

Genotyping-by-Sequencing Strategy for Integrating Genomic Structure, Diversity and Performance of Various Japanese Quail (Coturnix japonica) Breeds
 ,
,  , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Birds and Performance Data
2.2. Sampling and DNA Isolation
2.3. Sequencing, Genotyping and Quality Control of SNPs
2.4. Genetic Diversity Assessment
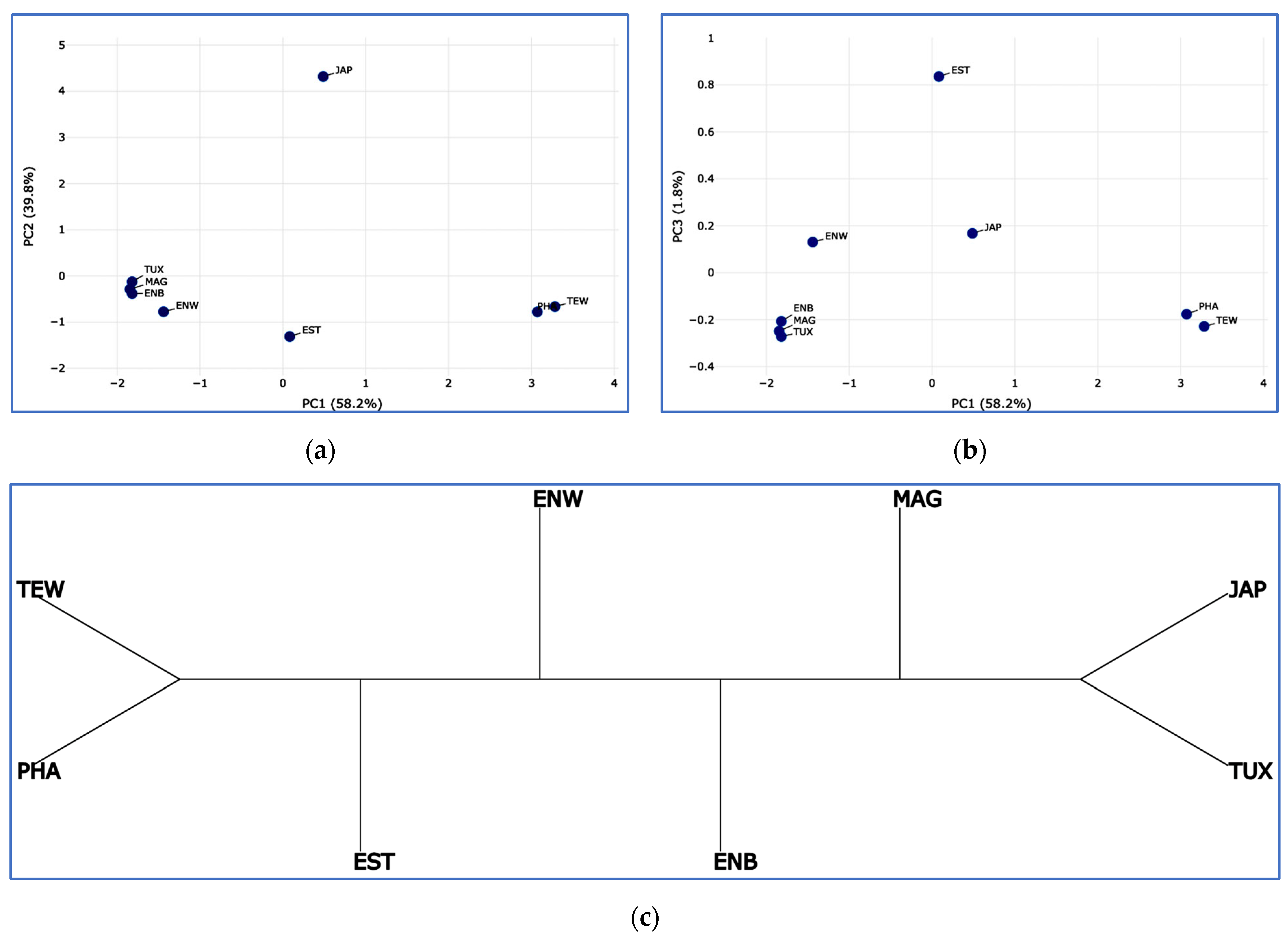
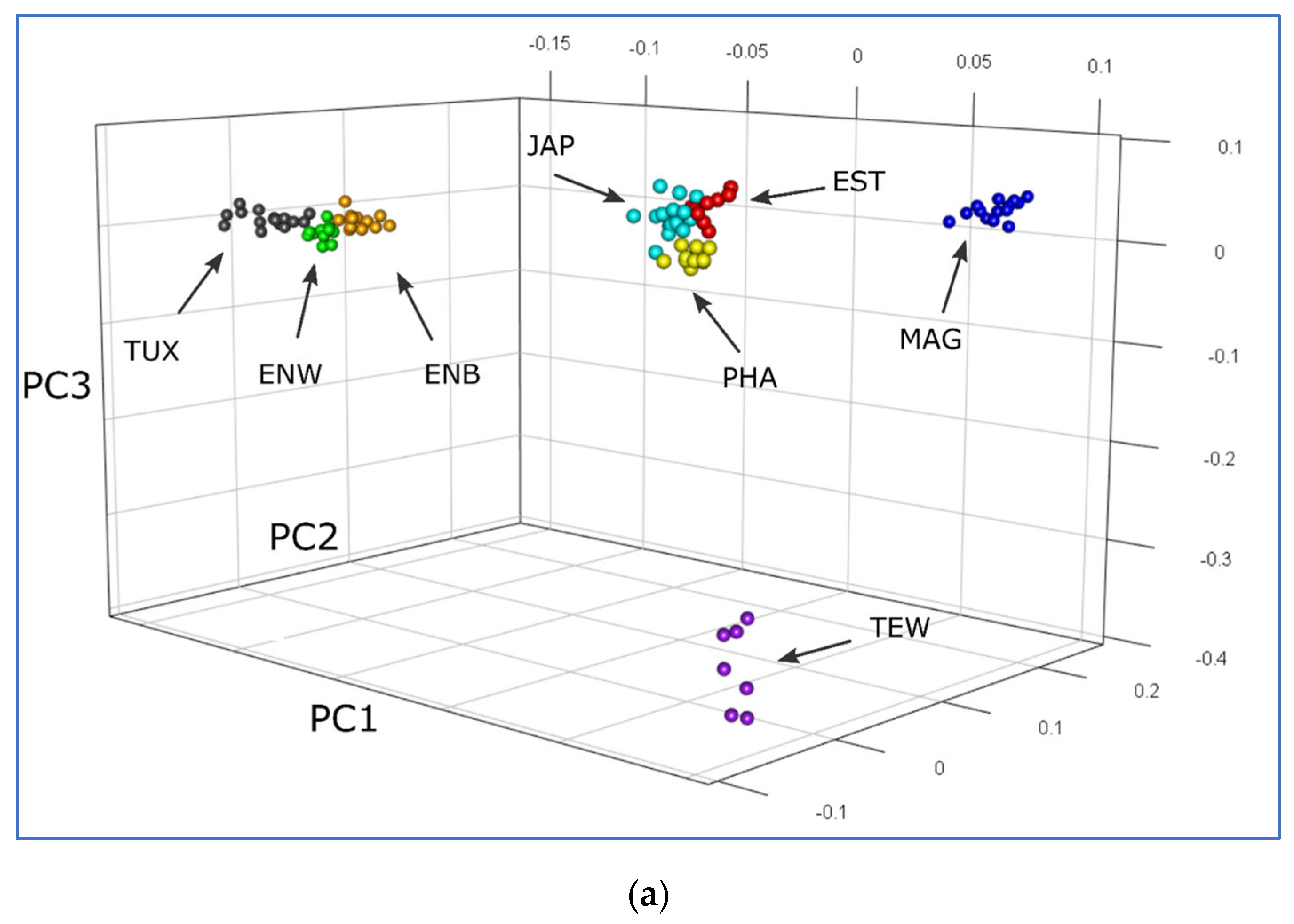
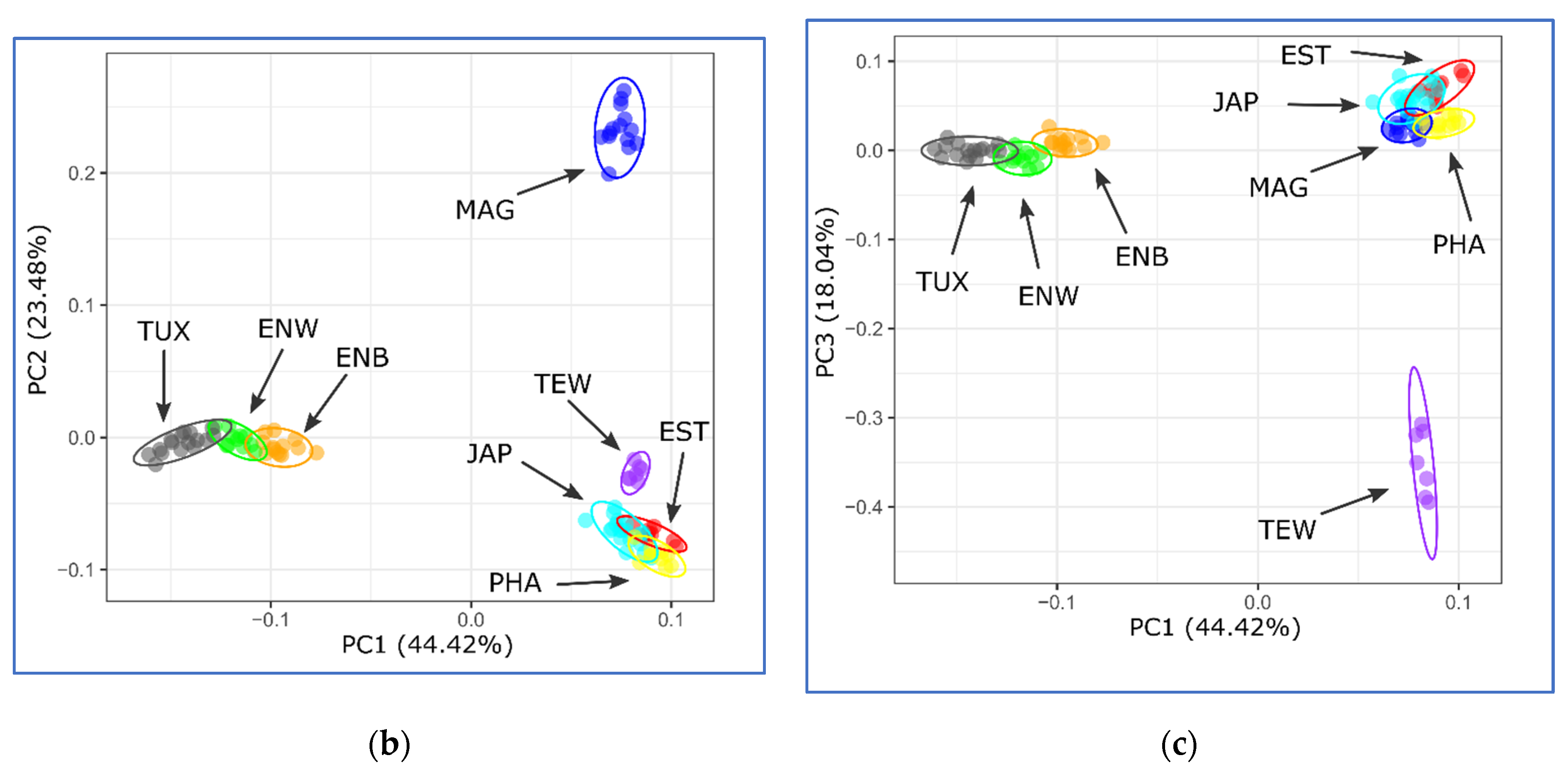
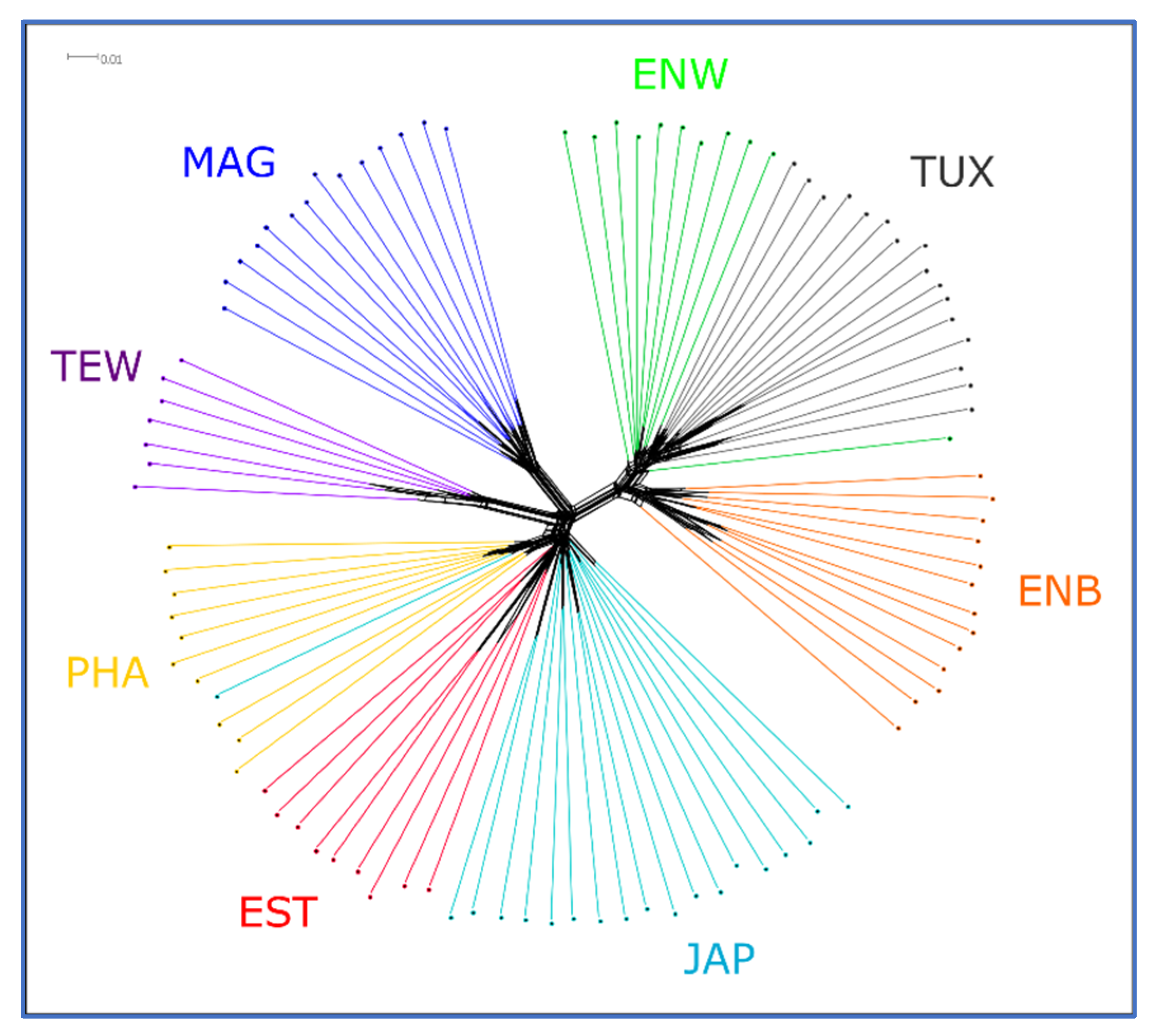
2.5. PCA, Neighbor-Net and Admixture Procedures
3. Results
3.1. Breed Performance
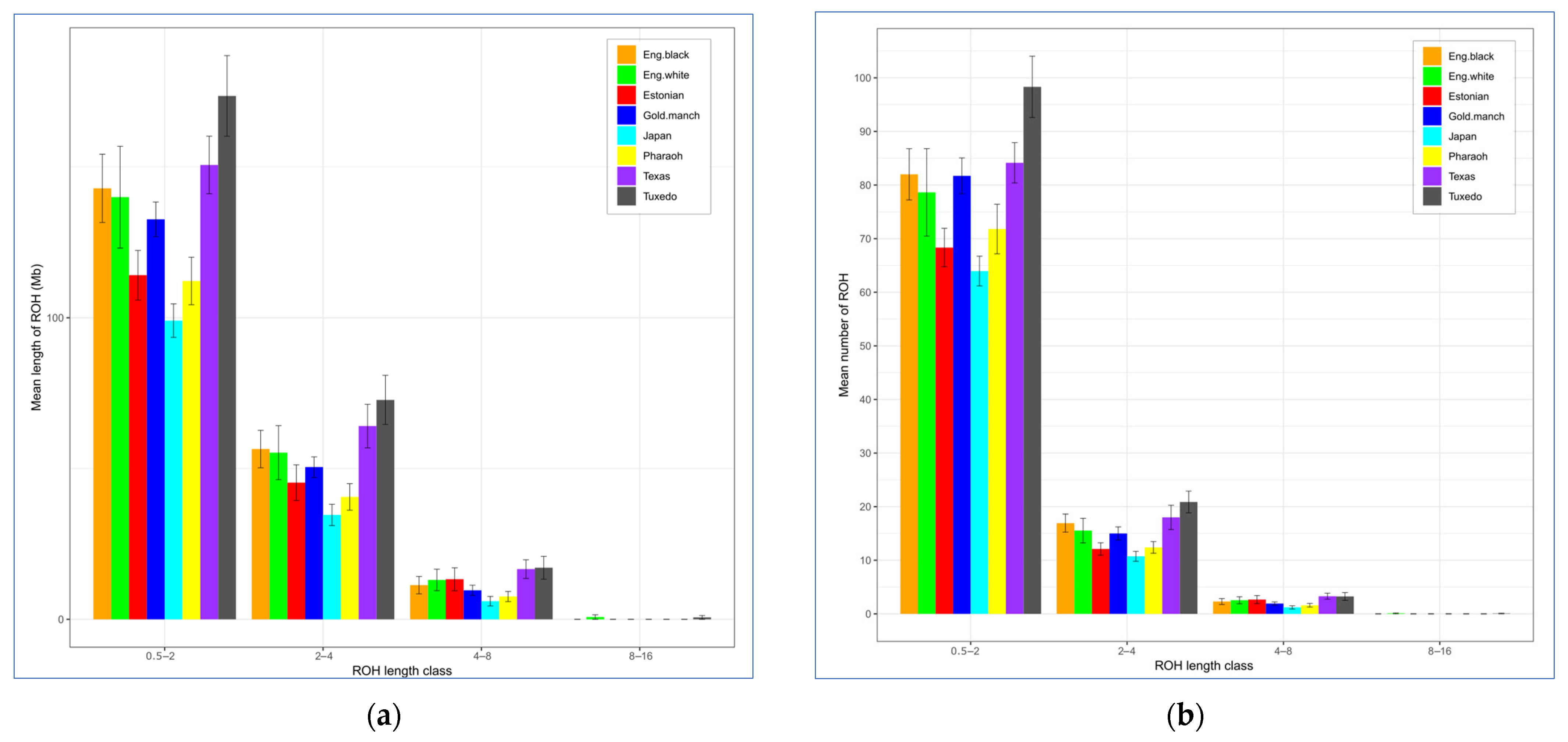
3.2. Analysis of Genetic Diversity
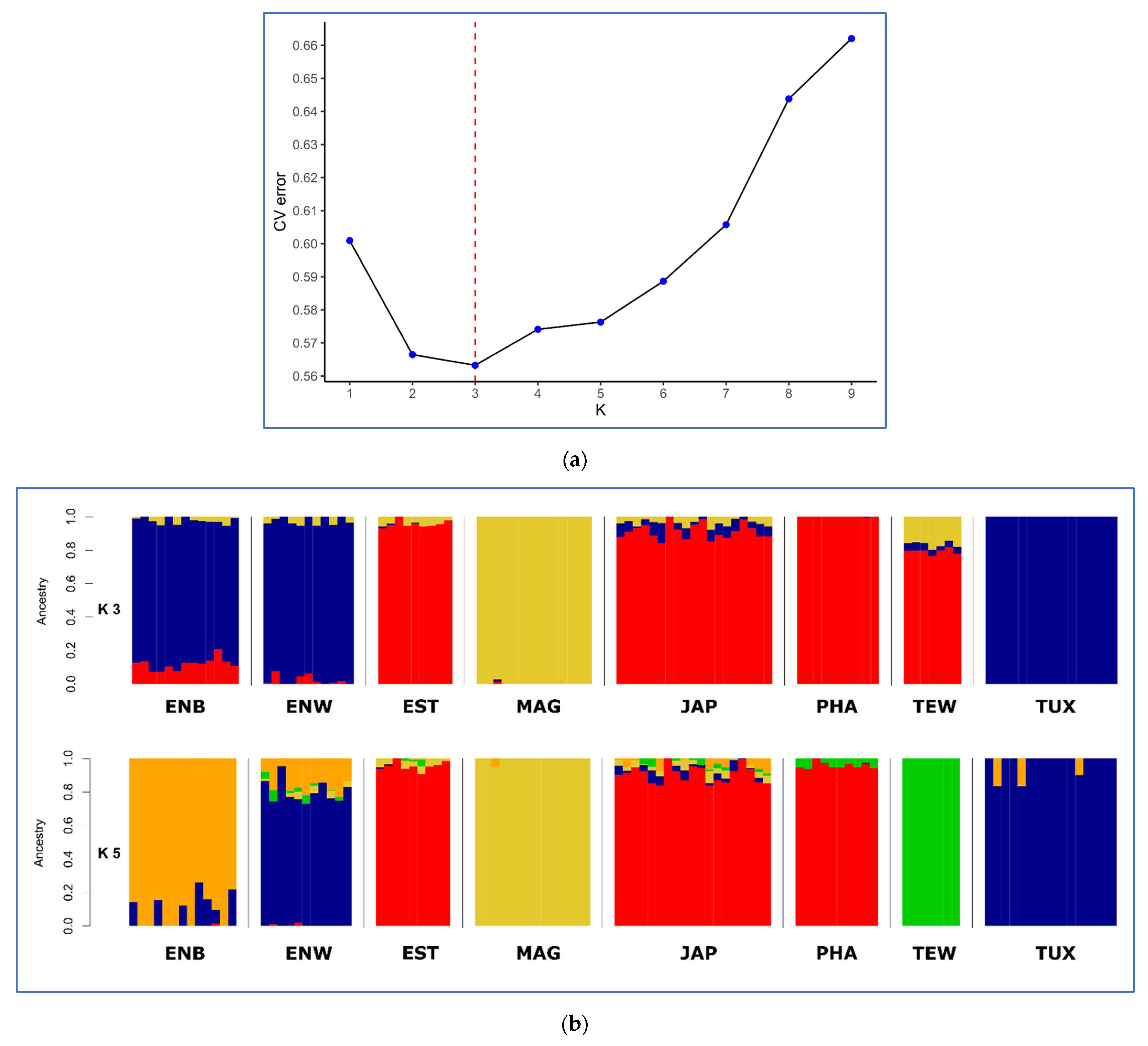
