 Jose Carballo1
Jose Carballo1 Andrés Martin Bellido1
Andrés Martin Bellido1 Juan Pablo Selva1,2
Juan Pablo Selva1,2 Diego Zappacosta1,3
Diego Zappacosta1,3 Cristian Andres Gallo1
Cristian Andres Gallo1 Emidio Albertini4
Emidio Albertini4 Mario Caccamo5
Mario Caccamo5 Viviana Echenique1,3*
Viviana Echenique1,3*- 1Centro de Recursos Naturales Renovables de la Zona Semiárida (CERZOS), Universidad Nacional del Sur-Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Bahía Blanca, Argentina
- 2Departamento de Biología, Bioquímica y Farmacia, Universidad Nacional del Sur (UNS), Bahía Blanca, Argentina
- 3Departamento de Agronomía, Universidad Nacional del Sur (UNS), Bahía Blanca, Argentina
- 4Dipartimento di Scienze Agrarie, Alimentari e Ambientali, Università degli Studi di Perugia, Perugia, Italy
- 5NIAB, Cambridge, United Kingdom
Introduction: In Eragrostis curvula, commonly known as weeping lovegrass, a synthetic diploidization event of the facultative apomictic tetraploid Tanganyika INTA cv. originated from the sexual diploid Victoria cv. Apomixis is an asexual reproduction by seeds in which the progeny is genetically identical to the maternal plant.
Methods: To assess the genomic changes related to ploidy and to the reproductive mode occurring during diploidization, a mapping approach was followed to obtain the first E. curvula pangenome assembly. In this way, gDNA of Tanganyika INTA was extracted and sequenced in 2x250 Illumina pair-end reads and mapped against the Victoria genome assembly. The unmapped reads were used for variant calling, while the mapped reads were assembled using Masurca software.
Results: The length of the assembly was 28,982,419 bp distributed in 18,032 contigs, and the variable genes annotated in these contigs rendered 3,952 gene models. Functional annotation of the genes showed that the reproductive pathway was differentially enriched. PCR amplification in gDNA and cDNA of Tanganyika INTA and Victoria was conducted to validate the presence/absence variation in five genes related to reproduction and ploidy. The polyploid nature of the Tanganyika INTA genome was also evaluated through the variant calling analysis showing the single nucleotide polymorphism (SNP) coverage and allele frequency distribution with a segmental allotetraploid pairing behavior.
Discussion: The results presented here suggest that the genes were lost in Tanganyika INTA during the diploidization process that was conducted to suppress the apomictic pathway, affecting severely the fertility of Victoria cv.
1 Introduction
In the last few years, the genome assembly paradigm, by which one reference genome was considered enough to represent the whole diversity of a given species, is shifting to a new method, called pangenome, to represent the genetic diversity within a single species. The requirement of researchers to cover more diversity and the decreasing cost of genome sequencing were the two key components for the emergence of the pangenome concept (https://www.genome.gov/, accessed 12 November 2022). A pangenome assembly is defined by the core genome, which is shared by all the individuals analyzed, and the variable or dispensable genome, which is only present in some of the individuals (Tettelin et al., 2005).
The methodological procedure to assemble a pangenome is not standardized yet. However, the most used pipeline so far is the Iterative mapping approach (Golicz et al., 2016b; Gao et al., 2019; Ruperao et al., 2021). Through this process, DNA sequencing reads from different genotypes are mapped against a reference genome assembly. The mapped reads are used to call variants, while the unmapped ones are assembled in new genomic fragments in order to annotate genes not present in the reference genome. After that, the newly assembled sequences are merged to the reference genome and the process starts again by mapping another sample (Golicz et al., 2016a). Once all the individuals are processed, the obtained pangenome contains the reference genome plus the newly assembled sequences.
In plant biology, the pangenome model is especially important, since the majority of the reference genomes sequenced were selected to represent an entire species in one genome and to study individual genomes as models to translate the results to orphan species. For example, Arabidopsis thaliana and rice (Oryza sativa) are the models used to study dicots and monocots species, respectively (Rensink and Buell, 2004). These species are also used as proofs of concept and for functional validation. In wheat, the first near complete genome assembly was obtained from the Chinese spring cultivar, which is distantly related to modern cultivars (Zimin et al., 2017). Therefore, a wheat pangenome was later sequenced including modern varieties, finding 12,150 variables genes that were absent in Chinese Spring and 225,064 non-synonymous single nucleotide polymorphisms (SNPs) (Montenegro et al., 2017). Other modern wheat varieties were sequenced by the 10+ Genome Project, with the aim of characterizing the wheat pangenome and annotating high-confidence gene models (Walkowiak et al., 2020). Recent publications indicate that in economically important crops like barley and rice, the pangenome approach is displacing the genome reference one (Jayakodi et al., 2020; Qin et al., 2021). Pangenomic assemblies of major crops successfully increase the number of genes related to agronomically important traits. In soybean (Glycine max), the pangenome assembly of seven individuals allowed to discover gene presence/absence variations (PAVs) and mutations related to biotic resistance, seed composition, flowering and maturity time, organ size, and final biomass not previously recorded (Li et al., 2014). Another interesting example comes from the Brassica oleracea pangenome, obtained from nine varieties that allowed to discover 11,484 genes not annotated in the reference genome, including those associated with disease resistance, flowering time, glucosinolate metabolism, and vitamin biosynthesis (Golicz et al., 2016b). Finally, in cotton, a pangenome assembled with genomic data from 1,961 individuals evidenced the presence of several genes of agronomic relevance that were lost during domestication (Li et al., 2021a).
Hundreds of plants were sequenced since the genome assembly of Arabidopsis thaliana two decades ago, showing the complexity and diversity of plant genomes (Arabidopsis Genome Initiative, 2000). From many evolutionary studies, it is already known that a paleoploidization event occurred 160 million of years ago affecting all the flowering plants (Callaway, 2013). After that, the majority of the species recovered the diploid level by two processes, namely, cytological diploidization and gene diploidization. Cytological diploidization was the result of fission, fusion, and other large-scale chromosomal rearrangements, while in gene diploidization, many duplicated genes were evolutionary lost, and only their subset was retained (Li et al., 2021b). Both processes eventually produced diploid-like chromosome pairing during meiosis, directing the formation of a diploid genotype. Anthers culture is another mechanism of synthetic diploidization in which the haploid pollen is regenerated into a plant. When the maternal plant is diploid, a duplication is required since the pollen is haploid, while in tetraploids, viable plants can be obtained from diploid pollen (Cho and Zapata, 1988; Christensen et al., 1997; Chen et al., 2016).
In Eragrostis curvula, a perennial grass commonly known as weeping lovegrass, a diploidization event was obtained synthetically from a facultative apomictic tetraploid plant. Briefly, young inflorescences from Tanganyika INTA cv. (2n=4x=40) were in vitro cultured passing through a callus stage, and after 4 months, the regenerated plants were transferred to pots with soil. One of the plants, the 23 R0, was shown to have 20 chromosomes instead of the normal 40 of the maternal plant, and it was fully sexual (Cardone et al., 2006). This plant was registered as Victoria cv. The hypothesis of this diploidization event was that a reduced microspore of Tanganyika INTA gave rise to Victoria. Since Tanganyika INTA is facultative apomictic, the probability that Victoria has arisen from a reduced megasporocyte is very low because the percentage of sexual embryos in this genotype is normally below 5% (Rodrigo et al., 2017).
Apomixis is an asexual reproduction by seeds in which the offspring is genetically identical to the maternal plant. Apomictic plants of E. curvula have their own type of embryo sac development, called Eragrostis type (Crane, 2001), instead of the Polygonum type found in sexual plants of the species. In this type of diplosporous apomixis, meiosis is completely circumvented, the embryo arises without fertilization, and one sperm cell of the pollen fertilizes the central cell to form the endosperm. These three processes are defined as apomeiosis, parthenogenesis, and pseudogamy, respectively. Even though many studies of the group at transcriptomic, genomic, and epigenomic levels were taken and several advances were made to elucidate the apomixis mechanisms in E. curvula, the key molecular components regulating this pathway have not yet been resolved (Cervigni et al., 2008a; Cervigni et al., 2008b; Garbus et al., 2017; Selva et al., 2017; Carballo et al., 2019; Garbus et al., 2019; Zappacosta et al., 2019; Selva et al., 2020; Carballo et al., 2021a; Pasten et al., 2022).
The assembled genome of the sexual diploid Victoria is already available at chromosome level (Carballo et al., 2019). Our aim is to use this genome as reference to follow a pangenomic approach to assess the genomic changes that occurred during the recent diploidization of the tetraploide apomictic Tanganyika INTA that produced the genotype Victoria, a sexual diploid genotype. Thus, the results presented here suggest that the genes lost in Tanganyika INTA produced the modification of the reproductive mode from apomictic to sexual, and as a consequence, the options in terms of reproductive mechanism were reduced, affecting seriously the fertility of Victoria cv.
2 Materials and methods
2.1 Plant material and sequencing
DNA was obtained from a Tanganyika INTA plant growing in the greenhouse at CERZOS, CCT-CONICET Bahía Blanca, Argentina. The cetyltrimethyl ammonium bromide (CTAB)-based method, previously tested in E. curvula, was used for DNA extraction starting from 80 mg of fresh leaf tissue (Meier et al., 2011). After library preparation, the DNA was sequenced in pair-end 2x250 bp in a HiSeq 1500 Illumina machine rendering ~118 Gbp. These data were deposited in NCBI database under the bioproject PRJNA914706.
