
Effects of the Cytoplasm and Mitochondrial Specific Hydroxyl Radical Scavengers TA293 and mitoTA293 in Bleomycin-Induced Pulmonary Fibrosis Model Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Mice
2.3. Cell Culture and Drug Treatment
2.4. Measurement of Senescence-Associated β-Galactosidase Activity
2.5. RNA Isolation and Quantitative RT-PCR
2.6. Western Blotting
2.7. In Vivo Imaging Analysis
2.8. Histological Analysis and Detection of Fibrosis
2.9. siRNA Transfection
2.10. Caspase-1 Activity Assay
2.11. Detection of Mitophagy
2.12. Subcellular Fractionation
2.13. Measurement of Lipid Peroxide, H2O2, and •OH in Mitochondrial and Cytosolic Fractions
2.14. Measurement of 8-hydroxy-2′-deoxyguanosine
2.15. Quantitation of Cytosol and Mitochondrial Glutathione
2.16. Measurement of Mitochondrial Membrane Potential
2.17. Hydroxyproline Assay
2.18. Quantitation of Intracellular Heme
2.19. Statistical Analysis
3. Results
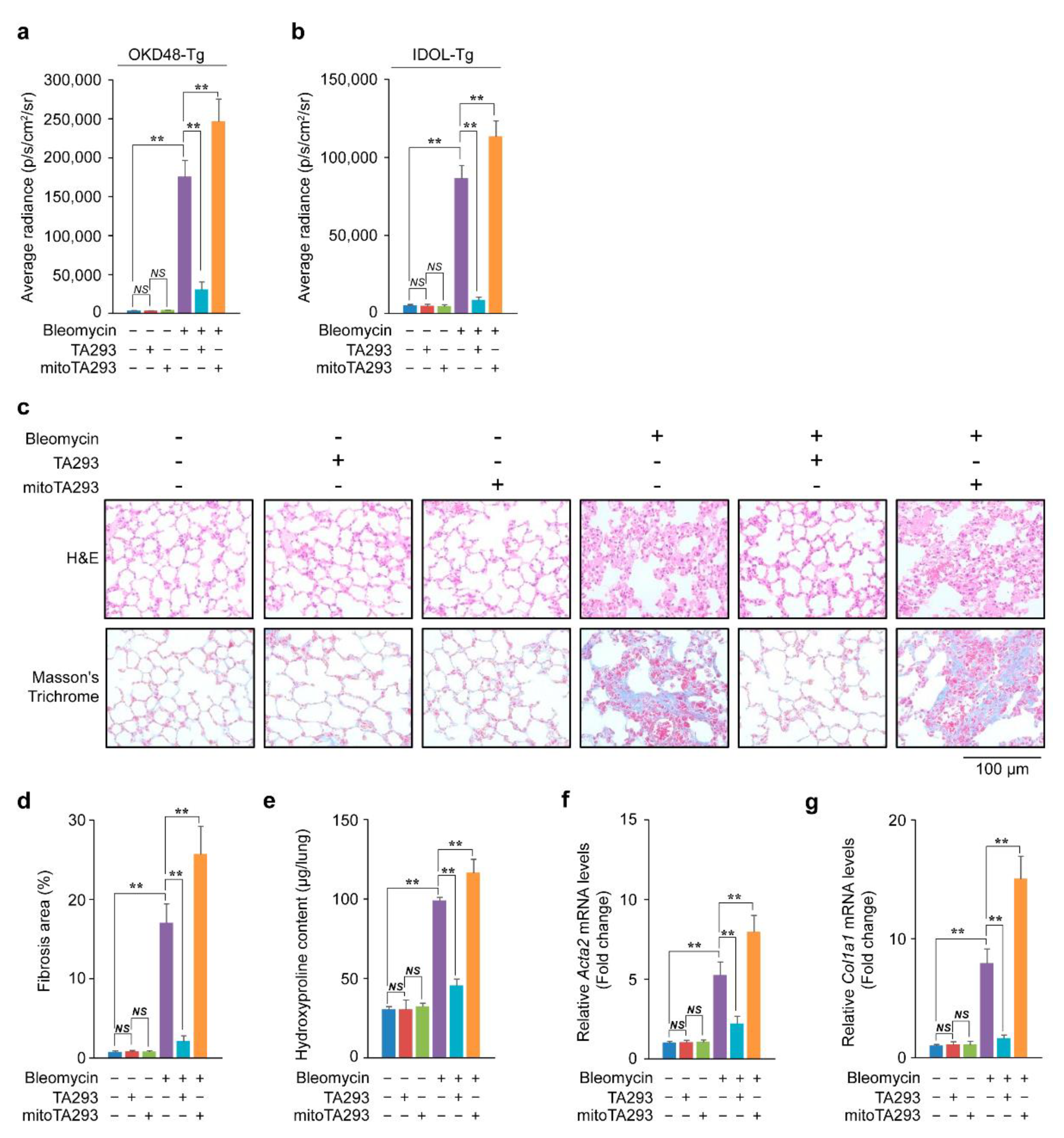
3.1. TA293 Suppresses Induction of Oxidative Stress, Inflammation, and Fibrosis, but mitoTA293 Exacerbates These Conditions in BLM-Induced Pulmonary Fibrosis
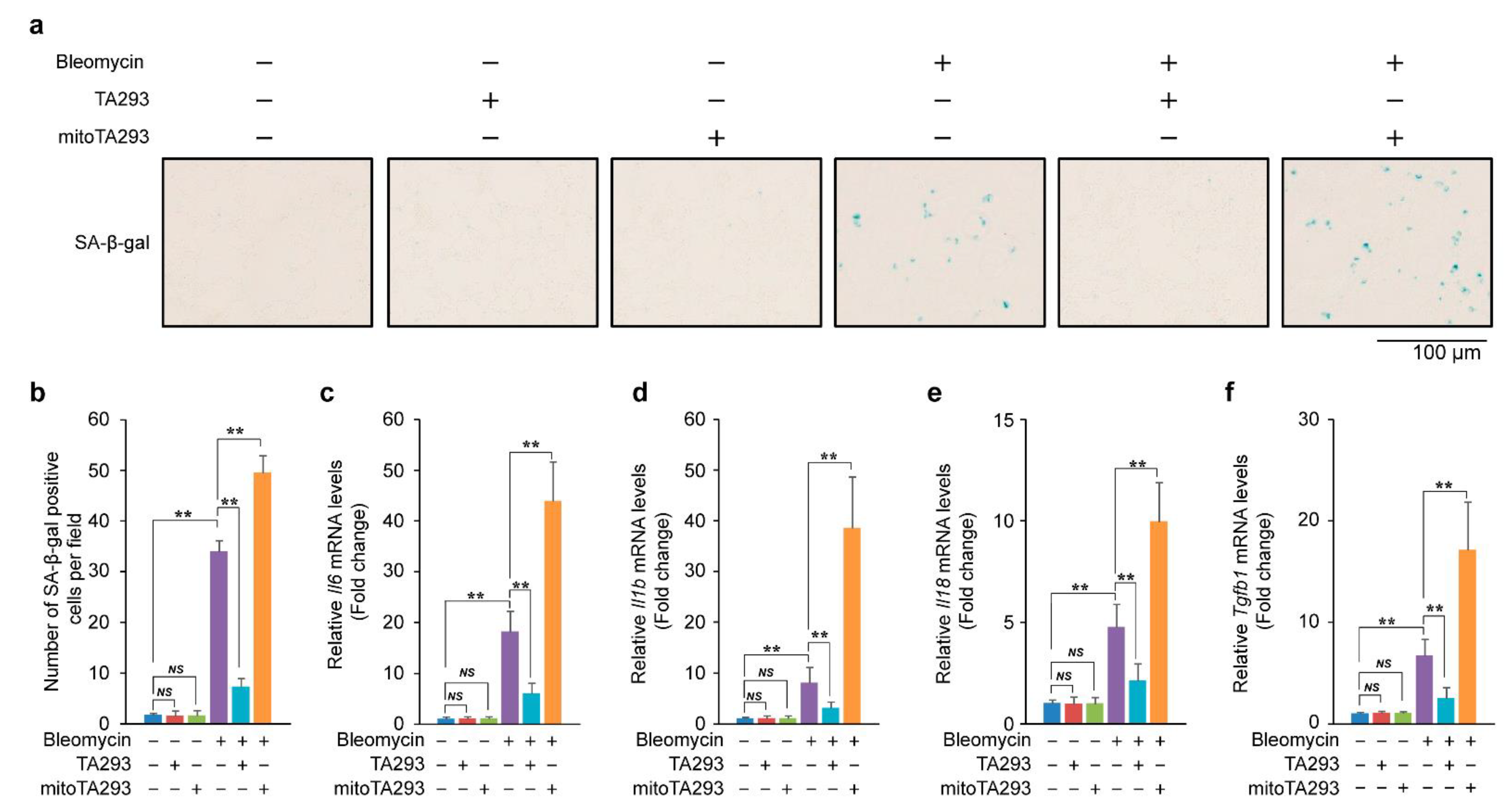
3.2. In the Lungs of BLM-Induced Pulmonary Fibrosis Mice, TA293 Suppresses and mitoTA293 Exacerbates Cellular Senescence
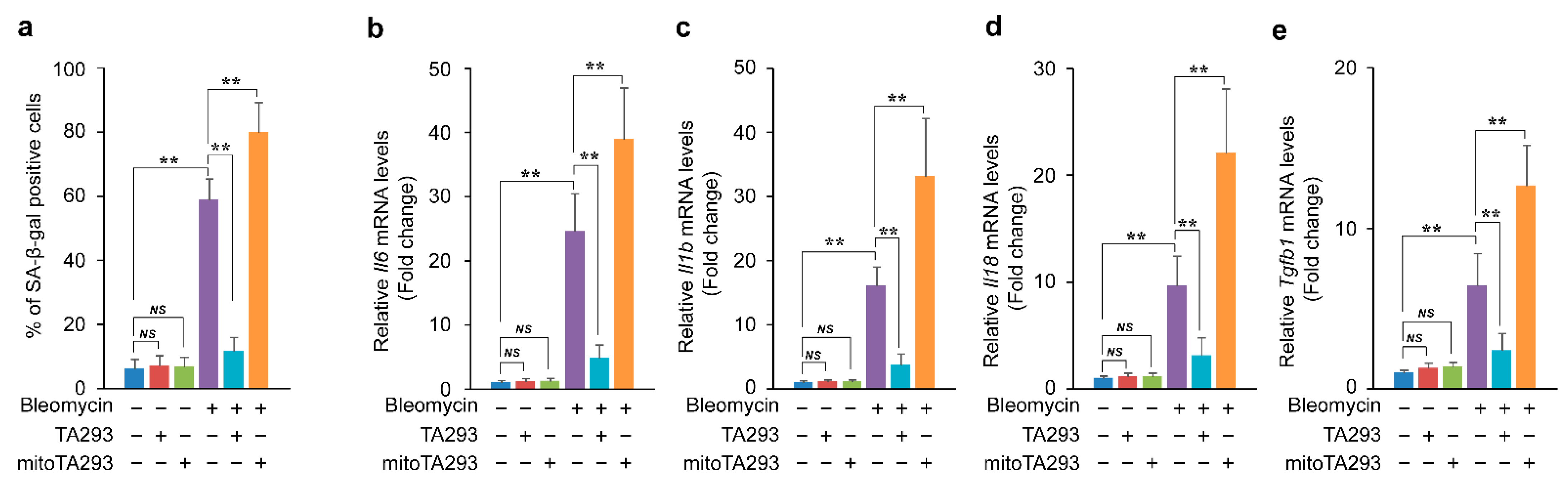
3.3. In BLM-Treated PAECs, TA293 Suppresses and mitoTA293 Exacerbates Cellular Senescence
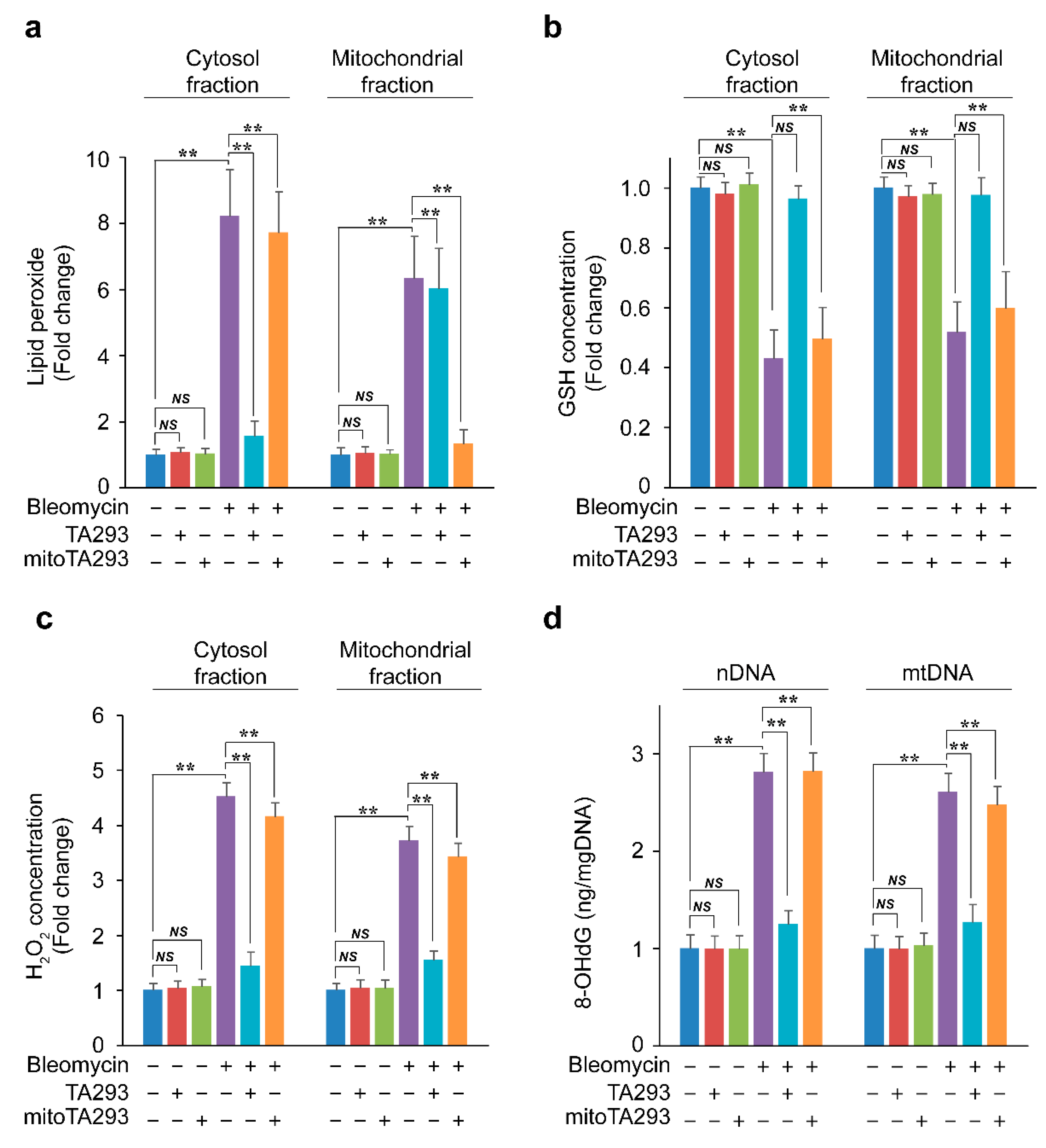
3.4. In BLM-Treated PAECs, TA293 Suppresses the Reduction of GSH in Cytoplasmic and Mitochondrial Fractions and Thereby, the Oxidative Damage to nDNA and mtDNA
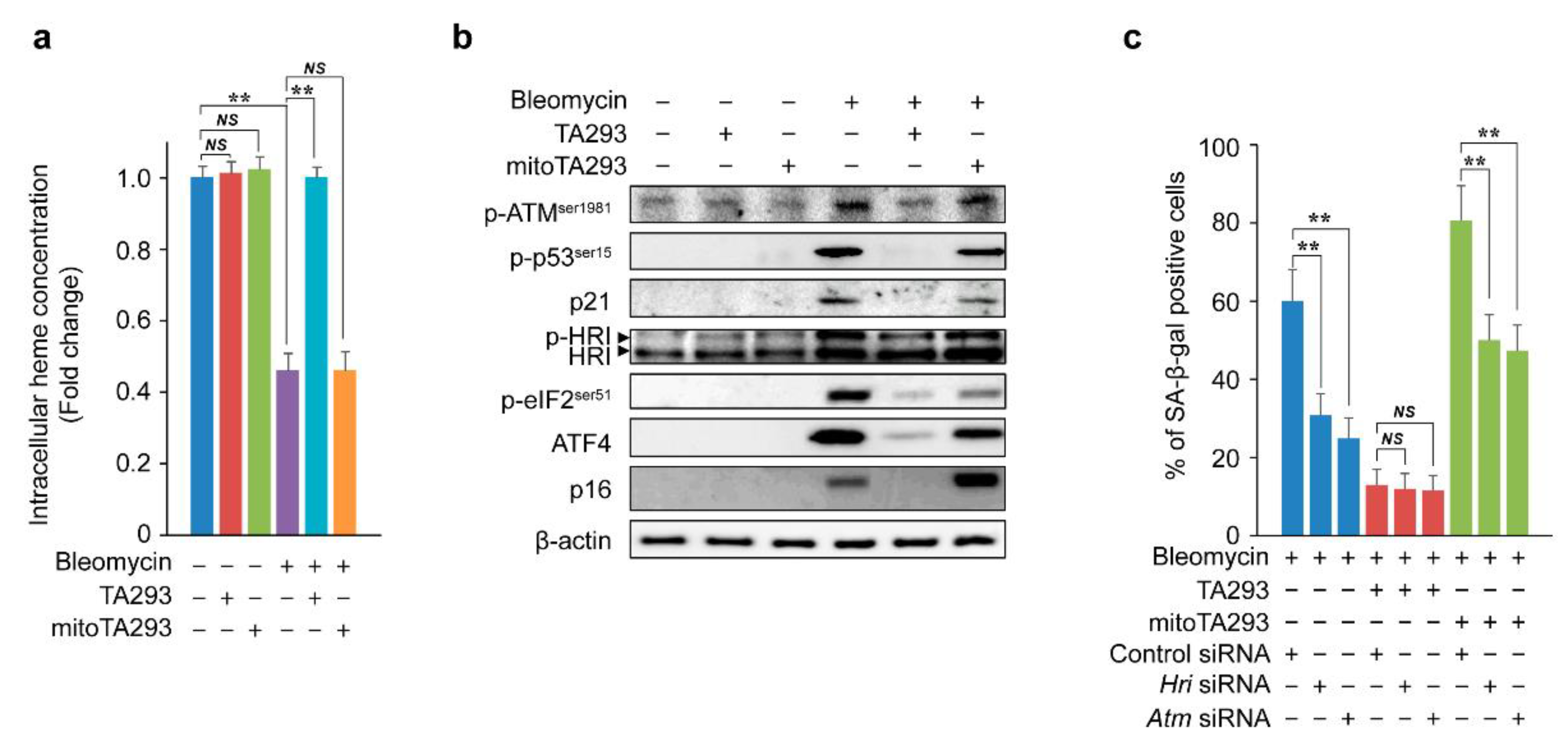
3.5. In BLM-Treated PAECs, TA293 Suppresses and mitoTA293 Exacerbates Cellular Senescence by Different Pathways
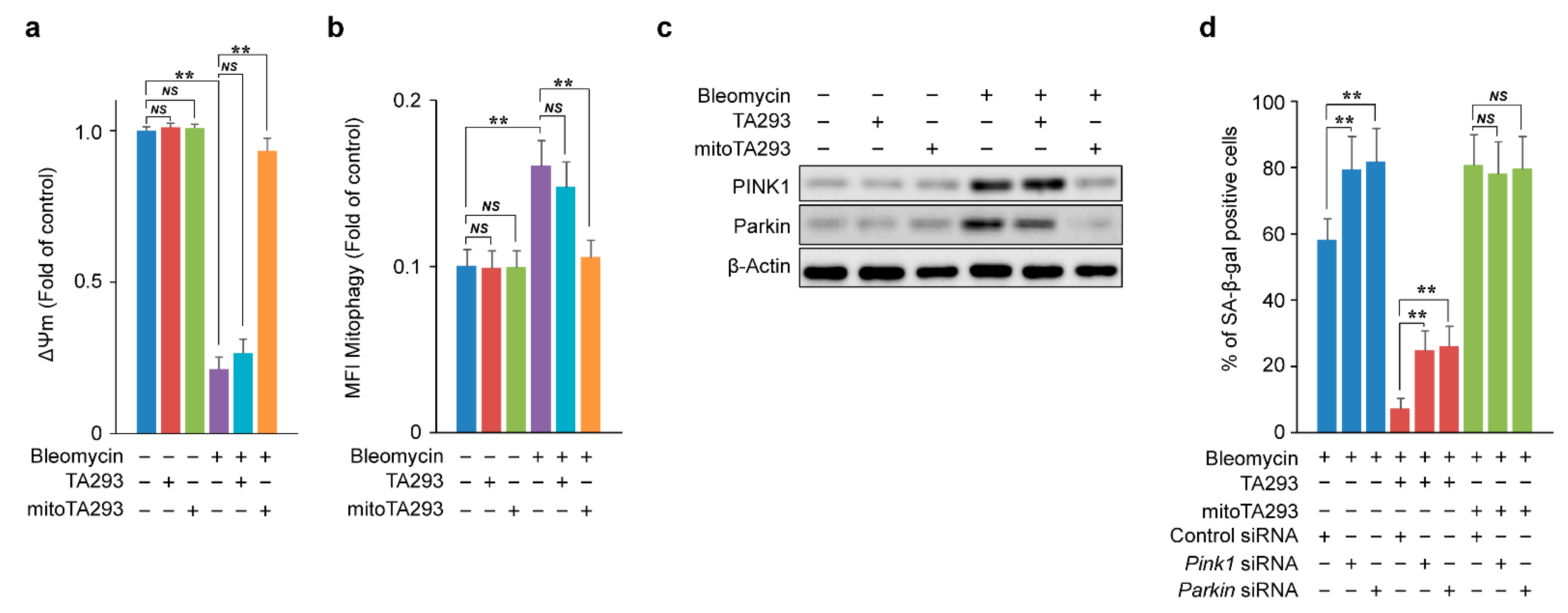
3.6. In BLM-Treated PAECs, mitoTA293 Suppresses the Induction of Mitophagy and Exacerbates Cellular Senescence
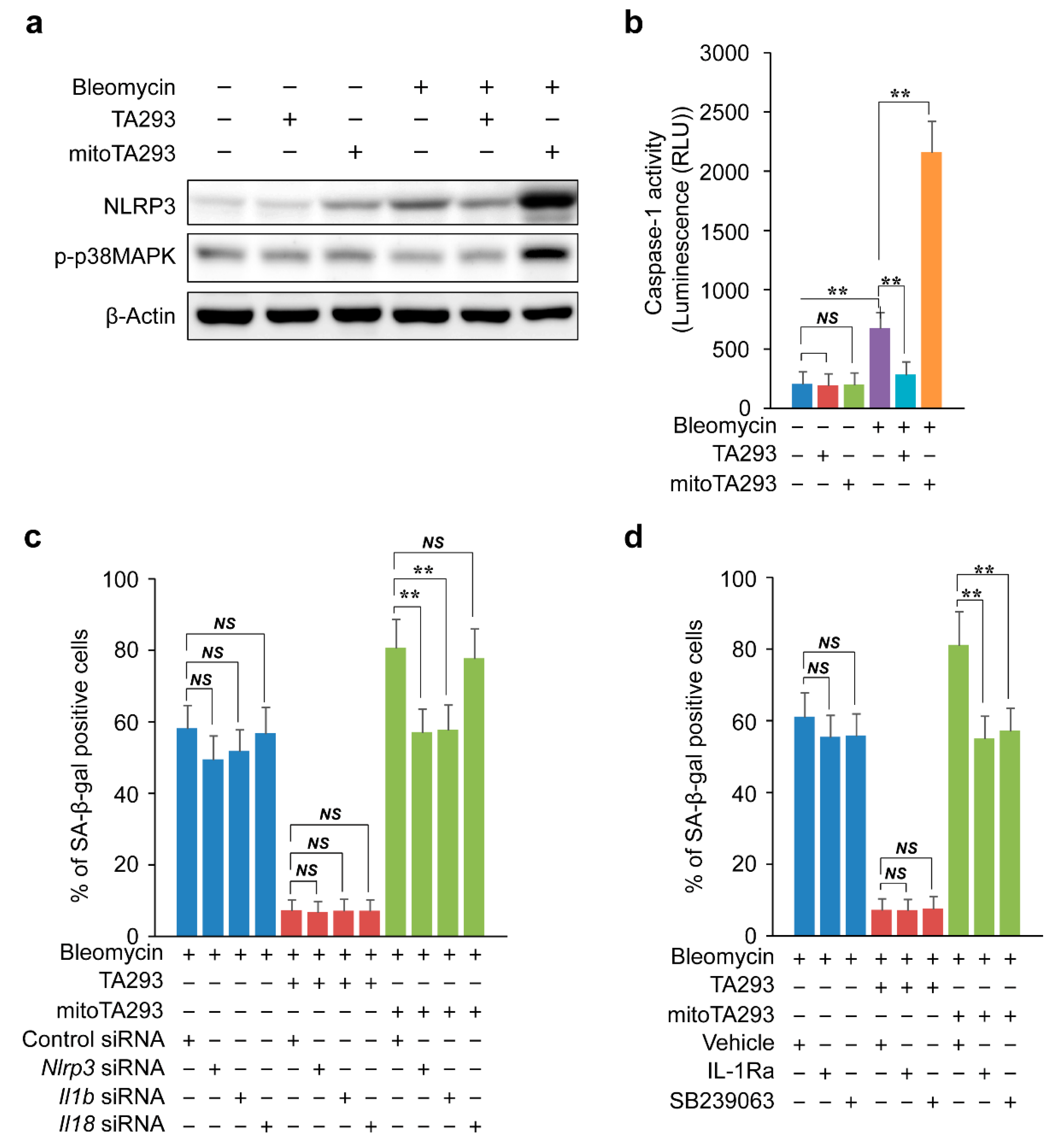
