
Genome-Wide Analysis of Milk Production Traits and Selection Signatures in Serbian Holstein-Friesian Cattle
 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Sampling
2.2. DNA Extraction and Genotyping
2.3. Quality Control of SNP Chip Data
2.4. GWAS
2.5. Identification of ROH Segments
2.6. Genomic Inbreeding Coefficient Based on ROH
2.7. Detection of ROH Islands
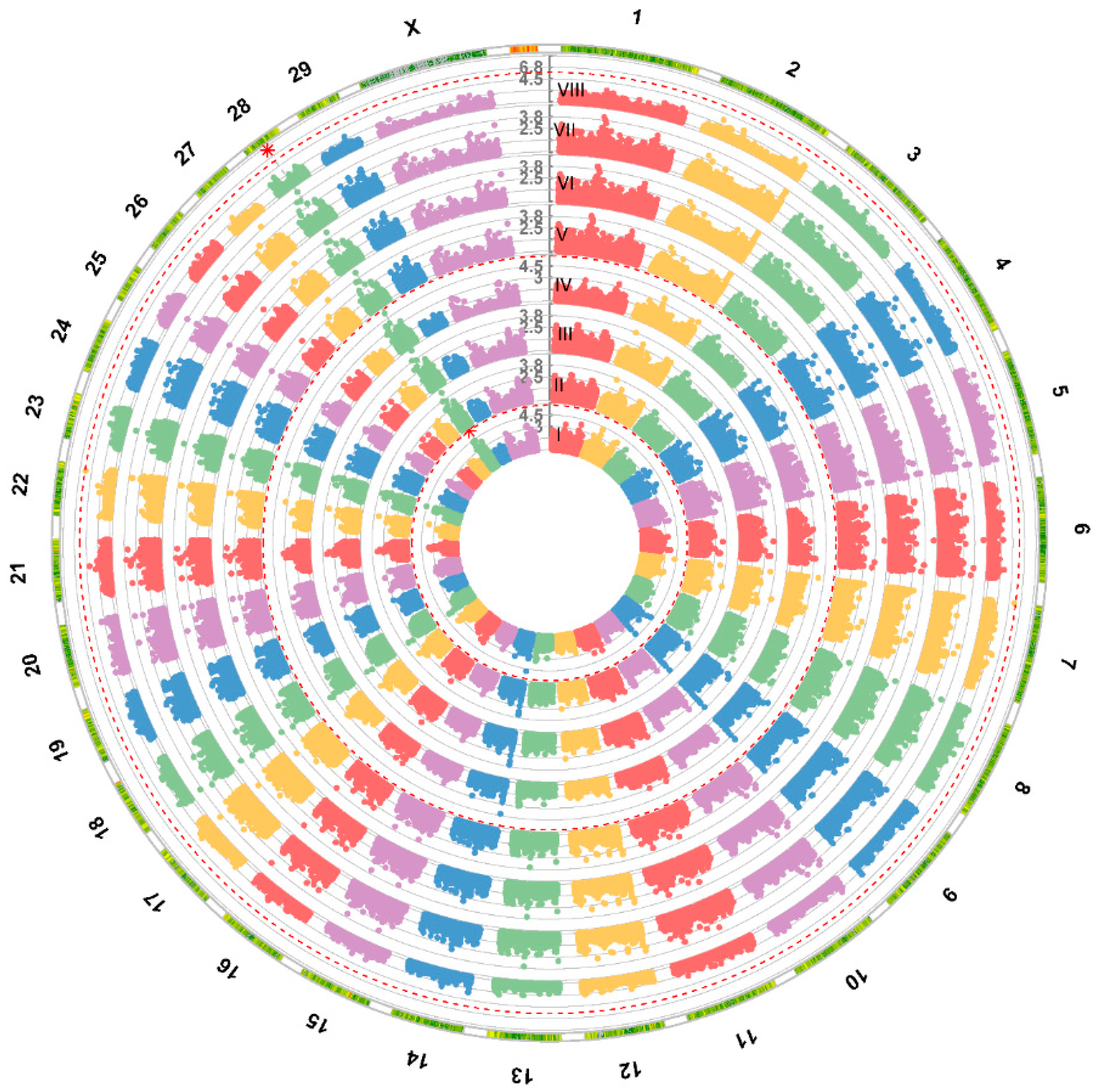
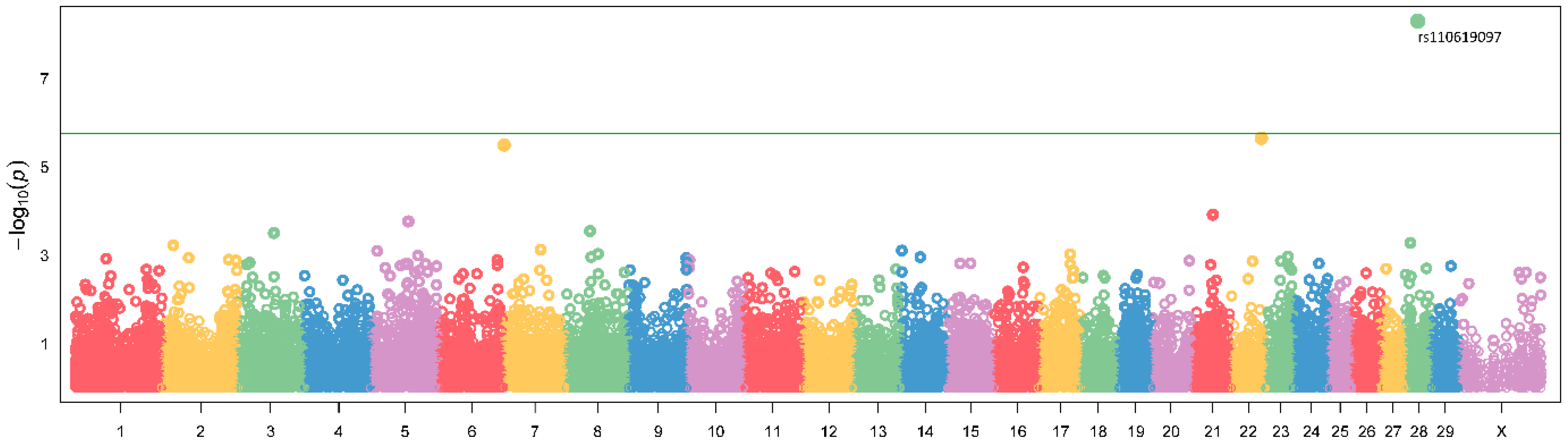
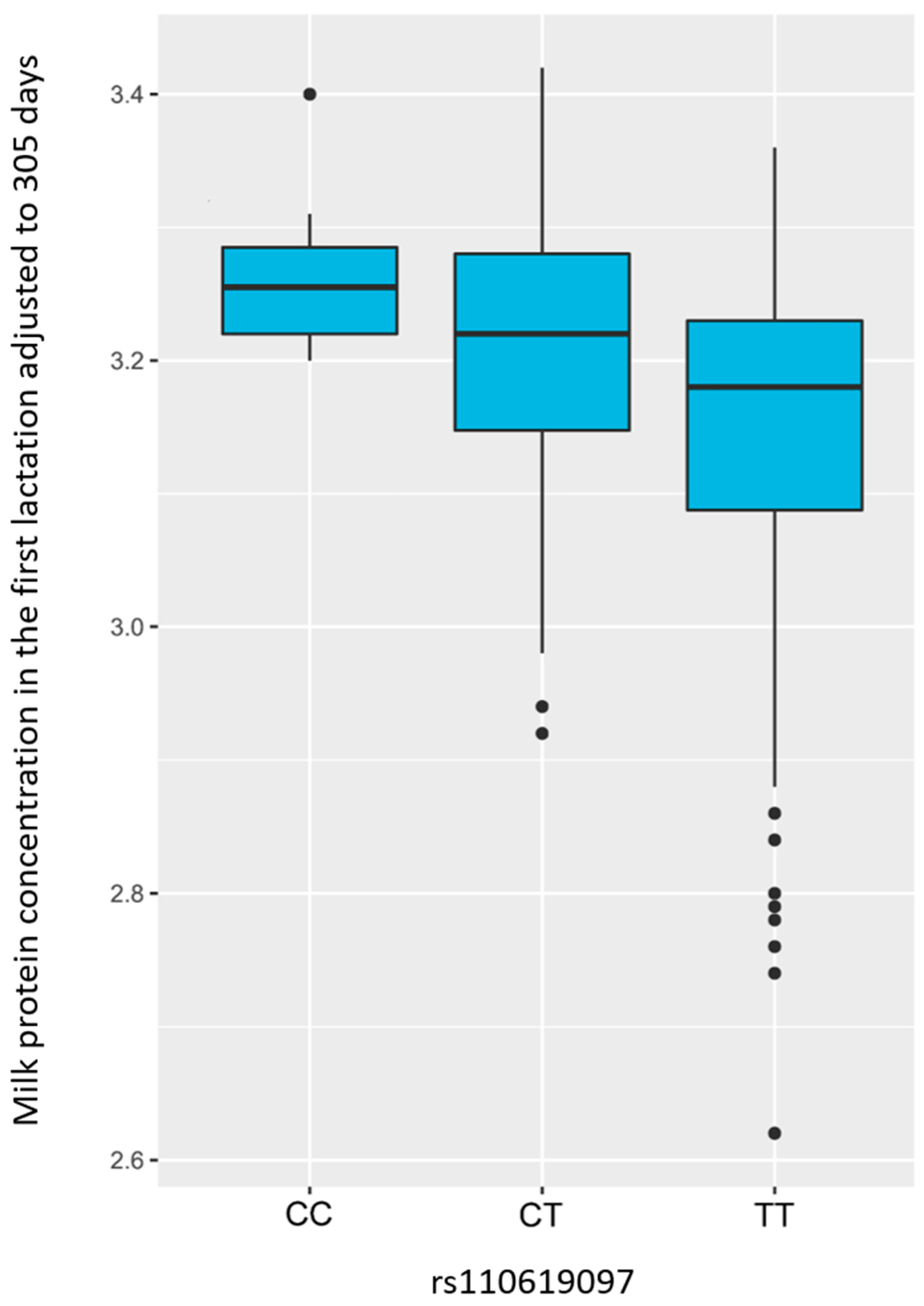
3. Results
3.1. Genome-Wide Association Study—GWAS
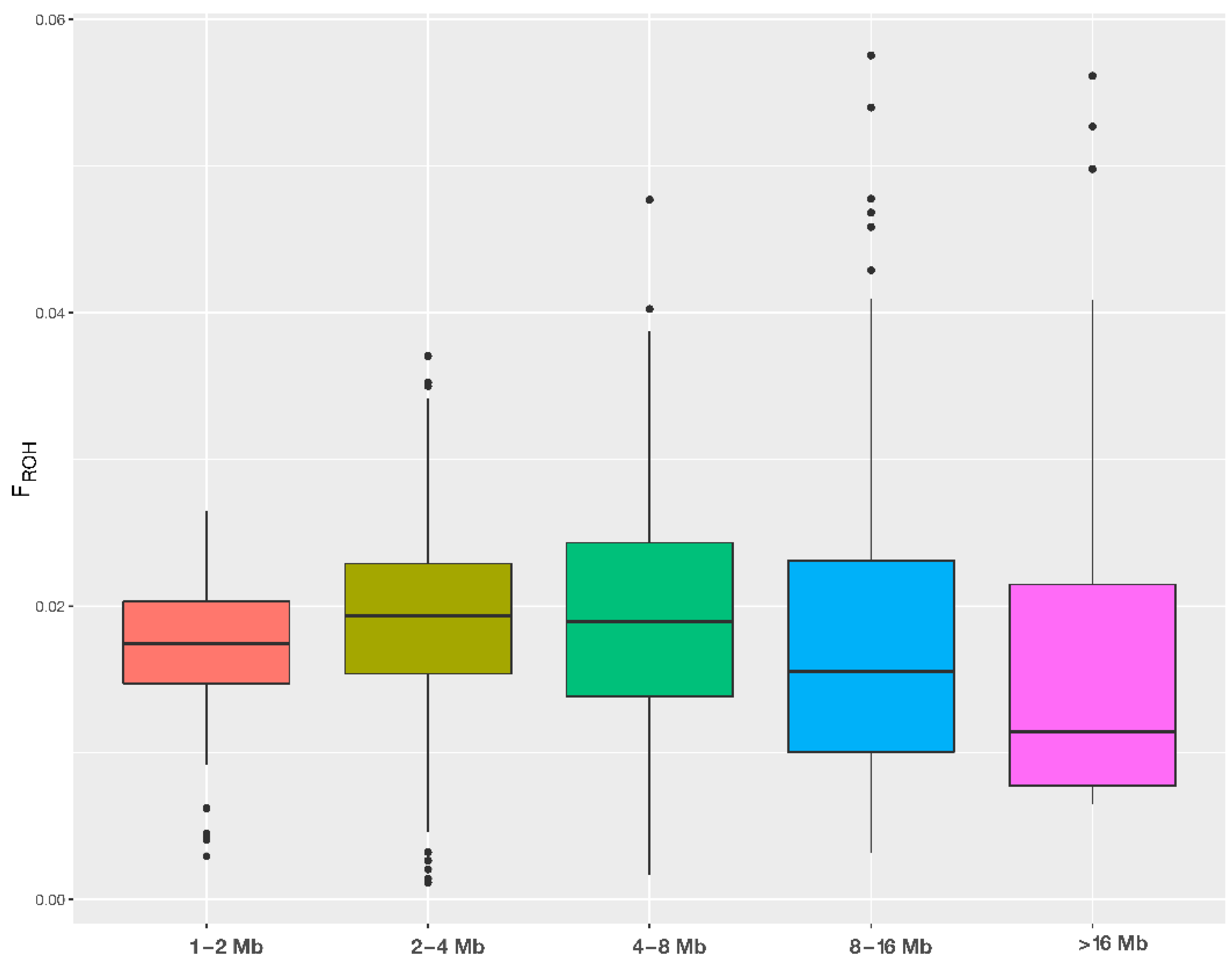
3.2. Runs of Homozygosity—ROH
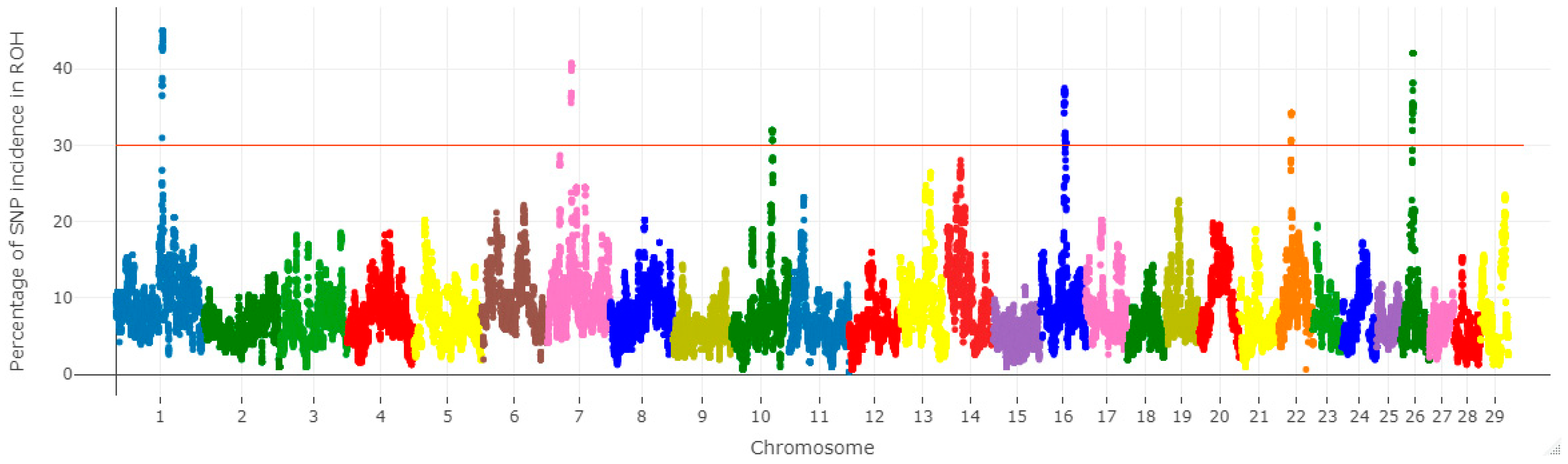
3.3. Selection Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ristanic, M.; Stanisic, L.; Maletic, M.; Glavinic, U.; Draskovic, V.; Aleksic, N.; Stanimirovic, Z. Bovine foetal sex determination—Different DNA extraction and amplification approaches for efficient livestock production. Reprod. Domest. Anim. 2018, 53, 947–954. [Google Scholar] [CrossRef]
- Ristanić, M.; Glavinić, U.; Vejnović, B.; Maletić, M.; Kirovski, D.; Teodorović, V.; Stanimirović, Z. Beta-casein gene polymorphism in Serbian Holstein-Friesian cows and its relationship with milk production traits. Acta Vet. 2020, 70, 497–510. [Google Scholar]
- Pedrosa, V.B.; Schenkel, F.S.; Chen, S.Y.; Oliveira, H.R.; Casey, T.M.; Melka, M.G.; Brito, L.F. Genomewide association analyses of lactation persistency and milk production traits in Holstein cattle based on imputed whole-genome sequence data. Genes 2021, 12, 1830. [Google Scholar] [CrossRef] [PubMed]
- Bekele, R.; Taye, M.; Abebe, G.; Meseret, S. Genomic regions and candidate genes associated with milk production traits in Holstein and its crossbred cattle: A review. Int. J. Genom. 2023, 2023, 8497453. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Z.; Wang, S.; Li, H. Progress of genome wide association study in domestic animals. J. Anim. Sci. Biotechnol. 