
The Antifungal Antibiotic Filipin as a Diagnostic Tool of Cholesterol Alterations in Lysosomal Storage Diseases and Neurodegenerative Disorders


{kind=link}
{kind=link}
Abstract
:1. Cholesterol Metabolism in the Brain
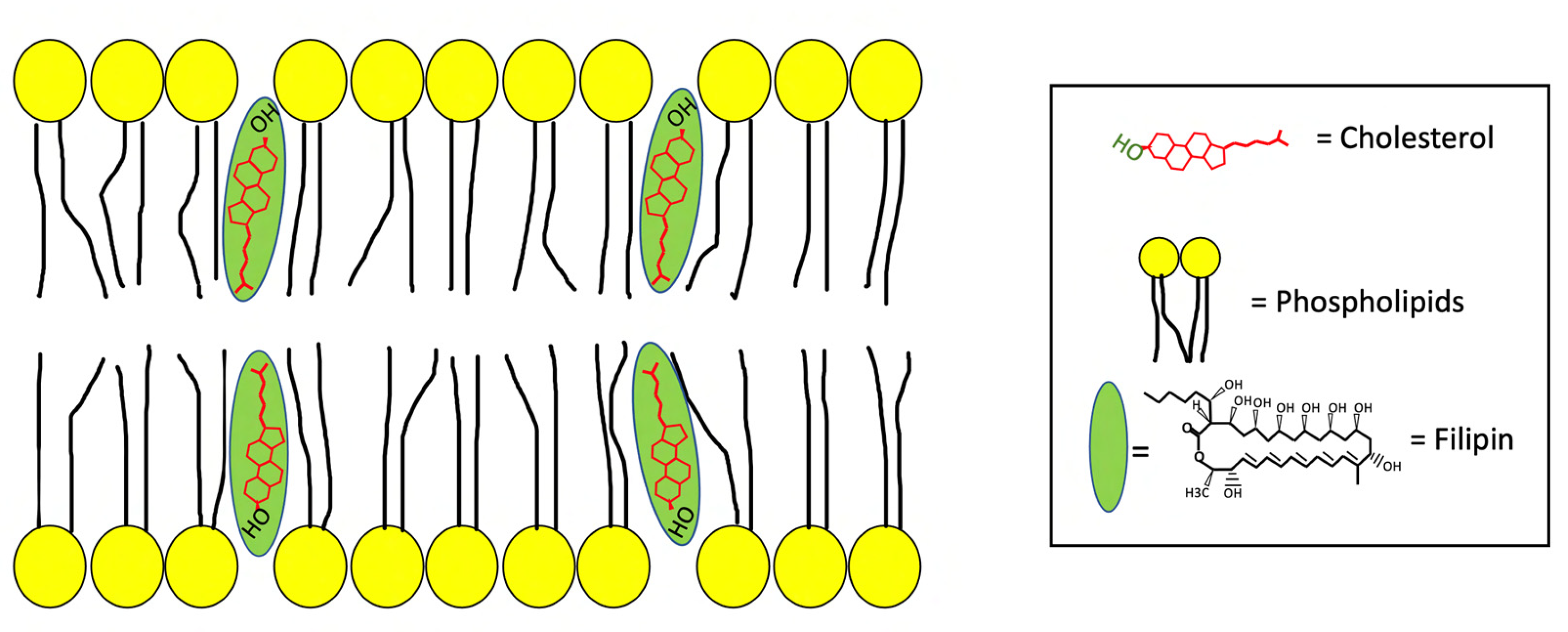
2. The Antifungal Antibiotic Filipin
3. Filipin as a Diagnostic Tool for Lysosomal Storage Diseases
3.1. Niemann Pick Type C Disease
3.2. GM1 Gangliosidosis
4. Filipin as a Diagnostic Tool for Common Neurodegenerative Diseases
4.1. Alzheimer’s Disease
4.2. Huntington Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Albuquerque, H.M.T.; Santos, C.M.M.; Silva, A.M.S. Cholesterol-Based Compounds: Recent Advances in Synthesis and Applications. Molecules 2018, 24, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, J.E. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 2006, 580, 5518–5524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Martìn, M.G.; Pfrieger, F.; Dotti, C.G. Cholesterol in brain disease: Sometimes determinant and frequently implicated. EMBO Rep. 2014, 15, 1036–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.L.; Strauss, J.F., 3rd. Molecular pathology and mechanism of action of the steroidogenic acute regulatory protein, StAR. J. Steroid. Biochem. Mol. Biol. 1999, 69, 131–141. [Google Scholar] [CrossRef]
- Kosters, A.; Jirsa, M.; Groen, A.K. Genetic background of cholesterol gallstone disease. Biochim. Biophys. Acta 2003, 1637, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Ikonen, E. Mechanisms for cellular cholesterol transport: Defects and human disease. Physiol. Rev. 2006, 86, 1237–1261. [Google Scholar] [CrossRef]
- Formanowicz, D.; Radom, M.; Rybarczyk, A.; Tanaś, K.; Formanowicz, P. Control of Cholesterol Metabolism Using a Systems Approach. Biology 2022, 11, 430. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Benarroch, E.E. Brain cholesterol metabolism and neurologic disease. Neurology 2008, 71, 1368–1373. [Google Scholar] [CrossRef]
- Beffert, U.; Danik, M.; Krzywkowski, P.; Ramassamy, C.; Berrada, F.; Poirier, J. The neurobiology of apolipoproteins and their receptors in the CNS and Alzheimer’s disease. Brain Res. Brain Res. Rev. 1998, 27, 119–142. [Google Scholar] [CrossRef]
- Karten, B.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Expression of ABCG1, but not ABCA1, correlates with cholesterol release by cerebellar astroglia. J. Biol. Chem. 2006, 281, 4049–4057. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Olsen, R.H.; Merkens, L.S.; DeBarber, A.; Steiner, R.D.; Sullivan, P.M.; Maeda, N.; Raber, J. Apolipoprotein E-low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol. Dis. 2014, 64, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Infante, R.E.; Wang, M.L.; Radhakrishnan, A.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 15287–15292. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.L.; Motamed, M.; Infante, R.E.; Abi-Mosleh, L.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 2010, 12, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Abi-Mosleh, L.; Infante, R.E.; Radhakrishnan, A.; Goldstein, J.L.; Brown, M.S. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19316–19321. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Shumate, J.L.; Caldwell, J.G.; Elghobashi-Meinhardt, N.; Lu, A.; Zhang, L.; Olsson, N.E.; Elias, J.E.; Pfeffer, S.R. Inter-domain dynamics drive cholesterol transport by NPC1 and NPC1L1 proteins. Elife 2020, 9, e57089. [Google Scholar] [CrossRef]
