
Next-Generation Sequencing of Four Mitochondrial Genomes of Dolichovespula (Hymenoptera: Vespidae) with a Phylogenetic Analysis and Divergence Time Estimation of Vespidae

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Gathering and DNA Isolation
2.2. Mitochondrial Genome Sequencing and Assembly
2.3. Mitochondrial Genome Annotation and Analysis
2.4. Phylogenetic Analysis
2.5. Divergence Time Estimation
3. Results
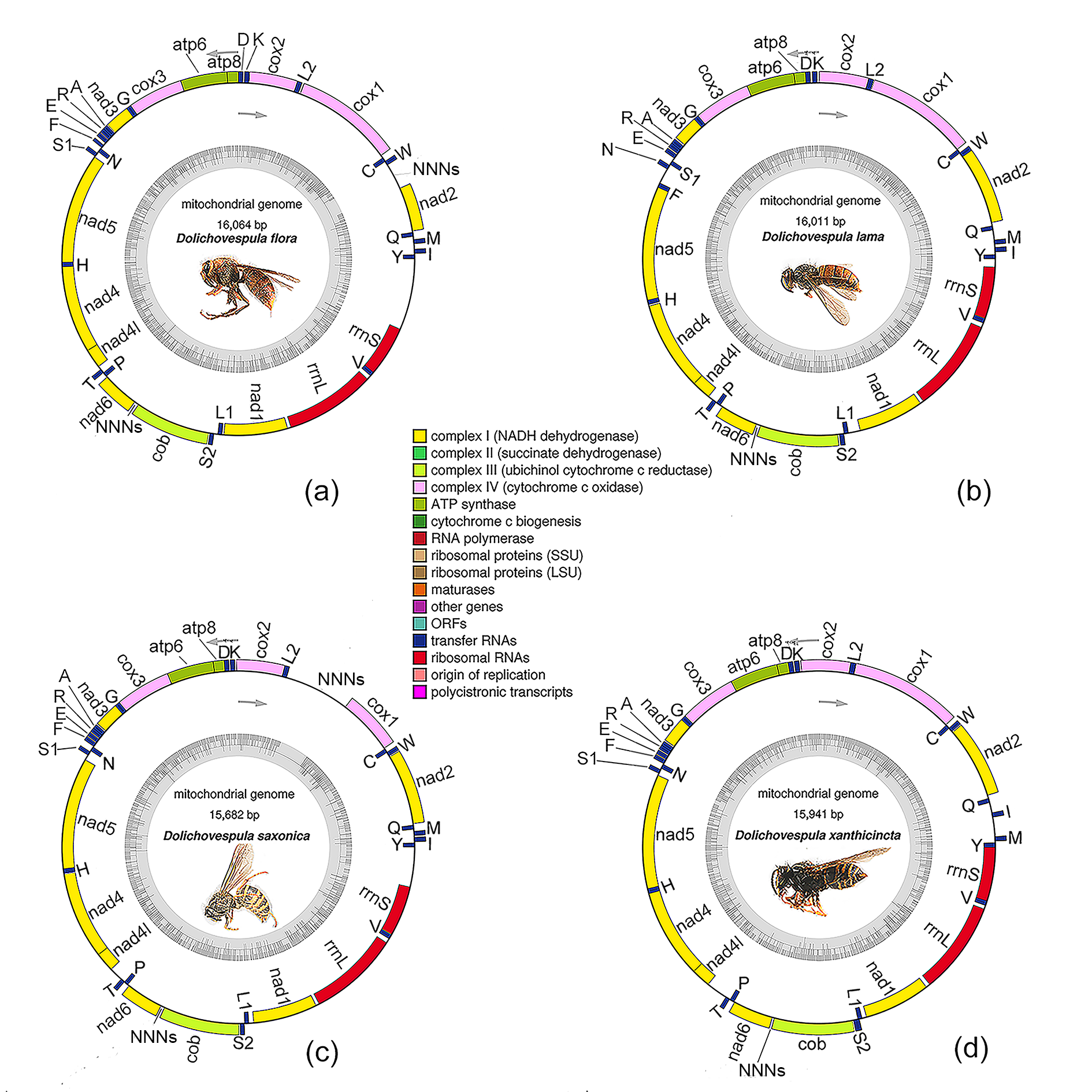
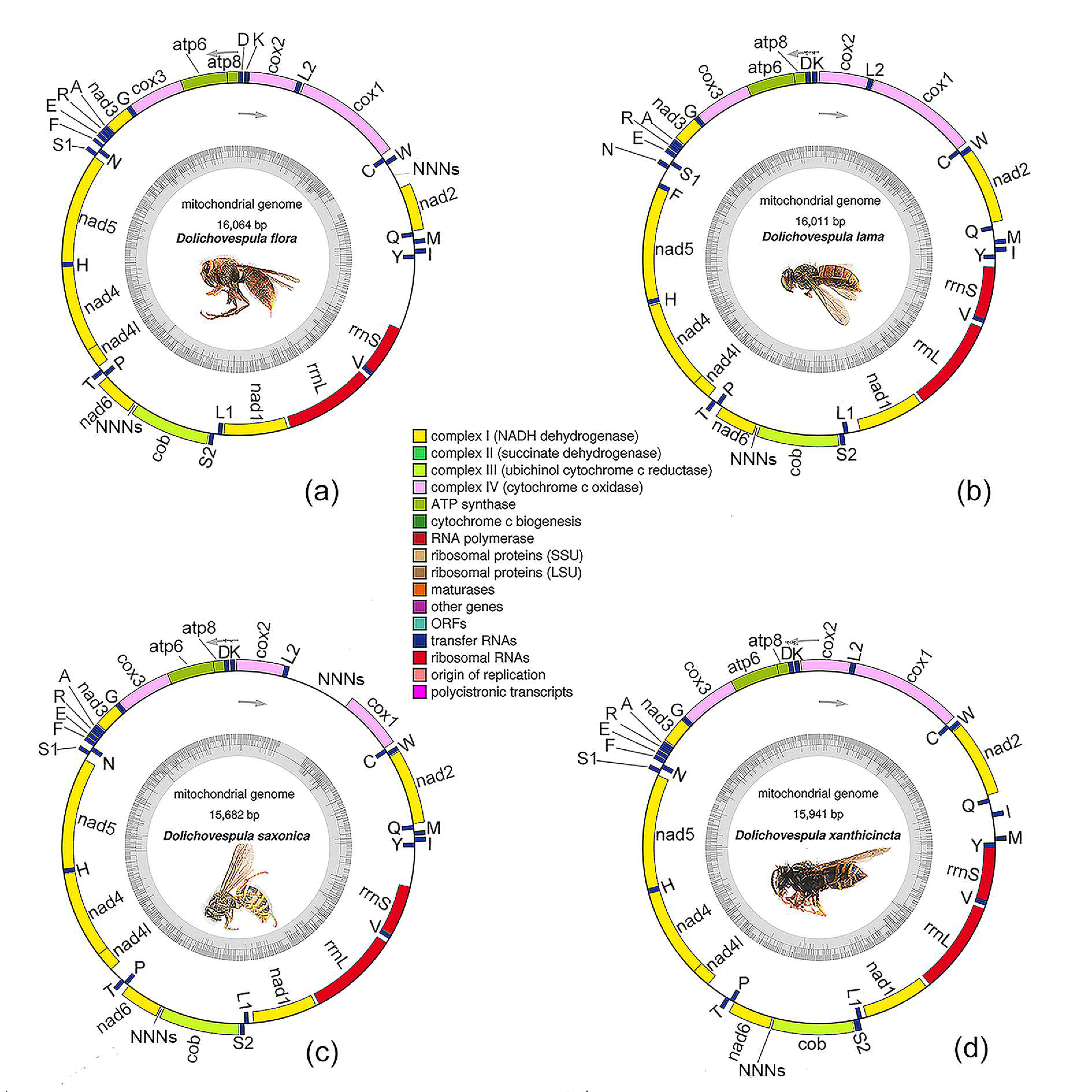
3.1. General Features of the Genomes
3.2. Protein-Coding Genes and Codon Usage
3.3. tRNA and rRNA Genes
3.4. Gene Rearrangements
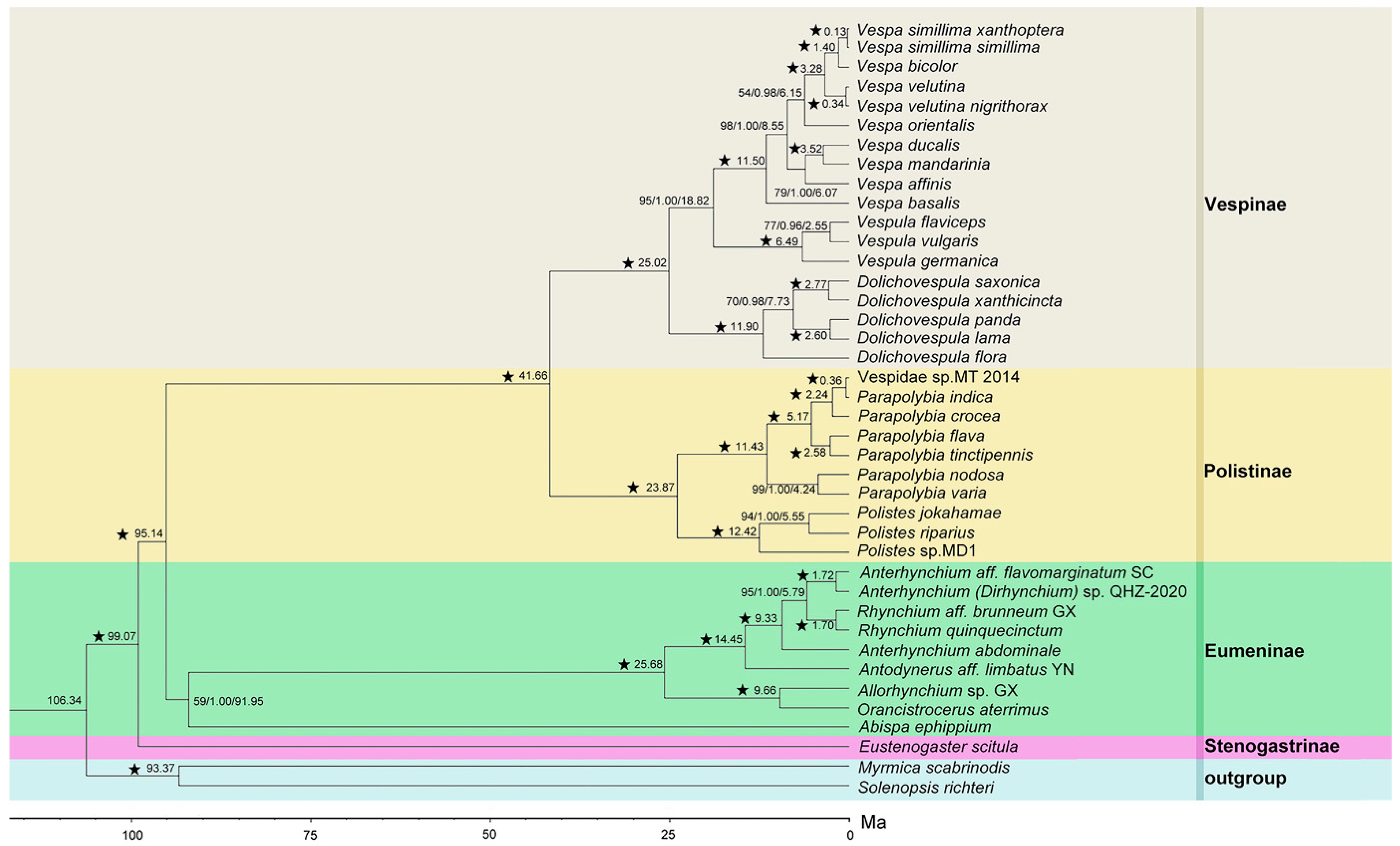
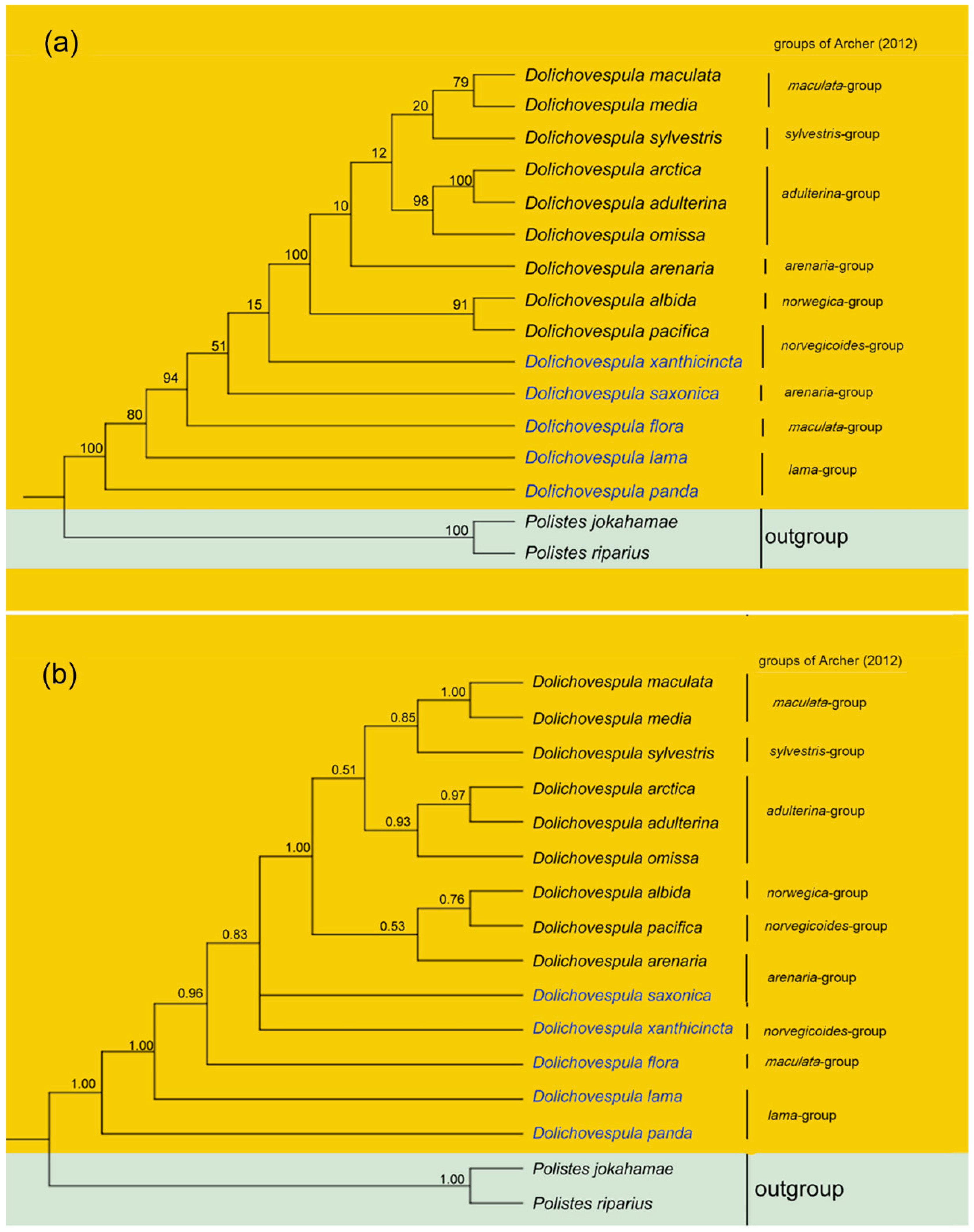
3.5. Phylogenetic Relationships
3.6. Estimated Time of Divergence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Archer, M.E. Taxonomy and world distribution of the Euro-Asian species of Dolichovespula (Hym., Vespinae). Entomol. Mon. Mag. 1999, 135, 153–160. [Google Scholar]
- Archer, M.E. Vespine Wasp of the World, Behavior, Ecology, Taxonomy of the Vespinae; Siri Scientific Press: Manchester, UK, 2012. [Google Scholar]
- Tan, J.L.; van Achterberg, C.; Chen, X.X. Potentially Lethal Social Wasps, Fauna of the Chinese Vespinae (Hymenoptera: Vespidae); Science Press: Beijing, China, 2015. [Google Scholar]
- Tan, J.L.; Zhou, T.; Tan, Q.Q.; van Achterberg, C.; Carpenter, J.M. Nest structure and all stages of the long-cheeked yellow jacket Dolichovespula stigma Lee (Hymenoptera: Vespidae), with a new synonym. J. Nat. Hist. 2017, 51, 793–806. [Google Scholar] [CrossRef]
- Tan, J.L.; Carpenter, J.M.; van Achterberg, C. Most northern Oriental distribution of Zethus Fabricius (Hymenoptera, Vespidae, Eumeninae), with a new species from China. J. Hymenopt. Res. 2018, 62, 1–13. [Google Scholar] [CrossRef]
- Tan, J.L.; Chen, X.X.; van Achterberg, C. Description of the male Dolichovespula flora Archer (Hymenoptera: Vespidae). Entomotaxonomia 2014, 36, 75–80. [Google Scholar]