2012, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Saowaphak, P.; Duangjinda, M.; Plaengkaeo, S.; Suwannasing, R.; Boonkum, W. Genetic correlation and genome-wide association study (GWAS) of the length of productive life, days open, and 305-days milk yield in crossbred Holstein dairy cattle. Genet. Mol. Res. 2017, 16, 1–11. [Google Scholar] [CrossRef]
- Sorbolini, S.; Bongiorni, S.; Cellesi, M.; Gaspa, G.; Dimauro, C.; Valentini, A.; Macciotta, N.P.P. Genome wide association study on beef production traits in Marchigiana cattle breed. J. Anim. Breed. Genet. 2017, 134, 43–48. [Google Scholar] [CrossRef]
- Sermyagin, A.A.; Gladyr, E.A.; Plemyashov, K.V.; Kudinov, A.A.; Dotsev, A.V.; Deniskova, T.E.; Zinovieva, N.A. Genome-wide association studies for milk production traits in Russian population of Holstein and black-and-white cattle. In Proceedings of the Scientific-Practical Conference “Research and Development—2016”, Moscow, Russia, 14–15 December 2016; Springer: Berlin/Heidelberg, Germany, 2018; pp. 591–599. [Google Scholar]
- Zhou, J.; Liu, L.; Chen, C.J.; Zhang, M.; Lu, X.; Zhang, Z.; Huang, X.; Shi, Y. Genome-wide association study of milk and reproductive traits in dual-purpose Xinjiang Brown cattle. BMC Genom. 2019, 20, 827. [Google Scholar] [CrossRef]
- Paiva, J.T.; Peixoto, M.G.C.D.; Bruneli, F.A.T.; Alvarenga, A.B.; Oliveira, H.R.; Silva, A.A.; Silva, D.A.; Veroneze, R.; Silva, F.F.; Lopes, P.S. Genetic parameters, genome-wide association and gene networks for milk and reproductive traits in Guzerá cattle. Livest. Sci. 2020, 242, 104273. [Google Scholar] [CrossRef]
- Pegolo, S.; Momen, M.; Morota, G.; Rosa, G.J.; Gianola, D.; Bittante, G.; Cecchinato, A. Structural equation modeling for investigating multi-trait genetic architecture of udder health in dairy cattle. Sci. Rep. 2020, 10, 7751. [Google Scholar] [CrossRef]
