
The Inflammatory Response in Human Keratinocytes Exposed to Cinnamaldehyde Is Regulated by Nrf2

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Invalidation of Nrf2
2.3. Cell Toxicity
2.4. Western Blot Analysis
2.5. Quantitative Reverse Transcription-Polymerase Chain Reaction
2.6. Cytokines Production Assessment
2.7. Targeted Liquid Chromatography–Mass Spectrometry Metabolites Analyses
2.8. Analysis of Oxygen Consumption and Extracellular Acidification Rates
2.9. Statistical Analyses
3. Results
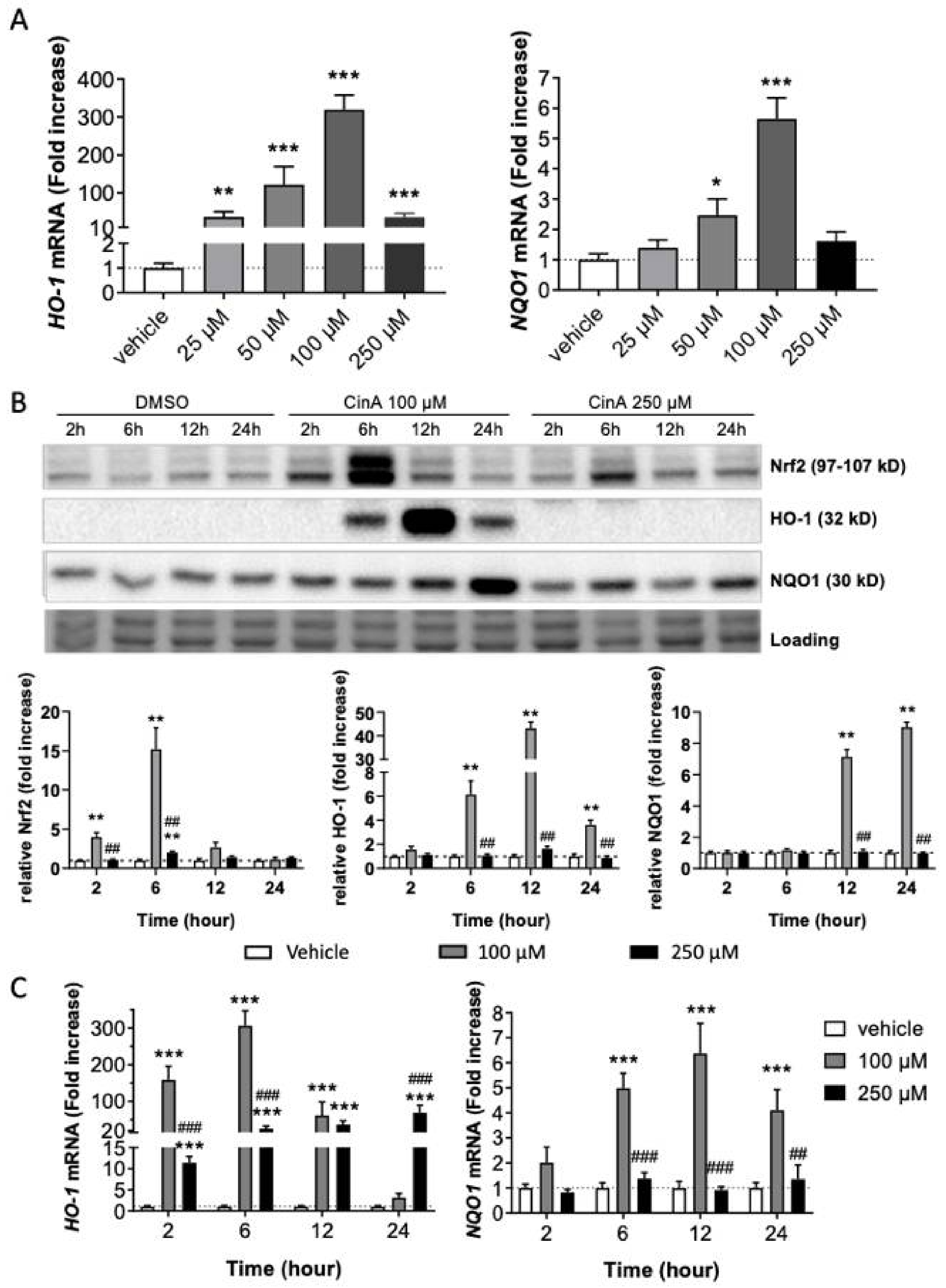
3.1. Nrf2 Pathway Activation in Response to CinA
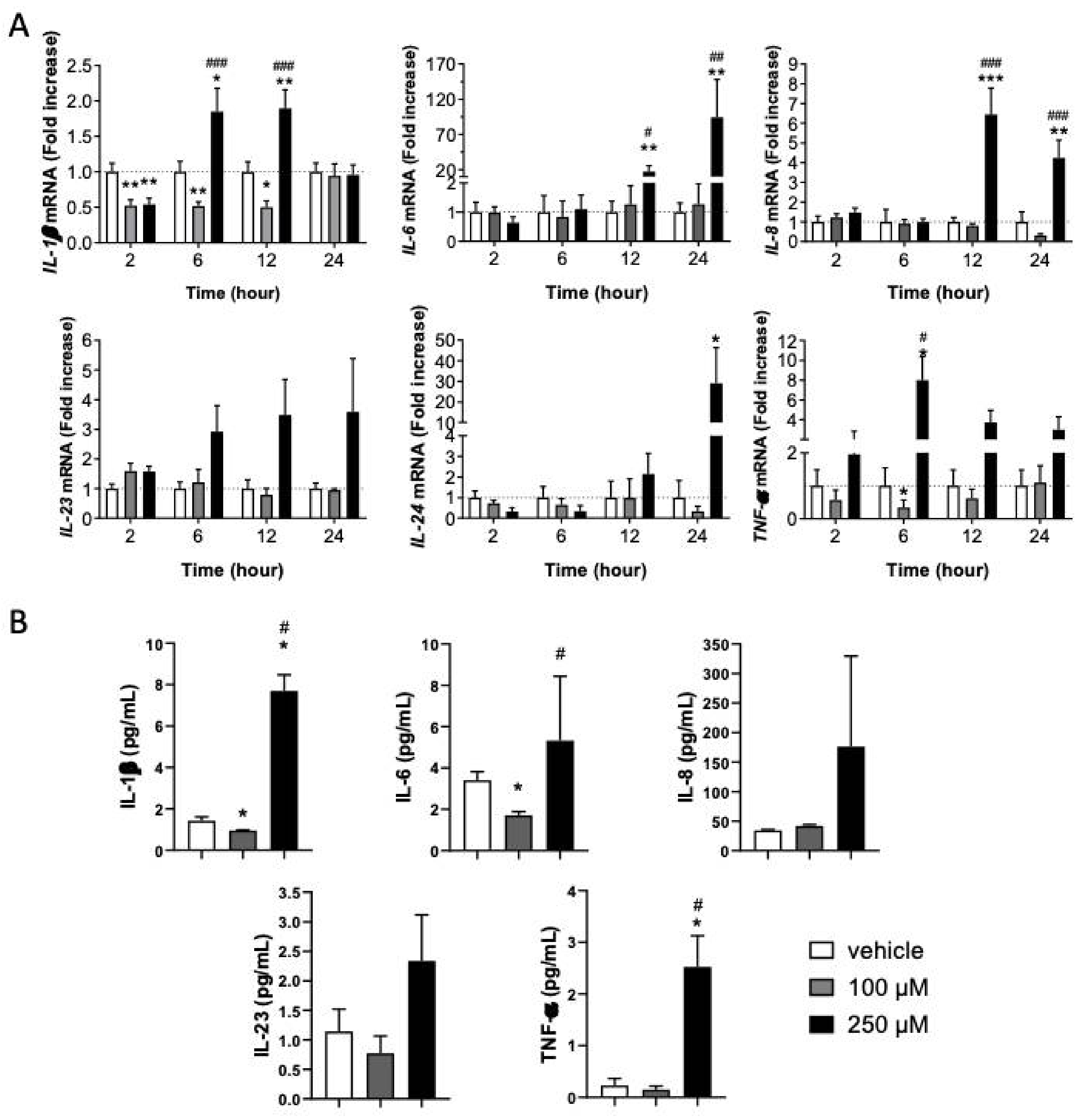
3.2. Inflammatory Response in KC Is Dependent on CinA Concentration
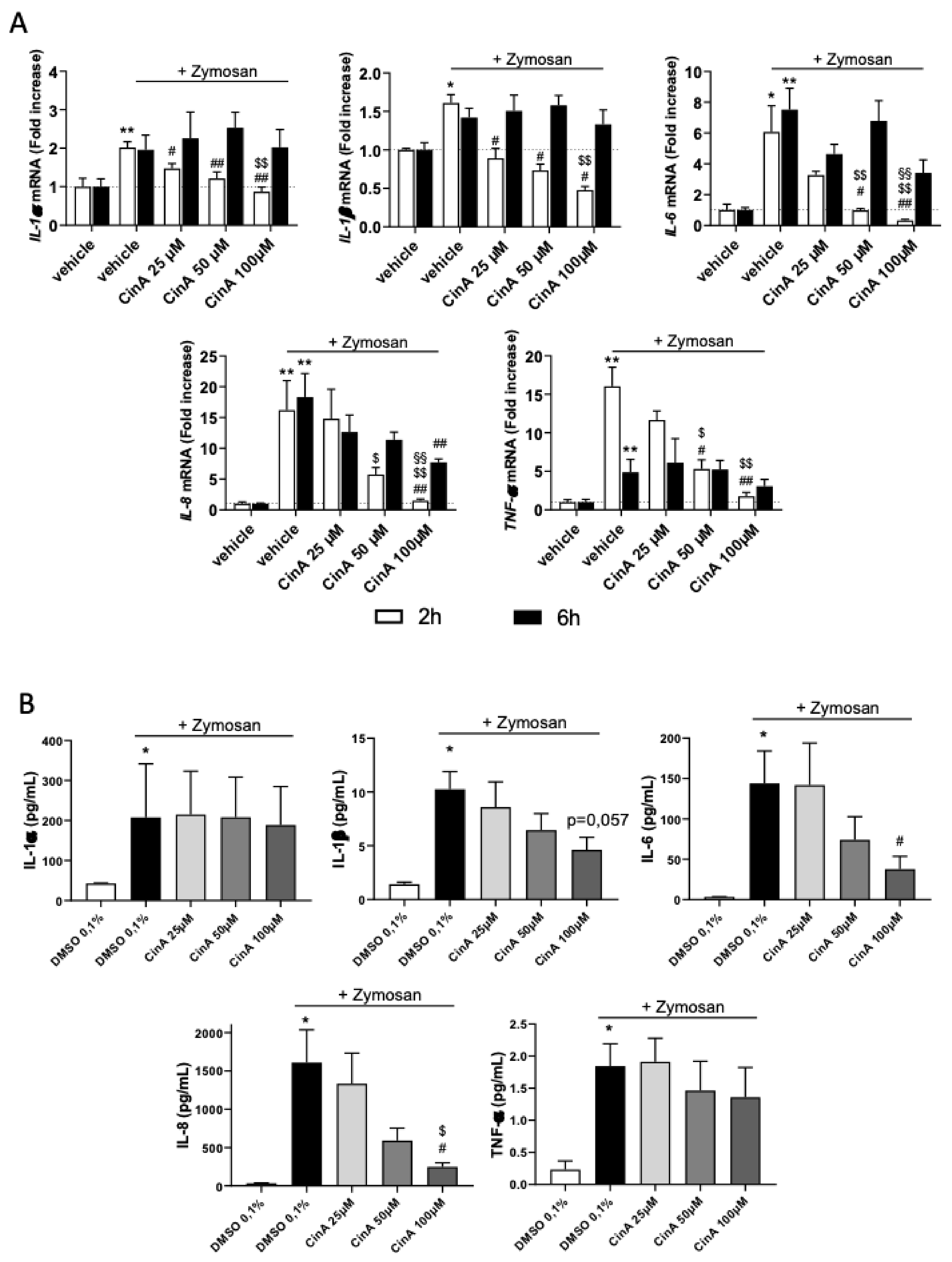
3.3. Concentration of CinA inducing Nrf2 Controls Zymosan-Induced KC Inflammation
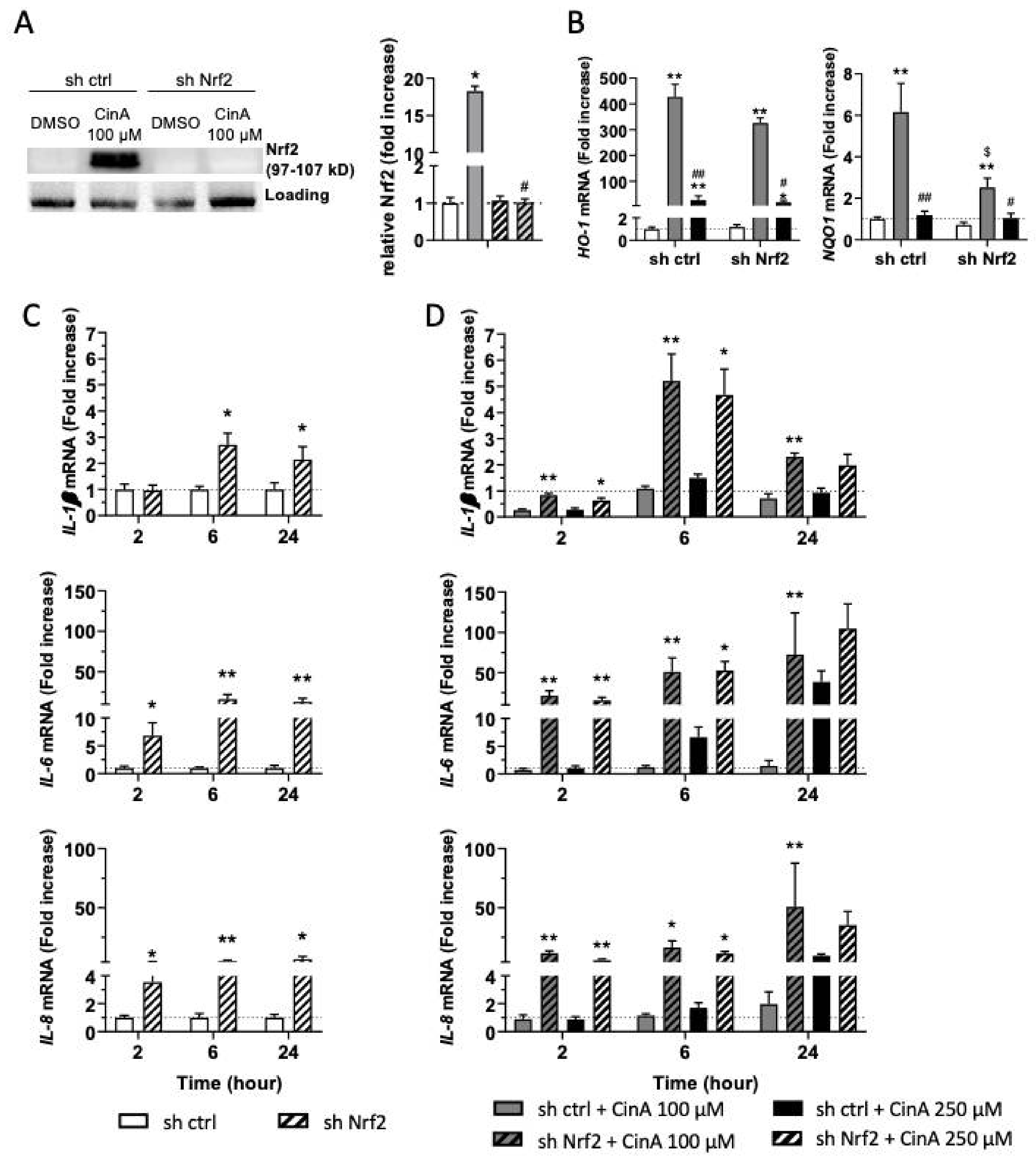
3.4. The Inflammatory Response in KC Induced by CinA Is Nrf2-Dependent
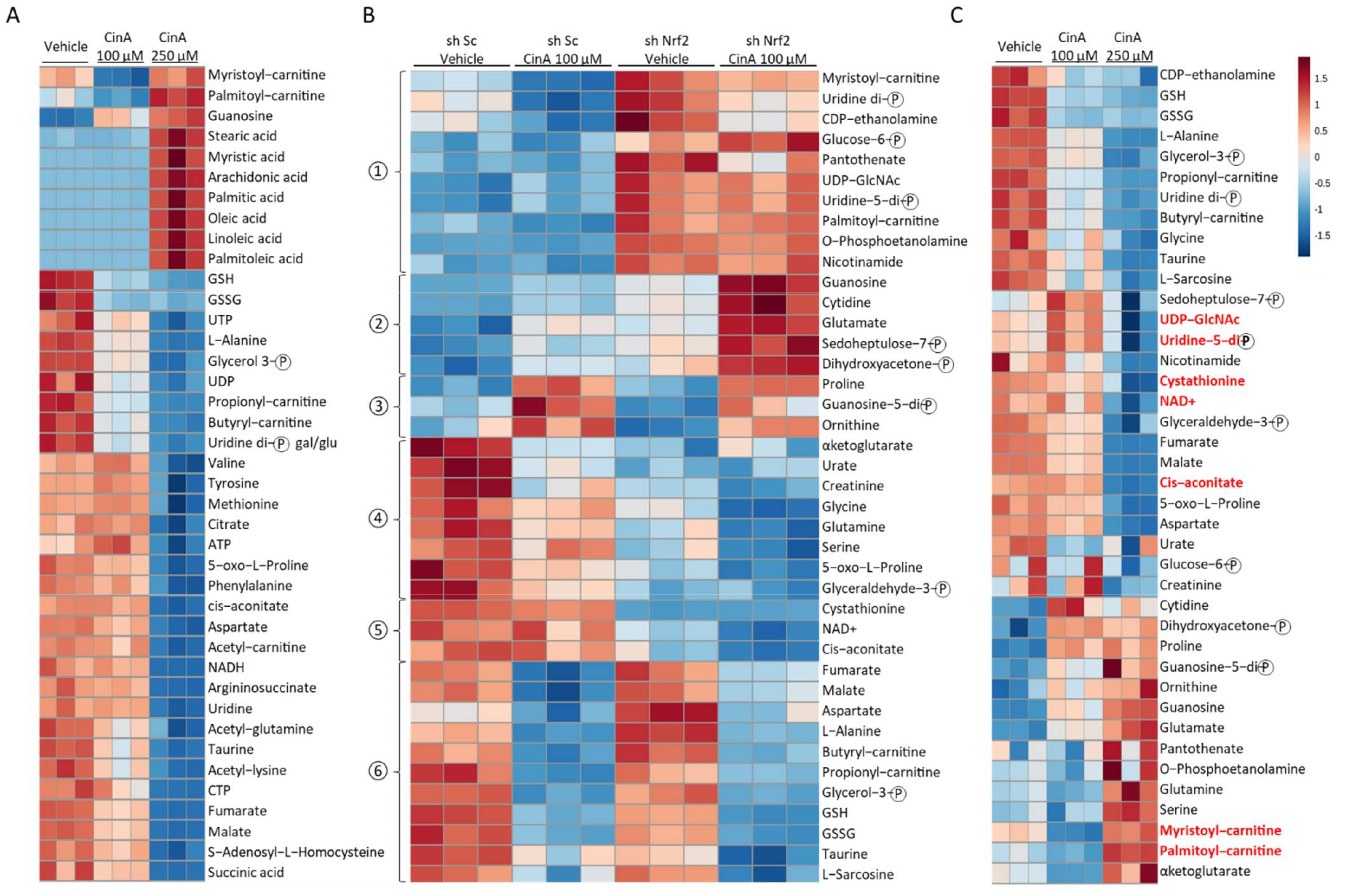
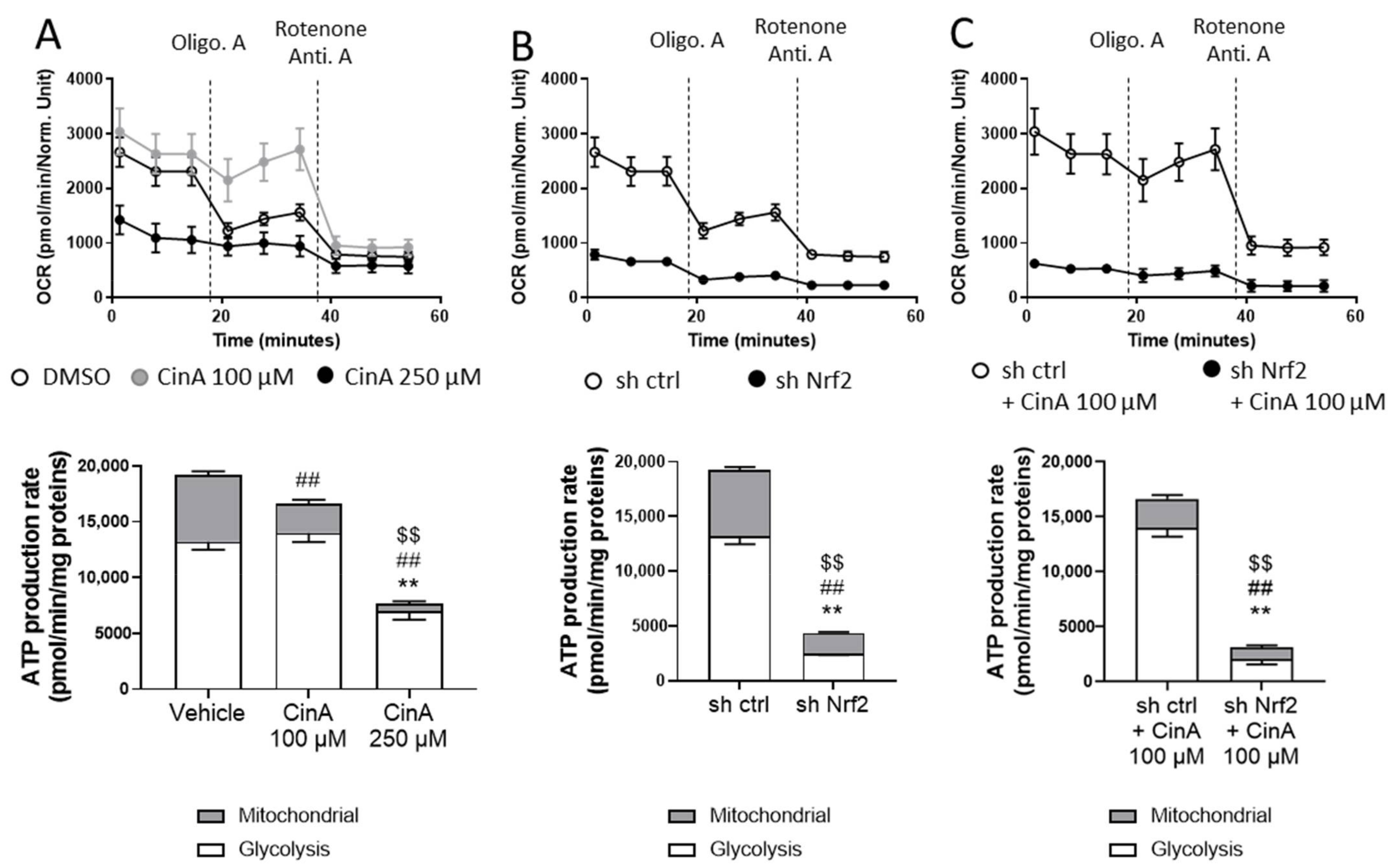
3.5. Concentration of CinA inducing Nrf2 Leads to Nrf2-Dependent Metabolic Reprogramming
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rawlings, A.V.; Harding, C.R. Moisturization and Skin Barrier Function. Dermatol. Ther. 2004, 17, 43–48. [Google Scholar] [CrossRef]
- Jiang, Y.; Tsoi, L.C.; Billi, A.C.; Ward, N.L.; Harms, P.W.; Zeng, C.; Maverakis, E.; Michelle Kahlenberg, J.; Gudjonsson, J.E. Cytokinocytes: The Diverse Contribution of Keratinocytes to Immune Responses in Skin. JCI Insight 2020, 5, e142067. [Google Scholar] [CrossRef]
- Silvestre, M.C.; Sato, M.N.; dos Reis, V.M.S. Innate Immunity and Effector and Regulatory Mechanisms Involved in Allergic Contact Dermatitis. Bras Derm. 2018, 93, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.H.; Igyártó, B.Z.; Gaspari, A.A. Early Immune Events in the Induction of Allergic Contact Dermatitis. Nat. Rev. Immunol. 