- Archer, M.E. A study of the male genitalia of the Vespinae (Hymenoptera: Vespidae). Entomol. Mon. Mag. 2016, 152, 43–48. [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.L.; Gong, Y.J.; Chen, P.Y.; Tan, Q.Q.; Tan, J.L.; Wei, S.J. Next-generation sequencing of the mitochondrial genome of Dolichovespula panda (Hymenoptera: Vespidae) with a phylogenetic analysis of Vespidae. J. Asia-Pac. Entomol. 2017, 20, 971–976. [Google Scholar] [CrossRef]
- Avise, J.C. Mitochondrial DNA and the evolutionary genetics of higher animals. Philos. Trans. R. Soc. B-Biol. Sci. 1986, 312, 325–342. [Google Scholar]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Knudsen, B.; Kohn, A.B.; Nahir, B.; McFadden, C.S.; Moroz, L.L. Complete DNA sequence of the mitochondrial genome of the sea-slug, Aplysia californica: Conservation of the gene order in Euthyneura. Mol. Phylogenet. Evol. 2006, 38, 459–469. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, S.; Fekra, L.; Jiang, F.; Khathutshelo, M.; Li, Z. The first two complete mitochondrial genome of Dacus bivittatus and Dacus ciliatus (Diptera: Tephritidae) by next-generation sequencing and implications for the higher phylogeny of Tephritidae. Int. J. Biol. Macromol. 2019, 140, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Gotzek, D.; Clarke, J.; Shoemaker, D. Mitochondrial genome evolution in fire ants (Hymenoptera: Formicidae). BMC Evol. Biol. 2010, 10, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowton, M.; Castro, L.R.; Campbell, S.L.; Bargon, S.D.; Austin, A.D. Frequent mitochondrial gene rearrangements at the Hymenopteran nad3-nad5 junction. J. Mol. Evol. 2003, 56, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Logse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Crampton-Platt, A.; Timmermans, M.J.; Gimmel, M.L.; Kutty, S.N.; Cockerill, T.D.; Khen, C.V.; Vogler, A.P. Soup to tree: The phylogeny of beetles inferred by mitochondrial metagenomics of a Bornean rainforest sample. Mol. Biol. Evol. 2015, 32, 2302–2316. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Dowton, M.; Austin, A.D. Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol. Biol. Evol. 1999, 16, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multigene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. Partitionfinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, B.D.; Moreau, C.S. Defensive traits exhibit an evolutionary trade-off and drive diversification in ants. Evolution 2017, 71, 315–328. [Google Scholar] [CrossRef]
- Huang, P.; Carpenter, J.M.; Chen, B.; Li, T.J. The first divergence time estimation of the subfamily Stenogastrinae (Hymenoptera: Vespidae) based on mitochondrial phylogenomics. Int. J. Biol. Macromol. 2019, 137, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Zardoya, R.; Suarez, M. Sequencing and phylogenomic analysis of whole mitochondrial genomes of animals. Methods Mol. Biol. 2008, 422, 185–200. [Google Scholar] [PubMed]
- Chen, Z.T.; Du, Y.Z. Rearrangement of mitochondrial genome in insects. J. Environ. Entomol. 2016, 38, 843–851. [Google Scholar]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.J.; Chen, X.X. Advances in comparative mitochondrial genomics of insects. Chin. J. Appl. Entomol. 2011, 48, 1573–1585. [Google Scholar]
- Zhang, Q.H.; Huang, P.; Chen, B.; Li, T.J. The complete mitochondrial genome of Orancistrocerus aterrimus aterrimus and comparative analysis in the family Vespidae (Hymenoptera, Vespidae, Eumeninae). ZooKeys 2018, 790, 127–144. [Google Scholar] [CrossRef]
- Takahashi, R.; Okuyama, H.; Minoshima, Y.N.; Takahashi, J. Complete mitochondrial DNA sequence of the alien hornet Vespa velutina (Insecta: Hymenoptera) invading Kyushu Island, Japan. Mitochondrial DNA Part B-Resour. 2018, 3, 179–181. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, J.M. Phylogenetic relationships and classification of the Vespinae (Hymenoptera: Vespidae). Syst. Entomol. 1987, 12, 413–431. [Google Scholar] [CrossRef]