- Da Cruz, A.S.; Silva, D.C.; Minasi, L.B.; de Farias Teixeira, L.K.; Rodrigues, F.M.; da Silva, C.C.; do Carmo, A.S.; da Silva, M.V.G.B.; Utsunomiya, Y.T.; Garcia, J.F.; et al. Single-Nucleotide Polymorphism variations associated with specific genes putatively identified enhanced genetic predisposition for 305-day milk yield in the Girolando crossbreed. Front. Genet. 2021, 11, 573344. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lim, B.; Cho, J.; Lee, S.; Dang, C.G.; Jeon, J.H.; Kim, J.M.; Lee, J. Genome-wide identification of candidate genes for milk production traits in Korean Holstein Cattle. Animals 2021, 11, 1392. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Arbab, A.A.I.; Abdalla, I.M.; Liu, D.; Zhang, Z.; Xu, T.; Su, G.; Yang, Z. Genetic Parameter Estimation and Genome-Wide Association Study-Based Loci Identification of Milk-Related Traits in Chinese Holstein. Front. Genet. 2022, 12, 799664. [Google Scholar] [CrossRef]
- de Souza Fonseca, P.A.; Caldwell, T.; Mandell, I.; Wood, K.; Cánovas, A. Genome-wide association study for meat tenderness in beef cattle identifies patterns of the genetic contribution in different post-mortem stages. Meat Sci. 2022, 186, 108733. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Gloria, L.S.; Pedrosa, V.B.; Doucette, J.; Boerman, J.P.; Brito, L.F. Unravelling the genomic background of resilience based on variability in milk yield and milk production levels in North American Holstein cattle through GWAS and Mendelian randomization analyses. J. Dairy Sci. 2024, 107, 1035–1053. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.A.; Alam, M.Z.; Iqbal, A.; Lee, Y.M.; Dang, C.G.; Kim, J.J. Genome-Wide Association Studies for Body Conformation Traits in Korean Holstein Population. Animals 2023, 13, 2964. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.B.; Wiggans, G.R.; Ma, L.; Sonstegard, T.S.; Lawlor, T.J.; Crooker, B.A.; Van Tassell, C.P.; Yang, J.; Wang, S.; Matukumalli, L.K.; et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary US Holstein cows. BMC Genom. 2011, 12, 408. [Google Scholar] [CrossRef] [PubMed]
