
Abstract
Chronic wasting disease (CWD) is a fatal, progressive disease that affects cervid species, including Rocky mountain elk (Cervus elaphus nelsoni). There are 2 allelic variants in the elk prion protein gene: L132 (leucine) and M132 (methionine). Following experimental oral challenge with the CWD agent incubation periods are longest in LL132 elk, intermediate in ML132 elk, and shortest in MM132 elk. In order to ascertain whether such CWD-infected elk carry distinct prion strains, groups of Tg12 mice that express M132 elk prion protein were inoculated intracranially with brain homogenate from individual CWD-infected elk of various genotypes (LL132, LM132, or MM132). Brain samples were examined for microscopic changes and assessment of the biochemical properties of disease-associated prion protein (PrPSc). On first passage, mice challenged with LL132 elk inoculum had prolonged incubation periods and greater PrPSc fibril stability compared to mice challenged with MM132 or LM132 inoculum. On second passage, relative incubation periods, western blot profiles, and neuropathology were maintained. These results suggest that the CWD prion isolated from LL132 elk is a novel CWD strain and that M132 PrPC is able to propagate some biophysical properties of the L132 PrPSc conformation.
Similar content being viewed by others

Introduction
Chronic wasting disease (CWD) is a fatal, progressive, neurodegenerative disease that has been reported in a number of cervid species including Rocky Mountain elk (Cervus elaphus nelsoni) and white-tailed deer (Odocoileus virginianus). Chronic wasting disease belongs to a group of diseases known as the transmissible spongiform encephalopathies (TSEs), or prion diseases, that are a group of neurodegenerative diseases in which a key feature is the accumulation of disease-associated prion protein (PrPSc) in the the brain.
The prion protein gene (PRNP) of Rocky Mountain elk encodes for either methionine (M) or leucine (L) at codon 1321,2. The amino acid expressed at codon 132 greatly affects incubation periods in elk after experimental oral inoculation with the chronic wasting disease agent: elk expressing prion protein homozygous for leucine at codon 132 (hereafter referred to as LL132 elk) incubate approximately 1.5 times longer than LM132 elk and 3 times longer than MM132 elk3,4. We recently demonstrated that these variations in incubation period may be influenced by genotype-associated differences in the relative amount of PrPSc in the brain and biochemical properties of PrPSc, including fibril stability and amyloid formation rate5.
TSE strains may be differentiated by host range, incubation period, clinical presentation, patterns of immunoreactivity or microscopic lesions in the brain, or the physiochemical properties of the PrPSc itself (reviewed in6). Prions lack genomic DNA to propagate strain information, and the strain properties are believed to be enciphered by the structure of the PrPSc conformer7,8,9,10,11. Two different strains of CWD have been identified when CWD from white-tailed deer was passaged into Syrian hamsters12, ferrets13, and transgenic mice expressing cervid PrP14,15. Natural CWD isolates from elk may contain either one12,16,17 or two14 strains.
To further explore the diversity of CWD strains in elk of different genotypes, brain homogenates from experimentally CWD-infected elk of the MM132, LM132 and LL132 genotypes were inoculated into transgenic mice expressing M132 prion protein18. Using this mouse model, we demonstrate that biological properties and biochemical characteristics of PrPSc in mice inoculated with brain homogenate from MM132 and LL132 elk are distinct from each other and similar to that in homologous donor elk. These differences are maintained on second passage suggesting that CWD-infected 132LL elk propagate a novel CWD strain that is distinct from the classic CWD strain of 132MM and 132 ML elk.
Results
To investigate the properties of PrPSc from experimentally infected elk of different PRNP genotypes, brain material from CWD-infected elk was bioassayed in Tg12 mice that express the M132 elk prion protein. When presenting bioassay results, the PrPSc label applied to mice (e.g. MM, LM, LL) refers to the prion protein genotype of the original donor elk, not the mouse itself.
Incubation periods
On first passage, mean incubation periods for MM-PrPSc (from MM132 elk) and LM-PrPSc (from LM132 elk) in Tg12 mice (referred to as MM-P1 mice or LM-P1 mice) were 174 and 164 days post-inoculation (dpi), respectively. The mean incubation time for LL-PrPSc (from LL132 elk) in Tg12 mice (termed LL-P1) was significantly longer (241 dpi, Log-rank test p < 0.0001) (Table 1, Fig. 1). On second passage, mean incubation periods (263 dpi) for LM-PrPSc in Tg12 mice (LM-P2a mice) were 20 days shorter than LL-PrPSc in Tg12 mice (LL-P2a mice; 283 dpi, Log-rank test p = 0.0387) and 101 days longer than MM-PrPSc (MM-P2 mice; 162 dpi, Log-rank test p < 0.0001) (Table 1, Fig. 1).

Survival curves for first (a) and second (b) passage of the agent of chronic wasting disease from elk of different prion protein genotypes. All transgenic mice express M132 elk prion protein. In mouse group names LL/LM/MM indicates the prion protein genotype of the elk donor for the first passage study i.e. LL132, LM132 and MM132 respectively.
The results of the LM-P2a bioassay were unexpected since generally incubation periods for second passage are similar to or shorter than first passage. On revisiting the first passage data, it was discovered that the mouse from the LM-P1 group that had been used as a donor for the LM-P2a study had a low EIA optical density (OD) result (OD = 1.8). The low EIA OD result indicates a relatively lower amount of PrPSc in the brain that we hypothesized could be the cause of the unexpectedly longer mean incubation period for the LM-P2a group. Therefore, we determined the amount of PrPSc in the brain homogenate from each donor mouse using EIA (Table 1). For studies that the donor mouse of the original passage had a low EIA OD (LL-P2a and LM-P2a), additional studies were performed using donors with OD = 4.0 (LL-P2b and LM-P2b) that is the highest measurable value in this test. In both additional studies the mean incubation periods were shorter than the first passage studies (Log-rank test, both p < 0.0001) (Table 1). Therefore, the LM-P2b and LL-P2b data are more comparable to that of MM-P2.
Amount of PrPSc versus incubation period in Tg12 mice
We observed a correlation between group mean incubation period and the amount of PrPSc present in the donor inoculum. Higher amounts of PrPSc in the inoculum was associated with shorter incubation periods and vice versa (Fig. S1). The relative amount of PrPSc in elk brain homogenate used to inoculate mice in the first passage studies was approximately 10 fold higher (Fig. S1a) than that from Tg12 mouse donors used to inoculate mice in the second passage studies (Fig. S1b).
