 Chao Fang1†
Chao Fang1† John P. Hamilton2,3†
John P. Hamilton2,3† Brieanne Vaillancourt2
Brieanne Vaillancourt2 Yi-Wen Wang2Joshua C. Wood2Natalie C. Deans2Taylor Scroggs4
Yi-Wen Wang2Joshua C. Wood2Natalie C. Deans2Taylor Scroggs4 Lemor Carlton2Kathrine Mailloux2
Lemor Carlton2Kathrine Mailloux2 David S. Douches5
David S. Douches5 Satya Swathi Nadakuduti6,7
Satya Swathi Nadakuduti6,7 Jiming Jiang1,8*
Jiming Jiang1,8* C. Robin Buell2,3,9*
C. Robin Buell2,3,9*- 1Department of Plant Biology, Michigan State University, East Lansing, MI, United States
- 2Center for Applied Genetic Technologies, University of Georgia, Athens, GA, United States
- 3Department of Crop & Soil Sciences, University of Georgia, Athens, GA, United States
- 4Department of Genetics, University of Georgia, Athens, GA, United States
- 5Department of Plant, Soil & Microbial Sciences, Michigan State University, East Lansing, MI, United States
- 6Department of Environmental Horticulture, University of Florida, Gainesville, FL, United States
- 7Plant Molecular and Cellular Biology Program, University of Florida, Gainesville, FL, United States
- 8Department of Horticulture, Michigan State University, East Lansing, MI, United States
- 9Institute of Plant Breeding, Genetics & Genomics, University of Georgia, Athens, GA, United States
Camelina sativa (L.) Crantz, a member of the Brassicaceae, has potential as a biofuel feedstock which is attributable to the production of fatty acids in its seeds, its fast growth cycle, and low input requirements. While a genome assembly is available for camelina, it was generated from short sequence reads and is thus highly fragmented in nature. Using long read sequences, we generated a chromosome-scale, highly contiguous genome assembly (644,491,969 bp) for the spring biotype cultivar ‘Suneson’ with an N50 contig length of 12,031,512 bp and a scaffold N50 length of 32,184,682 bp. Annotation of protein-coding genes revealed 91,877 genes that encode 133,355 gene models. We identified a total of 4,467 genes that were significantly up-regulated under cold stress which were enriched in gene ontology terms associated with “response to cold” and “response to abiotic stress”. Coexpression analyses revealed multiple coexpression modules that were enriched in genes differentially expressed following cold stress that had putative functions involved in stress adaptation, specifically within the plastid. With access to a highly contiguous genome assembly, comparative analyses with Arabidopsis thaliana revealed 23,625 A. thaliana genes syntenic with 45,453 Suneson genes. Of these, 24,960 Suneson genes were syntenic to 8,320 A. thaliana genes reflecting a 3 camelina homeolog to 1 Arabidopsis gene relationship and retention of all three homeologs. Some of the retained triplicated homeologs showed conserved gene expression patterns under control and cold-stressed conditions whereas other triplicated homeologs displayed diverged expression patterns revealing sub- and neo-functionalization of the homeologs at the transcription level. Access to the chromosome-scale assembly of Suneson will enable both basic and applied research efforts in the improvement of camelina as a sustainable biofuel feedstock.
1 Introduction
Camelina sativa (L.) Crantz, also known as false flax or gold-of-pleasure, is a low-cost renewable crop with multiple uses in food, feed, and bio-based applications. It has a broad environmental adaptability with a short life cycle of 85 to 100 days and can be grown in marginalized conditions with minimal agricultural inputs (Vollmann and Eynck, 2015; Malik et al., 2018; Zanetti et al., 2021). Camelina is a member of the Brassicaceae family and produces seeds with up to 40% oil by weight (Rodríguez-Rodríguez et al., 2013; Berti et al., 2016). If blended with conventional jet A fuel in equal proportions, camelina-based biofuel has been shown to reduce particle number and mass emissions by 50–70% (Moore et al., 2017). Key to its use as a sustainable biofuel is the development of camelina cultivars that are adapted to different climates and have favorable seed oil yield and fatty acid profiles. At the biochemical level, the deep knowledge of lipid metabolism in Arabidopsis thaliana (hereafter Arabidopsis) has been leveraged to camelina resulting in editing of fatty acid desaturase genes to alter the fatty acid profile in seed oil (Jiang et al., 2017; Morineau et al., 2017; Lee et al., 2021). However, in addition to serving as a storage molecule in seeds, fatty acids are integral components of membranes in which the composition of fatty acids (saturated vs unsaturated) impacts membrane fluidity.
To date, field studies on the impact of climate on camelina have shown that temperature, moisture, and soil type can impact seed yield and fatty acid profiles (Obour et al., 2017; Raziei et al., 2018). Furthermore, a controlled growth chamber experiment revealed that altered temperature resulted in significant changes in seed oil fatty acid profiles (Brock et al., 2020). In acclimation experiments in which spring and winter biotypes were exposed to low and then freezing temperatures, both physiological and gene expression changes were apparent between the biotypes reflecting differential responses to temperature (Anderson et al., 2022; Soorni et al., 2022). Gene expression differences were also observed between a spring and winter biotype following an 8-week cold acclimation period (Wang et al., 2022). In a limited study of genes involved in lipid metabolism, a cold stress treatment (4°C) induced expression of CsPDAT1-A and CsPDAT1-C that encode phospholipid:diacylglycerol acyltransferases which catalyze the final acylation step in triacylglycerol (TAG) biosynthesis (Yuan et al., 2017). Obtaining a better understanding of the impacts of climate on not only fatty acid and lipid profiles across organs but also other key agronomic traits is critical to developing camelina as a biofuel crop with resilience to climate variation.
The genome sequence of the doubled haploid DH55 C. sativa accession (641 Mb assembly) was published in 2014 and encodes ~89,000 genes (Kagale et al., 2014). Comparative genome analyses are consistent with a recent whole genome triplication in camelina that resulted in a highly undifferentiated hexaploid genome structure (Kagale et al., 2014). This is supported by recent chromosome painting, genome in situ hybridization, and phylogenetic analyses, which suggests that C. sativa is derived from an auto-allotetraploid C. neglecta‐like species and the diploid species C. hispida (Mandáková et al., 2019). As a polyploid, genome fractionation has occurred in camelina along with subgenome dominance (Kagale et al., 2014). Conserved as well as sub- and neo-functionalization of gene expression among the homeologs has been reported (Kagale et al., 2014; Heydarian et al., 2018; Gomez-Cano et al., 2022). In addition to genomic and a suite of transcriptomic resources (Kagale et al., 2014; Abdullah et al., 2016; Abdullah et al., 2018; Gomez-Cano et al., 2020; Gomez-Cano et al., 2022), population genetics studies have been performed that associate agronomic traits with genomic loci (King et al., 2019; Luo et al., 2019; Chaudhary et al., 2020; Li et al., 2021).
While the DH55 reference genome has been highly useful to the community and a new version (v2) of the reference genome has been released (http://cruciferseq.ca), both genome assemblies are highly fragmented due to exclusive use of short read sequences in the assembly process. The use of long read sequencing platforms, coupled with significantly improved algorithms for genome assembly, permit the construction of a chromosome-scale, high quality camelina genome assembly that can enable an improved understanding gene function including regulation of homeologs. In this study, we generated a chromosome-scale, high quality reference genome sequence and annotation for the spring biotype cultivar ‘Suneson’ which has been widely used by the research community (Na et al., 2018; Ozseyhan et al., 2018; King et al., 2019; Na et al., 2019; Lhamo et al., 2020; Gomez-Cano et al., 2022; Bengtsson et al., 2023). To further our understanding of the impact of gene regulation, we examined gene expression in camelina leaves exposed to cold stress revealing conserved as well as differential gene expression among retained homeologs. The Suneson genome resource will be of value to researchers interested in engineering camelina as a biofuel crop and in understanding genome evolution in polyploids.
2 Materials and methods
2.1 Plant material
For generation of high molecular weight DNA, Camelina sativa cv Suneson seeds were subjected to one round of single seed descent, planted in soil, and grown at 25.5°C day/18°C night for 19 days under a 15 hr photoperiod with 500 μE·m−2·s−1 of light. Plants were stored in the dark for 24 hours, leaves were harvested, and flash frozen in liquid nitrogen. For the Illumina whole genome shotgun (WGS) library, immature leaves were harvested from plants grown for 22 days in soil at 22°C day/18°C night under 400 μE·m−2·s−1 of light with a 15 hr photoperiod, and then dark treated for 24 hours prior to DNA isolation. For generation of transcript data to support genome annotation, we harvested mature leaf, immature seed stage 1, immature seed stage 2, stem, open flower, and root without the dark treatment and flash froze tissues in liquid nitrogen prior to RNA isolation.
To examine the response of Suneson to cold treatment, bulk seeds were sterilized three times with 75% ethyl alcohol for 5 minutes and then plated on Murashige and Skoog plates for 4 days prior to transfer to soil. Potted seedlings were grown in a growth chamber at 22°C day/18°C night under a 16 hr photoperiod. After three weeks, plants were exposed to cold temperature (10°C/day, 6°C/night) for 48 hrs; plants not exposed to cold stress were used as a control.