- Carpenter, J.M.; Perera, E. Phylogenetic relationships among yellowjackets and the evolution of social parasitism (Hymenoptera: Vespidae, Vespinae). Am. Mus. Novit. 2006, 31, 375–386. [Google Scholar] [CrossRef]
- Pickett, K.M.; Carpenter, J.M. Simultaneous analysis and the origin of eusociality in the Vespidae (Insecta: Hymenoptera). Arthropod Syst. Phylogeny 2010, 68, 3–33. [Google Scholar]
- Perrard, A.; Lopez-Osorio, F.; Carpenter, J.M. Phylogeny, landmark analysis and the use of wing venation to study the evolution of social wasps (Hymenoptera: Vespidae: Vespinae). Cladistics 2016, 32, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.M. The phylogenetic relationships and natural classificition of the Vespoidea (Hymenoptera). Syst. Entomol. 1981, 7, 11–38. [Google Scholar] [CrossRef]
- Carpenter, J.M. On “Molecular phylogeny of vespidae (Hymenoptera) and the evolution of sociality in wasps”. Am. Mus. Novit. 2003, 3389, 1–20. [Google Scholar] [CrossRef]
- Schmitz, J.; Moritz, R.F.A. Molecular phylogeny of Vespidae (Hymenoptera) and the evolution of sociality in wasps. Mol. Phylogenet. Evol. 1998, 9, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Hines, H.M.; Hunt, J.H.; O’Connor, T.K.; Gillespie, J.J.; Cameron, S.A. Multigene phylogeny reveals eusociality evolved twice in vespid wasps. Proc. Natl. Acad. Sci. USA 2007, 104, 3295–3299. [Google Scholar] [CrossRef] [Green Version]
- Peters, R.S.; Krogmann, L.; Mayer, C.; Donath, A.; Gunkel, S.; Meusemann, K.; Kozlov, A.; Podsiadlowski, L.; Petersen, M.; Lanfear, R.; et al. Evolutionary history of the Hymenoptera. Curr. Biol. 2017, 27, 1013–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bank, S.; Sann, M.; Mayer, C.; Meusemann, K.; Donath, A.; Podsiadlowski, L.; Kozlov, A.; Petersen, M.; Krogmann, L.; Meier, R. Transcriptome and target DNA enrichment sequence data