
Pharmacokinetics of Antimicrobials in Children with Emphasis on Challenges Faced by Low and Middle Income Countries, a Clinical Review

 , , , , and
, , , , and 
Abstract
:1. Introduction
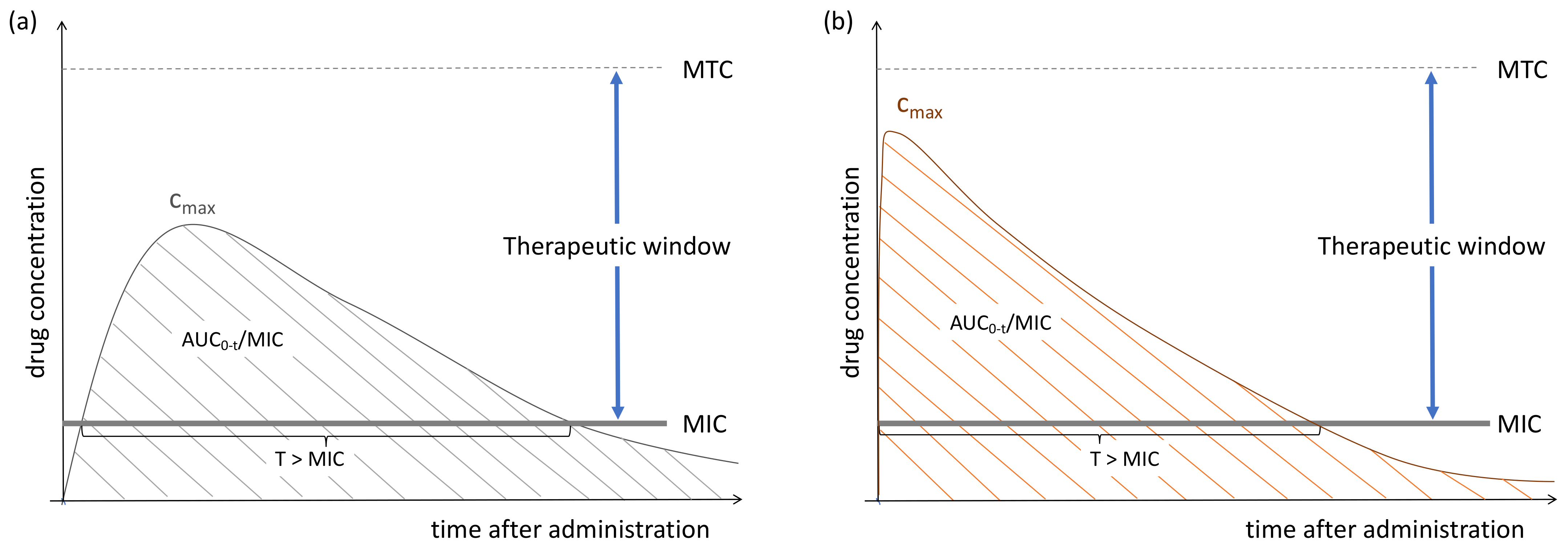
2. Pharmacokinetic–Pharmacodynamic Interaction of Antimicrobials
3. Pharmacokinetics: Drug Transport through Cell Membranes
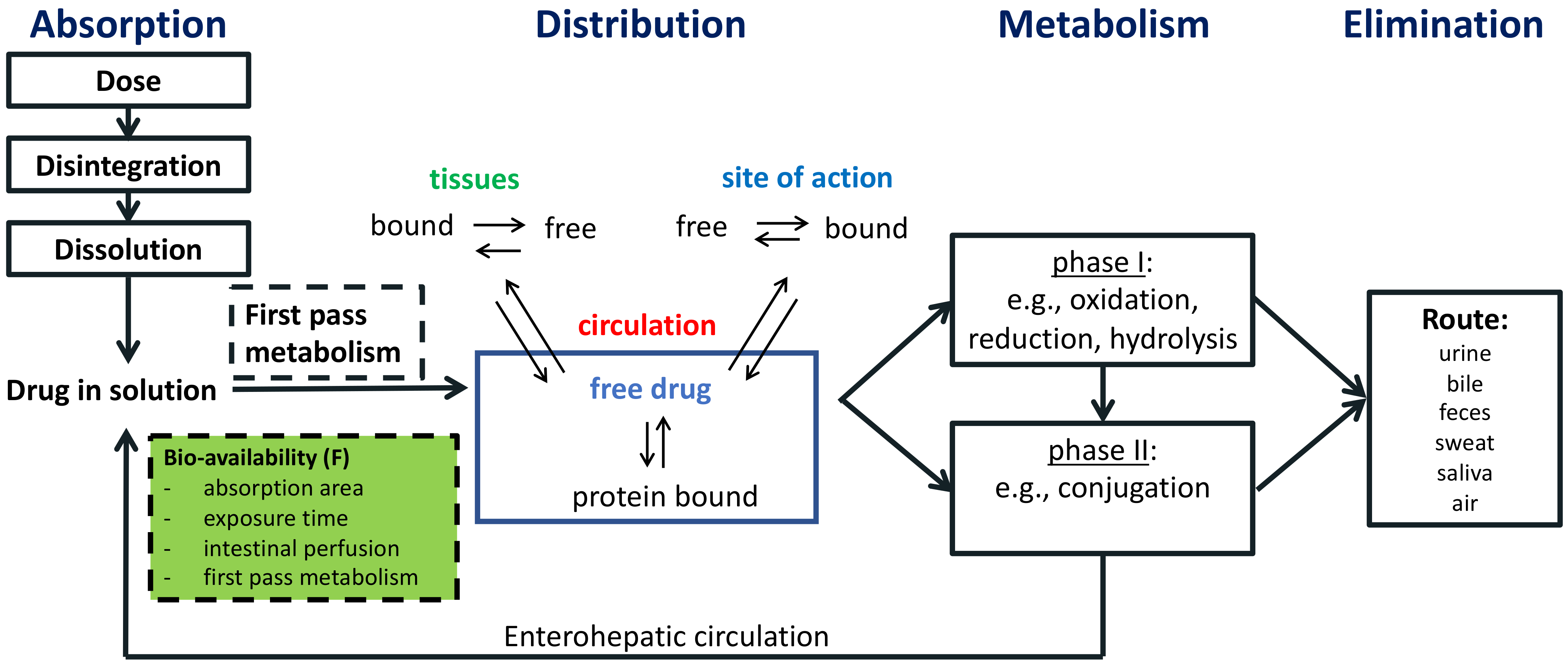
4. Pharmacokinetic Processes
4.1. Absorption
4.2. Distribution

{kind=link}
{kind=link}
| Agent | Cerebrospinal Fluid (CSF) Penetration |
|---|---|
| Aminoglycosides | |
| Amikacin Gentamicin Tobramycin | Systemic amikacin, gentamicin and tobramycin penetrate the CSF of inflamed meninges to a limited extent. Their clinical use for CNS infections is restricted by toxicities if administered intravenously. Intrathecal doses of amikacin, gentamicin, and tobramycin have been reported to be effective and well tolerated [90,91]. No information available for other aminoglycosides. |
| Antimycobacterials | |
| Ethambutol | Limited data suggest poor to moderate CSF penetration of inflamed meninges [92]. |
| Isoniazid | CSF concentration comparable with plasma concentration in inflamed meninges [92]. |
| Pyrazinamide | CSF concentration comparable with plasma concentration in inflamed meninges [92]. |
| Rifabutin | Higher CSF penetration than rifampicin, but toxicities may restrict its use in CNS infections [93].] |
| Rifampicin | Moderate CSF penetration at standard doses, therefore higher doses may be necessary for adequate CSF penetration [91,92]. |
| Bedaquiline | Bedaquiline penetrated freely into the CSF of adults under treatment with pulmonary tuberculosis [94]. |
| Clofazamine | Poor CSF penetration, which may be improved by chemical modification [95]. |
| Cycloserine | Good CSF penetration of inflamed meninges [92,96]. |
| Ethionamide | Good CSF penetration [90]. |
| Delamanid | Very limited clinical data available, low total CSF levels [97]. |
| Beta-lactamase inhibitors | |
| Avibactam | No data available. |
| Clavulanic acid | Very limited data suggest that amoxicillin-clavulanate may be effective for the treatment of bacterial meningitis [98,99]. |
| Sulbactam | Very high CSF:plasma concentrations in combination with ampicillin [91]. However, clinical experience with this agent for meningitis is limited. |
| Tazobactam | No clinical data available. |
| Vaborbactam | No clinical data available. |
| Carbapenems | |
| Doripenem | No clinical data available. |
| Ertapenem | No clinical data available. |
| Imipenem | Measurable CSF penetrations, but high proconvulsive activity may restrict its use [100]. |
| Meropenem | CSF concentrations adequate for treating meningitis [91]. |
| Cephalosporins | |
| Cephalexin | Usually ineffective due to lower CSF:serum concentrations [91]. |
| Cefazolin | CSF concentrations of uninflamed meninges close to the MIC of moderately susceptible bacteria [90]. |
| Cefadroxil | Usually ineffective due to lower CSF:serum concentrations [91]. |
| Cefaclor | No clinical data available. |
| Cefotetan | No clinical data available. |
| Cefoxitin | No clinical data available. |
| Cefprozil | No clinical data available. |
| Cefuroxime | Reaches CSF concentrations in excess of MIC [91]. |
| Cephamycin | No clinical data available. |
| Cefdinir | No clinical data available. |
| Cefepime | Adequate CSF penetration for treatment of meningitis [90]. |
| Cefixime | Cefixime crosses the blood brain barrier of inflamed meninges, but at limited concentrations and should therefore not be used to treat meningitis [91]. |
| Cefotaxime | Adequate CSF penetration [90,91] |
| Ceftriaxone | Ceftriaxone has an adequate CSF penetration of inflamed meninges. CSF concentrations are lower compared with cefotaxime, most likely given the higher degree of protein binding of ceftriaxone. Nevertheless, ceftriaxone is an adequate agent for treatment of meningitis [90,91]. |
| Ceftaroline | Different case studies reported that ceftaroline attained CSF concentration above MIC [101,102,103]. |
| Ceftazidime | CSF attains therapeutic levels in CSF [90,91]. |
| Ceftizoxime | Limited clinical data available suggest that ceftizoxime penetrates CSF [91]. |
| Ceftobiprole | No clinical data available, clinical study ongoing (NCT04178629). |
| Cefiderocol | Very limited clinical data available in humans suggests that cefiderocol CSF concentrations in meningitis exceed MIC of gram negative organisms [104]. |
| Fluoroquinolones | |
| Ciprofloxacin Delafloxacin Gatifloxacin Gemifloxacin Levofloxacin Moxifloxacin Norfloxacin Ofloxacin | As a group, fluoroquinolones demonstrate excellent CSF penetration. Clinical data are only available for ciprofloxacin, ofloxacin, levofloxacin and moxifloxacin [90]. |
| Glycopeptides | |
| Teicoplanin | The high protein binding of teicoplanin restricts CSF penetration after IV administration [100]. |
| Vancomycin | Vancomycin is highly hydrophilic and may reach sub therapeutic CSF concentration at conventional doses, but adequate concentrations at increased doses [91]. |
| Dalbavancin | No clinical data available. |
| Telavancin | No clinical data available. |
| Glycylcycline | |
| Tigecycline | Limited clinical data available suggest that tigecycline reaches adequate concentrations of inflamed meninges [90]. |
| Lincosamides | |
| Clindamycin Lincomycin | Lincomycin and its derivative Clindamycin is considered to have poor CSF penetration [91]. |
| Monobactams | |
| Aztreonam | Scant clinical data available suggest that aztreonam reaches sufficient CSF concentrations after systemic administration in inflamed meninges [45]. |
| Macrolides | |
| Azithromycin Clarithromycin Erythromycin Fidaxomicin | Macrolides have been unable to reach therapeutic CSF concentrations in adults [91]. |
| Nitroimidazoles | |
| Metronidazole | Good CSF penetration in both inflamed and no inflamed meninges [90,91]. |
| Tinidazole | No clinical data available. |
| Oxazolidinones | |
| Linezolid | CSF concentrations above the MIC of susceptible pathogens both with inflamed and uninflamed meninges [90]. |
| Tedizolid | No clinical data available. |
| Penicillins | |
| Penicillin G Penicillin V | Good CSF concentrations after intravenous administration [91]. |
| Temocillin | Very limited clinical data available suggest that temocillin may reach therapeutic concentrations in the CSF of patients with gram negative meningitis, but more data are necessary to assess this [105]. |
| Amoxicillin Ampicillin | Good CSF penetrations after IV administration [91]. |