2.2 Nucleic acid isolation, library construction, and sequencing
High molecular weight genomic DNA was isolated from dark-treated immature leaves using the Takara Bio Nucleobond HMW DNA Kit (Takara Bio USA, San Jose CA); short fragments were eliminated using the Short Read Eliminator kit (Pacific Biosciences, Menlo Park, CA). Oxford Nanopore Technologies (ONT) libraries were constructed using the Ligation Sequencing Kit (SQK-LSK114, Q20+ chemistry) and sequenced on FLO-MIN114 flow cells as described previously (Li et al., 2023b) (Supplementary Table 1). Bases were called using Guppy v6.3.7 in super high accuracy mode (https://nanoporetech.com/community). DNA for error correction was isolated from dark-treated immature leaves using the Qiagen genomic tip method (Vaillancourt and Buell, 2019) and WGS libraries were constructed using the PerkinElmer NEXTFLEX Rapid XP DNA-Seq Kit HT (Perkin Elmer, Waltham, MA) (Supplementary Table 2). Libraries (five in total) were sequenced on an Illumina NovaSeq 6000 in paired-end mode generating 150 nt reads.
2.3 Genome assembly and chromosome scaffolding
Jellyfish (v2.2.10) (Marçais and Kingsford, 2011) was used to count k-mers (k = 21) in the WGS reads which were analyzed with GenomeScope (v2.0) (Vurture et al., 2017) to determine the extent of heterozygosity and shared k-mers among the homeologs. In addition, a smudgeplot was generated using Smudgeplot (v0.2.5) (Ranallo-Benavidez et al., 2020) with k-mers (k = 21) counted with KMC (v3.1.1) (Kokot et al., 2017). ONT gDNA reads were filtered using seqtk (v1.3) (https://github.com/lh3/seqtk) to remove reads less than 15 kb. The genome was assembled using Flye (v2.9.1) (Kolmogorov et al., 2019) with the genome size set to .785g, zero polishing iterations, and asm-coverage 60. The initial assembly was error-corrected through two rounds of Medaka (v1.7.2; https://github.com/nanoporetech/medaka) with all of the ONT genomic DNA reads using the model r1041_e82_400bps_sup_g615. This was followed by two rounds of Pilon (v1.24) (Walker et al., 2014) using the alignments from Cudadapt-trimmed reads (v4.1) (Martin, 2011) that were aligned to the assembly using bwa-mem2 (v2.2.1) (Li, 2013). Contigs less than 50 kb were filtered out using seqkit (v2.3.0) (Shen et al., 2016). Two rounds of RagTag (v2.1.0) (Alonge et al., 2022) was used to generate a chromosome-scale assembly using the reference genome DH55 v2.0 (http://cruciferseq.ca). Kraken 2 (v2.1.2) (Wood et al., 2019) was used to check all reads and the final assembly for contamination. KAT (v 2.4.1) (Mapleson et al., 2017) was used to determine the representation of k-mers in the final assembly. Genome completeness was assessed using Benchmarking Universal Single Copy Orthologs (BUSCO, v5.4.3) (Waterhouse et al., 2018) with the embryophyta_odb10 database.
2.4 Preparation of RNA-seq and full-length cDNA libraries
To support high quality gene annotation, RNA was isolated from a diverse set of tissues using either the hot borate method (Wan and Wilkins, 1994) (mature leaf and stem) or Purelink RNA isolation kit (Thermo Fisher Scientific, Waltham MA) (immature seed stage 1, immature seed stage 2, open flower, and root). Total RNA was treated with Turbo DNAse (Thermo Fisher Scientific, Waltham MA) following the manufacturer’s directions. ONT cDNA libraries were constructed using the SQK-PCB109 library preparation kit (Oxford Nanopore Technologies, Oxford UK) and sequenced on FLO-MIN106 flow cells (Supplementary Table 1). Bases were called using Guppy v6.3.7 (https://nanoporetech.com/community) in the super high accuracy mode with barcode trimming disabled.
For cold stress experiments, two biological replicates of leaf tissue were collected from control and cold-treated plants and ground into a fine powder in liquid nitrogen. Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Germantown MD) and RNA-seq libraries were constructed using the KAPA mRNA HyperPrep Kit protocol (KAPA Biosystems, Wilmington, MA). RNA-seq libraries were sequenced in paired-end mode generating 150 nt reads on an Illumina NovaSeq 6000 (Supplementary Table 1). ONT cDNA libraries were constructed, sequenced, and bases called as described above.
2.5 Genome annotation
The Suneson genome was annotated for protein-coding genes as described previously (Pham et al., 2020). In brief, repetitive sequences were identified using RepeatModeler (v2.0.3) (Flynn et al., 2020) from which protein-coding genes were removed using Protex (v1.2)(Campbell et al., 2014). These filtered repeat sequences were added to the Repbase Viriplantae repeat dataset (v20150807) to construct a final repeat library. Prior to annotation, RepeatMasker (v4.1.2-p1) (Chen, 2004) was used to mask the genome using the parameters -s -nolow -no_is -gff. RNA-seq reads were cleaned of low quality sequences and adapters using Cutadapt (v2.10) (Martin, 2011) with a quality cutoff of 10 and a minimum length of 100nt (Supplementary Table 2). Cleaned reads were aligned to the Suneson genome using HISAT2 (v2.1.0) (Kim et al., 2019) with a maximum intron length of 5000 and genome-guided transcript assemblies were generated using Stringtie 2 (v2.2.1) (Kovaka et al., 2019). The BRAKER2 pipeline (v2.1.6) (Hoff et al., 2019) was used to predict gene models using the RNA-Seq alignments as hints. Gene models were refined through two rounds of PASA (v2.5.2) (Haas et al., 2003; Campbell et al., 2006) using the RNA-seq and ONT cDNA reads resulting in a set of 145,971 working gene models. To identify high confidence gene models, gene expression data were generated using Kallisto (v0.46.2) (Bray et al., 2016) with the mRNAseq reads and Stringtie (v2.2.1) (Kovaka et al., 2019) with the ONT cDNA reads. Predicted proteins were searched against the Arabidopsis v11 predicted proteome (Araport.org) using Diamond (v0.9.36) (Buchfink et al., 2015) and Pfam domains were identified using the PFAM database (v32.0) (El-Gebali et al., 2019) with HMMER (v3.3) (Mistry et al., 2013). High confidence gene models were determined based on gene expression (TPM > 0) and/or protein match to Arabidopsis and/or presence of a Pfam domain. Functional annotation was assigned to the gene models using matches to Arabidopsis, the presence of Pfam domains, and expression evidence. Transcription factors were predicted using iTAK v1.7 (Zheng et al., 2016) with the high-confidence representative peptide sequences.
2.6 Gene expression abundances, differential gene expression, and gene coexpression analyses
Gene expression abundance estimations were calculated for cold-stressed and control leaves (this study) along with publicly available data that was downloaded from the National Center for Biotechnology Information Sequence Read Archive. First, reads were cleaned using Cutadapt (v4.1) (Martin, 2011) with a minimum read length of 40, 3’ end quality cutoff of 30, flanking N base removal, and 3’ adapter sequence trimming. The Kallisto quant algorithm (v0.48.0) (Bray et al., 2016) was used to quantify expression with a k-mer size of 21; libraries that were sequenced in single end mode were run with two additional parameters, a fragment length of 200 and standard deviation of 20. Libraries that were sequenced in paired end mode were run with the –rf-stranded parameter. Gene coexpression networks were constructed using Simple Tidy GeneCoEx in R (Li et al., 2023a) with genes that had a TPM > 1.
To detect differential gene expression, RNA-seq reads were mapped to Suneson genome using HISAT2 (version 2.0.0-beta) (Kim et al., 2019) and expression abundances were calculated by StringTie (v1.3.3b) (Pertea et al., 2016) to determine gene expression abundances. Significantly differentially expressed homeologous genes among a triplet (p < 0.01) or between cold and control samples (|log2FC| > 1; p < 0.01) were identified using EdgeR (Robinson et al., 2010).
2.7 Homeologous gene identification
Genome annotation for Arabidopsis lyrata and A. thaliana (Araport11) was downloaded from Phytozome (v13) (Goodstein et al., 2012). The GENESPACE pipeline (Lovell et al., 2022) was run with the Suneson genome and A. thaliana using GENESPACE v0.9.3 to identify triplicated homeologs within Suneson relative to A. thaliana. For A. lyrata, GENESPACE v1.1.4 was used with the representative gene model annotations to identify syntelogs between Suneson and A. lyrata. The default pipeline options were used except for the ploidy option which was set to 1,3. The syntelogs were exported from the pan-genome databases using the query_pangenes GENESPACE function.
2.8 Gene expression of triplicated homeologous genes
We first classified variation in expression across triplicated homeologs under control conditions by ranking the three homeologous genes based on their average FPKM value. If the highest expressed gene in a triplet of homeologs showed a significantly higher expression level (p < 0.01 and fold change of FPKM > 2) than the other two copies, this homeolog was classified as a Class 1 homeolog. If two genes in the triplet showed a significantly higher expression level (p < 0.01 and fold change of FPKM > 2) than the third copy, this homeolog was classified as a Class 2 homeolog. If all of the copies of a triplet showed similar expression levels (fold change of FPKM between every two copies < 1.5), this homeolog was classified as a Class 3 homeolog. Gene ontology analyses of the three classes of homeologs were performed and displayed using TBtools (Chen et al., 2020).
In the response to cold stress, if a triplicated homeolog had a significantly higher expression level following cold stress relative to the control sample, this gene was termed a cold-induced gene. A cold-induced homeolog was classified as a Type 1 triplet if all three homeologs were cold-induced; a homeolog was classified as a Type 2 homeolog if two of the three copies were cold-induced; and a homeolog was classified as a Type 3 homeolog if one of the three copies was cold-induced. For every homeolog in a Type 1 triplet, we calculated the fold change of its FPKM value between cold-treated and control samples and used the fold change to represent the cold response level of this homeolog. We ranked the three homeologous genes based on their cold inducibility with the copy exhibiting the highest level ranked first and the copy with the lowest level ranked third. The cold inducibility of the first and third copies were compared to detect the divergence of their response to cold stress.