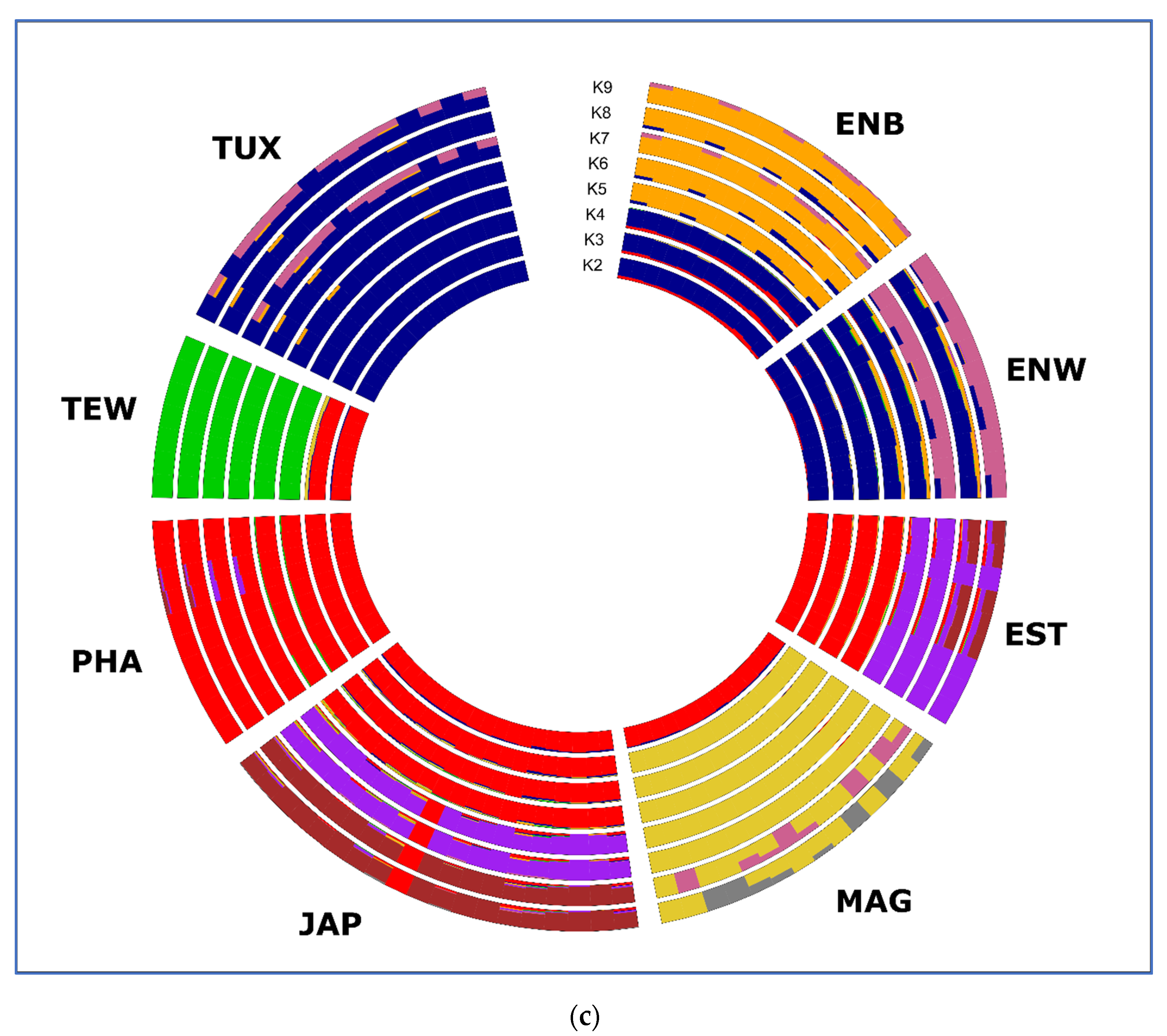
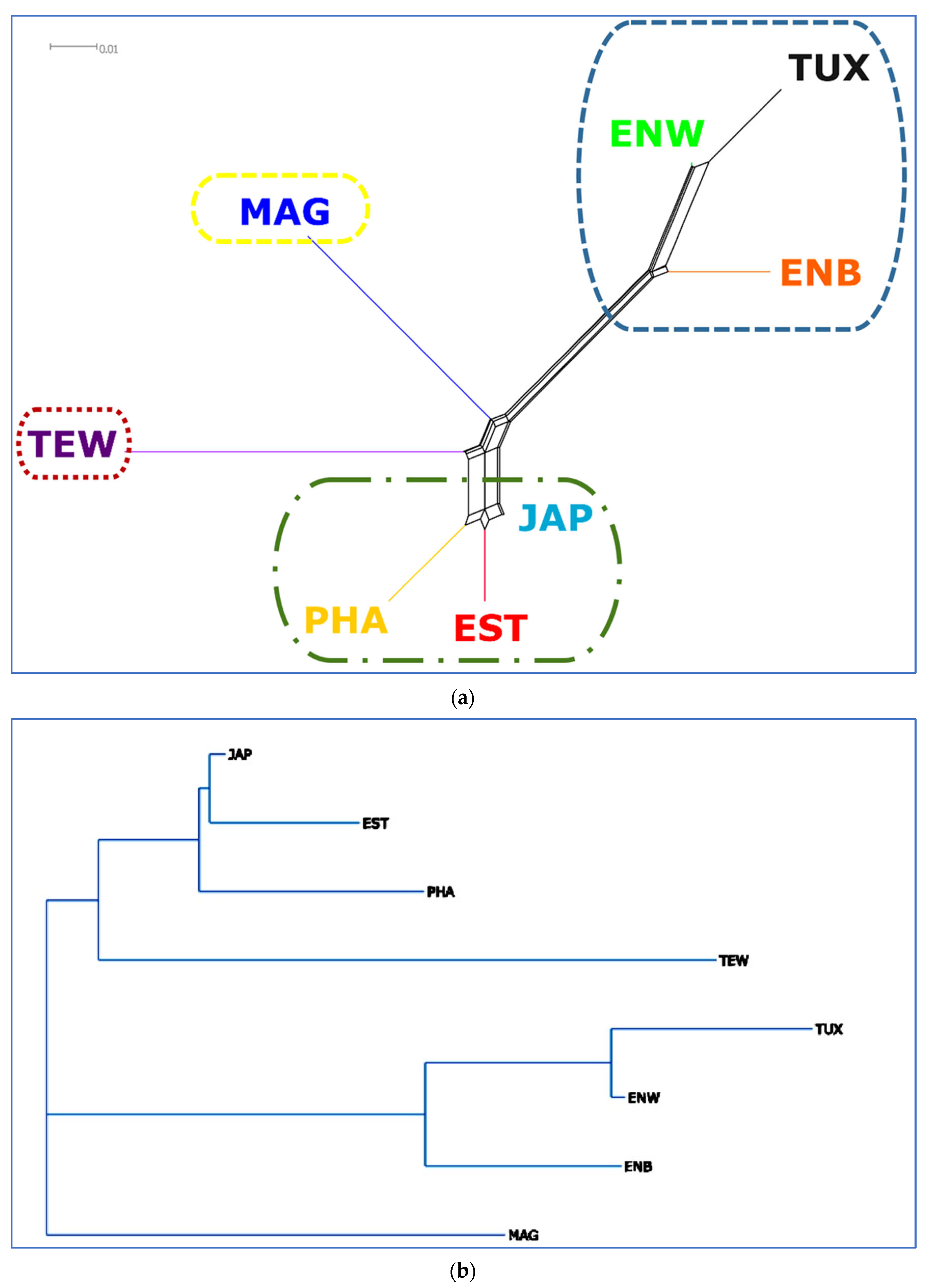
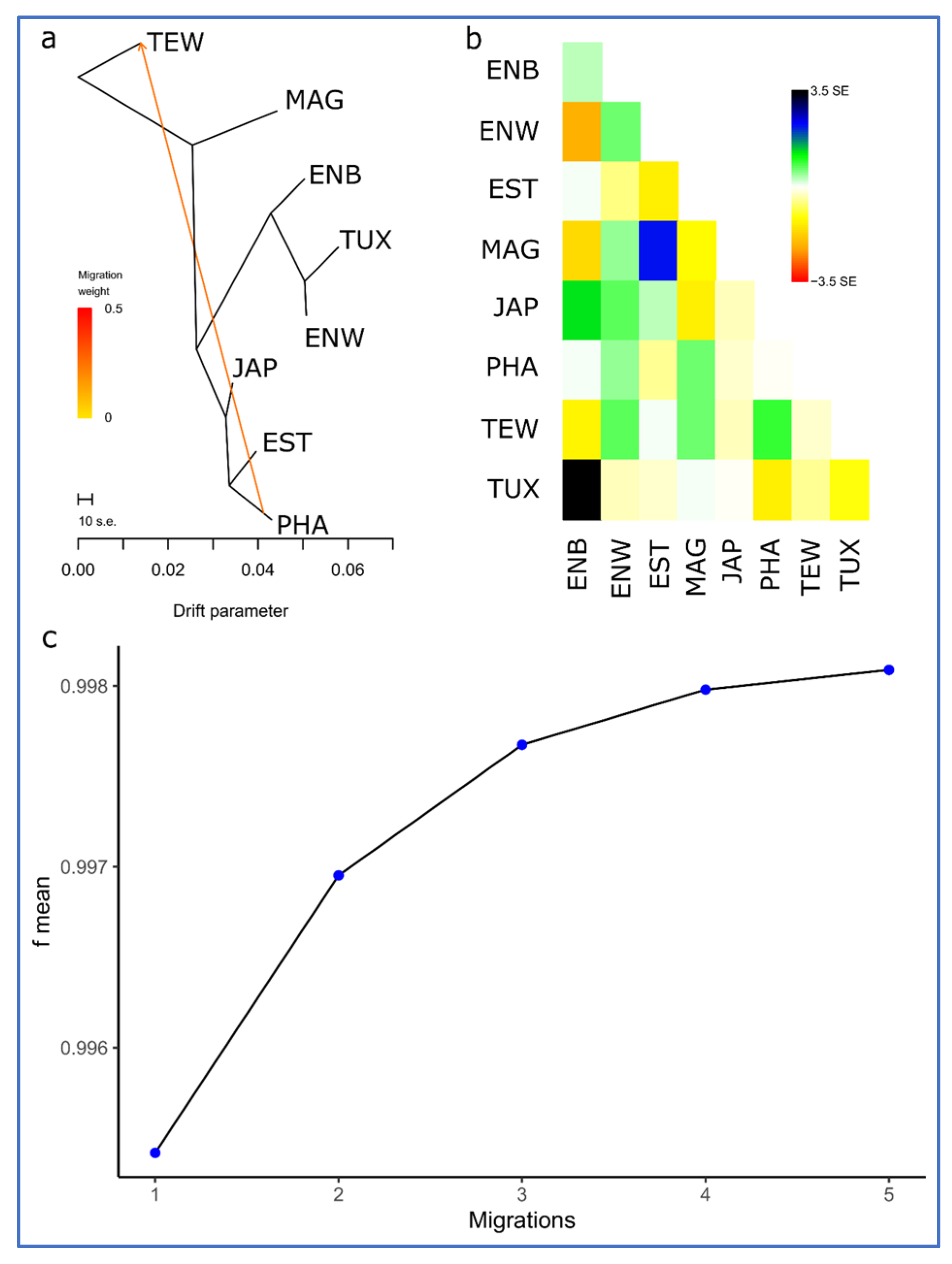
3.3. Between-Breed Genetic Relationships and Model-Based Clustering
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Venkitanarayanan, K.; Thakur, S.; Ricke, S.C. (Eds.) Food Safety in Poultry Meat Production; Springer International Publishing: Cham, Switzerland, 2019; ISBN 978-3-030-05010-8/978-3-030-05011-5. [Google Scholar] [CrossRef]
- Vorotnikov, V. Russia Puts Focus on ‘Non-Traditional’ Poultry; FoodNavigator Europe; William Reed Ltd.: Crawley, UK, 2014; Available online: https://www.foodnavigator.com/Article/2014/01/16/Russia-s-non-traditional-poultry-sees-planned-boost (accessed on 25 September 2023).