- Yoon, H.J.; Jeong, H.; Lee, H.H.; Jang, S. Molecular dynamics study with mutation shows that N-terminal domain structural re-orientation in Niemann-Pick type C1 is required for proper alignment of cholesterol transport. J. Neurochem. 2021, 156, 967–978. [Google Scholar] [CrossRef]
- Castanho, M.A.; Prieto, M.; Jameson, D.M. The pentaene macrolide antibiotic filipin prefers more rigid DPPC bilayers: A fluorescence pressure dependence study. Biochim. Biophys. Acta 1999, 1419, 1–14. [Google Scholar] [CrossRef]
- Ammann, A.; Gottlieb, D. Paper chromatography of antifungal antibiotics. Appl. Microbiol. 1955, 3, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Brock, T.D. The effect of oils and fatty acids on the production of filipin. Appl. Microbiol. 1956, 4, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Gimpl, G.; Gehrig-Burger, K. Cholesterol reporter molecules. Biosci. Rep. 2007, 27, 335–358. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.D.; Comly, M.E.; Kruth, H.S.; Vanier, M.; Filling-Katz, M.; Fink, J.; Barton, N.; Weintroub, H.; Quirk, J.M.; Tokoro, T.; et al. Niemann-pick variant disorders: Comparison of errors of cellular cholesterol homeostasis in group D and group C fibroblasts. Proc. Natl. Acad. Sci. USA 1987, 84, 556–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, V.; Watanabe, R.; Dominguez, M.; Sun, X.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat. Cell Biol. 1999, 1, 386–388. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Wüstner, D. Analysis of cholesterol trafficking with fluorescent probes. Methods Cell Biol. 2012, 108, 367–393. [Google Scholar]
- Bittman, R. Sterol-polyene antibiotic complexation: Probe of membrane structure. Lipids 1978, 13, 686–691. [Google Scholar] [CrossRef]
- Wilhelm, L.P.; Voilquin, L.; Kobayashi, T.; Tomasetto, C.; Alpy, F. Intracellular and Plasma Membrane Cholesterol Labeling and Quantification Using Filipin and GFP-D4. Methods Mol. Biol. 2019, 1949, 137–152. [Google Scholar]
- Bergy, M.E.; Eble, T.E. The filipin complex. Biochemistry 1968, 7, 653–659. [Google Scholar] [CrossRef]
- Norman, A.W.; Demel, R.A.; de Kruyff, B.; van Deenen, L.L. Studies on the biological properties of polyene antibiotics. Evidence for the direct interaction of filipin with cholesterol. J. Biol. Chem. 1972, 247, 1918–1929. [Google Scholar] [CrossRef]
- Verkleij, A.J.; Kruijff, B.; Gerritsen, W.F.; Demel, R.A.; van Deenen, L.L.; Ververgaert, P.H. Freeze-etch electron microscopy of erythrocytes, Acholeplasma laidlawii cells and liposomal membranes after the action of filipin and amphotericin B. Biochim. Biophys. Acta 1973, 291, 577–581. [Google Scholar] [CrossRef]
- Orci, L.; Montesano, R.; Meda, P.; Malaisse-Lagae, F.; Brown, D.; Perrelet, A.; Vassalli, P. Heterogeneous distribution of filipin—Cholesterol complexes across the cisternae of the Golgi apparatus. Proc. Natl. Acad. Sci. USA 1981, 78, 293–297. [Google Scholar] [CrossRef]
- Tillack, T.W.; Kinsky, S.C. A freeze.etch study of the effects of filipin on liposomes and human erythrocyte membranes. Biochim. Biophys. Acta 1973, 323, 43–54. [Google Scholar] [CrossRef]
- Robinson, J.M.; Karnovsky, M.J. Evaluation of the polyene antibiotic filipin as a cytochemical probe for membrane cholesterol. J. Histochem. Cytochem. 1980, 28, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.D.; Blanchette-Mackie, J.; Goldin, E.; O’Neill, R.R.; Carstea, G.; Roff, C.F.; Patterson, M.C.; Patel, S.; Comly, M.E.; Cooney, A.; et al. Progesterone block cholesterol translocation from lysosomes. J. Biol. Chem. 1992, 267, 23797–23805. [Google Scholar] [CrossRef]