2012, 12, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Esser, P.R.; Wölfle, U.; Dürr, C.; von Loewenich, F.D.; Schempp, C.M.; Freudenberg, M.A.; Jakob, T.; Martin, S.F. Contact Sensitizers Induce Skin Inflammation via ROS Production and Hyaluronic Acid Degradation. PLoS ONE 2012, 7, e41340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Gaide, O.; Pétrilli, V.; Martinon, F.; Contassot, E.; Roques, S.; Kummer, J.A.; Tschopp, J.; French, L.E. Activation of the IL-1β-Processing Inflammasome Is Involved in Contact Hypersensitivity. J. Investig. Dermatol. 2007, 127, 1956–1963. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.; Mraz, V.; Geisler, C.; Skov, L.; Bonefeld, C.M. The Role of Interleukin-1β in the Immune Response to Contact Allergens. Contact Dermat. 2021, 85, 387–397. [Google Scholar] [CrossRef]
- Cumberbatch, M.; Dearman, R.J.; Antonopoulos, C.; Groves, R.W.; Kimber, I. Interleukin (IL)-18 Induces Langerhans Cell Migration by a Tumour Necrosis Factor-Alpha- and IL-1beta-Dependent Mechanism. Immunology 2001, 102, 323–330. [Google Scholar] [CrossRef]
- Antonopoulos, C.; Cumberbatch, M.; Dearman, R.J.; Daniel, R.J.; Kimber, I.; Groves, R.W. Functional Caspase-1 Is Required for Langerhans Cell Migration and Optimal Contact Sensitization in Mice. J. Immunol. 2001, 166, 3672–3677. [Google Scholar] [CrossRef] [Green Version]
- Shornick, L.P.; De Togni, P.; Mariathasan, S.; Goellner, J.; Strauss-Schoenberger, J.; Karr, R.W.; Ferguson, T.A.; Chaplin, D.D. Mice Deficient in IL-1beta Manifest Impaired Contact Hypersensitivity to Trinitrochlorobenzone. J. Exp. Med. 1996, 183, 1427–1436. [Google Scholar] [CrossRef]