- Kolbehdari, D.; Wang, Z.; Grant, J.R.; Murdoch, B.; Prasad, A.; Xiu, Z.; Marques, E.; Stothard, P.; Moore, S.S. A whole genome scan to map QTL for milk production traits and somatic cell score in Canadian Holstein bulls. J. Anim. Breed. Genet. 2009, 126, 216–227. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, L.; Prakapenka, D.; VanRaden, P.M.; Cole, J.B.; Da, Y. A large-scale genome-wide association study in US Holstein cattle. Front. Genet. 2019, 10, 412. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, J.; Chen, C.J.; Zhang, J.; Wen, W.; Tian, J.; Zhang, Z.; Gu, Y. GWAS-based identification of new loci for milk yield, fat, and protein in Holstein cattle. Animals 2020, 10, 2048. [Google Scholar] [CrossRef]
- Schopen, G.; Visker, M.; Koks, P.; Mullaart, E.; van Arendonk, J.; Bovenhuis, H. Whole-genome association study for milk protein composition in dairy cattle. J. Dairy Sci. 2011, 94, 3148–3158. [Google Scholar] [CrossRef] [PubMed]
- Fontanesi, L.; Calò, D.G.; Galimberti, G.; Negrini, R.; Marino, R.; Nardone, A.; Ajmone-Marsan, P.; Russo, V. A candidate gene association study for nine economically important traits in Italian Holstein cattle. Anim. Genet. 2014, 45, 576–580. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, L.; Gao, Y.; Shi, L.; Li, Y.; Liang, W.; Sun, D. Determination of genetic associations between indels in 11 candidate genes and milk composition traits in Chinese Holstein population. BMC Genet. 2019, 20, 48. [Google Scholar] [CrossRef] [PubMed]
- Murgiano, L.; Jagannathan, V.; Benazzi, C.; Bolcato, M.; Brunetti, B.; Muscatello, L.V.; Dittmer, K.; Piffer, C.; Gentile, A.; Drögemüller, C. Deletion in the EVC2 gene causes chondrodysplastic dwarfism in Tyrolean Grey cattle. PLoS ONE 2014, 9, e94861. [Google Scholar] [CrossRef] [PubMed]
- Tiezzi, F.; Parker-Gaddis, K.L.; Cole, J.B.; Clay, J.S.; Maltecca, C. A genome-wide association study for clinical mastitis in first parity US Holstein cows using single-step approach and genomic matrix re-weighting procedure. PLoS ONE 2015, 10, e0114919. [Google Scholar] [CrossRef]