| Cloxacillin | Penetrates in CSF of inflamed meninges to a limited extent, therefore higher doses may be necessary to attain therapeutic targets [106]. Furthermore, therapy failure has been described in patients under treatment for Staphylococcus meningitis [107]. |
| Flucloxacillin | Penetrates in CSF of inflamed meninges to a limited extent, therefore higher doses may be necessary to attain therapeutic targets [106]. |
| Nafcillin | Insufficient CSF penetration for treatment of meningitis [91]. |
| Oxacillin | Limited CSF diffusion at conventional doses [108]. |
| Piperacillin | Crosses the inflamed and non-inflamed blood-brain barrier but in unpredictable amounts [109]. |
| Ticarcillins | Very limited data available, rather low and variable CSF concentrations after administration of ticarcillin-clavulanate [110]. |
| Polymyxins | |
| Polymyxin B Polymyxin E (Colistin) | Limited clinical data available suggest very low CSF penetration after systemic administration [111]. |
| Sulfonamides | |
| Sulfamethoxazole | High doses achieve good CSF concentrations both with inflamed and uninflamed meninges [90]. |
| Tetracyclines | |
| Doxycycline | Limited clinical data, same CSF penetration in both inflamed and uninflamed meninges [90]. |
| Minocycline | No clinical data available. |
| Miscellaneous | |
| Chloramphenicol | Chloramphenicol penetrates well into CSF, but significant toxicities prohibit the clinical use [90,91]. |
| Daptomycin | Limited PK data available on CSF penetration. Some case reports described the successful use of daptomycin in meningitis. |
| Fosfomycin | Enters the CSF in the presence and absence of meningeal inflammation [90]. |
4.3. Metabolism
4.4. Elimination
5. Therapeutic Drug Monitoring
6. Challenges in Attaining Effective Drug Concentrations in Children Living in LMIC
6.1. Co-Morbidities
6.2. Altered Polymorphisms
6.3. Challenges with Healthcare Administration
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Strong, K.L.; Pedersen, J.; Johansson, E.W.; Cao, B.; Diaz, T.; Guthold, R.; You, D.; Requejo, J.; Liu, L. Patterns and Trends in Causes of Child and Adolescent Mortality 2000–2016: Setting the Scene for Child Health Redesign. BMJ Glob. Health 2021, 6, e004760. [Google Scholar] [CrossRef] [PubMed]
- Perin, J.; Mulick, A.; Yeung, D.; Villavicencio, F.; Lopez, G.; Strong, K.L.; Prieto-Merino, D.; Cousens, S.; Black, R.E.; Liu, L. Global, Regional, and National Causes of under-5 Mortality in 2000–19: An Updated Systematic Analysis with Implications for the Sustainable Development Goals. Lancet Child Adolesc. Health 2022, 6, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Le Doare, K.; Bielicki, J.; Heath, P.T.; Sharland, M. Systematic Review of Antibiotic Resistance Rates among Gram-Negative Bacteria in Children with Sepsis in Resource-Limited Countries. J. Pediatr. Infect. Dis. Soc. 2015, 4, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Copana, R.; Morales, R.; Anugulruengkitt, S.; Puthanakit, T.; Maramba-Lazarte, C.; Williams, P.; Musembi, J.; Boga, M.; Issack, M.; et al. Keeping It Real: Antibiotic Use Problems and Stewardship Solutions in Low- and Middle-Income Countries. Pediatr. Infect. Dis. J. 2022, 41, S18–S25. [Google Scholar] [CrossRef]
- Villanueva, P.; Coffin, S.E.; Mekasha, A.; McMullan, B.; Cotton, M.F.; Bryant, P.A. Comparison of Antimicrobial Stewardship and Infection Prevention and Control Activities and Resources between Low-/Middle- and High-Income Countries. Pediatr. Infect. Dis. J. 2022, 41, S3–S9. [Google Scholar] [CrossRef]
- Gebretekle, G.B.; Haile Mariam, D.; Abebe Taye, W.; Mulu Fentie, A.; Amogne Degu, W.; Alemayehu, T.; Beyene, T.; Libman, M.; Gedif Fenta, T.; Yansouni, C.P.; et al. Half of Prescribed Antibiotics Are Not Needed: A Pharmacist-Led Antimicrobial Stewardship Intervention and Clinical Outcomes in a Referral Hospital in Ethiopia. Front. Public Health 2020, 8, 109. [Google Scholar] [CrossRef] [Green Version]
- Guiastrennec, B.; Ramachandran, G.; Karlsson, M.O.; Kumar, A.K.H.; Bhavani, P.K.; Gangadevi, N.P.; Swaminathan, S.; Gupta, A.; Dooley, K.E.; Savic, R.M. Suboptimal Antituberculosis Drug Concentrations and Outcomes in Small and HIV-Coinfected Children in India: Recommendations for Dose Modifications. Clin. Pharmacol. Ther. 2018, 104, 733–741. [Google Scholar] [CrossRef]
- Odenholt, I.; Gustafsson, I.; Löwdin, E.; Cars, O. Suboptimal Antibiotic Dosage as a Risk Factor for Selection of Penicillin-Resistant Streptococcus pneumoniae: In Vitro Kinetic Model. Antimicrob. Agents Chemother. 2003, 47, 518–523. [Google Scholar] [CrossRef] [Green Version]
- European Committee on Antimicrobial Susceptibility Testing. Available online: https://www.eucast.org/clinical_breakpoints/ (accessed on 6 June 2022).
- Onufrak, N.J.; Forrest, A.; Gonzalez, D. Antibiotics PK/PD. Clin. Ther. 2016, 38, 1930–1947. [Google Scholar] [CrossRef]
- Turfus, S.C.; Delgoda, R.; Picking, D.; Gurley, B.J. Chapter 25—Pharmacokinetics. In Pharmacognosy; Badal, S., Delgoda, R., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 495–512. [Google Scholar] [CrossRef]
- Rodríguez-Gascón, A.; Solinís, M.Á.; Isla, A. The Role of Pk/Pd Analysis in the Development and Evaluation of Antimicrobials. Pharmaceutics 2021, 13, 833. [Google Scholar] [CrossRef]
- Mansoor, A.; Mahabadi, N. Volume of Distribution. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK545280/ (accessed on 2 September 2022).
- Wanat, K. Biological Barriers, and the Influence of Protein Binding on the Passage of Drugs across Them. Mol. Biol. Rep. 2020, 47, 3221–3231. [Google Scholar] [CrossRef] [Green Version]
- Gaohua, L.; Miao, X.; Dou, L. Crosstalk of Physiological PH and Chemical pKa under the Umbrella of Physiologically Based Pharmacokinetic Modeling of Drug Absorption, Distribution, Metabolism, Excretion, and Toxicity. Expert Opin. Drug Metab. Toxicol. 2021, 17, 1103–1124. [Google Scholar] [CrossRef]
- Deb, P.K.; Al-Attraqchi, O.; Prasad, M.R.; Tekade, R.K. Protein and Tissue Binding: Implication on Pharmacokinetic Parameters. Implication on Pharmacokinetic Parameters; Elsevier: Amsterdam, The Netherlands, 2018; ISBN 9780128144244. [Google Scholar]
- Gao, Y.; Gesenberg, C.; Zheng, W. Oral Formulations for Preclinical Studies: Principle, Design, and Development Considerations; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780128024478. [Google Scholar]
- Manallack, D.T. The pKa Distribution of Drugs: Application to Drug Discovery. Perspect. Med. Chem. 2007, 1, 1177391X0700100. [Google Scholar] [CrossRef]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 4 September 2022).