3 Results and discussion
3.1 Genome assembly of C. sativa cv Suneson
As the camelina genome is a hexaploid, we assessed the number of unique k-mers in the Suneson genome using Illumina WGS reads. The k-mer distribution plot (Supplementary Figure 1) is consistent with a diploidized genome in which the majority of k-mers (k = 21) were present in single copy with a subset present in two copies and an even smaller subset in three copies. We also examined the pattern of near-identical k-mers using SmudgePlot (Supplementary Figure 2) in which 49% of the k-mer pairs were present as AAB, 46% present as AB, and 5% as AAAB, consistent with the hypothesized origin of hexaploid camelina being derived from an auto-allotetraploid C. neglecta‐like species and the diploid species C. hispida (Mandáková et al., 2019). Using ~42× coverage ONT genomic reads greater than 15 kb and the Flye assembler software, we assembled 647,473,868 bp of the Suneson genome into 551 contigs with an N50 contig length of 12,024,690 bp (Supplementary Table 3). Two rounds of error correction with Medaka followed by two rounds of Pilon were performed. The assembly was filtered to remove contigs less than 50 kb, yielding a 644,482,469 bp assembly contained in 157 contigs with an N50 length of 12,031,512 bp (Supplementary Table 3). To assemble to the 20 camelina chromosomes, Ragtag was used with the DH55 reference assembly resulting in 98.3% of the Suneson assembly anchored to the chromosomes (Supplementary Table 4). To validate the assembly, we used the KAT program to determine the representation of WGS-derived k-mers in the final assembly. As shown in Supplementary Figure 3, the majority of k-mers were present in single copy within the assembly with limited numbers present at two copies and a small set of k-mers present in three copies. To assess the representation of genic sequences in the genome assembly, we ran BUSCO with the embryophyta_odb10 database. A total of 1,606 of the 1,614 (99.5%) BUSCO orthologs were complete in the Suneson assembly, of which, 1,581 (98.0%) are duplicated as expected due to the hexaploid nature of the camelina genome; a mere 0.2% and 0.3% were fragmented or missing, respectively (Supplementary Table 5).
3.2 Genome annotation
Repetitive sequences in the Suneson genome were identified using a combination of de novo repeat identification and sequence similarity to existing Viridiplantae repetitive sequences. In total, 44.2% of the genome was annotated as repetitive (Supplementary Table 6), which is substantially higher than the percentage (25%) identified in the DH55 assembly which is attributable to the short-read-derived DH55 genome sequence. Retroelements (25%) dominated the annotated repetitive sequences relative to DNA transposons (2.85%). Annotation of protein-coding genes using five mRNA-seq libraries and full-length cDNA sequences derived from eight different tissues (Supplementary Table 7) resulted in 145,971 working gene models from 103,435 loci (Supplementary Table 8). Of these working models, 133,355 were high confidence models derived from 91,877 loci. While the number of total genes was similar between the Suneson and DH55 v2 annotation, access to substantial full-length cDNA sequence data permitted annotation of more gene models in the Suneson assembly compared to the DH55 assembly. In the Suneson annotation, the average number of gene models per locus in the working and high confidence gene sets to 1.41 and 1.45, respectively, which is higher compared to the average of 1.06 gene models per locus in the DH55 annotation (Supplementary Table 8). To assess the quality of the genome annotation, we examined the representation of BUSCO orthologs. Within the representative high confidence gene model set, 98.4% of the BUSCO orthologs were present with 92.8% present as duplicated, consistent with the hexaploid nature of the camelina genome (Supplementary Table 5).
Kagale et al. (2014) reported a high degree of synteny between C. sativa and A. lyrata. In the Suneson genome, 59,645 genes were syntenic to 20,934 A. lyrata genes as shown in the riparian plot of synteny between these two species (Figure 1A). To understand the relationship of the subgenomes within Suneson, we identified syntelogs between A. thaliana and Suneson using GENESPACE resulting in 23,625 A. thaliana genes syntenic with 45,453 Suneson genes (Supplementary Table 9). The riparian plot of the three subgenomes relative to the five chromosomes of A. thaliana (Figure 1B) highlights the significant degree of conservation between A. thaliana and the three subgenomes in Suneson. Of these, 24,960 Suneson genes were syntenic to 8,320 A. thaliana genes reflecting a 3 camelina homeolog to 1 A. thaliana relationship and retention of all three homeologs (Supplementary Table 10). In addition to fully retained homeologs, 2,829 A. thaliana genes were syntenic to 5,658 C. sativa genes (1:2 ratio) while 7,253 A. thaliana genes were syntenic to 7,253 C. sativa genes (1:1 ratio).
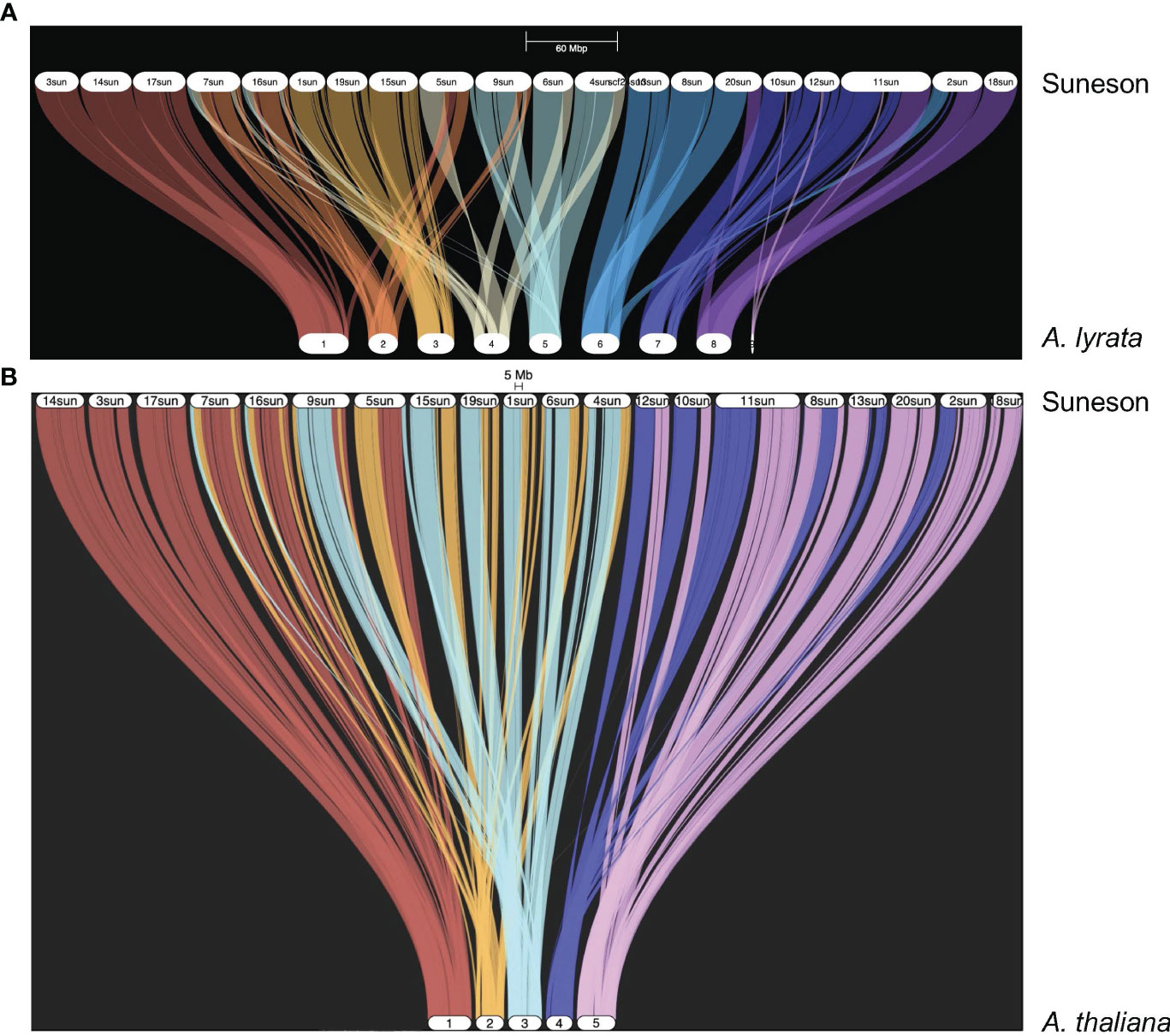
Figure 1 Syntelogs between Arabidopsis species and Camelina sativa cv Suneson. GENESPACE was used to identify syntelogs (A) between Arabidopsis lyrata and Camelina sativa cv Suneson and (B) between Arabidopsis thaliana and Camelina sativa cv Suneson.
3.3 Response of camelina leaves to cold stress
We performed RNA-seq using leaf tissue collected from cold-treated plants to gain insight on the impact of short-term cold stress on leaf tissue. A total of 4,467 genes showed a significantly higher expression level in cold-treated samples compared to their expression under control temperature; 4,851 were down-regulated under cold stress (Supplementary Tables 11, 12). Gene ontology terms related to stress response were enriched in cold-inducible genes including “response to cold”, “response to temperature stimulus”, and “cold acclimation” (Figure 2A; Supplementary Table 13).