2.2 RNA extraction
For RNA extraction, the Promega SV Total RNA Isolation System© was used. In brief, up to 60 mg of tissue was processed, and the RNA was separated, precipitated, and bounded using the spin baskets containing a silica membrane. Later, RNA was treated with a RNase-Free DNase I and eluted with nuclease-free water. The quality and quantity of the RNA was assessed using the DeNovix DS-11 Spectrophotometer and an Agarose Gel protocol (Aranda et al., 2012). Afterwards, first-strand cDNA was performed using the NEB M-MuVL Reverse Transcriptase©. Finally, PCRs were conducted through the PB-L TAQ Pegasus© protocol, and amplicons were analyzed by electrophoresis in 1% (m/v) agarose gel.
2.3 Flow cytometry
Flow cytometry was used for genome size determinations of plants of cvs. Victoria and Tanganyika INTA using the service of the Instituto de Genética, INTA Castelar (Castelar, Buenos Aires, Argentina) according to the protocol of Galbraith et al. (1983). Fresh leaves from each genotype were stained with propidium iodide, and the samples were examined by a Partec PA II flow cytometer. For genome size estimation in base pairs, the fluorescence of each cultivar in relative fluorescence units (RFUs) was contrasted against the Secale cereale genome (7.917 Mb), since the flow cytometry peak of this species did not overlap with that expected for E. curvula (Doležel et al., 1998).
2.4 Pangenome assembly
For the pangenome assembly, the iterative mapping method was used (Figure 1) (Golicz et al., 2016b; Zhao et al., 2020; Ruperao et al., 2021). Tanganyika INTA reads were mapped against the Victoria reference genome (Carballo et al., 2019; NCBI bioproject PRJNA508722) using Bowtie2 software (parameters -I 0 -X 1,000) (Langmead and Salzberg, 2012). The Victoria cultivar was obtained from in vitro culture of Tanganyika INTA young inflorescences (Echenique et al., 1996), and the genome sequencing was made from DNA of the original R1 plant. The unmapped reads were extracted from the Bowtie2 output and assembled with Masurca V3.2.8 software (Zimin et al., 2013). The assembled contigs were aligned against the NCBI Viridiplantae plastid database in order to discard possible contaminations using the blast alignment tool (Altschul et al., 1990). Then, to remove redundancy, the assembly was self-compared with nucmer aligner filtering sequences >90% identity and >90% coverage (Marçais et al., 2018). The remaining contigs with more than 1,000 bp were included in the assembly.
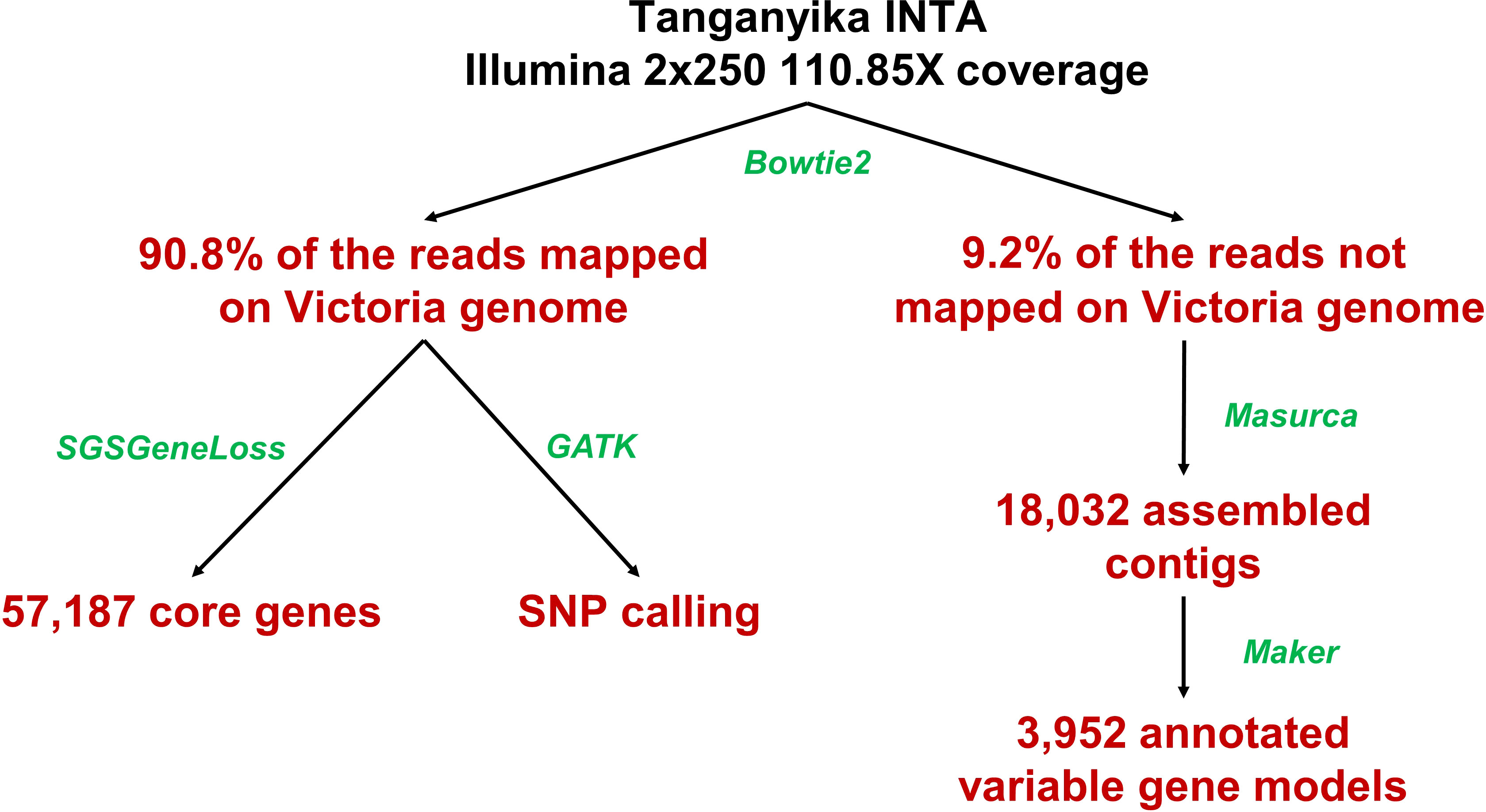
Figure 1 Pipeline used to assemble the Eragrostis curvula pangenome. Programs used are highlighted in green and the result in red.
2.5 Pangenome annotation
Repetitive elements included in the newly assembled contigs were annotated and classified using the EDTA software (Ou et al., 2019). For gene annotation, the software maker (Holt and Yandell, 2011) was implemented using RNA-seq data from E. curvula and E. tef and protein sequences from related species such as Eragrostis nindensis, Setaria italica, Panicum halli, and Oropetium thomaeum (Zhang et al., 2012; Lovell et al., 2018; VanBuren et al., 2018; Garbus et al., 2019; Pardo et al., 2020; Selva et al., 2020; VanBuren et al., 2020). The gene models obtained through this process were functionally annotated using as reference the UniProt database (Ruch et al., 2021). For this task, Blastp software was used with an e-value cutoff of 0.001 (Altschul et al., 1990). Gene Ontology (GO) was based on Interproscan 5 annotation (Jones et al., 2014), and the GO enrichment analysis of the variable genes was performed using the R package clusterProfiler (Yu et al., 2012) with a p-value adjusted cutoff of 0.05.
2.6 Gene presence/absence variations
Genomic sequence data from Tanganyika INTA and transcriptomic data from E. curvula cv. Don Walter (Selva et al., 2020) were mapped against the pangenome using Bowtie2 (Langmead and Salzberg, 2012). Gene presence/absence variations were assessed with the SGSGeneLoss software (Golicz et al., 2015) in order to perform phylogenetic analysis. After that, a matrix was constructed to perform phylogenetic analysis using R software (Team, R. Core, 2018).
2.7 Variant calling
SNP calling was performed following the GATK best practice protocol (McKenna et al., 2010). Briefly, Tanganyika INTA reads were converted to bam, and the adapters were marked with Picard tools (https://broadinstitute.github.io/picard/). Then, the resulted bam was converted back to fastq and mapped against the genome with BWA-MEM software. Finally, SNP calling and filtering was performed based on mapping quality and coverage.
2.8 Gene presence/absence validations
Genes present in Tanganyika INTA and absent in Victoria were selected based on its functional annotation (Supplementary Table 1) and its expression during reproductive stages in A. thaliana (Klepikova et al., 2016; Mergner et al., 2020) and in E. curvula (Garbus et al., 2017). Thus, genes related to ploidy, expressed during floral development and genes previously described as involved in apomixis were used as templates for primers design. For this task, IDT Primer Quest online tool (https://www.idtdna.com/Primerquest/Home/Index) was used.
3 Results
3.1 Genome size determinations
As was expected, Victoria (2n=2x=20) showed half of the Tanganyika INTA (2n=4x=40) genome size (Figure 2). In terms of fluorescence, Victoria and Tanganyika displayed 2,487.59 and 4,833.95 RFU, respectively. The genome size in base pairs, estimated by using the S. cereale reference standard (7,917 Mb), was on average 604.3 (CV=6.33) and 1,247.5 (CV=6.7) Mb for the haploid genome for Victoria and Tanganyika INTA genomes, respectively.