- DrugBank Online. Available online: https://go.drugbank.com/ (accessed on 7 September 2022).
- Asín-prieto, E.; Rodríguez-gasc, A. Applications of the Pharmacokinetic/Pharmacodynamic (PK/PD) Analysis of Antimicrobial Agents. J. Infect. Chemother. 2015, 21, 319–329. [Google Scholar] [CrossRef]
- PubChem. Neomycin (Compound). Available online: https://pubchem.ncbi.nlm.nih.gov/compound/8378 (accessed on 22 September 2022).
- Gumbo, T.; Angulo-barturen, I.; Ferrer-bazaga, S. Pharmacokinetic-Pharmacodynamic and Dose-Response Relationships of Antituberculosis Drugs: Recommendations and Standards for Industry and Academia. J. Infect. Dis. 2015, 211, S96–S106. [Google Scholar] [CrossRef] [Green Version]
- Sekaggya-wiltshire, C.; Dooley, K.E. Expert Opinion on Drug Metabolism & Toxicology Pharmacokinetic and Pharmacodynamic Considerations of Rifamycin Antibiotics for the Treatment of Tuberculosis. Expert Opin. Drug Metab. Toxicol. 2019, 15, 615–618. [Google Scholar] [CrossRef] [Green Version]
- WHO. Guideline Updates on the Management of Severe Acute Malnutrition in Infants and Children; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- PubChem. Cycloserine (Compound). Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cycloserine#section=Absorption-Distribution-and-Excretion (accessed on 3 October 2022).
- Deshpande, D.; Pasipanodya, J.G.; Mpagama, S.G.; Srivastava, S.; Bendet, P.; Koeuth, T.; Lee, P.S.; Heysell, S.K.; Gumbo, T. Ethionamide Pharmacokinetics/Pharmacodynamics- Derived Dose, the Role of MICs in Clinical Outcome, and the Resistance Arrow of Time in Multidrug-Resistant Tuberculosis. Clin. Infect. Dis. 2018, 67, S317–S326. [Google Scholar] [CrossRef]
- Adembri, C.; Novelli, A.; Nobili, S. Some Suggestions from PK/PD Principles to Contain Resistance in the Clinical Setting—Focus on ICU Patients and Gram-Negative Strains. Antibiotics 2020, 9, 676. [Google Scholar] [CrossRef]
- Iyer, R.N. Beta Lactam. In Comprehensive Pharmacology; Elsevier: Amsterdam, The Netherlands, 2022; pp. 3–63. [Google Scholar]
- DrugBank. Online Monograph Vaborbactam. Available online: https://go.drugbank.com/drugs/DB12107 (accessed on 27 September 2022).
- DrugBank. Online Monograph Doripenem. Available online: https://go.drugbank.com/drugs/DB06211 (accessed on 27 September 2022).
- Landersdorfer, C.B.; Nation, R.L. Limitations of Antibiotic MIC-Based PK-PD Metrics: Looking Back to Move Forward. Front. Pharmacol. 2021, 12, 770518. [Google Scholar] [CrossRef]
- Cohen, R.; Grimprel, E. Antibiotic Pharmacokinetic and Pharmacodynamic Parameters in Pediatric Clinical Practice. Arch. Pédiatrie 2017, 24, S6–S8. [Google Scholar] [CrossRef] [PubMed]
- Dellamonica, P. Cefuroxime Axetil. Int. J. Antimicrob. Agents 1994, 4, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, C.; Muller, A.E.; Van Den Broek, P.; Cruz De Almeida, B.D.M.; Van Den Berg, C.; Van Oldenrijk, J.; Bos, P.K.; Koch, B.C.P. An Overview of the Protein Binding of Cephalosporins in Human Body Fluids: A Systematic Review. Front. Pharmacol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Cefmetazole. Available online: http://www.antimicrobe.org/drugpopup/Cefmetazole.htm (accessed on 29 September 2022).
- Padda, I.S.; Nagalli, S. Cefotaxime. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560653/ (accessed on 29 September 2022).
- Ramón, J.; Perea, A. Ceftobiprole Review Ceftobiprole: Pharmacokinetics and PK/PD Profile. Rev. Española Quimioter. 2019, 32, 11–16. [Google Scholar]
- Prats, G.; Rossi, V.; Salvatori, E.; Mirelis, B. Prulifloxacin: A New Antibacterial Fluoroquinolone. Expert Rev. Anti-Infect. Ther. 2006, 4, 27–41. [Google Scholar] [CrossRef]
- Wenzler, E.; Liao, S.; Rodvold, K.A. Pharmacodynamics of Lipoglycopeptides. In Antibiotic Pharmacodynamics; Humana: Towota, NJ, USA, 2016; pp. 285–315. [Google Scholar]
- DrugBank Online. Teicoplanin. Available online: https://go.drugbank.com/drugs/DB06149 (accessed on 1 October 2022).
- Dalbavancin for Injection, Product Monograph. Available online: https://pdf.hres.ca/dpd_pm/00060897.PDF (accessed on 1 October 2022).
- DrugBank Online. Clindamycin. Available online: https://go.drugbank.com/drugs/DB01190 (accessed on 1 October 2022).
- Ramsey, C.; Macgowan, A.P. A Review of the Pharmacokinetics and Pharmacodynamics of Aztreonam. J. Antimicrob. Chemother. 2016, 71, 2704–2712. [Google Scholar] [CrossRef] [Green Version]
- Child, J.; Chen, X.; Mistry, R.D.; Somme, S.; Macbrayne, C.; Anderson, P.L.; Jones, R.N.; Parker, S.K. Pharmacokinetic and Pharmacodynamic Properties of Metronidazole in Pediatric Patients with Acute Appendicitis: A Prospective Study. J. Pediatr. Infect. Dis. Soc. 2019, 8, 297–302. [Google Scholar] [CrossRef]
- Ampicillin. Available online: https://web.archive.org/web/20150712001731/http:/www.drugs.com/monograph/ampicillin.html#r9 (accessed on 10 October 2022).
- Fidaxomicin (DificidTM) Monograph. Available online: https://www.pbm.va.gov/PBM/clinicalguidance/drugmonographs/FidaxomicinMonograph.doc (accessed on 12 October 2022).
- Oxacillin—Drug Summary. Available online: https://www.pdr.net/drug-summary/Oxacillin-oxacillin-3066 (accessed on 10 October 2022).
- Alexandre, K.; Fantin, B. Pharmacokinetics and Pharmacodynamics of Temocillin. Clin. Pharmacokinet. 2018, 57, 287–296. [Google Scholar] [CrossRef]
- Al-Shaer, M.H.; Alghamdi, W.A.; Graham, E.; Peloquin, C.A. Meropenem, Cefepime, and Piperacillin Protein Binding in Patient Samples. Ther. Drug Monit. 2020, 42, 129–132. [Google Scholar] [CrossRef]
- Sulfadiazine. Available online: http://www.antimicrobe.org/drugpopup/sulfadiazine.htm (accessed on 6 October 2022).
- Sulfadoxine. Available online: http://www.antimicrobe.org/drugpopup/sulfadoxine.htm (accessed on 6 October 2022).
- Agwuh, K.N.; Macgowan, A. Pharmacokinetics and Pharmacodynamics of the Tetracyclines Including Glycylcyclines. J. Antimicrob. Chemother. 2006, 58, 256–265. [Google Scholar] [CrossRef]
- Lee, B.L.; Sachdeva, M.; Chambers, H.F. Effect of Protein Binding of Daptomycin on MIC and Antibacterial Activity. Antimicrob. Agents Chemother. 1991, 35, 2505–2508. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Saw, S. Daptomycin. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470407/ (accessed on 8 October 2022).