- Zhou, S.; Davidson, C.; McGlynn, R.; Stephney, G.; Dobrenis, K.; Vanier, M.T.; Walkley, S.U. Endosomal/lysosomal processing of gangliosides effects neuronal cholesterol sequestration in Niemann-Pick disease type C. Am. J. Pathol. 2011, 179, 890–902. [Google Scholar] [CrossRef]
- Camuso, S.; La Rosa, P.G.; Fiorenza, M.T.; Canterini, S. Pleiotropic effects of BDNF on the cerebellum and hippocampus: Implications for neurodevelopmental disorders. Neurobiol. Dis. 2022, 163, 105606. [Google Scholar] [CrossRef]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef]
- Panigrahi, I.; Dhanorkar, M.; Suthar, R.; Kumar, C.; Baalaaji, M.; Thapa, B.R.; Kalra, J. Niemann-Pick Disease: An Underdiagnosed Lysosomal Storage Disorder. Case Rep. Genet. 2019, 2019, 3108093. [Google Scholar] [CrossRef] [Green Version]
- Breiden, B.; Sandhoff, K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int. J. Mol. Sci. 2020, 21, 2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liscum, L. Niemann-Pick type C mutations cause lipid traffic jam. Traffic 2000, 1, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.A.; Gal, M.R.; Bragshaw, R.D.; Mohr, A.J.; Chue, B.; Richardson, T.; Callahan, J.W. A comparative study of cytoplasmic granules imaged by the real-time microscope, Nile Red and Filipin in fibroblasts from patients with lipid storage diseases. J. Inherit. Metab. Dis. 2005, 28, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Pentchev, P.G.; Comly, M.E.; Kruth, H.S.; Vanier, M.T.; Wenger, D.A.; Patel, S.; Brady, R.O. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc. Natl. Acad. Sci. USA 1985, 82, 8247–8251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Peake, K.B.; Vance, J.E. Defective cholesterol trafficking in Niemann-Pick C-deficient cells. FEBS Lett. 2010, 584, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Oddi, S.; Caporali, P.; Dragotto, J.; Totaro, A.; Maiolati, M.; Scipioni, L.; Angelucci, C.B.; Orsini, C.; Canterini, S.; Rapino, C.; et al. The endocannabinoid system is affected by cholesterol dyshomeostasis: Insights from a murine model of Niemann Pick type C disease. Neurobiol. Dis. 2019, 130, 104531. [Google Scholar] [CrossRef]
- Sokol, J.; Blanchette-Mackie, J.; Kruth, H.S.; Dwyer, N.K.; Amende, L.M.; Butler, J.D.; Robinson, E.; Patel, S.; Bradi, R.O.; Comly, M.E.; et al. Type C Niemann-Pick disease. Lysosomal accumulation and defective intracellular mobilization of low density lipoprotein cholesterol. J. Biol. Chem. 1988, 263, 3411–3417. [Google Scholar] [CrossRef]
- Neufeld, E.B.; Cooney, A.M.; Pitha, J.; Dawidowicz, E.A.; Dwyer, N.K.; Pentchev, P.G.; Blanchette-Mackie, E.J. Intracellular trafficking of cholesterol monitored with a cyclodextrin. J. Biol. Chem. 1996, 271, 21604–21613. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, M.; Di Pietro, C.; La Sala, G.; Fiorenza, M.T.; Marazziti, D.; Canterini, S. Anomalies in Dopmaine Transporter Expression and Primary Cilium Distribution in the Dorsal Striatum of a Mouse Model of Niemann-Pick C1 Disease. Front. Cell. Neurosci. 2019, 13, 226. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Akaboshi, S.; Ishida, G.; Takeshima, T.; Yano, T.; Taniguchi, M.; Ohno, K.; Nakashima, K. Increased levels of GM2 ganglioside in fibroblasts from a patient with juvenile Niemann-Pick disease type C. Brain Dev. 1998, 20, 95–97. [Google Scholar] [CrossRef]