Western blotting
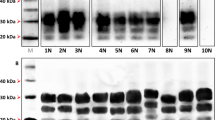
Western blot migration patterns were consistent across first and second passages of each elk inoculum (Fig. 2). No differences in western blot migration pattern were observed between LL-P2a and LL-P2b groups, or LM-P2a and LM-P2b groups (data not shown). PrPSc migration patterns for LM and MM mice were similar with the lower (unglycosylated) band migrating at approximately 20 kDa. PrPSc from LL mice ran somewhat lower, with the lower band at approximately 18 kDa. The relative positions of the lower bands in LL and MM/LM mice are consistent with the differences in banding pattern observed between LL132 and MM/LM132 elk3.

Western blot analysis of brain homogenates from mice challenged with brain homogenate from elk with chronic wasting disease. Lanes 1 and 11, markers. All mice express M132 elk prion protein. LL/LM/MM indicates the prion protein genotype of the elk donor for the first passage study i.e. LL132, LM132 and MM132 respectively. P1, first passage (elk to mouse); P2, second passage (mouse to mouse). Anti-prion protein antibody 6H4.
Histopathology
To investigate whether the inoculum source influenced brain pathology, we scored the severity of spongiform change in defined grey and white matter neuroanatomical areas for each mouse and calculated mean vacuolation scores for each group of mice to create lesion profiles.
Lesion profiles for all groups were broadly similar for all inoculation groups, although some minor differences were observed (data not shown). Amongst first passage groups, the severity of vacuolation in the neocortex at the level of the thalamus was somewhat lower in mice inoculated with LL132 elk brain homogenate. Amongst second passage groups, the overall severity of vacuolation was greatest in MM-P2 mice, particularly in the medulla, cerebellum, hippocampus and interpeduncular nucleus. Compared to other second passage groups, LM-P2a mice showed less severe vacuolation in the neocortex at the level of the thalamus and caudate putamen.
Although the overall vacuolation scores for the hippocampus were similar for all inoculation groups, differences were observed in the distribution of vacuolation within the hippocampus (Fig. 3a–c). To investigate this further, the severity of vacuolation in 3 hippocampal regions (dentate gyrus, CA3 and CA1) was scored using the same 0–5 scale as for vacuolation lesion profiling in other brain areas. In LL-PrPSc mice, vacuolation was more severe in the CA1 area and less severe in the dentate gyrus (Fig. 3a,g). This pattern was reversed in LM-PrPSc and MM-PrPSc mice such that vacuolation was more severe in the dentate gyrus and less severe in the CA1 area (Fig. 3b,c,h). Subjectively, the severity of vacuolation in the CA1 area was higher in LL-PrPSc mice as compared to LM-PrPSc and MM-PrPSc mice. However, these differences were only statistically significant for the LL-P2b group (ANOVA Kruskal-Wallis, p < 0.04). The severity of vacuolation in the CA3 area was similar across all groups (ANOVA Kruskal-Wallis, all p > 0.8). These results were maintained across first and second passage.

Microscopic changes in the hippocampus (a–c, g,h) and cerebellum (d–f, i) of cervidized mice inoculated with brain material from LL132 (a,d), LM132 (b,e) or MM132 (c,f) CWD-affected elk. Graphs depict the severity of vacuolation in the CA1 area (g) and dentate gyrus (h) of the hippocampus, and magnitude of reduction of granule cells in the cerebellum (i). Error bars reflect the SEM (standard error of the mean). Statistically significant differences are indicated by column labels that do not share the same letters. All mice express M132 elk prion protein; LL/LM/MM indicates prion protein genotype of the elk donors for the first passage study i.e. LL132, LM132 and MM132 respectively; P1, first passage (elk to mouse); P2, second passage (mouse to mouse). All photomicrographs original magnification 10×.
While performing vacuolation lesion profiling, neuronal loss in the granular layer of the cerebellar cortex was noted in some animals. To investigate this further, the severity of granule cell reduction was scored on a scale of 0–4 to reflect the surface area affected: 0, normal cellularity; score 1, 2, 3, 4, reduced cellularity in 1–25%, 26–50%, 51–75% and 76–100% of the section, respectively. Loss of granule cells was absent to mild (mean score = 0.6) in LL-PrPSc mice (Fig. 3d,i) and moderate to severe in LM-PrPSc (mean score = 3.4) and MM-PrPSc (mean score = 2.9) mice (Fig. 3e,f and i). Loss of granule cells was less severe in MM-P1 mice (mean score = 2.0) compared to MM-P2 mice (mean score = 3.7) (Fig. 3i). The Purkinje cells and cells in the molecular layer were normal.
Immunohistopathology
PrPSc immunoreactivity was present throughout the brain of all mice examined. The pattern of PrPSc deposition in the brain was similar for MM-PrPSc (MM-P1, MM-P2) and LM-PrPSc (LM-P1, LM-P2a, LM-P2b) mice and different for LL-PrPSc (LL-P1, LL-P2a, LL-P2b) mice (Fig. 4) and these patterns were consistent across first and second passage.

Patterns of PrPSc immunoreactivity in brains from mice from the second passage of CWD-affected elk brain homogenate from LL132 (a), LM132 (b) and MM132 (c) elk. In LL mice (a) immunoreactivity is present as diffuse immunolabeling of the neuropil while in LM (b) and MM (c) mice there are widespread medium to large aggregates in addition to diffuse immunolabeling of the neuropil. PrPSc detected with mAb 6H4.
In LL-PrPSc mice (Fig. 4a), immunoreactivity was present as diffuse immunolabeling of the neuropil with scattered aggregates in the molecular layer, and occasionally the granular cell layer, of the cerebellum. In the hippocampus, immunoreactivity was more severe in the CA1 and CA3 areas and less severe in the dentate gyrus.
In MM-PrPSc (Fig. 4b) and LM-PrPSc (Fig. 4c) mice, the PrPSc deposition pattern was characterized by widespread medium to large aggregates and diffuse immunolabeling of the neuropil. In the cerebellum, immunoreactivity was intense in the granular cell layer and mild to moderate in the molecular layer. In the hippocampus, immunoreactivity was moderate to severe in the dentate gyrus and CA3 area, and mild to moderate in the CA1 area. Moderate to intense immunoreactivity was present in the ependymal cells of the mesencephalic aqueduct and third ventricle.