Figure 2 Gene expression in Camelina sativa cv Suneson following cold stress. (A) Gene ontology (GO) terms enriched in cold induced genes. Only the top 10 enriched GO terms are shown here. A complete list of enriched GO terms is included in Supplementary Table 13. Sizes of symbols reflect numbers of genes, color reflects log10 p-value and symbols reflect GO categories (BP: Biological process; CC: Cellular compartment; MF: Molecular function). (B) Gene expression in C. sativa cv Suneson leaf and developing seeds. Gene expression abundances for 1,452 genes involved in fatty acid and lipid metabolism were calculated using Kallisto for control and cold stressed leaves (this study) and two sets of seed development series obtained from the National Center for Biotechnology Information Sequence Read Archive. Numbers for seed samples reflect days post anthesis. (C) Modules of coexpressed genes using gene expression abundances from control and cold-stressed leaves (this study) and two seed development studies. Numbers for seed samples reflect days post anthesis.
As temperature impacts fatty acid and lipid composition, we examined the expression of genes involved in lipid and fatty acid metabolism under cold stress in Suneson leaves. Using 552 A. thaliana genes previously associated with lipid and fatty acid metabolism (Nguyen et al., 2013), we identified 1,474 genes in the Suneson assembly involved in lipid and fatty acid metabolism (Supplementary Table 14). Expression of these genes were strikingly different between leaf and developing seeds as shown in Figure 2B consistent with the diverged function of lipid metabolism in these tissues. Of these, 60 were differentially up-regulated in cold stressed leaves while 111 were down-regulated. Multiple genes involved in remodeling membrane lipids were upregulated (Supplementary Table 11) including lipid transfer proteins functioning in phospholipid transfer between cell membranes. Also up-regulated were genes encoding phosphoinositide-specific phospholipase C which is associated with hormone signaling, abiotic stresses, and pathogen responses (Rupwate and Rajasekharan, 2012). Notably, a DIACYLGLYCEROL KINASE (DGK) gene was upregulated when exposed to cold temperatures (Supplementary Table 11). As plants balance the levels of phosphatidic acid, diacylglycerol, and triacylglycerol during cold stress, DGKs plays a major role in remodeling cold-responsive lipids (Tan et al., 2018). We did not observe up-regulation of phospholipid:diacylglycerol acyltransferases which have been reported to be induced by cold stress (Yuan et al., 2017) and shown to enhance fitness under cold stress in A. thaliana (Demski et al., 2020). This may be attributable to the warmer and shorter cold stress conditions employed in this study which may not have been sufficient to induce gene expression. In contrast, different classes of lipoxygenases (LOX gene family) involved in lipid catabolism and the formation of oxylipins including the defense-related hormone jasmonic acid were downregulated under cold stress (Supplementary Table 12) consistent with a previous study (Zhu et al., 2018). Oxylipins have been reported to play a role in cold stress through jasmonic acid-mediated regulation of Inducer of CBF (ICE) – C-Repeat Binding Factor (CBF)/DRE Binding Factor 1 through the alleviation of oxidative damage in cells (Hu et al., 2013).
To identify genes that are co-regulated under cold stress, we constructed gene coexpression networks. After filtering for the top 20% variable genes and an r > 0.7, 14,765 genes with 13,803,988 edges were used to construct coexpression modules (Figure 2C; Supplementary Table 15). With respect to gene expression following cold stress, four modules were of interest (Modules 5, 23, 124, 167). Module 5 contained 233 genes, of which, 62 were up-regulated and 22 were down-regulated following cold stress. Genes in Module 5 were enriched in GO terms associated with response to light, abiotic stress, and environmental stimuli as well as regulation of genes involved in photosynthesis (Supplementary Table 16). With respect to cellular compartment, Module 5 genes were associated with the plastid and the peroxisome. Module 23 (93 genes) had 35 and 4 genes up-regulated and down-regulated, respectively, in response to cold stress; GO terms associated with Module 23 were associated with response to abiotic stress, cold, and temperature stimulus and were associated with plastic cellular compartment (Supplementary Table 16). Similar to Module 5, Module 124 included 99 genes, of which, 24 were up-regulated and 29 were down-regulated and associated with GO terms involved in response to light and abiotic stimuli with localization within the plastid compartment (Supplementary Table 16). While only containing 38 genes, Module 167 had 23 genes up-regulated and a single gene down-regulated following cold-stress; GO associations suggest this module was associated with DNA repair (Supplementary Table 16).
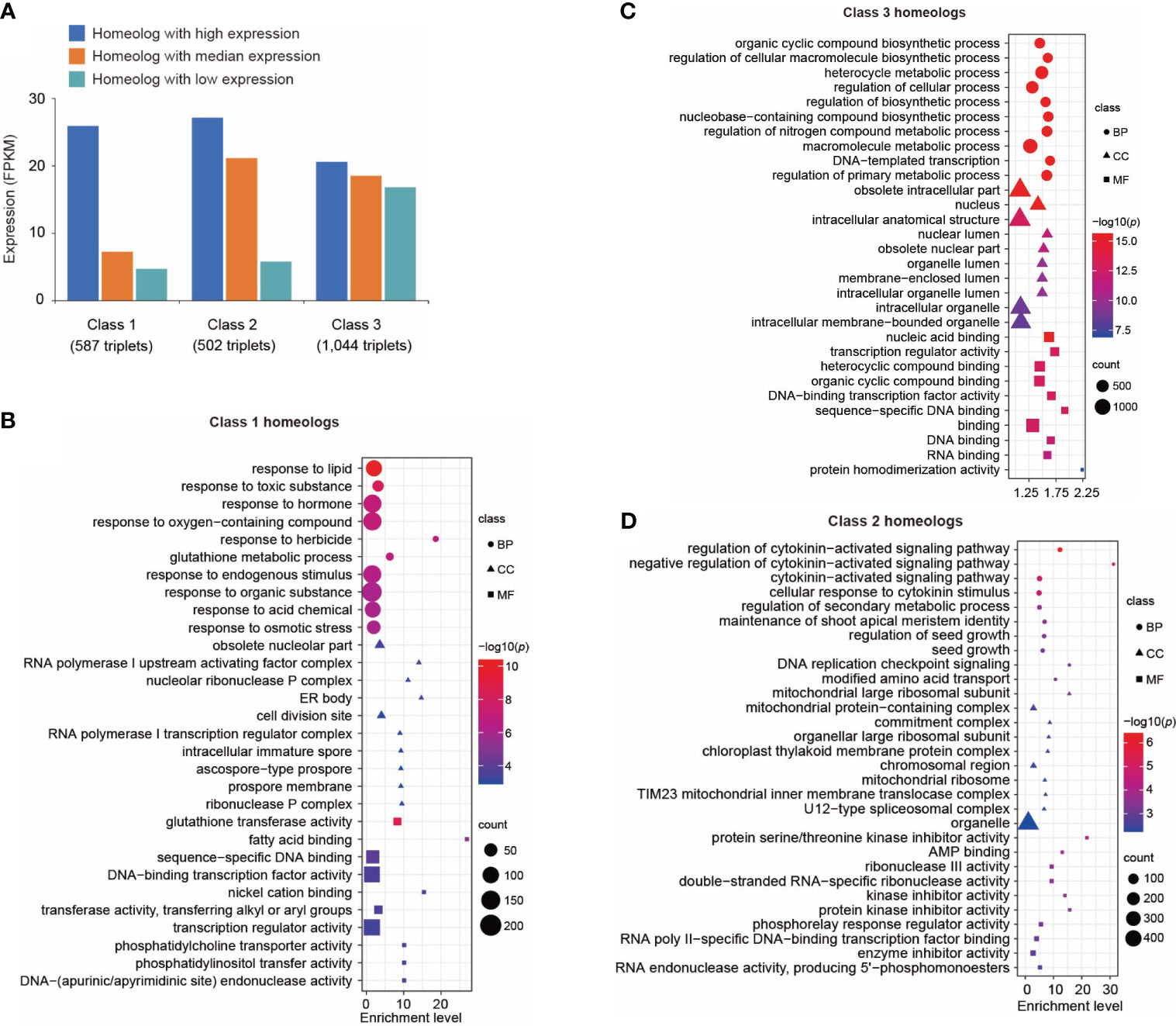
Figure 3 Transcriptional divergence of triplicated homeologous genes in control leaves. (A) Classification of three classes of triplicated homeologous genes based on gene expression. Class 1 represents the homeolog in which the highest expressed gene showed a significantly higher expression level (p < 0.01 and fold change of FPKM > 2) than the other two copies. Class 2 represents the homeolog in which two genes showed a significantly higher expression level (p < 0.01 and fold change of FPKM > 2) than the third copy. Class 3 represents the homeologs in which three copies showed similar expression levels (fold change of FPKM between every two copies < 1.5). (B) Gene ontology (GO) terms enriched in Class 1 genes. Only the top 10 enriched GO terms are shown. A complete list of enriched GO terms is included in Supplementary Table 17. (C) GO terms enriched in Class 3 genes. Only the top 10 enriched GO terms are shown here. A complete list of enriched GO terms is included in Supplementary Table 18. (D) GO terms enriched in Class 2 genes. Only the top 10 enriched GO terms are shown here. A complete list of enriched GO terms is included in Supplementary Table 19. Sizes of symbols reflect numbers of genes, color reflects log10 p-value and symbols reflect GO categories (BP, Biological process; CC, Cellular compartment; MF, Molecular function).
