 Ming Lei
Ming Lei Samantha C. Salvage
Samantha C. Salvage Antony P. Jackson
Antony P. Jackson Christopher L.-H. Huang
Christopher L.-H. Huang- 1Department of Pharmacology, University of Oxford, Oxford, United Kingdom
- 2Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom
- 3Physiological Laboratory, University of Cambridge, Cambridge, United Kingdom
Cardiac arrhythmias cause significant morbidity and mortality and pose a major public health problem. They arise from disruptions in the normally orderly propagation of cardiac electrophysiological activation and recovery through successive cardiomyocytes in the heart. They reflect abnormalities in automaticity, initiation, conduction, or recovery in cardiomyocyte excitation. The latter properties are dependent on surface membrane electrophysiological mechanisms underlying the cardiac action potential. Their disruption results from spatial or temporal instabilities and heterogeneities in the generation and propagation of cellular excitation. These arise from abnormal function in their underlying surface membrane, ion channels, and transporters, as well as the interactions between them. The latter, in turn, form common regulatory targets for the hierarchical network of diverse signaling mechanisms reviewed here. In addition to direct molecular-level pharmacological or physiological actions on these surface membrane biomolecules, accessory, adhesion, signal transduction, and cytoskeletal anchoring proteins modify both their properties and localization. At the cellular level of excitation–contraction coupling processes, Ca2+ homeostatic and phosphorylation processes affect channel activity and membrane excitability directly or through intermediate signaling. Systems-level autonomic cellular signaling exerts both acute channel and longer-term actions on channel expression. Further upstream intermediaries from metabolic changes modulate the channels both themselves and through modifying Ca2+ homeostasis. Finally, longer-term organ-level inflammatory and structural changes, such as fibrotic and hypertrophic remodeling, similarly can influence all these physiological processes with potential pro-arrhythmic consequences. These normal physiological processes may target either individual or groups of ionic channel species and alter with particular pathological conditions. They are also potentially alterable by direct pharmacological action, or effects on longer-term targets modifying protein or cofactor structure, expression, or localization. Their participating specific biomolecules, often clarified in experimental genetically modified models, thus constitute potential therapeutic targets. The insights clarified by the physiological and pharmacological framework outlined here provide a basis for a recent modernized drug classification. Together, they offer a translational framework for current drug understanding. This would facilitate future mechanistically directed therapeutic advances, for which a number of examples are considered here. The latter are potentially useful for treating cardiac, in particular arrhythmic, disease.
1 Introduction: normal and abnormal cardiac rhythms
1.1 Clinical importance of cardiac arrhythmias
The co-ordinated and effective mechanical function of the heart requires orderly initiation and conduction of electrophysiological action potential (AP, Figure 1A) signals through its participating excitable cells. This process involves successively the atria, atrioventricular node (AVN), Purkinje conducting tissue, and ventricular endocardial and epicardial myocardium (Figure 1B). The breakdown of this process constitutes the underlying cause of cardiac arrhythmias, the major contributors to clinical mortality and morbidity. Autopsies of 5–35-year-old patients implicate cardiac causes in 56.4% of non-traumatic sudden deaths. Of these, ∼30% may be arrhythmic, with many stemming from ischemic heart disease (Behr et al., 2003). However, ∼4% of sudden cardiac deaths (SCDs) show no accompanying structural abnormalities (Tung et al., 1994; Clancy and Kass, 2005; Martin et al., 2011d; Martin et al., 2012; Martin et al., 2013), implicating underlying channelopathy (Tester and Ackerman, 2007; Bezzina et al., 2015). Arrhythmias also arise as adverse effects of pharmacotherapeutic agents (Killeen, 2009).
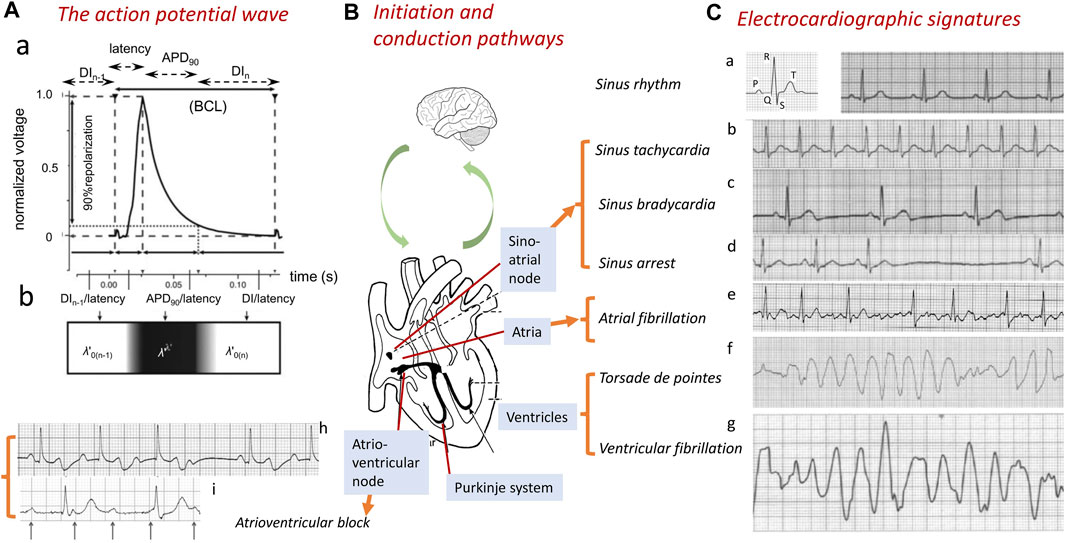
FIGURE 1. Normal and abnormal cardiac conduction. Generation of the normal wave of cardiac excitation, exemplified by (A) a propagating action potential (AP) in the right ventricle of a murine heart beating at 600 bpm. The AP comprises a depolarization phase following a conduction latency, followed by repolarization recovery and a refractory period (a). The AP waveform can be characterized by its BCL, APD90, latency, and the DI of the current (nth) and preceding [(n-1)th] action potential. The latter spatially map onto a traveling wave characterized by its conduction velocity θ and active and resting wavelengths λ′ and λ0’ and basic cycle distance, BCD’ = λ’ + λ0’ (b). The AP is (B) transmitted through successive initiating, sino-atrial node (SAN), conducting, atrioventricular (AV) and Purkinje, tissue, and contractile atrial and ventricular structures. These processes are subject to modulation at the systems, including the nervous system level. (C) Abnormalities in this communication disrupt the normal cardiac electrocardiographic signal (a), resulting in abnormal electrical patterns. These are exemplified by sinus rhythm tachy- or bradycardic abnormalities (a–c) or arrest (d) and breakdown in orderly excitation resulting in atrial fibrillation (e) or ventricular torsade de pointes (f) or fibrillation (g). AVN or Purkinje tissue conduction block is exemplified here by second-degree Mobitz I heart block (h) and third-degree complete heart block (i) [(A) from Figure 1 of the work of Matthews et al. (2013); (C) adapted from Figure 14.1 of the work of Huang (2021)].
Different arrhythmias show specific electrocardiographic (ECG) signatures varying with the cardiac region involved (Figure 1C) (Huang, 2021). Sinus node disorders manifest as tachycardic conditions, as well as, contrastingly, sinus pauses/arrests, chronotropic incompetence, and other bradycardic conditions. The latter constitute indications for ∼50% of the 106/y permanent pacemaker implants worldwide (de Marneffe et al., 1993; Dobrzynski et al., 2007; Dobrev, 2009). Requirements for their implantation increases with age, reaching 1:600 at age >65 years. The most common sustained arrhythmia, atrial fibrillation (AF), similarly age-related (Dun and Boyden, 2009; Wakili et al., 2011), affects 1%–2% of the general population (Stewart et al., 2002; Lloyd-Jones et al., 2004; Andrade et al., 2014), predisposing to further major cardiac and cerebrovascular morbidity and mortality (Stewart et al., 2002), including stroke (Wolf et al., 1991). Ventricular arrhythmias, exemplified by ventricular tachycardia (VT), potentially leading to ventricular fibrillation (VF) and SCD (Rubart and Zipes, 2005; Turakhia and Tseng, 2007), are implicated in >300,000 and ∼70,000 deaths/year in the United States (Kannel et al., 1987) and United Kingdom, respectively (Colquitt et al., 2014). Some degree of rhythmic cardiac activity persists with AVN or Purkinje conduction blocks. Of the different extents of AVN conduction block, first-degree block results in electrocardiographic (ECG) complexes showing consistent PR interval prolongations (>0.20 s). Second-degree blocks Mobitz type 1 (Wenckebach) and Mobitz type 2 both manifest as repeated ECG cycles with dropped QRS complexes, The latter are preceded by cycles showing progressively increasing (Figure 1Ch) and constant PR intervals, respectively. Third-degree heart block (Figure 1Ci) contrastingly manifests as complete P wave and QRS complex dissociation. Finally, Purkinje tissue right and left bundle branch blocks result in broadened (>120 ms duration) QRS complexes with abnormal kinetic features varying with right- or left-sided ECG leads and the site of the block.