- Van Heeswijk, R.P.G.; Dannemann, B.; Hoetelmans, R.M.W. Bedaquiline: A Review of Human Pharmacokinetics and Drug–Drug Interactions. J. Antimicrob. Chemother. 2014, 69, 2310–2318. [Google Scholar] [CrossRef]
- Hoffmann, M.; DeMaio, W.; Jordan, R.A.; Talaat, R.; Harper, D.; Speth, J.; Scatina, J. Metabolism, Excretion, and Pharmacokinetics of [14C]Tigecycline, a First-in-Class Glycylcycline Antibiotic, after Intravenous Infusion to Healthy Male Subjects. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1543–1553. [Google Scholar] [CrossRef] [Green Version]
- Doogue, M.P.; Polasek, T.M. The ABCD of Clinical Pharmacokinetics. Ther. Adv. Drug Saf. 2013, 4, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, J.M.; Bouzom, F.; Hugues, C.; Ungell, A.L. Oral Drug Absorption in Pediatrics: The Intestinal Wall, Its Developmental Changes and Current Tools for Predictions. Biopharm. Drug Dispos. 2017, 38, 209–230. [Google Scholar] [CrossRef] [Green Version]
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-Based Mechanistic Prediction of Oral Drug Absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Debotton, N.; Dahan, A. A Mechanistic Approach to Understanding Oral Drug Absorption in Pediatrics: An Overview of Fundamentals. Drug Discov. Today 2014, 19, 1322–1336. [Google Scholar] [CrossRef]
- Kearns, G.L.; Abdel-Rahman, S.M.; Alander, S.W.; Blowey, D.L.; Leeder, J.S.; Kauffman, R.E. Developmental Pharmacology—Drug Disposition, Action, and Therapy in Infants and Children. New Engl. J. Med. 2003, 349, 1157–1167. [Google Scholar] [CrossRef]
- Reiter, P.D. Neonatal Pharmacology and Pharmacokinetics. NeoReviews 2002, 3, e229–e236. [Google Scholar] [CrossRef]
- Strolin, M.; Whomsley, R.; Baltes, E.L. Differences in Absorption, Distribution, Metabolism and Excretion of Xenobiotics between the Paediatric and Adult Populations. Expert Opin. Drug Metab. Toxicol. 2005, 1, 447–471. [Google Scholar] [CrossRef]
- Deng, J.; Zhu, X.; Chen, Z.; Fan, C.H.; Kwan, H.S.; Wong, C.H.; Shek, K.Y.; Zuo, Z.; Lam, T.N. A Review of Food–Drug Interactions on Oral Drug Absorption. Drugs 2017, 77, 1833–1855. [Google Scholar] [CrossRef] [PubMed]
- Mooij, M.G.; De Koning, B.A.; Huijsman, M.L.; De Wildt, S.N. Ontogeny of Oral Drug Absorption Processes in Children. Expert Opin. Drug Metab. Toxicol. 2012, 8, 1293–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keij, F.M.; Kornelisse, R.F.; Hartwig, N.G.; Reiss, I.K.M.; Allegaert, K.; Tramper-Stranders, G.A. Oral Antibiotics for Neonatal Infections: A Systematic Review and Meta-Analysis. J. Antimicrob. Chemother. 2019, 74, 3150–3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulds, G.; Luke, D.R.; Teng, R.; Willavize, S.A.; Friedman, H.; Curatolo, W.J. The Absence of an Effect of Food on the Bioavailability of Azithromycin Administered as Tablets, Sachet or Suspension. J. Antimicrob. Chemother. 1996, 37, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshelman, F.N.; Spyker, D.A. Pharmacokinetics of Amoxicillin and Ampicillin: Crossover Study of the Effect of Food. Antimicrob. Agents Chemother. 1978, 14, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Augmentin® (Amoxicillin/Clavulanate Potassium) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/050564s051lbl.pdf (accessed on 5 October 2022).
- Fassbender, M.; Lode, H.; Schaberg, T.; Borner, K.; Koeppe, P. Pharmacokinetics of New Oral Cephalosporins, Including a New Carbacephem. Clin. Infect. Dis. 1993, 16, 646–653. [Google Scholar] [CrossRef]
- Williams, P.E.O.; Harding, S.M. The Absolute Bioavailability of Oral Cefuroxime Axetil in Male and Female Volunteers after Fasting and after Food. J. Antimicrob. Chemother. 1984, 13, 191–196. [Google Scholar] [CrossRef]
- Sommers, D.; Wyk, M.; Moncrieff, J.; Schoeman, H. Influence of Food and Reduced Gastric Acidity on the Bioavailability of Bacampicillin and Cefuroxime Axetil. Br. J. Clin. Pharmacol. 1984, 18, 535–539. [Google Scholar] [CrossRef] [Green Version]
- Neuvonen, P.J.; Kivistö, K.T.; Lehto, P. Interference of Dairy Products with the Absorption of Ciprofloxacin. Clin. Pharmacol. Ther. 1991, 50, 498–502. [Google Scholar] [CrossRef]
- Spenard, J.; Aumais, C.; Massicotte, J.; Brunet, J.-S.; Tremblay, C.; Grace, M.; Lefebvre, M. Effects of Food and Formulation on the Relative Bioavailability of Bismuth Biskalcitrate, Metronidazole, and Tetracycline given for Helicobacter Pylori Eradication. Br. J. Clin. Pharmacol. 2005, 60, 374–377. [Google Scholar] [CrossRef]
- Peloquin, C.A.; Namdar, R.; Singleton, M.D.; Nix, D.E. Pharmacokinetics of Rifampin Under Fasting Conditions, with Food, and with Antacids. Chest 1999, 115, 12–18. [Google Scholar] [CrossRef]
- Bushra, R.; Aslam, N.; Khan, A. Food Drug Interactions. Oman Med. J. 2011, 26, 77–83. [Google Scholar] [CrossRef]
- Rutter, N. Percutaneous Drug Absorption in the Newborn: Hazards and Uses. Clin. Perinatol. 1987, 14, 911–930. [Google Scholar] [CrossRef]
- Brunton, L.L.; Hilal-Dandan, R.; Knollmann, B.C. Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 13th ed.; Shanahan, J.F., Lebowitz, H., Eds.; McGraw-Hill Education: New York, NY, USA, 2018; ISBN 978-1-25-958473-9. [Google Scholar]
- Shah, S.; Barton, G.; Fischer, A. Pharmacokinetic Considerations and Dosing Strategies of Antibiotics in the Critically Ill Patient. J. Intensive Care Soc. 2015, 16, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Katzung, B.G. Basic and Clinical Pharmacology, 14th ed.; McGraw-Hill: New York, NY, USA, 2018; ISBN 978-1-259-64115-2. [Google Scholar]
- Batchelor, H.K.; Marriott, J.F. Paediatric Pharmacokinetics: Key Considerations. Br. J. Clin. Pharmacol. 2015, 79, 395–404. [Google Scholar] [CrossRef] [Green Version]
- McNamara, P.J.; Alcorn, J. Protein Binding Predictions in Infants. AAPS PharmSci 2002, 4, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Ulldemolins, M.; Roberts, J.A.; Rello, J.; Paterson, D.L.; Lipman, J. The Effects of Hypoalbuminaemia on Optimizing Antibacterial Dosing in Critically Ill Patients. Clin. Pharmacokinet. 2011, 50, 99–110. [Google Scholar] [CrossRef]
- Saunders, N.R.; Liddelow, S.A.; Dziegielewska, K.M. Barrier Mechanisms in the Developing Brain. Front. Pharmacol. 2012, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- Verscheijden, L.F.M.; van Hattem, A.C.; Pertijs, J.C.L.M.; de Jongh, C.A.; Verdijk, R.M.; Smeets, B.; Koenderink, J.B.; Russel, F.G.M.; de Wildt, S.N. Developmental Patterns in Human Blood–Brain Barrier and Blood–Cerebrospinal Fluid Barrier ABC Drug Transporter Expression. Histochem. Cell Biol. 2020, 154, 265–273. [Google Scholar] [CrossRef]
- Lam, J.; Baello, S.; Iqbal, M.; Kelly, L.E.; Shannon, P.T.; Chitayat, D.; Matthews, S.G.; Koren, G. The Ontogeny of P-Glycoprotein in the Developing Human Blood–Brain Barrier: Implication for Opioid Toxicity in Neonates. Pediatr. Res. 2015, 78, 417–421. [Google Scholar] [CrossRef]
- Haritova, A. A role of p-glycoprotein in modulation of antibiotic pharmacokinetics. Trakia J. Sci. 2008, 6, 1–6. [Google Scholar]
- Nau, R.; Seele, J.; Djukic, M.; Eiffert, H. Pharmacokinetics and Pharmacodynamics of Antibiotics in Central Nervous System Infections. Curr. Opin. Infect. Dis. 2018, 31, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Sullins, A.K.; Abdel-Rahman, S.M. Pharmacokinetics of Antibacterial Agents in the CSF of Children and Adolescents. Pediatr. Drugs 2013, 15, 93–117. [Google Scholar] [CrossRef] [PubMed]
- Donald, P.R. Cerebrospinal Fluid Concentrations of Antituberculosis Agents in Adults and Children. Tuberculosis 2010, 90, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Crabol, Y.; Catherinot, E.; Veziris, N.; Jullien, V.; Lortholary, O. Rifabutin: Where Do We Stand in 2016? J. Antimicrob. Chemother. 2016, 71, 1759–1771. [Google Scholar] [CrossRef] [Green Version]
- Upton, C.M.; Steele, C.I.; Maartens, G.; Diacon, A.H.; Wiesner, L.; Dooley, K.E. Pharmacokinetics of Bedaquiline in Cerebrospinal Fluid (CSF) in Patients with Pulmonary Tuberculosis (TB). J. Antimicrob. Chemother. 2022, 77, 1720–1724. [Google Scholar] [CrossRef]