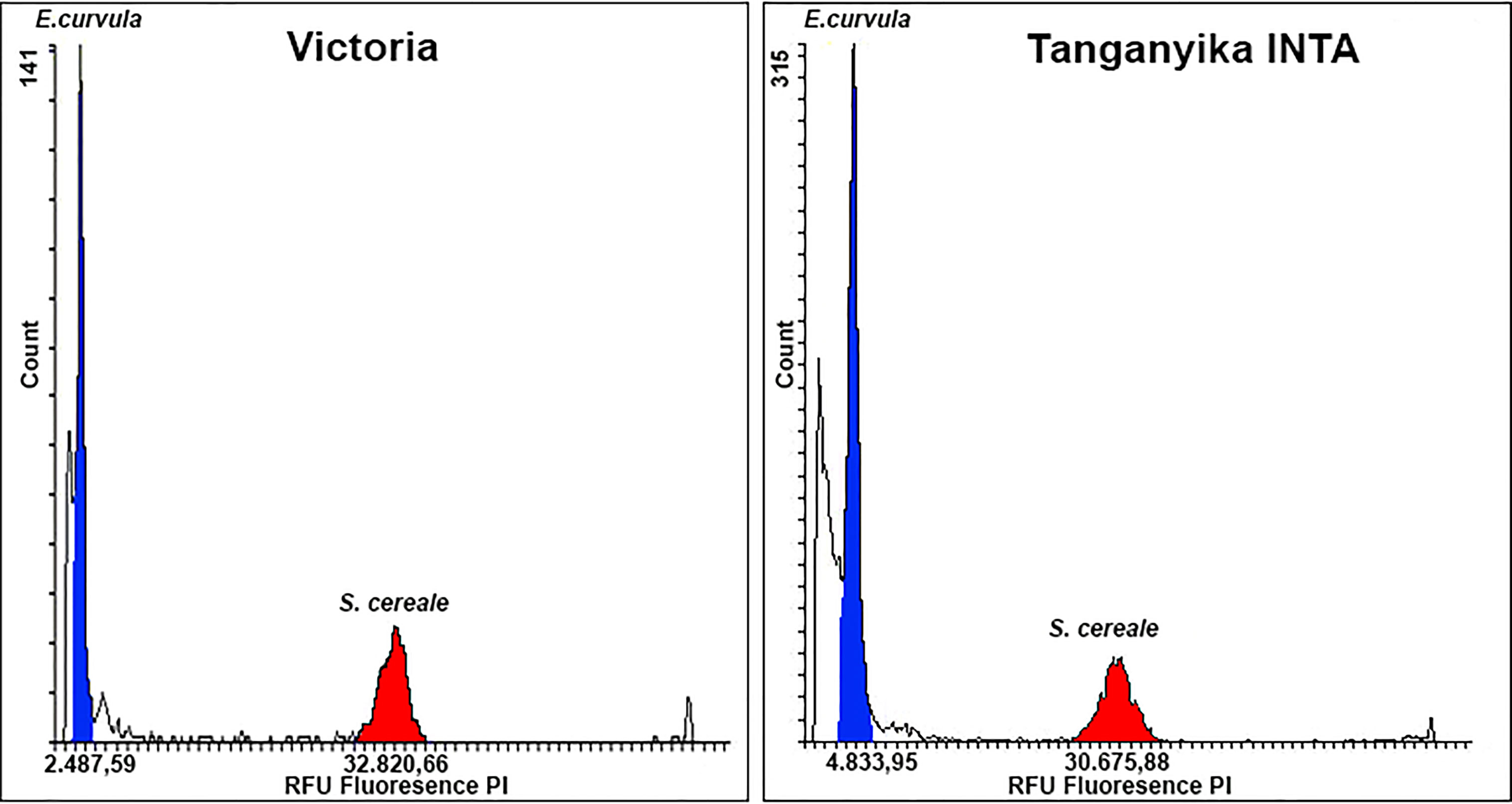
Figure 2 Flow cytometry histogram to assess E. curvula genome size. Y-axis shows nuclei count, while Y-axis display propidium iodide (PI) fluorescence. E. curvula and S. cereale are highlighted in blue and red, respectively. Victoria genotype presented half of the Tanganyika INTA RFU obtaining a genome length of 604.3 and 1,247.5 Mb, respectively.
3.2 Pangenome assembly and annotation
Tanganyika INTA whole genome sequencing rendered 266,042,616 2x250 bp Illumina reads (~118 Gbp). These reads were mapped against the Victoria diploid genome assembly using Bowtie2 (Figure 1) (Langmead and Salzberg, 2012). A total of 24,473,069 (9.2%) reads did not map and were de novo assembled with Masurca software (Zimin et al., 2013). This assembly resulted in 18,102 contigs, which were filtrated based on contaminations and identities (see Materials and Methods) in 18,032 contigs. The total length of the assembly was 28,982,419 bp, and that of the N50 was 2,176 bp. The annotation rendered 3,952 gene models with an N50 of 1,158 bp. These genes are considered the variable genes, since they are not present in Victoria. This accounts for ~7% of the total annotated genes (56,469) from the Victoria genome assembly. As in other studied species (Golicz et al., 2016b; Ruperao et al., 2021), the variable genes are significatively shorter and have less exons than the core genes based on t-tests (Figure 3).
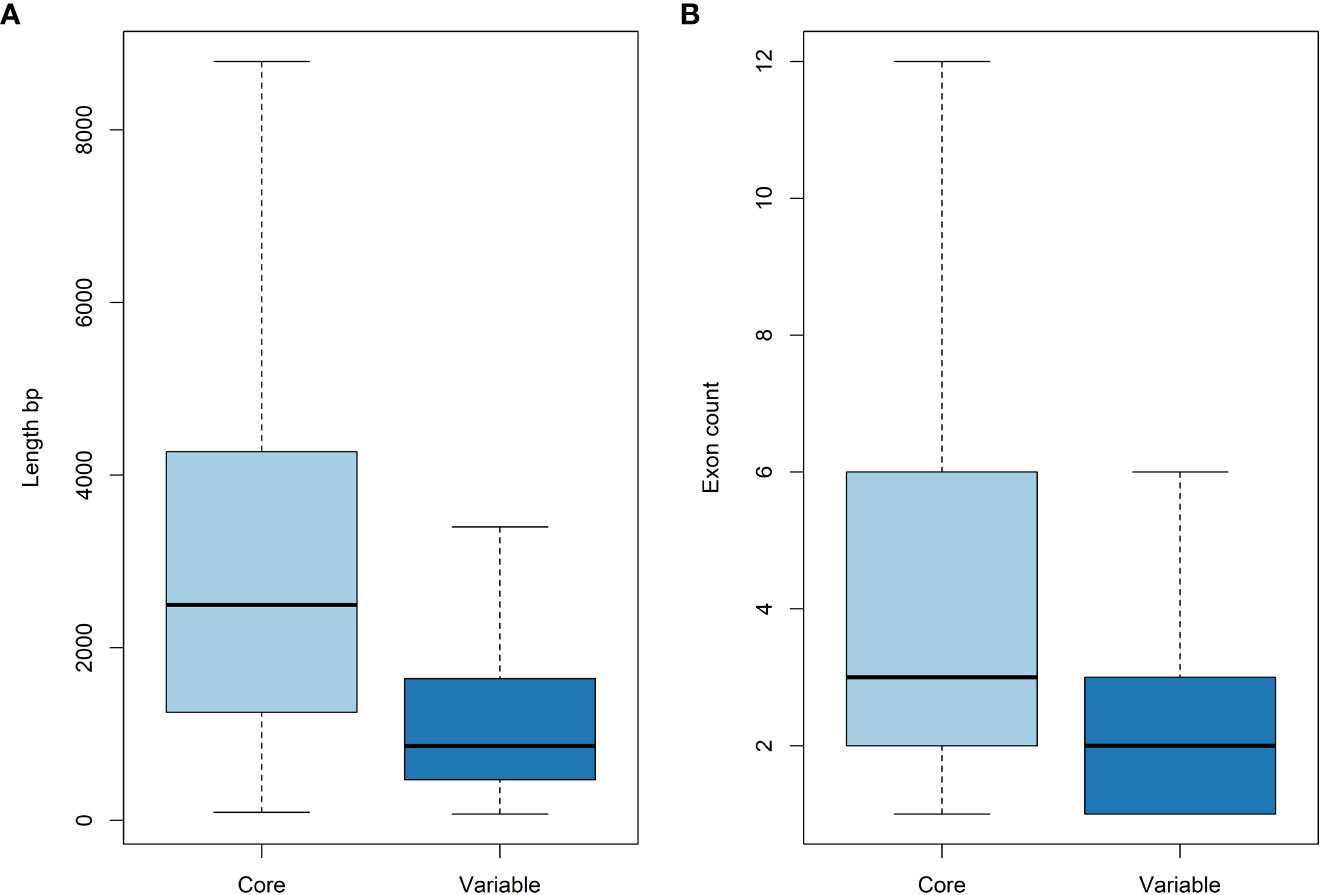
Figure 3 Gene length (A) and number of exons (B) of core and variable genes. Variable genes are shorter and have less exons than core genes.
In order to detect terms related to ploidy or reproduction, GO enrichment analysis was performed over the 3,952 variable genes (Figure 4) (Supplementary Table 2). Interestingly, the differentially enriched terms included those genes related to cell division such as DNA repair, telomere maintenance, and DNA helicase activity, suggesting that within the lost genes, genes associated with reproductive pathways are included.
