
Cervid Prion Protein Polymorphisms: Role in Chronic Wasting Disease Pathogenesis

 and
and
Abstract
:1. Introduction
2. Prnp Polymorphisms in Cervids
3. Cervid PrP Polymorphisms and Effects on PrPC Structure
4. Prion Protein Polymorphisms and PrPCWD Strains
5. PrPCWD Shedding in Cervid Species
6. Transmission Barriers and Zoonotic Potential of CWD
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gilch, S.; Chitoor, N.; Taguchi, Y.; Stuart, M.; Jewell, J.E.; Schätzl, H.M. Chronic Wasting Disease. In Topics in Current Chemistry; Springer International Publishing: Cham, Switzerland, 2011; Volume 305, pp. 51–77. [Google Scholar]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathiason, C.K.; Powers, J.G.; Dahmes, S.J.; Osborn, D.A.; Miller, K.V.; Warren, R.J.; Mason, G.L.; Hays, S.A.; Hayes-Klug, J.; Seelig, D.M.; et al. Infectious Prions in the Saliva and Blood of Deer with Chronic Wasting Disease. Science 2006, 314, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.E.; Brown, J.; Kreeger, T.; Williams, E.S. Prion protein in cardiac muscle of elk (Cervus elaphus nelsoni) and white-tailed deer (Odocoileus virginianus) infected with chronic wasting disease. J. Gen. Virol. 2006, 87, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Angers, R.C.; Browning, S.R.; Seward, T.S.; Sigurdson, C.J.; Miller, M.W.; Hoover, E.A. Prions in Skeletal Muscles of Deer with Chronic Wasting Disease. Science 2006, 311, 1117. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G.; Lessard, P.; Tamgüney, G.; Freyman, Y.; Deering, C.; Letessier, F.; de Armond, S.J.; Prusiner, S.B. Transmission and Detection of Prions in Feces. J. Infect. Dis. 2008, 198, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Tamgüney, G.; Miller, M.W.; Wolfe, L.L.; Sirochman, T.M.; Glidden, D.V.; Palmer, C.G.S.; Lemus, A.; de Armond, S.J.; Prusiner, S.B. Asymptomatic deer excrete infectious prions in faeces. Nat. Cell Biol. 2009, 461, 529–532. [Google Scholar] [CrossRef] [Green Version]
- Haley, N.J.; Mathiason, C.K.; Carver, S.; Zabel, M.D.; Telling, G.C.; Hoover, E.A. Detection of Chronic Wasting Disease Prions in Salivary, Urinary, and Intestinal Tissues of Deer: Potential Mechanisms of Prion Shedding and Transmission. J. Virol. 2011, 85, 6309–6318. [Google Scholar] [CrossRef] [Green Version]
- John, T.R.; Schätzl, H.M.; Gilch, S. Early detection of chronic wasting disease prions in urine of pre-symptomatic deer by real-time quaking-induced conversion assay. Prion 2013, 7, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Henderson, D.M.; Denkers, N.D.; Hoover, C.E.; Garbino, N.; Mathiason, C.K.; Hoover, E.A. Longitudinal Detection of Prion Shedding in Saliva and Urine by Chronic Wasting Disease-Infected Deer by Real-Time Quaking-Induced Conversion. J. Virol. 2015, 89, 9338–9347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.C.; Hannaoui, S.; John, T.R.; Dudas, S.; Czub, S.; Gilch, S. Early and Non-Invasive Detection of Chronic Wasting Disease Prions in Elk Feces by Real-Time Quaking Induced Conversion. PLoS ONE 2016, 11, e0166187. [Google Scholar] [CrossRef]
- Kramm, C.; Pritzkow, S.; Lyon, A.; Nichols, T.; Morales, R.; Soto, C. Detection of Prions in Blood of Cervids at the Asymptomatic Stage of Chronic Wasting Disease. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, D.M.; Tennant, J.M.; Haley, N.J.; Denkers, N.D.; Mathiason, C.K.; Hoover, E.A. Detection of chronic wasting disease prion seeding activity in deer and elk feces by real-time quaking-induced conversion. J. Gen. Virol. 2017, 98, 1953–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tennant, J.M.; Li, M.; Henderson, D.M.; Tyer, M.L.; Denkers, N.D.; Haley, N.J.; Mathiason, C.K.; Hoover, E.A. Shedding and stability of CWD prion seeding activity in cervid feces. PLoS ONE 2020, 15, e0227094. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.J.; Phillips, K.E.; Schramm, P.T.; McKenzie, D.; Aiken, J.M.; Pedersen, J.A. Prions Adhere to Soil Minerals and Remain Infectious. PLoS Pathog. 2006, 2, e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.J.; Pedersen, J.A.; Chappell, R.J.; McKenzie, D.; Aiken, J.M. Oral Transmissibility of Prion Disease Is Enhanced by Binding to Soil Particles. PLoS Pathog. 2007, 3, e93. [Google Scholar] [CrossRef]
- Bartelt-Hunt, S.L.; Bartz, J.C. Behavior of Prions in the Environment: Implications for Prion Biology. PLoS Pathog. 2013, 9, e1003113. [Google Scholar] [CrossRef] [Green Version]
- Pritzkow, S.; Morales, R.; Moda, F.; Khan, U.; Telling, G.C.; Hoover, E.; Soto, C. Grass Plants Bind, Retain, Uptake, and Transport Infectious Prions. Cell Rep. 2015, 11, 1168–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, A.; McKenzie, D.; Cullingham, C.; Aiken, J.M. Long-Term Incubation PrPCWD with Soils Affects Prion Recovery but Not Infectivity. Pathogens 2020, 9, 311. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.W.; Williams, E.S.; Hobbs, N.T.; Wolfe, L.L. Environmental Sources of Prion Transmission in Mule Deer. Emerg. Infect. Dis. 2004, 10, 1003–1006. [Google Scholar] [CrossRef]
- Yuan, Q.; Telling, G.; Bartelt-Hunt, S.L.; Bartz, J.C. Dehydration of Prions on Environmentally Relevant Surfaces Protects Them from Inactivation by Freezing and Thawing. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Mathiason, C.K.; Hays, S.A.; Powers, J.; Hayes-Klug, J.; Langenberg, J.; Dahmes, S.J.; Osborn, D.A.; Miller, K.V.; Warren, R.J.; Mason, G.L.; et al. Infectious Prions in Pre-Clinical Deer and Transmission of Chronic Wasting Disease Solely by Environmental Exposure. PLoS ONE 2009, 4, e5916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgsson, G.; Sigurdarson, S.; Brown, P. Infectious agent of sheep scrapie may persist in the environment for at least 16 years. J. Gen. Virol. 2006, 87, 3737–3740. [Google Scholar] [CrossRef]
- Williams, E.S.; Young, S. Chronic Wasting Diease of Captive Mule Deer: A Spongifom Encephalopathy. J. Wildl. Dis. 1980, 16, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.S.; Young, S. Spongiform Encephalopathy of Rpcky Mountain Elk. J. Wildl. Dis. 1982, 18, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Miller, M.W. Chronic wasting disease in deer and elk in North America: -EN- -FR- -ES-. Rev. Sci. Tech. OIE 2002, 21, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Sohn, H.-J.; Kim, J.-H.; Choi, K.-S.; Nah, J.-J.; Joo, Y.-S.; Jean, Y.-H.; Ahn, S.-W.; Kim, O.-K.; Kim, D.-Y.; Balachandran, A. A Case of Chronic Wasting Disease in an Elk Imported to Korea from Canada. J. Veter. Med. Sci. 2002, 64, 855–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
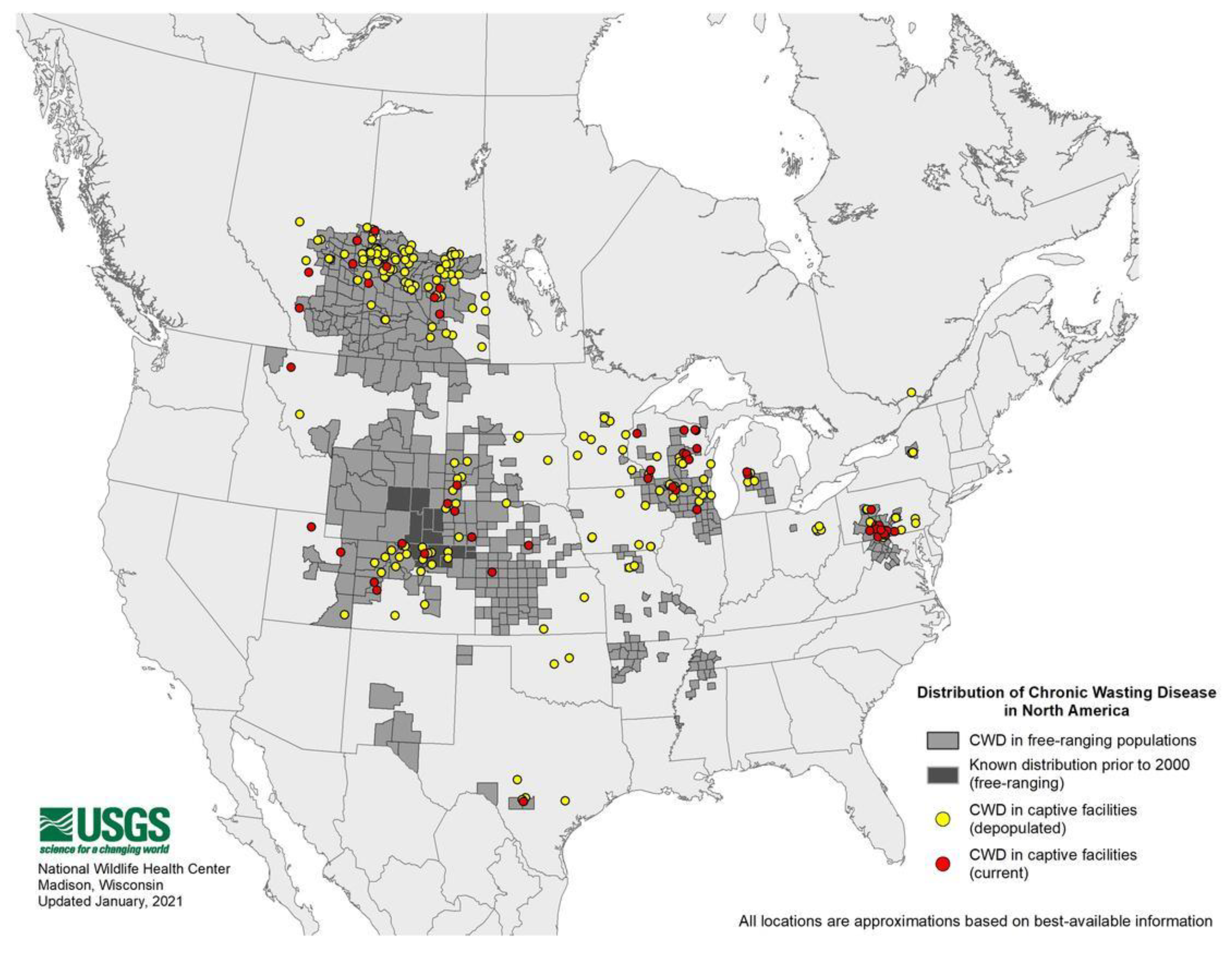
- Expanding Distribution of Chronic Wasting Disease. Available online: https://www.usgs.gov/centers/nwhc/science/expanding-distribution-chronic-wasting-disease?qt-science_center_objects=0#qt-science_center_objects (accessed on 15 December 2020).