- de Castro, R.R.; do Carmo, F.A.; Martins, C.; Simon, A.; de Sousa, V.P.; Rodrigues, C.R.; Cabral, L.M.; Sarmento, B. Clofazimine Functionalized Polymeric Nanoparticles for Brain Delivery in the Tuberculosis Treatment. Int. J. Pharm. 2021, 602, 120655. [Google Scholar] [CrossRef]
- Kempker, R.R.; Smith, A.G.C.; Avaliani, T.; Gujabidze, M.; Bakuradze, T.; Sabanadze, S.; Avaliani, Z.; Collins, J.M.; Blumberg, H.M.; Alshaer, M.H.; et al. Cycloserine and Linezolid for Tuberculosis Meningitis: Pharmacokinetic Evidence of Potential Usefulness. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2022, 75, 682–689. [Google Scholar] [CrossRef]
- Tucker, E.W.; Pieterse, L.; Zimmerman, M.D.; Udwadia, Z.F.; Peloquin, C.A.; Gler, M.T.; Ganatra, S.; Tornheim, J.A.; Chawla, P.; Caoili, J.C.; et al. Delamanid Central Nervous System Pharmacokinetics in Tuberculous Meningitis in Rabbits and Humans. Antimicrob. Agents Chemother. 2019, 63, e00913-19. [Google Scholar] [CrossRef] [Green Version]
- Bakken, J.S.; Bruun, J.N.; Gaustad, P.; Tasker, T.C. Penetration of Amoxicillin and Potassium Clavulanate into the Cerebrospinal Fluid of Patients with Inflamed Meninges. Antimicrob. Agents Chemother. 1986, 30, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Münch, R.; Lüthy, R.; Blaser, J.; Siegenthaler, W. Human Pharmacokinetics and CSF Penetration of Clavulanic Acid. J. Antimicrob. Chemother. 1981, 8, 29–37. [Google Scholar] [CrossRef]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of Drugs through the Blood-Cerebrospinal Fluid/Blood-Brain Barrier for Treatment of Central Nervous System Infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [Green Version]
- Cies, J.J.; Moore, W.S.; Enache, A.; Chopra, A. Ceftaroline Cerebrospinal Fluid Penetration in the Treatment of a Ventriculopleural Shunt Infection: A Case Report. J. Pediatr. Pharmacol. Ther. JPPT Off. J. PPAG 2020, 25, 336–339. [Google Scholar] [CrossRef]
- Balouch, M.A.; Bajwa, R.J.; Hassoun, A. Successful Use of Ceftaroline for the Treatment of MRSA Meningitis Secondary to an Infectious Complication of Lumbar Spine Surgery. J. Antimicrob. Chemother. 2015, 70, 624–625. [Google Scholar] [CrossRef] [Green Version]
- Kuriakose, S.S.; Rabbat, M.; Gallagher, J.C. Ceftaroline CSF Concentrations in a Patient with Ventriculoperitoneal Shunt-Related Meningitis. J. Antimicrob. Chemother. 2015, 70, 953–954. [Google Scholar] [CrossRef] [Green Version]
- Kufel, W.D.; Abouelhassan, Y.; Steele, J.M.; Gutierrez, R.L.; Perwez, T.; Bourdages, G.; Nicolau, D.P. Plasma and Cerebrospinal Fluid Concentrations of Cefiderocol during Successful Treatment of Carbapenem-Resistant Acinetobacter baumannii Meningitis. J. Antimicrob. Chemother. 2022, 77, 2737–2741. [Google Scholar] [CrossRef]
- Lupia, T.; De Benedetto, I.; Stroffolini, G.; Di Bella, S.; Mornese Pinna, S.; Zerbato, V.; Rizzello, B.; Bosio, R.; Shbaklo, N.; Corcione, S.; et al. Temocillin: Applications in Antimicrobial Stewardship as a Potential Carbapenem-Sparing Antibiotic. Antibiotics 2022, 11, 493. [Google Scholar] [CrossRef]
- Schievink, H.I.; Mattie, H.; Thomeer, R.T.; Van Strijen, E. The Passage of Cloxacillin into Cerebrospinal Fluid in the Absence of Meningitis. Br. J. Clin. Pharmacol. 1993, 36, 57–60. [Google Scholar] [CrossRef] [Green Version]
- Le Turnier, P.; Gregoire, M.; Deslandes, G.; Lakhal, K.; Deschanvres, C.; Lecomte, R.; Talarmin, J.-P.; Dubée, V.; Bellouard, R.; Boutoille, D.; et al. Should We Reconsider Cefazolin for Treating Staphylococcal Meningitis? A Retrospective Analysis of Cefazolin and Cloxacillin Cerebrospinal Fluid Levels in Patients Treated for Staphylococcal Meningitis. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2020, 26, 1415.e1–1415.e4. [Google Scholar] [CrossRef]
- Grégoire, M.; Gaborit, B.; Deschanvres, C.; Lecomte, R.; Deslandes, G.; Dailly, É.; Ambrosi, X.; Bellouard, R.; Asseray, N.; Lakhal, K.; et al. High-Dosage Cefazolin Achieves Sufficient Cerebrospinal Diffusion to Treat an External Ventricular Drainage-Related Staphylococcus aureus Ventriculitis. Antimicrob. Agents Chemother. 2019, 63, e01844-18. [Google Scholar] [CrossRef] [Green Version]
- Pacifici, G.M. Clinical Pharmacology of Piperacillin-Tazobactam Combination in Infants and Children. Clin. Med. Investig. 2019. [Google Scholar] [CrossRef]
- Franz, P.; von Rosen, F.; Garner, C.; Swozil, U.; Schmiedek, P.; Einhäupl, K.; Adam, D. Cerebrospinal Fluid Penetration after Single or Multiple Dosage with Ticarcillin/Clavulanate. J. Antimicrob. Chemother. 1989, 24 (Suppl. B), 107–110. [Google Scholar] [CrossRef] [PubMed]
- Markantonis, S.L.; Markou, N.; Fousteri, M.; Sakellaridis, N.; Karatzas, S.; Alamanos, I.; Dimopoulou, E.; Baltopoulos, G. Penetration of Colistin into Cerebrospinal Fluid. Antimicrob. Agents Chemother. 2009, 53, 4907–4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyberth, H.W.; Kauffman, R.E. Basics and Dynamics of Neonatal and Pediatric Pharmacology. In Pediatric Clinical Pharmacology; Springer: Berlin/Heidelberg, Germany, 2011; Volume 258, pp. 3–58. ISBN 354022565X. [Google Scholar]
- Susa, S.T.; Preuss, C.V. Drug Metabolism. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK442023/ (accessed on 10 October 2022).
- De Wildt, S.N.; Tibboel, D.; Leeder, J.S. Drug Metabolism for the Paediatrician. Arch. Dis. Child. Educ. Pract. Ed. 2014, 99, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Barreto, E.F.; Larson, T.R.; Koubek, E.J. Drug Excretion. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1–18. [Google Scholar] [CrossRef]
- Krishna, D.R.; Klotz, U. Extrahepatic Metabolism of Drugs in Humans. Clin. Pharmacokinet. 1994, 26, 144–160. [Google Scholar] [CrossRef]
- Lynch, T.; Price, A. The Effect of Cytochrome P450 Metabolism on Drug Response, Interactions, and Adverse Effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar]
- Stavropoulou, E.; Pircalabioru, G.G.; Bezirtzoglou, E. The Role of Cytochromes P450 in Infection. Front. Immunol. 2018, 9, 89. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Rosenbaum, S. Developmental Pharmacokinetics in Pediatric Populations. J. Pediatr. Pharmacol. Ther. 2014, 19, 262–276. [Google Scholar] [CrossRef]
- Kiss, M.; Mbasu, R.; Nicolaï, J.; Barnouin, K.; Kotian, A.; Mooij, M.G.; Kist, N.; Wijnen, R.M.H.; Ungell, A.L.; Cutler, P.; et al. Ontogeny of Small Intestinal Drug Transporters and Metabolizing Enzymes Based on Targeted Quantitative ProteomicsS. Drug Metab. Dispos. 2021, 49, 1038–1046. [Google Scholar] [CrossRef]
- Krasinski, K.; Perkin, R.; Rutledge, J. Gray Baby Syndrome Revisited. Clin. Pediatr. 1982, 21, 571–572. [Google Scholar] [CrossRef]
- Cascorbi, I. Drug Interactions—Principles, Examples and Clinical Consequences. Dtsch. Arztebl. Int. 2012, 109, 546–556. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and Induction of CYP Enzymes in Humans: An Update; Springer: Berlin/Heidelberg, Germany, 2020; Volume 94, ISBN 0123456789. [Google Scholar]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Relevance of Half-Life in Drug Design. J. Med. Chem. 2018, 61, 4273–4282. [Google Scholar] [CrossRef]
- van den Anker, J.; Reed, M.D.; Allegaert, K.; Kearns, G.L. Developmental Changes in Pharmacokinetics and Pharmacodynamics. J. Clin. Pharmacol. 2018, 58, S10–S25. [Google Scholar] [CrossRef] [Green Version]
- Rodieux, F.; Wilbaux, M.; van den Anker, J.N.; Pfister, M. Effect of Kidney Function on Drug Kinetics and Dosing in Neonates, Infants, and Children. Clin. Pharmacokinet. 2015, 54, 1183–1204. [Google Scholar] [CrossRef] [Green Version]
- Gattineni, J.; Baum, M. Developmental Changes in Renal Tubular Transport—An Overview. Pediatr. Nephrol. 2015, 30, 2085–2098. [Google Scholar] [CrossRef] [Green Version]
- Quigley, R. Developmental Changes in Renal Function. Curr. Opin. Pediatr. 2012, 24, 184–190. [Google Scholar] [CrossRef]
- Baptista, J.P. Augmented renal clearance. In Antibiotic Pharmacokinetic/Pharmacodynamic Considerations in the Critically III; Adis Singapore: Singapore, 2017; Volume 49, pp. 125–150. [Google Scholar] [CrossRef]
- Rhoney, D.H.; Metzger, S.A.; Nelson, N.R. Scoping Review of Augmented Renal Clearance in Critically Ill Pediatric Patients. Pharmacotherapy 2021, 41, 851–863. [Google Scholar] [CrossRef]