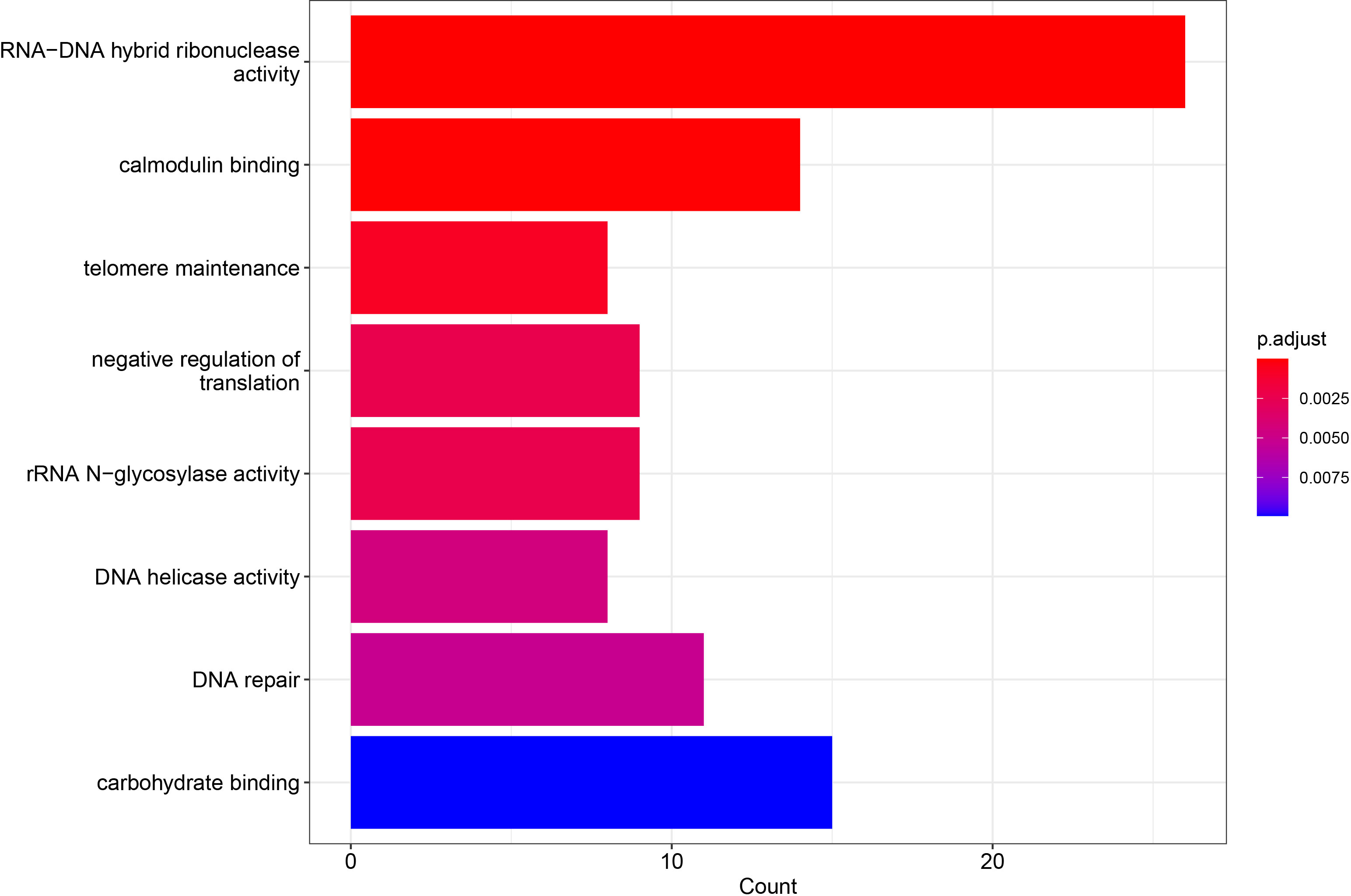
Figure 4 Differentially enriched GO terms of E. curvula pangenome variable genes. p-value cutoff was set in 0.05. The GO terms in the were sorted based on p-value. Three terms related to reproduction were found enriched.
3.3 Gene presence/absence
The gene presence/absence variations (PAVs) analysis was made with SGSGeneLoss software (Golicz et al., 2015), mapping Tanganyika INTA reads against the pangenome. In theory, all Victoria’s genes should be present in Tanganyika INTA, since it originated from this genotype. Nevertheless, 958 gene models (1.64%) of the Victoria genes were not found in Tanganyika INTA. Out of this, only 67 gene models aligned against the UniProt database and were not enriched in any pathway (Supplementary Table 3). PAVs were also assessed using transcriptomic reads from Don Walter cv., which is apomictic, finding that 53,208 genes of the pangenome present in Victoria, Tanganyika INTA, and Don Walter genotypes (core genes). A phylogenetic tree was constructed with these data being Tanganyika INTA and Victoria closer than Don Walter (Figure 5).
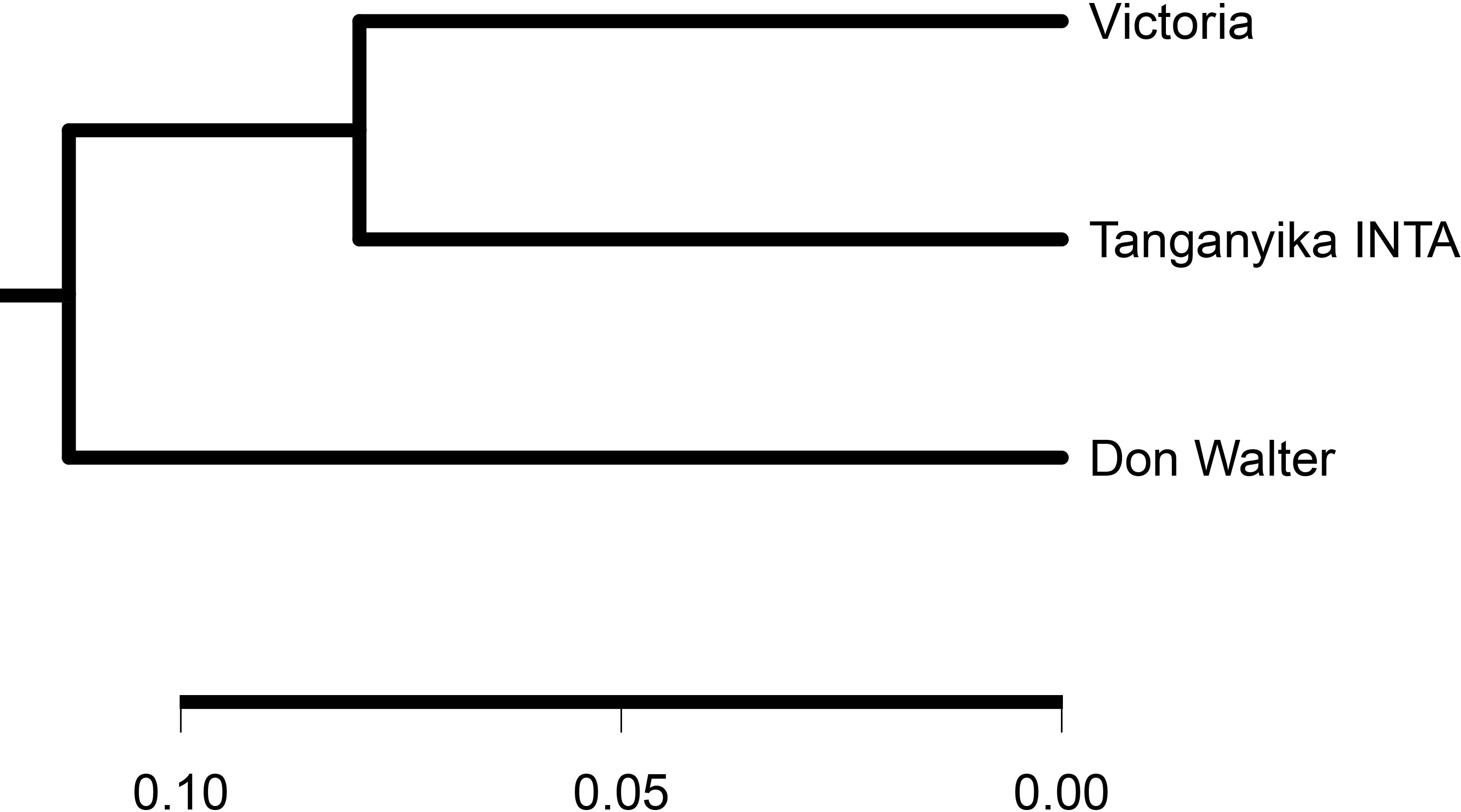
Figure 5 Phylogenetic tree of Eragrostis curvula including Victoria, Tanganyika INTA, and Don Walter INTA cvs. As expected, Tanganyika INTA is closer than Don Walter genotype.
3.4 Variant calling
The variant calling was performed by mapping Tanganyika INTA reads against the reference genome assembly. After filtering, a total of 6,679,446 SNPs were called using GATK software (McKenna et al., 2010). The coverage of the SNPs and the allele frequency were especially analyzed in order to understand the representation of the tetraploid genome onto the diploid one. Interestingly, the coverage of Tanganyika’s SNPs over the Victoria chromosomes displayed two peaks, representing the diploid and haploid coverage (Figure 6A). However, the coverage was not equally distributed in all the chromosomes, with contig12, contig6, contig1, contig28, and contig30 being predominantly haploid and contig10, contig38, and contig3 diploid. The allele frequency shows a peak mainly at one-fourth and three-fourths of the distribution, even though in contigs with increased haploid coverage, the frequency of one-half was significant. From these data, it seems that Tanganyika INTA has both allotetraploid and autotetraploid behavior. Another phenomenon affecting these plots is that a certain degree of heterozygosity is expected between alleles, so it was not possible to distinguish between sub-genome differences and heterozygosity.