- Minvielle, F. What are quail good for in a chicken-focused world? Worlds Poult. Sci. J. 2009, 65, 601–608. [Google Scholar] [CrossRef]
- Romanov, M.N.; Sazanov, A.A.; Moiseyeva, I.G.; Smirnov, A.F. Poultry. In Genome Mapping and Genomics in Animals, Vol. 3: Genome Mapping and Genomics in Domestic Animals; Cockett, N.E., Kole, C., Eds.; Springer: Berlin/Heidelberg, Gemany; New York, NY, USA, 2009; pp. 75–141. ISBN 978-3-540-73834-3/978-3-540-73835-0. [Google Scholar] [CrossRef]
- Podstreshnyi, O.P.; Tereshchenko, O.V.; Katerynych, O.O.; Tkachyk, T.E.; Podstreshna, I.O. Production of Quail Eggs and Meat: Methodical Recommendations, 2nd ed.; Tereshchenko, O.V., Ed.; Poultry Research Institute, NAAS of Ukraine: Birky, Ukraine, 2010; Available online: https://www.researchgate.net/publication/342802513_Podstresnij_OP_Teresenko_OV_Katerinic_OO_Tkacik_TE_Podstresna_IO_Virobnictvo_perepelinih_aec_ta_m’asa_metodicni_rekomendacii_Uklad_OV_Teresenko_ta_in_pid_red_OV_Teresenka_-_2-e_vid_pererob_ta_dop_-_Bi (accessed on 25 September 2023). (In Ukrainian)
- Podstreshnyi, O.; Tereshchenko, O. Maintenance of adult quails. Ahrar. Krayina 2012, 6, 8–9. Available online: https://www.researchgate.net/publication/342832587_Podstresnij_O_Teresenko_O_Utrimanna_doroslih_perepeliv_Agrarna_kraina_-_2012_-_Cerven-_S_8-9 (accessed on 25 September 2023). (In Ukrainian).
