
Pardaxin Activates Excessive Mitophagy and Mitochondria-Mediated Apoptosis in Human Ovarian Cancer by Inducing Reactive Oxygen Species

 , , and
, , and
Abstract
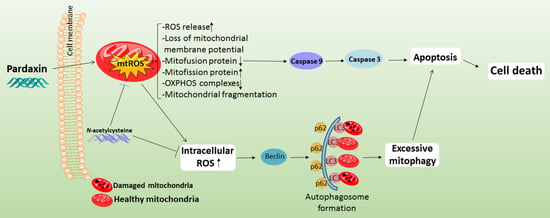
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Maintanance
2.3. MTT-Based Cell Viability Determination
2.4. Terminal Deoxynucleotidyl Transferase-Mediated dUTP (TUNEL) Assay
2.5. Staining of FITC-Annexin V/Propidium Iodide (PI)
2.6. Detection of Cellular ROS
2.7. Detection of Mitochondrial ROS
2.8. CellROX® Green Staining
2.9. JC-1 Staining for Measuring Mitochondrial Membran Potential
2.10. Measurement of Acidic Vesicular Organelles (AVOs)
2.11. Western Blot Analysis
2.12. Measurement of Mitochondrial Respiratory Functions
2.13. Mitochondrial Morphology
2.14. Statistical Analysis
3. Results
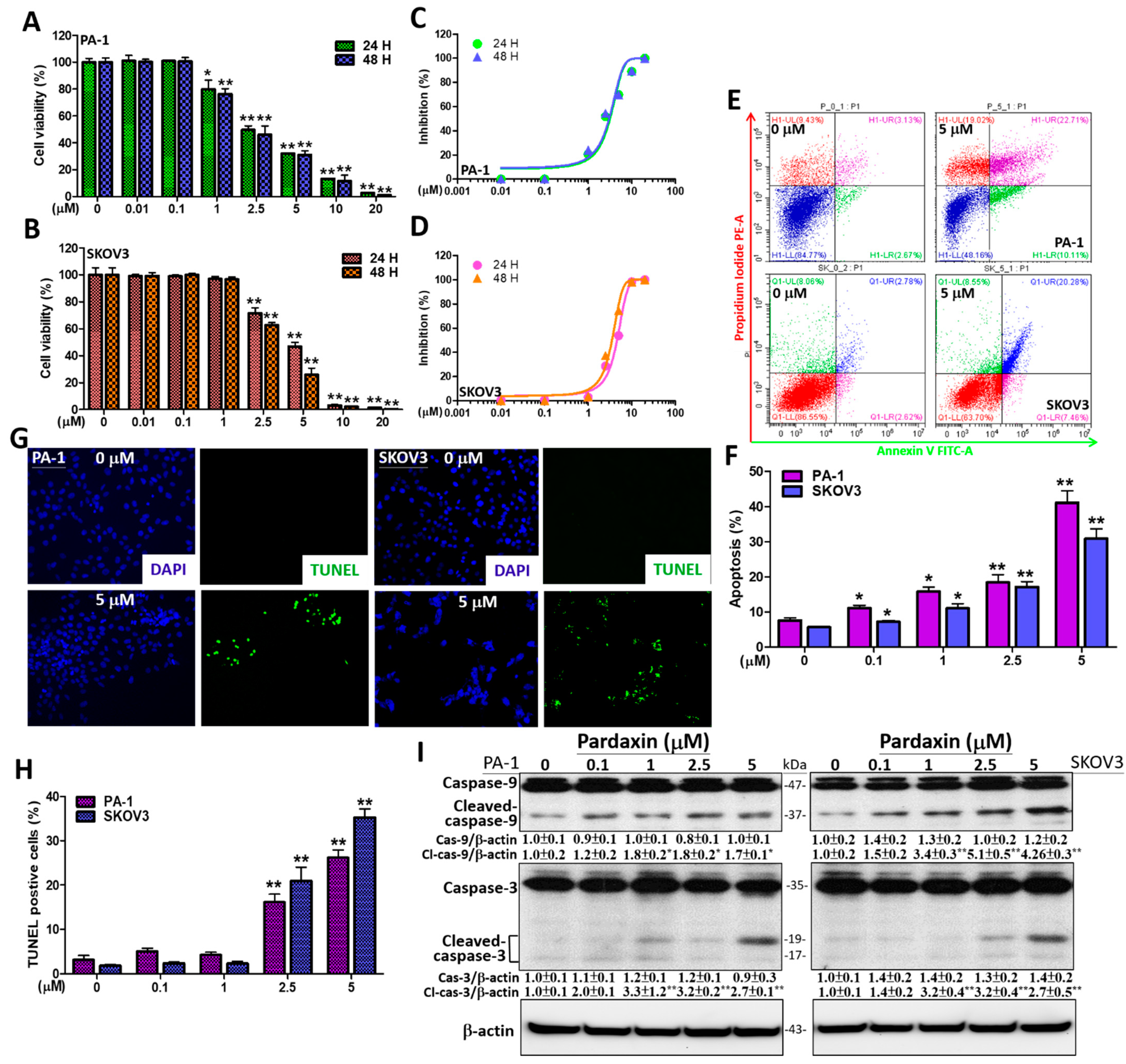
3.1. Pardaxin Significantly Induces Apoptosis via DNA Fragmentation and Caspase Activation
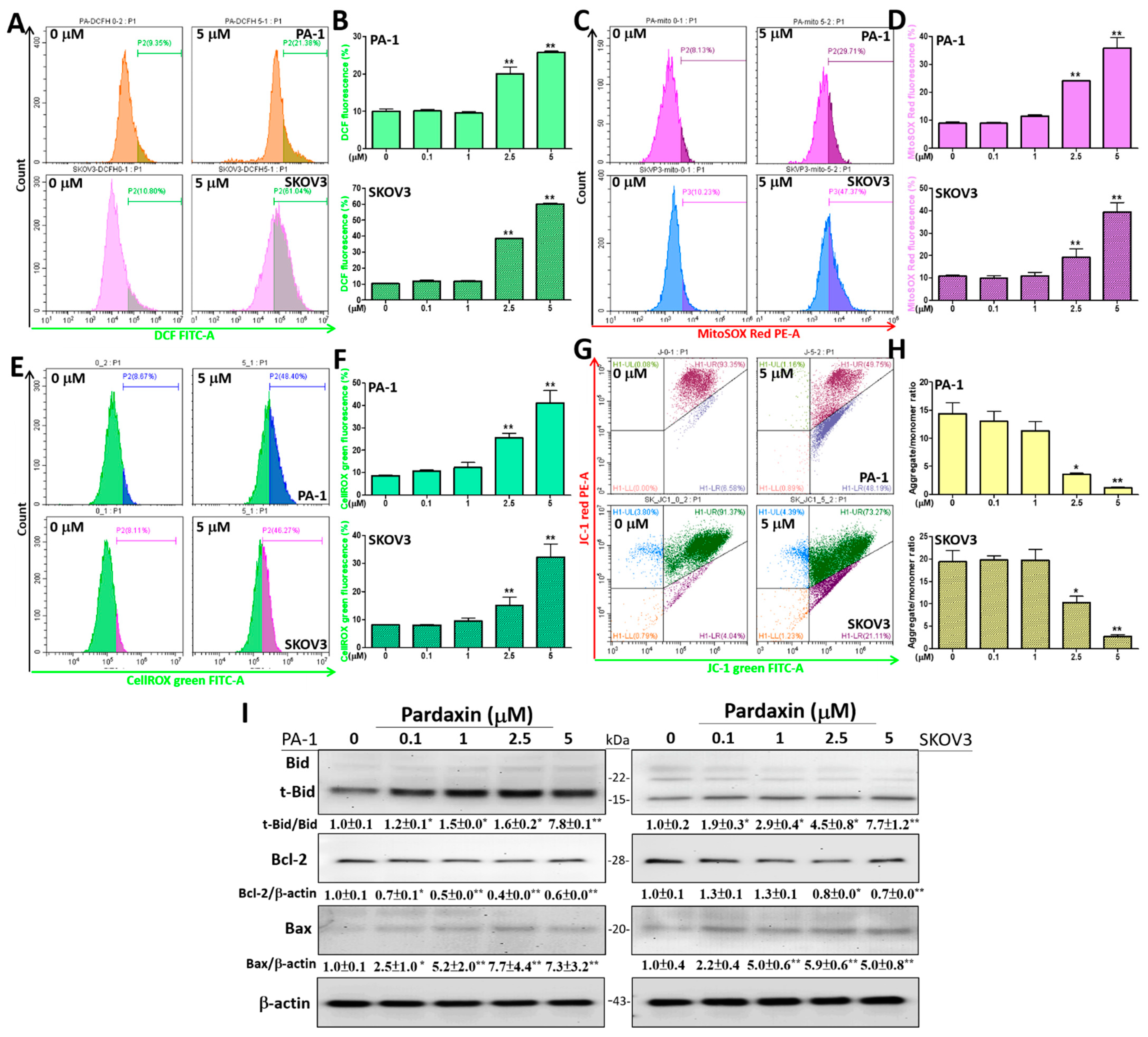
3.2. Intracellular and Mitochondrial ROS (mtROS) Levels Are Augmented, Whereas the Mitochondrial Membrane Potential (ΔΨ) Is Reduced with Altered Expression Profiles of Bcl-2 Family Proteins after Pardaxin Treatment
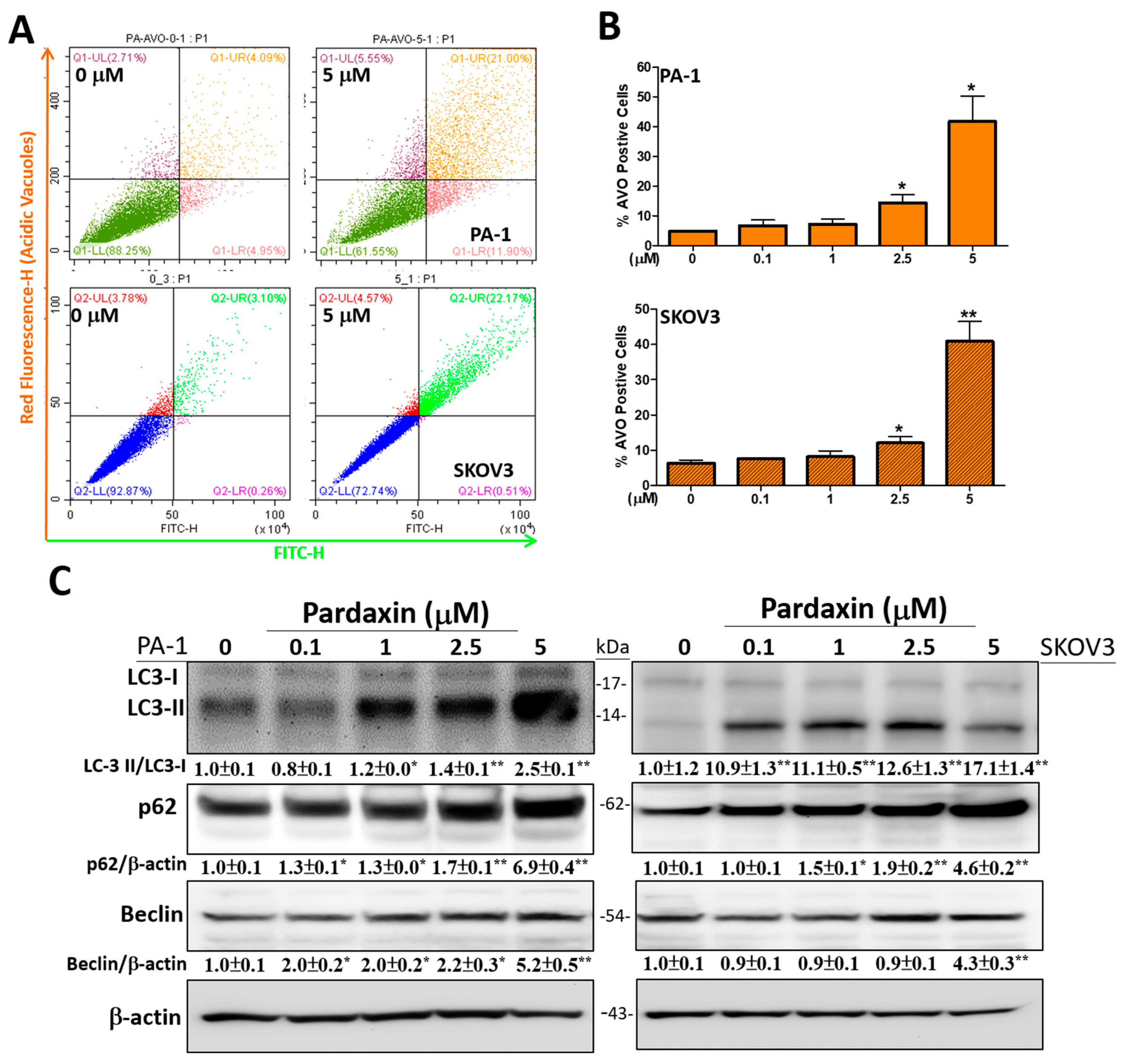
3.3. Autophagy-Related Acidic Vesicular Organelles Were Increasingly Detected with Upregulated Expression Levels of Autophagic Proteins after Pardaxin Treatment
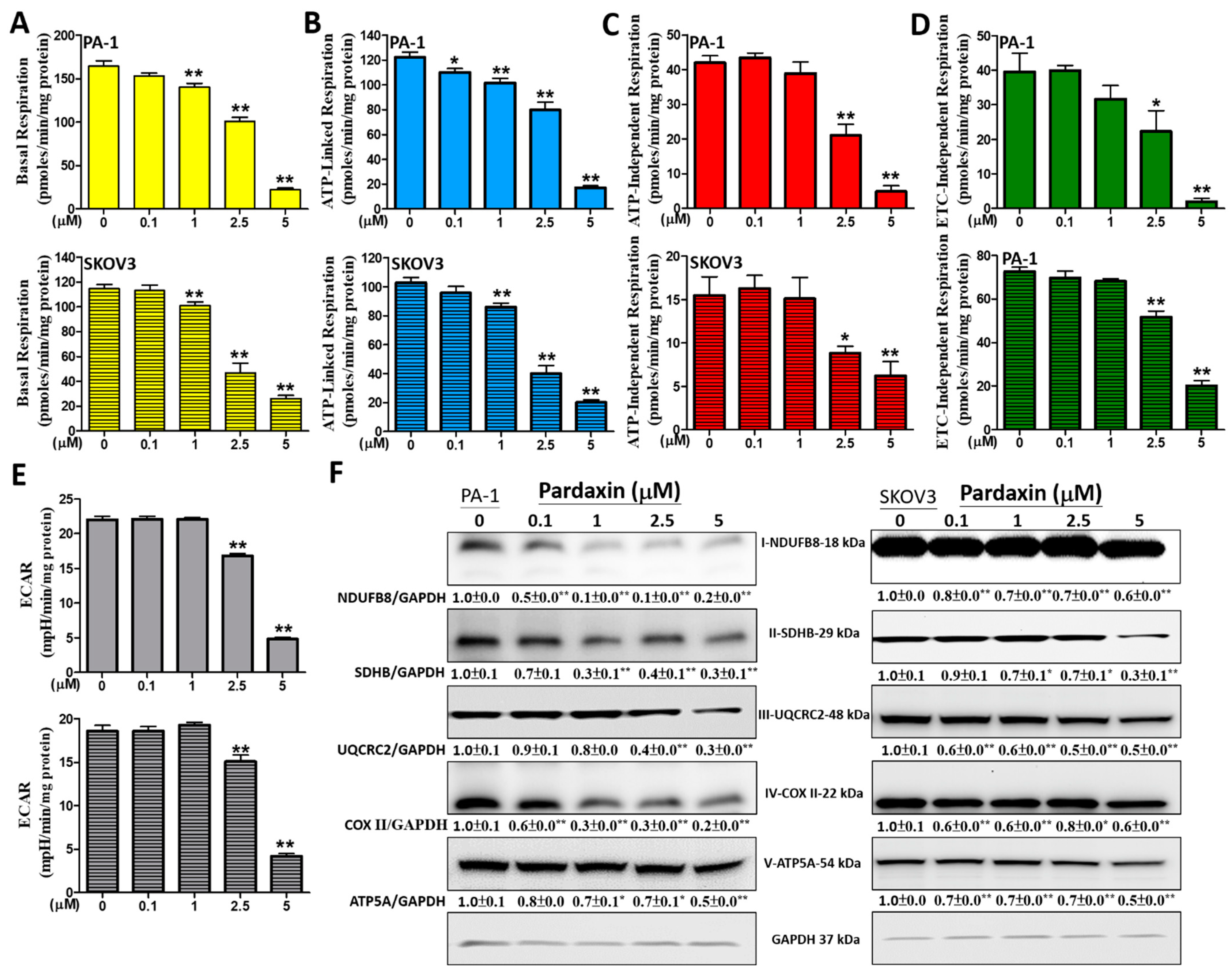
3.4. The Effects of Pardaxin on Oxygen Consumption Rate, Extracellular Acidification Rate, and ETC Complex I–V Proteins of Mitochondria in PA-1 and SKOV3 Cells
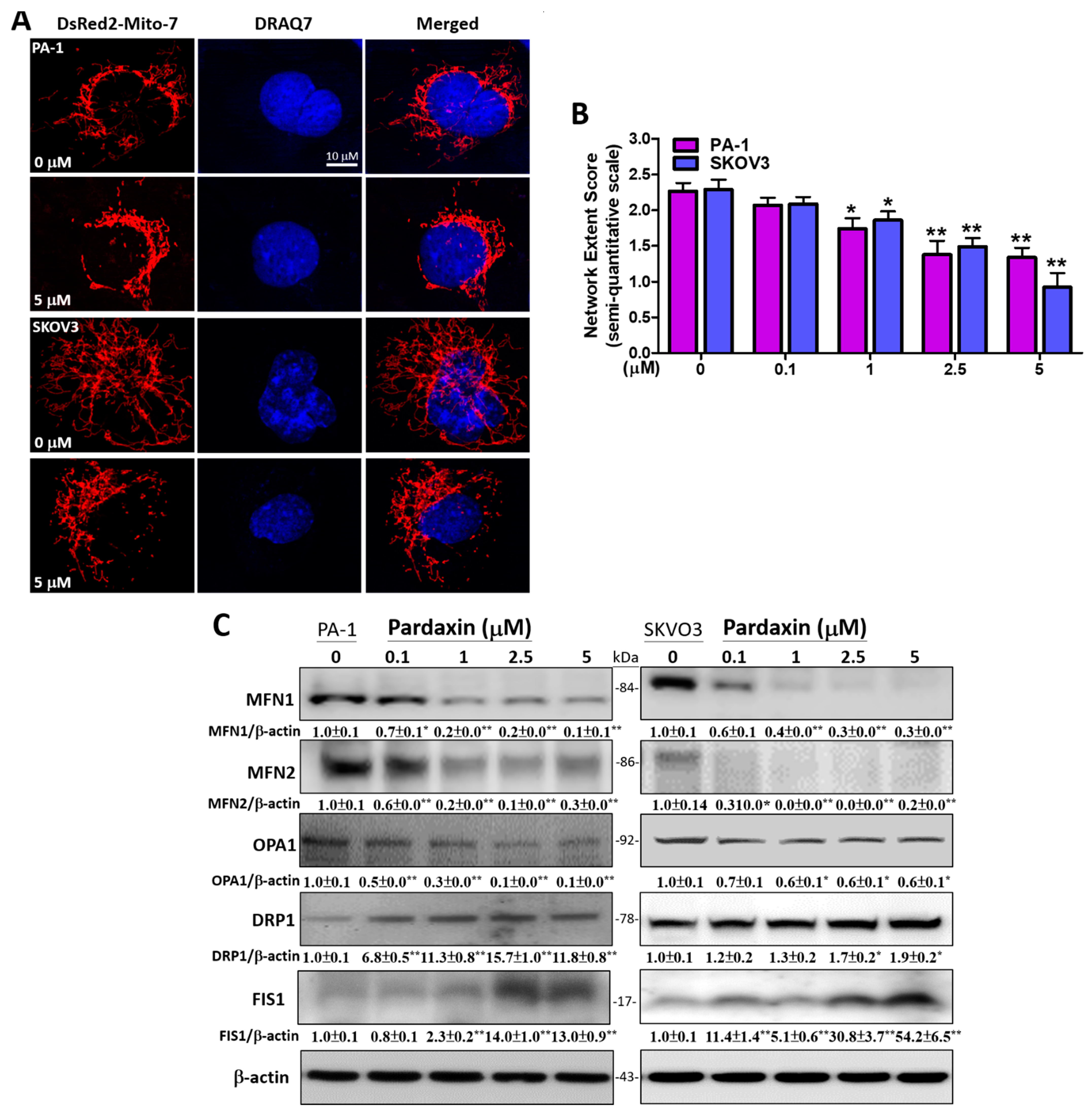