1.2 Normal cardiac activity
Cardiac excitation is initiated and paced by repetitive cycles of sino-atrial node excitation (Lei et al., 2007; Donald and Lakatta, 2023). These trigger periodic waves of APs that then propagate through successive cardiac structures (Figure 1B) (Draper and Weidmann, 1951; Matthews et al., 2013). Each AP begins with rapid phase 0 depolarization from the resting membrane potential. Subsequent completion of the slower recovery over action potential duration (APD) and effective refractory period (ERP) permits further excitation cycles. Atrial APs show triangular waveforms with prominent initial phase 1 recoveries (Figure 2A). Human ventricular APs (Figure 2B) similarly begin with rapid (∼400 V s−1) phase 0 depolarization from the (∼-90 mV) resting potential to a +40 to +60 mV overshoot voltage. The subsequent phase 1 initial rapid repolarization is followed by a prolonged phase 2 plateau, lengthening the APD and ERP. It is terminated by phase 3 repolarization leading to phase 4 electrical diastole (Weidmann, 1951). APs initiate the physiologically important cellular events mediating cardiac systole/diastole. They also drive cell-to-cell axial currents through cytoplasmic and gap junction resistances. They also possibly activate cell–cell ephaptic couplings. The latter events enable their propagation into hitherto quiescent cells, forming a coherent, advancing electrical wavefront through conducting or myocardial tissue, while leaving a trailing, recovering refractory region. Such excitation wavefronts span the thickness of the atrial wall. Ventricular propagation involves excitation proceeding from the transmural epicardium to endocardium and from the base to apex.
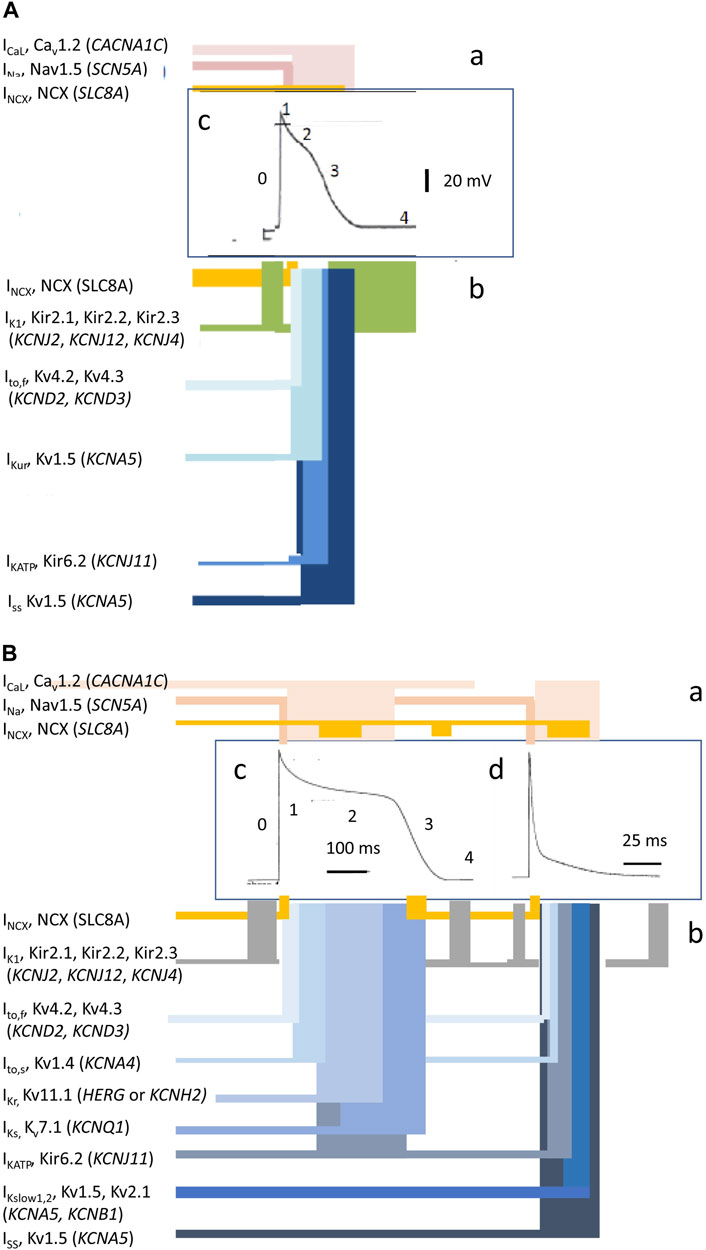
FIGURE 2. Ion current contributions to basic features of cardiac electrophysiological excitation. Human atrial (A) and human and murine ventricular (B) inward (a) and outward ionic current contributions (b), atiributable to surface membrane ion channels, to human (c) and mouse (d) AP waveforms [from Figure 1 of the work of Huang (2017) and Figure 1 of the work of Huang et al. (2020)].
2 Electrophysiological basis of arrhythmogenesis
2.1 Spatial stability and instability in the propagated AP wave
This normal propagation of activation waves through continuous electrically coupled three-dimensional cardiomyocyte networks (Kucera et al., 1998; Kléber and Rudy, 2004) can be disrupted by pro-arrhythmic ectopic triggering events. In contrast, compromised spatial and temporal coherence and stability in the wavefronts generate an arrhythmic substrate. The spatial extent and stability of this excitation depend on its wavelength, λ, within which further premature excitation cannot occur. This normally ensures that only one such generated and propagated wave can occur per heartbeat. The term λ is determined by the propagation velocity θ and recovery ERP or APD: λ = θ×ERP or θ×APD (Huang, 2017). Figure 1Aa illustrates a typical basic cycle length (BCL) containing a murine ventricular AP. It defines its critical temporal properties of latency, APD, and succeeding diastolic interval, DIn, at 90% repolarization (APD90 and DI90). It then maps these temporal properties onto their corresponding spatial properties, assuming a constant AP conduction velocity θ. In the cardiac tissue through which the AP propagates, the latter form active and resting wavelengths λ′ and λ0’, making up the basic cycle distance, BCD’ = λ’ + λ0’, of the propagated AP waveform (Figure 1Ab).
Larger λ reduces the likelihood that areas of depolarization and repolarization collide to produce obstacles causing unidirectional conduction blocks. In the latter event, APs have to take a slow conducting pathway to traverse this non-conducting myocardium. Such blocks result from depolarization abnormalities (Figure 3, path 1; dark gray) (King et al., 2013a). Collision would involve previously excited adjacent regions of the normal myocardium (path 2; white). However, should the latter possess APs with sufficient λ (yellow region), the collision would simply involve refractory tissue, precluding re-excitation in path 2 (Figure 3Aa). Similar outcomes result from impulses from ectopic triggering immediately following the normal AP (Figure 3Ab). The traveling wave then passes undisrupted over the heterogeneity (Weiss et al., 2005). Contrastingly, an AP conducting retrogradely along path 1 entering the beginning of path 2 with λ reduced to values smaller than the dimensions of the available circuits would initiate persistent re-entrant excitation into the recovered myocardial regions (Figure 3Bc) (King et al., 2013a; Huang, 2017).
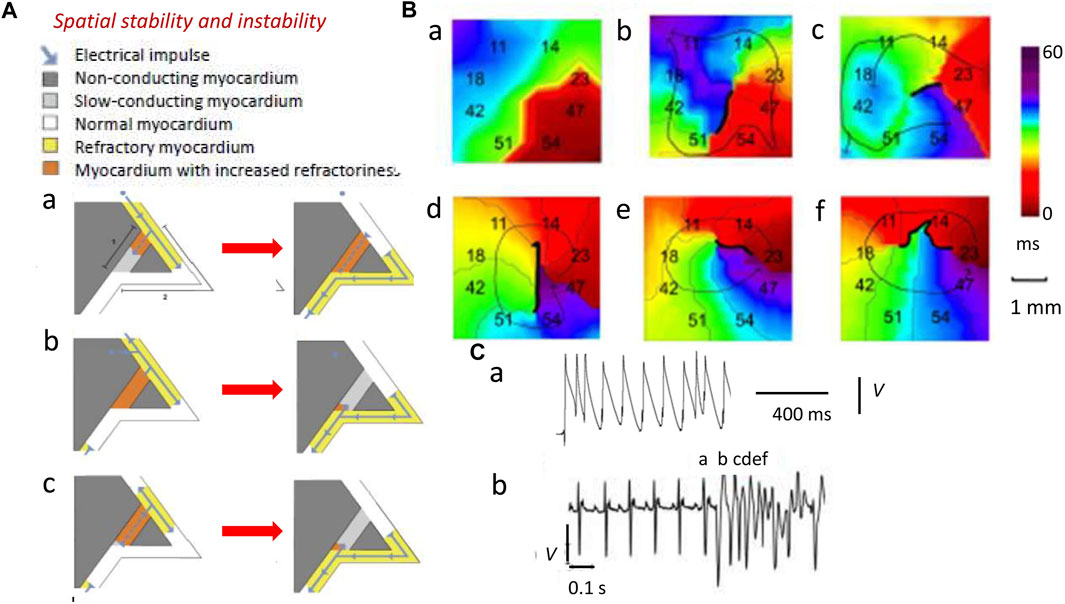
FIGURE 3. Spatial instabilities in the genesis of re-entrant arrhythmia. (A) Pro-arrhythmic mechanisms involving the conducting pathway with compromised conduction [dark gray path 1, (a)] and a second normally conducting pathway [white path 2, (a)]. A normal AP conduction with velocity θ and ERP has wavelength λ = θ × ERP (yellow region) in path 2 (b) (blue arrow). If the latter initiates a slow-conducting AP, this travels along path 1 (a) but under normal circumstances (b), this collides with refractory tissue within path 2 and then cannot re-enter the circuit (b). Thus, an abnormal triggered impulse immediately following the normal AP (a) cannot enter the refractory path 2 (b). However, a retrogradely conducting AP with reduced wavelength Λ shorter than the dimensions of the propagation pathways, due to reduced θ and/or ERP, along path 1 (a) entering the beginning of path 2 (b) causes self-perpetuating re-entrant excitation (c). (B) Isochronal AP propagation multi-electrode array mapping of re-entrant circuit initiation of ventricular tachycardia (VT) in the right ventricular epicardium of isolated Langendorff-perfused murine Scn5a+/− hearts following flecainide challenge. Thick black lines denote conduction block. Thin arrows denote lines of propagation. (a) Crowded isochronal lines in the last sinus beat, demonstrating area of conduction slowing. (b) Superimposed premature ventricular beat leads to line of block with impulse propagation flowing around it. (c) A second ventricular ectopic event causes the formation of a re-entrant circuit. (d) The circuit continues into the next beat (e, f) to initiate VT. Migration of line of block causing non-stationary vortex resulting in polymorphic arrhythmia should be noted. (C) Premature ventricular ectopic beat occurring as (a) isolated monophasically recorded event and (b) resulting in the initiation of electrocardiographically recorded sustained polymorphic VT [(A) Reproduced from King et al. (2013a), Figure 3; licensed under CC-BY 4.0 (B), (Cb) Adapted from Martin et al. (2011c), Figure 4; (Ca) Reproduced from Killeen et al. (2007), Figure 5C].