3.7. TA293 and mitoTA293 Have Opposite Effects on the NLRP3 Inflammasome/Caspase-1/IL1β/ILR/p-p38/p16 and p21 Pathways and Cellular Senescence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raghu, G. Idiopathic pulmonary fibrosis: Lessons from clinical trials over the past 25 years. Eur. Respir. J. 2017, 50, 1701209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buendía-Roldá, I.; Mejía, M.; Navarro, C.; Selman, M. Idiopathic pulmonary fibrosis: Clinical behavior and aging associated comorbidities. Respir. Med. 2017, 129, 46–52. [Google Scholar] [CrossRef]
- Liu, Y.M.; Nepali, K.; Liou, J.P. Idiopathic pulmonary fibrosis: Current status, recent progress, and emerging targets. J. Med. Chem. 2017, 60, 527–553. [Google Scholar] [CrossRef]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Pérez, E.R.F.; Daniels, C.E.; Schroeder, D.R.; St Sauver, J.; Hartman, T.E.; Bartholmai, B.J.; Yi, E.S.; Ryu, J.H. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: A population-based study. Chest 2010, 137, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Shimizu, T.; Shinkai, Y.; Hirata, Y.; Inagaki, H.; Takeda, K.; Azuma, A.; Yamamoto, M.; Kawada, T. Nrf2 regulates the risk of a diesel exhaust inhalation-induced immune response during bleomycin lung injury and fibrosis in mice. Int. J. Mol. Sci. 2017, 18, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, A.; Ask, K.; Warburton, D.; Gauldie, J.; Kolb, M. The bleomycin animal model: A useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 2008, 40, 362–382. [Google Scholar] [CrossRef] [Green Version]
- Highfield, J.A.; Mehta, L.K.; Parrick, J.; Wardman, P. Synthesis, hydroxyl radical production and cytotoxicity of analogues of bleomycin. Bioorg. Med. Chem. 2000, 8, 1065–1073. [Google Scholar] [CrossRef]
- Allawzi, A.; Elajaili, H.; Redente, E.F.; Nozik-Grayck, E. Oxidative toxicology of bleomycin: Role of the extracellular redox environment. Curr. Opin. Toxicol. 2019, 13, 68–73. [Google Scholar] [CrossRef]