- Yasukawa, S.; Miyazaki, Y.; Yoshii, C.; Nakaya, M.; Ozaki, N.; Toda, S.; Kuroda, E.; Ishibashi, K.; Yasuda, T.; Natsuaki, Y.; et al. An ITAM-Syk-CARD9 Signalling Axis Triggers Contact Hypersensitivity by Stimulating IL-1 Production in Dendritic Cells. Nat. Commun. 2014, 5, 3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottrez, F.; Boitel, E.; Auriault, C.; Aeby, P.; Groux, H. Genes Specifically Modulated in Sensitized Skins Allow the Detection of Sensitizers in a Reconstructed Human Skin Model. Development of the SENS-IS Assay. Toxicol. In Vitro 2015, 29, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.S.; Wright, S.C.; Nicholson, W.V.; Morrison, K.D.; Prescott, A.R.; Ten Have, S.; Whitfield, P.D.; Lamond, A.I.; Brown, S.J. Functional and Proteomic Analysis of a Full Thickness Filaggrin-Deficient Skin Organoid Model. Wellcome Open Res. 2019, 4, 134. [Google Scholar] [CrossRef] [Green Version]
- Elias, M.S.; Long, H.A.; Newman, C.F.; Wilson, P.A.; West, A.; McGill, P.J.; Wu, K.C.; Donaldson, M.J.; Reynolds, N.J. Proteomic Analysis of Filaggrin Deficiency Identifies Molecular Signatures Characteristic of Atopic Eczema. J. Allergy Clin. Immunol. 2017, 140, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Ali, Z.; Gerbeix, C.; Hemon, P.; Esser, P.R.; Martin, S.F.; Pallardy, M.; Kerdine-Römer, S. Allergic Skin Inflammation Induced by Chemical Sensitizers Is Controlled by the Transcription Factor Nrf2. Toxicol. Sci. 2013, 134, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Van der Veen, J.W.; Gremmer, E.R.; Vermeulen, J.P.; van Loveren, H.; Ezendam, J. Induction of Skin Sensitization Is Augmented in Nrf2-Deficient Mice. Arch. Toxicol. 2013, 87, 763–766. [Google Scholar] [CrossRef]
- Ahn, S.-G.; Jin, Y.-H.; Yoon, J.-H.; Kim, S.-A. The Anticancer Mechanism of 2′-Hydroxycinnamaldehyde in Human Head and Neck Cancer Cells. Int. J. Oncol. 2015, 47, 1793–1800. [Google Scholar] [CrossRef] [Green Version]