provide new insights into the phylogeny of vespid wasps (Hymenoptera: Aculeata: Vespidae). Mol. Phylogenet. Evol. 2017, 116, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Piekarski, P.K.; Carpenter, J.M.; Lemon, A.R.; Lemmon, E.M.; Sharanowski, B.J. Phylogenomic evidence overturns current conceptions of social evolution in wasps (Vespidae). Mol. Biol. Evol. 2018, 35, 2097–2109. [Google Scholar] [CrossRef] [Green Version]
- Piekarski, P.K.; Longair, R.W.; Rogers, S.M. Monophyly of eusocial wasps (Hymenoptera: Vespidae): Molecules and morphology tell opposing histories. J. Undergrad. Res. Alta. 2014, 4, 11–14. [Google Scholar]
- Lopez-Osorio, F.; Pickett, K.M.; Carpenter, J.M.; Ballif, B.A.; Agnarsson, I. Phylogenomic analysis of yellowjackets and hornets (Hymenoptera: Vespidae, Vespinae). Mol. Phylogenet. Evol. 2017, 107, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Saito, F.; Kojima, J. Phylogenetic analysis and biogeography of the nocturnal hornets, Provespa (Insecta: Hymenoptera: Vespidae: Vespinae). Spec. Divers. 2011, 16, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Pantera, B.; Hoffman, D.R.; Carresi, L.; Cappugi, G.; Turillazzi, S.; Manao, G.; Severino, M.; Spadolini, I.; Orsomando, G.; Moneti, G.; et al. Characterization of the major allergens purified from the venom of the paper wasp Polistes gallicus. Biochim. Biophys. Acta-Gen. Subj. 2003, 1623, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.M.; Rasnitsyn, A.P. Mesozoic Vespidae. Psyche 1990, 97, 1–20. [Google Scholar] [CrossRef]
- Perrard, A.; Grimaldi, D.; Carpenter, J.M. Early lineages of Vespidae (Hymenoptera) in Cretaceous amber. Syst. Entomol. 2017, 42, 379–386. [Google Scholar] [CrossRef] [Green Version]
- van der Vecht, J. The Vespine of the Indo-Malayan and Papuan areas (Hymenoptera, Vespidae). Zool. Verh. 1957, 34, 1–83. [Google Scholar]