- Brown, A.; Ojango, J.; Gibson, J.; Coffey, M.; Okeyo, M.; Mrode, R. Genomic selection in a crossbred cattle population using data from the dairy genetics East Africa project. J. Dairy Sci. 2016, 99, 7308–7312. [Google Scholar] [CrossRef]
- Boison, S.A.; Utsunomiya, A.T.H.; Santos, D.J.A.; Neves, H.H.R.; Carvalheiro, R.; Mészáros, G.; Utsunomiya, Y.T.; do Carmo, A.S.; Verneque, R.S.; Machado, M.A.; et al. Accuracy of genomic predictions in Gyr (Bos indicus) dairy cattle. J. Dairy Sci. 2017, 100, 5479–5490. [Google Scholar] [CrossRef]
- Meuwissen, T.; Goddard, M. The use of family relationships and linkage disequilibrium to impute phase and missing genotypes in up to whole-genome sequence density genotypic data. Genetics 2010, 185, 1441–1449. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef]
- Zhang, Q.; Calus, M.P.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015, 16, 88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef] [PubMed]
- Makanjuola, B.O.; Maltecca, C.; Miglior, F.; Marras, G.; Abdalla, E.A.; Schenkel, F.S.; Baes, C.F. Identification of unique ROH regions with unfavorable effects on production and fertility traits in Canadian Holsteins. Genet. Sel. Evol. 2021, 53, 68. [Google Scholar] [CrossRef] [PubMed]
- Rothammer, S.; Seichter, D.; Förster, M.; Medugorac, I. A genome-wide scan for signatures of differential artificial selection in ten cattle breeds. BMC Genom. 2013, 14, 908. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, J.J.D.S.; Silva, M.V.G.B.D.; Paiva, S.R.; Oliveira, S.M.P.D. Identification of selection signatures in livestock species. Genet. Mol. Biol. 2014, 37, 330–342. [Google Scholar] [CrossRef]
- Ristanic, M. Comparative Analyses of β-Casein Gene Polymorphism (A1/A2 Genotype) and Its Effect on Qualitative Composition of Milk in Lactating and Autochthonous Cattle Breeds. Ph.D. Thesis, Faculty of Veterinary Medicine, University of Belgrade, Belgrade, Serbia, 2022. [Google Scholar]