3.5. Pardaxin Alters Mitochondrial Morpholgy by Regulating the Expression Levels of Mitochondrial Fission and Fusion Proteins
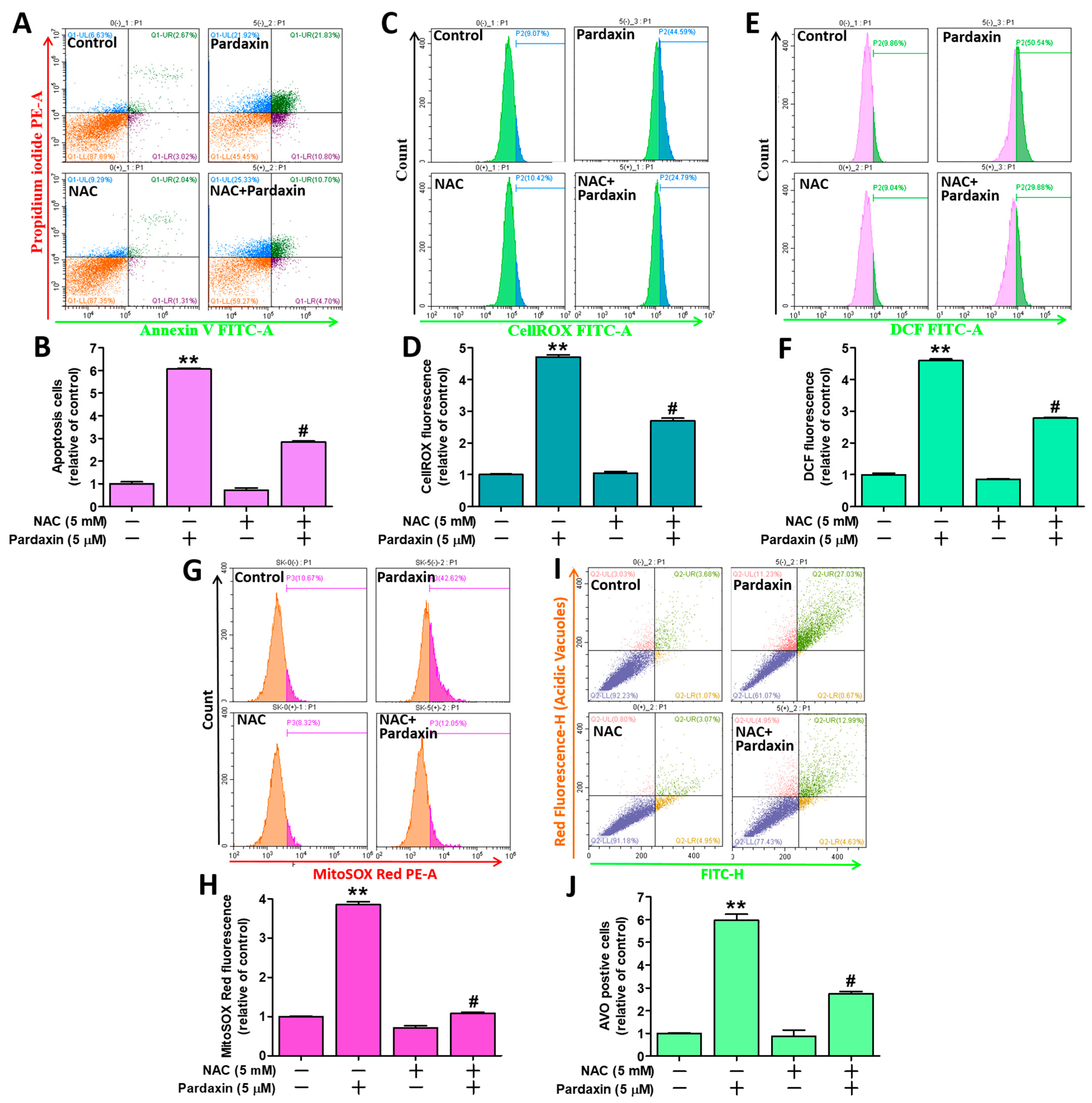
3.6. Pardaxin-Induced Apoptosis, ROS Generation, and Autophagy Is Reversed by Pretreatment with the Antixoidant N-acetylcysteine
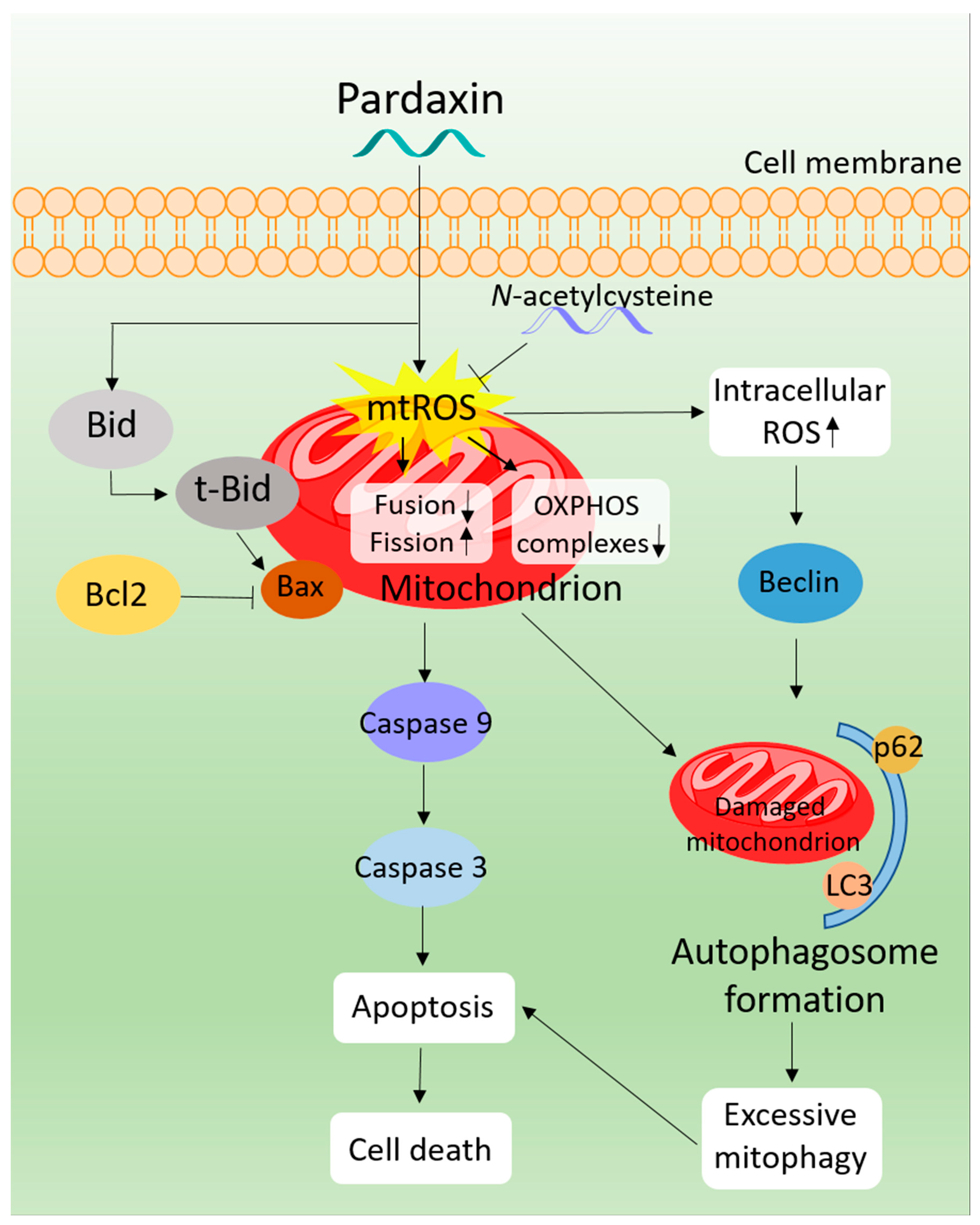
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, H.M.; Fang, Z.; Sun, W.; Clark, L.H.; Stine, J.E.; Tran, A.Q.; Sullivan, S.A.; Gilliam, T.P.; Zhou, C.; Bae-Jump, V.L. Atorvastatin exhibits anti-tumorigenic and anti-metastatic effects in ovarian cancer in vitro. Am. J. Cancer Res. 2017, 7, 2478. [Google Scholar]
- Coleman, R.L.; Spirtos, N.M.; Enserro, D.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Kim, J.-W.; Park, S.-Y.; Kim, B.-G.; Nam, J.-H.; et al. Secondary Surgical Cytoreduction for Recurrent Ovarian Cancer. N. Engl. J. Med. 2019, 381, 1929–1939. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A.; Raja, F.A.; Fotopoulou, C.; Gonzalez-Martin, A.; Colombo, N.; Sessa, C. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi24–vi32. [Google Scholar] [CrossRef]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.J.; Kim, K.I.; Kim, G.; Moon, E.; Yang, S.S.; Lee, J.-S. Atmospheric-Pressure Plasma Jet Induces Apoptosis Involving Mitochondria via Generation of Free Radicals. PLoS ONE 2011, 6, e28154. [Google Scholar] [CrossRef] [Green Version]
- Borek, C. Antioxidant health effects of aged garlic extract. J. Nutr. 2001, 131, 1010S–1015S. [Google Scholar] [CrossRef] [Green Version]
- Barrera, G. Oxidative Stress and Lipid Peroxidation Products in Cancer Progression and Therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef] [Green Version]