- Podstreshnyi, O.; Tereshchenko, O. Feeding young quails. Ahrar. Krayina 2012, 7, 6. Available online: https://www.researchgate.net/publication/342832583_Podstresnij_O_Teresenko_O_Godivla_molodnaka_perepeliv_Agrarna_kraina_-_2012_-_Lipen_-_S_6_httpagrokrainacomuapoultry_farming268-godvlya-molodnyaka-perepelvhtml (accessed on 25 September 2023). (In Ukrainian).
- Shimakura, K. Notes on the genetics of the Japanese quail: I. The simple, Mendelian, autosomal, recessive character, “brown-splashed white”, of its plumage. Jpn. J. Genet. 1940, 16, 106–112, (In Japanese with English Summary). [Google Scholar] [CrossRef]
- Huss, D.; Poynter, G.; Lansford, R. Japanese quail (Coturnix japonica) as a laboratory animal model. Lab. Anim. 2008, 37, 513–519. [Google Scholar] [CrossRef]
- Chang, G.B.; Chang, H.; Liu, X.P.; Xu, W.; Wang, H.Y.; Zhao, W.M.; Olowofeso, O. Developmental research on the origin and phylogeny of quails. Worlds Poult. Sci. J. 2005, 61, 105–112. [Google Scholar] [CrossRef]
- Romanov, M.N.; Wezyk, S.; Cywa-Benko, K.; Sakhatsky, N.I. Poultry genetic resources in the countries of Eastern Europe—History and current state. Poult. Avian Biol. Rev. 1996, 7, 1–29. Available online: https://www.researchgate.net/publication/255710929_Poultry_genetic_resources_in_the_countries_of_Eastern_Europe_-_history_and_current_state (accessed on 25 September 2023).
- Volkovoy, S.; Bondarenko, Y. Japanese quail plumage rainbow. Priusadebnoye Khozyaystvo 1989, 5, 14–15. Available online: https://yablonka.net/world/zh/686-raduga-opereniya-yaponskogo-perepela.html (accessed on 25 September 2023). (In Russian).
- Genofond. Catalogue of Breeds: Quails; Official Site of the Company Genofond LLC: Sergiev Posad, Russia, 2015; Available online: http://www.genofond-sp.ru/quail.html (accessed on 25 September 2023). (In Russian)
- Baumgartner, J.; Bondarenko, Y.V. Search for Autosexing Strains and Crosses in Japanese Quail. In Proceedings of the 8th International Symposium on Actual Problems of Avian Genetics, Smolenice, Czechoslovakia, 3–6 April 1989; Slovak Society for Agriculture, Forestry, Food and Veterinary Sciences of Slovak Academy of Sciences: Bratislava, Czechoslovakia; Poultry Research and Production Institute: Ivanka pri Dunaji, Bratislava, Czechoslovakia; Czechoslovak Branch of WPSA: Smolenice, Czechoslovakia, 1989; pp. 262–265. [Google Scholar]
- Bondarenko, Y.V. Contemporary Methods for Determining the Sex of Young Domestic and Ornamental Birds, 4th ed.; NTUL: Sumy, Ukraine, 2020; Available online: https://web.archive.org/web/20220325163425/https:/repo.snau.edu.ua/bitstream/123456789/8319/1/%D0%9A%D0%BD%D0%B8%D0%B3%D0%B0%20%D0%91%D0%BE%D0%BD%D0%B4%D0%B0%D1%80%D0%B5%D0%BD%D0%BA%D0%BE_%2B17.12.%202019%20%D0%A0%D0%BE%D0%B7%D0%B1%D0%BB%D0%BE%D0%BA%D0%BE%D0%B2%D0%B0%D0%BD%D0%B0.pdf (accessed on 25 September 2023). (In Russian)
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Gurgul, A.; Miksza-Cybulska, A.; Szmatoła, T.; Jasielczuk, I.; Piestrzyńska-Kajtoch, A.; Fornal, A.; Semik-Gurgul, E.; Bugno-Poniewierska, M. Genotyping-by-sequencing performance in selected livestock species. Genomics 2019, 111, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Sonah, H.; Bastien, M.; Iquira, E.; Tardivel, A.; Légaré, G.; Boyle, B.; Normandeau, É.; Laroche, J.; Larose, S.; Jean, M.; et al. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE 2013, 8, e54603. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Clarke, S.; Faville, M.; Griffiths, A.; Cao, M.; Tan, R.; Van Stijn, T.; Anderson, R.; Ashby, R.; Rowe, S.; et al. Genotyping-by-sequencing Applications in Biology. In Proceedings of the Plant and Animal Genome XXV Conference, San Diego, CA, USA, 13–18 January 2017; Scherago International: Surfside, FL, USA, 2017. Abstract P0128. [Google Scholar] [CrossRef]
- De Donato, M.; Peters, S.O.; Mitchell, S.E.; Hussain, T.; Imumorin, I.G. Genotyping-by-sequencing (GBS): A novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS ONE 2013, 8, e62137. [Google Scholar] [CrossRef] [PubMed]
- Larson, W.A.; Seeb, L.W.; Everett, M.V.; Waples, R.K.; Templin, W.D.; Seeb, J.E. Genotyping by sequencing resolves shallow population structure to inform conservation of Chinook salmon (Oncorhynchus tshawytscha). Evol. Appl. 2014, 7, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Ogada, S.; Otecko, N.O.; Moraa Kennedy, G.; Musina, J.; Agwanda, B.; Obanda, V.; Lichoti, J.; Peng, M.S.; Ommeh, S. Demographic history and genetic diversity of wild African harlequin quail (Coturnix delegorguei delegorguei) populations of Kenya. Ecol. Evol. 2021, 11, 18562–18574. [Google Scholar] [CrossRef] [PubMed]
- Ravagni, S.; Sanchez-Donoso, I.; Jiménez-Blasco, I.; Andrade, P.; Puigcerver, M.; Chorão Guedes, A.; Godinho, R.; Gonçalves, D.; Leitão, M.; Leonard, J.A.; et al. Evolutionary history of an island endemic, the Azorean common quail. Mol. Ecol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Prituzhalova, A.O.; Volkova, N.A.; Kuzmina, T.I.; Vetokh, A.N.; Dzhagaev, A.Y. Monitoring of indicators of chromatin status in quails ovarian follicles granulosa cells of different directions of productivity. Agrar. Nauka 2023, 368, 53–57, (In Russian with English Summary). [Google Scholar] [CrossRef]
- Mills, A.D.; Crawford, L.L.; Domjan, M.; Faure, J.M. The behavior of the Japanese or domestic quail Coturnix japonica. Neurosci. Biobehav. Rev. 1997, 21, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Ryabokon, Y.O.; Pabat, V.O.; Mykytyuk, D.M.; Frolov, V.V.; Katerynych, O.O.; Bondarenko, Y.V.; Mosyakina, T.V.; Gadyuchko, O.T.; Kovalenko, G.T.; Gritsenko, D.M.; et al. Catalog of Poultry Breeding Resources of Ukraine; Ryabokon, Y.O., Ed.; Poultry Research Institute: Kharkiv, Ukraine, 2005; Available online: http://avianua.com/archiv/plevreestr/per.pdf (accessed on 25 September 2023). (In Ukrainian)
- Domesticfutures. Quail Breeds: Characteristics with Photos; domesticfutures.com. 2021. Available online: https://domesticfutures.com/porody-perepelov-harakteristiki-s-fotografiyami-4457 (accessed on 25 September 2023).
- Genchev, A. Egg production potential of Manchurian Golden quail breeders. Agric. Sci. Technol. 2011, 3, 73–80. Available online: http://agriscitech.eu/wp-content/uploads/2014/05/GB_02.pdf (accessed on 25 September 2023).
- Purely Poultry. Gold Coturnix Quail Set; Purely Poultry: Durand, WI, USA, 2023; Available online: https://www.purelypoultry.com/index.php?main_page=product_info&products_id=1267 (accessed on 25 September 2023).
- German, N.Y.; Vetokh, A.N.; Dzhagaev, A.Y.; Ilyina, E.R.; Kotova, T.O. Morphometric parameters of eggs from breeds quail for meat. Vet. Kormlenie 2023, 2, 20–23, (In Russian with English Summary). [Google Scholar] [CrossRef]
- Vakhrameev, A.B.; Narushin, V.G.; Larkina, T.A.; Barkova, O.Y.; Peglivanyan, G.K.; Dysin, A.P.; Dementieva, N.V.; Makarova, A.V.; Shcherbakov, Y.S.; Pozovnikova, M.V.; et al. Disentangling clustering configuration intricacies for divergently selected chicken breeds. Sci. Rep. 2023, 13, 3319. [Google Scholar] [CrossRef] [PubMed]
- GraphPad Software. Dotmatics. Available online: https://www.graphpad.com/ (accessed on 18 October 2023).
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; Anderson, R.M.; van Stijn, T.C.; Kristjánsson, T.; Clarke, S.M. Construction of relatedness matrices using genotyping-by-sequencing data. BMC Genom. 2015, 16, 1047. [Google Scholar] [CrossRef] [PubMed]
- AgResearch. DECONVQC; GitHub, Inc. 2016. Available online: https://github.com/AgResearch/DECONVQC (accessed on 25 September 2023).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Version 0.10.1; Bioinformatics Group, Babraham Institute: Cambridge, UK, 2012; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 25 September 2023).