- Wang, J.; Zhang, Z. GAPIT version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; The R Foundation: Vienna, Austria, 2013. [Google Scholar]
- Gorssen, W.; Meyermans, R.; Janssens, S.; Buys, N. A publicly available repository of ROH islands reveals signatures of selection in different livestock and pet species. Genet. Sel. Evol. 2021, 53, 2. [Google Scholar] [CrossRef]
- Sahana, G.; Cai, Z.; Sanchez, M.P.; Bouwman, A.C.; Boichard, D. Invited review: Good practices in genome-wide association studies to identify candidate sequence variants in dairy cattle. J. Dairy Sci. 2023, 106, 5218–5241. [Google Scholar] [CrossRef]
- Prylipko, T.; Kuzminska, I.; Sheludchenko, L.; Semenyshena, R.; Koval, T. Control of the Quality and Safety of Dairy Products in Ukraine: International and Legal Aspects. Eur. Food Feed Law Rev. 2023, 18, 22. [Google Scholar]
- Rahimzadeh Barzoki, H.; Faraji, H.; Beirami, S.; Keramati, F.Z.; Nayik, G.A.; Izadi Yazdanaabadi, Z.; Mozaffari Nejad, A.S. Seasonal Study of Aflatoxin M1 Contamination in Cow Milk on the Retail Dairy Market in Gorgan, Iran. Dairy 2023, 4, 571–580. [Google Scholar] [CrossRef]
- Cattle QTLdb. Available online: https://www.animalgenome.org/cgi-bin/QTLdb/BT/qdetails?QTL_ID=65988 (accessed on 10 December 2023).
- Dadousis, C.; Pegolo, S.; Rosa, G.J.M.; Gianola, D.; Bittante, G.; Cecchinato, A. Pathway-based genome-wide association analysis of milk coagulation properties, curd firmness, cheese yield, and curd nutrient recovery in dairy cattle. J. Dairy Sci. 2017, 100, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Inoue, K.; Ogawa, S.; Ichinoseki, K.; Arakawa, A.; Fukuzawa, Y.; Okamura, T.; Kobayashi, E.; Taniguchi, M.; Oe, M.; et al. Comparing pedigree and genomic inbreeding coefficients, and inbreeding depression of reproductive traits in Japanese Black cattle. BMC Genom. 2023, 24, 376. [Google Scholar] [CrossRef]
- Bjelland, D.W.; Weigel, K.A.; Vukasinovic, N.; Nkrumah, J.D. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J. Dairy Sci. 2013, 96, 4697–4706. [Google Scholar] [CrossRef]