- Suryadevara, V.; Ramchandran, R.; Kamp, D.W.; Natarajan, V. Lipid mediators regulate pulmonary fibrosis: Potential mechanisms and signaling pathways. Int. J. Mol. Sci. 2020, 21, 4257. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, W.; Cao, Q.; Wang, Z.; Zhao, M.; Xu, L.; Zhuang, Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 2020, 6, 80. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowler, R.P.; Nicks, M.; Warnick, K.; Crapo, J.D. Role of extracellular superoxide dismutase in bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L719–L726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortijo, J.; Cerdá-Nicolás, M.; Serrano, A.; Bioque, G.; Estrela, J.M.; Santangelo, F.; Esteras, A.; Llombart-Bosch, A.; Morcillo, E.J. Attenuation by oral N-acetylcysteine of bleomycin-induced lung injury in rats. Eur. Respir. J. 2001, 17, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Anathy, V.; Lahue, K.G.; Chapman, D.G.; Chia, S.B.; Casey, D.T.; Aboushousha, R.; van der Velden, J.L.J.; Elko, E.; Hoffman, S.M.; McMillan, D.H.; et al. Reducing protein oxidation reverses lung fibrosis. Nat. Med. 2018, 24, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Imai, J.; Ito, T.; Takagaki, H.; Ui, M.; Hatta, S. The novel antioxidant TA293 reveals the role of cytoplasmic hydroxyl radicals in oxidative stress-induced senescence and inflammation. Biochem. Biophys. Res. Commun. 2017, 482, 1183–1189. [Google Scholar] [CrossRef]
- Ohsawa, I.; Ishikawa, M.; Takahashi, M.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.I.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694. [Google Scholar] [CrossRef]
- Kayar, S.R.; Axley, M.J.; Homer, L.D.; Harabin, A.L. Hydrogen gas is not oxidized by mammalian tissues under hyperbaric conditions. Undersea Hyperb. Med. 1994, 21, 265–275. [Google Scholar]
- Turnbull, S.; Tabner, B.J.; El-Agnaf, O.M.; Moore, S.; Davies, Y.; Allsop, D. alpha-Synuclein implicated in Parkinson’s disease catalyses the formation of hydrogen peroxide in vitro. Free Radic. Biol. Med. 2001, 30, 1163–1170. [Google Scholar] [CrossRef]