- Morris, K.M.; Hindle, M.M.; Boitard, S.; Burt, D.W.; Danner, A.F.; Eory, L.; Forrest, H.L.; Gourichon, D.; Gros, J.; Hillier, L.W.; et al. The quail genome: Insights into social behaviour, seasonal biology and infectious disease response. BMC Biol. 2020, 18, 14. [Google Scholar] [CrossRef]
- Szpak, M. Ensembl 104 Has Been Released; Ensembl Blog. 2021. Available online: https://www.ensembl.info/2021/05/05/ensembl-104-has-been-released/ (accessed on 25 September 2023).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt. Version 3.4; GitHub, Inc. 2021. Available online: https://github.com/marcelm/cutadapt (accessed on 25 September 2023).
- Kang, J.; Dodds, K.; Byrne, S.; Faville, M.; Black, M.; Hess, A.; Hess, M.; McCulloch, A.; Jacobs, J.; Milbourne, D.; et al. snpGBS: A Simple and Flexible Bioinformatics Workflow to Identify SNPs from Genotyping-by-sequencing Data. In Exploiting Genetic Diversity of Forages to Fulfil Their Economic and Environmental Roles, Proceedings of the 34th Meeting of the EUCARPIA Fodder Crops and Amenity Grasses Section in Cooperation with the EUCARPIA Festulolium Working Group, Freising, Germany, 6–8 September 2021; Hartmann, S., Bachmann-Pfabe, S., Byrne, S., Feuerstein, U., Julier, B., Kölliker, R., Kopecky, D., Roldan-Ruiz, I., Ruttink, T., Sampoux, J.-P., et al., Eds.; Palacký University Press: Olomouc, Czech Republic, 2021; pp. 67–70. ISBN 978-80-244-5967-7/978-80-244-5968-4/978-80-244-5969-1. [Google Scholar] [CrossRef]
- AgResearch. snpGBS; GitHub, Inc. 2021. Available online: https://github.com/AgResearch/snpGBS (accessed on 25 September 2023).
- Langmead, B. bowtie2: A Fast and Sensitive Gapped Read Aligner. Version 2.4.4; GitHub, Inc. 2021. Available online: https://github.com/BenLangmead/bowtie2 (accessed on 25 September 2023).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.r-project.org/ (accessed on 25 September 2023).
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodohl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Kalinowski, S.T. Counting alleles with rarefaction: Private alleles and hierarchical sampling designs. Conserv. Genet. 2004, 5, 539–543. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98141-3. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Milanesi, M.; Capomaccio, S.; Vajana, E.; Bomba, L.; Garcia, J.F.; Ajmone-Marsan, P.; Colli, L. BITE: An R package for biodiversity analyses. bioRxiv 2017, 181610. [Google Scholar] [CrossRef]
- Zenkova, D.; Kamenev, V.; Sablina, R.; Artyomov, M.; Sergushichev, A. Phantasus: Visual and Interactive Gene Expression Analysis. 2018. Available online: https://ctlab.itmo.ru/phantasus (accessed on 25 September 2023). [CrossRef]
- Boc, A.; Diallo, A.B.; Makarenkov, V. T-REX: A web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res. 2012, 40, W573–W579. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef] [PubMed]
- Fitak, R.R. OptM: Estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protoc. 2021, 6, bpab017. [Google Scholar] [CrossRef] [PubMed]
- Biscarini, F.; Paolo Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes; R Package Version 0.9.6; The Comprehensive R Archive Network (CRAN); Institute for Statistics and Mathematics, Vienna University of Economics and Business: Vienna, Austria, 2019; Available online: https://CRAN.R-project.org/package=detectRUNS (accessed on 25 September 2023).
- Abdelmanova, A.S.; Dotsev, A.V.; Romanov, M.N.; Stanishevskaya, O.I.; Gladyr, E.A.; Rodionov, A.N.; Vetokh, A.N.; Volkova, N.A.; Fedorova, E.S.; Gusev, I.V.; et al. Unveiling comparative genomic trajectories of selection and key candidate genes in egg-type Russian White and meat-type White Cornish chickens. Biology 2021, 10, 876. [Google Scholar] [CrossRef]
- Rostamzadeh Mahdabi, E.; Esmailizadeh, A.; Ayatollahi Mehrgardi, A.; Asadi Fozi, M. A genome-wide scan to identify signatures of selection in two Iranian indigenous chicken ecotypes. Genet. Sel. Evol. 2021, 53, 72. [Google Scholar] [CrossRef]
- Cendron, F.; Mastrangelo, S.; Tolone, M.; Perini, F.; Lasagna, E.; Cassandro, M. Genome-wide analysis reveals the patterns of genetic diversity and population structure of 8 Italian local chicken breeds. Poult. Sci. 2021, 100, 441–451. [Google Scholar] [CrossRef]
- Moiseyeva, I.G.; Bannikova, L.V.; Altukhov, Y.P. State of poultry breeding in Russia: Genetic monitoring. Mezhdunar. S-kh. Zh. 1993, 5–6, 66–69. (In Russian) [Google Scholar]
- Bondarenko, Y.V.; Kutnyuk, P.I. Some Results of Genetic Monitoring of Embryonic Defects in Poultry Populations. In Gene Pool of Animal Breeds and Methods of its Use, Proceedings of the Materials of the International Scientific and Practical Conference Dedicated to the 110th Anniversary of the Birth of Academician N.D. Potemkin, Kharkov, Ukraine, 5–6 December 1995; Ministry of Agriculture and Food of Ukraine, Kharkov Zooveterinary Institute, RIO KhZVI: Kharkov, Ukraine, 1995; pp. 63–64. (In Russian) [Google Scholar]
- Bondarenko, Y.V.; Podstreshny, A.P. Genetic Monitoring of Chicken Populations. In Proceedings of the 2nd International Conference on Molecular Genetic Markers of Animals, Kiev, Ukraine, 15–17 May 1996; Agrarna Nauka: Kiev, Ukraine, 1996; pp. 47–48. (In Russian). [Google Scholar]
- Zakharov-Gesekhus, I.A.; Stolpovsky, Y.A.; Ukhanov, S.V.; Moiseyeva, I.G.; Sulimova, G.E. Monitoring the gene pools of animal populations in connection with selection tasks and the study of phylogeny. In Farm Animals; Russian Academy of Sciences: Moscow, Russia, 2007; pp. 122–124. (In Russian) [Google Scholar]
- Heffelfinger, C.; Fragoso, C.A.; Moreno, M.A.; Overton, J.D.; Mottinger, J.P.; Zhao, H.; Tohme, J.; Dellaporta, S.L. Flexible and scalable genotyping-by-sequencing strategies for population studies. BMC Genom. 2014, 15, 979. [Google Scholar] [CrossRef] [PubMed]
- Mischler, C.; Veale, A.; Van Stijn, T.; Brauning, R.; McEwan, J.C.; Maloney, R.; Robertson, B.C. Population connectivity and traces of mitochondrial introgression in New Zealand black-billed gulls (Larus bulleri). Genes 2018, 9, 544. [Google Scholar] [CrossRef] [PubMed]
- Rexer-Huber, K.; Veale, A.J.; Catry, P.; Cherel, Y.; Dutoit, L.; Foster, Y.; McEwan, J.C.; Parker, G.C.; Phillips, R.A.; Ryan, P.G.; et al. Genomics detects population structure within and between ocean basins in a circumpolar seabird: The white-chinned petrel. Mol. Ecol. 2019, 28, 4552–4572. [Google Scholar] [CrossRef]
- Wold, J.R.; Robertson, C.J.; Chambers, G.K.; Van Stijn, T.; Ritchie, P.A. Genetic connectivity in allopatric seabirds: Lack of inferred gene flow between Northern and Southern Buller’s albatross populations (Thalassarche bulleri ssp.). Emu-Austral Ornithol. 2021, 121, 113–123. [Google Scholar] [CrossRef]
- Foster, Y.; Dutoit, L.; Grosser, S.; Dussex, N.; Foster, B.J.; Dodds, K.G.; Brauning, R.; Van Stijn, T.; Robertson, F.; McEwan, J.C.; et al. Genomic signatures of inbreeding in a critically endangered parrot, the kākāpō. G3 2021, 11, jkab307. [Google Scholar] [CrossRef] [PubMed]
- Tixier-Boichard, M.; Coquerelle, G.; Vilela-Lamego, C.; Weigend, S.; Barre-Dirrie, A.; Groenen, M.; Crooijmans, R.; Vignal, A.; Hillel, J.; Freidlin, P.; et al. Contribution of Data on History, Management and Phenotype to the Description of the Diversity between Chicken Populations Sampled within the AVIANDIV Project. In Proceedings of the Poultry Genetics Symposium, Mariensee, Germany, 6–8 October 1999; Preisinger, R., Ed.; Working Group 3 of WPSA, Lohmann Tierzucht: Cuxhaven, Germany, 1999; pp. 15–21. Available online: https://jukuri.luke.fi/handle/10024/446389 (accessed on 25 September 2023).
- Nunome, M.; Nakano, M.; Tadano, R.; Kawahara-Miki, R.; Kono, T.; Takahashi, S.; Kawashima, T.; Fujiwara, A.; Nirasawa, K.; Mizutani, M.; et al. Genetic divergence in domestic Japanese quail inferred from mitochondrial DNA D-loop and microsatellite markers. PLoS ONE 2017, 12, e0169978. [Google Scholar] [CrossRef]
- Smith, S.; Fusani, L.; Boglarka, B.; Sanchez-Donoso, I.; Marasco, V. Lack of introgression of Japanese quail in a captive population of common quail. Eur. J. Wildl. Res. 2018, 64, 51. [Google Scholar] [CrossRef]
- Amaral, A.J.; Silva, A.B.; Grosso, A.R.; Chikhi, L.; Bastos-Silveira, C.; Dias, D. Detection of hybridization and species identification in domesticated and wild quails using genetic markers. Folia Zool. 2007, 56, 285–300. Available online: https://www.ivb.cz/wp-content/uploads/56_285-300.pdf (accessed on 25 September 2023).