- Curtin, J.; Donovan, M.; Cotter, T.G. Regulation and measurement of oxidative stress in apoptosis. J. Immunol. Methods 2002, 265, 49–72. [Google Scholar] [CrossRef] [Green Version]
- Wiemerslage, L.; Lee, D. Quantification of mitochondrial morphology in neurites of dopaminergic neurons using multiple parameters. J. Neurosci. Methods 2016, 262, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.R.; Lewis, W.H. Mitochondria in Tissue Culture. Science 1914, 39, 330–333. [Google Scholar] [CrossRef]
- Suen, D.-F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Köritzer, J.; Boxhammer, V.; Schäfer, A.; Shimizu, T.; Klämpfl, T.G.; Li, Y.-F.; Welz, C.; Schwenk-Zieger, S.; Morfill, G.E.; Zimmermann, J.L.; et al. Restoration of Sensitivity in Chemo—Resistant Glioma Cells by Cold Atmospheric Plasma. PLoS ONE 2013, 8, e64498. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, M.; Robert, E.; Lerondel, S.; Sarron, V.; Ries, D.; Dozias, S.; Sobilo, J.; Gosset, D.; Kieda, C.; Legrain, B.; et al. ROS implication in a new antitumor strategy based on non-thermal plasma. Int. J. Cancer 2012, 130, 2185–2194. [Google Scholar] [CrossRef]
- Green, D.R.; Amarante-Mendes, G.P. The Point of No Return: Mitochondria, Caspases, and the Commitment to Cell Death. Apoptosis Mech. Role Dis. 1998, 24, 45–61. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular selfdigestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, H.-N.; Wang, K.; Zhang, L.; Huang, Z.; Liu, J.; Zhang, Z.; Luo, M.; Lei, Y.; Peng, Y.; et al. Ketoconazole exacerbates mitophagy to induce apoptosis by downregulating cyclooxygenase-2 in hepatocellular carcinoma. J. Hepatol. 