- Kobayashi, T.; Beuchat, M.H.; Lindsay, M.; Frias, S.; Palmiter, R.D.; Sakuraba, H.; Parton, R.G.; Gruenberg, J. Late endosomal membranes rich in lysobisphosphanatidic acid regulate cholesterol transport. Nat. Cell Biol. 1999, 1, 113–118. [Google Scholar] [CrossRef]
- Liscum, L.; Munn, N.J. Intracellular cholesterol transport. Biochim. Biophys. Acta 1999, 1438, 19–37. [Google Scholar] [CrossRef]
- Zervas, M.; Somers, K.L.; Thrall, M.A.; Walkley, S.U. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 2001, 11, 1283–1287. [Google Scholar] [CrossRef] [Green Version]
- te Vruchte, D.; Lloyd-Evans, E.; Veldman, R.J.; Neville, D.C.; Dwek, R.A.; Platt, F.M.; Blitterswijk, W.J.; Sillence, D.J. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J. Biol. Chem. 2004, 279, 26167–26175. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.P.; Levy, B.; Ioannou, Y.A. Evidence for a Niemann-pick C (NPC) gene family: Identification and characterization of NPC1L1. Genomics 2000, 65, 137–145. [Google Scholar] [CrossRef]
- Sun, X.; Marks, D.L.; Park, W.D.; Wheatley, C.L.; Puri, V.; O’ Brien, J.F.; Kraft, D.L.; Lundquist, P.A.; Patterson, M.C.; Pagano, R.E.; et al. Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am. J. Hum. Genet. 2001, 68, 1361–1372. [Google Scholar] [CrossRef] [Green Version]
- Vanier, M.T.; Gissen, P.; Bauer, P.; Coll, M.J.; Burlina, A.; Hendriksz, C.J.; Latour, P.; Goizet, C.; Welford, R.W.; Marquardt, T.; et al. Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Mol. Genet. Metab. 2016, 118, 244–254. [Google Scholar] [CrossRef]
- Burton, B.K.; Ellis, A.G.; Orr, B.; Chatlani, S.; Yoon, K.; Shoaff, J.R.; Gallo, D. Estimating the prevalence of Niemann-Pick disease type C (NPC) in the United States. Mol. Genet. Metab. 2021, 134, 182–187. [Google Scholar] [CrossRef]
- Vanier, M.T. Phenotypic and genetic heterogeneity in Niemann-Pick disease type C: Current knowledge and practical implications. Wien. Klin. Wochenschr. 1997, 109, 68–73. [Google Scholar]
- Wraith, J.E.; Guffon, N.; Rohrbach, M.; Hwu, W.L.; Korenke, G.C.; Bembi, B.; Luzy, C.; Giorgino, R.; Sedel, F. Natural history of Niemann-Pick disease type C in a multicentre observational retrospective cohort study. Mol. Genet. Metab. 2009, 98, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Jurìckovà, K.; Karimzadeh, P.; Kolnikovà, M.; Malinovà, V.; Torres, J.; Kolb, S.A. Evaluation of different suspicion indices in identifying patients with Niemann-Pick disease Type C in clinical practice: A post hoc analysis of a retrospective chart review. Orphanet J. Rare Dis. 2019, 14, 161. [Google Scholar] [CrossRef] [PubMed]
- Harzer, K.; Kustermann-Kuhn, B. Quantified increases of cholesterol, total lipid and globotriaosylceramide in filipin-positive Niemann-Pick type C fibroblasts. Clin. Chim. Acta 2001, 305, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Canterini, S.; Dragotto, J.; Dardis, A.; Zampieri, S.; De Stefano, M.E.; Mangia, F.; Erickson, R.P.; Fiorenza, M.T. Shortened primary cilium lenght and dysregulated Sonic hedgehog signaling in Niemann-Pick C1 disease. Hum. Mol. Genet. 2017, 26, 2277–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, M.C.; Clayton, P.; Gissen, P.; Anheim, M.; Bauer, P.; Bonnot, O.; Dardis, A.; Dionisi-Vici, C.; Klünemann, H.H.; Latour, P.; et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: An update. Neurol. Clin. Pract. 2017, 7, 499–511. [Google Scholar] [CrossRef]