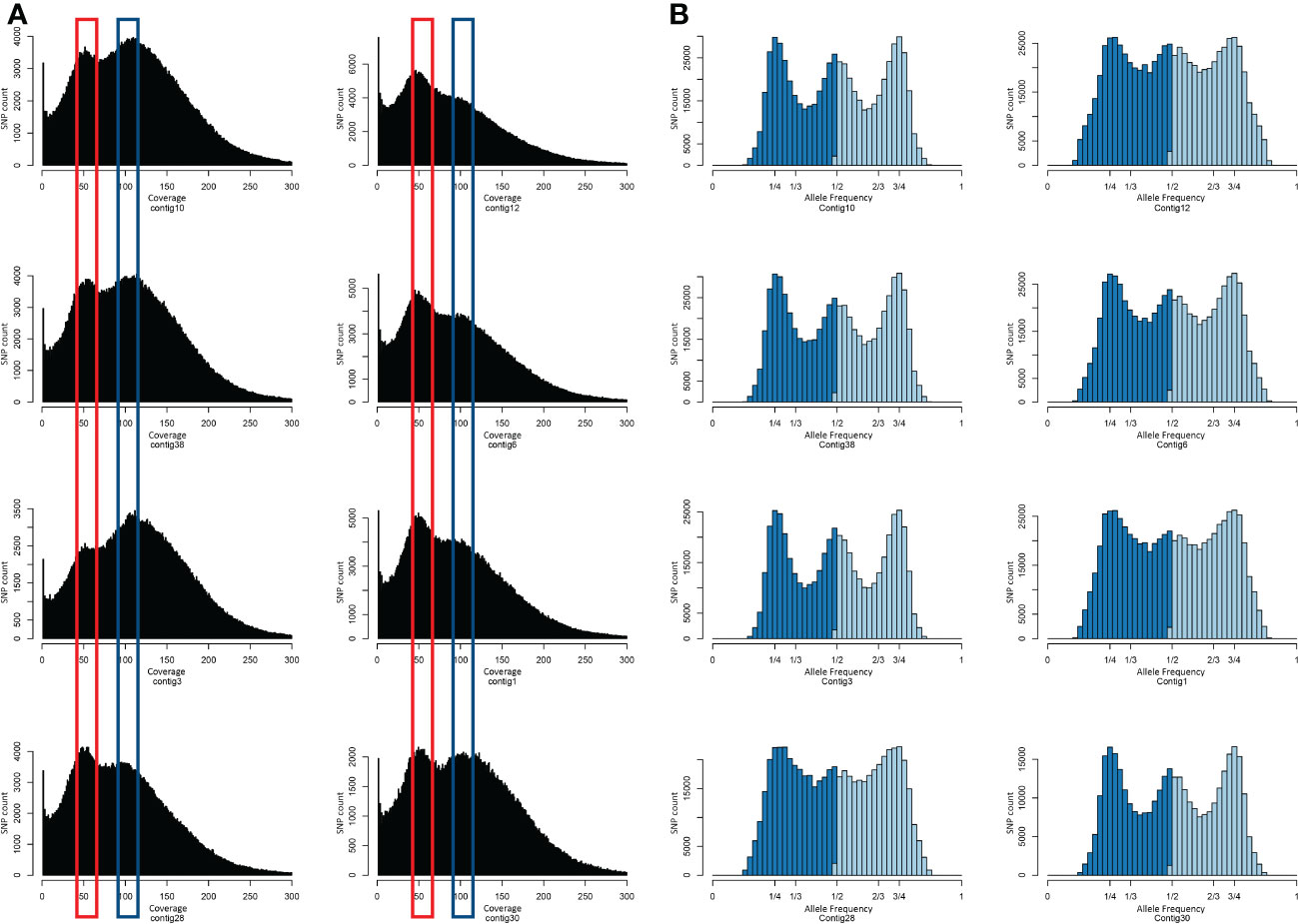
Figure 6 Tanganyika INTA SNP coverage (A) and allele frequency distribution (B) on the eight longest chromosomes of the Victoria genome assembly. (A) The y-axis represents the number of SNP and the x-axis the SNP coverage. The red and blue shapes highlight the two peaks around 50× and 100× showing the SNPs with diploid and tetraploid coverage. (B) The y-axis represents the number of SNP and the x-axis the allele frequency.
3.5 Variable gene validation
The variable gene models were functionally annotated using Uniprot database. In this way, five genes associated with reproduction and ploidy were selected for validation (Supplementary Table 1). Thus, PCR analysis was conducted using cDNA from leaves and spikelets and gDNA from spikelets extracted from Tanganyika INTA and Victoria genotypes (Figure 7).
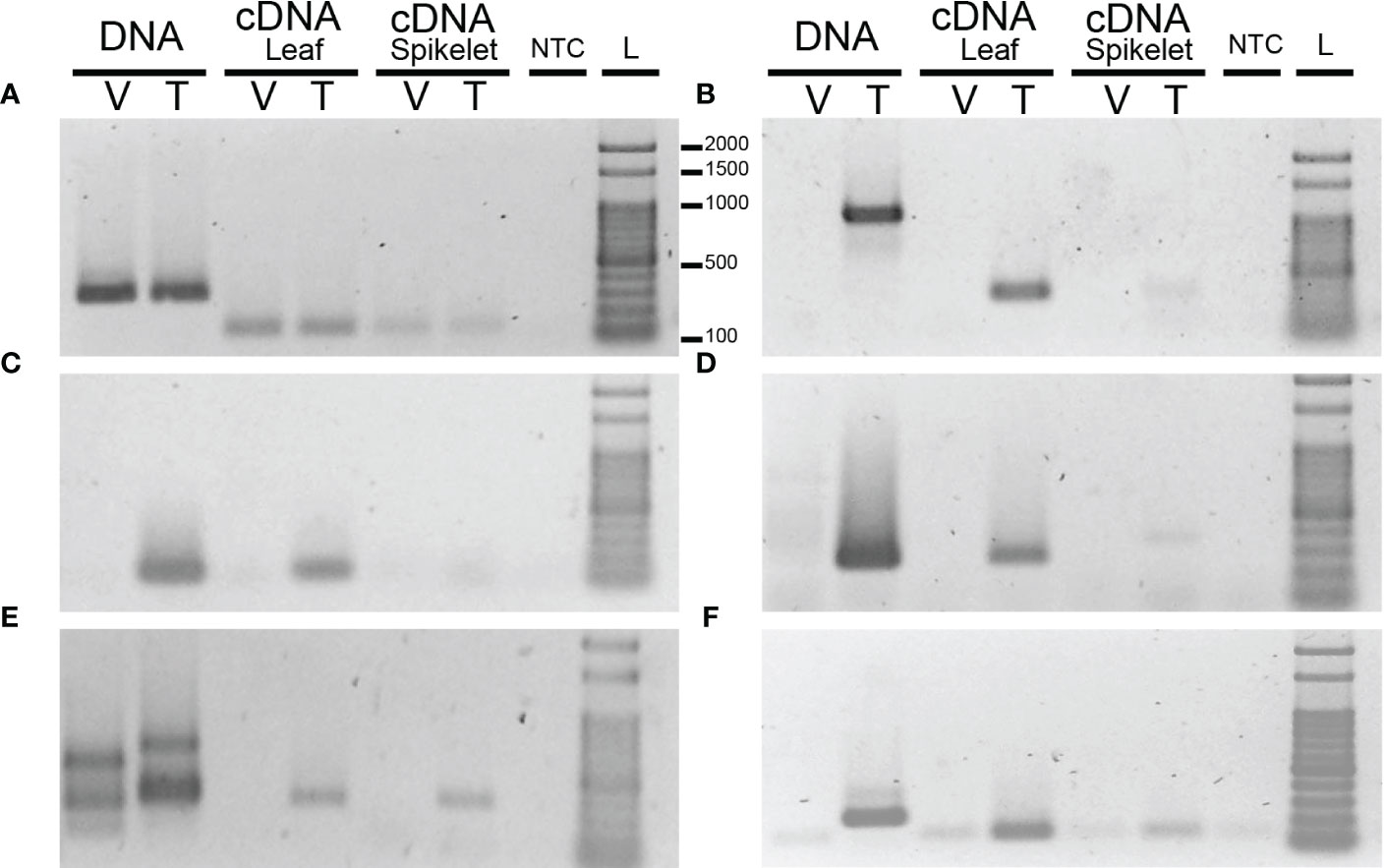
Figure 7 PCR amplification using DNA and cDNA from leaves and spikelets of E. curvula, Victoria (V), and Tanganyika (T) cvs. UBICE (ubiquitin conjugating enzyme transcript) (A) was used as positive control, and primers were designed over the annotated genes (B) ABD1, (C) PVA12, (D) MEE66, (E) ATXR2, and (F) RECQ4A. NTC (negative control) and L (Ladder). Presence/absence of the five genes was validated.
Degradation via ubiquitination is a pathway associated with reproduction (Okada et al., 2013; Zühl et al., 2019; Selva et al., 2020). In this way, the gene ABD1 (ABA-hypersensitive DCAF1, AT4G38480), which serves as a CUL4 E3 ligase substrate receptor, is only present in Tanganyika INTA and expressed in both leaves and spikelets (Figure 7B). Even though during the functional characterization of ABD1, the reproductive development was not assessed (Seo et al., 2014), this gene is involved in the regulation of abscisic acid (ABA), which was previously associated with embryo sac development (Chen et al., 2020). Therefore, the absence of ABD1 could change the concentration of ABA during embryo sac development, altering the reproductive performance.