- van der Vecht, J. The geographical distribution of the social wasps (Hymenoptera, Vespidae). In Proceedings of the XII International Congress of Entomology, London, UK, 8–16 July 1964; Royal Entomological Society of London: London, UK, 1965; pp. 440–441. [Google Scholar]
- Matsuura, M.; Yamane, S. Biology of the Vespine Wasps; Springer: Berlin, Germany, 1990. [Google Scholar]
- Richards, O.W. The biology of the social wasps (Hymenoptera, Vespidae). Biol. Rev. 1971, 46, 483–528. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | D. flora | D. lama | D. saxonica | D. xanthicincta |
|---|---|---|---|---|
| Whole length (bp) | 16,064 | 16,011 | 15,682 | 15,941 |
| Protein-coding genes | 13 | 13 | 13 | 13 |
| tRNA genes | 22 | 22 | 22 | 22 |
| rRNA genes | 2 | 2 | 2 | 2 |
| Majority strand | 23 | 22 | 23 | 24 |
| Minority strand | 14 | 15 | 14 | 13 |
| Whole (A + T)% | 82.78 | 82.85 | 83.05 | 82.21 |
| A% | 41.51 | 41.8 | 41.83 | 41.35 |
| T% | 41.27 | 41.05 | 41.23 | 40.86 |
| G% | 5.94 | 5.58 | 5.92 | 6.10 |
| C% | 11.27 | 11.57 | 11.03 | 11.54 |
| AT-skew | 0.00 | 0.01 | 0.01 | 0.01 |
| GC-skew | −0.30 | −0.35 | −0.30 | −0.31 |
| PCGs length (bp) | 10,831 | 11,204 | 10,528 | 11,222 |
| PCGs (A + T)% | 80.31 | 81.03 | 80.70 | 79.96 |
| tRNA length (bp) | 1492 | 1497 | 1495 | 1485 |
| tRNA (A + T)% | 86.48 | 86.32 | 85.03 | 85.62 |
| rRNA length (bp) | 2129 | 2149 | 2132 | 2147 |
| rRNA (A + T)% | 84.40 | 84.69 | 83.35 | 83.28 |
| A + T-rich region length(bp) | 1069 | 114 | 593 | 309 |
| A + T-rich region (A + T)% | 93.08 | 91.23 | 92.41 | 92.88 |
| Gene overlap region | 8 | 7 | 7 | 7 |
| Range (bp) | 1–8 | 1–7 | 1–7 | 1–7 |
| Size (bp) | 27 | 27 | 28 | 27 |
| intergenic region | 24 | 25 | 23 | 23 |
| Range (bp) | 570 | 1074 | 962 | 1114 |
| Size (bp) | 1–766 | 1–224 | 3–286 | 2–309 |
| Adjacent genes | 4 | 4 | 6 | 6 |
| Dolichovespula flora | Dolichovespula lama | ||||||||||
| Gene | T% | C% | A% | G% | A + T% | Gene | T% | C% | A% | G% | A + T% |
| nad2 | 46.13 | 11.16 | 40.18 | 2.53 | 86.31 | nad2 | 47.96 | 9.50 | 39.89 | 2.66 | 87.84 |