- Marlatt, M.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Sources and mechanisms of cytoplasmic oxidative damage in Alzheimer’s disease. Acta Neurobiol. Exp. 2004, 64, 81–87. [Google Scholar]
- Schaar, C.E.; Dues, D.J.; Spielbauer, K.K.; Machiela, E.; Cooper, J.F.; Senchuk, M.; Hekimi, S.; van Raamsdonk, J.M. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015, 11, e1004972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, T.; Imai, J.; Takagaki, H.; Ui, M.; Hatta, S. Cytoplasmic OH scavenger TA293 attenuates cellular senescence and fibrosis by activating macrophages through oxidized phospholipids/TLR4. Life Sci. 2019, 221, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Akai, R.; Tokuda, M.; Iwawaki, T. A transgenic mouse model for monitoring oxidative stress. Sci. Rep. 2012, 2, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwawaki, T.; Akai, R.; Oikawa, D.; Toyoshima, T.; Yoshino, M.; Suzuki, M.; Takeda, N.; Ishikawa, T.; Kataoka, Y.; Yamamura, K.I. Transgenic mouse model for imaging of interleukin-1β-related inflammation in vivo. Sci. Rep. 2015, 5, 17205. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, N.I.; Schnapp, A.; Park, J.E. Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am. J. Respir. Crit. Care Med. 2006, 173, 69–776. [Google Scholar] [CrossRef]
- Aoshiba, K.; Tsuji, T.; Nagai, A. Bleomycin induces cellular senescence in alveolar epithelial cells. Eur. Respir. J. 2003, 22, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Anåker, A.; von Koch, L.; Sjöstrand, C.; Bernhardt, J.; Elf, M.A. comparative study of patients’ activities and interactions in a stroke unit before and after reconstruction-The significance of the built environment. PLoS ONE 2017, 12, e0177477. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.I.; Lau, L.F. Cellular senescence controls fibrosis in wound healing. Aging 2010, 2, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Crapo, J.D.; Young, S.L.; Fram, E.K.; Pinkerton, K.E.; Barry, B.E.; Crapo, R.O. Morphometric characteristics of cells in the alveolar region of mammalian lungs. Am. Rev. Respir. Dis. 1983, 128, S42–S46. [Google Scholar] [CrossRef]