- Chao, L.K.; Hua, K.-F.; Hsu, H.-Y.; Cheng, S.-S.; Lin, I.-F.; Chen, C.-J.; Chen, S.-T.; Chang, S.-T. Cinnamaldehyde Inhibits Pro-Inflammatory Cytokines Secretion from Monocytes/Macrophages through Suppression of Intracellular Signaling. Food Chem. Toxicol. 2008, 46, 220–231. [Google Scholar] [CrossRef]
- Clouet, E.; Bechara, R.; Raffalli, C.; Damiens, M.-H.; Groux, H.; Pallardy, M.; Ferret, P.-J.; Kerdine-Römer, S. The THP-1 Cell Toolbox: A New Concept Integrating the Key Events of Skin Sensitization. Arch. Toxicol. 2019, 93, 941–951. [Google Scholar] [CrossRef]
- Ade, N.; Leon, F.; Pallardy, M.; Peiffer, J.-L.; Kerdine-Romer, S.; Tissier, M.-H.; Bonnet, P.-A.; Fabre, I.; Ourlin, J.-C. HMOX1 and NQO1 Genes Are Upregulated in Response to Contact Sensitizers in Dendritic Cells and THP-1 Cell Line: Role of the Keap1/Nrf2 Pathway. Toxicol. Sci. 2009, 107, 451–460. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helou, D.G.; Martin, S.F.; Pallardy, M.; Chollet-Martin, S.; Kerdine-Römer, S. Nrf2 Involvement in Chemical-Induced Skin Innate Immunity. Front. Immunol. 2019, 10, 1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 Signaling Pathway: Pivotal Roles in Inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-F.; Kuo, H.-P.; Liu, M.; Chou, C.-K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.-T.; Huo, L.; Hsu, M.-C.; et al. KEAP1 E3 Ligase-Mediated Downregulation of NF-KappaB Signaling by Targeting IKKbeta. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a Regulator of Cell Metabolism and Inflammation in Cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 Regulatory Network Provides an Interface between Redox and Intermediary Metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of Inflammation by the Antioxidant Haem Oxygenase 1. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef]
- Vallion, R.; Divoux, J.; Glauzy, S.; Ronin, E.; Lombardi, Y.; Lubrano di Ricco, M.; Grégoire, S.; Nemazanyy, I.; Durand, A.; Fradin, D.; et al. Regulatory T Cell Stability and Migration Are Dependent on MTOR. J. Immunol. 2020, 205, 1799–1809. [Google Scholar] [CrossRef]