Decreased λs, thus, increase likelihoods of regenerative wave breakup into multiple wavelets, forming scroll waves (Davidenko et al., 1995; Zaitsev et al., 2000; Pandit and Jalife, 2013) along chaotic conduction pathways (Krogh-Madsen et al., 2012; Matthews et al., 2013; Spector, 2013). Contact multi-electrode (0.5 mm) array isochronal AP mapping techniques visualized the initiation of such a sequence. These studies were performed in right ventricular (RV) epicardia of intact beating flecainide-challenged pro-arrhythmic murine Scn5a+/− hearts (Figures 3Ba–f) (Martin et al., 2011c). Scn5a+/− ventricle APs show slowed conduction and increased activation and recovery dispersions. Here, following a delayed epicardial activation in the last normal beat reflected in the close isochronal contours (Figure 3Ba) (Killeen et al., 2007), a superimposed premature ventricular activation (Figure 3Ca) produces a line of block with AP propagation flowing around it (Figure 3Bb). A ventricular ectopic event then initiates an anticlockwise running circuit (Figure 3Bc) persistent through the following beat (Figure 3Bd). A consequent continually changing line of block now produces a non-stationary vortex (Figures 3Be,f) causing the polymorphic VT, apparent in the accompanying electrocardiographic trace (Figure 3Cb).
In addition to θ, recovery ERPs and APDs also affect λ and wave stability (Kléber and Rudy, 2004; Sabir et al., 2008a; Killeen et al., 2008b). ERPs and APDs vary across normal mammalian ventricular myocardium, causing spatial, epicardium-to-endocardium, transmural repolarization gradients, preserving orderly electrophysiological activation/recovery sequences and reducing likelihoods of repolarization abnormalities. Finally, differences between ERP and APD leaving abnormal re-excitation windows shorten λ, predisposing to pro-arrhythmic ectopic events (Sabir et al., 2007; Ashino et al., 2010; Martin et al., 2011a).
2.2 Temporal stability and instability of the AP wave
Temporal electrophysiological heterogeneities are typically observed as alternating variations in AP amplitude or APD between successive beats. Such alternans classically presages the breakdown of the regular pattern of electrophysiological activity and appearance of clinical (Nearing et al., 1991; Rosenbaum et al., 1994; Armoundas et al., 1998) or experimental arrhythmias (Pastore et al., 1999). Alternans in APD at a given heart rate, and therefore BCL, is thought to arise from variations in the timecourse of AP recovery or restitution to the resting potential, quantifiable as the APD. This variation, in turn, affects the subsequent diastolic interval (DI) prior to the AP corresponding to the following beat (Nolasco and Dahlen, 1968; Hayashi et al., 2007). The DI, thus, defines the period over which the membrane potential of its preceding AP is repolarized to its resting level. This recovery time, in turn, potentially affects the recovery properties, specifically the APD, of the subsequent AP. For example, shortened recovery times, DI, may preclude full recovery of the ion channel activity that underlies AP generation prior to the following beat. Typically, reductions or increases in DI have been reported to result, respectively, in reductions or increases in the APD of the subsequent AP. This effect follows a relationship termed the A-curve (Figure 4A) (Huang, 2021). However, at a fixed BCL, the resulting variations in APD would, in turn, alter the succeeding DI (Figure 4B). This latter effect is described by a linear D-line given by DI = BCL–APD (Figure 4C). These effects together would drive an interaction between DI and APD through successive cardiac cycles. Thus, the altered DI would, in turn, cause alterations in the APD of the subsequent AP and its DI that follows. This interaction can be described by superimposing the respective A-curve and D-lines.
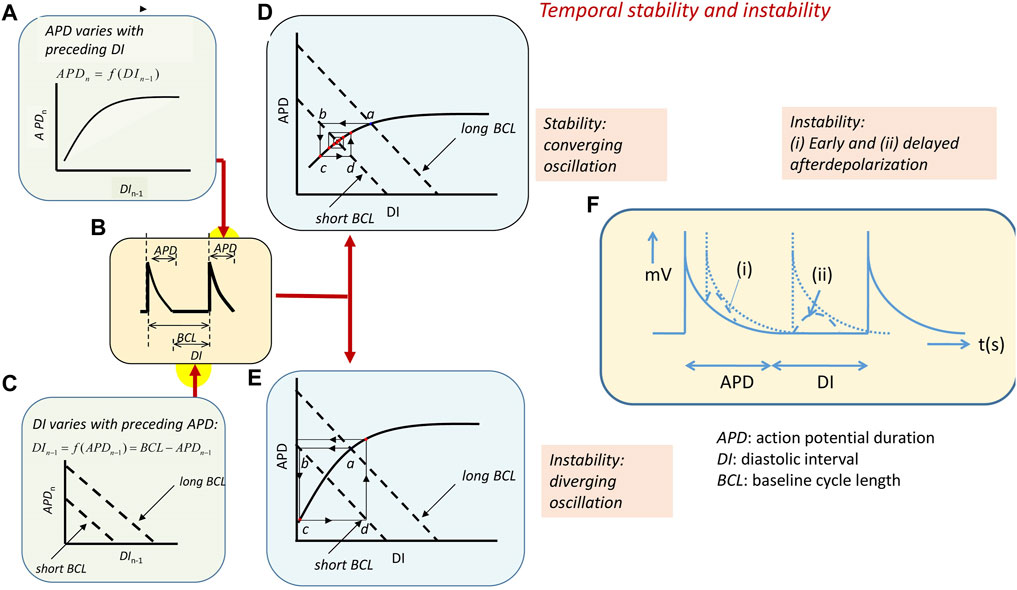
FIGURE 4. Arrhythmic substrate arising from temporal electrophysiological instabilities. (A) The nth action potential in a series has duration APDn dependent on its preceding diastolic interval DIn−1 along an A-curve. Symbols clarified in (B) showing action potential with a BCL made up of the APD and succeeding diastolic interval DI. (C) D-line showing the linear effect of APDn variations on DIn−1 at different BCLs: DI = BCL–APD. (D, E) Intersection between the A-curve and D-line gives steady-state APD and DI at any BCL (a). Increasing heart rate decreases the BCL and alters the D-line (b). This alters the APD (c) and, consequently, the succeeding DI (d). (D) A converging cycle resulting in declining oscillations results where APDn depends on DIn−1 with less than unity slope. The cycle remains constant with a unity slope and (E) diverges with a greater than unity slope. APD giving unstable waxing oscillations. (F) Transient instabilities in the form of (i) early and (ii) delayed afterdepolarizations in the course and following full recovery of the AP waveform [(A–E) from Figure 14.4 of the work of Huang (2021)].
The intersection between the A-curve and D-lines defines the steady-state APD and DI at any given BCL (Figure 4, Figures 4Da, Ea) (Sabir et al., 2008b). However, the interaction between the processes that they represent will vary with the heart rate as the latter, in turn, alters the BCL. Alterations in BCLs shift the D-line (Figures 4Db, Eb). This, in turn, alters the intersection point between the A-curve and D-line and, therefore, the DI corresponding to this intersection. The heart will then transition to a new APD as determined by the projection of this point to the A-curve (Figures 4Dc, Ec). This, in turn, alters the DI of the succeeding AP as given by the subsequent projection to the D-line (Figures 4Dd, Ed). The result is an iterative cycle of oscillating APD through successive heartbeats determined by the alternating projections to and from the A-curve and D-line.
Whether these oscillations converge, resulting in stable, persistent activity, or diverge, giving rise to unstable activity, is determined by the slope of the A-curves around the intersection region. For example, zero A-curve slope at the intersection results in an immediate attainment of a final steady-state APD and DI without oscillations. Where the A-curve slope falls between zero and unity (Figure 4D), the successive projection lines map a convergence back to the set point, giving a transient alternans ultimately stabilizing with a waning in the amplitude of the oscillation. The unity slope corresponding to the critical DI, DIcrit, gives projection lines that neither converge nor diverge, yielding stable alternans. Intersections where the A-curve slope exceeds unity result in diverging projection lines. This corresponds to a progressively increasing instability and a waxing oscillation in which the projection lines ultimately veer away from the left-hand limit of the A-curve. This causes a pro-arrhythmic conduction block and potential re-entry (Figure 4E) (Matthews et al., 2010; Matthews et al., 2012; Matthews et al., 2013).