2019, 70, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Shike, H.; Lauth, X.; Westerman, M.E.; Ostland, V.E.; Carlberg, J.M.; Van Olst, J.C.; Shimizu, C.; Bulet, P.; Burns, J.C. Bass hepcidin is a novel antimicrobial peptide induced by bacterial challenge. Eur. J. Biochem. 2002, 269, 2232–2237. [Google Scholar] [CrossRef]
- Silphaduang, U.; Noga, E.J. Peptide antibiotics in mast cells of fish. Nature 2001, 414, 268–269. [Google Scholar] [CrossRef]
- Sung, W.S.; Park, S.H.; Lee, D.G. Antimicrobial effect and membrane-active mechanism of Urechistachykinins, neuropeptides derived from Urechis unicinctus. FEBS Lett. 2008, 582, 2463–2466. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-F.; Huang, S.-Y.; Liao, C.-Y.; Sung, C.-S.; Chen, J.-Y.; Wen, Z.-H. The use of the antimicrobial peptide piscidin (PCD)-1 as a novel anti-nociceptive agent. Biomaterials 2015, 53, 1–11. [Google Scholar] [CrossRef]
- Narayana, J.L.; Huang, H.-N.; Wu, C.-J.; Chen, J.-Y. Epinecidin-1 antimicrobial activity: In vitro membrane lysis and In vivo efficacy against Helicobacter pylori infection in a mouse model. Biomaterials 2015, 61, 41–51. [Google Scholar] [CrossRef]
- Ting, C.-H.; Chen, Y.-C.; Wu, C.-J.; Chen, J.-Y. Targeting FOSB with a cationic antimicrobial peptide, TP4, for treatment of triple-negative breast cancer. Oncotarget 2016, 7, 40329–40347. [Google Scholar] [CrossRef]