The gene PVA12 was also not found in Victoria (Figure 7C). This gene interacts specifically with DC1 domain-containing protein, which produce vacuoleless gametophytes (D'ippólito et al., 2017). The lack of the vacuole in both male and female gametophytes produce arrest before the first round of mitosis after meiosis. As in Victoria, normal gametophyte development was found when PVA12 was mutated, suggesting that other genes could have the same function.
Maternal effect embryo (MEE) arrest are genes required for embryo development and embryogenesis. MEE66 was found in gDNA and cDNA of Tanganyika INTA and absent in Victoria (Figure 7D). The mutation of this gene in A. thaliana produces arrest at the two-cell embryo stage (Pagnussat et al., 2005).
Another gene not present in Victoria was ATXR2. This gene was only amplified in cDNA, since the primers were specifically designed on the splice junction (Figure 7E). ATXR2 repress de novo shoot organogenesis activating the expression of ARR5 and ARR7 via H3K36me3 marks in their promoters (Lee et al., 2021). When ATXR2 is mutated, ARR5 and ARR7 are not expressed and shoot regeneration is improved, since these genes activate WUS expression. Even more, during embryogenesis, ARR genes are also activated (Müller and Sheen, 2008), suggesting that the lack of ATXR2 in Victoria could distort the normal embryo development.
A key gene related to reproduction and probably with ploidy was RECQ4A, a DNA helicase that repairs double-stranded breaks during DNA replication. Although faint bands, product of dimers, were found in gDNA and cDNA of Victoria, the expected amplicon size for RECQ4A was only observed in Tanganyika INTA cDNA and gDNA (Figure 7F). In meiosis, double-stranded breaks are catalyzed by SPO22-1 and SPO11-2 genes, which are part of the MiMe complex used to produce mitosis instead of meiosis in rice and A. thaliana (d'Erfurth et al., 2009; Mieulet et al., 2016). Loss of function of RECQ4A in A. thaliana produces chromatin bridges between non-homologous chromosomes producing defective pollen and reduced fertility, whereas non-alterations during recombination were observed (Higgins et al., 2011). Even though it was not very frequent, recq4a mutant showed fragments of chromosomes due to the tension of the chromatin bridges between non-homologous chromosomes.
4 Discussion
In this study, a pangenomic approach was adopted to unravel genome diversity in E. curvula species. Even though this grass is studied as an apomictic model (Carballo et al., 2021b), here, we focused on the alterations produced by a recent diploidization event (Echenique et al., 1996) of the tetraploid cv. Tanganyika INTA to produce the Victoria genotype, obtained by in vitro culture of inflorescences (Cardone et al., 2006). The ploidy reduction brought about the change in reproductive mode from facultative apomictic in Tanganyika INTA to sexual in the diploid Victoria.
Using flow cytometry, the genome size of both the diploid Victoria and the tetraploid Tanganyika INTA genotypes were measured, resulting in ~600 and ~1,250 Mb, respectively. These values were expected for tetraploid and diploid E. curvula genotypes and were in concordance with previous measurements (Zappacosta et al., 2019). Interestingly, the genome size of Tanganyika INTA was bigger than that of the allotetraploid E. tef (~800 Mb) and closer to that of E. nindensis (1,035 Mb) (Hundera et al., 2000; Pardo et al., 2020).
As in other species, a mapping approach was used to assemble the new contigs (Golicz et al., 2016b; Gao et al., 2019; Ruperao et al., 2021). The mapping rate of Tanganyika INTA was close to 92%, and the new pangenome increased the length up to 28,982,419 bp. The newly annotated gene models classified as variable were 3,952. The SGSGeneLoss software also found that only 958 of the gene models (1.64%) present in Victoria were not found in Tanganyika INTA. Only 64 out of 958 genes found a match against UniProt database and were not particularly enriched in any term or pathway. Since Victoria originated from Tanganyika INTA, in theory, all the genes present in the tetraploid genome should be present in the diploid. However, it is known that some biases are produced during the sequencing, especially in polyploid genomes, that could produce loss of coverage in certain genomic regions (Schatz et al., 2012). Even more, as in most of the genome assemblies, small contaminations that are hard to detect could be inserted in the Victoria genome assembly responsible for the genes absent in Tanganyika INTA (Steinegger and Salzberg, 2020).
There are two main forces responsible for natural diploidization in plant species: cytological diploidization and genic diploidization (Ma and Gustafson, 2005). Cytological diploidization produces chromosomal rearrangements that eventually produce homologous pairing and exclude homologous pairing. On the other hand, genic diploidization is the consequence of gene deletions during evolution that lead to failures in chromosome pairing (Li et al., 2021b). Syntenic analysis between Victoria and E. tef showed that, although there are some rearrangements between genomes, most of the chromosomes are well conserved (Carballo et al., 2019). Given these results, it is logical to think that Victoria was produced by the regeneration of a haploid microspore. It is also important to note that even though Tanganyika INTA female gametophyte has approximately 95% of apomeiosis, the male meiosis is completely normal (Rodrigo et al., 2017). Therefore, probably, Victoria was regenerated from a male gamete that, after recombination, lost the genes responsible for the apomictic development. Even more, the sexual pathway seems to be affected as well, since Victoria has decreased fertility and yield. The external genetic and epigenetic changes applied by the tissue culture could also be responsible for the variations in the reproductive pathway (Madlung and Comai, 2004; Bednarek and Orłowska, 2020). A plausible explanation for the decrease in the reproductive performance in Victoria is that the sexual reproduction could not overcome the loos of the apomictic pathway.
The SNP coverage over the chromosome length contigs in Victoria showed a binomial distribution with one peak around 50× (representing haploid coverage) and other close to 100×. This supports the idea that Tanganyika INTA is a segmental allotetraploid with a sub-genome coverage peak at 50×, and both alleles are covered at the 100× peak (Figure 6A). Interestingly, not all the chromosomes show the same distribution; Contig12, Contig6, Contig1, Contig30, and Contig28 are predominantly covered by one sub-genome, whereas Contig10, Contig38, and Contig3 are fully covered. This is in agreement with the statement that allopolyploidy and autopolyploidy show continuous variation in sub-genome divergence rather than being independent processes (Stebbins, 1947; Madlung and Comai, 2004). In fact, in many different species, it was observed that both auto- and allopolyploids present certain degree of multi- and bivalent pairing during meiosis (Li et al., 2021b). Allele frequency correlates with the genome coverage, showing diploid-like trend in contigs with predominantly 50× of coverage and tetraploid like distribution in the 100× covered contigs (Figure 6B). A similar behavior was shown in Spartina pectinate, where genotypes classified as allotetraploid displayed a polysomic inheritance (Crawford et al., 2016). The results obtained in this work suggest that Tanganyika INTA is segmental allotetraploid, whereas in other E. curvula cultivars, they are classified as both allotetraploid and segmental allotetraploid, based on cytological observations of the meiosis pairing (Poverene, 1988).
Out of the 3,952 gene models, ABD1, PVA12, MEE66, ATXR2, and RECQ4A were selected to be used as templates for PCR primer design. These genes were validated in both gDNA and cDNA from leaves and spikelets from Tanganyika INTA and Victoria. Even though the presence/absence of ATXR2 in cDNA is clear, several non-specific bands were amplified in gDNA of Victoria and Tanganyika INTA, suggesting that these primers also amplify in repetitive regions that are hard to sequence and assembly not present in E. curvula databases. All the genes assessed here are, at some point, associated with both reproduction and ploidy level. Regarding the genes that are absent in Victoria, it is important to note that after diploidization, the reproductive performance of Victoria was seriously affected (Carballo et al., 2021b). The lack of some genes such as RECQ4A, MEE66, and PVA12, which are directly involved in the reproductive pathway, could be the cause of the reduction in fertility. Particularly, RECQ4A is a helicase, and its mutation in A. thaliana produces defective pollen and reduces fertility (Higgins et al., 2011). Moreover, RECQ4A mutants showed chromatin bridges between non-homologous chromosomes and enhanced homologous recombination in somatic cells (Hartung et al., 2007; Higgins et al., 2011).
In conclusion, the pangenome approach used here provides evidence about the E. curvula ploidy nature and complements cytological observations in previous works (Cardone et al., 2006), indicating that Tanganyika INTA is segmental allotetraploid. In addition, it gives information about the genes related to both reproduction and ploidy. The loose of the pathways involved in apomixis in Victoria could be associated with the disruption or lack of the genetic region linked to apomixis and/or the presence of epigenetic marks that are characteristics of a certain ploidy level. Moreover, the loose of genes involved in apomixis reduce the mechanisms available for reproduction, decreasing the fertility of Victoria.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, BioProject: PRJNA914706, SRA: SRR22846089.