Finally, intrinsic instabilities in the AP waveform itself or following its full recovery to resting potential, if large enough to reach re-excitation threshold, potentially cause isolated triggered beats and, in the presence of arrhythmic substrate, persistent arrhythmia (January and Riddle, 1989). In the ventricles, early afterdepolarizations (EADs) interrupting AP recovery timecourses reflect events continuing from the ventricular phase 2 plateau (Figure 4Fi). Transient, delayed afterdepolarizations (DADs) follow full repolarization (Figure 4F(ii)) (Killeen et al., 2008a). Such abnormal triggering in the pulmonary or the superior caval veins may precipitate AF.
3 Ion channels underlying normal and abnormal rhythmic activity
3.1 Ion channels mediating action potential excitation
Membrane-level physiological processes can each be identified with specific surface membrane ion channels and transporters, each constituting disease and potential clinical therapeutic targets. Ion channels function and interact through both their sensitivity to and effects on membrane potential, constructing the electrophysiological events found in normal activity or disease states. Membrane transporters mediating metabolically coupled transport or ion exchanges also exert electrogenic effects dependent on the relative stoichiometry and charges of their translocated ions. These targets variously participate in activating rapid depolarizing (phase 0) (exemplified for human atrial (A) and human and murine ventricular (B) cardiomyocytes in Figures 2Aa, Ba) as well as recovery, early repolarizing (phase 1), plateau and late repolarization (phases 2 and 3), and electrically diastolic phases (phase 4) stages of AP waves (Figures 2Ab, Bb).
First, overall cardiac pacing is normally driven by SAN and in heart block, AVN and Purkinje tissue automaticity. Among inward pacemaker currents (Huang et al., 2016) are inward, hyperpolarization-induced cyclic-nucleotide-activated channel (HCN)-mediated If and electrogenic 3Na+/1Ca2+ exchange (NCX) currents driven by sarcoplasmic reticular (SR) Ca2+ release (Lakatta et al., 2010; Donald and Lakatta, 2023). This results in a time-dependent phase 4 depolarization from the background resting potential. This process is modulated by autonomic, adrenergic, or cholinergic stimulation or inhibition. It results in the membrane potential attaining the Ca2+ (ICa) and then the Na+ current (INa) excitation thresholds. This initiates the SAN AP (Lei et al., 2007). Clinical automaticity abnormalities arise from abnormal ionic current activity or altered background diastolic or resting potentials. They can manifest as sinus node disorder (SND), abnormal AVN or Purkinje tissue pacing, and spontaneous impulses in pathologically partially depolarized atrial and ventricular muscle.
Second, Na+ channels typified by Nav1.5, activated by prior depolarization, carry rapidly developing inward depolarizing transmembrane INa. This drives the rapid depolarizing activating phase 0 of the consequent atrial or ventricular cardiomyocyte APs. Nav1.5 shows complex functional and distribution patterns among subcellular cardiomyocyte subdomains; cardiomyocytes may further express additional Nav1.x subtypes (Remme, 2023). Subsequent INa inactivation contributes to AP recovery alterable by pathological persistent, potentially pro-arrhythmic, late, INaL, currents (Chadda et al., 2017; Yu et al., 2018; Liu et al., 2023). Nav1.5 is also involved in SAN pacemaking through AP propagation from SAN to surrounding atrial muscle (Lei et al., 2005). Genetic loss or gain of INa function causes distinct pro-arrhythmic human Brugada (BrS) and long QT3 syndromes (LQTS3), respectively, experimentally recapitulated in loss (Martin et al., 2010; Martin et al., 2011b) or gain of Nav1.5 function murine models affecting AP activation and recovery (Killeen et al., 2008a; Sabir et al., 2008a). Both experimental and clinical BrS and LQTS3 aberrations and their arrhythmic tendency were accentuated/relieved by flecainide; quinidine produced the reverse effects (Benhorin et al., 2000; Stokoe et al., 2007; Martin et al., 2011b; Sabir et al., 2013). Nav1.5 mutations may also be involved in the impaired cardiac pacemaking in SND (Lei et al., 2005).
Third, similarly inward and depolarization-activated Cav1.2-mediated ICa may initiate APs in the SAN and AVN besides prolonging atrial and ventricular phase 2 plateau activation. Their loss-of-function mutations, recently exemplified by CACNB2b-S143F and CACNA1C-G37R (Zeng et al., 2023), resemble those involving Nav1.5 with pro-arrhythmic phenotypes accompanied by J wave syndromes (Huang, 2017).
Fourth, diverse, similarly depolarization-activated voltage-dependent K+ channels contrastingly carrying repolarizing outward currents (Smith and Cain, 2004; Tamargo et al., 2004; Nerbonne and Kass, 2005; Enyedi and Czirjak, 2010; Schmitt et al., 2014; Huang, 2017; Jeevaratnam et al., 2017) contribute to AP recovery. Transient outward Kv4.3 and Kv4.2, Ito, mediate early phase 1 repolarization and, along with atrial-specific Kv1.5 (KCNA5) carrying ultra-rapid IKur acetylcholine-sensitive GIRK1 and GIRK4 carrying IKACh, shorten atrial relative to ventricular APDS. Gain-of-function Kv4.3 and Kv4.2 mutations are implicated in AF (Alrabghi et al., 2023). Ventricular Kv11.1 (HERG or KCNH2) produce rapidly activating phase 1 rapid IKr and then inactivate through AP phases 0–2 (Sanguinetti and Tristani-Firouzi, 2006; Vandenberg et al., 2012) but re-activate over phase 3 and early phase 4 to terminate the plateau. IKr may actually peak during terminal repolarization phases of normally paced AP waveforms, potentially suppressing pro-arrhythmic actions of early afterdepolarizations or premature beats (Lu et al., 2001; Vandenberg et al., 2006). Kv7.1 (KCNQ1)-mediated IKs contrastingly activates more slowly over phase 2 producing a persistent phase 3 K+ conductance.
Fifth, voltage-independent inward rectifying K+, Kir2.1, Kir2.2, and Kir2.3 (KCNJ2, KCNJ12, and KCNJ4), channels produce a reduced inward IK1 at the phases 0–2 depolarized voltages but outward current with phase 3 repolarization. Along with background K2P2.1 (KCNK2)-mediated K2P currents and normally small adenosine triphosphate (ATP)-sensitive Kir6.2 (KCNJ11)-mediated IKATP (Foster and Coetzee, 2016), they also stabilize phase 4 diastolic resting potentials. Loss and gain of K+ channel function involving IKr, IKs, and IK1 can produce pro-arrhythmic long (LQTS) and short QT syndrome (SQTS), both predisposing to atrial and ventricular arrhythmias (Hancox et al., 2023). Similarly, protein expressional changes recently exemplified in IKs were associated with ventricular arrhythmias. The ubiquitin-like-modifier leukocyte antigen F-associated transcript 10 (FAT10) may reduce Kv7.1 ubiquitination, decreasing IKs (Chen et al., 2023).
3.2 Ion channels mediating action potential conduction
AP propagation between successive cardiomyocytes underlying coherent waves of excitation (King et al., 2013a; Huang et al., 2020) depends on local circuit current flow driven by INa through low-resistance (Verheule et al., 1997) gap junctions (GJs) comprising connexins Cx40 and/or Cx43 (Moreno et al., 1994; Traub et al., 1994; Rohr, 2004). It is determined by maximum AP depolarization rates (dV/dt)max dependent on INa, membrane capacitance, and cytosolic and connexin resistances (Jeevaratnam et al., 2011; King et al., 2013a). Adjacent cardiomyocytes are joined end-to-end at specialized perinexal regions including closely apposed adjacent cell membranes (Rhett and Gourdie, 2012; Rhett et al., 2013; Veeraraghavan et al., 2014) with locally elevated Cx expression and Nav1.5 densities (Veeraraghavan et al., 2015; Hichri et al., 2018; Struckman et al., 2021) increased by Nav1.5 clustering (Salvage et al., 2020b). Mathematical modeling suggests such structural organization is additionally or alternatively compatible with a direct cell-to-cell ephaptic propagation of excitation (Sperelakis and Mann, 1977; Sperelakis, 2002; Mori et al., 2008; Carmeliet, 2019; Morris et al., 2024). An albeit slower propagation of excitation persists even with compromised gap junction coupling under pathological conditions following ischemic insult (Rohr, 2004), atrial fibrillation (Chaldoupi et al., 2009), and experimental loss-of-function Cx43 (Eloff et al., 2001; Gutstein et al., 2001) or Cx40 genetic platforms (Hagendorff et al., 1999; Bagwe et al., 2005). Both propagation mechanisms are potentially modifiable by the longer-term tissue fibrotic or inflammatory processes discussed below (Jeevaratnam et al., 2011).
4 A hierarchy of mechanisms for ion channel modification
The immediate cause of pro-arrhythmic events involves actions of, and interactions between, cell membrane ion channels within individual or between adjacent cardiomyocytes. However, these are influenced by a hierarchy of mechanisms. These extend over a 12-decade, logarithmic timescale. This ranges from ns/μs (∼10–6 s) timescales shown by molecular events, through cellular events extending over ms/s within each or multiple cardiac cycles, to hours/days or even weeks/months (∼106 s) shown by remodeling processes (Figure 5A). Thus, there are (Figure 5B) (a) cell surface membrane ion channels and transporters underlying automaticity and AP excitation, propagation, and recovery and the (b) cellular feedforward and feedback effects of excitation–contraction coupling and its triggering by Ca2+. These latter overlap over microsecond/millisecond time scales. In contrast, the (c) systems-level G-protein signaling-dependent autonomic inputs and their related central nervous system circadian rhythms extend over second/minute/hour time scales. Finally, (d) longer-term upstream mechanisms related to metabolic feedback and other upstream modulators extend over days/weeks/months. Nevertheless, each of these processes can be grouped (Figure 5C) by their participating biomolecules (Figure 5D) (Lei et al., 2018). The scheme complements and prompts extensions of existing modeling attempts that extend to full electromechanical coupling at the systems level. These had integrated local and transmural ventricular myocyte surface electrical (ten Tusscher et al., 2004) with the cytosolic and SR Ca2+ transport and storage properties outlined below. They also modeled the consequent Ca2+–troponin binding to cross-bridge cycling activity and myofilament mechanics (Rice et al., 2008). These finally extended to mechanical activity in realistic two- and three-dimensional ventricular cardiac tissue models (Pathmanathan and Whiteley, 2009; Adeniran et al., 2013; Huang, 2015).