- Hefny, F.; Stuart, A.; Kung, J.Y.; Mahmoud, S.H. Prevalence and Risk Factors of Augmented Renal Clearance: A Systematic Review and Meta-Analysis. Pharmaceutics 2022, 14, 445. [Google Scholar] [CrossRef]
- Wacharachaisurapol, N.; Sukkummee, W.; Anunsittichai, O.; Srisan, P.; Sangkhamal, S.; Chantharit, P.; Vandepitte, W.P.; Wattanavijitkul, T.; Puthanakit, T. Dose Recommendations for Intravenous Colistin in Pediatric Patients from a Prospective, Multicenter, Population Pharmacokinetic Study. Int. J. Infect. Dis. 2021, 109, 230–237. [Google Scholar] [CrossRef]
- Avedissian, S.N.; Rohani, R.; Bradley, J.; Le, J.; Rhodes, N.J. Optimizing Aminoglycoside Dosing Regimens for Critically Ill Pediatric Patients with Augmented Renal Clearance: A Convergence of Parametric and Nonparametric Population Approaches. Antimicrob. Agents Chemother. 2021, 65, e02629-20. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.H.; Nicolau, D.P. Augmented Renal Clearance and How to Augment Antibiotic Dosing. Antibiotics 2020, 9, 393. [Google Scholar] [CrossRef] [PubMed]
- Margineanu, I.; Akkerman, O.; Cattaneo, D.; Goletti, D.; Marriott, D.J.E.; Migliori, G.B.; Mirzayev, F.; Peloquin, C.A.; Stienstra, Y.; Alffenaar, J.-W. Practices of Therapeutic Drug Monitoring in Tuberculosis: An International Survey. Eur. Respir. J. 2022, 59, 2102787. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, S.; Bolhuis, M.S.; Sturkenboom, M.G.G.; Akkerman, O.W.; de Lange, W.C.M.; van der Werf, T.S.; Alffenaar, J.-W.C. Incorporating Therapeutic Drug Monitoring into the World Health Organization Hierarchy of Tuberculosis Diagnostics. Eur. Respir. J. 2016, 47, 1867–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolhuis, M.S.; Akkerman, O.W.; Sturkenboom, M.G.G.; de Lange, W.C.M.; van der Werf, T.S.; Alffenaar, J.-W.C. Individualized Treatment of Multidrug-Resistant Tuberculosis Using Therapeutic Drug Monitoring. Int. J. Mycobacteriology 2016, 5, S44–S45. [Google Scholar] [CrossRef] [Green Version]
- Rybak, M.J.; Le, J.; Lodise, T.P.; Levine, D.P.; Bradley, J.S.; Liu, C.; Mueller, B.A.; Pai, M.P.; Wong-Beringer, A.; Rotschafer, J.C.; et al. Therapeutic Monitoring of Vancomycin for Serious Methicillin-Resistant Staphylococcus Aureus Infections: A Revised Consensus Guideline and Review by the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, the Pediatr. Clin. Infect. Dis. 2020, 71, 1361–1364. [Google Scholar] [CrossRef]
- Ewoldt, T.M.J.; Abdulla, A.; Rietdijk, W.J.R.; Muller, A.E.; de Winter, B.C.M.; Hunfeld, N.G.M.; Purmer, I.M.; van Vliet, P.; Wils, E.-J.; Haringman, J.; et al. Model-Informed Precision Dosing of Beta-Lactam Antibiotics and Ciprofloxacin in Critically Ill Patients: A Multicentre Randomised Clinical Trial. Intensive Care Med. 2022, 48, 1760–1771. [Google Scholar] [CrossRef]
- Katona, P.; Katona-Apte, J. The Interaction between Nutrition and Infection. Clin. Infect. Dis. 2008, 46, 1582–1588. [Google Scholar] [CrossRef]
- WHO. Fact Sheet Malnutrition. Available online: https://www.who.int/news-room/fact-sheets/detail/malnutrition (accessed on 6 October 2022).
- Oshikoya, K.A.; Sammons, H.M.; Choonara, I. A Systematic Review of Pharmacokinetics Studies in Children with Protein-Energy Malnutrition. Eur. J. Clin. Pharmacol. 2010, 66, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Verrest, L.; Wilthagen, E.A.; Beijnen, J.H.; Huitema, A.D.R.; Dorlo, T.P.C. Influence of Malnutrition on the Pharmacokinetics of Drugs Used in the Treatment of Poverty-Related Diseases: A Systematic Review. Clin. Pharmacokinet. 2021, 60, 1149–1169. [Google Scholar] [CrossRef]
- Lazzerini, M.; Tickell, D. Antibiotics in Severely Malnourished Children: Systematic Review of Efficacy, Safety and Pharmacokinetics. Bull. World Health Organ. 2011, 89, 594–607. [Google Scholar] [CrossRef]
- Standing, J.F.; Ongas, M.O.; Ogwang, C.; Kagwanja, N.; Murunga, S.; Mwaringa, S.; Ali, R.; Mturi, N.; Timbwa, M.; Manyasi, C.; et al. Dosing of Ceftriaxone and Metronidazole for Children with Severe Acute Malnutrition. Clin. Pharmacol. Ther. 2018, 104, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Dipasquale, V.; Cucinotta, U.; Romano, C. Acute Malnutrition in Children: Pathophysiology, Clinical Effects and Treatment. Nutrients 2020, 12, 2413. [Google Scholar] [CrossRef]
- Otiti, M.I.; Allen, S.J. Severe Acute Malnutrition in Low- and Middle-Income Countries. Paediatr. Child Health 2021, 31, 301–307. [Google Scholar] [CrossRef]
- Bolme, P.; Margareta, E. Influence of Diarrhea on the Oral Absorption of Penicillin V and Ampicillin in Children. Scand. J. Infect. Dis. 1975, 7, 141–145. [Google Scholar] [CrossRef]
- Justesen, U.S.; Andersen, A.B.; Klitgaard, N.A.; Brosen, K.; Gerstoft, J.; Pedersen, C. Pharmacokinetic Interaction between Rifampin and the Combination of Indinavir and Low-Dose Ritonavir in HIV-Infected Patients. Clin. Infect. Dis. 2004, 38, 426–429. [Google Scholar] [CrossRef]
- de Paula, R.P.; Nascimento, A.F.; Sousa, S.M.B.; Bastos, P.R.V.; Barbosa, A.A.L. Glomerular Filtration Rate Is Altered in Children with Sickle Cell Disease: A Comparison between Hb SS and Hb SC. Rev. Bras. Hematol. Hemoter. 2013, 35, 349–351. [Google Scholar] [CrossRef]
- Hirschberg, R. Glomerular Hyperfiltration in Sickle Cell Disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 748–749. [Google Scholar] [CrossRef] [Green Version]
- Chami, N.; Kabyemera, R.; Masoza, T.; Ambrose, E.; Kimaro, F.; Kayange, N.; Hokororo, A.; Furia, F.F.; Peck, R. Prevalence and Factors Associated with Renal Dysfunction in Children Admitted to Two Hospitals in Northwestern Tanzania. BMC Nephrol. 2019, 20, 79. [Google Scholar] [CrossRef] [Green Version]
- Halle, M.P.; Lapsap, C.T.; Barla, E.; Fouda, H.; Djantio, H.; Moudze, B.K.; Akazong, C.A.; Priso, E.B. Epidemiology and Outcomes of Children with Renal Failure in the Pediatric Ward of a Tertiary Hospital in Cameroon. BMC Pediatr. 2017, 17, 202. [Google Scholar] [CrossRef]
- Denti, P.; Garcia-Prats, A.J.; Draper, H.R.; Wiesner, L.; Winckler, J.; Thee, S.; Dooley, K.E.; Savic, R.M.; McIlleron, H.M.; Schaaf, H.S.; et al. Levofloxacin Population Pharmacokinetics in South African Children Treated for Multidrug-Resistant Tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e01521-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredrick, F.; Francis, J.M.; Ruggajo, P.J.; Maro, E.E. Renal Abnormalities among HIV Infected Children at Muhimbili National Hospital (MNH)—Dar Es Salaam, Tanzania. BMC Nephrol. 2016, 17, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, G.A.; Fabian, L.; Gomez, M.; Gonzalez, C.D.; Diez, R.A.; Girolamo, G. Di Age-Distribution and Genotype-Phenotype Correlation for N-Acetyltransferase in Argentine Children under Isoniazid Treatment. Int. J. Clin. Pharmacol. Ther. 2014, 52, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, H.S.; Parkin, D.P.; Seifart, H.I.; Werely, C.J.; Hesseling, P.B.; Van Helden, P.D.; Maritz, J.S.; Donald, P.R. Isoniazid Pharmacokinetics in Children Treated for Respiratory Tuberculosis. Arch. Dis. Child. 2005, 90, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Kiser, J.J.; Seifart, H.I.; Werely, C.J.; Mitchell, C.D.; D’Argenio, D.Z.; Fletcher, C.V. The Pharmacogenetics of NAT2 Enzyme Maturation in Perinatally HIV Exposed Infants Receiving Isoniazid. J. Clin. Pharmacol. 2012, 52, 511–519. [Google Scholar] [CrossRef]
- Holgate, S.L.; Bekker, A.; Pillay-Fuentes Lorente, V.; Dramowski, A. Errors in Antimicrobial Prescription and Administration in Very Low Birth Weight Neonates at a Tertiary South African Hospital. Front. Pediatr. 2022, 10, 838153. [Google Scholar] [CrossRef]
- Oshikoya, K.A.; Oreagba, I.A.; Ogunleye, O.O.; Senbanjo, I.O.; MacEbong, G.L.; Olayemi, S.O. Medication Administration Errors among Paediatric Nurses in Lagos Public Hospitals: An Opinion Survey. Int. J. Risk Saf. Med. 2013, 25, 67–78. [Google Scholar] [CrossRef]
- Nwobodo, N. Therapeutic Drug Monitoring in a Developing Nation: A Clinical Guide. JRSM Open 2014, 5, 205427041453112. [Google Scholar] [CrossRef] [Green Version]
- Greybe, L. Newsletter of the African Society for Paediatric Infectious Diseases. Available online: https://cct.mycpd.co.za/FIDSSA/AfSPID_Bulletin_Dec_2020.pdf (accessed on 9 September 2022).