- Huang, T.-C.; Chen, J.-Y. Proteomic analysis reveals that pardaxin triggers apoptotic signaling pathways in human cervical carcinoma HeLa cells: Cross talk among the UPR, c-Jun and ROS. Carcinogenesis 2013, 34, 1833–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.J.; Hong, Y.K.; Kim, K.H.; Han, C.H.; Cho, S.H.; Choi, J.S.; Kim, B.-W. Methanolic extract of Pterocarpus santalinus induces apoptosis in HeLa cells. J. Ethnopharmacol. 2006, 105, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Y.; Wen, H.; Lin, Y.; Hu, Y.; Zhang, Y.; Xia, Q.; Lin, Z. QSAR modeling and design of cationic antimicrobial peptides based on structural properties of amino acids. Comb. Chem. High Throughput Screen. 2012, 15, 347–353. [Google Scholar] [CrossRef]
- Huang, P.-H.; Chen, J.-Y.; Kuo, C.-M. Three different hepcidins from tilapia, Oreochromis mossambicus: Analysis of their expressions and biological functions. Mol. Immunol. 2007, 44, 1922–1934. [Google Scholar] [CrossRef]
- Oren, Z.; Shai, Y. A Class of Highly Potent Antibacterial Peptides Derived from Pardaxin, A Pore-Forming Peptide Isolated from Moses Sole Fish Pardachirus marmoratus. Eur. J. Biochem. 1996, 237, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y.; Bach, D.; Yanovsky, A. Channel formation properties of synthetic pardaxin and analogues. J. Biol. Chem. 1990, 265, 20202–20209. [Google Scholar] [CrossRef]
- Chen, Y.F.; Shih, P.-C.; Kuo, H.M.; Yang, S.N.; Lin, Y.Y.; Chen, W.F.; Tzou, S.J.; Liu, H.T.; Chen, N.F. TP3, an antimicrobial peptide, inhibits infiltration and motility of glioblastoma cells via modulating the tumor microenvironment. Cancer Med. 2020, 9, 3918–3931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, C.-H.; Ma, Y.-L.; Shih, P.-C.; Chen, C.-T.; Cheng, S.-Y.; Pan, C.-Y.; Jean, Y.-H.; Chu, Y.-M.; Lin, S.-C.; Lai, Y.-C.; et al. The antimicrobial peptide tilapia piscidin 3 induces mitochondria-modulated intrinsic apoptosis of osteosarcoma cells. Biochem. Pharmacol. 