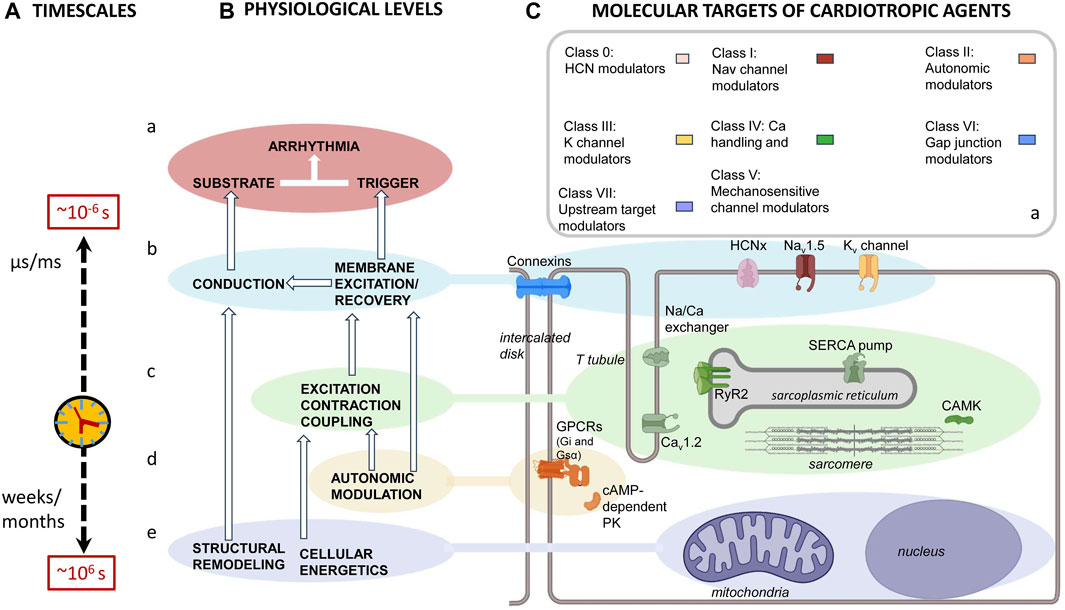
FIGURE 5. Mapping of physiological mechanisms, molecular targets, and therapeutic agents related to arrhythmia. (A) Pro-arrhythmic processes taking place over successively longer timescales running from ns/μs, ∼10–6 s timescale, molecular events, through ms/s events within each cardiac cycle or multiple cardiac cycles, to hours/days or even week/month, ∼106 s, timescales resulting from (B) an interleaving hierarchy of physiological processes involving interactions (a) at the molecular/membrane level, interactions between surface membrane ion channels underlying automaticity and AP excitation, propagation and recovery. (b) At the cellular level, excitation contraction coupling processes mediate Ca2+ signaling. (c) At the systems level, autonomic inputs and their related G-protein signalling, modulated by central nervous system circadian rhythms. (d) Longer-term electrophysiological and structural remodeling effects related to metabolic feedback and other upstream modulators. These map onto (C) recently reclassified (a) anti-arrhythmic drugs that, in turn, act upon (b–e) the molecular targets mediating these physiological processes. These include (b) surface membrane ion channels contributing inward depolarizing hyperpolarization-activated cyclic nucleotide gate (HCN); cardiac Na+ (Nav1.5) and Ca2+ (Cav1.2) channel, outward K+ channel (Kvx.x), and conducting connexin (CX) mediated currents. (c) Excitation contraction coupling-related Cav1.2, cardiac ryanodine receptor type 2 (RyR2) and transient receptor potential channel (TRPx), and their regulatory calcium/calmodulin kinase II (CaMKII); (d) autonomic signaling through inhibitory Gi, and stimulatory G proteins, Gs, involving PKA signaling. Longer-term modulation involves (e) energetic and modeling processes.
Many clarifications of these relationships arise from monogenically modified murine models permitting single cardiomyocyte, tissue, whole organ, and systems-level experimental study (Huang, 2017). Murine hearts translate to human hearts in their two-sided atrial and ventricular circulations, pacing, or conducting SAN, AVN, and atrioventricular (AV) components. They differ in size, heart rates, some ion channel types (Figures 2Ba, b), and consequent detailed AP waveforms (Figures 2c, d). Nevertheless, they share major, depolarization and conduction, AP features and regional heterogeneities (Huang, 2017). Particular genetic variants recapitulate corresponding human clinical arrhythmic and pharmacological phenotypes. Genetically induced pluripotent stem cell-derived cardiomyocyte (iPSC-CM) platforms also show promise as cellular rather than systems models in not recapitulating in vivo conducting, Purkinje, and contractile tissue organization. However, many human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) show immature embryonic-like rather than adult atrial/ventricular phenotypes (Lee et al., 2012). Nevertheless, recent reports describe hiPSC-CMs with atrial AP properties and acetylcholine-activated K+ current expression (Ahmad et al., 2023). There are recent iPSC models for normal and disease changes in ion channel expression, Ca2+ homeostasis, neurocardiac interactions, and hypertrophy (Li et al., 2023; Chen et al., 2023; Langa et al., 2023; Zhou et al., 2023). Theoretical reconstructions remain useful in predicting physiological end effects (Alrabghi et al., 2023; Hancox et al., 2023). Ultimate translatability of such results still requires direct human clinical electrophysiological studies with recently innovated electrocardiographic (Van Der Werf and Lambiase, 2021) and electrical mapping capacities (Taggart et al., 2022).
5 Ion channel modulation at the level of the cell membrane
A number of associated proteins modify function and expression of surface membrane ion channels in short local feedback loops. First, auxiliary β-subunits modulate function in their various associated Na+, Ca2+, and K+ channel subtypes. Na+ channel β-subunits (Navβ) each comprise an amino-terminal immunoglobulin (Ig) connected to a single transmembrane domain and a short largely disordered intracellular region. They enhance channel trafficking to the surface membrane modifying peak INa, modulate steady-state and kinetic inactivation (Namadurai et al., 2015), and ion channel clustering in the plasma membrane (Salvage et al., 2020).
The Navβ1 and Navβ3 isoforms associate non-covalently with α-subunits, mostly with the Ig domain binding to the domain III voltage-sensing module (VSM); the Navβ2 and Navβ4 Ig domains covalently associate via a disulfide bond to an extracellular loop region of domain II (Salvage et al., 2020a). However, Nav1.5 constitutes an interesting exception to this rule. Here, the Navβ1 and Navβ3 subunit Ig domains cannot bind to domain III VSM because the binding site is likely blocked by glycosylation. Similarly, Navβ2 and Navβ4 Ig domains cannot form disulfide bonds because the necessary free cysteine residues are absent in the Nav1.5 α-subunit. Consequently, Nav1.5-associated β-subunit Ig domains may be free to form extended cis and trans cross-links. Here, Navβ1-subunits may interact in trans influencing cell proximity. In contrast, cis-Navβ3 interactions may enhance local clustering of Nav1.5 (Salvage et al., 2020a). These may enhance ephaptic cell-to-cell AP conduction at intercalated disc perinexal membranes (Salvage et al., 2020a; Salvage et al., 2023). Genetic β-subunit mutations produce pro-arrhythmic phenotypes resembling conduction defects shown by Scn5a ± arrhythmic models (Hakim et al., 2008; Hakim et al., 2010).
Of K+-channel-associated β-subunits, KCNE2-encoded minK-related protein 1 (MiRP1) is associated with many K+ channel α-subunits. It modifies steady-state and kinetic activation and deactivation properties of HERG K+ channel-mediated IKr (Lu et al., 2003). Similarly, KCNE1-encoded β-subunits are associated with IKs channels. Their genetic ablation causes abnormal AP recovery, increasing APDs and inverting transmural APD gradients (Thomas et al., 2007; Hothi et al., 2009), and pro-arrhythmic phenotypes including early afterdepolarizations (EADs) and spontaneous VT in murine hearts recapitulating human Jervell and Lange–Nielsen syndrome (LQTS5) (Balasubramaniam et al., 2003).
Second, ion channels interact with cell adhesion, signal transduction, and cytoskeletal anchoring proteins. Nav1.5 interacting proteins may also function in specific subcellular Nav1.5 localizations producing regional variations in Nav1.5 expression, INa, density, and kinetics between subcellular microdomains. At lateral cardiomyocyte membranes, Nav1.5 interacts with dystrophin–syntrophin complexes, calcium/calmodulin (CaM)-dependent serine protein kinase, and caveolin-3. At intercalated disks, Nav1.5 associates with N-cadherin, connexin-43, βIV-spectrin, and desmosomal proteins including plakophilin-2 and desmoglein-2. Gene mutations involving some of these proteins are associated with arrhythmic disorders suggesting Nav1.5 dysfunction (Shy et al., 2013; Remme, 2023).
