondrial Dysfunction in Brain Endothelial Cells: A Potential Role of Heme Oxygenase 2" Antioxidants 12, no. 1: 160. https://doi.org/10.3390/antiox12010160