- Kahn, S.; Dubé, C.; Bates, L.; Balachandran, A. Chronic wasting disease in Canada: Part 1. Can. Veter. J. 2004, 45, 397–404. [Google Scholar]
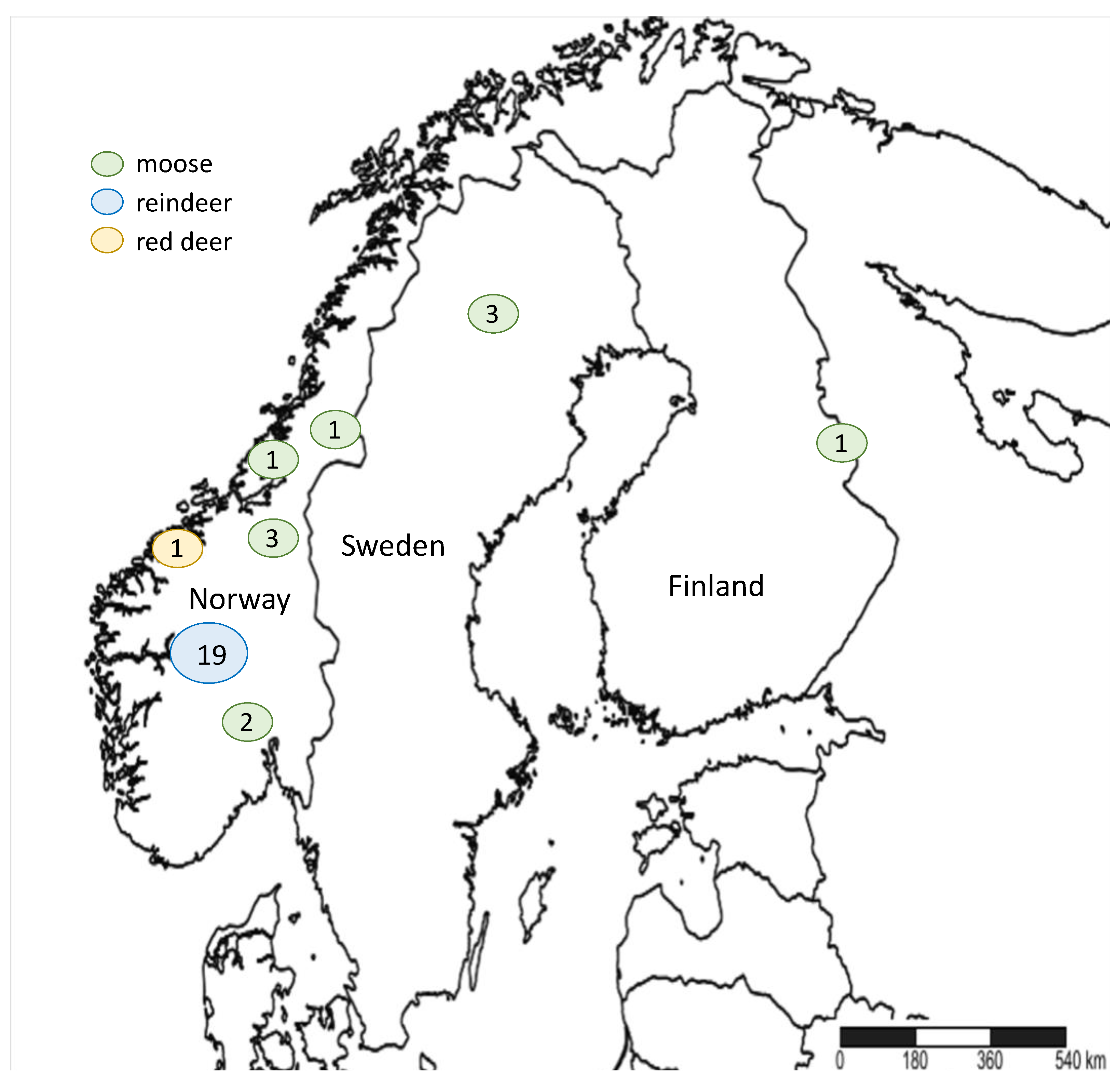
- Vikøren, T.; Våge, J.; Madslien, K.I.; Røed, K.H.; Rolandsen, C.M.; Tran, L.; Hopp, P.; Veiberg, V.; Heum, M.; Moldal, T.; et al. First Detection of Chronic Wasting Disease in a Wild Red Deer (Cervus elaphus) in Europe. J. Wildl. Dis. 2019, 55, 970–972. [Google Scholar] [CrossRef]
- Benestad, S.L.; Mitchell, G.; Simmons, M.; Ytrehus, B.; Vikøren, T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Veter. Res. 2016, 47, 1–7. [Google Scholar] [CrossRef]
- Pirisinu, L.; Tran, L.; Chiappini, B.; Vanni, I.; di Bari, M.A.; Vaccari, G.; Vikøren, T.; Madslien, K.I.; Våge, J.; Spraker, T.; et al. Novel Type of Chronic Wasting Disease Detected in Moose (Alces alces), Norway. Emerg. Infect. Dis. 2018, 24, 2210–2218. [Google Scholar] [CrossRef] [Green Version]
- ProMED-Mail Chronic Wasting Disease, Cervid—FINLAND: First Case, Moose. ProMED-mail. 2018. Available online: https://promedmail.org/promed-post/?id=5684473 (accessed on 15 December 2020).
- ProMED-Mail Chronic Wasting Disease—Sweden: (Norrbotten) Moose, First Case. ProMED-mail. 2019. Available online: https://promedmail.org/promed-post/?id=6390297 (accessed on 15 December 2020).