Fibril stability of PrPSc from Tg12 mice
The fibril stability of PrPSc in homogenized brain from selected mice from each inoculum x passage combination was measured using an EIA-based stability assay. This assay is based on the use of the IDEXX HerdChek EIA platform to recognize misfolded protein; as a result, PK digestion is not involved, and the assay can detect both PK-sensitive and PK-resistant PrPSc populations (therefore retaining a broader pool of PrPSc for analysis). For samples from the first passage of elk brain into the mice, the LL-P1 mice exhibited a large increase in signal between 0.25 and 1 M GdnHCl conditions, and a relatively low absolute signal at the 0.25 M GdnHCl condition. This likely reflects increased binding of PrPSc to the capture surface or increased epitope exposure over the GdnHCl range in question, either of which would result an increased signal in the assay. It is worth noting that this may also influence determination of EIA signal in addition to differences in total PrPSc concentration in brain tissue. We address this in the determination of fibril stability by analyzing the data in two ways: using normalization of the EIA signal to the 0.25 M point (Fig. 5a,c), and using normalization to the 1 M point (Fig. 5b,d).

Fibril stability of PrPSc from transgenic mice expressing elk M132 prion protein. (a,b) Stability of PrPSc from first passage mice, normalized to either the 0.25 M data point (a) or the 1 M data point (b). Curves are averaged across 4 (MM, LL groups) or 5 (LM group) mice per group with the curve for each mouse reflecting the mean across technical replicates. (c,d) Stability of PrPSc from second passage mice, normalized to either the 0.25 M data point (c) or the 1 M data point (d). Curves are averaged across 5 (LM-P2a, MM-P2 groups), 6 (LM-P2b), or 8 (LL-P2b) mice, with the curve for each mouse reflecting the mean across technical replicates. [GdnHCl]1/2 = Fraction of PrPSc remaining (0.5). In each case, error bars reflect the SEM (standard error of the mean). (e,f) Comparison of [GdnHCl]1/2 values of PrPSc across first and second passage mice, normalized to either the 0.25 M data point (e) or the 1 M data point (f), as displayed in Table 2. LM values represent those collected from LM-P2b mice. Letters were added to correspond to the assessment of statistical significance described in Table 2.
On first passage, the curves (Fig. 5a,b) and [GdnHCl]1/2 values (Table 2) were similar for the MM-P1 and the LM-P1 mice, and distinct for the LL-P1 mice. [GdnHCl]1/2 values were significantly different between the MM-PrPSc and LL-PrPSc mice (p = 0.0007 in an unpaired t-test assuming unequal variances) (Fig. 5e,f). Therefore, the relative stability of PrPSc from MM132 versus LL132 elk brain was propagated on first passage in transgenic M132 elk mice.
Next, we examined the fibril stability of PrPSc in the second passage mouse brains, using analysis with 0.25 M (Fig. 5c) and 1 M (Fig. 5d) normalization. Due to the relatively low starting signals in the LL-P2a brains, only the LL-P2b samples were used to assess LL-P2 stability. The resulting [GdnHCl]1/2 values (Table 2) demonstrate that upon the second passage into transgenic M132 mice, the fibril stability profiles of the mice converge (Fig. 5c–f). This suggests that although the PK cleavage properties of PrPSc are maintained across first and second passage (Fig. 2), the fibril stability of PrPSc undergoes conformational changes on second passage resulting in convergence of stability curves across all experimental groups, despite the retention of the western blotting pattern. The LM-P2b fibril stability was lower than observed for the MM-P2 group that could correlate with the faster incubation time observed for this group.
Real time quaking-induced conversion
To determine if RT-QuIC can be used to detect PrPSc in CWD infected mouse brains and to investigate potential differences in prion seeding activity, RT-QuIC reactions using mature length recombinant 132 L or 132 M elk prion protein substrates were seeded with CWD infected and non-infected mouse brain homogenates.
Seed prepared from brains from first passage mice showed an increase in ThT fluorescence in both substrates (132 L or 132 M). Normal brain homogenates did not result in an increase in ThT fluorescence. Using 132 L substrate (Fig. 6a), the lag time for reactions seeded with LL-P1 mouse brain (18 hours) was shorter than that for LM-P1 (24 hours) and MM-P1 (30 hours) samples. Using 132 M substrate (Fig. 6b), assays seeded with MM-P1 mouse brain homogenate produced the shortest lag time (10 hours) while lag times for LM-P1 and LL-P1 samples were longer (both 15 hours).

RT-QuIC detection of seeding activity in brain samples from transgenic mice expressing M132 elk prion protein, first passage (a,b) and second passage (c,d). Substrate is full-length recombinant elk prion protein 132 L (a,c) or 132 M (b,d). Dashed horizontal line indicates the positive threshold above which a sample is considered positive. (a,b) Mice were inoculated with brain homogenate from CWD-affected elk of different genotypes: MM132 (green), LM132 (red), LL132 (blue); negative seed (purple). (c,d) Mice were inoculated with brain homogenate from first passage mice that were inoculated with brain homogenate from CWD-affected elk of different genotypes: MM132 (yellow, black), LM132 (green, purple), LL132 (blue, red); negative seed (tan). Each curve represents the mean ThT fluorescence of eight technical replicates for one mouse. RT-QuIC reaction mixtures were seeded with 10−3 dilutions of normalized brain homogenate. A final concentration of 300 mM NaCl was used with each substrate. The positive threshold was calculated as ~7,000 relative fluorescence units of brain homogenate from normal mice.
When RT-QuIC reactions were seeded with brain homogenate from second passage mice, samples from all mice showed an increase in ThT fluorescence in both substrates (132 L or 132 M). Using 132 L substrate (Fig. 6c), LL-P2a samples resulted in shorter lag times (12 and 15 hour) and higher fibril formation compared to LM-P2a (20 and 24 hours) and MM-P2 samples (25 and 27 hours). Using the 132 M substrate (Fig. 6d), the lag times for all mice were similar.