- Wculek, S.K.; Khouili, S.C.; Priego, E.; Heras-Murillo, I.; Sancho, D. Metabolic Control of Dendritic Cell Functions: Digesting Information. Front. Immunol. 2019, 10, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, E.H.; Verway, M.J.; Johnson, R.M.; Roy, D.G.; Steadman, M.; Hayes, S.; Williams, K.S.; Sheldon, R.D.; Samborska, B.; Kosinski, P.A.; et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8+ T Cells. Immunity 2019, 51, 856–870.e5. [Google Scholar] [CrossRef]
- Raffalli, C.; Clouet, E.; Kuresepi, S.; Damiens, M.-H.; Lepoittevin, J.-P.; Pallardy, M.; Ferret, P.-J.; Giménez-Arnau, E.; Kerdine-Römer, S. Editor’s Highlight: Fragrance Allergens Linalool and Limonene Allylic Hydroperoxides in Skin Allergy: Mechanisms of Action Focusing on Transcription Factor Nrf2. Toxicol. Sci. 2018, 161, 139–148. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter Metabolic Analysis Reveals a Close Link between Attenuated Mitochondrial Bioenergetic Function and Enhanced Glycolysis Dependency in Human Tumor Cells. Am. J. Physiol. Cell Physiol. 2007, 292, C125–C136. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, M.; Dütsch, S.; auf dem Keller, U.; Navid, F.; Schwarz, A.; Johnson, D.A.; Johnson, J.A.; Werner, S. Nrf2 Establishes a Glutathione-Mediated Gradient of UVB Cytoprotection in the Epidermis. Genes Dev. 2010, 24, 1045–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pareek, T.K.; Belkadi, A.; Kesavapany, S.; Zaremba, A.; Loh, S.L.; Bai, L.; Cohen, M.L.; Meyer, C.; Liby, K.T.; Miller, R.H.; et al. Triterpenoid Modulation of IL-17 and Nrf-2 Expression Ameliorates Neuroinflammation and Promotes Remyelination in Autoimmune Encephalomyelitis. Sci. Rep. 2011, 1, 201. [Google Scholar] [CrossRef] [PubMed]