Author contributions
Conception: JC, VE, DZ, and JS. Experimental design: JC, VE, and JS. Materials: JC, JS, and DZ. Experimental procedures: JC, AB, JS, and DZ. Bioinformatic data processing: JC and AG. Analysis and interpretation: JC, VE, MC, and DZ. Writing: JC, VE, MC, and DZ. Critical review: JC, EA, MC, and VE. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the following: H2020-MSCA-RISE-2019, “Mechanisms of apomictic Developments (MAD),” ID 872417; H2020-MSCA-RISE-2020, “The polyploidy paradigm and its role in plant breeding (POLYPLOID),” ID 101007438; CONICET, “Estudio de la región determinante de la apomixis en Eragrostis curvula utilizando herramientas de secuenciación de última generación,” ID PIP 11220200101905CO; and BBSRC, “An evolutionary approach to optimising synthetic apomixis in cereal crops,” ID BB/V016466/1.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie Grant Agreement No 101007438. This project has received funding from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie Grant Agreement No 872417.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1133986/full#supplementary-material
References
Altschul, S., Gish, W., Miller, W., Myers, E., Lipman, D. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. doi: 10.1038/35048692
Aranda, P., LaJoie, D., Jorcyk, C. (2012). Bleach gel: A simple agarose gel for analyzing RNA quality. Electrophoresis 33, 366–369. doi: 10.1002/elps.201100335
Bednarek, P. T., Orłowska, R. (2020). Plant tissue culture environment as a switch-key of (epi) genetic changes. Plant Cell Tiss Org 140, 245–257. doi: 10.1007/s11240-019-01724-1
Callaway, E. (2013). Shrub genome reveals secrets of flower power. Nat. News. doi: 10.1038/nature.2013.14426
Carballo, J., Santos, B., Zappacosta, D., Garbus, I., Selva, J. P., Gallo, C. A., et al. (2019). A high-quality genome of Eragrostis curvula grass provides insights into poaceae evolution and supports new strategies to enhance forage quality. Sci. Rep-UK 9, 1–15. doi: 10.1038/s41598-019-46610-0
Carballo, J., Zappacosta, D., Marconi, G., Gallardo, J., Di Marsico, M., Gallo, C. A., et al. (2021a). Differential methylation patterns in apomictic vs. sexual genotypes of the diplosporous grass Eragrostis curvula. Plants 10, 946. doi: 10.3390/plants10050946
Carballo, J., Zappacosta, D., Selva, J. P., Caccamo, M., Echenique, V. (2021b). Eragrostis curvula, a model species for diplosporous apomixis. Plants 10, 1818. doi: 10.3390/plants10091818
Cardone, S., Polci, P., Selva, J. P., Mecchia, M., Pessino, S., Hermann, P., et al. (2006). Novel genotypes of the subtropical grass Eragrostis curvula for the study of apomixis (diplospory). Euphytica 151, 263–272. doi: 10.1007/s10681-006-9156-x
Cervigni, G., Paniego, N., Díaz, M., Selva, J. P., Zappacosta, D., Zanazz, D., et al. (2008a). Expressed sequence tag analysis and development of gene associated markers in a near-isogenic plant system of Eragrostis curvula. Plant Mol. Biol. 67, 1–10. doi: 10.1007/s11103-007-9282-4
Cervigni, G., Paniego, N., Pessino, S., Selva, J. P., Díaz, M., Spangenberg, G., et al. (2008b). Gene expression in diplosporous and sexual eragrostis curvula genotypes with differing ploidy levels. Plant Mol. Biol. 67, 11–23. doi: 10.1007/s11103-008-9305-9
Chen, K., Li, G. J., Bressan, R. A., Song, C. P., Zhu, J. K., Zhao, Y. (2020). Abscisic acid dynamics, signaling, and functions in plants. J. Integr. Plant Biol. 62, 25–54. doi: 10.1111/jipb.12899
Chen, X., Nilanthi, D., Yang, Y., Wu, H. (2016). Anther culture and plant regeneration of tetraploid purple coneflower (Echinacea purpurea l.). J. Biosci. Medicines 4, 89–96. doi: 10.4236/jbm.2016.412013
Cho, M. S., Zapata, F. (1988). Callus formation and plant regeneration in isolated pollen culture of rice (Oryza sativa l. cv. Taipei 309). Plant Sci. 58, 239–244. doi: 10.1016/0168-9452(88)90014-3
Christensen, J. R., Borrino, E., Olesen, A., Andersen, S. B. (1997). Diploid, tetraploid, and octoploid plants from anther culture of tetraploid orchard grass, Dactylis glomerata l. Plant Breed 116, 267–270. doi: 10.1111/j.1439-0523.1997.tb00994.x
Crane, C. (2001). “Classification of apomictic mechanisms,” in The flowering of apomixis: From mechanisms to genetic engineering. Eds. Savidan, Y., Carman, J. G., Dresselhaus, T. (Mexico: ClMMYT, IRD, European Commission OC VI (FAIR), 24–43. Mexico.
Crawford, J., Brown, P., Voigt, T., Lee, D. K. (2016). Linkage mapping in prairie cordgrass (Spartina pectinata link) using genotyping-by-sequencing. Mol. Breed. 36, 1–12. doi: 10.1007/s11032-016-0484-9
d'Erfurth, I., Jolivet, S., Froger, N., Catrice, O., Novatchkova, M., Mercier, R. (2009). Turning meiosis into mitosis. PloS Biol. 7, e1000124. doi: 10.1371/journal.pbio.1000124
D'ippólito, S., Arias, L., Casalongué, C., Pagnussat, G., Fiol, D. (2017). The DC 1-domain protein VACUOLELESS GAMETOPHYTES is essential for development of female and male gametophytes in arabidopsis. Plant J. 90, 261–275. doi: 10.1111/tpj.13486
Doležel, J., Greilhuber, J., Lucretti, S., Meister, A., Lysák, M., Nardi, L., et al. (1998). Plant genome size estimation by flow cytometry: inter-laboratory comparison. Ann. Bot-London 82, 17–26. doi: 10.1093/oxfordjournals.aob.a010312
Echenique, V., Polci, P., Mroginski, L. (1996). Plant regeneration in weeping lovegrass (Eragrostis curvula) through inflorescence culture. Plant Cell Tissue Org 46, 123–130. doi: 10.1007/BF00034845
Galbraith, D., Harkins, K., Maddox, J., Ayres, N., Sharma, D., Firoozabady, E. (1983). Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 220, 1049–1051. doi: 10.1126/science.220.4601.1049
Gao, L., Gonda, I., Sun, H., Ma, Q., Bao, K., Tieman, D., et al. (2019). The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 51, 1044–1051. doi: 10.1038/s41588-019-0410-2
Garbus, I., Romero, J. R., Selva, J. P., Pasten, M. C., Chinestra, C., Carballo, J., et al. (2017). De novo transcriptome sequencing and assembly from apomictic and sexual Eragrostis curvula genotypes. PloS One 12, e0185595. doi: 10.1371/journal.pone.0185595
Garbus, I., Selva, J. P., Pasten, M. C., Bellido, A., Carballo, J., Albertini, E., et al. (2019). Characterization and discovery of miRNA and miRNA targets from apomictic and sexual genotypes of eragrostis curvula. BMC Genomics 20, 1–13. doi: 10.1186/s12864-019-6169-0
Golicz, A., Batley, J., Edwards, D. (2016a). Towards plant pangenomics. Plant Biotechnol. J. 14, 1099–1105. doi: 10.1111/pbi.12499
Golicz, A., Bayer, P., Barker, G., Edger, P., Kim, H.-R., Martinez, P., et al. (2016b). The pangenome of an agronomically important crop plant brassica oleracea. Nat. Commun. 7, 1–8. doi: 10.1038/ncomms13390
Golicz, A., Martinez, P., Zander, M., Patel, D. A., Van De Wouw, A., Visendi, P., et al. (2015). Gene loss in the fungal canola pathogen leptosphaeria maculans. Funct. Integr. Genomic 15, 189–196. doi: 10.1007/s10142-014-0412-1
Hartung, F., Suer, S., Puchta, H. (2007). Two closely related RecQ helicases have antagonistic roles in homologous recombination and DNA repair in Arabidopsis thaliana. P Natl. Acad. Sci. U.S.A. 104, 18836–18841. doi: 10.1073/pnas.070599810
Higgins, J., Ferdous, M., Osman, K., Franklin, F. C. (2011). The RecQ helicase AtRECQ4A is required to remove inter-chromosomal telomeric connections that arise during meiotic recombination in arabidopsis. Plant J. 65, 492–502. doi: 10.1111/j.1365-313X.2010.04438.x
Holt, C., Yandell, M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinf. 12, 1–14. doi: 10.1186/1471-2105-12-491
Hundera, F., Arumuganathan, K., Baenziger, P. S. (2000). Determination of relative nuclear DNA content of tef [Eragrostis tef (Zucc.) trotter] using flow cytometry. J. Genet. Breed 54, 165–168.