- Millat, G.; Baïlo, N.; Molinero, S.; Rodriguez, C.; Chikh, K.; Vanier, M.T. Niemann-Pick C disease: Use of denaturing high performance liquid chromatography for the detection of NPC1 and NPC2 genetic variations and impact on management of patients and families. Mol. Genet. Metab. 2005, 86, 220–232. [Google Scholar] [CrossRef]
- McKay Bounford, K.; Gissen, P. Genetic and laboratory diagnostic approach in Niemann Pick disease type C. J. Neurol. 2014, 261 (Suppl. 2), S569–S575. [Google Scholar] [CrossRef] [Green Version]
- Patterson, M.C.; Hendriksdz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F.; NP-C Guidelines Working Group. Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef]
- Alobaidy, H. Recent advances in the diagnosis and treatment of niemann-pick disease type C in children: A guide to early diagnosis for the general pediatrician. Int. J. Pediatr. 2015, 2015, 816593. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Sakai, N.; Shinpoo, M.; Noguchi, A.; Takahashi, T.; Matsuda, S.; Yamamoto, M.; Narita, A.; Ohno, K.; Ohashi, T.; et al. The useful preliminary diagnosis of Niemann-Pick disease type C by filipin test in blood smear. Mol. Genet. Metab. 2013, 110, 401–404. [Google Scholar] [CrossRef]
- Hammerschmidt, T.G.; de Oliveira Schmitt Ribas, G.; Saraiva-Pereira, M.L.; Bonatto, M.P.; Kessler, R.G.; Souza, F.T.S.; Trapp, F.; Michelin-Tirelli, K.; Burin, M.G.; Giugliani, R.; et al. Molecular and biochemical biomarkers for diagnosis and therapy monitorization of Niemann-Pick type C patients. Int. J. Dev. Neurosci. 2018, 66, 18–23. [Google Scholar] [CrossRef]
- Vanier, M.T.; Latour, P. Laboratory diagnosis of Niemann-Pick disease type C: The filipin staining test. Methods Cell Biol. 2015, 126, 357–375. [Google Scholar]
- Weismann, C.M.; Ferreira, J.; Keeler, A.M.; Su, Q.; Qui, L.; Shaffer, S.A.; Xu, Z.; Gao, G.; Sena-Esteves, M. Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum. Mol. Genet. 2015, 24, 4353–4364. [Google Scholar] [CrossRef] [Green Version]
- Haddad, M.R.; Choi, E.Y.; Donsante, A.; Zerfas, P.M.; Kaler, S.G. 382. Infrared Fluorescent Protein (iRFP) as a Reporter for Monitoring and Modulating Neurons. Mol. Ther. 2013, 21 (Suppl. 1), 147. [Google Scholar]
- Broekman, M.L.D.; Baek, R.C.; Comer, L.A.; Fernandez, J.L.; Seyfried, T.N.; Sena-Esteves, M. Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus-mediated gene delivery. Mol. Ther. 2007, 15, 30–37. [Google Scholar] [CrossRef]
- Arthur, J.R.; Heinecke, K.A.; Seyfried, T.N. Filipin recognizes both GM1 and cholesterol in GM1 gangliosidosis mouse brain. J. Lipid Res. 2011, 52, 1345–1351. [Google Scholar] [CrossRef] [Green Version]
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 2009, 4, e6951. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Ramirez, C.M.; Miller, A.M.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 2010, 51, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Series Introduction: Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Castello, A.M.; Howard, K.D.; Castaneda, A.J.; Soriano, S. Filipin Levels as a Potential Predictors of Alzheimer’s Disease Risk. Adv. Alzheimers Dis. 2014, 3, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Gonzàlez-Guevara, E.; Càrdenas, G.; Pèrez-Severiano, F.; Martìnez-Lazcano, J.C. Dysregulated Brain Cholesterol Metabolism Is Linked to Neuroinflammation in Huntington’s Disease. Mov. Disord. 