- Maicas, N.; Ferrándiz, M.L.; Brines, R.; Ibáñez, L.; Cuadrado, A.; Koenders, M.I.; van den Berg, W.B.; Alcaraz, M.J. Deficiency of Nrf2 Accelerates the Effector Phase of Arthritis and Aggravates Joint Disease. Antioxid. Redox. Signal. 2011, 15, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, J.S.; Zaidi, S.F.; Shaharyar, S.; Refaat, A.; Usmanghani, K.; Saiki, I.; Sugiyama, T. Anti-Inflammatory Effect of Cinnamaldehyde in Helicobacter Pylori Induced Gastric Inflammation. Biol. Pharm. Bull. 2015, 38, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.-X.; Zhong, S.; Meng, X.-B.; Zheng, N.-Y.; Zhang, P.; Wang, Y.; Qin, L.; Wang, X.-L. Cinnamaldehyde Inhibits Inflammation of Human Synoviocyte Cells Through Regulation of Jak/Stat Pathway and Ameliorates Collagen-Induced Arthritis in Rats. J. Pharm. Exp. 2020, 373, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Basketter, D.A.; Balikie, L.; Dearman, R.J.; Kimber, I.; Ryan, C.A.; Gerberick, G.F.; Harvey, P.; Evans, P.; White, I.R.; Rycroft, R.J. Use of the Local Lymph Node Assay for the Estimation of Relative Contact Allergenic Potency. Contact Dermat. 2000, 42, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Corsini, E.; Galbiati, V.; Mitjans, M.; Galli, C.L.; Marinovich, M. NCTC 2544 and IL-18 Production: A Tool for the Identification of Contact Allergens. Toxicol. In Vitro 2013, 27, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Uchi, H.; Yasumatsu, M.; Morino-Koga, S.; Mitoma, C.; Furue, M. Inhibition of Aryl Hydrocarbon Receptor Signaling and Induction of NRF2-Mediated Antioxidant Activity by Cinnamaldehyde in Human Keratinocytes. J. Derm. Sci. 2017, 85, 36–43. [Google Scholar] [CrossRef]