Discussion
In this study, we demonstrate that intracranial inoculation of transgenic mice expressing M132 elk prion protein (Tg12) with brain homogenate from elk of different PRNP genotypes that were experimentally infected with CWD results in clinical disease and accumulation of misfolded prion protein (PrPSc). Compared to mice inoculated with MM132 elk brain homogenate, LL132 homogenate results in disease with a longer incubation time, lower migration pattern on western blot, more stable PrPSc fibrils as determined by an EIA-based stability assay, and fewer PrPSc aggregates in the brain as observed by immunohistochemistry (IHC). The disease phenotype in mice inoculated with LM132 elk brain homogenate is largely similar to that in mice inoculated with MM132 elk brain homogenate. Therefore, the phenotype of disease in mice appears to be strongly influenced by the PRNP genotype of the elk donor, although other factors, for example the amount of PrPSc in the donor inoculum, also play a role.
We have previously reported PRNP genotype-driven differences in disease expression in elk experimentally inoculated with the agent of CWD3,4,5. Compared to MM132 elk, disease in LL132 elk is characterized by prolonged incubation periods, lower migration pattern on western blot, higher fibril stability, and differences in the pattern of PrPSc accumulation in the brain3,5. With the exception of fibril stability, these phenotypic features were maintained when brain homogenate from CWD-infected elk was bioassayed in Tg12 mice, suggesting that the LL132 isolate has novel CWD strain characteristics.
All three donor elk for the first passage mouse studies were inoculated with brain homogenate prepared from pooled brain material from one MM132 and one LM132 elk (equal parts MM132 and LM132 donor tissue)5 . Therefore it is possible that a mixture of prion strains was propagated in the donor elk. Although the sample size was small, the disease phenotype was consistent across multiple animals of the same PRNP genotype which makes the de novo emergence of a prion strain, as described previously for white-tailed deer, unlikely15. End-point titration of the all three elk donor inocula in Tg12 mice could help to tease out individual strains from a mixture.
Similar patterns of PrPSc immunoreactivity and microscopic lesions were observed in the brains of Tg12 mice inoculated with brain homogenate from CWD-infected elk of the MM132 or LM132 genotypes (hereafter, referred to as MM-PrPSc and LM-PrPSc mice, respectively), while a different pattern was observed in the brains of mice inoculated with LL132 elk brain homogenate (LL-PrPSc mice). These results are consistent with genotype-associated differences in pathology seen in donor elk5. For example, large aggregates and plaque-like PrPSc accumulations were observed throughout the brain of MM-PrPSc and LM-PrPSc mice, while immunoreactivity in LL-PrPSc mice was mostly diffuse. In donor elk, PrPSc accumulation in the brains of MM132 and LM132 elk was primarily neuropil-associated, while in LL132 elk, there was prominent PrPSc immunoreactivity associated with neurons and astrocytes4,5. Therefore, although the morphologic types of PrPSc immunolabeling are different in mice and elk, the PRNP genotype-associated grouping of PrPSc deposition patterns – that is, similar patterns in MM-PrPSc and LM-PrPSc mice and a different pattern in LL-PrPSc mice – are consistent across both elk and Tg12 mice.
Using standardized vacuolation lesion profiling techniques19, we were not able to detect any significant differences between mouse groups. However, in the hippocampus, spongiform change was more severe in the dentate gyrus in MM-PrPSc and LM-PrPSc mice, while in LL-PrPSc mice, the CA1 area was more affected. A second microscopic change was observed in the cerebellum, where there was a marked reduction in the number of granule cells in MM-PrPSc and LM-PrPSc mice, while this change was mild to absent in LL-PrPSc mice. These results indicate that prion isolates from elk of different genotypes target and cause pathology in different populations of cells in the brain.
In donor elk, similarities in disease expression in MM132 and LM132 elk are thought to be due to a high proportion of the PrPSc in LM132 elk being M1325. This hypothesis is extrapolated from studies in scrapie-affected PRNP heterozygous sheep demonstrating that although expression of the host cellular form of the prion protein (PrPC) is equal for each PRNP allele, conversion of PrPC to the disease-associated form (PrPSc) is more efficient for the PrPC moiety that confers a greater susceptibility to disease20,21. Since Tg12 mice express PrPC-132M, patterns of disease expression on first passage are likely driven by sequence similarities or differences between the PrPSc in the elk donor inoculum and the PrPC in the Tg12 mice22. At second passage, all donor mice are Tg12, so all donor PrPSc is M132. Therefore, patterns of disease expression on second passage are probably associated with sequence-independent differences in PrPSc conformation7,8,9,10,11.
The relationship between PrPSc fibril stability in guanidinium hydrochloride (GdnHCl) and incubation time varies between host species and prions strains23,24,25,26,27,28,29,30. In CWD-affected elk, longer incubation periods were associated with higher fibril stability and fibril stabilities of PrPSc from MM132 and LM132 elk were similar5. These relationships were maintained on first passage (P1) of elk brain homogenate in Tg12 mice and indicate that the M132 elk prion protein (as expressed in mice) can adopt the higher stability PrPSc conformation. The relative fibril stabilities in our first passage mice are in contrast to those reported for first passage of elk CWD isolates in Tg(CerPrP-M132)1536+/− mice, where fibrils from mice inoculated with LM132 elk brain homogenate were more stable and fibrils from mice inoculated with MM132 or LL132 were less stable and similar to each other17. Another point of difference is in the relationship between PrPSc fibril stability and the morphology of PrPSc accumulation in the brain. Previously, studies in mice using mouse-adapted prion strains reported an association between higher PrPSc fibril stabilities and aggregated and plaque-like PrPSc accumulations, while isolates with lower fibril stabilities produce diffuse PrPSc deposits25,27. These results are in contrast to our findings where higher fibril stability of PrPSc from mice inoculated with CWD prions from LL132 elk brain is associated with diffuse PrPSc accumulation. The discrepancy between these results and our findings could be due to prion isolate and/or recipient mouse line related factors.
On second passage (P2) there was a significant reduction in the fibril stability of PrPSc from LL-P2 mice as compared to LL-P1 mice such that the fibril stability was similar to MM-P2 mice. In spite of this, the western blot migration patterns for LL-P1 and LL-P2 mice were identical which indicates that the conformational changes leading to changes in fibril stability are independent of PK cleavage properties. This convergence in fibril stability properties is likely due to adaptation in the Tg12 mouse model and/or prion strain selection.