- Chen, K.J.; Li, Q.; Wen, C.M.; Duan, Z.X.; Zhang, J.Y.; Xu, C.; Wang, J.M. Bleomycin (BLM) induces epithelial-to-mesenchymal transition in cultured A549 cells via the TGF-β/Smad signaling pathway. J. Cancer 2016, 11, 1557–1564. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Chiang, J.Y.; Choi, J.Y.; Xiong, Z.M.; Mao, X.; Collins, F.S.; Hodes, R.J.; Cao, K. Transient induction of telomerase expression mediates senescence and reduces tumorigenesis in primary fibroblasts. Proc. Natl. Acad. Sci. USA 2019, 38, 18983–18993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enami, S.; Sakamoto, Y.; Colussi, A.J. Fenton chemistry at aqueous interfaces. Proc. Natl. Acad. Sci. USA 2014, 111, 623–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Cho, C.S. Blot-based detection of dehydroalanine-containing glutathione peroxidase with the use of biotin-conjugated cysteamine. Methods Enzymol. 2010, 474, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Shakra, A.; Zeiger, E. Formation of 8-hydroxy-2′-deoxyguanosine following treatment of 2′-deoxyguanosine or DNA by hydrogen peroxide or glutathione. Mutat. Res. 1997, 390, 45–50. [Google Scholar] [CrossRef]
- Han, A.P.; Yu, C.; Lu, L.; Fujiwara, Y.; Browne, C.; Chin, G.; Fleming, M.; Leboulch, P.; Orkin, S.H.; Chen, J.J. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001, 20, 6909–6918. [Google Scholar] [CrossRef] [Green Version]