- Mysterud, A.; Benestad, S.L.; Rolandsen, C.M.; Våge, J. Policy implications of an expanded chronic wasting disease universe. J. Appl. Ecol. 2020, 58, 281–285. [Google Scholar] [CrossRef]
- Barria, M.A.; Balachandran, A.; Morita, M.; Kitamoto, T.; Barron, R.; Manson, J.; Knight, R.; Ironside, J.W.; Head, M.W. Molecular Barriers to Zoonotic Transmission of Prions. Emerg. Infect. Dis. 2014, 20, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caughey, B. Prion protein conversions: Insight into mechanisms, TSE transmission barriers and strains. Br. Med. Bull. 2003, 66, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.-M.; Espinosa, J.-C.; Aguilar-Calvo, P.; Herva, M.-E.; Relaño-Ginés, A.; Villa-Diaz, A.; Morales, M.; Parra, B.; Alamillo, E.; Brun, A.; et al. Elements Modulating the Prion Species Barrier and Its Passage Consequences. PLoS ONE 2014, 9, e89722. [Google Scholar] [CrossRef] [PubMed]
- Raymon, G.J.; Bossers, A.; Raymond, L.D.; O’Rourke, K.I.; McHolland, L.E.; Bryant, P.K. Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J. 2000, 19, 4425–4430. [Google Scholar] [CrossRef] [Green Version]
- Davenport, K.A.; Henderson, D.M.; Bian, J.; Telling, G.C.; Mathiason, C.K.; Hoover, E.A. Insights into Chronic Wasting Disease and Bovine Spongiform Encephalopathy Species Barriers by Use of Real-Time Conversion. J. Virol. 2015, 89, 9524–9531. [Google Scholar] [CrossRef] [Green Version]
- Bruce, M.; Chree, A.; McConnell, I.; Foster, J.; Pearson, G.; Fraser, H. Transmission of bovine spongiform encephalopathy and scrapie to mice: Strain variation and the species barrier. Philos. Trans. R. Soc. B Biol. Sci. 1994, 343, 405–411. [Google Scholar] [CrossRef]
- Angers, R.; Christiansen, J.; Nalls, A.V.; Kang, H.-E.; Hunter, N.; Hoover, E.; Mathiason, C.K.; Sheetz, M.; Telling, G.C. Structural effects of PrP polymorphisms on intra- and interspecies prion transmission. Proc. Natl. Acad. Sci. USA 2014, 111, 11169–11174. [Google Scholar] [CrossRef] [Green Version]
- Schätzl, H.M.; da Costa, M.; Taylor, L.; Cohen, F.E.; Prusiner, S.B. Prion Protein Gene Variation Among Primates. J. Mol. Biol. 1995, 245, 362–374. [Google Scholar] [CrossRef]
- Lee, L.Y.-L.; Chen, R.P.-Y. Quantifying the Sequence-Dependent Species Barrier between Hamster and Mouse Prions. J. Am. Chem. Soc. 2007, 129, 1644–1652. [Google Scholar] [CrossRef]
- Sharma, A.; Bruce, K.L.; Chen, B.; Gyoneva, S.; Behrens, S.H.; Bommarius, A.S.; Chernoff, Y.O. Contributions of the Prion Protein Sequence, Strain, and Environment to the Species Barrier. J. Biol. Chem. 2016, 291, 1277–1288. [Google Scholar] [CrossRef] [Green Version]
- Hill, A.F.; Joiner, S.; Linehan, J.; Desbruslais, M.; Lantos, P.L.; Collinge, J. Species-barrier-independent prion replication in apparently resistant species. Proc. Natl. Acad. Sci. USA 2000, 97, 10248–10253. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.; Foster, D.; Mirenda, C.; Serban, D.; Coufal, F.; Wälchli, M.; Torchia, M.; Groth, D.; Carlson, G.; DeArmond, S.J.; et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 1989, 59, 847–857. [Google Scholar] [CrossRef]
- Horiuchi, M.; Priola, S.A.; Chabry, J.; Caughey, B. Interactions between heterologous forms of prion protein: Binding, inhibition of conversion, and species barriers. Proc. Natl. Acad. Sci. USA 2000, 97, 5836–5841. [Google Scholar] [CrossRef] [Green Version]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA molecules stimulate prion protein conversion. Nat. Cell Biol. 2003, 425, 717–720. [Google Scholar] [CrossRef]
- Baron, G.S.; Caughey, B.; Starke, D.W.; Chock, P.B.; Mieyal, J.J. Effect of Glycosylphosphatidylinositol Anchor-dependent and -independent Prion Protein Association with Model Raft Membranes on Conversion to the Protease-resistant Isoform. J. Biol. Chem. 2003, 278, 14883–14892. [Google Scholar] [CrossRef] [Green Version]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, X.; Yuan, C.-G.; Ma, J. Generating a Prion with Bacterially Expressed Recombinant Prion Protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [Green Version]
- Ma, J. The Role of Cofactors in Prion Propagation and Infectivity. PLoS Pathog. 2012, 8, e1002589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarava, N.; Baskakov, I.V. The Evolution of Transmissible Prions: The Role of Deformed Templating. PLoS Pathog. 2013, 9, e1003759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Montalban, N.; Lee, Y.J.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Changes in prion replication environment cause prion strain mutation. FASEB J. 2013, 27, 3702–3710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katorcha, E.; Gonzalez-Montalban, N.; Makarava, N.; Kovacs, G.G.; Baskakov, I.V. Prion replication environment defines the fate of prion strain adaptation. PLoS Pathog. 2018, 14, e1007093. [Google Scholar] [CrossRef]
- Collinge, J. Prion Strain Mutation and Selection. Science 2010, 328, 1111–1112. [Google Scholar] [CrossRef] [PubMed]
- Barrio, T.; Filali, H.; Otero, A.; Sheleby-Elías, J.; Marín, B.; Vidal, E.; Béringue, V.; Torres, J.M.; Groschup, M.; Andréoletti, O.; et al. Mixtures of prion substrains in natural scrapie cases revealed by ovinised murine models. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational variations in an infectious protein determine prion strain differences. Nat. Cell Biol. 2004, 428, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Collins, S.R.; Toyama, B.H.; Weissman, J.S. The physical basis of how prion conformations determine strain phenotypes. Nature 2006, 442, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Savtchenko, R.; Lasch, P.; Beekes, M.; Baskakov, I.V. Preserving prion strain identity upon replication of prions in vitro using recombinant prion protein. Acta Neuropathol. Commun. 2018, 6, 92. [Google Scholar] [CrossRef]
- Solforosi, L.; Milani, M.; Mancini, N.; Clementi, M.; Burioni, R. A closer look at prion strains. Prion 2013, 7, 99–108. [Google Scholar] [CrossRef]
- Makarava, N.; Kovacs, G.G.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Stabilization of a Prion Strain of Synthetic Origin Requires Multiple Serial Passages. J. Biol. Chem. 2012, 287, 30205–30214. [Google Scholar] [CrossRef] [Green Version]
- Morales, R.; Abid, K.; Soto, C. The prion strain phenomenon: Molecular basis and unprecedented features. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Bruce, M.E.; McConnell, I.; Fraser, H.; Dickinson, A.G. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: Implications for the nature of the agent and host control of pathogenesis. J. Gen. Virol. 1991, 72, 595–603. [Google Scholar] [CrossRef]
- Bruce, M.E.; Dickinson, A.G. Biological Evidence that Scrapie Agent Has an Independent Genome. J. Gen. Virol. 1987, 68, 79–89. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Characteristics of a Short Incubation Model of Scrapie in the Golden Hamster. J. Gen. Virol. 1977, 34, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Lasmézas, C.I.; Deslys, J.-P.; Demaimay, R.; Adjou, K.T.; Lamoury, F.; Dormont, D.; Robain, O.; Ironside, J.; Hauw, J.-J. BSE transmission to macaques. Nat. Cell Biol. 1996, 381, 743–744. [Google Scholar] [CrossRef]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Desbruslais, M.; Joiner, S.; Sidle, K.C.L.; Gowland, I.; Collinge, J.; Doey, L.J.; Lantos, P. The same prion strain causes vCJD and BSE. Nat. Cell Biol. 1997, 389, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Bencsik, A.; Debeer, S.; Petit, T.; Baron, T. Possible Case of Maternal Transmission of Feline Spongiform Encephalopathy in a Captive Cheetah. PLoS ONE 2009, 4, e6929. [Google Scholar] [CrossRef] [Green Version]
- Zanusso, G.; Nardelli, E.; Rosati, A.; Fabrizi, G.; Ferrari, S.; Carteri, A.; de Simone, F.; Rizzuto, N.; Monaco, S. Simultaneous occurrence of spongiform encephalopathy in a man and his cat in Italy. Lancet 1998, 352, 1116–1117. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Miller, M.W. Other animal prion diseases. Br. Med. Bull. 2003, 66, 199–212. [Google Scholar] [CrossRef]