2020, 178, 114064. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.-Y.; Shih, P.-C.; Huang, P.-W.; Lee, Y.-H.; Chen, Y.-F.; Tai, M.-H.; Liu, C.-H.; Wen, Z.-H.; Kuo, H.-M. Sinularin, an Anti-Cancer Agent Causing Mitochondria-Modulated Apoptosis and Cytoskeleton Disruption in Human Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3946. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-Protocol 2019, 9, e3128. [Google Scholar] [CrossRef]
- Stankiewicz, M.; Jonas, W.; Hadas, E.; Cabaj, W.; Douch, P.G.C. Supravital staining of eosinophils. Int. J. Parasitol. 1996, 26, 445–446. [Google Scholar] [CrossRef]
- Traganos, F.; Darzynkiewicz, Z. Lysosomal Proton Pump Activity: Supravital Cell Staining with Acridine Orange Differentiates Leukocyte Subpopulations. Methods Cell Biol. 1994, 41, 185–194. [Google Scholar] [PubMed]
- Youle, R.J.; Van DerBliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free. Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Kehoe, S.T.; Kumar, L.; Friedlander, M. Cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynecol. Obstet. 2018, 143, 59–78. [Google Scholar] [CrossRef] [PubMed]
- Koroth, J.; Nirgude, S.; Tiwari, S.; Gopalakrishnan, V.; Mahadeva, R.; Kumar, S.; Karki, S.S.; Choudhary, B. Investigation of anti-cancer and migrastatic properties of novel curcumin derivatives on breast and ovarian cancer cell lines. BMC Complementary Altern. Med. 2019, 19, 273. [Google Scholar] [CrossRef]
- Shaw, T.J.; Senterman, M.K.; Dawson, K.; Crane, C.A.; Vanderhyden, B.C. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol. Ther. 2004, 10, 1032–1042. [Google Scholar] [CrossRef]