For LL-P2a and MM-P2 mice the relationship between elk donor PRNP genotype and incubation period was similar across first and second passages. However, variability was observed between the LL-P2a and LL-P2b groups, and the LM-P2a and LM-P2b groups.
For the LL-P2a and LL-P2b groups, amyloid formation rates, western blot profiles, neuropathology and fibril stability results were similar. Therefore the prolonged mean incubation period of the LL-P2a group compared to the LL-P2b group can be attributed to a lower PrPSc titer in the LL-P2a inoculum as indicated by a low EIA optical density (OD) result.
The explanation for the differences observed between the LM-P2a and LM-P2b groups is less clear. Mice from both groups had similar amyloid formation rates, western blot profiles and neuropathology results and, consistent with the situation in the LL-P2 groups, the lower PrPSc titer of the LM-P2a inoculum likely contributed to the longer mean incubation periods observed in this group. However, the fibril stability of PrPSc from mice in the LM-P2a group was higher than both the LL-P2 and MM-P2 groups, while the fibril stability of PrPSc from mice in the LM-P2b group was lower.
When Tg(CerPrP-M132)1536+/− mice were inoculated with brain homogenates from CWD-affected MM132 elk, different patterns of neuropathology were observed in mice that developed disease early in the incubation period (up to 250 days post-inoculation) as compared to mice that developed disease later (250–400 days post-inoculation), although all mice in the study had similar western blot migration patterns and glycoform ratios14. The incubation period of the donor mouse for the LM-P2a study was approximately 50% of the mean incubation period for the LM-P1 group, so perhaps the prion strain isolated from this mouse was a short incubation period CWD strain with different biological characteristics, in particular fibril stability, compared to long incubation period strain(s) present in mice that developed disease later. Furthermore, it is probable that the original LM132 elk brain homogenate contained a mix of PrPSc conformations (PrPSc-132M and PrPSc-132L) that may have been differentially propagated on second passage. Unfortunately, the brain of the mouse selected for passage was not fixed for microscopic examination and the PrPSc titer was too low to perform the fibril stability assay, but further experiments are underway to investigate whether these apparent strain characteristics are maintained on subsequent passages.
We also utilized RT-QuIC to investigate the influence of donor elk genotype on PrPSc characteristics in first and second passage in transgenic mice expressing M132 elk prion protein. Using recombinant mature length elk prion protein (132 L and 132 M) as substrates, seed prepared from brains from mice inoculated with MM132 elk inoculum converted 132 M substrate more readily compared to LM132 or LL132 seeds. In contrast, the 132 L substrate was converted most efficiently by seed prepared from brains from mice inoculated with LL132 elk inoculum. Seed homogenates were normalized prior to testing so the observed differences in conversion efficiencies are not due to variation in the amount of PrPSc in the seed. These patterns of conversion efficiencies in first passage mice are similar to those observed in donor elk5.
On second passage, results using 132 L substrate were similar to first passage results. However, using 132 M substrate, lag times for PrPSc from mice from all challenge groups (MM-P2, LM-P2, LL-P2) were similar. Transgenic Tg12 mice express M132 cellular prion protein (PrPC), so at second passage, both donor mouse PrPSc and recipient mouse PrPC are homologous. This could be a possible explanation for the convergence of lag times for samples from all challenge groups when the 132 M substrate was used. However, the fact that elk donor genotype-associated differences in lag time are still observed when 132 L substrate is used, and that western blot profiles for LL-PrPSc mice are consistent across first and second passages and different from LM-PrPSc and MM-PrPSc mice, suggests that the M132 PrPSc in LL-P2 mice still maintains distinct biochemical characteristics compared to M132 PrPSc from LM-P2 and MM-P2 mice. Bioassay of elk brain homogenate in mice expressing L132 PrPSc would help to clarify the role of the L132 polymorphism in selection and emergence of prion strains in elk.
The current study lends evidence to the fact that there are multiple distinct strains of the CWD agent in elk. It cannot be entirely ruled out that the inoculum used in the original elk transmission study contained multiple strains. The inoculum used for the oral dosing was comprised of pooled material from two clinically ill elk: one elk of each the MM132 and ML132 genotypes. The infectous material came from a single premises and western blotting of the inoculum resulted in a single migration pattern. However, further analysis of the inoculum was not performed at the time of the original study and none of this inoculum is currently available for testing. Since results from the original transmission study in elk indicate that biochemical properties of of PrPSc from MM132 and LM132 elk are similar5, it seems likely that the second strain, if present in the original inoculum, was present as a minor component. Although two strains of the CWD agent have been previously described in naturally infected elk14, the lower western blot migration pattern of the LL132 strain described in the experimentally challenged elk reported here clearly distinguishes it from previously described strains. Thus it would appear that elk are able to amplify a total of at least three strains of CWD.
In previous studies of naturally occurring CWD in elk, one study31 reported similar western blot migration patterns for MM132 and LL132 elk and a different pattern for LM132 elk, while in another study no genotype-associated differences were observed between MM132, LM132 and LL132 elk17. Given this variation in western blot migration pattern in naturally infected elk it is not possible to be certain whether the lower migration pattern observed in our donor elk is an additional naturally occurring variant or represents an experimental epiphenomenon related to e.g. experimental inoculation with a high dose of pooled CWD infectivity. Bioassay of additional CWD isolates from naturally infected LL132 elk may help to improve our understanding of naturally occurring LL132 elk CWD strains.
To summarize, we have demonstrated that inoculation of mice expressing elk prion protein with brain homogenate from CWD-affected elk produces disease with different biological and biochemical characteristics depending on elk donor PRNP genotype. The phenotype of disease in mice inoculated with brain homogenate from MM132, LM132 and LL132 elk is similar to that observed in donor elk with regards to relative incubation period, PrPSc fibril stability, and western blot migration patterns. On second passage we observed a convergence in incubation time, fibril stability, and amyloid formation in 132 M substrate for MM and LL groups, presumably due to prion strain selection by the Tg12 mouse model. However, differences in western blot profiles and neuropathology were maintained across first and second passages indicating that CWD from experimentally inoculated MM132 and LL132 elk represent distinct strains of CWD. Second passage of the LM132 isolate resulted in two distinct fibril stability profiles. These results provide further evidence of the existence of multiple strains of CWD in elk. If the novel LL132 strain that we have isolated from experimentally infected elk is recapitulated in naturally elk, the presence of this third strain of CWD would have the potential to complicate CWD management strategies in free-ranging and captive populations.