- Lozada-Soto, E.A.; Maltecca, C.; Lu, D.; Miller, S.; Cole, J.B.; Tiezzi, F. Trends in genetic diversity and the effect of inbreeding in American Angus cattle under genomic selection. Genet. Sel. Evol. 2021, 53, 50. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.T.; Pryce, J.E.; Baes, C.; Maltecca, C. Invited review: Inbreeding in the genomics era: Inbreeding, inbreeding depression, and management of genomic variability. J. Dairy Sci. 2017, 100, 6009–6024. [Google Scholar] [CrossRef] [PubMed]
- Ablondi, M.; Malacarne, M.; Cipolat-Gotet, C.; van Kaam, J.T.; Sabbioni, A.; Summer, A. Genome-wide scan reveals genetic divergence in Italian Holstein cows bred within PDO cheese production chains. Sci. Rep. 2021, 15, 12601. [Google Scholar] [CrossRef]
- Doekes, H.P.; Veerkamp, R.F.; Bijma, P.; de Jong, G.; Hiemstra, S.J.; Windig, J.J. Inbreeding depression due to recent and ancient inbreeding in Dutch Holstein–Friesian dairy cattle. Genet. Sel. Evol. 2019, 51, 54. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Forutan, M.; Ansari Mahyari, S.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef] [PubMed]
- Makanjuola, B.O.; Miglior, F.; Abdalla, E.A.; Maltecca, C.; Schenkel, F.S.; Baes, C.F. Effect of genomic selection on rate of inbreeding and coancestry and effective population size of Holstein and Jersey cattle populations. J. Dairy Sci. 2020, 103, 5183–5199. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef]
- Gurgul, A.; Szmatoła, T.; Topolski, P.; Jasielczuk, I.; Żukowski, K.; Bugno-Poniewierska, M. The use of runs of homozygosity for estimation of recent inbreeding in Holstein cattle. J. Appl. Genet. 2016, 57, 527–530. [Google Scholar] [CrossRef]
- Serão, N.V.; González-Peña, D.; Beever, J.E.; Faulkner, D.B.; Southey, B.R.; Rodriguez-Zas, S.L. Single nucleotide polymorphisms and haplotypes associated with feed efficiency in beef cattle. BMC Genet. 2013, 14, 94. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ROH Length Category | ||||||