3.4 Expression patterns of retained homeologous genes in Camelina sativa
We analyzed the transcriptional divergence of triplicated homeologous genes in the Suneson genome. We identified 8,320 sets of triplicated homeologous genes (see Methods) and analyzed their expression in leaf tissue. We first investigated the expression patterns of these homeologs under control conditions. Among these homeologs, over 76% (6,323/8,320) exhibited expression of all three homeologous genes, while 6.0% (499/8,320) and 6.4% (529/8,320) of the triplets displayed expression of only one and two of the three homeologous copies, respectively. All three copies of the remaining 969 (11.6%) triplets were not expressed in control leaf tissue.
The triplicated homeologs were then cataloged into Class 1, 2, and 3 based on the expression levels of the three homeologs. We identified 587 triplets (Class 1) in which the expression level of one copy is significantly higher (p < 0.01) than both of the two other copies (Figure 3A). We performed GO analysis on this group of genes. Interestingly, genes responsive to stimuli such as chemicals, stress, and endogenous stimuli were highly enriched in this group (Figure 3B; Supplementary Table 17). These data suggest neofunctionalization at the expression level among these homeologs. A similar result was reported in A. thaliana in which one copy of duplicated genes tends to retain their ancestral stress responses following gene duplication (Zou et al., 2009). We identified 1,044 triplicated homeologs (Class 3) in which the three copies showed a similar level of expression (Figure 3A); specifically, the fold change in expression between any two of the three copies was less than 1.5. Gene ontology analysis of this group of genes revealed enrichment of genes related to fundamental processes including biosynthetic processes, metabolic processes, cellular processes, developmental processes, and rhythmic processes (Figure 3C; Supplementary Table 18). This result indicates that it is favorable to retain expression of all three copies of genes related to fundamental biological processes. For comparison, we identified a set of 502 triplicated homeologs (Class 2) in which the expression levels of two copies were significantly higher (p < 0.01) than the third copy (Figure 3A); GO analysis revealed that signaling pathways were highly enriched in this class of genes (Figure 3D; Supplementary Table 19).
In allopolyploids, genes from one subgenome were often preferentially retained or achieved a higher level of expression than those from other subgenomes, which is known as subgenome dominance and has been documented in an increasing number of plant species (Thomas et al., 2006; Schnable et al., 2011; Alger and Edger, 2020). If one of the parental progenitors of an allopolyploid is highly adapted to the environment where the polyploid species originated, then genes responsible for environmental adaptation from this progenitor may be preferentially retained. For example, disease resistance genes were found to be preferentially retained and associated with subgenome dominance in strawberry (Barbey et al., 2019; Edger et al., 2019) and Brassica napus (de Jong and Adams, 2023). As noted above, in A. thaliana one copy of duplicated genes tends to retain their ancestral stress responses following gene duplication (Zou et al., 2009). Here, we show in C. sativa that a specific homeolog of genes responsive to stimuli tends to gain dominance in transcription in comparison to other homeologs. These results suggest that stress responsive genes have a distinct evolutionary trajectory in the evolution of allopolyploid species.
3.5 Diverged responses to cold stress among retained homeologous genes
We investigated how the expression of retained triplicated homeologous genes evolved in their response to an environmental cue, that of cold stress. Of the 4,467 genes up-regulated in response to cold stress, 36.5% (1,632/4,467) belong to 935 triplicated homeologs. We classified the 935 homeologs into three types. Type 1: all three homeologous copies were cold-inducible; Type 2: two of the three copies were cold-inducible; Type 3: only one copy was cold inducible. Our analysis revealed that 75% of the triplicated homeologous genes displayed diverged cold responses as at least one homoeologous copy was not induced after cold treatment (Type 2 or 3) (Figure 4A). Examples of Type 1 triplicated homeologs with retained expression are Camsa.SUN.04G044570, Camsa.SUN.04G044580, Camsa.SUN.05G006530, Camsa.SUN.05G006520, Camsa.SUN.06G040820, and Camsa.SUN.06G040830 which are syntelogs with the A. thaliana cold-regulated (COR) genes AT2G42530 (COR15b) and AT2G42540 (COR15a) present in tandem on A. thaliana chromosome 2 (Figure 4B). Arabidopsis COR15a and COR15b are small chloroplast-targeted polypeptides induced under cold stress, localized in the chloroplast stroma which function in freezing tolerance (Lin and Thomashow, 1992a; Lin and Thomashow, 1992b; Wilhelm and Thomashow, 1993; Artus et al., 1996; Thomashow, 1998; Thalhammer and Hincha, 2013). While all six Suneson genes are up-regulated in response to cold stress, the triplicated homeologs differ in basal gene expression levels and in the extent of up-regulation (Figure 4C). Syntelogs of AT2G42530 had lower basal expression but higher log2 fold-change relative to the AT2G42540 syntelogs which had a higher basal expression but lower log2 fold-change (Figure 4C). In addition, the extent of cold-induction within each set of the triplicated homeologs differed. For example, the log2 fold-change of Camsa.SUN.04G044570 is lower than Camsa.SUN.05G006530 and Camsa.SUN.06G040820 (Figure 4C). Similar cold-specific expression of the Wcor15 homeolog has been documented in allopolyploid wheat and suggested to play an important role in cold hardiness in wheat and barley (Takumi et al., 2003).
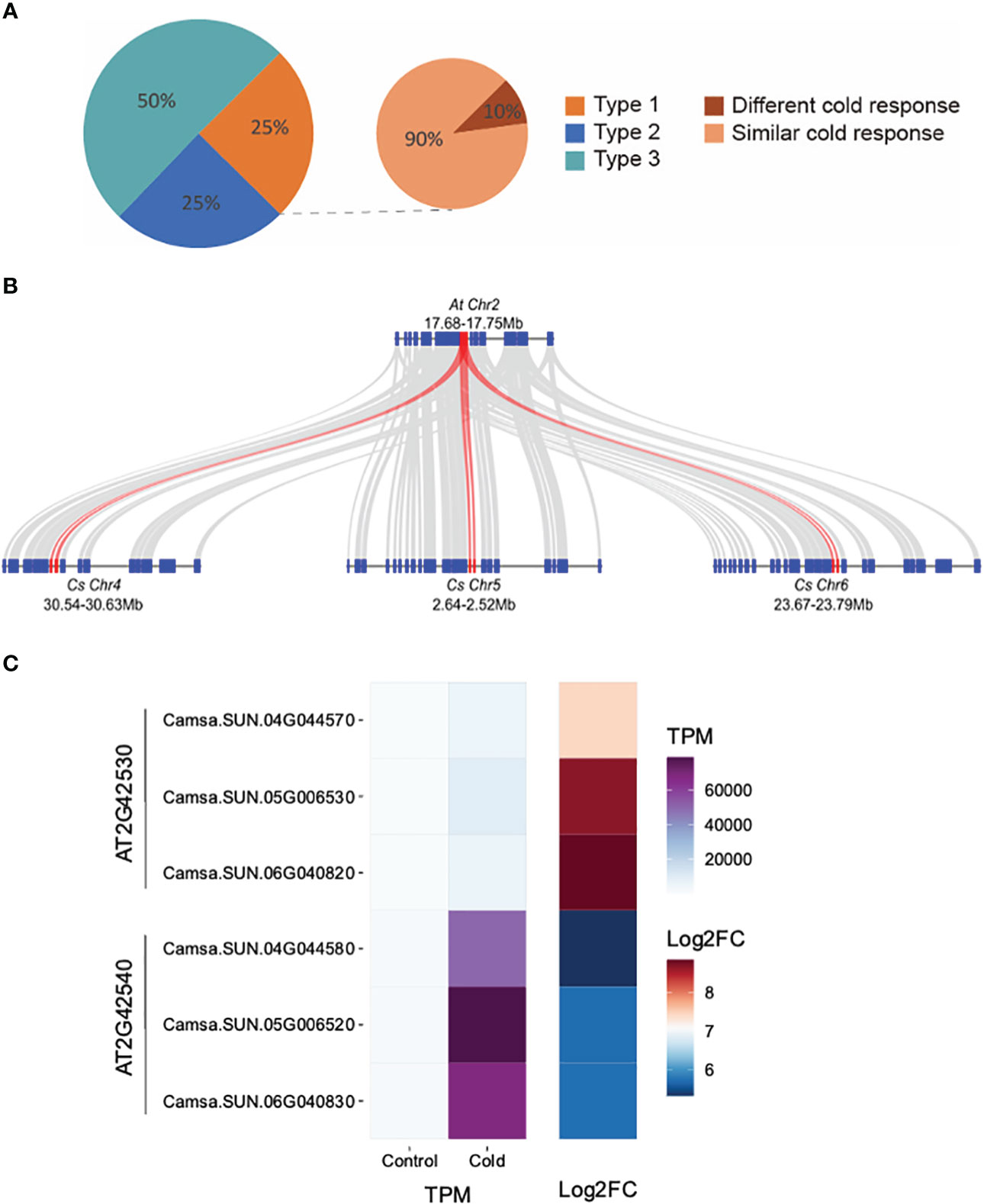
Figure 4 Transcriptional divergence of triplicated homeologous genes in C. sativa leaves following cold stress. (A) Classification of the three types of cold-response triplicated homeologs. The three homeologous genes of Type 1 triplets were all cold-induced; Two of the three homeologs from Type 2 triplets were cold-induced; Only one of the three homeologs from Type 3 triplets was cold-induced. “Different cold response” indicates the Type 1 triplets in which one homeolog showed at least two-times higher cold inducibility than the two other homeologs. The remaining Type 1 triplets are termed as “similar cold response”. (B) McScan was used to display the systemic relationship of C. sativa homeologs of Arabidopsis thaliana COR15 genes (red) and flanking genes. (C) Gene expression of COR15 homeologs in control and cold-treated leaves.