2020, 35, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Altomari, N.; Bruno, F.; Laganà, V.; Smirne, N.; Colao, R.; Curcio, S.; Di Lorenzo, R.; Frangipane, F.; Maletta, R.; Puccio, G.; et al. A Comparison of Behavioral and Psychological Symptoms of Dementia (BPSD) and BPSD Sub-Syndromes in Early-Onset and Late-Onset Alzheimer’s Disease. J. Alzheimers Dis. 2022, 85, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Laganà, V.; Bruno, F.; Altomari, N.; Bruni, G.; Smirne, N.; Curcio, S.; Mirabelli, M.; Colao, R.; Puccio, G.; Frangipane, F.; et al. Neuropsychiatric or Behavioral and Psychological Symptoms of Dementia (BPSD): Focus on Prevalence and Natural History in Alzheimer’s Disease and Frontotemporal Dementia. Front. Neurol. 2022, 13, 832199. [Google Scholar] [CrossRef] [PubMed]
- Abondio, P.; Sarno, S.; Giuliani, C.; Laganà, V.; Maletta, R.; Bernardi, L.; Bruno, F.; Colao, R.; Puccio, G.; Frangipane, F.; et al. Amyloid Precursor Protein A713T Mutation in Calabrian Patients with Alzheimer’s Disease: A Population Genomics Approach to Estimate Inheritance from a Common Ancestor. Biomedicines 2021, 10, 20. [Google Scholar] [CrossRef]
- Bruno, F.; Malvaso, A.; Canterini, S.; Bruni, A.C. Antimicrobial Peptides (AMPs) in the Pathogenesis of Alzheimer’s Disease: Implications for Diagnosis and Treatment. Antibiotics 2022, 11, 726. [Google Scholar] [CrossRef]
- Palladino, G.; Nicolia, V.; Kovacs, G.G.; Canterini, S.; Ciraci, V.; Fuso, A.; Mangia, F.; Scarpa, S.; Fiorenza, M.T. Sexually Dimorphic Expression of Reelin in the Brain of a Mouse Model of Alzheimer Disease. J. Mol. Neurosci. 2017, 61, 359–367. [Google Scholar] [CrossRef]
- Nudelman, K.N.H.; McDonald, B.C.; Lahiri, D.K.; Saykin, A.J. Biological Hallmarks of Cancer in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7173–7187. [Google Scholar] [CrossRef]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Gamba, P.; Testa, G.; Sottero, B.; Gargiulo, S.; Poli, G.; Leonarduzzi, G. The link between altered cholesterol metabolism and Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2012, 1259, 54–64. [Google Scholar] [CrossRef]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta-and gamma-secretase activities and Abeta production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Paris, D.; Town, T.; Rojiani, A.M.; Sparks, D.L.; Delledonne, A.; Crawford, F.; Abdullah, L.I.; Humphrey, J.A.; Dickson, D.W.; et al. Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP (SW) mice. J. Neuropathol. Exp. Neurol. 2001, 60, 778–785. [Google Scholar] [CrossRef] [Green Version]
- Panchal, M.; Loeper, J.; Cossec, J.C.; Perruchini, C.; Lazar, A.; Pompon, D.; Duyckaerts, C. Enrichment of cholesterol in microdissected Alzheimer’s disease senile plaques as assessed by mass spectrometry. J. Lipid Res. 2010, 51, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Montesinos, J.; Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Agrawal, R.R.; Velasco, K.R.; Yun, T.D.; Stavrovskaya, I.G.; Xu, Y.; Koo, S.Y.; et al. The Alzheimer’s disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. EMBO J. 2020, 39, e103791. [Google Scholar] [CrossRef]
- Marquer, C.; Laine, J.; Dauphinot, L.; Hanbouch, L.; Lemercier-Neuillet, C.; Pierrot, N.; Bossers, K.; Le, M.; Corlier, F.; Benstaali, C.; et al. Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer’s disease early phenotypes. Mol. Neurodegener. 2014, 9, 60. [Google Scholar] [CrossRef]