- Kirkwood, J.K.; Cunningham, A.A.; Wells, G.A.; Wilesmith, J.W.; Barnett, J.E. Spongiform encephalopathy in a herd of greater kudu (Tragelaphus strepsiceros): Epidemiological observations. Veter. Rec. 1993, 133, 360–364. [Google Scholar] [CrossRef]
- Kirkwood, J.K.; Cunningham, A.A. Epidemiological observations on spongiform encephalopathies in captive wild animals in the British Isles. Veter. Rec. 1994, 135, 296–303. [Google Scholar] [CrossRef]
- Imran, M.; Mahmood, S. An overview of animal prion diseases. Virol. J. 2011, 8, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, T.; Belli, P.; Madec, J.Y.; Moutou, F.; Vitaud, C.; Savey, M. Spongiform encephalopathy in an imported cheetah in France. Veter. Rec. 1997, 141, 270–271. [Google Scholar] [CrossRef]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian Evolution of Prions in Cell Culture. Science 2009, 327, 869–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartz, J.C.; Bessen, R.A.; McKenzie, D.; Marsh, R.F.; Aiken, J.M. Adaptation and Selection of Prion Protein Strain Conformations following Interspecies Transmission of Transmissible Mink Encephalopathy. J. Virol. 2000, 74, 5542–5547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, P.; de Pace, A.H.; Collins, S.R.; Weissman, J.S. Generation of prion transmission barriers by mutational control of amyloid conformations. Nat. Cell Biol. 2003, 424, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Peretz, D.; Williamson, R.; Legname, G.; Matsunaga, Y.; Vergara, J.; Burton, D.R.; de Armond, S.J.; Prusiner, S.B.; Scott, M.R. A Change in the Conformation of Prions Accompanies the Emergence of a New Prion Strain. Neuron 2002, 34, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Igel-Egalon, A.; Laferrière, F.; Tixador, P.; Moudjou, M.; Herzog, L.; Reine, F. Crossing Species Barriers Relies on Structurally Distinct Prion Assemblies and Their Complementation. Mol. Neurobiol. 2020, 57, 2572–2587. [Google Scholar] [CrossRef]
- Le Dur, A.; Laï, T.L.; Stinnakre, M.-G.; Laisné, A.; Chenais, N.; Rakotobe, S.; Passet, B.; Reine, F.; Soulier, S.; Herzog, L.; et al. Divergent prion strain evolution driven by PrPC expression level in transgenic mice. Nat. Commun. 2017, 8, 14170. [Google Scholar] [CrossRef] [Green Version]
- Sigurdson, C.J.; Nilsson, K.P.R.; Hornemann, S.; Heikenwalder, M.; Manco, G.; Schwarz, P.; Ott, D.; Rülicke, T.; Liberski, P.P.; Julius, C.; et al. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc. Natl. Acad. Sci. USA 2008, 106, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Sigurdson, C.J.; Nilsson, K.P.R.; Hornemann, S.; Manco, G.; Fernández-Borges, N.; Schwarz, P.; Castilla, J.; Wüthrich, K.; Aguzzi, A. A molecular switch controls interspecies prion disease transmission in mice. J. Clin. Investig. 2010, 120, 2590–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigurdson, C.J.; Joshi-Barr, S.; Bett, C.; Winson, O.; Manco, G.; Schwarz, P. Spongiform Encephalopathy in Transgenic Mice Expressing a Point Mutation in the β2–α2 Loop of the Prion Protein. J. Neurosci. 2011, 31, 13840–13847. [Google Scholar] [CrossRef] [Green Version]
- Kurt, T.D.; Bett, C.; Fernández-Borges, N.; Joshi-Barr, S.; Hornemann, S.; Rülicke, T. Prion Transmission Prevented by Modifying the β2-α2 Loop Structure of Host PrPC. J. Neurosci. 2014, 34, 1022–1027. [Google Scholar] [CrossRef] [Green Version]
- Gossert, A.D.; Bonjour, S.; Lysek, D.A.; Fiorito, F.; Wüthrich, K. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc. Natl. Acad. Sci. USA 2005, 102, 646–650. [Google Scholar] [CrossRef] [Green Version]
- Harrathi, C.; Fernández-Borges, N.; Eraña, H.; Elezgarai, S.R.; Venegas, V.; Charco, J.M.; Castilla, J. Insights into the Bidirectional Properties of the Sheep–Deer Prion Transmission Barrier. Mol. Neurobiol. 2018, 56, 5287–5303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, C. Constraining the loop, releasing prion infectivity. Proc. Natl. Acad. Sci. USA 2008, 106, 10–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, L.M.; John, T.R.; Schätzl, H.M.; Lewis, R.V. Introducing a Rigid Loop Structure from Deer into Mouse Prion Protein Increases Its Propensity for Misfolding In Vitro. PLoS ONE 2013, 8, e66715. [Google Scholar] [CrossRef] [Green Version]
- Hannaoui, S.; Amidian, S.; Cheng, Y.C.; Velásquez, C.D.; Dorosh, L.; Law, S.; Telling, G.; Stepanova, M.; McKenzie, D.; Wille, H.; et al. Destabilizing polymorphism in cervid prion protein hydrophobic core determines prion conformation and conversion efficiency. PLoS Pathog. 2017, 13, e1006553. [Google Scholar] [CrossRef]
- Velásquez, C.D.; Kim, C.; Herbst, A.; Daude, N.; Garza, M.C.; Wille, H.; Aiken, J.M.; McKenzie, D. Deer Prion Proteins Modulate the Emergence and Adaptation of Chronic Wasting Disease Strains. J. Virol. 2015, 89, 12362–12373. [Google Scholar] [CrossRef] [Green Version]
- Hagenaars, T.J.; Melchior, M.B.; Windig, J.J.; Bossers, A.; Davidse, A.; van Zijderveld, F.G. Modelling of strategies for genetic control of scrapie in sheep: The importance of population structure. PLoS ONE 2018, 13, e0195009. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, P.; Rasero, R.; Ru, G.; Aiassa, E.; Colussi, S.; Ingravalle, F.; Peletto, S.; Perrotta, M.G.; Sartore, S.; Soglia, D.; et al. Predicting the impact of selection for scrapie resistance on PRNP genotype frequencies in goats. Veter. Res. 2018, 49, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ricci, A.; Allende, A.; Bolton, D.; Chemaly, M.; Davies, R.; Escámez, P.S.F.; Gironés, R.; Herman, L.; Koutsoumanis, K.; Lindqvist, R.; et al. Genetic resistance to transmissible spongiform encephalopathies (TSE) in goats. EFSA J. 2017, 15, e04962. [Google Scholar] [CrossRef]
- Nodelijk, G.; van Roermund, H.J.; van Keulen, L.J.; Engel, B.; Vellema, P.; Hagenaars, T.J. Breeding with resistant rams leads to rapid control of classical scrapie in affected sheep flocks. Veter. Res. 2011, 42, 5. [Google Scholar] [CrossRef] [Green Version]
- Wik, L.; Mikko, S.; Klingeborn, M.; Stéen, M.; Simonsson, M.; Linné, T. Polymorphisms and variants in the prion protein sequence of European moose (Alces alces), reindeer (Rangifer tarandus), roe deer (Capreolus capreolus) and fallow deer (Dama dama) in Scandinavia. Prion 2012, 6, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.J.; Samuel, M.D.; O’Rourke, K.I.; Johnson, C.J. The role of genetics in chronic wasting disease of North American cervids. Prion 2012, 6, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Sigurdson, C.J. A prion disease of cervids: Chronic wasting disease. Veter. Res. 2008, 39, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Haley, N.J.; Merrett, K.; Stein, A.B.; Simpson, D.; Carlson, A.; Mitchell, G.; Staskevicius, A.; Nichols, T.; Lehmkuhl, A.D.; Thomsen, B.V. Estimating relative CWD susceptibility and disease progression in farmed white-tailed deer with rare PRNP alleles. PLoS ONE 2019, 14, e0224342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.; Johnson, J.; Clayton, M.; McKenzie, D.; Aiken, J. Prion Protein Gene Heterogeneity in Free-Ranging White-Taikde Deer Within the Chronic Wasting Disease Affected Region of Wisconsin. J. Wildl. Dis. 2003, 39, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Johnson, J.; Vanderloo, J.P.; Keane, D.; Aiken, J.M.; McKenzie, D. Prion protein polymorphisms in white-tailed deer influence susceptibility to chronic wasting disease. J. Gen. Virol. 2006, 87, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.J.; Herbst, A.; Duque-Velasquez, C.; Vanderloo, J.P.; Bochsler, P.; Chappell, R.; McKenzie, D. Prion Protein Polymorphisms Affect Chronic Wasting Disease Progression. PLoS ONE 2011, 6, e17450. [Google Scholar] [CrossRef]
- Otero, A.; Velásquez, C.D.; Johnson, C.; Herbst, A.; Bolea, R.; Badiola, J.J.; Aiken, J.; McKenzie, D. Prion protein polymorphisms associated with reduced CWD susceptibility limit peripheral PrPCWD deposition in orally infected white-tailed deer. BMC Veter. Res. 2019, 15, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, K.I.; Spraker, T.R.; Hamburg, L.K.; Besser, T.E.; Brayton, K.A.; Knowles, D.P.; Mercier, S.; Verhaagh, S.; Goudsmit, J.; Lemckert, A.; et al. Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J. Gen. Virol. 2004, 85, 1339–1346. [Google Scholar] [CrossRef]