- Barilani, M.; Deregnaucourt, S.; Gallego, S.; Galli, L.; Mucci, N.; Piombo, R.; Puigcerver, M.; Rimondi, S.; Rodríguez-Teijeiro, J.D.; Spanò, S.; et al. Detecting hybridization in wild (Coturnix c. coturnix) and domesticated (Coturnix c. japonica) quail populations. Biol. Conserv. 2005, 126, 445–455. [Google Scholar] [CrossRef]
- Chazara, O.; Minvielle, F.; Roux, D.; Bed’hom, B.; Feve, K.; Coville, J.-L.; Kayang, B.B.; Lumineau, S.; Vignal, A.; Boutin, J.-M.; et al. Evidence for introgressive hybridization of wild common quail (Coturnix coturnix) by domesticated Japanese quail (Coturnix japonica) in France. Conserv. Genet. 2010, 11, 1051–1062. [Google Scholar] [CrossRef]
- Sanchez-Donoso, I.; Vilà, C.; Puigcerver, M.; Butkauskas, D.; de la Calle, J.R.C.; Morales-Rodríguez, P.A.; Rodríguez-Teijeiro, J.D. Are farm-reared quails for game restocking really common quails (Coturnix coturnix): A genetic approach. PLoS ONE 2012, 7, e39031. [Google Scholar] [CrossRef] [PubMed]
- Oyun, N.Y.; Moiseyeva, I.G.; Sevastianova, A.A.; Vakhrameev, A.B.; Alexandrov, A.V.; Kuzevanova, A.Y.; Alimov, A.A.; Sulimova, G.E. Mitochondrial DNA polymorphism in different populations of Spangled Orloff chickens. Genetika 2015, 51, 1057–1065, (In Russian with English Summary). [Google Scholar] [CrossRef]
- Oyun, N.Y.; Moiseyeva, I.G.; Sevastianova, A.A.; Vakhrameev, A.B.; Alexandrov, A.V.; Kuzevanova, A.Y.; Alimov, A.A.; Sulimova, G.E. Mitochondrial DNA polymorphism in different populations of Orloff Spangled chicken breed. Russ. J. Genet. 2015, 51, 908–915. [Google Scholar] [CrossRef]
- Kowalczyk, M.; Staniszewski, A.; Kamiñska, K.; Domaradzki, P.; Horecka, B. Advantages, possibilities, and limitations of mitochondrial DNA analysis in molecular identification. Folia Biol. 2021, 69, 101–111. [Google Scholar] [CrossRef]
- Romanov, M.N.; Weigend, S. Genetic Diversity in Chicken Populations Based on Microsatellite Markers. In Proceedings of the Conference “From Jay Lush to Genomics: Visions for Animal Breeding and Genetics”, Ames, IA, USA, 16–18 May 1999; Dekkers, J.C.M., Lamont, S.J., Rothschild, M.F., Eds.; Iowa State University, Department of Animal Science: Ames, IA, USA, 1999; p. 174. Available online: https://web.archive.org/web/20050314091227/http://www.agbiotechnet.com/proceedings/jaylush.asp#34 (accessed on 25 September 2023).
- Mohammadabadi, M.R.; Nikbakhti, M.; Mirzaee, H.R.; Shandi, M.A.; Saghi, D.A.; Romanov, M.N.; Moiseyeva, I.G. Genetic variability in three native Iranian chicken populations of the Khorasan province based on microsatellite markers. Genetika 2010, 46, 572–576. Available online: https://www.researchgate.net/publication/44661596_Genetic_variability_in_three_native_Iranian_chicken_populations_of_the_Khorasan_province_based_on_microsatellite_markers (accessed on 25 September 2023). [CrossRef]
- Weigend, S.; Romanov, M.N.; Ben-Ari, G.; Hillel, J. Overview on the Use of Molecular Markers to Characterize Genetic Diversity in Chickens. In Proceedings of the XXII World’s Poultry Congress & Exhibition, Participant List & Full Text CD + Book of Abstracts, Istanbul, Turkey, 8–13 June 2004; WPSA—Turkish Branch: Istanbul, Turkey, 2004; p. 192. Available online: https://www.researchgate.net/publication/372751440_Overview_on_the_use_of_molecular_markers_to_characterize_genetic_diversity_in_chickens (accessed on 25 September 2023).
- Dementieva, N.V.; Shcherbakov, Y.S.; Tyshchenko, V.I.; Terletsky, V.P.; Vakhrameev, A.B.; Nikolaeva, O.A.; Ryabova, A.E.; Azovtseva, A.I.; Mitrofanova, O.V.; Peglivanyan, G.K.; et al. Comparative analysis of molecular RFLP and SNP markers in assessing and understanding the genetic diversity of various chicken breeds. Genes 2022, 13, 1876. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Wang, X.; Zhang, C.; Tu, Y.; Lv, W.; Cai, X.; Xu, Z.; Yao, J.; Yang, C. Genomic analysis of GBS data reveals genes associated with facial pigmentation in Xinyang blue-shelled layers. Arch. Anim. Breed. 2020, 63, 483–491. [Google Scholar] [CrossRef]
- Zhu, F.; Cui, Q.Q.; Hou, Z.C. SNP discovery and genotyping using Genotyping-by-Sequencing in Pekin ducks. Sci. Rep. 2016, 6, 36223. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, Q.; Lin, R.; Chen, H.; Zhang, M.; Jiang, B.; Wang, Y.; Xue, P.; Gan, Q.; Shen, Y.; et al. Genome-wide association study for the primary feather color trait in a native Chinese duck. Front. Genet. 2023, 14, 1065033. [Google Scholar] [CrossRef] [PubMed]
- Grzegorczyk, J.; Gurgul, A.; Oczkowicz, M.; Szmatoła, T.; Fornal, A.; Bugno-Poniewierska, M. Single nucleotide polymorphism discovery and genetic differentiation analysis of geese bred in Poland, using genotyping-by-sequencing (GBS). Genes 2021, 12, 1074. [Google Scholar] [CrossRef]
- Deng, M.T.; Zhu, F.; Yang, Y.Z.; Yang, F.X.; Hao, J.P.; Chen, S.R.; Hou, Z.C. Genome-wide association study reveals novel loci associated with body size and carcass yields in Pekin ducks. BMC Genom. 2019, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Mathur, S.; DeWoody, J.A. Genetic load has potential in large populations but is realized in small inbred populations. Evol. Appl. 2021, 14, 1540–1557. [Google Scholar] [CrossRef]
- Lake, J.A.; Dekkers, J.C.; Abasht, B. Genetic basis and identification of candidate genes for wooden breast and white striping in commercial broiler chickens. Sci. Rep. 2021, 11, 6785. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, I. Chicken genetic resources in Russia. Ptitsevodstvo 1995, 5, 12–15. (In Russian) [Google Scholar]
- Moiseyeva, I.G. The state of poultry genetic resources in Russia. Anim. Genet. Resour. 1996, 17, 73–86. [Google Scholar] [CrossRef]
- Weigend, S.; Romanov, M.N.; Rath, D. Methodologies to Identify, Evaluate and Conserve Poultry Genetic Resources. In Proceedings of the XXII World’s Poultry Congress & Exhibition, Participant List & Full Text CD + Book of Abstracts, Istanbul, Turkey, 8–13 June 2004; WPSA—Turkish Branch: Istanbul, Turkey, 2004; p. 84. Available online: https://www.researchgate.net/publication/250917228_Methodologies_to_identify_evaluate_and_conserve_poultry_genetic_resources (accessed on 25 September 2023).
- Sulimova, G.E.; Stolpovsky, Y.A.; Kashtanov, S.N.; Moiseeva, I.G.; Zakharov, I.A. Methods of managing the genetic resources of domesticated animals. In Fundamentals of Biological Resource Management: Collection of Scientific Articles; Rysin, L.P., Ed.; Partnership of Scientific Publications KMK LLC: Moscow, Russia, 2005; pp. 331–342. ISBN 5-87317-254-4. Available online: https://elibrary.ru/item.asp?id=50435256 (accessed on 25 September 2023). (In Russian)
- Tagirov, M.T.; Tereshchenko, L.V.; Tereshchenko, A.V. Substantiation of the possibility of using primary germ cells as material for the preservation of poultry genetic resources. Ptakhivnytstvo 2006, 58, 464–473. Available online: https://www.researchgate.net/publication/342751269_Tagirov_MT_Teresenko_LV_Teresenko_AV_Obosnovanie_vozmoznosti_ispolzovania_pervicnyh_zarodysevyh_kletok_v_kacestve_materiala_dla_sohranenia_geneticeskih_resursov_ptic_Ptahivnictvo_Mizvid_temat_nauk_zb_ (accessed on 25 September 2023). (In Russian with English Summary).