To further compare the “cold inducibility” of each gene within all of the 233 Type 1 triplicated homeologs, we calculated the fold change in expression levels between control and cold treatment sample. We then ranked the three homeologous copies of each triplicated homeolog based on their cold inducibility, with the copy exhibiting the highest expression level ranked first and the copy with the lowest level ranked third (Figure 4A). Our analysis revealed that >10% of the Type 1 homeologs exhibited a two-fold or greater difference in cold inducibility between the first and third-ranked copies, suggesting divergence in the degree of cold inducibility among the homeologous genes. Such homeolog expression bias, where one homeolog is preferentially expressed relative to the other, has been reported in multiple other allopolyploid species including Gossypium (Hovav et al., 2008; Flagel and Wendel, 2010), Triticum (Bottley et al., 2006; Wei et al., 2019), Brassica (Auger et al., 2009; Wu et al., 2018; Lee and Adams, 2020), and other species (Grover et al., 2012). Abiotic stress conditions, especially cold stress, considerably impacts expression bias of homeologs involved in physiological responses. Homeologs with differential gene expression are involved in the CBF-COR signaling pathway, fatty acid metabolism which impacts plasma membrane fluidity and stabilization, scavenging reactive oxygen species, sucrose metabolism, and accumulation of secondary metabolites, all which contribute to cold tolerance (Combes et al., 2013; Lee and Adams, 2020; Park and Jang, 2020; Wu et al., 2022). Therefore, studying homeolog gene expression and their sub-functionalization provides foundational knowledge that can be utilized in engineering cold tolerant camelina.
4 Conclusions
Access to chromosome-scale genome assemblies and high quality annotation have been foundational resources for genomics-enabled improvement of crop plants. These data have facilitated the understanding of genetic diversity, population structure, structural variation, and quantitative genetics across many crop species. For camelina to be an adaptable biofuel feedstock, improvements in agronomic performance and optimization of seed oil composition and yield will be required. This will entail both conventional breeding and biotechnological approaches that will be enabled by tapping into genetic diversity (Luo et al., 2019; Li et al., 2021) and facile transformation via floral dip (Liu et al., 2012). Access to a chromosome-scale, highly contiguous genome assembly for the widely used spring biotype Suneson was generated in this study and will enable not only basic research on molecular, physiological, and biochemical traits but also breeding cultivars with improved agronomic and biofuel traits. In addition to generation of a chromosome-scale assembly of Suneson and classification of syntelogs with two Arabidopsis species, we documented the transcriptional response to cold stress in vegetative leaves including identification of differentially expressed genes, generation of coexpression modules, and characterization of conserved/diverged expression of homeologous genes. These datasets provide a foundation for more detailed interrogation of gene function and regulation in camelina as well as how these diverged from the model species, A. thaliana.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Raw sequence reads for all generated data are available through the National Center for Biotechnology Information Sequence Read Archive under BioProject ID PRJNA927321. The genome assembly, annotation, gene expression abundances and GENESPACE results are available on Figshare (https://figshare.com/s/6f95ce23f7c4eded54d6).
Author contributions
CF: Formal Analysis, Methodology, Writing – original draft, Writing – review & editing, Data curation, Investigation, Software, Visualization. JH: Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. BV: Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. Y-WW: Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing – review & editing. JW: Investigation, Methodology, Writing – review & editing, Supervision. ND: Investigation, Writing – review & editing, Formal Analysis, Software, Visualization. TS: Writing – review & editing, Methodology. LC: Methodology, Writing – review & editing. KM: Methodology, Writing – review & editing, Supervision. DD: Supervision, Writing – review & editing, Conceptualization, Funding acquisition. SN: Conceptualization, Funding acquisition, Writing – review & editing, Formal Analysis, Investigation, Resources, Writing – original draft. JJ: Conceptualization, Funding acquisition, Investigation, Writing – review & editing, Supervision. CRB: Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Formal Analysis, Methodology, Project administration, Resources, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by funds from the University of Georgia, Georgia Seed Development, and the Georgia Research Alliance to CRB, the USDA National Institute of Food and Agriculture Biotechnology Risk Assessment Grant Program (2018-33522-28736) to CRB, SN and DD, and startup funds from Michigan State University to JJ.
Acknowledgments
We acknowledge the expertise of the RTSF Genomics Core at Michigan State University and the Texas A&M AgriLife Research: Genomics and Bioinformatics Service in providing sequencing services.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1271625/full#supplementary-material
References
Abdullah, H. M., Akbari, P., Paulose, B., Schnell, D., Qi, W., Park, Y., et al. (2016). Transcriptome profiling of Camelina sativa to identify genes involved in triacylglycerol biosynthesis and accumulation in the developing seeds. Biotechnol. Biofuels 9, 136. doi: 10.1186/s13068-016-0555-5
Abdullah, H. M., Chhikara, S., Akbari, P., Schnell, D. J., Pareek, A., Dhankher, O. P. (2018). Comparative transcriptome and metabolome analysis suggests bottlenecks that limit seed and oil yields in transgenic Camelina sativa expressing diacylglycerol acyltransferase 1 and glycerol-3-phosphate dehydrogenase. Biotechnol. Biofuels 11, 335. doi: 10.1186/s13068-018-1326-2
Alger, E. I., Edger, P. P. (2020). One subgenome to rule them all: underlying mechanisms of subgenome dominance. Curr. Opin. Plant Biol. 54, 108–113. doi: 10.1016/j.pbi.2020.03.004
Alonge, M., Lebeigle, L., Kirsche, M., Jenike, K., Ou, S., Aganezov, S., et al. (2022). Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 23, 258. doi: 10.1186/s13059-022-02823-7
Anderson, J. V., Neubauer, M., Horvath, D. P., Chao, W. S., Berti, M. T. (2022). Analysis of Camelina sativa transcriptomes identified specific transcription factors and processes associated with freezing tolerance in a winter biotype. Ind. Crops Prod. 177, 114414. doi: 10.1016/j.indcrop.2021.114414
Artus, N. N., Uemura, M., Steponkus, P. L., Gilmour, S. J., Lin, C., Thomashow, M. F. (1996). Constitutive expression of the cold-regulated Arabidopsis thaliana COR15a gene affects both chloroplast and protoplast freezing tolerance. Proc. Natl. Acad. Sci. U. S. A. 93, 13404–13409. doi: 10.1073/pnas.93.23.13404
Auger, B., Baron, C., Lucas, M.-O., Vautrin, S., Bergès, H., Chalhoub, B., et al. (2009). Brassica orthologs from BANYULS belong to a small multigene family, which is involved in procyanidin accumulation in the seed. Planta 230, 1167–1183. doi: 10.1007/s00425-009-1017-0
Barbey, C. R., Lee, S., Verma, S., Bird, K. A., Yocca, A. E., Edger, P. P., et al. (2019). Disease resistance genetics and genomics in octoploid strawberry. G3 9, 3315–3332. doi: 10.1534/g3.119.400597
Bengtsson, J. D., Wallis, J. G., Bai, S., Browse, J. (2023). The coexpression of two desaturases provides an optimized reduction of saturates in camelina oil. Plant Biotechnol. J. 21, 497–505. doi: 10.1111/pbi.13966
Berti, M., Gesch, R., Eynck, C., Anderson, J., Cermak, S. (2016). Camelina uses, genetics, genomics, production, and management. Ind. Crops Prod. 94, 690–710. doi: 10.1016/j.indcrop.2016.09.034
Bottley, A., Xia, G. M., Koebner, R. M. D. (2006). Homoeologous gene silencing in hexaploid wheat. Plant J. 47, 897–906. doi: 10.1111/j.1365-313X.2006.02841.x
Bray, N. L., Pimentel, H., Melsted, P., Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Brock, J. R., Scott, T., Lee, A. Y., Mosyakin, S. L., Olsen, K. M. (2020). Interactions between genetics and environment shape Camelina seed oil composition. BMC Plant Biol. 20, 423. doi: 10.1186/s12870-020-02641-8
Buchfink, B., Xie, C., Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M., Buell, C. R. (2006). Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7, 327. doi: 10.1186/1471-2164-7-327
Campbell, M. S., Law, M., Holt, C., Stein, J. C., Moghe, G. D., Hufnagel, D. E., et al. (2014). MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164, 513–524. doi: 10.1104/pp.113.230144
Chaudhary, R., Koh, C. S., Kagale, S., Tang, L., Wu, S. W., Lv, Z., et al. (2020). Assessing Diversity in the Camelina Genus Provides Insights into the Genome Structure of Camelina sativa. G3 10, 1297–1308. doi: 10.1534/g3.119.400957
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 5, 4.10 11–14.10. 14.. doi: 10.1002/0471250953.bi0410s05
Combes, M.-C., Dereeper, A., Severac, D., Bertrand, B., Lashermes, P. (2013). Contribution of subgenomes to the transcriptome and their intertwined regulation in the allopolyploid Coffea arabica grown at contrasted temperatures. New Phytol. 200, 251–260. doi: 10.1111/nph.12371
de Jong, G. W., Adams, K. L. (2023). Subgenome-dominant expression and alternative splicing in response to Sclerotinia infection in polyploid Brassica napus and progenitors. Plant J. 114, 142–158. doi: 10.1111/tpj.16127
Demski, K., Łosiewska, A., Jasieniecka-Gazarkiewicz, K., Klińska, S., Banaś, A. (2020). Phospholipid:Diacylglycerol acyltransferase1 overexpression delays senescence and enhances post-heat and cold exposure fitness. Front. Plant Sci. 11, 611897. doi: 10.3389/fpls.2020.611897
Edger, P. P., Poorten, T. J., VanBuren, R., Hardigan, M. A., Colle, M., McKain, M. R., et al. (2019). Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51, 541–547. doi: 10.1038/s41588-019-0356-4
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Flagel, L. E., Wendel, J. F. (2010). Evolutionary rate variation, genomic dominance and duplicate gene expression evolution during allotetraploid cotton speciation. New Phytol. 186, 184–193. doi: 10.1111/j.1469-8137.2009.03107.x
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U. S. A. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Gomez-Cano, F., Carey, L., Lucas, K., García Navarrete, T., Mukundi, E., Lundback, S., et al. (2020). CamRegBase: a gene regulation database for the biofuel crop, Camelina sativa. Database 2020, baaa075. doi: 10.1093/database/baaa075
Gomez-Cano, F., Chu, Y.-H., Cruz-Gomez, M., Abdullah, H. M., Lee, Y. S., Schnell, D. J., et al. (2022). Exploring Camelina sativa lipid metabolism regulation by combining gene co-expression and DNA affinity purification analyses. Plant J. 110, 589–606. doi: 10.1111/tpj.15682
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Grover, C. E., Gallagher, J. P., Szadkowski, E. P., Yoo, M. J., Flagel, L. E., Wendel, J. F. (2012). Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 196, 966–971. doi: 10.1111/j.1469-8137.2012.04365.x
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K., Jr., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Heydarian, Z., Yu, M., Gruber, M., Coutu, C., Robinson, S. J., Hegedus, D. D. (2018). Changes in gene expression in Camelina sativa roots and vegetative tissues in response to salinity stress. Sci. Rep. 8, 9804. doi: 10.1038/s41598-018-28204-4
Hoff, K. J., Lomsadze, A., Borodovsky, M., Stanke, M. (2019). “Whole-genome annotation with BRAKER,” in Gene Prediction: Methods and Protocols. Ed. Kollmar, M. (New York, NY: Springer New York), 65–95.