- Bowman, S.E.J.; Bren, K.L. The chemistry and biochemistry of heme c: Functional bases for covalent attachment. Nat. Prod. Rep. 2008, 25, 1118–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, T.; Kurokawa, R.; Hirano, S.I.; Imai, J. Hydrogen indirectly suppresses increases in hydrogen peroxide in cytoplasmic hydroxyl radical-induced cells and suppresses cellular senescence. Int. J. Mol. Sci. 2019, 20, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertin, G.; Averbeck, D. Cadmium: Cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review). Biochimie 2006, 88, 1549–1559. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Núñez, G. Intracellular sensors of microbes and danger. Immunol. Rev. 2011, 243, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Bello-Irizarry, S.N.; Wang, J.; Olsen, K.; Gigliotti, F.; Wright, T.W. The alveolar epithelial cell chemokine response to pneumocystis requires adaptor molecule MyD88 and interleukin-1 receptor but not toll-like receptor 2 or 4. Infect. Immun. 2012, 80, 3912–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, A.; Han, J.; Cato, A.C.B.; Akira, S.; Li, J.D. Glucocorticoids synergize with IL-1beta to induce TLR2 expression via MAP Kinase Phosphatase-1-dependent dual Inhibition of MAPK JNK and p38 in epithelial cells. BMC Mol. Biol. 2004, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, N.; Xiang, R.; Sun, P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, R.M.; Peisach, J.; Blumberg, W.E.; Horwitz, S.B. Iron-bleomycin interactions with oxygen and oxygen analogues. Effects on spectra and drug activity. J. Biol. Chem. 1979, 254, 10906–10912. [Google Scholar] [CrossRef]
- Dong, W.W.; Zhang, Y.Q.; Zhu, X.Y.; Mao, Y.F.; Sun, X.J.; Liu, Y.J.; Jiang, L. Protective effects of hydrogen-rich saline against lipopolysaccharide-induced alveolar epithelial-to-mesenchymal transition and pulmonary fibrosis. Med. Sci. Monit. 2017, 23, 2357–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuhara, K.; Nakashima, T.; Abe, M.; Masuda, T.; Hamada, H.; Iwamoto, H.; Fujitaka, K.; Kohno, N.; Hattori, N. Suplatast tosilate protects the lung against hyperoxic lung injury by scavenging hydroxyl radicals. Free Radic. Biol. Med. 2017, 106, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, M.J.; Haak, A.J.; Tschumperlin, D.J.; LeBrasseur, N.K. Targeting senescent cells in fibrosis: Pathology, paradox, and practical considerations. Curr. Rheumatol. Rep. 2018, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50, 1602367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.G.; Cheng, W.H.; McClung, J.P. Metabolic regulation and function of glutathione peroxidase-1. Annu. Rev. Nutr. 2007, 27, 41–61. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Dansen, T.B. Reactive oxygen species induced p53 activation: DNA damage, redox signaling, or both? Antioxid. Redox Signal. 2020, 33, 839–859. [Google Scholar] [CrossRef]
- Semchyshyn, H.M. Reactive carbonyl species in vivo: Generation and dual biological effects. Sci. World J. 2014, 2014, 417842. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yang, J.R.; Chen, X.M.; Cai, G.Y.; Lin, L.R.; He, Y.N. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am. J. Physiol. Cell. Physiol. 2015, 308, C621–C630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Thornton, T.M.; Rincon, M. Non-classical p38 map kinase functions: Cell cycle checkpoints and survival. Int. J. Biol. Sci. 2009, 5, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Piguet, P.F.; Vesin, C.; Grau, G.E.; Thompson, R.C. Interleukin 1 receptor antagonist (IL-1ra) prevents or cures pulmonary fibrosis elicited in mice by bleomycin or silica. Cytokine 1993, 5, 57–61. [Google Scholar] [CrossRef]
- Borthwick, L.A. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin. Immunopathol. 2016, 38, 517–534. [Google Scholar] [CrossRef] [Green Version]
- Underwood, D.C.; Osborn, R.R.; Bochnowicz, S.; Webb, E.F.; Rieman, D.J.; Lee, J.C.; Romanic, A.M.; Adams, J.L.; Hay, D.W.; Griswold, D.E. SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L895–L902. [Google Scholar] [CrossRef]