- Jacobs, J.; Hardy, L.; Semret, M.; Lunguya, O.; Phe, T.; Affolabi, D.; Yansouni, C.; Vandenberg, O. Diagnostic Bacteriology in District Hospitals in Sub-Saharan Africa: At the Forefront of the Containment of Antimicrobial Resistance. Front. Med. 2019, 6, 205. [Google Scholar] [CrossRef]


| (A) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Class | Agent | PK/PD Index | Molecular Weight (g/mol) [20] | pKa [21] | LogP [20] | Fraction Protein Binding (%) [20] | Metabolism [21] | Alternative Route of Elimination [21] |
| Aminoglycosides | Amikacin | Cmax/MIC [22] | 585.6 | 8.1–12.1 | −8.8–−7.4 | <10% | Aminoglycosides are not significantly metabolized. | |
| Gentamicin | 477.6 | 10.1–12.6 | −4.1–−1.9 | 0–30% | ||||
| Kanamycin | 484.5 | 9.5–12.1 | −6.9–−6.3 | N/A | ||||
| Neomycin | 614.6 | 12.9 [23] | −9–−3.7 | N/A | ||||
| Streptomycin | 581.6 | 11.1–11.6 | −8–−2.5 | N/A | ||||
| Spectinomycin | 332.4 | 7.0–9.2 | −3.1–−2.3 | Not significant | ||||
| Tobramycin | 467.5 | 9.7–12.5 | −6.2–−5.8 | Not significant | ||||
| First line anti- mycobacterials | Isoniazid | AUC/MIC [24] | 137.1 | 1.8–13.6 | −0.8–−0.7 | 0–10% | Hepatic | |
| Pyrazinamide | AUC/MIC [24] | 123.1 | −0.5–13 | −1–−0.6 | ~10% | Mainly hepatic | ||
| Rifabutin | AUC/MIC, Cmax/MIC [25] | 847.0 | 6.9–9.0 | 4.1–4.7 | 85% | Hepatic | Feces | |
| Second line antimycobacterials | Cycloserine | T > MIC [26] | 102.1 | 4.2–8.4 | −1.5–−0.9 | N/A | Hepatic [27] | |
| Ethionamide | AUC/MIC [28] | 166.3 | 5–11.9 | 0.4–1.1 | ~30% | Extensive hepatic metabolism | ||
| Beta-lactamase inhibitors | Clavulanic acid | T > MIC [29] | 199.2 | −2.6–3.2 | −2.3–−1.2 | ~25% for amoxicillin-clavulanic acid | Hepatic | Feces, exhaled air |
| Sulbactam | 233.2 | −3.8–3.1 | −1 | ~38% | <25% is metabolized by the liver [30] | |||
| Tazobactam | 300.3 | 0.8–2.9 | −2 | ~30% | Hepatic | |||
| Avibactam | 265.3 | −3.9–−2 | −1.8 | 5.7–8.2% | Not significant | |||
| Vaborbactam | 297.1 | −2.6–3.8 | 1.0–1.9 [31] | ~33% | Not significant | |||
| Relebactam | 348.4 | −2–10 | −3.6 | ~22% | Not significant | |||
| Carbapenems | Doripenem | T > MIC [22] | 420.5 | 3.3–9.5 | −5.6, −1.3 [32] | 8.1% | Limited hepatic metabolism | |
| Ertapenem | 475.5 | 3.2–9.0 | 0.3–1.5 | 85–95% | Limited hepatic metabolism | |||
| Imipenem | 299.4 | 3.2–10.9 | −0.7 | 20% | Renal metabolism | |||
| Meropenem | 383.5 | 3.3–9.4 | −2.4–−0.6 | ~2% | <30% of a dose undergoes hepatic metabolism | |||
| First generation cephalosporins | Cephalexin | T > MIC [33,34] | 347.4 | 3.3–7.2 | 0.6–0.7 | 10–15% | Not significant | |
| Cefazolin | 454.5 | 0.3–2.8 | −0.6 | 74–86% | Not significant | |||
| Cefadroxil | 363.4 | 3.3–7.2 | −2.1–−0.4 | 28.1% | Not significant | |||
| Second generation cephalosporins | Cefaclor | 367.8 | 2.8–7.2 | −2.3–0.9 | 23.5% | Not significant | ||
| Cefuroxime | 424.4 | −1.1–3.0 | −0.8–−0.2 | 50% | Not significant | |||
| Cefuroxime axetil | 510.5 | −1.2–10.9 | 0.9 | 28–38% [35] | Axetil is metabolized by the liver | |||
| Cefotetan | 575.6 | −1.5–3.0 | 0.1 | 88% | Not significant | |||
| Cefoxitin | 427.5 | −3.8–3.4 | 0 | 31–54% [36] | Minimal hepatic metabolism | |||
| Cefprozil | 389.4 | 3.3–7.2 | −1.4–0.6 | 36% | Not significant | |||
| Cefmetazole | 471.5 | −1.7–3.2 | −2.2–−0.6 | 85% [37] | Not significant | |||
| Third generation cephalosporins | Cefdinir | 395.4 | 2.7–9.7 | −3.5–0 | 60–70% | Not significant | ||
| Cefditoren | 506.6 | 2.3–3.7 | 0.7 | 88% | Not significant | |||
| Cefixime | 453.5 | 2.5–4.0 | −0.7–−0.4 | 65% | Hepatic | |||
| Cefpodoxime | 427.5 | 2.8–3.6 | −1.4 | 21–33% | Minimal hepatic metabolism | |||
| Ceftazidime | 546.6 | 2.4–4.0 | −1.6–0.4 | 5–23% | Not significant | |||
| Ceftizoxime | 383.4 | 2.7–3.6 | 0 | 30% | Not significant | |||
| Ceftibuten | 410.4 | 2.9–4.7 | −0.3 | 65% | ~10% is metabolized by the liver | |||
| Ceftriaxone | 554.6 | 2.7–3.4 | −1.7–−1.3 | 95% | Negligible | Bile | ||
| Cefotaxime | 455.5 | 2.7–3.6 | −1.4–−0.5 | 8–41% [36] | Partially (15–20%) by the liver [38] | |||
| Ceftolozane | 666.7 | 2.5–9.1 | −6.2–−3.2 | 16–21% | Not significant | |||
| Fourth generation cephalosporins | Cefepime | 480.6 | 2.8–3.6 | −0.1 | 20% | <1% is metabolized by the liver | ||
| Fifth generation cephalosporins | Ceftobiprole | 534.6 | 2.9–10.4 | −2.4 | <16% [39] | Minimal hepatic metabolism [39] | ||
| Ceftaroline | 684.7 | 0.4–1.8 | 2.3 | ~20% | Minimal hepatic metabolism | Feces | ||
| Siderophore cephalosporins | Cefiderocol | 752.2 | 2.6–4.0 | −2.3–1 | 40–60% | Minimal hepatic metabolism | ||
| Fluoroquinolones | Ciprofloxacin | AUC/MIC [34] | 331.3 | 5.6–8.8 | −1.1–2.3 | 20–40% | Up to 15% hepatic metabolism | Feces |
| Delafloxacin | 440.8 | −1.3–5.6 | 2.7 | 84% | Hepatic | Feces | ||
| Gatifloxacin | 375.4 | 5.5–8.8 | −0.7–2.6 | 20% | Limited hepatic metabolism | |||
| Levofloxacin | 361.4 | 5.4–6.7 | −0.4–2.1 | 24–38% | Very limited metabolism | Feces | ||
| Norfloxacin | 319.3 | 5.6–8.8 | −1.0–2.1 | 10–15% | Hepatic and renal | Feces | ||
| Ofloxacin | 361.4 | 5.4–6.7 | −0.4–2.1 | 32% | Hepatic | Feces | ||
| Prulifloxacin | 461.5 | 5.2–6.0 | 1.0 | 41–59% [40] | Hepatic | Feces | ||
| Glycopeptides | Teicoplanin | AUC/MIC [41] | 1879.7 | 3.0–7.1 | 0.5 | 90–95% [42] | Minimal hepatic metabolism | |
| Vancomycin | 1449.3 | 3.0–9.9 | −3.1–−2.6 | ~50% | Not significant | |||
| Lipoglyco- peptides | Dalbavancin | 1816.7 | 1.7–9.9 [43] | 3.8 | 93% | Unlikely to have significant metabolism | Feces | |
| Telavancin | 1755.6 | 1.6–10.0 | −2.1 | >90% | Unknown | |||
| Oritavancin | 1793.1 | 2.2–10.0 | 1.5–4.1 | 85% | Not significant | Feces | ||
| Lincosamides | Clindamycin | AUC/MIC [22,34] | 425.0 | 7.6–12.4 | 2.2 | 60–94% [44] | Hepatic | Feces |
| Lincomycin | 406.5 | 8.0–12.4 | 0.2–0.6 | 28–86% | Hepatic | Bile | ||
| Monobactams | Aztreonam | T > MIC [45] | 435.4 | −1.5–3.9 | 0.3 | 43–56% | 6–16% is metabolized by the liver | |
| Nitroimidazoles | Metronidazole | AUC/MIC, Cmax/MIC [22,46] | 171.2 | 2.6–15.4 | −0.1–0 | <20% | Hepatic | Feces |
| Secnidazole | Undefined | 185.2 | 3.1–15.2 | 0.2 | <5–15% | N/A | ||
| Tinidazole | Undefined | 247.2 | 3.3 | −0.4–0.7 | 12% | Hepatic | Feces | |
| Oxazolidones | Linezolid | AUC/MIC [22] | 337.4 | −1.2–14.9 | 0.7–1.3 | ~31% | Hepatic | |
| Natural penicillins | Penicillin G | T > MIC [22] | 334.4 | −2.8–3.5 | 1.5–1.8 | 45–68% | Hepatic | Bile |
| Aminopenicillins | Amoxicillin | 365.4 | 3.2–7.2 | −2–0.9 | 17% | Hepatic | ||
| Ampicillin | 349.4 | 3.2–7.2 | −1.1–1.4 | 8–25% [47] | Hepatic | |||
| Semi-synthetic penicillins | Cloxacillin | 435.9 | −0.4–3.8 | 2.4–3 | ~94% | Intestinal | Bile | |
| Dicloxacillin | 470.3 | −0.7–3.8 | 2.9–3.7 | 96–97% [48] | Hepatic | |||