6 Feedforward and feedback between excitation–contraction coupling and surface membrane processes
Excitation–contraction coupling refers to the feedforward events connecting Nav1.5-mediated surface and transverse (T-) tubular membrane depolarization by AP initiation and propagation to initiation of contraction. Tubular Cav1.2 channels sense triggering surface membrane voltage changes, transducing these into ryanodine receptor (RyR2)-mediated SR Ca2+ store release by Ca2+-induced Cav1.2-RyR2 coupling (Endo, 2009). This likely occurs at T-SR junctional regions where the relevant membranes assume geometrically close ∼100–400 nm parallel alignment while remaining electrically separate (Martin et al., 2003; Dulhunty, 2006). Nav1.5, Cav1.2, and RyR2 come into close proximity within the resulting restricted diffusion spaces permitting local ion accumulation or depletion (Bardsley et al., 2021). Local Ca2+ release might then significantly elevate [Ca2+]TSR (Saucerman and Bers, 2012; Despa et al., 2014; Sanchez et al., 2021), in turn elevating bulk cytosolic [Ca2+]i-activating troponin, thereby triggering myofilament action and cardiomyocyte contraction. The background cytosolic [Ca2+]i is ultimately restored by SR Ca2+-ATPase (SERCA2), which mediates the Ca2+ return to its SR store and expulsion into the extracellular space by sarcolemmal NCX and, to a lesser extent, sarcolemmal Ca2+-ATPase. Further intracellular organelles, including lysosomes and mitochondria, are often regulated by further signaling molecules, such as cADP-ribose, nicotinic acid adenine dinucleotide phosphate (NAADP), and inositol tris-phosphate (IP3), which also modulate cellular Ca2+ homeostasis (Terrar, 2023).
Feedback actions of consequently altered intracellular Ca2+ fit its strategic second messenger function through widespread cellular processes and cell types (Figure 6A) (Huang, 2017). Released Ca2+ itself exerts a short-loop slow RyR inactivation (Laver and Curtis, 1996). Further longer-loop feedback modulation may modify surface membrane channel or transporter activity with pro- or anti-arrhythmic effects (Figures 6B–D), acting on either arrhythmic triggering or substrate (Figure 6E). Of voltage-sensitive channels mediating inward depolarizing current, Na+ and Ca2+ channel C-terminal domains contain both direct and indirect Ca2+ regulatory binding sites in the form of one or more EF hand motifs and an isoleucine–glutamine (IQ) CaM-binding domain, respectively. Their III–IV loops also contain Ca2+-CaM-binding sites. The channels additionally possess multiple kinase, including CaM kinase II (CaMKII) and phosphokinase C (PKC), and phosphorylatable regulatory sites. One or more of these sites have been implicated in Ca2+-mediated feedback inhibition of Nav1.5 in intact cardiac myocytes and implicated in arrhythmogenic phenotypes in both pharmacological (King et al., 2013b; Valli et al., 2018b) and genetic disease models (King et al., 2013c; Zhang et al., 2013; Salvage et al., 2021; Salvage et al., 2023). Similar mechanisms may operate for skeletal muscle Nav1.4 (Matthews et al., 2019; Sarbjit-Singh et al., 2020; Liu et al., 2021). Ca2+, likely through CaMKII, conversely increases INaL with potential pro-arrhythmic effects (Liu et al., 2023).
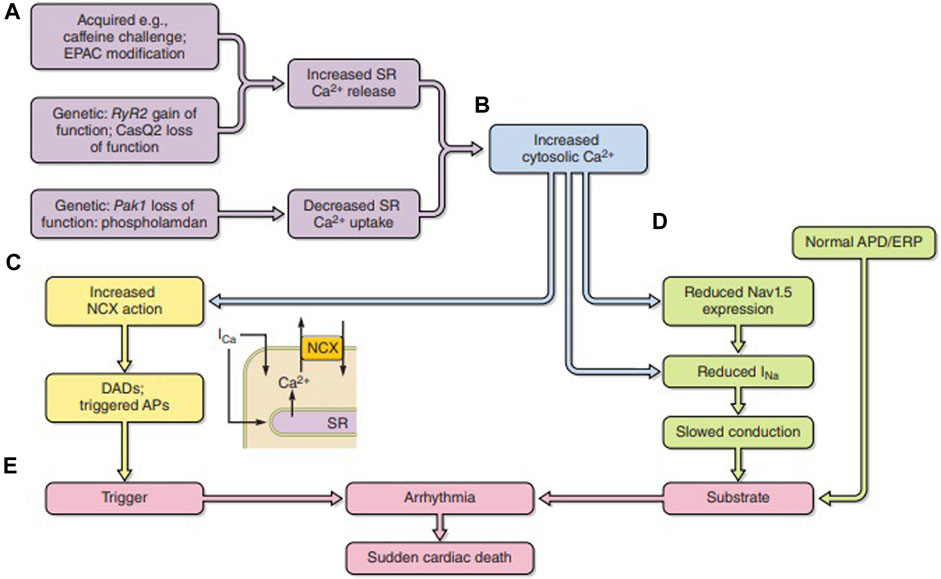
FIGURE 6. Pro-arrhythmic feedback relationships between excitation contraction coupling and surface membrane ionic channel function. (A) Acquired or genetic perturbations increasing sarcoplasmic reticular (SR) Ca2+ release or decreasing SERCA-mediated Ca2+ reuptake both alter cytosolic [Ca2+] (B). This alters (C) 3Na+/1Ca2+ exchange (NCX) electrogenic activity causing triggering DADs. It may also (D) reduce Nav1.5 synthesis or membrane trafficking or directly alter Nav1.5 function, slowing down AP conduction. The trigger/substrate combination (C, D) causes (E) sustained arrhythmia [from Figure 18 of the work of Huang (2017)].
Of ion exchange processes, NCX stoichiometry produces inward, potentially pro-arrhythmic INCX under conditions of elevated [Ca2+]i in polarized membranes (Shattock and Bers, 1989).
Of voltage-sensitive channels mediating outward hyperpolarizing current, Ca2+-activated K+ channel opening contrastingly hyperpolarizes, thereby reducing membrane excitability. In addition to membrane depolarization, large conductance Big K+ (BK) channel opening requires μM local [Ca2+] binding to two distinct high-affinity intracellular C-terminal domain Ca2+-binding sites (Zeng et al., 2005; Yang et al., 2015; Sancho and Kyle, 2021), modifying sino-atrial node firing rate and cardiac pacing (Meredith et al., 2014; Pineda et al., 2021). Voltage-insensitive small conductance (SK) SK1, SK2, and SK3 K+ channels (Adelman et al., 2012; Weisbrod, 2020) respond to RyR-induced [Ca2+]i through a regulatory C-terminal CaM-binding domain (Neelands et al., 2001; Li et al., 2023). SK2 channels, in particular (Schmitt et al., 2014), are clinically implicated in atrial fibrillation (Healey et al., 2005) and arrhythmia and hypertrophy accompanying clinical and experimental ventricular failure (Terentyev et al., 2014; Gu et al., 2018). SK2 modified AP waveform in pro-hypertrophic Pak-1-deficient murine experimental platforms predisposing to ventricular hypertrophy and failure following angiotensin II challenge (Yang et al., 2021). Finally, of anion channels, Ca2+-activated Cl−, TMEM16A, channels are [Ca2+] sensitive (Yang and Colecraft, 2016).
7 Autonomic feedforward and feedback
Autonomic, sympathetic and parasympathetic, modification of pacing, ion current activation, and excitation–contraction coupling involves transmitter and co-transmitter binding to G-protein-coupled receptors (GPCRs). G-protein-linked activation cascades show significant amplification through their constituent signaling molecules exerting multiple inotropic, chronotropic, and lusitropic effects on cardiac function (Bers, 2002). Both levels of and balance between sympathetic and parasympathetic activity may affect arrhythmogenicity: autonomic dysregulation is involved in multiple cardiac arrhythmias. Thus, both increased sympathetic and vagal rebound activity amplify ventricular arrhythmic risk. Its modulation may offer novel therapeutic options.
The sympathetic transmitters adrenaline and noradrenaline are, respectively, released into the circulation by adrenal medullary release and locally by widely distributed cardiac sympathetic nerve terminals. They, respectively, preferentially bind surface membrane β1- and β2-adrenergic receptors. Cardiomyocytes express β1-adrenergic receptors, activating the stimulatory Gs protein. Its Gα subunit then binds guanosine triphosphate (GTP), dissociates from the GPCR and its βγ-subunit, and activates adenylyl cyclase, enhancing cellular cyclic 3′,5’ adenosine monophosphate (cAMP) levels, with diverse compartmented cellular actions (Zaccolo, 2023). Of nerve–cardiomyocyte communication mechanisms under current study, ventricular Dbh+ catecholaminergic cardiomyocytes (Dbh+ Cate-CMs) expressing dopamine β-hydroxylase occur in close structural relationship with the sympathetic innervation. Their catecholaminergic-type vesicles suggest endocrine functions. They may also be involved in embryonic development and maturation of the cardiac conduction system (Wang et al., 2017).
At the membrane level, sympathetic activity increases heart rate and modifies AP generation and waveform. cAMP binds to and opens SAN HCN, increasing If and heart rates. It also activates protein kinase A (PKA) whose widespread phosphorylation actions excite Nav1.5, Kv11.1, Kv7.1, and Cav1.2, increasing rapid inward INa and slow outward IKr and IKs and ICaL, increasing ventricular AP amplitudes but shortening the AP plateau durations. It also accelerates SAN pacemaker potentials (Huang and Lei, 2023). In excitation–contraction coupling, sympathetic activity increases ICaL and net cellular Ca2+ entry increasing rates and force of subsequent muscle contraction. PKA-mediated RyR2 phosphorylation also reduces regulatory FKBP12 ligand binding otherwise stabilizing RyR2 closure, thereby increasing RyR2 Ca2+ sensitivity and Ca2+-induced Ca2+ release. It also phosphorylates phospholamban (PLN), relieving its SERCA2 inhibition, enhancing diastolic SR Ca2+ store.