- Haley, N.J.; Siepker, C.; Walter, W.D.; Thomsen, B.V.; Greenlee, J.J.; Lehmkuhl, A.D.; Richt, J.A. Antemortem Detection of Chronic Wasting Disease Prions in Nasal Brush Collections and Rectal Biopsy Specimens from White-Tailed Deer by Real-Time Quaking-Induced Conversion. J. Clin. Microbiol. 2016, 54, 1108–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, D.P.; Barr, D.J.; Bochsler, P.N.; Hall, S.M.; Gidlewski, T.; O’Rourke, K.I.; Spraker, T.R.; Samuel, M.D. Chronic Wasting Disease in a Wisconsin White-Tailed Deer Farm. J. Veter. Diagn. Investig. 2008, 20, 698–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.A.; Nakada, S.M.; Bollinger, T.K.; Pybus, M.J.; Merrill, E.H.; Coltman, D.W. Polymorphisms at the PRNP Gene Influence Susceptibility to Chronic Wasting Disease in Two Species of Deer (Odocoileus Spp.) in Western Canada. J. Toxicol. Environ. Health Part A 2009, 72, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.E.; Conner, M.M.; Wolfe, L.L.; Miller, M.W.; Williams, E.S. Low frequency of PrP genotype 225SF among free-ranging mule deer (Odocoileus hemionus) with chronic wasting disease. J. Gen. Virol. 2005, 86, 2127–2134. [Google Scholar] [CrossRef]
- Fox, K.A.; Jewell, J.E.; Williams, E.S.; Miller, M.W. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J. Gen. Virol. 2006, 87, 3451–3461. [Google Scholar] [CrossRef]
- Schätzl, H.M.; Wopfner, F.; Gilch, S.; von Brunn, A.; Jäger, G. Is codon 129 of prion protein polymorphic in human beings but not in animals? Lancet 1997, 349, 1603–1604. [Google Scholar] [CrossRef]
- O’Rourke, K.I.; Besser, T.E.; Miller, M.W.; Cline, T.F.; Spraker, T.R.; Jenny, A.L.; Wild, M.A.; Zebarth, G.L.; Williams, E.S. PrP genotypes of captive and free-ranging Rocky Mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J. Gen. Virol. 1999, 80. [Google Scholar] [CrossRef]
- Hamir, A.N.; Gidlewski, T.; Spraker, T.R.; Miller, J.M.; Creekmore, L.; Crocheck, M.; Cline, T.; O’Rourke, K.I. Preliminary Observations of Genetic Susceptibility of elk (Cervus Elaphus Nelsoni) to Chronic Wasting Disease by Experimental Oral Inoculation. J. Veter. Diagn. Investig. 2006, 18, 110–114. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, K.I.; Spraker, T.R.; Zhuang, D.; Greenlee, J.J.; Gidlewski, T.E.; Hamir, A.N. Elk with a long incubation prion disease phenotype have a unique PrPd profile. NeuroReport 2007, 18, 1935–1938. [Google Scholar] [CrossRef] [PubMed]
- Perucchini, M.; Griffin, K.; Miller, M.W.; Goldmann, W. PrP genotypes of free-ranging wapiti (Cervus elaphus nelsoni) with chronic wasting disease. J. Gen. Virol. 2008, 89, 1324–1328. [Google Scholar] [CrossRef] [PubMed]
- Monello, R.J.; Galloway, N.L.; Powers, J.G.; Madsen-Bouterse, S.A.; Edwards, W.H.; Wood, M.E.; O’Rourke, K.I.; Wild, M.A. Pathogen-mediated selection in free-ranging elk populations infected by chronic wasting disease. Proc. Natl. Acad. Sci. USA 2017, 114, 12208–12212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.J.; Vrentas, C.E.; Hwang, S.; Greenlee, M.H.W.; Nicholson, E.M.; Greenlee, J.J. Pathologic and biochemical characterization of PrPSc from elk with PRNP polymorphisms at codon 132 after experimental infection with the chronic wasting disease agent. BMC Veter. Res. 2018, 14, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervenáková, L.; Rohwer, R.; Williams, S.; Brown, P.; Gajdusek, D.C. High sequence homology of the PrP gene in mule deer and Rocky Mountain elk. Lancet 1997, 350, 219–220. [Google Scholar] [CrossRef]
- Bian, J.; Christiansen, J.R.; Moreno, J.A.; Kane, S.J.; Khaychuk, V.; Gallegos, J. Primary structural differences at residue 226 of deer and elk PrP dictate selection of distinct CWD prion strains in gene-targeted mice. Proc. Natl. Acad. Sci. USA 2019, 2019, 03947. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.L.; Williamson, H.; Güere, M.E.; Tharaldsen, H.; Baker, K.; Smith, S.L.; Pérez-Espona, S.; Krojerová-Prokešová, J.; Pemberton, J.M.; Goldmann, W.; et al. Variation in the prion protein gene (PRNP) sequence of wild deer in Great Britain and mainland Europe. Veter. Res. 2019, 50, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hamir, A.; Kunkle, R.; Nicholson, E.; Miller, J.; Hall, S.; Schoenenbruecher, H.; Brunelle, B.; Richt, J. Preliminary Observations on the Experimental Transmission of Chronic Wasting Disease (CWD) from Elk and White-Tailed Deer to Fallow Deer. J. Comp. Pathol. 2008, 138, 121–130. [Google Scholar] [CrossRef]
- Rhyan, J.C.; Miller, M.W.; Spraker, T.R.; Mccollum, M.; Nol, P.; Wolfe, L.L.; Davis, T.R.; Creekmore, L.; O’Rourke, K.I. Failure of Fallow Deer (Dama dama) to Develop Chronic Wasting Disease When Exposed to a Contaminated Environment and Infected Mule Deer (Odocoileus hemionus). J. Wildl. Dis. 2011, 47, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Hamir, A.N.; Greenlee, J.J.; Nicholson, E.M.; Kunkle, R.A.; Richt, J.A.; Miller, J.M.; Hall, M. Experimental transmission of chronic wasting disease (CWD) from elk and white-tailed deer to fallow deer by intracerebral route: Final report. Can. J. Veter. Res. 2011, 75, 152–156. [Google Scholar]
- Roh, I.S.; Kim, H.J.; Kim, H.J.; Suh, T.Y.; Han, J.H.; Kang, H.E.; Sohn, H.J. Polymorphisms in the Prion Protein Gene, Associated with Chronic Wasting Disease, in the Korean Water Deer (Hydropotes inermis argyropus). J. Veter. Sci. Technol. 2018, 9, 1–4. [Google Scholar] [CrossRef]
- Jeong, H.-J.; Lee, J.-B.; Park, S.-Y.; Chang-Seon, S.; Kim, B.-S.; Rho, J.-R. Single-nucleotide polymorphisms in prion protein gene of the Korean subspecies of Chinese water deer (Hydropotes inermis argyropus). Korean J. Vet. Res. 2009, 49, 59–62. [Google Scholar]
- Cullingham, C.I.; Peery, R.M.; Dao, A.; McKenzie, D.I.; Coltman, D.W. Predicting the spread-risk potential of chronic wasting disease to sympatric ungulate species. Prion 2020, 14, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreeger, T.J.; Montgomery, D.L.; Jewell, J.E.; Schultz, W.; Williams, E.S. Oral Transmission of Chronic Wasting Disease in Captive Shira’s Moose. J. Wildl. Dis. 2006, 42, 640–645. [Google Scholar] [CrossRef] [Green Version]
- Huson, H.J.; Happ, G.M. Polymorphisms of the prion protein gene (PRNP) in Alaskan moose (Alces alces gigas). Anim. Genet. 2006, 37, 425–426. [Google Scholar] [CrossRef] [Green Version]
- Happ, G.M.; Huson, H.J.; Beckmen, K.B.; Kennedy, L.J. Prion Protein Genes in Caribou from Alaska. J. Wildl. Dis. 2007, 43, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, G.B.; Sigurdson, C.J.; O’Rourke, K.I.; Algire, J.; Harrington, N.P.; Walther, I.; Spraker, T.R.; Balachandran, A. Experimental Oral Transmission of Chronic Wasting Disease to Reindeer (Rangifer tarandus tarandus). PLoS ONE 2012, 7, e39055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.C.; Musiani, M.; Cavedon, M.; Gilch, S. High prevalence of prion protein genotype associated with resistance to chronic wasting disease in one Alberta woodland caribou population. Prion 2017, 11, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Arifin, M.I.; Staskevicius, A.; Shim, S.Y.; Huang, Y.; Fenton, H.; McLoughlin, P.D.; Mitchell, G.; Cullingham, C.I.; Gilch, S. Large-scale prion protein genotyping in Canadian caribou populations and potential impact on chronic wasting disease susceptibility. Mol. Ecol. 2020, 29, 3830–3840. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Kunkle, R.; Greenlee, M.H.W.; Nicholson, E.; Richt, J.; Hamir, A.; Waters, W.R.; Greenlee, J. Horizontal Transmission of Chronic Wasting Disease in Reindeer. Emerg. Infect. Dis. 2016, 22, 2142–2145. [Google Scholar] [CrossRef] [Green Version]
- Güere, M.E.; Våge, J.; Tharaldsen, H.; Benestad, S.L.; Vikøren, T.; Madslien, K.; Hopp, P.; Rolandsen, C.M.; Røed, K.H.; Tranulis, M.A. Chronic wasting disease associated with prion protein gene (PRNP) variation in Norwegian wild reindeer (Rangifer tarandus). Prion 2020, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorberg, I.; Chan, K.; Priola, S.A. Deletion of β-Strand and α-Helix Secondary Structure in Normal Prion Protein Inhibits Formation of Its Protease-Resistant Isoform. J. Virol. 2001, 75, 10024–10032. [Google Scholar] [CrossRef] [Green Version]