| Flucloxacillin | 453.9 | −0.9–3.8 | 2.6–3.2 | 95–96% [48] | Hepatic | |||
| Oxacillin | 401.4 | −0.1–3.8 | 2.4 | 92–96% | 45–50% hepatic [49] | |||
| Temocillin [50] | 414.5 | −4.3–3.1 | 1.1 | ~80% [48] | N/A | |||
| Ureidopenicillins | Piperacillin | 517.6 | −4.3–3.5 | 0.3–0.5 | 39.4–71.3% [51] | Not significant | Bile | |
| Carboxy- penicillins | Ticarcillin | 384.4 | −6.3–3.1 | 0.8 | 45% | N/A | ||
| Polymixins * | Polymyxin B | AUC/MIC [33] | 1203.5 | 8.9–11.6 | −2.5 | 79–92% | N/A | |
| Sulfonamides | Sulfadiazine | Cmax/MIC, AUC/MIC [22] | 250.3 | 2.0–6.4 | −0.2–−0.1 | 20–25% [52] | Hepatic | |
| Sulfadoxine | 310.3 | 3.4–6.1 | 0.7 | ~94% [53] | Hepatic | |||
| Sulfamethoxazole | 253.3 | 2.0–6.2 | 0.7–0.9 | ~70% | Hepatic | |||
| Tetracyclines | Doxycycline | AUC/MIC [54] | 444.4 | 3.1–8.3 | −0.7–0.6 | >90% | Hepatic | Feces |
| Tetracycline | 444.4 | 3.3–9.3 | −2–−1.3 | 20–67% | Not significant | Feces | ||
| Miscellaneous | Chloramphenicol | Cmax/MIC, AUC/MIC [22] | 323.1 | −2.8–8.7 | 0.7–1.1 | 50–60% in adults, 32% in premature neonates | Extensive hepatic metabolism | |
| Daptomycin | AUC/MIC [22] | 1619.7 | 3.0–9.6 | −5.1 | 90–94% [55] | Minimum extent, metabolism site unknown [56] | Feces | |
| Fosfomycin | AUC/MIC [34] | 138.1 | −4.3–1.3 | −1.6–−1.4 | No plasma binding | Not significant | ||
| Trimethoprim | Cmax/MIC, AUC/MIC [22] | 290.3 | 7.1–17.3 | 0.6–0.9 | 44% | Hepatic | ||
| Nitrofurantoin | Undefined | 238.2 | −2.2–8.3 | −0.5 | <90% | Hepatic | ||
| (B) | ||||||||
| Class | Agent | PK/PD Index | Molecular Weight (g/mol) [20] | pKa [21] | LogP [20] | Fraction Protein Binding (%) [20] | Metabolism [21] | Alternative Route of Elimination [21] |
| First line anti- mycobacterials | Ethambutol | Cmax/MIC, AUC/MIC [24] | 204.3 | 9.7–14.8 | −0.4–0.4 | 20–30% | Hepatic | Urine |
| Rifabutin | AUC/MIC, Cmax/MIC [25] | 847.0 | 6.9–9.0 | 4.1–4.7 | 85% | Hepatic | Urine | |
| Third line antimycobacterials | Bedaquiline | AUC/MIC, Cmax/MIC [57] | 555.5 | 8.9–13.6 | 7.7 | >99.9% | Hepatic | |
| Clofazimine | Not identified [26] | 473.4 | 6.6–16.2 | 7–7.7 | N/A | N/A | ||
| Delamanid | Not Identified [26] | 534.5 | 5.5 | 5.6 | >99.5% | Hepatic | ||
| Beta-lactamase inhibitors | Clavulanic acid | T > MIC [29] | 199.2 | −2.6–3.2 | −2.3–−1.2 | ~25% for amoxicillin-clavulanic acid | Significant hepatic metabolism | Urine, exhaled air |
| Fluoroquinolones | Gemifloxacin | AUC/MIC [34] | 389.4 | 5.4–9.4 | −0.7–2.3 | 60–70% | Limited hepatic metabolism | Urine |
| Moxifloxacin | 401.4 | 5.5–9.5 | 0.6–2.9 | 50% | <50% hepatic metabolism | Urine | ||
| Macrolides | Clarithromycin | AUC/MIC, T > MIC [34] | 748.0 | 9–12.5 | 1.7–3.2 | ~70% | Hepatic | Urine |
| Fidaxomicin | 1058 | −1.4–5.9 | 6.4 | 31% [48] | Intestinal | |||
| Oxazolidones | Tedizolid | AUC/MIC [22] | 370.3 | −1.7–14.6 | 1.4 | 70–90% | Hepatic | Urine |
| Tetracyclines | Eravacycline | AUC/MIC [54] | 558.6 | 3.0–9.0 | 1 | 79–90% | Hepatic | Urine |
| Omadacycline | 556.6 | 2.9–10.5 | 3 | ~20% | Not significant | Urine | ||
| Tigecycline | 585.7 | 3.2–9.0 | −0.2–1.1 | 71–89% | Hepatic [58] | Urine | ||
| Daptomycin | AUC/MIC [22] | 1619.7 | 3.0–9.6 | −5.1 | 90–94% [55] | Metabolism site unknown [56] | Urine | |
| (C) | ||||||||
| Class | Agent | PK/PD Index | Molecular Weight (g/mol) [20] | pKa [21] | LogP [20] | Fraction Protein Binding (%) [20] | Metabolism [21] | Alternative Route of Elimination [21] |
| First line Antimyco- bacterials | Rifampicin | AUC/MIC, Cmax/MIC [25] | 822.9 | 1.7–7.4 | 2.7–4.9 | 90% | Hepatic | Urine |
| Third generation cephalosporins | Cefoperazone | T > MIC [33,34] | 645.7 | −1.7–3.2 | −0.7 | 82–93% | Not significant | |
| Lincosamides | Lincomycin | AUC/MIC [22,34] | 406.5 | 8.0–12.4 | 0.2–0.6 | 28–86% | Hepatic | Urine |
| Macrolides | Azithromycin | AUC/MIC, T > MIC [34] | 749.0 | 8.5–12.4 | 3.0–4.0 | 7–51% | Hepatic | Urine |
| Erythromycin | 733.9 | 9–12.5 | 2.6–3.1 | 70–93% | Hepatic | Urine | ||
| Natural penicillins | Penicillin V | T > MIC [22] | 350.4 | −4.9–3.4 | 1.4–2.1 | 50–80% | Hepatic | Urine |
| Penicillin G | 334.4 | −2.8–3.5 | 1.5–1.8 | 45–68% | Hepatic | Urine | ||
| Semi-synthetic penicillins | Cloxacillin | 435.9 | −0.4–3.8 | 2.4–3 | ~94% | Intestinal | Urine | |
| Nafcillin | 414.5 | −1.9–3.3 | 2.9–3.3 | 88.4–91.4% | Hepatic | |||
| Ureidopenicillins | Piperacillin | 517.6 | −4.3–3.5 | 0.3–0.5 | 39.4–71.3% [51] | Not significant | Urine | |
| Tetracyclines | Minocycline | AUC/MIC [54] | 457.5 | 3.2–8.8 | −0.6–0.1 | 76% | Hepatic | Urine |
| Agent | Food Effect on Absorption |
|---|---|
| Amoxicillin | No effect of fasting status for infants, children and adults [69,70]. |
| Amoxicillin/clavulanate | Concomitant food ingestion may enhance absorption and reduce gastric upset [71]. |
| Ampicillin | Impaired when taken with food. Therefore, if administered PO, ampicillin should be administered 1 h before or 2 h after meals [70]. |
| Azithromycin | Tablets and suspension present no food effect [69]. |
| Cephalexin, cefadroxil, cefaclor, cefprozil, cefixime | Not affected by food intake [72]. |
| Cefuroxime axetil | Absorption and dissolution into active form are improved when taken with food [73,74]. |
| Ciprofloxacin | Impaired by dairy products, Ca2+ and Mg2+ supplements [75]. |
| Metronidazole | Food may decrease the rate but not the extent of absorption. However, food may reduce gastric upset [76]. |
| Rifampicin | Impaired when taken with food, therefore should be taken on an empty stomach [77]. |
| Tetracycline | Impaired when taken with food or with divalent metal cations, such as Fe+2 and Ca+2 [78]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meesters, K.; Alemayehu, T.; Benou, S.; Buonsenso, D.; Decloedt, E.H.; Pillay-Fuentes Lorente, V.; Downes, K.J.; Allegaert, K. Pharmacokinetics of Antimicrobials in Children with Emphasis on Challenges Faced by Low and Middle Income Countries, a Clinical Review. Antibiotics 2023, 12, 17. https://doi.org/10.3390/antibiotics12010017
Meesters K, Alemayehu T, Benou S, Buonsenso D, Decloedt EH, Pillay-Fuentes Lorente V, Downes KJ, Allegaert K. Pharmacokinetics of Antimicrobials in Children with Emphasis on Challenges Faced by Low and Middle Income Countries, a Clinical Review. Antibiotics. 2023; 12(1):17. https://doi.org/10.3390/antibiotics12010017
Chicago/Turabian StyleMeesters, Kevin, Tinsae Alemayehu, Sofia Benou, Danilo Buonsenso, Eric H. Decloedt, Veshni Pillay-Fuentes Lorente, Kevin J. Downes, and Karel Allegaert. 2023. "Pharmacokinetics of Antimicrobials in Children with Emphasis on Challenges Faced by Low and Middle Income Countries, a Clinical Review" Antibiotics 12, no. 1: 17. https://doi.org/10.3390/antibiotics12010017