- German, N.Y.; Volkova, N.A.; Larionova, P.V.; Vetokh, A.N.; Volkova, L.A.; Sermyagin, A.A.; Shakhin, A.V.; Anshakov, D.V.; Fisinin, V.I.; Zinovieva, N.A. Genome-wide association studies of growth dynamics in quails Coturnix coturnix. Sel’skokhozyaistvennaya Biol. 2022, 57, 1136–1146, (In Russian with English Summary). [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Code | n1 | Origin | Refs |
|---|---|---|---|---|
| Egg type | ||||
Japanese | JAP | 19 | Japan; domesticated in Japan and China in 12th century or earlier; selected in the 1st half of the 20th century, brought to the USSR from Japan in the mid-20th century and/or from Yugoslavia in 1964 | [10,11,12,13,25,26,27] |
English (British) White | ENW | 11 | England; a mutant from JAP quails; imported to the USSR from Hungary in 1987 | [12,13,24,27] |
English (British) Black | ENB | 13 | England; a mutant from JAP quails; imported to the USSR from Hungary in 1971 | [13,27] |
Tuxedo | TUX | 16 | from crossing ENW and ENB | [12,13,27] |
Manchurian (Manchu) Golden (or Golden Phoenix) | MAG | 14 | Marsh Farms, CA, USA, 1960s; bred by Albert Marsh as a natural mutant in a flock of brown-colored quails | [12,13,27,28,29] |
| Dual purpose (or universal) | ||||
Estonian (or Kitevers) | EST | 9 | Estonia, 1988; from crossing JAP (a Moscow line), ENW and Pharaoh | [11,13,27] |
| Meat type | ||||
Pharaoh | PHA | 10 | USA; wild-type plumage; an imported French fattening line used in this study | [12,13,26,27,30] |
Texas White (or Texas Pharaoh, White Pharaoh, Snowy) | TEW | 7 | Texas, USA; from crossing PHA and ENW | [27,30] |
| Breed 2 | n | EN | EW | BW | IPI | |
|---|---|---|---|---|---|---|
| 6 Weeks | 6 Months | |||||
| Egg type | ||||||
| JAP (a) | 41 | 165.4 ± 14.7 a | 11.0 ± 0.9 a | 146.7 ± 13.6 a | 149.0 ± 13.2 a | 12.2 ± 1.2 a |
| ENW (b) | 11 | 134.8 ± 8.5 a,b | 10.2 ± 1.0 a,b | 157.6 ± 12.7 a,b | 166.6 ± 9.0 a,b | 8.3 ± 1.2 a,b |
| ENB (c) | 11 | 133.4 ± 8.0 a,c | 10.4 ± 0.9 c | 151.5 + 15.0 c | 159.5 ± 14.0 a,c | 8.8 ± 1.4 a,c |
| TUX (d) | 11 | 131.1 ± 7.4 a,d | 10.2 ± 0.9 a,d | 141.4 ± 10.5 b,d | 149.0 ± 12.6 b,d | 9.0 ± 0.8 a,d |
| MAG (e) | 12 | 147.5 ± 4.5 a,b,c,d,e | 10.6 ± 1.6 e | 168.2 ± 17.2 a,c,d,e | 180.1 ± 18.9 a,b,c,d,e | 8.8 ± 2.1 a,e |
| Dual purpose | ||||||
| EST (f) | 18 | 148.9 ± 9.7 a,b,c,d,f | 11.8 ± 1.4 a,b,c,d,e,f | 248.2 ± 10.6 a,b,c,d,e,f | 247.3 ± 15.4 a,b,c,d,e,f | 7.1 ± 1.1 a,b,c,d,e,f |
| Meat type | ||||||
| PHA (g) | 12 | 118.2 ± 8.2 a,b,c,d,e,f | 12.6 ± 0.8 a,b,c,d,e,f | 292.3 ± 16.2 a,b,c,d,e,f,g | 294.3 ± 19.5 a,b,c,d,e,f,g | 5.1 ± 0.6 a,b,c,d,e,f |
| TEW (h) | 23 | 121.4 ± 18.7 a,b,c,d,e,f | 12.7 ± 1.0 a,b,c,d,e,f | 305.5 + 21.3 a.b,c,d,e,f,g | 317.7 ± 25.9 a,b,c,d,e,f,g | 4.9 ± 1.0 a,b,c,d,e,f |
| Breed 2 | HO (M ± SE) | HE (M ± SE) | UHE (M ± SE) | AR (M ± SE) | FIS [CI 95%] | UFIS [Cl 95%] |
|---|---|---|---|---|---|---|
| Egg type | ||||||
| JAP | 0.303 ± 0.001 | 0.310 ± 0.001 | 0.319 ± 0.001 | 1.864 ± 0.001 | 0.020 [0.016; 0.024] | 0.046 [0.043; 0.049] |
| ENW | 0.281 ± 0.001 | 0.273 ± 0.001 | 0.287 ± 0.001 | 1.778 ± 0.002 | −0.029 [−0.033; −0.025] | 0.020 [0.016; 0.024] |
| ENB | 0.282 ± 0.001 | 0.276 ± 0.001 | 0.287 ± 0.001 | 1.774 ± 0.002 | −0.019 [−0.023; −0.015] | 0.020 [0.016; 0.024] |
| TUX | 0.265 ± 0.001 | 0.263 ± 0.001 | 0.271 ± 0.001 | 1.730 ± 0.002 | −0.010 [−0.014; −0.006] | 0.022 [0.018; 0.026] |
| MAG | 0.286 ± 0.001 | 0.285 ± 0.001 | 0.295 ± 0.001 | 1.790 ± 0.002 | −0.005 [−0.009; −0.001] | 0.031 [0.027; 0.035] |
| Dual purpose | ||||||
| EST | 0.302 ± 0.001 | 0.295 ± 0.001 | 0.313 ± 0.001 | 1.839 ± 0.002 | −0.025 [−0.030; −0.020] | 0.032 [0.028; 0.036] |
| Meat type | ||||||
| PHA | 0.290 ± 0.001 | 0.286 ± 0.001 | 0.301 ± 0.001 | 1.815 ± 0.002 | −0.017 [−0.021; −0.013] | 0.035 [0.031; 0.039] |
| TEW | 0.282 ± 0.002 | 0.264 ± 0.001 | 0.284 ± 0.001 | 1.757 ± 0.003 | −0.067 [−0.072; −0.062] | 0.011 [0.006; 0.016] |
| Breed 2 | ROH Length, Mb (M ± SE) | ROH No. (M ± SE) | FROH (M ± SE) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Average | Min | Max | Average | Min | Max | Average | Min | Max | |
| Egg type | |||||||||
| JAP | 99.04 ± 5.55 | 48.71 | 138.58 | 75.89 ± 3.38 | 49 | 104 | 0.119 ± 0.007 | 0.06 | 0.17 |
| ENW | 140.00 ± 16.88 | 8.43 | 216.10 | 96.82 ± 10.55 | 7 | 133 | 0.169 ± 0.020 | 0.01 | 0.26 |
| ENB | 142.92 ± 11.32 | 58.47 | 201.15 | 101.23 ± 6.37 | 54 | 137 | 0.172 ± 0.014 | 0.07 | 0.24 |
| TUX | 173.54 ± 13.36 | 36.79 | 237.00 | 122.50 ± 7.81 | 31 | 164 | 0.209 ± 0.016 | 0.04 | 0.29 |
| MAG | 132.63 ± 5.77 | 91.63 | 174.35 | 98.64 ± 3.93 | 69 | 121 | 0.160 ± 0.007 | 0.11 | 0.21 |
| Dual purpose | |||||||||
| EST | 114.11 ± 8.23 | 56.47 | 137.53 | 83.11 ± 4.83 | 49 | 98 | 0.137 ± 0.010 | 0.07 | 0.17 |
| Meat type | |||||||||
| PHA | 112.18 ± 7.86 | 62.22 | 157.96 | 85.80 ± 5.01 | 54 | 108 | 0.135 ± 0.009 | 0.07 | 0.19 |
| TEW | 150.66 ± 9.54 | 115.13 | 191.32 | 105.43 ± 4.74 | 87 | 122 | 0.181 ± 0.011 | 0.14 | 0.23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volkova, N.A.; Romanov, M.N.; Abdelmanova, A.S.; Larionova, P.V.; German, N.Y.; Vetokh, A.N.; Shakhin, A.V.; Volkova, L.A.; Anshakov, D.V.; Fisinin, V.I.; et al. Genotyping-by-Sequencing Strategy for Integrating Genomic Structure, Diversity and Performance of Various Japanese Quail (Coturnix japonica) Breeds. Animals 2023, 13, 3439. https://doi.org/10.3390/ani13223439
Volkova NA, Romanov MN, Abdelmanova AS, Larionova PV, German NY, Vetokh AN, Shakhin AV, Volkova LA, Anshakov DV, Fisinin VI, et al. Genotyping-by-Sequencing Strategy for Integrating Genomic Structure, Diversity and Performance of Various Japanese Quail (Coturnix japonica) Breeds. Animals. 2023; 13(22):3439. https://doi.org/10.3390/ani13223439
Chicago/Turabian StyleVolkova, Natalia A., Michael N. Romanov, Alexandra S. Abdelmanova, Polina V. Larionova, Nadezhda Yu. German, Anastasia N. Vetokh, Alexey V. Shakhin, Ludmila A. Volkova, Dmitry V. Anshakov, Vladimir I. Fisinin, and et al. 2023. "Genotyping-by-Sequencing Strategy for Integrating Genomic Structure, Diversity and Performance of Various Japanese Quail (Coturnix japonica) Breeds" Animals 13, no. 22: 3439. https://doi.org/10.3390/ani13223439