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer’s Disease; From Risk Genes to Pathological Effects. Front. Aging Neurosci. 2021, 13, 690372. [Google Scholar] [CrossRef]
- Nicholson, A.M.; Ferreira, A. Increased membrane cholesterol might render mature hippocampal neurons more susceptible to beta-amyloid-induced calpain activation and tau toxicity. J. Neurosci. 2009, 29, 4640–4651. [Google Scholar] [CrossRef] [Green Version]
- Björkhem, I.; Heverin, M.; Leoni, V.; Meaney, S.; Diczfalusy, U. Oxysterols and Alzheimer’s disease. Acta Neurol. Scand. Suppl. 2006, 185, 43–49. [Google Scholar] [CrossRef]
- Björkhem, I.; Cedazo-Minguez, A.; Leoni, V.; Meaney, S. Oxysterols and neurodegenerative diseases. Mol. Asp. Med. 2009, 30, 171–179. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef]
- Lütjohann, D.; Papassotiropoulos, A.; Björkhem, I.; Locatelli, S.; Bagli, M.; Oehring, R.D.; Schlegel, U.; Jessen, F.; Rao, M.L.; von Bergmann, K.; et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J. Lipid Res. 2000, 41, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zeng, F.; Liu, Y.H.; Li, H.Y.; Dong, S.Y.; Peng, Z.Y.; Wang, Y.J.; Zhou, H.D. CYP46A1 and the APOEε4 Allele Polymorphisms Correlate with the Risk of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8179–8187. [Google Scholar] [CrossRef] [PubMed]
- Papassotiropoulos, A.; Lütjohann, D.; Bagli, M.; Locatelli, S.; Jessen, F.; Rao, M.L.; Maier, W.; Björkhem, I.; von Bergmann, K.; Heun, R. Plasma 24S-hydroxycholesterol: A peripheral indicator of neuronal degeneration and potential state marker for Alzheimer’s disease. Neuroreport 2000, 11, 1959–1962. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Vitet, H.; Brandt, V.; Saudou, F. Traffic signaling: New functions of huntingtin and axonal transport in neurological disease. Curr. Opin. Neurobiol. 2020, 63, 122–130. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neuropathology of Huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar]
- Blumenstock, S.; Dudanova, I. Cortical and Striatal Circuits in Huntington’s Disease. Front. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- McAllister, B.; Gusella, J.F.; Landwehrmeyer, G.B.; Lee, J.M.; MacDonald, M.E.; Orth, M.; Rosser, A.E.; Williams, N.M.; Holmans, P.; Jones, L.; et al. Timing and Impact of Psychiatric, Cognitive, and Motor Abnormalities in Huntington Disease. Neurology 2021, 96, e2395–e2406. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. The impairment of cholesterol metabolism in Huntington disease. Biochim. Biophys. Acta 2015, 1851, 1095–1105. [Google Scholar] [CrossRef]
- Kacher, R.; Mounier, C.; Caboche, J.; Betuing, S. Altered Cholesterol Homeostasis in Huntington’s Disease. Front. Aging Neurosci. 2022, 14, 797220. [Google Scholar] [CrossRef]
- Trushina, E.; Singh, R.D.; Dyer, R.B.; Cao, S.; Shah, V.H.; Parton, R.G.; Pagano, R.E.; McMurray, C.T. Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum. Mol. Genet. 2006, 15, 3578–3591. [Google Scholar] [CrossRef] [Green Version]
- Fielding, C.J.; Fielding, P.E. Caveolae and intracellular trafficking of cholesterol. Adv. Drug Deliv. Rev. 2001, 49, 251–264. [Google Scholar] [CrossRef]
- Del Toro, D.; Xifró, X.; Pol, A.; Humbert, S.; Saudou, F.; Canals, J.M.; Alberch, J. Altered cholesterol homeostasis contributes to enhanced excitotoxicity in Huntington’s disease. J. Neurochem. 2010, 115, 153–167. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef] [Green Version]