- Collinge, J.; Palmer, M.; Dryden, A. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 1991, 337, 1441–1442. [Google Scholar] [CrossRef]
- Palmer, M.S.; Dryden, A.J.; Hughes, J.T.; Collinge, J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt–Jakob disease. Nat. Cell Biol. 1991, 352, 340–342. [Google Scholar] [CrossRef]
- O’Rourke, K.I.; Baszler, T.V.; Miller, J.M.; Spraker, T.R.; Sadler-Riggleman, I.; Knowles, D.P. Monoclonal Antibody F89/160.1.5 Defines a Conserved Epitope on the Ruminant Prion Protein. J. Clin. Microbiol. 1998, 36, 1750–1755. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.; Tatum, T.; Hwang, S.; Vrentas, C.; Greenlee, M.H.W.; Kong, Q.; Nicholson, E.; Greenlee, J. Novel Strain of the Chronic Wasting Disease Agent Isolated from Experimentally Inoculated Elk with LL132 Prion Protein. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Brayton, K.A.; O’Rourke, K.I.; Lyda, A.K.; Miller, M.W.; Knowles, D.P. A processed pseudogene contributes to apparent mule deer prion gene heterogeneity. Gene 2004, 326, 167–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, L.L.; Fox, K.A.; Miller, M.W. “Atypical” Chronic Wasting Disease in PRNP Genotype 225FF Mule Deer. J. Wildl. Dis. 2014, 50, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, M.P.; Leymaster, K.A.; Freking, B.A.; Hawk, D.A.; Smith, T.P.L.; Keele, J.W.; Snelling, W.M.; Fox, J.M.; Chitko-McKown, C.G.; Laegreid, W.W. Prion gene sequence variation within diverse groups of U.S. sheep, beef cattle, and deer. Mamm. Genome 2003, 14, 765–777. [Google Scholar] [CrossRef]
- Robinson, S.J.; Samuel, M.D.; Johnson, C.J.; Adams, M.; McKenzie, D.I. Emerging prion disease drives host selection in a wildlife population. Ecol. Appl. 2012, 22, 1050–1059. [Google Scholar] [CrossRef] [Green Version]
- Kelly, A.C.; Mateus-Pinilla, N.E.; Diffendorfer, J.; Jewell, E.; Ruiz, M.O.; Killefer, J.; Shelton, P.; Beissel, T.; Novakofski, J. Prion sequence polymorphisms and chronic wasting disease resistance in Illinois white-tailed deer (Odocoileus virginianus). Prion 2008, 2, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Herbst, A.; Velásquez, C.D.; Triscott, E.; Aiken, J.M.; McKenzie, D. Chronic Wasting Disease Prion Strain Emergence and Host Range Expansion. Emerg. Infect. Dis. 2017, 23, 1598–1600. [Google Scholar] [CrossRef] [Green Version]
- Duque-Velásquez, C.; Kim, C.; Haldiman, T.; Kim, C.; Herbst, A.; Aiken, J.M. Chronic wasting disease (CWD) prion strains evolve via adaptive diversification of conformers in hosts expressing prion protein polymorphisms. J. Biol. Chem. 2020, 15, 4985–5001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meade-White, K.; Race, B.; Trifilo, M.; Bossers, A.; Favara, C.; Lacasse, R.; Miller, M.; Williams, E.; Oldstone, M.; Race, R.; et al. Resistance to Chronic Wasting Disease in Transgenic Mice Expressing a Naturally Occurring Allelic Variant of Deer Prion Protein. J. Virol. 2007, 81, 4533–4539. [Google Scholar] [CrossRef] [Green Version]
- Race, B.; Meade-White, K.; Miller, M.W.; Fox, K.A.; Chesebro, B. In Vivo Comparison of Chronic Wasting Disease Infectivity from Deer with Variation at Prion Protein Residue 96. J. Virol. 2011, 85, 9235–9238. [Google Scholar] [CrossRef] [Green Version]
- Chabry, J.; Caughey, B.; Chesebro, B. Specific Inhibition of in Vitro Formation of Protease-resistant Prion Protein by Synthetic Peptides. J. Biol. Chem. 1998, 273, 13203–13207. [Google Scholar] [CrossRef] [Green Version]
- Hölscher, C.; Delius, H.; Bürkle, A. Overexpression of Nonconvertible PrPcΔ114–121 in Scrapie-Infected Mouse Neuroblastoma Cells Leads to trans-Dominant Inhibition of Wild-Type PrPSc Accumulation. J. Virol. 1998, 72, 1153–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
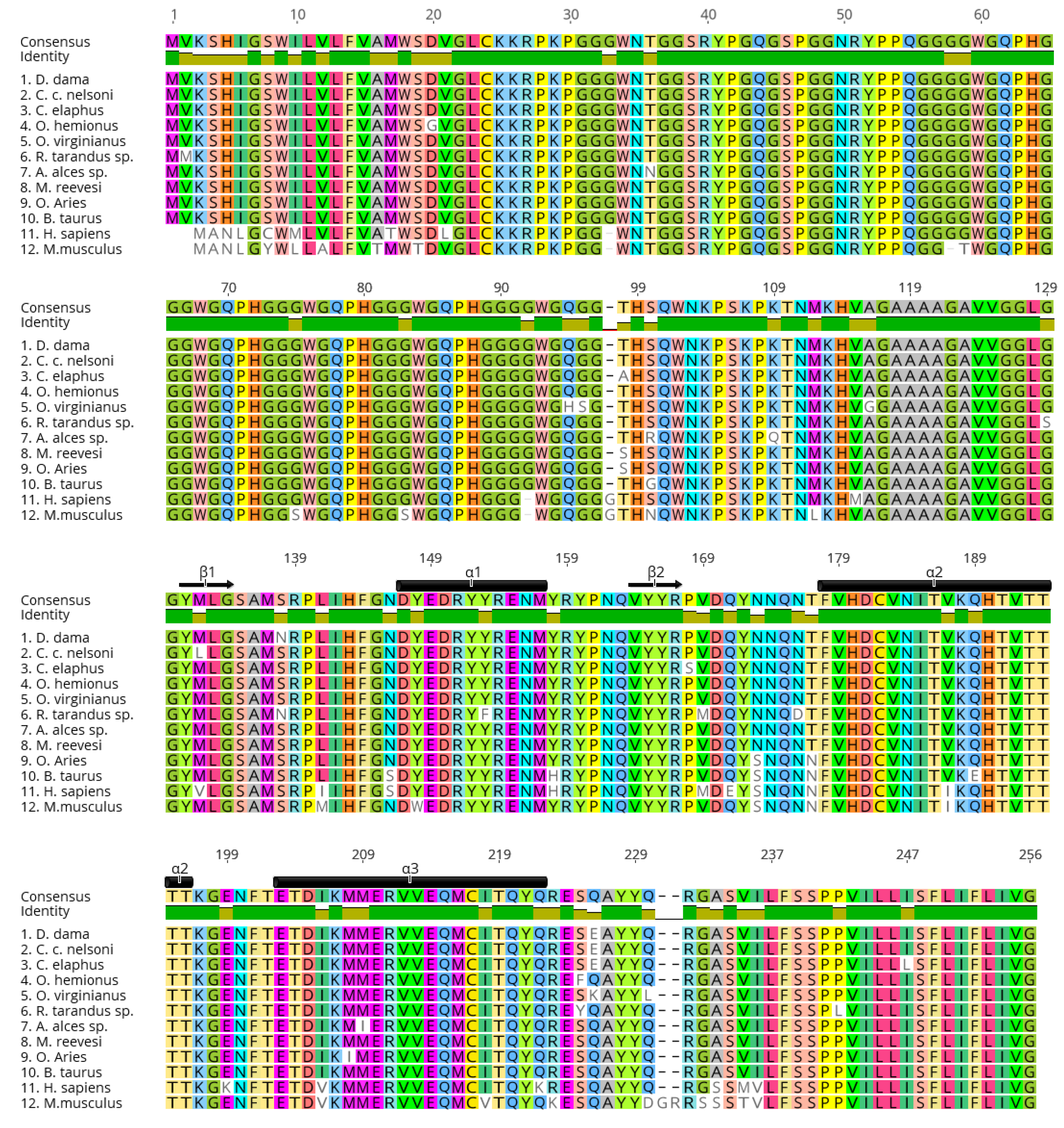
- Wopfner, F.; Weidenhöfer, G.; Schneider, R.; von Brunn, A.; Gilch, S.; Schwarz, T.F. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein 11Edited by A. R. Fersht. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef]
- Wang, F.; Yin, S.; Wang, X.; Zha, L.; Sy, M.-S.; Ma, J. Role of the Highly Conserved Middle Region of PrP in PrP-Lipid Interaction. Biochemistry 2010, 49, 8169–8176. [Google Scholar] [CrossRef] [Green Version]
- Hegde, R.S.; Mastrianni, J.A.; Scott, M.R.; DeFea, K.A.; Tremblay, P.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. A Transmembrane Form of the Prion Protein in Neurodegenerative Disease. Science 1998, 279, 827–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurt, T.D.; Telling, G.C.; Zabel, M.D.; Hoover, E.A. Trans-species amplification of PrPCWD and correlation with rigid loop 170N. Virology 2009, 387, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Kurt, T.D.; Jiang, L.; Fernández-Borges, N.; Bett, C.; Liu, J.; Yang, T. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J. Clin. Investig. 2015, 125, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Pérez, D.R.; Damberger, F.F.; Wüthrich, K. Horse Prion Protein NMR Structure and Comparisons with Related Variants of the Mouse Prion Protein. J. Mol. Biol. 2010, 400, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, R.; Satoh, K.; Sano, K.; Fuse, T.; Yamaguchi, N.; Ishibashi, D.; Matsubara, T.; Nakagaki, T.; Yamanaka, H.; Shirabe, S.; et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat. Med. 2011, 17, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Haley, N.J.; Rielinger, R.; Davenport, K.A.; O’Rourke, K.; Mitchell, G.; Richt, J.A. Estimating chronic wasting disease susceptibility in cervids using real-time quaking-induced conversion. J. Gen. Virol. 2017, 98, 2882–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scialò, C.; de Cecco, E.; Manganotti, P.; Legname, G. PRION 2019 emerging concepts. Prion 2019, 13 (Suppl. 1), 1–141. [Google Scholar]
- Pattison, I.; Millson, G. Scrapie Produced Experimentally in Goats with Special Reference to the Clinical Syndrome. J. Comp. Pathol. Ther. 1961, 71, 101–110. [Google Scholar] [CrossRef]
- Aguzzi, A.; Heikenwalder, M.; Polymenidou, M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 2007, 8, 552–561. [Google Scholar] [CrossRef]
- Angers, R.C.; Kang, H.-E.; Napier, D.; Browning, S.; Seward, T.; Mathiason, C.; Balachandran, A.; McKenzie, D.; Castilla, J.; Soto, C.; et al. Prion Strain Mutation Determined by Prion Protein Conformational Compatibility and Primary Structure. Science 2010, 328, 1154–1158. [Google Scholar] [CrossRef] [Green Version]
- Collinge, J.; Clarke, A.R. A General Model of Prion Strains and Their Pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angers, R.C.; Seward, T.S.; Napier, D.; Green, M.; Hoover, E.; Spraker, T.; O’Rourke, K.; Balachandran, A.; Telling, G.C. Chronic Wasting Disease Prions in Elk Antler Velvet. Emerg. Infect. Dis. 2009, 15, 696–703. [Google Scholar] [CrossRef]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP Sc molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef]
- Bian, J.; Kang, H.-E.; Telling, G.C. Quinacrine promotes replication and conformational mutation of chronic wasting disease prions. Proc. Natl. Acad. Sci. USA 2014, 111, 6028–6033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, S.R.; Mason, G.L.; Seward, T.; Green, M.; Eliason, G.A.J.; Mathiason, C. Transmission of Prions from Mule Deer and Elk with Chronic Wasting Disease to Transgenic Mice Expressing Cervid PrP. J. Virol. 2004, 78, 13345–13350. [Google Scholar] [CrossRef] [Green Version]
- Hoover, C.E.; Davenport, K.A.; Henderson, D.M.; Denkers, N.D.; Mathiason, C.K.; Soto, C.; Zabel, M.D.; Hoover, E.A. Pathways of Prion Spread during Early Chronic Wasting Disease in Deer. J. Virol. 2017, 91, e00077-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shearin, H.; Bessen, R.A. Axonal and Transynaptic Spread of Prions. J. Virol. 2014, 88, 8640–8655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathiason, C.K.; Hayes-Klug, J.; Hays, S.A.; Powers, J.; Osborn, D.A.; Dahmes, S.J.; Miller, K.V.; Warren, R.J.; Mason, G.L.; Telling, G.C.; et al. B Cells and Platelets Harbor Prion Infectivity in the Blood of Deer Infected with Chronic Wasting Disease. J. Virol. 2010, 84, 5097–5107. [Google Scholar] [CrossRef] [Green Version]