- Luo, Y.; Zou, P.; Zou, J.; Wang, J.; Zhou, D.; Liu, L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp. Gerontol. 2011, 46, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.M.; Zhang, J.; Zhang, Y.; Fei, C.; Wang, L.; Yi, Z.W.; Zhang, Z.Q. Interleukin-18 promotes fibroblast senescence in pulmonary fibrosis through down-regulating Klotho expression. Biomed. Pharmacother. 2019, 113, 108756. [Google Scholar] [CrossRef]
- Girard-Guyonvarc, C.; Palomo, J.; Martin, P.; Rodriguez, E.; Troccaz, S.; Palmer, G.; Gabay, C. Unopposed IL-18 signaling leads to severe TLR9-induced macrophage activation syndrome in mice. Blood 2018, 131, 1430–1441. [Google Scholar] [CrossRef]
- Weiss, E.S.; Girard-Guyonvarc’h, C.; Holzinger, D.; de Jesus, A.A.; Tariq, Z.; Picarsic, J.; Schiffrin, E.J.; Foell, D.; Grom, A.A.; Ammann, S.; et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018, 131, 1442–1455. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in cell senescence: Is mitophagy the weakest link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araya, J.; Tsubouchi, K.; Sato, N.; Ito, S.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 2019, 15, 510–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakai, T.; Takagaki, H.; Yamagiwa, N.; Ui, M.; Hatta, S.; Imai, J. Effects of the Cytoplasm and Mitochondrial Specific Hydroxyl Radical Scavengers TA293 and mitoTA293 in Bleomycin-Induced Pulmonary Fibrosis Model Mice. Antioxidants 2021, 10, 1398. https://doi.org/10.3390/antiox10091398
Sakai T, Takagaki H, Yamagiwa N, Ui M, Hatta S, Imai J. Effects of the Cytoplasm and Mitochondrial Specific Hydroxyl Radical Scavengers TA293 and mitoTA293 in Bleomycin-Induced Pulmonary Fibrosis Model Mice. Antioxidants. 2021; 10(9):1398. https://doi.org/10.3390/antiox10091398
Chicago/Turabian StyleSakai, Takahiro, Hidetsugu Takagaki, Noriyuki Yamagiwa, Michio Ui, Shinichi Hatta, and Jun Imai. 2021. "Effects of the Cytoplasm and Mitochondrial Specific Hydroxyl Radical Scavengers TA293 and mitoTA293 in Bleomycin-Induced Pulmonary Fibrosis Model Mice" Antioxidants 10, no. 9: 1398. https://doi.org/10.3390/antiox10091398