Hovav, R., Udall, J. A., Chaudhary, B., Rapp, R., Flagel, L., Wendel, J. F. (2008). Partitioned expression of duplicated genes during development and evolution of a single cell in a polyploid plant. Proc. Natl. Acad. Sci. U. S. A. 105, 6191–6195. doi: 10.1073/pnas.0711569105
Hu, Y., Jiang, L., Wang, F., Yu, D. (2013). Jasmonate regulates the INDUCER OF CBF EXPRESSION–C-REPEAT BINDING FACTOR/DRE BINDING FACTOR1 cascade and freezing tolerance in arabidopsis. Plant Cell 25, 2907–2924. doi: 10.1105/tpc.113.112631
Jiang, W. Z., Henry, I. M., Lynagh, P. G., Comai, L., Cahoon, E. B., Weeks, D. P. (2017). Significant enhancement of fatty acid composition in seeds of the allohexaploid, Camelina sativa, using CRISPR/Cas9 gene editing. Plant Biotechnol. J. 15, 648–657. doi: 10.1111/pbi.12663
Kagale, S., Koh, C., Nixon, J., Bollina, V., Clarke, W. E., Tuteja, R., et al. (2014). The emerging biofuel crop Camelina sativa retains a highly undifferentiated hexaploid genome structure. Nat. Commun. 5, 3706. doi: 10.1038/ncomms4706
Kim, D., Paggi, J. M., Park, C., Bennett, C., Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
King, K., Li, H., Kang, J., Lu, C. (2019). Mapping quantitative trait loci for seed traits in Camelina sativa. Theor. Appl. Genet. 132, 2567–2577. doi: 10.1007/s00122-019-03371-8
Kokot, M., Dlugosz, M., Deorowicz, S. (2017). KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761. doi: 10.1093/bioinformatics/btx304
Kolmogorov, M., Yuan, J., Lin, Y., Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kovaka, S., Zimin, A. V., Pertea, G. M., Razaghi, R., Salzberg, S. L., Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278. doi: 10.1186/s13059-019-1910-1
Lee, J. S., Adams, K. L. (2020). Global insights into duplicated gene expression and alternative splicing in polyploid Brassica napus under heat, cold, and drought stress. Plant Genome 13, e20057. doi: 10.1002/tpg2.20057
Lee, K. R., Jeon, I., Yu, H., Kim, S. G., Kim, H. S., Ahn, S. J., et al. (2021). Increasing monounsaturated fatty acid contents in hexaploid Camelina sativa seed oil by FAD2 gene knockout using CRISPR-cas9. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.702930
Lhamo, D., Shao, Q., Tang, R., Luan, S. (2020). Genome-wide analysis of the five phosphate transporter families in camelina sativa and their expressions in response to low-P. Int. J. Mol. Sci. 21, 8365. doi: 10.3390/ijms21218365
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. doi: 10.48550/arXiv.1303.3997
Li, C., Deans, N. C., Buell, C. R. (2023a). “Simple Tidy GeneCoEx”: A gene co-expression analysis workflow powered by tidyverse and graph-based clustering in R. Plant Genome 16, e20323. doi: 10.1002/tpg2.20323
Li, H., Hu, X., Lovell, J. T., Grabowski, P. P., Mamidi, S., Chen, C., et al. (2021). Genetic dissection of natural variation in oilseed traits of camelina by whole-genome resequencing and QTL mapping. Plant Genome 14, e20110. doi: 10.1002/tpg2.20110
Li, C., Wood, J. C., Vu, A. H., Hamilton, J. P., Rodriguez Lopez, C. E., Payne, R. M. E., et al. (2023b). Single-cell multi-omics enabled discovery of alkaloid biosynthetic pathway genes in the medical plant Catharanthus roseus. Nat. Chem. Biol. 19, 1031–1041. doi: 10.1038/s41589-023-01327-0
Lin, C., Thomashow, M. F. (1992a). A cold-regulated Arabidopsis gene encodes a polypeptide having potent cryoprotective activity. Biochem. Biophys. Res. Commun. 183, 1103–1108. doi: 10.1016/S0006-291X(05)80304-3
Lin, C., Thomashow, M. F. (1992b). DNA sequence analysis of a complementary DNA for cold-regulated arabidopsis gene cor15 and characterization of the COR 15 polypeptide. Plant Physiol. 99, 519–525. doi: 10.1104/pp.99.2.519
Liu, X., Brost, J., Hutcheon, C., Guilfoil, R., Wilson, A., Leung, S., et al. (2012). Transformation of the oilseed crop Camelina sativa by Agrobacterium-mediated floral dip and simple large-scale screening of transformants. In Vitro Cell. Dev. Biol. - Plant 48, 462–468. doi: 10.1007/s11627-012-9459-7
Lovell, J. T., Sreedasyam, A., Schranz, M. E., Wilson, M., Carlson, J. W., Harkess, A., et al. (2022). GENESPACE tracks regions of interest and gene copy number variation across multiple genomes. Elife 11, e78526. doi: 10.7554/eLife.78526
Luo, Z., Tomasi, P., Fahlgren, N., Abdel-Haleem, H. (2019). Genome-wide association study (GWAS) of leaf cuticular wax components in Camelina sativa identifies genetic loci related to intracellular wax transport. BMC Plant Biol. 19, 187. doi: 10.1186/s12870-019-1776-0
Malik, M. R., Tang, J., Sharma, N., Burkitt, C., Ji, Y., Mykytyshyn, M., et al. (2018). Camelina sativa, an oilseed at the nexus between model system and commercial crop. Plant Cell Rep. 37, 1367–1381. doi: 10.1007/s00299-018-2308-3
Mandáková, T., Pouch, M., Brock, J. R., Al-Shehbaz, I. A., Lysak, M. A. (2019). Origin and evolution of diploid and allopolyploid Camelina genomes were accompanied by chromosome shattering. Plant Cell 31 (11), 2596–2612. doi: 10.1105/tpc.19.00366
Mapleson, D., Garcia Accinelli, G., Kettleborough, G., Wright, J., Clavijo, B. J. (2017). KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576. doi: 10.1093/bioinformatics/btw663
Marçais, G., Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. doi: 10.14806/ej.17.1.200
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121. doi: 10.1093/nar/gkt263
Moore, R. H., Thornhill, K. L., Weinzierl, B., Sauer, D., D’Ascoli, E., Kim, J., et al. (2017). Biofuel blending reduces particle emissions from aircraft engines at cruise conditions. Nature 543, 411–415. doi: 10.1038/nature21420
Morineau, C., Bellec, Y., Tellier, F., Gissot, L., Kelemen, Z., Nogue, F., et al. (2017). Selective gene dosage by CRISPR-Cas9 genome editing in hexaploid Camelina sativa. Plant Biotechnol. J. 15, 729–739. doi: 10.1111/pbi.12671
Na, G., Aryal, N., Fatihi, A., Kang, J., Lu, C. (2018). Seed-specific suppression of ADP-glucose pyrophosphorylase in Camelina sativa increases seed size and weight. Biotechnol. Biofuels 11, 330. doi: 10.1186/s13068-018-1334-2
Na, G., Mu, X., Grabowski, P., Schmutz, J., Lu, C. (2019). Enhancing microRNA167A expression in seed decreases the α-linolenic acid content and increases seed size in Camelina sativa. Plant J. 98, 346–358. doi: 10.1111/tpj.14223
Nguyen, H. T., Silva, J. E., Podicheti, R., Macrander, J., Yang, W., Nazarenus, T. J., et al. (2013). Camelina seed transcriptome: a tool for meal and oil improvement and translational research. Plant Biotechnol. J. 11, 759–769. doi: 10.1111/pbi.12068
Obour, A. K., Obeng, E., Mohammed, Y. A., Ciampitti, I. A., Durrett, T. P., Aznar-Moreno, J. A., et al. (2017). Camelina seed yield and fatty acids as influenced by genotype and environment. Agron. J. 109, 947–956. doi: 10.2134/agronj2016.05.0256
Ozseyhan, M. E., Kang, J., Mu, X., Lu, C. (2018). Mutagenesis of the FAE1 genes significantly changes fatty acid composition in seeds of Camelina sativa. Plant Physiol. Biochem. 123, 1–7. doi: 10.1016/j.plaphy.2017.11.021
Park, Y. C., Jang, C. S. (2020). Molecular dissection of two homoeologous wheat genes encoding RING H2-type E3 ligases: TaSIRFP-3A and TaSIRFP-3B. Planta 252, 26. doi: 10.1007/s00425-020-03431-0