- Hamir, A.N.; Kunkle, R.A.; Miller, J.M.; Hall, S.M. Abnormal Prion Protein in Ectopic Lymphoid Tissue in a Kidney of an Asymptomatic White-tailed Deer Experimentally Inoculated with the Agent of Chronic Wasting Disease. Veter. Pathol. 2006, 43, 367–369. [Google Scholar] [CrossRef]
- Kaatz, M.; Fast, C.; Ziegler, U.; Balkema-Buschmann, A.; Hammerschmidt, B.; Keller, M.; Oelschlegel, A.; McIntyre, L.; Groschup, M.H. Spread of Classic BSE Prions from the Gut via the Peripheral Nervous System to the Brain. Am. J. Pathol. 2012, 181, 515–524. [Google Scholar] [CrossRef]
- Seelig, D.M.; Mason, G.L.; Telling, G.C.; Hoover, E.A. Pathogenesis of Chronic Wasting Disease in Cervidized Transgenic Mice. Am. J. Pathol. 2010, 176, 2785–2797. [Google Scholar] [CrossRef]
- Bartz, J.C.; de Joia, C.; Tucker, T.; Kincaid, A.E.; Bessen, R.A. Extraneural Prion Neuroinvasion without Lymphoreticular System Infection. J. Virol. 2005, 79, 11858–11863. [Google Scholar] [CrossRef] [Green Version]
- Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Rapid Prion Neuroinvasion following Tongue Infection. J. Virol. 2003, 77, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Glatzel, M.; Aguzzi, A. PrPC expression in the peripheral nervous system is a determinant of prion neuroinvasion. J. Gen. Virol. 2000, 81, 2813–2821. [Google Scholar] [CrossRef] [PubMed]
- Mammadova, N.; Cassmann, E.; Greenlee, J.J. Successful transmission of the chronic wasting disease (CWD) agent to white-tailed deer by intravenous blood transfusion. Res. Veter. Sci. 2020, 133, 304–306. [Google Scholar] [CrossRef]
- Nalls, A.V.; McNulty, E.; Hoover, C.E.; Pulscher, L.A.; Hoover, E.A.; Mathiason, C.K. Infectious Prions in the Pregnancy Microenvironment of Chronic Wasting Disease-Infected Reeves’ Muntjac Deer. J. Virol. 2017, 91, e00501-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, D.M.; Manca, M.; Haley, N.J.; Denkers, N.D.; Nalls, A.V.; Mathiason, C.K.; Caughey, B.; Hoover, E.A. Rapid Antemortem Detection of CWD Prions in Deer Saliva. PLoS ONE 2013, 8, e74377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, I.H.; Wright, S.D.; Johnson, C.J.; Pedersen, J.A.; Samuel, M.D. Temporal patterns of chronic wasting disease prion excretion in three cervid species. J. Gen. Virol. 2017, 98, 1932–1942. [Google Scholar] [CrossRef]
- Kramm, C.; Soto, P.; Nichols, T.A.; Morales, R. Chronic wasting disease (CWD) prion detection in blood from pre-symptomatic white-tailed deer harboring PRNP polymorphic variants. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Bruce, M.; Chree, A.; Williams, E.; Fraser, H. Perivascular PrP amyloid in the brains of mice infected with chronic wasting disease. Brain Pathol. 2000, 10, 662–663. [Google Scholar]
- Raymond, G.J.; Raymond, L.D.; Meade-White, K.D.; Hughson, A.G.; Favara, C.; Gardner, D.; Williams, E.S.; Miller, M.W.; Race, R.E.; Caughey, B. Transmission and Adaptation of Chronic Wasting Disease to Hamsters and Transgenic Mice: Evidence for Strains. J. Virol. 2007, 81, 4305–4314. [Google Scholar] [CrossRef] [Green Version]
- Sigurdson, C.J.; Manco, G.; Schwarz, P.; Liberski, P.; Hoover, E.A.; Hornemann, S.; Polymenidou, M.; Miller, M.W.; Glatzel, M.; Aguzzi, A. Strain Fidelity of Chronic Wasting Disease upon Murine Adaptation. J. Virol. 2006, 80, 12303–12311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heisey, D.M.; Mickelsen, N.A.; Schneider, J.R.; Johnson, C.J.; Johnson, C.J.; Langenberg, J.A. Chronic Wasting Disease (CWD) Susceptibility of Several North American Rodents That Are Sympatric with Cervid CWD Epidemics. J. Virol. 2010, 84, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bari, M.A.; Nonno, R.; Castilla, J.; D’Agostino, C.; Pirisinu, L.; Riccardi, G.; Conte, M.; Richt, J.; Kunkle, R.; Langeveld, J.; et al. Chronic Wasting Disease in Bank Voles: Characterisation of the Shortest Incubation Time Model for Prion Diseases. PLoS Pathog. 2013, 9, e1003219. [Google Scholar] [CrossRef] [PubMed]
- Perrott, M.R.; Sigurdson, C.J.; Mason, G.L.; Hoover, E.A. Evidence for distinct chronic wasting disease (CWD) strains in experimental CWD in ferrets. J. Gen. Virol. 2012, 93, 212–221. [Google Scholar] [CrossRef]
- Sigurdson, C.; Mathiason, C.; Perrott, M.; Eliason, G.; Spraker, T.; Glatzel, M.; Manco, G.; Bartz, J.; Miller, M.; Hoover, E. Experimental Chronic Wasting Disease (CWD) in the Ferret. J. Comp. Pathol. 2008, 138, 189–196. [Google Scholar] [CrossRef]
- Bartz, J.C.; Marsh, R.F.; McKenzie, D.I.; Aiken, J.M. The Host Range of Chronic Wasting Disease Is Altered on Passage in Ferrets. Virology 1998, 251, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Mathiason, C.K.; Nalls, A.V.; Seelig, D.M.; Kraft, S.L.; Carnes, K.; Anderson, K.R.; Hayes-Klug, J.; Hoover, E.A. Susceptibility of Domestic Cats to Chronic Wasting Disease. J. Virol. 2012, 87, 1947–1956. [Google Scholar] [CrossRef] [Green Version]
- Seelig, D.M.; Nalls, A.V.; Flasik, M.; Frank, V.; Eaton, S.; Mathiason, C.K.; Hoover, E.A. Lesion profiling and subcellular prion localization of cervid chronic wasting disease in domestic cats. Veter. Pathol. 2014, 52, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Waddell, L.; Greig, J.; Mascarenhas, M.; Otten, A.; Corrin, T.; Hierlihy, K. Current evidence on the transmissibility of chronic wasting disease prions to humans-A systematic review. Transbound. Emerg. Dis. 2017, 65, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Haley, N.J.; Hoover, E.A. Chronic Wasting Disease of Cervids: Current Knowledge and Future Perspectives. Annu. Rev. Anim. Biosci. 2015, 3, 305–325. [Google Scholar] [CrossRef]
- Czub, S.; Schulz-Schaeffer, W.; Stahl-Hennig, C.; Beekes, M.; Schaetzl, H.; Motzkus, D. First evidence of intracranial and peroral transmission of chronic wasting disease (CWD) into cynomolgus macaques: A work in progress. In Proceedings of the (Abstract) from Prion 2017: Deciphering Neurodegenerative Disorders, Edinburgh, Scotland, 23–26 May 2017. [Google Scholar]
- Moore, S.J.; Greenlee, M.H.W.; Kondru, N.; Manne, S.; Smith, J.D.; Kunkle, R.A.; Kanthasamy, A.; Greenlee, J.J. Experimental Transmission of the Chronic Wasting Disease Agent to Swine after Oral or Intracranial Inoculation. J. Virol. 2017, 91, e00926-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamir, A.N.; Kunkle, R.A.; Cutlip, R.C.; Miller, J.M.; O’Rourke, K.I.; Williams, E.S.; Miller, M.W.; Stack, M.J.; Chaplin, M.J.; Richt, J.A. Experimental Transmission of Chronic Wasting Disease Agent from Mule Deer to Cattle by the Intracerebral Route. J. Veter. Diagn. Investig. 2005, 17, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Hamir, A.; Kunkle, R.; Miller, J.; Greenlee, J.; Richt, J. Experimental Second Passage of Chronic Wasting Disease (CWDmule deer) Agent to Cattle. J. Comp. Pathol. 2006, 134, 63–69. [Google Scholar] [CrossRef]
- Hamir, A.N.; Miller, J.M.; Kunkle, R.A.; Hall, S.M.; Richt, J.A. Susceptibility of Cattle to First-passage Intracerebral Inoculation with Chronic Wasting Disease Agent from White-tailed Deer. Veter. Pathol. 2007, 44, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamir, A.N.; Kehrli, M.E.; Kunkle, R.A.; Greenlee, J.J.; Nicholson, E.M.; Richt, J.A. Experimental interspecies transmission studies of the transmissible spongiform encephalopathies to cattle: Comparison to bovine spongiform encephalopathy in cattle. J. VET Diagn. Invest. 2011, 23, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, E.S.; O’Toole, D.; Miller, M.W.; Kreeger, T.J.; Jewell, J.E. Cattle (Bos Taurus) Resist Chronic Wasting Disease Following Oral Inoculation Challenge or Ten Years’ Natural Exposure in Contaminated Eeniroments. J. Wildl. Dis. 2018, 54, 460–470. [Google Scholar] [CrossRef]
- Tamgüney, G.; Giles, K.; Bouzamondo-Bernstein, E.; Bosque, P.J.; Miller, M.W.; Safar, J.; DeArmond, S.J.; Prusiner, S.B. Transmission of Elk and Deer Prions to Transgenic Mice. J. Virol. 2006, 80, 9104–9114. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, M.K.; Al-Doujaily, H.; Sigurdson, C.J.; Glatzel, M.; O’Malley, C.; Powell, C.; Asante, E.A.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.F.; et al. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J. Gen. Virol. 2010, 91, 2651–2657. [Google Scholar] [CrossRef]