Methods
Ethics statement
This experiment was carried out in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Academy of Sciences, Washington, DC). The protocol was approved by the Institutional Animal Care and Use Committee at the National Animal Disease Center (protocol numbers: ARS 2729 and ARS-2017-628).
Inoculum preparation and animal procedures
Infectivity in brain tissue from selected elk was assayed via intracranial inoculation of Tg(ElkPrP-132M)Prnp0/0 mice (hereafter referred to as Tg12 mice) that express M132 elk prion protein at approximately twofold the expression level of brain PrP in wild-type FVB mice18. CWD inoculum was prepared from the brains of experimentally challenged CWD-affected elk of the MM132, LM132, and LL132 genotypes. These elk had been challenged intracranially with pooled brain material from one MM132 and one LM132 elk, both of which had showed clinical signs of CWD4. The results of the elk challenge study have been reported previously3,4,5.
Mice were inoculated intracranially with 20 µL of 1% w/v brain homogenate from a single elk as described previously32. Mice were monitored daily and euthanized when they displayed unequivocal neurological signs (poor coordination, difficulty moving, unable to move, anorexia) or at the time of study termination (700 days post-inoculation). The incubation period was calculated as the number of days between inoculation and euthanasia.
Mouse brains were sectioned longitudinally, and 2/3 of the brain was fixed for microscopic examination and 1/3 was frozen at −80 C for biochemical assays. Tissue for microscopic examination were fixed in 10% buffered formalin, embedded in paraffin wax, sectioned at 5 μm, and stained with hematoxylin and eosin. Frozen tissue was tested for the presence of PrPSc using enzyme immunoassay and western blotting as described below.
For second passage, brain samples from mice were prepared as 10% w/v brain homogenates in phosphate buffered saline as described previously33.
Histopathology
For each mouse, the severity of spongiform change was semi-quantified on a scale of 0–5 or 0–3 for 9 gray matter areas and 3 white matter areas, respectively, as described previously19. Lesion profiles were obtained by two observers scoring 7–13 positive (by enzyme immunoassay, see below) mice per inoculation group.
Immunohistochemistry
For each mouse paraffin-embedded brain sections were immunostained manually for detection of PrPSc using the mouse anti-PrP monoclonal antibody 6C2 (CVI-WUR, Lelystad, Netherlands) as described previously34 with the following modifications: for antigen retrieval, rehydrated sections were incubated with 98% formic acid for 15 minutes and then autoclaved for 30 minutes in an antigen retrieval solution (DAKO Target Retrieval Solution, Dako Corp., Carpinteria, CA). Tissues were then blocked for 20 minutes at room temperature (Background Buster; Innovex, Lincoln, RI).
Western blotting
Western blotting was performed on homogenate prepared from frozen brain samples from 5–6 mice per group. Western blots were performed on PK-digested acetone-precipitated PrPSc samples. After centrifugation pellets were re-suspended in 1X LDS loading dye (Life Technologies) + 5% β-mercaptoethanol, boiled for 5 minutes, loaded onto a 12% NuPAGE Bis-Tris gel, and run in 1x MOPS Buffer (Life Technologies). Proteins were transferred to PVDF by tank blotting at 25 V for 45 minutes. The membrane was then preblocked for 30 minutes with TBST (Tris-Buffered Saline + 0.1% Tween-20) + 3% BSA (Bovine Serum Albumin) before the addition of anti-PrP mouse monoclonal antibodies 6H4 (Prionics) at a 1:10,000 dilution either for 1 hr at room temperature (RT) or overnight (O/N) at 4 C. A secondary biotinylated sheep anti-mouse antibody and an HRP-conjugated streptavidin incubation step were performed for 40–60 minutes each, each in TBST + 3% BSA, and blots were washed 3 times for 5 minutes each time between antibody and streptavidin steps. Blots were developed with ECL Plus (Pierce) and imaged by fluorescence on a GBOX (Synoptics, Cambridge, UK).
Antigen-capture enzyme immunoassay (EIA)
The IDEXX HerdChek BSE-Scrapie Ag EIA plate (Westbrook, ME) was used with modifications for the EIA-based fibril stability assay and the determination of PrPSc levels. The capture surface of the IDEXX EIA is a proprietary ligand that is specific for misfolded protein with detection of bound protein by a PrP-specific antibody, and does not require protease digestion to distinguish PrPC from PrPSc. Brain samples were prepared as 20% w/v homogenates in 1X PBS (phosphate-buffered saline, lacking calcium and magnesium) and homogenized in a beadbeater.
Calculation of amount of PrPSc versus incubation period
The relative amount of PrPSc in donor elk inocula and donor mouse brains was determined using an EIA-based assay as described previously5. Briefly, 1% w/v brain homogenates were serially diluted in 1X PBS and tested using the EIA assay and diluted until the OD450 readings were in the linear range of detection. We have previously determined the assay range across which a dilution series of infected brain homogenate approximates a linear function, which was utilized for concentration estimation and the fibril stability assay. To provide a normalization metric across multiple samples, the 1% (w/v) homogenate was assigned a brain unit equivalent (BU) value of 100 and equivalent BU’s were calculated for each dilution, i.e. 1:2 dilution = 50 BU, 1:4 dilution = 25 BU. For each sample, the EIA OD reading in the linear range (minus the negative control value) was divided by the BU of the dilution at which the linear range OD was measured, to generate an OD/BU value. We then calculated the ratio of the OD/BU values for each sample as compared to the sample with the lowest OD/BU value. Ratio values were plotted against incubation period.