- Shih, P.C. Revisiting the development of small molecular inhibitors that directly target the signal transducer and activator of transcription 3 (STAT3) domains. Life Sci. 2020, 242, 117241. [Google Scholar] [CrossRef]
- Shih, P.-C.; Mei, K.-C. Role of STAT3 signaling transduction pathways in cancer stem cell-associated chemoresistance. Drug Discov. Today 2021, 26, 1450–1458. [Google Scholar] [CrossRef]
- Liu, L.; Wu, N.; Wang, Y.; Zhang, X.; Xia, B.; Tang, J.; Cai, J.; Zhao, Z.; Liao, Q.; Wang, J. TRPM7 promotes the epithelial-mesenchymal transition in ovarian cancer through the calcium-related PI3K/AKT oncogenic signaling. J. Exp. Clin. Cancer Res. 2019, 38, 106. [Google Scholar] [CrossRef]
- Yang, S.D.; Ahn, S.H.; Kim, J.I. 3-Oxoacid CoA transferase 1 as a therapeutic target gene for cisplatin-resistant ovarian cancer. Oncol. Lett. 2018, 15, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlikova, B.; Legrand, N.; Panning, J.; Dicato, M.; Diederich, M. Anti-Inflammatory and Anticancer Drugs from Nature. Cancer Treat. Res. 2014, 159, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Indumathy, S.; Dass, C.R. Finding chemo: The search for marine-based pharmaceutical drugs active against cancer. J. Pharm. Pharmacol. 2013, 65, 1280–1301. [Google Scholar] [CrossRef]
- Rocha, M.J.; Cruzeiro, C.R.; Reis, M.; Pardal, M.Â.; Rocha, E. Pollution by endocrine disruptors in a southwest European temperate coastal lagoon (Ria de Aveiro, Portugal). Environ. Monit. Assess. 2016, 188, 101. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Caino, M.C. Mitochondrial Dynamics in Type 2 Diabetes and Cancer. Front. Endocrinol. 