- Wadsworth, J.D.F.; Asante, E.A.; Desbruslais, M.; Linehan, J.M.; Joiner, S.; Gowland, I.; Welch, J.; Stone, L.; Lloyd, S.E.; Hill, A.F.; et al. Human Prion Protein with Valine 129 Prevents Expression of Variant CJD Phenotype. Science 2004, 306, 1793–1796. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Meikle, V.M.H. Host-genotype and agent effects in scrapie incubation: Change in allelic interaction with different strains of agent. Mol. Genet. Genom. 1971, 112, 73–79. [Google Scholar] [CrossRef]
- Benestad, S.L.; Arsac, J.-N.; Goldmann, W.; Nöremark, M. Atypical/Nor98 scrapie: Properties of the agent, genetics, and epidemiology. Veter. Res. 2008, 39, 19. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.A.; Bosque, P.; Filley, C.M.; Arciniegas, D.B.; Kleinschmidt-Demasters, B.K.; Pape, W.J.; Tyler, K.L. Colorado Surveillance Program for Chronic Wasting Disease Transmission to Humans: Lessons From 2 Highly Suspicious but Negative Cases. Arch Neurol. 2007, 64, 439. [Google Scholar] [CrossRef]
- Mawhinney, S.; Pape, W.J.; Forster, J.E.; Anderson, C.A.; Bosque, P.; Miller, M.W. Human Prion Disease and Relative Risk Associated with Chronic Wasting Disease. Emerg. Infect. Dis. 2006, 12, 1527–1535. [Google Scholar] [CrossRef]
- Belay, E.D.; Gambetti, P.; Schonberger, L.B.; Parchi, P.; Lyon, D.R.; Capellari, S.; McQuiston, J.H.; Bradley, K.; Dowdle, G.; Crutcher, J.M.; et al. Creutzfeldt-Jakob Disease in Unusually Young Patients Who Consumed Venison. Arch. Neurol. 2001, 58, 1673–1678. [Google Scholar] [CrossRef]
- Olszowy, K.; Lavelle, J.; Rachfal, K.; Hempstead, S.; Drouin, K.; Darcy, J.; Reiber, C.; Garruto, R. Six-year follow-up of a point-source exposure to CWD contaminated venison in an Upstate New York Community: Risk behaviours and health outcomes 2005–2011. Public Health 2014, 128, 860–868. [Google Scholar] [CrossRef]
- Barria, M.A.; Telling, G.C.; Gambetti, P.; Mastrianni, J.A.; Soto, C. Generation of a New Form of Human PrPSc in Vitro by Interspecies Transmission from Cervid Prions. J. Biol. Chem. 2011, 286, 7490–7495. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.; Huang, S.; Zou, W.; Vanegas, D.; Wang, M.; Wu, D.; Yuan, J.; Zheng, M.; Bai, H.; Deng, H.; et al. Chronic Wasting Disease of Elk: Transmissibility to Humans Examined by Transgenic Mouse Models. J. Neurosci. 2005, 25, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Race, B.; Williams, K.; Chesebro, B. Transmission studies of chronic wasting disease to transgenic mice overexpressing human prion protein using the RT-QuIC assay. Vet. Res. 2019, 50, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Race, B.; Meade-White, K.D.; Miller, M.W.; Barbian, K.D.; Rubenstein, R.; LaFauci, G.; Cervenáková, L.; Favara, C.; Gardner, D.; Long, D.; et al. Susceptibilities of Nonhuman Primates to Chronic Wasting Disease. Emerg. Infect. Dis. 2009, 15, 1366–1376. [Google Scholar] [CrossRef]
- Race, B.; Williams, K.; Orru, C.D.; Hughson, A.G.; Lubke, L.; Chesebro, B. Lack of Transmission of Chronic Wasting Disease to Cynomolgus Macaques. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Race, B.; Meade-White, K.D.; Phillips, K.; Striebel, J.; Race, R.; Chesebro, B. Chronic Wasting Disease Agents in Nonhuman Primates. Emerg. Infect. Dis. 2014, 20, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Osterholm, M.T.; Anderson, C.J.; Zabel, M.D.; Scheftel, J.M.; Moore, K.A.; Appleby, B.S. Chronic Wasting Disease in Cervids: Implications for Prion Transmission to Humans and Other Animal Species. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comoy, E.E.; Mikol, J.; Luccantoni-Freire, S.; Correia, E.; Lescoutra-Etchegaray, N.; Durand, V.; Dehen, C.; Andreoletti, O.; Casalone, C.; Richt, J.A.; et al. Transmission of scrapie prions to primate after an extended silent incubation period. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Species | PrP Codon | Allele | Effect of Polymorphism on CWD Pathogenesis (In Vivo) | Geographic Location | Ref. | |
|---|---|---|---|---|---|---|
| wt | var | |||||
| White-Tailed Deer (Odocoileus virginianus) | 95 | Q | H | Prolonged survival, reduced susceptibility and reduced peripheral prion spread | U.S.: WI, NE CA: AB, SK | [102,103,104,105,106,107] |
| 96 | G | S | Prolonged survival, reduced susceptibility, delayed lymphoreticular spread | U.S.: WI, NE CA: AB, SK | [13,102,103,104,106,107,108] | |
| 116 | A | G | Reduced susceptibility and lower infection rate | U.S.: NE CA: AB, SK | [101,106,109] | |
| 226 | Q | K | Reduced susceptibility and lower infection rate | U.S.: WI | [101,107,108] | |
| 226 | Q | R | N/A | CA: AB, SK | [109] | |
| 230 | Q | L | N/A | CA: AB, SK | [109] | |
| Mule Deer (O. hemionus) | 20 | D | G | Possibly more susceptible | U.S.: WY, CO CA: AB, SK | [109,110] |
| 225 | S | F | Slower disease progression | U.S.: WY, CO | [110,111] | |
| Elk (Cervus canadensis) | 132 | M | L | ML and LL have increased disease incubation periods, LL has lesser grey matter and greater white matter spongiform change, less PrPSc accumulation, and more stable fibrils, and 132L alleles are twice as frequent in herds known to be infected for >30 years than uninfected herds | U.S.: CO, WY, other mid-western/western states (e.g., SD, ND, MT, NE, MI) | [112,113,114,115,116,117,118] |
| 226 | E | - | More stable PrPCWD strains, but less conformational stability. | CA U.S. | [119,120] | |
| Red Deer (C. elaphus) | 98 | T | A | N/A | Britain, Czech Republic | [121] |
| 168 | P | S | N/A | Britain | [121] | |
| 226 | Q | E | N/A | Britain, Norway, Czech Republic | [121] | |
| 247 | I | L | N/A | Czech Republic | [121] | |
| Sika Deer (C. nippon) | 226 | Q | E | N/A | Britain | [121] |
| Fallow Deer (Dama dama) | 138 | N | - | Resistance to natural infection and prolonged incubation periods in intra-cerebrally infected animals | Britain | [122,123,124] |
| 226 | E | - | N/A | Britain | [122,123,124] | |
| Reeve’s Muntjac Deer (Muntiacus reevesii) | 98 | - | S | N/A | Britain | [121] |
| Chinese Water Deer (Hydropotes inermis inermis) | 100 | S | N | N/A | Britain | [121] |
| OR * | 5 | 4 | N/A | Britain | [121] | |
| Korean Water Deer (H. i. argyropus) | 96 | G | D | N/A | South Korea | [125] |
| 100 | N | S | N/A | South Korea | [125,126] | |
| 170 | D | G | N/A | South Korea | [125] | |
| Moose (Alces sp.) | 36 | T | N | N/A | Canada | [98] |
| 100 | S | R | N/A | Canada | [127] | |
| 109 | K | Q | N/A | Sweden | [98] | |
| 209 | M | I | N/A | U.S.: WY, WK | [128,129] | |
| Caribou/Reindeer (Rangifer tarandus sp.) | 2 | V | M | N/A | CA U.S.: AK | [130,131,132] |
| 129 | G | S | N/A | CA U.S.: AK | [130,131,133] | |
| 138 | S | N | 138SN and NN reindeer have prolonged incubation periods in oral transmission, and both also have no or limited PrPCWD distribution in the CNS | CA: BC, YK, NT, AB, SK U.S.: AK | [130,131,133,134] | |
| 153 | Y | F | N/A | CA | [133] | |
| 169 | V | M | N/A | CA U.S.: AK | [130,131,133] | |
| 176 | N | D | N/A | Sweden, Norway CA: NWT | [98,133] | |
| 225 | S | Y | Higher risk of infection | Sweden, Norway | [98,135] | |
| 242 | P | L | N/A | CA | [133] | |
| OR | 5 | 4 | Higher risk of infection | Norway | [135] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arifin, M.I.; Hannaoui, S.; Chang, S.C.; Thapa, S.; Schatzl, H.M.; Gilch, S. Cervid Prion Protein Polymorphisms: Role in Chronic Wasting Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 2271. https://doi.org/10.3390/ijms22052271
Arifin MI, Hannaoui S, Chang SC, Thapa S, Schatzl HM, Gilch S. Cervid Prion Protein Polymorphisms: Role in Chronic Wasting Disease Pathogenesis. International Journal of Molecular Sciences. 2021; 22(5):2271. https://doi.org/10.3390/ijms22052271
Chicago/Turabian StyleArifin, Maria Immaculata, Samia Hannaoui, Sheng Chun Chang, Simrika Thapa, Hermann M. Schatzl, and Sabine Gilch. 2021. "Cervid Prion Protein Polymorphisms: Role in Chronic Wasting Disease Pathogenesis" International Journal of Molecular Sciences 22, no. 5: 2271. https://doi.org/10.3390/ijms22052271