- Kim, B.H.; Lee, Y.G.; Lee, J.; Lee, J.Y.; Cho, J.Y. Regulatory Effect of Cinnamaldehyde on Monocyte/Macrophage-Mediated Inflammatory Responses. Mediat. Inflamm. 2010, 2010, 529359. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.F.; Muhammad, J.S.; Shahryar, S.; Usmanghani, K.; Gilani, A.-H.; Jafri, W.; Sugiyama, T. Anti-Inflammatory and Cytoprotective Effects of Selected Pakistani Medicinal Plants in Helicobacter Pylori-Infected Gastric Epithelial Cells. J. Ethnopharmacol. 2012, 141, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.; Vijayan, V.; Santoso, S.; Kietzmann, T.; Immenschuh, S. Inhibition and Genetic Deficiency of P38 MAPK Up-Regulates Heme Oxygenase-1 Gene Expression via Nrf2. J. Immunol. 2009, 182, 7048–7057. [Google Scholar] [CrossRef] [Green Version]
- Gaber, T.; Strehl, C.; Buttgereit, F. Metabolic Regulation of Inflammation. Nat. Rev. Rheumatol. 2017, 13, 267–279. [Google Scholar] [CrossRef]
- Cibrian, D.; de la Fuente, H.; Sánchez-Madrid, F. Metabolic Pathways That Control Skin Homeostasis and Inflammation. Trends Mol. Med. 2020, 26, 975–986. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T Cell Metabolism Drives Immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [Green Version]
- Wickersham, M.; Wachtel, S.; Wong Fok Lung, T.; Soong, G.; Jacquet, R.; Richardson, A.; Parker, D.; Prince, A. Metabolic Stress Drives Keratinocyte Defenses against Staphylococcus Aureus Infection. Cell Rep. 2017, 18, 2742–2751. [Google Scholar] [CrossRef]
- Jiang, J.; Emont, M.P.; Jun, H.; Qiao, X.; Liao, J.; Kim, D.-I.; Wu, J. Cinnamaldehyde Induces Fat Cell-Autonomous Thermogenesis and Metabolic Reprogramming. Metabolism 2017, 77, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, M.; Li, M.; Zhao, T.; Zhou, L. Cinnamaldehyde Inhibits the Growth of Phytophthora Capsici through Disturbing Metabolic Homoeostasis. PeerJ 2021, 9, e11339. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate Is an Anti-Inflammatory Metabolite That Activates Nrf2 via Alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.