2018, 9, 211. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Aaronson, S.A.; Abrams, J.; Alnemri, E.S.; Andrews, D.W.; Baehrecke, E.H.; Bazan, N.G.; Blagosklonny, M.V.; Blomgren, K.; Borner, C.; et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009, 16, 1093–1107. [Google Scholar] [CrossRef] [Green Version]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial Dysfunction in Atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; Gillies McKenna, W.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 Values (μM) | ||
|---|---|---|
| Time (h) | PA-1 | SKOV3 |
| 24 | 3.12 ± 0.21 | 4.60 ± 0.17 |
| 48 | 3.00 ± 0.22 | 3.51 ± 0.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-P.; Shih, P.-C.; Feng, C.-W.; Wu, C.-C.; Tsui, K.-H.; Lin, Y.-H.; Kuo, H.-M.; Wen, Z.-H. Pardaxin Activates Excessive Mitophagy and Mitochondria-Mediated Apoptosis in Human Ovarian Cancer by Inducing Reactive Oxygen Species. Antioxidants 2021, 10, 1883. https://doi.org/10.3390/antiox10121883
Chen Y-P, Shih P-C, Feng C-W, Wu C-C, Tsui K-H, Lin Y-H, Kuo H-M, Wen Z-H. Pardaxin Activates Excessive Mitophagy and Mitochondria-Mediated Apoptosis in Human Ovarian Cancer by Inducing Reactive Oxygen Species. Antioxidants. 2021; 10(12):1883. https://doi.org/10.3390/antiox10121883
Chicago/Turabian StyleChen, Yen-Po, Po-Chang Shih, Chien-Wei Feng, Chang-Cheng Wu, Kuan-Hao Tsui, You-Hsien Lin, Hsiao-Mei Kuo, and Zhi-Hong Wen. 2021. "Pardaxin Activates Excessive Mitophagy and Mitochondria-Mediated Apoptosis in Human Ovarian Cancer by Inducing Reactive Oxygen Species" Antioxidants 10, no. 12: 1883. https://doi.org/10.3390/antiox10121883