EIA-based fibril stability assay
PrPSc fibril stability was determined using an EIA-based assay as described previously24. Briefly, dilutions of mouse brain samples were incubated at concentrations of guanidine hydrochloride (GdnHCl) over a range from 0.25 M to 4.0 M. Brain homogenate was diluted up to 8X into PBS in order to bring the final EIA optical density (OD450) in the ELISA well to 1.0 or below. A final concentration of brain homogenate of ≤ 2% (w/v) was used in all samples to avoid exceeding the buffering capacity, as was observed to occur at higher concentrations of brain homogenate23. The relative amount of PrPSc remaining, as assessed by the OD450 after dilution to 0.25 M GdnHCl, was plotted against GdnHCl concentration. For each inoculation group, the assay was applied to 4–8 mice with 3–8 technical replicates per mouse. The mean EIA OD values for each GdnHCl concentration were calculated across biological replicates and used to generate a curve. The midpoint of the curve, or [GdnHCl]1/2, is defined as the concentration of GdnHCl at which the PrPSc signal was reduced by half of the signal at 0.25 M GdnHCl; PrPSc with a smaller [GdnHCl]1/2 is less stable. As described previously23, due to variations in the upper baseline shape, the Smooth Line function in Microsoft Excel was used to connect data points in each curve (for each individual animal) and visualize the midpoint. In order to assess statistical differences, Welch’s ANOVA (accounting for unequal variances) was used, with the Games-Howell test for (non-parametric) post-hoc analysis. Analysis was conducted in two batches: (a) across values normalized to 0.25 M, and (b) across values normalized to 1 M.
Recombinant prion protein production and purification
E. coli BL21(λDE3) was transformed with a pET28a vector containing the elk PrP gene corresponding to mature length PrP (amino acids 23–231, GenBank accession number AAC12860.2), and elk recombinant PrP was expressed and purified as described for bovine PrP35,36. The concentration of filtered protein eluent was determined by UV absorbance at 280 nm using an extinction coefficient of 59485 M−1cm−1 as calculated for mature length elk prion protein.
Real-time quaking induced conversion (RT-QuIC)
Brain homogenate from one mouse from each of the first passage groups and two mice from each of the second passage groups was used as seeds for the RT-QuIC reactions. A 98 µL volume of RT-QuIC reaction mixture consisting of 10 mM phosphate buffer (pH 7.4), 300 mM NaCl, 0.1 mg/ml recombinant elk prion protein (132 L or 132 M), 10 µM thioflavin T (ThT), and 1 mM ethylenediaminetetraacetic acid tetrasodium salt (EDTA) was loaded into each well of a black 96-well plate with a clear bottom (Nunc, Thermo Fisher Scientific), and 2 µL of normalized brain homogenate dilution was added. The plate was then sealed and incubated in a BMG FLUOstar Omega plate reader at 42 °C for 100 h with cycles of 15 min shaking (700 rpm, double orbital pattern) and 15 min rest. All reactions for each sample were performed in 8 replicates. To be considered positive, the ThT fluorescence of at least two replicate reactions must be positive and above the pre-defined positive threshold that was calculated as 10 standard deviations above the mean fluorescence of normal elk brain homogenates37,38,39.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
O’Rourke, K. I. et al. Monoclonal antibody F89/160.1.5 defines a conserved epitope on the ruminant prion protein. J. Clin. Microbiol. 36, 1750–1755 (1998).
Schätzl, H. M., Da Costa, M., Taylor, L., Cohen, F. E. & Prusiner, S. B. Prion protein gene variation among primates. J. Mol. Biol. 265, 257, https://doi.org/10.1006/jmbi.1994.0030 (1997).
O’Rourke, K. et al. Elk with a long incubation prion disease phenotype have a unique PrPd profile. Cellular. NeuroReport 18, 1935–1938, https://doi.org/10.1097/WNR.0b013e3282f1ca2f (2007).
Hamir, A. N. et al. Preliminary observations of genetic susceptibility of elk (Cervus elaphus nelsoni) to chronic wasting disease by experimental oral inoculation. J. Vet. Diagn. Invest. 18, 110–114, https://doi.org/10.1177/104063870601800118 (2006).
Moore, S. J. et al. Pathologic and biochemical characterization of PrP(Sc) from elk with PRNP polymorphisms at codon 132 after experimental infection with the chronic wasting disease agent. BMC Vet. Res. 14, 80, https://doi.org/10.1186/s12917-018-1400-9 (2018).
Gough, K. C., Rees, H. C., Ives, S. E. & Maddison, B. C. Methods for Differentiating Prion Types in Food-Producing Animals. Biol. 4, 785–813, https://doi.org/10.3390/biology4040785 (2015).
Bessen, R. A. & Marsh, R. F. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 68, 7859–7868 (1994).
Telling, G. C. et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Sci. 274, 2079–2082, https://doi.org/10.1126/science.274.5295.2079 (1996).
Collinge, J., Sidle, K. C., Meads, J., Ironside, J. & Hill, A. F. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nat. 383, 685–690, https://doi.org/10.1038/383685a0 (1996).
Peretz, D. et al. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 10, 854–863, https://doi.org/10.1110/ps.39201 (2001).
Green, K. M. et al. Accelerated high fidelity prion amplification within and across prion species barriers. PLoS Pathog. 4, e1000139, https://doi.org/10.1371/journal.ppat.1000139 (2008).
Raymond, G. J. et al. Transmission and adaptation of chronic wasting disease to hamsters and transgenic mice: evidence for strains. J. Virol. 81, 4305–4314, https://doi.org/10.1128/JVI.02474-06 (2007).
Perrott, M. R., Sigurdson, C. J., Mason, G. L. & Hoover, E. A. Evidence for distinct chronic wasting disease (CWD) strains in experimental CWD in ferrets. J. Gen. Virol. 93, 212–221, https://doi.org/10.1099/vir.0.035006-0 (2012).
Angers, R. C. et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Sci. 328, 1154–1158, https://doi.org/10.1126/science.1187107 (2010).
Duque Velasquez, C. et al. Deer prion proteins modulate the emergence and adaptation of chronic wasting disease strains. J. Virol. 89, 12362–12373, https://doi.org/10.1128/JVI.02010-15 (2015).
Tamguney, G. et al. Transmission of elk and deer prions to transgenic mice. J. Virol. 80, 9104–9114, https://doi.org/10.1128/JVI.00098-06 (2006).
Green, K. M. et al. The elk PRNP codon 132 polymorphism controls cervid and scrapie prion propagation. J. Gen. Virol. 89, 598–608, https://doi.org/10.1099/vir.0.83168-0 (2008).
Kong, Q. et al. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J. Neurosci. 25, 7944–7949, https://doi.org/10.1523/JNEUROSCI.2467-05.2005 (2005).
Fraser, H. & Dickinson, A. G. The sequential development of the brain lesion of scrapie in three strains of mice. J. Comp. Pathol. 78, 301–311, https://doi.org/10.1016/0021-9975(68)90006-6 (1968).