| cox1 | 39.73 | 14.14 | 33.78 | 12.35 | 73.51 | cox1 | 38.65 | 14.72 | 34.09 | 12.54 | 72.74 |
| cox2 | 39.43 | 13.24 | 38.84 | 8.48 | 78.27 | cox2 | 41.92 | 12.57 | 38.52 | 6.99 | 80.44 |
| atp8 | 41.11 | 10.56 | 42.78 | 5.56 | 83.89 | atp8 | 41.08 | 9.73 | 43.24 | 5.95 | 84.32 |
| atp6 | 44.74 | 12.46 | 36.94 | 5.86 | 81.68 | atp6 | 44.28 | 12.88 | 36.76 | 6.08 | 81.04 |
| cox3 | 42.26 | 16.67 | 33.93 | 7.14 | 76.19 | cox3 | 42.77 | 13.20 | 35.28 | 8.76 | 78.05 |
| nad3 | 44.75 | 16.30 | 32.87 | 6.08 | 77.62 | nad3 | 43.13 | 14.01 | 36.54 | 6.32 | 79.67 |
| nad5 | 50.74 | 6.55 | 30.80 | 11.90 | 81.55 | nad5 | 50.14 | 6.27 | 30.77 | 12.82 | 80.91 |
| nad4 | 49.85 | 5.95 | 32.14 | 12.05 | 81.99 | nad4 | 49.38 | 5.32 | 32.76 | 12.54 | 82.15 |
| nad4l | 49.52 | 2.22 | 36.19 | 12.06 | 85.71 | nad4l | 52.50 | 1.88 | 34.38 | 11.25 | 86.88 |
| nad6 | 50.17 | 10.20 | 35.79 | 3.85 | 85.95 | nad6 | 47.10 | 9.84 | 40.25 | 2.81 | 87.35 |
| cob | 42.26 | 13.24 | 33.18 | 11.31 | 75.45 | cob | 42.92 | 13.49 | 34.28 | 9.31 | 77.21 |
| nad1 | 45.54 | 5.80 | 35.12 | 13.54 | 80.65 | nad1 | 49.02 | 5.06 | 32.92 | 13.00 | 81.94 |
| rrnL | 42.28 | 10.82 | 42.28 | 4.60 | 84.56 | rrnL | 43.21 | 10.55 | 41.76 | 4.48 | 84.97 |
| rrnS | 44.20 | 11.08 | 39.90 | 4.82 | 84.09 | rrnS | 42.75 | 10.98 | 41.44 | 4.84 | 84.18 |
| Dolichovespula saxonica | Dolichovespula xanthicincta | ||||||||||
| Gene | T% | C% | A% | G% | A + T% | Gene | T% | C% | A% | G% | A + T% |
| nad2 | 48.31 | 8.57 | 40.30 | 2.82 | 88.61 | nad2 | 49.14 | 10.38 | 36.48 | 4.00 | 85.62 |
| cox1 | 39.42 | 14.17 | 33.88 | 12.52 | 73.30 | cox1 | 38.86 | 15.05 | 32.29 | 13.81 | 71.14 |
| cox2 | 39.91 | 13.79 | 38.03 | 8.27 | 77.94 | cox2 | 40.06 | 13.74 | 37.72 | 8.48 | 77.78 |
| atp8 | 43.21 | 10.49 | 43.21 | 3.09 | 86.42 | atp8 | 45.67 | 10.10 | 36.06 | 8.17 | 81.73 |
| atp6 | 45.32 | 12.33 | 36.26 | 6.09 | 81.58 | atp6 | 44.29 | 13.21 | 36.64 | 5.86 | 80.93 |