- Valenza, M.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Fenu, S.; Jamot, L.; Strand, A.; Tarditi, A.; Woodman, B.; Racchi, M.; et al. Dysfunction of the Cholesterol Biosynthetic Pathway in Huntington’s Disease. J. Neurosci. 2005, 25, 9932–9939. [Google Scholar] [CrossRef] [Green Version]
- Valenza, M.; Carroll, J.B.; Leoni, V.; Bertram, L.N.; Björkhem, I.; Singaraja, R.R.; Di Donato, S.; Lutjohann, D.; Hayden, M.R.; Cattaneo, E. Cholesterol biosynthesis pathway is disturbed in YAC128 mice and is modulated by huntingtin mutation. Hum. Mol. Genet. 2007, 16, 2187–2198. [Google Scholar] [CrossRef]
- Leoni, V.; Mariotti, C.; Tabrizi, S.J.; Valenza, M.; Wild, E.J.; Henley, S.M.D.; Hobbs, N.Z.; Mandelli, M.L.; Grisoli, M.; Björkhem, I.; et al. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain 2008, 131, 2851–2859. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Mariotti, C.; Nanetti, L.; Salvatore, E.; Squitieri, F.; Bentivoglio, A.; del Poggio, M.B.; Piacentini, S.; Monza, D.; Valenza, M.; et al. Whole body cholesterol metabolism is impaired in Huntington’s disease. Neurosci. Lett. 2011, 494, 245–249. [Google Scholar] [CrossRef]
- Maltese, W.A. Cholesterol synthesis in cultured skin fibroblasts from patients with Huntington’s disease. Biochem. Med. 1984, 32, 144–150. [Google Scholar] [CrossRef]
- Marullo, M.; Valenza, M.; Leoni, V.; Caccia, C.; Scarlatti, C.; De Mario, A.; Zuccato, C.; Di Donato, S.; Carafoli, E.; Cattaneo, E. Pitfalls in the detection of cholesterol in Huntington’s disease models. PLoS Curr. 2012, 4, e505886e9a1968. [Google Scholar] [CrossRef]
- Wang, C.; Scott, S.M.; Sun, S.; Zhao, P.; Hutt, D.M.; Shao, H.; Gestwicki, E.J.; Balch, E.W. Individualized management of genetic diversity in Niemann-Pick C1 through modulation of the Hsp70 chaperone system. Hum. Mol. Genet. 2019, 29, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bruno, F.; Conidi, M.E.; Puccio, G.; Frangipane, F.; Laganà, V.; Bernardi, L.; Smirne, N.; Mirabelli, M.; Colao, R.; Curcio, S.; et al. A Novel Mutation (D395A) in Valosin-Containing Protein Gene Is Associated With Early Onset Frontotemporal Dementia in an Italian Family. Front. Genet. 2021, 12, 795029. [Google Scholar] [CrossRef] [PubMed]
- Crawley, A.C.; Walkley, S.U. Developmental Analysis of CNS Pathology in the Lysosomal Storage Disease α-Mannosidosis. J. Neuropathol. Exp. Neurol. 2007, 66, 687–697. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, F.; Camuso, S.; Capuozzo, E.; Canterini, S. The Antifungal Antibiotic Filipin as a Diagnostic Tool of Cholesterol Alterations in Lysosomal Storage Diseases and Neurodegenerative Disorders. Antibiotics 2023, 12, 122. https://doi.org/10.3390/antibiotics12010122
Bruno F, Camuso S, Capuozzo E, Canterini S. The Antifungal Antibiotic Filipin as a Diagnostic Tool of Cholesterol Alterations in Lysosomal Storage Diseases and Neurodegenerative Disorders. Antibiotics. 2023; 12(1):122. https://doi.org/10.3390/antibiotics12010122
Chicago/Turabian StyleBruno, Francesco, Serena Camuso, Elisabetta Capuozzo, and Sonia Canterini. 2023. "The Antifungal Antibiotic Filipin as a Diagnostic Tool of Cholesterol Alterations in Lysosomal Storage Diseases and Neurodegenerative Disorders" Antibiotics 12, no. 1: 122. https://doi.org/10.3390/antibiotics12010122