Jacobs, J. G. et al. Proteinase K-resistant material in ARR/VRQ sheep brain affected with classical scrapie is composed mainly of VRQ prion protein. J. Virol. 85, 12537–12546, https://doi.org/10.1128/JVI.00448-11 (2011).
Morel, N., Andreoletti, O., Grassi, J. & Clement, G. Absolute and relative quantification of sheep brain prion protein (PrP) allelic variants by matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry. Rapid Commun. Mass. Spectrom. 21, 4093–4100, https://doi.org/10.1002/rcm.3317 (2007).
Prusiner, S. B. et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63, 673–686, https://doi.org/10.1016/0092-8674(90)90134-Z (1990).
Vrentas, C. E. et al. Stability properties of PrP(Sc) from cattle with experimental transmissible spongiform encephalopathies: use of a rapid whole homogenate, protease-free assay. BMC Vet. Res. 9, 167, https://doi.org/10.1186/1746-6148-9-167 (2013).
Vrentas, C. E., Greenlee, J. J., Tatum, T. L. & Nicholson, E. M. Relationships between PrPSc stability and incubation time for United States scrapie isolates in a natural host system. PLoS One 7, e43060, https://doi.org/10.1371/journal.pone.0043060 (2012).
Bett, C. et al. Biochemical properties of highly neuroinvasive prion strains. PLoS Pathog. 8, e1002522, https://doi.org/10.1371/journal.ppat.1002522 (2012).
Legname, G. et al. Strain-specified characteristics of mouse synthetic prions. Proc. Natl Acad. Sci. USA 102, 2168–2173, https://doi.org/10.1073/pnas.0409079102 (2005).
Legname, G. et al. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Natl Acad. Sci. USA 103, 19105–19110, https://doi.org/10.1073/pnas.0608970103 (2006).
Peretz, D. et al. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 34, 921–932, https://doi.org/10.1016/S0896-6273(02)00726-2 (2002).
Pirisinu, L. et al. A new method for the characterization of strain-specific conformational stability of protease-sensitive and protease-resistant PrP. PLoS One 5, e12723, https://doi.org/10.1371/journal.pone.0012723 (2010).
Wemheuer, W. M. et al. Similarities between forms of sheep scrapie and Creutzfeldt-Jakob disease are encoded by distinct prion types. Am. J. Pathol. 175, 2566–2573, https://doi.org/10.2353/ajpath.2009.090623 (2009).
Spraker, T. R., Balachandran, A., Zhuang, D. & O’Rourke, K. I. Variable patterns of distribution of PrP(CWD) in the obex and cranial lymphoid tissues of Rocky Mountain elk (Cervus elaphus nelsoni) with subclinical chronic wasting disease. Vet. Rec. 155, 295–302, https://doi.org/10.1136/vr.155.10.295 (2004).
Smith, J. D., Nicholson, E. M. & Greenlee, J. J. Evaluation of a combinatorial approach to prion inactivation using an oxidizing agent, SDS, and proteinase K. BMC Vet. Res. 9, 151, https://doi.org/10.1186/1746-6148-9-151 (2013).
Smith, J. D., Nicholson, E. M., Foster, G. H. & Greenlee, J. J. Exposure of RML scrapie agent to a sodium percarbonate-based product and sodium dodecyl sulfate renders PrPSc protease sensitive but does not eliminate infectivity. BMC Vet. Res. 9, 8, https://doi.org/10.1186/1746-6148-9-8 (2013).
West Greenlee, M. H. et al. Temporal resolution of misfolded prion protein transport, accumulation, glial activation, and neuronal death in the retinas of mice inoculated with scrapie. Am. J. Pathol. 186, 2302–2309, https://doi.org/10.1016/j.ajpath.2016.05.018 (2016).
Vrentas, C. E., Onstot, S. & Nicholson, E. M. A comparative analysis of rapid methods for purification and refolding of recombinant bovine prion protein. Protein Expr. Purif. 82, 380–388, https://doi.org/10.1016/j.pep.2012.02.008 (2012).
Hwang, S., Greenlee, J. J. & Nicholson, E. M. Use of bovine recombinant prion protein and real-time quaking-induced conversion to detect cattle transmissible mink encephalopathy prions and discriminate classical and atypical L- and H-Type bovine spongiform encephalopathy. PLoS One 12, e0172391, https://doi.org/10.1371/journal.pone.0172391 (2017).
Orru, C. D. et al. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio 6, https://doi.org/10.1128/mBio.02451-14 (2015).
Dassanayake, R. P. et al. Sensitive and specific detection of classical scrapie prions in the brains of goats by real-time quaking-induced conversion. J. Gen. Virol. 97, 803–812, https://doi.org/10.1099/jgv.0.000367 (2016).
Orru, C. D. et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N. Engl. J. Med. 371, 519–529, https://doi.org/10.1056/NEJMoa1315200 (2014).
Acknowledgements
The authors would like to thank Robyn Kokemuller for assistance with vacuolation lesion profiling and Joe Lesan, Leisa Mandell, Kevin Hassall and Semakelang Lebepe-Mazur for excellent technical support. This study was carried out under the guidelines of the institutional ACUC committee at NADC. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity employer.
Author information
Authors and Affiliations
Contributions
J.M., M.H.W.G. and J.G. conceived the experiment(s), Q.K. provided Tg12 mice. J.M., T.T., S.H., and J.G. conducted the experiment(s), J.M., S.H., C.V., E.N. and J.G. analysed the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moore, J., Tatum, T., Hwang, S. et al. Novel Strain of the Chronic Wasting Disease Agent Isolated From Experimentally Inoculated Elk With LL132 Prion Protein. Sci Rep 10, 3148 (2020). https://doi.org/10.1038/s41598-020-59819-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59819-1
This article is cited by
-
Identification of a novel risk factor for chronic wasting disease (CWD) in elk: S100G single nucleotide polymorphism (SNP) of the prion protein gene (PRNP)
Veterinary Research (2023)
-
Propagation of PrPSc in mice reveals impact of aggregate composition on prion disease pathogenesis
Communications Biology (2023)
-
Emergence of CWD strains
Cell and Tissue Research (2023)
-
Review on PRNP genetics and susceptibility to chronic wasting disease of Cervidae
Veterinary Research (2021)
-
Environmental and host factors that contribute to prion strain evolution
Acta Neuropathologica (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