The Epac2 isoform of cAMP-dependent exchange protein activates CaMKII (Takla et al., 2020; Terrar, 2023) and, therefore, RyR2-mediated SR Ca2+ release (Hothi et al., 2008). At further upstream levels discussed below, Epac1 activates Ca2+-dependent calcineurin signaling, driving hypertrophic, morphological, and cytoskeletal changes. Additionally, excitatory postganglionic sympathetic co-transmitters, adenine nucleotides, act on metabotropic P2Y receptors. Adenosine (A1) receptor activation activates phosphokinase C (PKC), which also acts on voltage-gated Na+ and K+ channels, L-type Ca2+ channels, and RyR2. Finally, sympathetic responses can differ among cardiomyocyte types, exemplified by the distinct pulmonary vein (PV) and left atrial cardiomyocyte responses to noradrenaline implicated in atrial ectopy (Bond et al., 2020).
The parasympathetic, inhibitory, transmitter, acetylcholine (ACh), acts on muscarinic (M2) receptors activating the Gi2 protein in the SAN, AVN or atria, in both the presence and absence of, pre-existing adrenergic challenge. It does so in ventricular tissue in the presence of such challenge. Its Gα subunit binds GTP and splits off from the receptor and its Gβγ-subunit. The latter opens GIRK1 and GIRK4 channel components, increasing inward rectifying IKACh or IKAdo, particularly in supraventricular tissue (Schmitt et al., 2014; Dascal and Kahanovitch, 2015; Lerman, 2015). The dissociated Giα inhibits adenylate cyclase (AC) and, therefore, cAMP production (DiFrancesco, 1993), increasing ICaL and If in pacemaker cells. Gi activation also increases protein phosphatase (PP2A) activity through cell division control protein 42 homolog (Cdc42)/Ras-related C3 botulinum toxin substrate 2 (rac2) and the cardioprotective p21-activated kinase PAK1. PP2A dephosphorylates PKA-phosphorylated proteins at the same serine/threonine phosphorylation sites, reversing their L-type Ca2+ channel, RyR2 and PLN effects. Parasympathetic action, thus, slows heart rates and decreases contractile force.
PAK1 may be cardioprotective through increasing PP2A activity (Ke et al., 2009; Wang et al., 2015) and its remodeling actions (Liu et al., 2011; Wang et al., 2014; He et al., 2023; Jung et al., 2023). It may also modify Cav1.2/Cav1.3 (ICaL)-mediated Ca2+ entry, RyR2-mediated SR Ca2+ release, and CaMKII-mediated SERCA2a and NCX transcriptional regulation (He et al., 2023) and promote SERCA activity (Ke et al., 2009; Wang et al., 2014; Wang et al., 2015). PAK1 deficiency may promote atrial arrhythmogenesis under adrenergic stress conditions through posttranslational and transcriptional modifications of key molecules including RyR2 and CaMKII (Jung et al., 2023).
Over longer systems-level timescales, autonomic innervation provides effector pathways for central nervous responses to environmental inputs. Normal circadian ion channel remodeling cycles explain SAN pacemaking changes, resulting in higher background heart rates during wakefulness than during periods of sleep (Jesus et al., 2021). These are likely driven by sympathetic, as opposed to parasympathetic, actions coupled to suprachiasmatic nuclear circadian rhythms. Rather than variations of autonomic transmitter activity (Black et al., 2019), recent evidence attributes these to periodic transcriptional cardiac remodeling cycles varying ion channel abundances and their consequent ionic current densities. The SAN ion channel transcriptome for many important cardiac ion channels, particularly HCN, displays circadian rhythms (Anderson et al., 2023; Black et al., 2019; D’Souza et al., 2021; Wang et al., 2021), sensitive to chronic but not acute pharmacological autonomic blockade (Pitzalis et al., 1996; Black et al., 2019). It possibly involves cAMP response element action on key Per1 and Per2.18 clock genes (Travnickova-Bendova et al., 2002). Critically needed are future studies of autonomic plasticity and age-related properties (Chadda et al., 2018; Takla et al., 2023).
8 Upstream modulatory targets: energetic feedback
Longer-term pro-arrhythmic upstream cellular energetic and tissue remodeling can accompany metabolic pathology in various conditions, including acute hypoxic/ischemia–reperfusion (Liu et al., 2020), chronic obesity, insulin resistance and type 2 diabetic conditions (Vianna et al., 2006; Sato et al., 2009; Kucharska-Newton et al., 2010), and cardiac failure, accompanying primary hypertrophic or fibrotic changes (Terentyev et al., 2008; Grivennikova et al., 2010; Belevych et al., 2012; Chouchani et al., 2014). First, maintenance of ionic gradients and Ca2+ cycling processes represents ∼30–40% of cardiomyocyte ATP utilization. Of the latter, ∼90% is replenished by their extensive mitochondrial network (Leone and Kelly, 2011; Nguyen et al., 2019; Wang et al., 2019).
Mitochondrial mass and function are regulated by transcriptional coactivators (Sonoda et al., 2007): peroxisome proliferator-activated receptor (PPAR) γ coactivator-1 (PGC-1) family members are highly expressed in oxidative tissues including the heart. Of these, mitochondrial promoter PGC-1α or PGC-1β expression increases with upstream signals including cold exposure, fasting, and exercise, likely signaling anticipated cellular energy requirements (Figure 7A) (Huang, 2017). PGC-1 proteins exert matching multi-level regulation of cellular mitochondrial function and metabolism. Of their activated nuclear receptor targets, PPARα regulates genes involved in mitochondrial fatty acid oxidation; estrogen-related receptor alpha (ERRα) regulates mitochondrial fatty acid oxidative and oxidative phosphorylative energy transduction pathways (Vega et al., 2000). Additionally, PCG-1α coactivation of nuclear respiratory factor-1 (NRF-1) and -2 (NRF-2) (Wu et al., 1999) modulates nuclear-encoded transcription factor Tfam expression required in replicating, maintaining, and transcribing mitochondrial DNA (Garesse and Vallejo, 2001) and mitochondrial biogenesis (Vega et al., 2000; Huss et al., 2004). These mechanisms may underlie the induction of nuclear genes encoding mitochondrial proteins in energetic pathways, including the tricarboxylic acid cycle, and nuclear and mitochondrial genes encoding electron transport chain and oxidative phosphorylation complex components in cultured cardiomyocytes with forced PGC-1 expression (Lehman et al., 2000). PCG-1s fall in obesity, insulin resistance, type II diabetes mellitus, and aging in parallel with mitochondrial dysfunction (Leone and Kelly, 2011; Dillon et al., 2012).
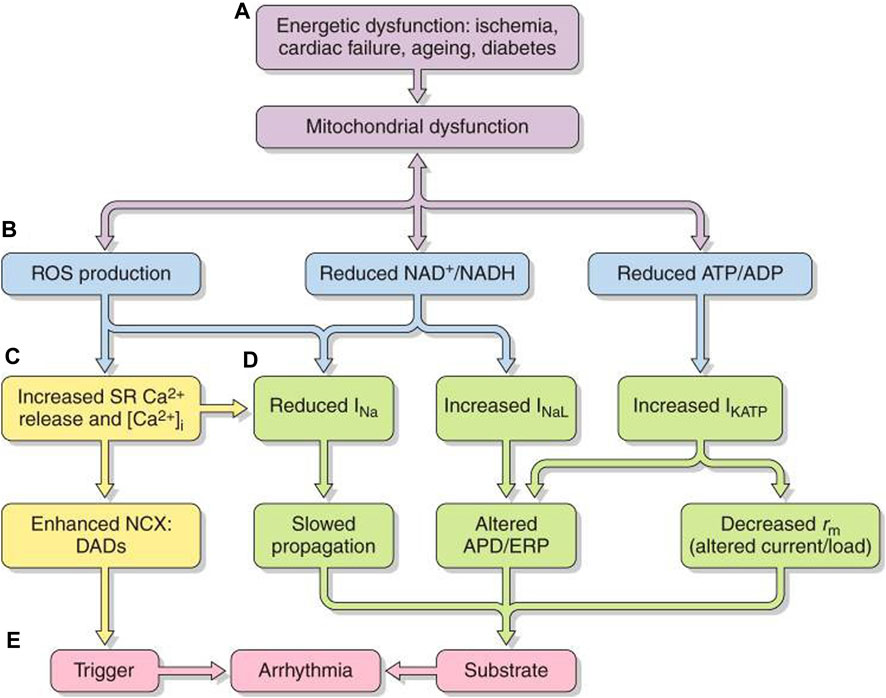
FIGURE 7. Pro-arrhythmic feedback relationships between energetic dysfunction and surface membrane ionic channel activity. Simplified scheme in which (A) energetic dysfunction in ischemia, cardiac failure, aging, or diabetes is reflected in (B) mitochondrial dysfunction causing ROS production, altered NAD+/NADH, and ATP/ADP. This leads to increased (C) RyR2-mediated SR Ca2+ release and increased [Ca2+]i NCX and consequent DAD triggering activity. (D) Altered Na+ and K+ channel function affects AP excitation, propagation, and recovery. The trigger/substrate combination (C, D) causes (E) sustained arrhythmia [from Figure 20 of the work of Huang (2017)].