| cox3 | 44.29 | 12.69 | 33.76 | 9.26 | 78.05 | cox3 | 41.78 | 14.24 | 35.50 | 8.48 | 77.28 |
| nad3 | 45.33 | 14.01 | 34.07 | 6.59 | 79.40 | nad3 | 46.26 | 13.85 | 33.80 | 6.09 | 80.06 |
| nad5 | 48.59 | 6.78 | 32.11 | 12.52 | 80.70 | nad5 | 49.05 | 6.57 | 31.52 | 12.86 | 80.57 |
| nad4 | 48.78 | 5.74 | 32.49 | 12.99 | 81.26 | nad4 | 47.62 | 5.90 | 33.71 | 12.76 | 81.33 |
| nad4l | 50.48 | 2.56 | 34.82 | 12.14 | 85.30 | nad4l | 50.48 | 2.89 | 35.05 | 11.58 | 85.53 |
| nad6 | 46.55 | 10.00 | 38.45 | 5.00 | 85.00 | nad6 | 46.26 | 9.15 | 41.26 | 3.33 | 87.52 |
| cob | 42.28 | 12.90 | 35.40 | 9.42 | 77.68 | cob | 42.76 | 12.38 | 34.76 | 10.10 | 77.52 |
| nad1 | 46.96 | 5.26 | 34.67 | 13.11 | 81.63 | nad1 | 47.90 | 5.13 | 33.95 | 13.03 | 81.85 |
| rrnL | 43.11 | 10.58 | 40.99 | 5.32 | 84.10 | rrnL | 43.72 | 10.07 | 41.59 | 4.63 | 85.30 |
| rrnS | 41.66 | 12.22 | 40.34 | 5.78 | 82.00 | rrnS | 41.35 | 11.83 | 38.42 | 5.34 | 79.77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Wen, Q.; Wang, T.; Ran, F.; Wang, M.; Fan, X.; Wei, S.; Li, Z.; Tan, J. Next-Generation Sequencing of Four Mitochondrial Genomes of Dolichovespula (Hymenoptera: Vespidae) with a Phylogenetic Analysis and Divergence Time Estimation of Vespidae. Animals 2022, 12, 3004. https://doi.org/10.3390/ani12213004
Wang H, Wen Q, Wang T, Ran F, Wang M, Fan X, Wei S, Li Z, Tan J. Next-Generation Sequencing of Four Mitochondrial Genomes of Dolichovespula (Hymenoptera: Vespidae) with a Phylogenetic Analysis and Divergence Time Estimation of Vespidae. Animals. 2022; 12(21):3004. https://doi.org/10.3390/ani12213004
Chicago/Turabian StyleWang, Hang, Qian Wen, Tongfei Wang, Fanrong Ran, Meng Wang, Xulei Fan, Shujun Wei, Zhonghu Li, and Jiangli Tan. 2022. "Next-Generation Sequencing of Four Mitochondrial Genomes of Dolichovespula (Hymenoptera: Vespidae) with a Phylogenetic Analysis and Divergence Time Estimation of Vespidae" Animals 12, no. 21: 3004. https://doi.org/10.3390/ani12213004