-H.; et al. Electrophilic Properties of Itaconate and Derivatives Regulate the IκBζ-ATF3 Inflammatory Axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Bambouskova, M.; Potuckova, L.; Paulenda, T.; Kerndl, M.; Mogilenko, D.A.; Lizotte, K.; Swain, A.; Hayes, S.; Sheldon, R.D.; Kim, H.; et al. Itaconate Confers Tolerance to Late NLRP3 Inflammasome Activation. Cell Rep. 2021, 34, 108756. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Peace, C.G.; O’Neill, L.A. The Role of Itaconate in Host Defense and Inflammation. J. Clin. Invest. 2022, 132, e148548. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Kim, H.-J.; Lee, M.; Jin, S.H.; Hong, S.H.; Ahn, S.; Kim, S.O.; Shin, D.W.; Lee, S.-T.; Noh, M. Cystathionine Metabolic Enzymes Play a Role in the Inflammation Resolution of Human Keratinocytes in Response to Sub-Cytotoxic Formaldehyde Exposure. Toxicol. Appl. Pharm. 2016, 310, 185–194. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallion, R.; Hardonnière, K.; Bouredji, A.; Damiens, M.-H.; Deloménie, C.; Pallardy, M.; Ferret, P.-J.; Kerdine-Römer, S. The Inflammatory Response in Human Keratinocytes Exposed to Cinnamaldehyde Is Regulated by Nrf2. Antioxidants 2022, 11, 575. https://doi.org/10.3390/antiox11030575
Vallion R, Hardonnière K, Bouredji A, Damiens M-H, Deloménie C, Pallardy M, Ferret P-J, Kerdine-Römer S. The Inflammatory Response in Human Keratinocytes Exposed to Cinnamaldehyde Is Regulated by Nrf2. Antioxidants. 2022; 11(3):575. https://doi.org/10.3390/antiox11030575
Chicago/Turabian StyleVallion, Romain, Kévin Hardonnière, Abderrahmane Bouredji, Marie-Hélène Damiens, Claudine Deloménie, Marc Pallardy, Pierre-Jacques Ferret, and Saadia Kerdine-Römer. 2022. "The Inflammatory Response in Human Keratinocytes Exposed to Cinnamaldehyde Is Regulated by Nrf2" Antioxidants 11, no. 3: 575. https://doi.org/10.3390/antiox11030575