|---|---|---|---|---|---|---|
| Parameter | 1–2 Mbp | 2–4 Mbp | 4–8 Mbp | 8–16 Mbp | >16 Mbp | |
| Total length of ROH per animal | Average Value | 43.848 | 48.119 | 48.145 | 44.038 | 39.332 |
| Standard Deviation | 9.979 | 15.504 | 20.138 | 25.655 | 24.867 | |
| Median | 43.817 | 48.553 | 47.651 | 39.144 | 28.769 | |
| Minimum | 7.416 | 2.927 | 4.244 | 8.013 | 16.282 | |
| Maximum | 66.538 | 93.096 | 119.858 | 144.577 | 141.065 | |
| Number of ROH per animal | Average Value | 30.140 | 17.505 | 8.679 | 3.992 | 1.726 |
| Standard Deviation | 6.723 | 5.488 | 3.550 | 2.286 | 0.933 | |
| Median | 30.000 | 18.000 | 8.000 | 4.000 | 1.000 | |
| Minimum | 6.000 | 1.000 | 1.000 | 1.000 | 1.000 | |
| Maximum | 45.000 | 35.000 | 21.000 | 12.000 | 5.000 | |
| Genomic inbreeding FROH | Average Value | 0.017 | 0.019 | 0.019 | 0.018 | 0.016 |
| Standard Deviation | 0.004 | 0.006 | 0.008 | 0.010 | 0.010 | |
| Median | 0.017 | 0.019 | 0.019 | 0.016 | 0.011 | |
| Minimum | 0.003 | 0.001 | 0.002 | 0.003 | 0.006 | |
| Maximum | 0.026 | 0.037 | 0.048 | 0.058 | 0.056 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ristanic, M.; Zorc, M.; Glavinic, U.; Stevanovic, J.; Blagojevic, J.; Maletic, M.; Stanimirovic, Z. Genome-Wide Analysis of Milk Production Traits and Selection Signatures in Serbian Holstein-Friesian Cattle. Animals 2024, 14, 669. https://doi.org/10.3390/ani14050669
Ristanic M, Zorc M, Glavinic U, Stevanovic J, Blagojevic J, Maletic M, Stanimirovic Z. Genome-Wide Analysis of Milk Production Traits and Selection Signatures in Serbian Holstein-Friesian Cattle. Animals. 2024; 14(5):669. https://doi.org/10.3390/ani14050669
Chicago/Turabian StyleRistanic, Marko, Minja Zorc, Uros Glavinic, Jevrosima Stevanovic, Jovan Blagojevic, Milan Maletic, and Zoran Stanimirovic. 2024. "Genome-Wide Analysis of Milk Production Traits and Selection Signatures in Serbian Holstein-Friesian Cattle" Animals 14, no. 5: 669. https://doi.org/10.3390/ani14050669