Jayakodi, M., Padmarasu, S., Haberer, G., Suresh Bonthala, V., Gundlach, H., Monat, C., et al. (2020). The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 588, 284–289. doi: 10.1038/s41586-020-2947-8
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Klepikova, A. V., Kasianov, A. S., Gerasimov, E. S., Logacheva, M. D., Penin, A. A. (2016). A high resolution map of the arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 88, 1058–1070. doi: 10.1111/tpj.13312
Langmead, B., Salzberg, S. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, K., Park, O.-S., Go, Y., Yu, J., Han, J. H., Kim, J., et al. (2021). Arabidopsis ATXR2 represses de novo shoot organogenesis in the transition from callus to shoot formation. Cell Rep. 7, 109980. doi: 10.1016/j.celrep.2021.109980
Li, J., Yuan, D., Wang, P., Wang, Q., Sun, M., Liu, Z., et al. (2021a). Cotton pan-genome retrieves the lost sequences and genes during domestication and selection. Genome Biol. 22, 1–26. doi: 10.1186/s13059-021-02351-w
Li, Y. H., Zhou, G., Ma, J., Jiang, W., Jin, L. G., Zhang, Z., et al. (2014). De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 32, 1045–1052. doi: 10.1038/nbt.2979
Li, Z., McKibben, M., Finch, G., Blischak, P., Sutherland, B., Barker, M. (2021b). Patterns and processes of diploidization in land plants. Ann. Rev. Plant Biol. 72, 387–410. doi: 10.1146/annurev-arplant-050718-100344
Lovell, J., Jenkins, J., Lowry, D., Mamidi, S., Sreedasyam, A., Weng, X., et al. (2018). The genomic landscape of molecular responses to natural drought stress in panicum hallii. Nat. Commun. 9, 1–10. doi: 10.1038/s41467-018-07669-x
Ma, X.-F., Gustafson, J. P. (2005). Genome evolution of allopolyploids: a process of cytological and genetic diploidization. Cytogenet. Genome Res. 109, 236–249. doi: 10.1159/000082406
Madlung, A., Comai, L. (2004). The effect of stress on genome regulation and structure. Ann. Bot-London 94, 481–495. doi: 10.1093/aob/mch172
Marçais, G., Delcher, A., Phillippy, A., Coston, R., Salzberg, S., Zimin, A. (2018). MUMmer4: A fast and versatile genome alignment system. PloS Comput. Biol. 14, e1005944. doi: 10.1371/journal.pcbi.1005944
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Meier, M., Zappacosta, D., Selva, J. P., Pessino, S., Echenique, V. (2011). Evaluation of different methods for assessing the reproductive mode of weeping lovegrass plants, Eragrostis curvula (Schrad.). Nees. Aust. J. Bot. 59, 253–261. doi: 10.1071/BT10267
Mergner, J., Frejno, M., List, M., Papacek, M., Chen, X., Chaudhary, A., et al. (2020). Mass-spectrometry-based draft of the arabidopsis proteome. Nature 579, 409–414. doi: 10.1038/s41586-020-2094-2
Mieulet, D., Jolivet, S., Rivard, M., Cromer, L., Vernet, A., Mayonove, P., et al. (2016). Turning rice meiosis into mitosis. Cell Res. 26, 1242–1254. doi: 10.1038/cr.2016.117
Montenegro, J., Golicz, A., Bayer, P., Hurgobin, B., Lee, H.-T., Kenneth Chan, C.-K., et al. (2017). The pangenome of hexaploid bread wheat. Plant J. 90, 1007–1013. doi: 10.1111/tpj.13515
Müller, B., Sheen, J. (2008). Cytokinin and auxin interaction in root stem-cell specification during early embryogenesis. Nature 453, 1094–1097. doi: 10.1038/nature06943
Okada, T., Hu, Y., Tucker, M., Taylor, J., Johnson, S., Spriggs, A., et al. (2013). Enlarging cells initiating apomixis in Hieracium praealtum transition to an embryo sac program prior to entering mitosis. Plant Physiol. 163, 216–231. doi: 10.1104/pp.113.219485
Ou, S., Su, W., Liao, Y., Chougule, K., Agda, J., Hellinga, A., et al. (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 1–18. doi: 10.1186/s13059-019-1905-y
Pagnussat, G., Yu, H.-J., Ngo, Q., Rajani, S., Mayalagu, S., Johnson, C., et al. (2005). Genetic and molecular identification of genes required for female gametophyte development and function in arabidopsis. Development 132, 603–614. doi: 10.1242/dev.01595
Pardo, J., Man Wai, C., Chay, H., Madden, C., Hilhorst, H., Farrant, J., et al. (2020). Intertwined signatures of desiccation and drought tolerance in grasses. P Natl. Acad. Sci. 117, 10079–10088. doi: 10.1073/pnas.2001928117
Pasten, M. C., Carballo, J., Gallardo, J., Zappacosta, D., Selva, J. P., Rodrigo, J. M., et al. (2022). A combined transcriptome-miRNAome approach revealed that a kinesin gene is differentially targeted by a novel miRNA in an apomictic genotype of eragrostis curvula. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1012682
Poverene, M. (1988). Contribución citogenética y quimiosistemática a la taxonomía del pasto llorón, eragrostis curvula (Schrad.) nees s. lat (Bahía Blanca, Argentina: Universidad Nacional del Sur). Ph.D. Thesis.
Qin, P., Lu, H., Du, H., Wang, H., Chen, W., Chen, Z., et al. (2021). Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 184, 3542–3558. doi: 10.1016/j.cell.2021.04.046
R Core Team. (2018). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing). Available at: https://www.R-project.org/.
Rensink, W. A., Buell, C. R. (2004). Arabidopsis to rice. applying knowledge from a weed to enhance our understanding of a crop species. Plant Physiol. 135, 622–629. doi: 10.1104/pp.104.040170
Rodrigo, J. M., Zappacosta, D., Selva, J. P., Garbus, I., Albertini, E., Echenique, V. (2017). Apomixis frequency under stress conditions in weeping lovegrass (Eragrostis curvula). PloS One 12, e0175852. doi: 10.1371/journal.pone.0175852
Ruch, P., Teodoro, D., UniProt Consortium. (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi: 10.1093/nar/gkaa1100
Ruperao, P., Thirunavukkarasu, N., Gandham, P., Selvanayagam, S., Govindaraj, M., Nebie, B., et al. (2021). Sorghum pan-genome explores the functional utility for genomic-assisted breeding to accelerate the genetic gain. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.666342
Schatz, M., Witkowski, J., McCombie, W. R. (2012). Current challenges in de novo plant genome sequencing and assembly. Genome Biol. 13, 1–7. doi: 10.1186/gb-2012-13-4-243
Selva, J. P., Siena, L., Rodrigo, J. M., Garbus, I., Zappacosta, D., Romero, J. R., et al. (2017). Temporal and spatial expression of genes involved in DNA methylation during reproductive development of sexual and apomictic eragrostis curvula. Sci. Rep-UK 7, 1–11. doi: 10.1038/s41598-017-14898-5
Selva, J. P., Zappacosta, D., Carballo, J., Rodrigo, J. M., Bellido, A., Gallo, C. A., et al. (2020). Genes modulating the increase in sexuality in the facultative diplosporous grass Eragrostis curvula under water stress conditions. Genes 11, 969. doi: 10.3390/genes11090969
Seo, K. I., Lee, J. H., Nezames, C. D., Zhong, S., Song, E., Byun, M. O., et al. (2014). ABD1 is an arabidopsis DCAF substrate receptor for CUL4-DDB1–based E3 ligases that acts as a negative regulator of abscisic acid signaling. Plant Cell 26, 695–711. doi: 10.1105/tpc.113.119974
Stebbins, G. (1947). Types of polyploids: their classification and significance. Adv. Genet. 1, 403–429. doi: 10.1016/S0065-2660(08)60490-3
Steinegger, M., Salzberg, S. L. (2020). Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol. 21, 1–2. doi: 10.1186/s13059-020-02023-1
Tettelin, H., Masignani, V., Cieslewicz, M., Donati, C., Medini, C., Ward, N., et al. (2005). Genome analysis of multiple pathogenic isolates of streptococcus agalactiae: implications for the microbial “pan-genome”. P Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
VanBuren, R., Man Wai, C., Keilwagen, J., Pardo, J. (2018). A chromosome-scale assembly of the model desiccation tolerant grass Oropetium thomaeum. Plant Direct 2, e00096. doi: 10.1002/pld3.96
VanBuren, R., Man Wai, C., Wang, X., Pardo, J., Yocca, A. E., Wang, H., et al. (2020). Exceptional subgenome stability and functional divergence in the allotetraploid Ethiopian cereal teff. Nat. Commun. 11, 1–11. doi: 10.1038/s41467-020-14724-z
Walkowiak, S., Gao, L., Monat, C., Haberer, G., Kassa, M. T., Brinton, J., et al. (2020). Multiple wheat genomes reveal global variation in modern breeding. Nature 588, 277–283. doi: 10.1038/s41586-020-2961-x
Yu, G., Wang, L.-G., Han, Y., He, Q.-Y. (2012). clusterProfiler: an r package for comparing biological themes among gene clusters. Omics 16, 284–287. doi: 10.1089/omi.2011.0118
Zappacosta, D., Gallardo, J., Carballo, J., Meier, M., Rodrigo, J. M., Gallo, C. A., et al. (2019). A high-density linkage map of the forage grass Eragrostis curvula and localization of the diplospory locus. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00918
Zhang, G., Liu, X., Quan, Z., Cheng, S., Xu, X., Pan, S., et al. (2012). Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 30, 549–554. doi: 10.1038/nbt.2195
Zhao, J., Bayer, P., Ruperao, P., Saxena, R., Khan, A., Golicz, A., et al. (2020). Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnol. J. 18, 1946–1954. doi: 10.1111/pbi.13354
Zimin, A., Marçais, G., Puiu, D., Roberts, M., Salzberg, S., Yorke, J. (2013). The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677. doi: 10.1093/bioinformatics/btt476
Zimin, A., Puiu, D., Hall, R., Kingan, S., Clavijo, B., Salzberg, S. (2017). The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. Gigascience 6, gix097. doi: 10.1093/gigascience/gix097
Keywords: diploidization, Eragrostis curvula, plant reproduction, ploidy, apomixis
Citation: Carballo J, Bellido AM, Selva JP, Zappacosta D, Gallo CA, Albertini E, Caccamo M and Echenique V (2023) From tetraploid to diploid, a pangenomic approach to identify genes lost during synthetic diploidization of Eragrostis curvula. Front. Plant Sci. 14:1133986. doi: 10.3389/fpls.2023.1133986
Received: 29 December 2022; Accepted: 24 February 2023;
Published: 13 March 2023.
Edited by:
Inna Lermontova, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), GermanyReviewed by:
Alexandre Berr, UPR2357 Institut de Biologie Moléculaire des Plantes (IBMP), FranceMartin Mau, University of Saskatchewan, Canada
Ahmet L. Tek, Niğde Ömer Halisdemir University, Türkiye
Copyright © 2023 Carballo, Bellido, Selva, Zappacosta, Gallo, Albertini, Caccamo and Echenique. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viviana Echenique, echeniq@criba.edu.ar