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Pham, G. M., Hamilton, J. P., Wood, J. C., Burke, J. T., Zhao, H., Vaillancourt, B., et al. (2020). Construction of a chromosome-scale long-read reference genome assembly for potato. Gigascience 9, giaa100. doi: 10.1093/gigascience/giaa100
Ranallo-Benavidez, T. R., Jaron, K. S., Schatz, M. C. (2020). GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432. doi: 10.1038/s41467-020-14998-3
Raziei, Z., Kahrizi, D., Rostami-Ahmadvandi, H. (2018). Effects of climate on fatty acid profile in Camelina sativa. Cell. Mol. Biol. 64, 91–96. doi: 10.14715/cmb/2018.64.5.15
Robinson, M. D., McCarthy, D. J., Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Rodríguez-Rodríguez, M. F., Sánchez-García, A., Salas, J. J., Garcés, R., Martínez-Force, E. (2013). Characterization of the morphological changes and fatty acid profile of developing Camelina sativa seeds. Ind. Crops Prod. 50, 673–679. doi: 10.1016/j.indcrop.2013.07.042
Rupwate, S. D., Rajasekharan, R. (2012). Plant phosphoinositide-specific phospholipase C: an insight. Plant Signal. Behav. 7, 1281–1283. doi: 10.4161/psb.21436
Schnable, J. C., Springer, N. M., Freeling, M. (2011). Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. U. S. A. 108, 4069–4074. doi: 10.1073/pnas.1101368108
Shen, W., Le, S., Li, Y., Hu, F. (2016). SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PloS One 11, e0163962. doi: 10.1371/journal.pone.0163962
Soorni, J., Kazemitabar, S. K., Kahrizi, D., Dehestani, A., Bagheri, N., Kiss, A., et al. (2022). Biochemical and transcriptional responses in cold-acclimated and non-acclimated contrasting camelina biotypes under freezing stress. Plants 11, 3178. doi: 10.3390/plants11223178
Takumi, S., Koike, A., Nakata, M., Kume, S., Ohno, R., Nakamura, C. (2003). Cold-specific and light-stimulated expression of a wheat (Triticum aestivum L.) Cor gene Wcor15 encoding a chloroplast-targeted protein. J. Exp. Bot. 54, 2265–2274. doi: 10.1093/jxb/erg247
Tan, W.-J., Yang, Y.-C., Zhou, Y., Huang, L.-P., Xu, L., Chen, Q.-F., et al. (2018). DIACYLGLYCEROL ACYLTRANSFERASE and DIACYLGLYCEROL KINASE modulate triacylglycerol and phosphatidic acid production in the plant response to freezing stress. Plant Physiol. 177, 1303–1318. doi: 10.1104/pp.18.00402
Thalhammer, A., Hincha, D. K. (2013). “The function and evolution of closely related COR/LEA (Cold-regulated/late embryogenesis abundant) proteins in Arabidopsis thaliana,” in Plant and Microbe Adaptations to Cold in a Changing World (New York: Springer), 89–105.
Thomas, B. C., Pedersen, B., Freeling, M. (2006). Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 16, 934–946. doi: 10.1101/gr.4708406
Thomashow, M. F. (1998). Role of cold-responsive genes in plant freezing tolerance. Plant Physiol. 118, 1–8. doi: 10.1104/pp.118.1.1
Vaillancourt, B., Buell, C. R. (2019). High molecular weight DNA isolation method from diverse plant species for use with Oxford Nanopore sequencing. BioRxiv. doi: 10.1101/783159
Vollmann, J., Eynck, C. (2015). Camelina as a sustainable oilseed crop: Contributions of plant breeding and genetic engineering. Biotechnol. J. 10, 525–535. doi: 10.1002/biot.201400200
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204. doi: 10.1093/bioinformatics/btx153
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9, e112963. doi: 10.1371/journal.pone.0112963
Wan, C. Y., Wilkins, T. A. (1994). A modified hot borate method significantly enhances the yield of high-quality RNA from cotton (Gossypium hirsutum L.). Anal. Biochem. 223, 7–12. doi: 10.1006/abio.1994.1538
Wang, H., Doğramacı, M., Anderson, J. V., Horvath, D. P., Chao, W. S. (2022). Transcript profiles differentiate cold acclimation-induced processes in a summer and winter biotype of Camelina. Plant Mol. Biol. Rep. 40, 359–375. doi: 10.1007/s11105-021-01324-4
Waterhouse, R. M., Seppey, M., Simão, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2018). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Wei, J., Cao, H., Liu, J.-D., Zuo, J.-H., Fang, Y., Lin, C.-T., et al. (2019). Insights into transcriptional characteristics and homoeolog expression bias of embryo and de-embryonated kernels in developing grain through RNA-Seq and Iso-Seq. Funct. Integr. Genomics 19, 919–932. doi: 10.1007/s10142-019-00693-0
Wilhelm, K. S., Thomashow, M. F. (1993). Arabidopsis thaliana cor15b, an apparent homologue of cor15a, is strongly responsive to cold and ABA, but not drought. Plant Mol. Biol. 23, 1073–1077. doi: 10.1007/BF00021822
Wood, D. E., Lu, J., Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. doi: 10.1186/s13059-019-1891-0
Wu, W., Guo, W., Ni, G., Wang, L., Zhang, H., Ng, W. L. (2022). Expression Level Dominance and Homeolog Expression Bias Upon Cold Stress in the F1 Hybrid Between the Invasive Sphagneticola trilobata and the Native S. calendulacea in South China, and Implications for Its Invasiveness. Front. Genet. 13. doi: 10.3389/fgene.2022.833406
Wu, J., Lin, L., Xu, M., Chen, P., Liu, D., Sun, Q., et al. (2018). Homoeolog expression bias and expression level dominance in resynthesized allopolyploid Brassica napus. BMC Genomics 19, 586. doi: 10.1186/s12864-018-4966-5
Yuan, L., Mao, X., Zhao, K., Ji, X., Ji, C., Xue, J., et al. (2017). Characterisation of phospholipid: diacylglycerol acyltransferases (PDATs) from Camelina sativa and their roles in stress responses. Biol. Open 6, 1024–1034. doi: 10.1242/bio.026534
Zanetti, F., Alberghini, B., Marjanović Jeromela, A., Grahovac, N., Rajković, D., Kiprovski, B., et al. (2021). Camelina, an ancient oilseed crop actively contributing to the rural renaissance in Europe. A review. Agron. Sustain. Dev. 41, 2. doi: 10.1007/s13593-020-00663-y
Zhu, J., Wang, X., Guo, L., Xu, Q., Zhao, S., Li, F., et al. (2018). Characterization and alternative splicing profiles of the lipoxygenase gene family in tea plant (Camellia sinensis). Plant Cell Physiol. 59, 1765–1781. doi: 10.1093/pcp/pcy091
Zou, C., Lehti-Shiu, M. D., Thomashow, M., Shiu, S. H. (2009). Evolution of stress-regulated gene expression in duplicate genes of Arabidopsis thaliana. PloS Genet. 5, e1000581. doi: 10.1371/journal.pgen.1000581
Keywords: camelina, cold stress, genome assembly, homeolog, lipid
Citation: Fang C, Hamilton JP, Vaillancourt B, Wang Y-W, Wood JC, Deans NC, Scroggs T, Carlton L, Mailloux K, Douches DS, Nadakuduti SS, Jiang J and Buell CR (2023) Cold stress induces differential gene expression of retained homeologs in Camelina sativa cv Suneson. Front. Plant Sci. 14:1271625. doi: 10.3389/fpls.2023.1271625
Received: 02 August 2023; Accepted: 26 October 2023;
Published: 16 November 2023.
Edited by:
Manohar Chakrabarti, The University of Texas Rio Grande Valley, United StatesReviewed by:
Mir Asif Iquebal, Indian Council of Agricultural Research, IndiaMunevver Dogramaci, Agricultural Research Service (USDA), United States
Copyright © 2023 Fang, Hamilton, Vaillancourt, Wang, Wood, Deans, Scroggs, Carlton, Mailloux, Douches, Nadakuduti, Jiang and Buell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiming Jiang, jiangjm@msu.edu; C. Robin Buell, robin.buell@uga.edu
†These authors have contributed equally to this work