Mitochondrial dysfunction, whether intrinsic or arising from excessive energetic demand, compromised vascular oxygen supply or pathological energetic disorders, destabilizes inner membrane potentials, driving their electron transport chain. At the cell membrane level, the consequently compromised ATP and/or rising adenosine diphosphate (ADP) increase sarcolemmal K-ATP (sarcKATP) channel opening probabilities (Akar and O’Rourke, 2011). This shortens APDs and ERPs and compromises cell excitability and AP propagation (Akar and O’Rourke, 2011). These events predispose to re-entrant arrhythmia (Fosset et al., 1988; Faivre and Findlay, 1990).
Increased reactive oxygen species (ROS) production accompanying oxidative stress also influences cardiomyocyte excitability (Figures 7B–D). The resulting atrial and ventricular arrhythmic tendency may result from reduced Nav1.5 expression (Liu et al., 2010), increased INaL(Liu et al., 2023), and reduced connexin-43 (Cx43) trafficking and function (Akar and O’Rourke, 2011; Yang et al., 2014; Yang et al., 2015) and cell–cell coupling (Smyth et al., 2010) and inhibited voltage-dependent IK (Wang et al., 2004), sarcolemmal KATP (Weiss et al., 1989), and ICa. These effects could affect AP conduction (Liu et al., 2010) and underlie the atrial ERP shortening and AF initiation with rapid pacing (Figure 7E) (Carnes et al., 2001; Dudley et al., 2005). Right atrial appendages of AF patients show increased oxidative stress markers (Emelyanova et al., 2016). Accompanying changes in reduced (NADH) or oxidized nicotinamide adenine dinucleotide (NAD+), reflecting cell oxidative state, also, respectively, inhibit and enhance activity even with normally expressed Nav1.5 (Liu et al., 2010).
At the excitation–contraction coupling level, increased ROS oxidize RyR2, increasing SR Ca2+, and reduce SERCA-mediated Ca2+ reuptake, increasing cytosolic [Ca2+]I (Kukreja et al., 1988). CaMKII oxidation may induce a kinase activity similar to auto-phosphorylated CaMKII (Erickson et al., 2008). ROS also oxidize and activate PKA (Eager and Dulhunty, 1998). ROS, thus, altered intracellular Ca2+ cycling (Terentyev et al., 2008; Brown and O’Rourke, 2010; Bovo et al., 2012) in aging rabbit ventricular myocytes, effects reversed by mitochondrial specific ROS scavengers (Terentyev et al., 2008). At the level of further upstream targets, ROS are implicated in fibroblast activation and transforming growth factor-β (TGF-β) production, leading to cardiac fibrosis (Friedrichs et al., 2012).
Mice deficient in either but not both PCG-1α or PGC-1β survive to adulthood. Pgc-1α−/− hearts had normal baseline function but failure with increased afterload (Arany et al., 2005). Pgc-1β−/− hearts showed normal background features but blunted heart rate responses to adrenergic challenge (Lelliott et al., 2006), increased arrhythmic propensity, and APD alternans and VT episodes (Gurung et al., 2011). Single cells showed altered ion channel expression and spontaneous diastolic Ca2+ transients. Chronic studies in young (12–16 weeks) and aged (>52 weeks) Pgc-1β−/− mice suggested sinus node and AVN dysfunction in intact animals following β1-adrenergic challenge (Ahmad et al., 2018b). The atria and ventricles of Langendorff-perfused Pgc-1β−/− hearts showed increased arrhythmic inducibility with age, accompanied by reduced (dV/dt)max, increased AP latencies, and reduced APD, together reducing λ and increasing arrhythmogenicity (Ahmad et al., 2017; Valli et al., 2017a; Ahmad et al., 2018a). These findings correlated with direct demonstrations of reduced INa but not IK (Valli et al., 2018a; Ahmad et al., 2019). Pgc-1β−/− hearts also showed accelerated age-related fibrotic change, discussed further in the next section (Ahmad et al., 2017; Valli et al., 2017c).
9 Upstream cardiac remodeling
Second, longer-term inflammatory and structural, fibrotic and hypertrophic, changes can pro-arrhythmically add to and influence the physiological processes above (Nattel et al., 2007; Dobrev and Nattel, 2010; Iwasaki et al., 2011). They involve inflammatory cell recruitment, cellular proliferation, angiogenesis, and extracellular matrix accumulation (Murphy et al., 2015). Accumulating evidence implicates inflammatory processes involving tumor necrosis factor (TNF)-α, macrophage migration inhibitory factor, interleukin (IL)-1β, IL-6, and JUN N-terminal kinases (JNKs) with pro-inflammatory M1 macrophages participating particularly in AF. Additionally, pro-inflammatory (M2) macrophages may enhance atrial fibroblast proliferation and differentiation. TNF-α, in turn, activates profibrotic transforming growth factor-β (TGF-β) signaling and matrix metalloproteinase secretion. TGF family members, particularly TGF-β1, regulate tissue homeostasis and repair, immune and inflammatory responses, extracellular matrix (ECM) deposition, and cell differentiation and growth (Yang et al., 2010). TGF-β1 is a key contributor to fibrosis (Dobaczewski et al., 2011). TGF-β1 signaling is itself activated by angiotensin II (Ang II) action on local cardiac renin–angiotensin system (RAS) angiotensin receptor type 1 ATR1s, which themselves directly activate TGF-β production.
TGF-β likely acts through Smad and synergistically with extracellular signal-regulated protein kinase ½ (ERK1/2), c-Jun NH2-terminal kinase (JNK), p38 mitogen-activated protein kinases (p38MAPK), all mitogen-activated protein kinases (MAPK) (Dobaczewski et al., 2011). It stimulates myofibroblast differentiation, key to the fibrotic process, and ECM protein synthesis (Desmouliere et al., 1993) through inhibiting matrix metalloproteinase (MMP) and inducing tissue inhibitor metalloproteinase (TIMP) synthesis (Mauviel, 2005). Ang II similarly acts in this, both itself, in synergy with TGF-β1 (Schnee and Hsueh, 2000; Murphy et al., 2015), and itself activating TGF-β1 signaling (Schultz et al., 2002; Dobaczewski et al., 2011). TGF-β1 administration-induced fibroblast proliferation causes atrial fibrosis (Dzeshka et al., 2015; Hu et al., 2015) and AF (Lifei et al., 2020).
ATR1s also activate G protein, Gαq/11, Gα12/13, and Gβy, and non-G protein-related pathways, modulating multiple oxidase and kinase signaling pathways. Among serine/threonine kinases, Ang II- or ROS-mediated CaMKII activation enhances phosphorylation of excitation–contraction coupling-related proteins and cell survival and transcription factors, driving hypertrophic and inflammatory gene expression (Swaminathan et al., 2012; Rusciano et al., 2019). PKC activates NAD(P) H oxidase, thence increasing ROS production and consequent cardiomyocyte hypertrophy (Nakamura et al., 1998; Sugden and Clerk, 1998). Activation of the MAPKs, including ERK1/2, JNK, and p38MAPK, is implicated in cell growth and hypertrophy (Wollert and Drexler, 1999), as well as cardiac fibrosis, through increased procollagen I, procollagen III, fibronectin, and TGF-β gene transcription. Epidermal growth factor receptor (EGFR) and platelet-derived growth factor (PDGF) insulin receptor signaling can also occur. Of non-receptor tyrosine kinases, Src, janus kinase/signal transducer and activator of transcription IL (JAK/STAT), and focal adhesion kinase (FAK) (Mehta and Griendling, 2007), JAK/STAT may promote pressure overload and ischemia-related hypertrophy (Booz et al., 2002). Finally, recent reports implicate upregulated Notch signaling in hypertrophic and dilated cardiomyopathy (Langa et al., 2023).
Cardiac remodeling first affects surface membrane ion channel function, excitation–contraction coupling, and conduction of excitation. Of inflammatory cytokines, TNF-α may reduce ICa,L, while increasing Ito, systolic Ca2+ transients, diastolic intracellular [Ca2+], and inward NCX and decreasing SR ATPase expression. These changes accentuate DAD-induced triggered activity (Lee et al., 2007). It may also alter Cx40 and Cx43 expression and distribution. JNK2 activates CaMKII, increasing diastolic SR Ca2+ leak, and downregulates Cx43 expression, compromising cell-to-cell communication and atrial AP conduction (Yan et al., 2018) Inflammasome NLRP3 activation upregulates RyR2 expression and CaMKII-dependent hyperphosphorylation and consequent pro-arrhythmic diastolic SR Ca2+ release, similarly inducing DADs and ectopic firing (Yao et al., 2018). It also increases IKur and IKACh and increases atrial hypertrophy and fibrosis (Heijman et al., 2020). PAK1 signaling may exert protective signaling actions through Cav1.2/Cav1.3 (ICaL)-mediated Ca2+ entry, RyR2-mediated SR Ca2+ release, and CaMKII-mediated regulation of SERCA2a and NCX transcription (He et al., 2023). Its deficiency may modify posttranslational and transcriptional processing in the key Ca2+ homeostatic molecules RyR2 and CaMKII with atrial pro-arrhythmic effects under adrenergic stress (Jung et al., 2023).
Second, fibrotic changes may contribute to both AF and ventricular arrhythmias. Increased fibrosis-related gene expression accompanying altered surface and Ca2+ homeostasis ion channel gene expression may contribute to SAN dysfunction in experimental rat pulmonary arterial hypertension (Logantha et al., 2023). Disruption of myocardial bundle continuity and gap junction formation due to increased extracellular matrix associated with fibrotic and/or hypertrophic change reduces and accentuates heterogeneities in AP conduction velocity, disrupting AP propagation wavefronts and promoting re-entry (Davies et al., 2014). Connexin-mediated electrotonic coupling between cardiomyocytes and inexcitable fibroblasts with relatively depolarized (∼-30 mV) resting potentials may depolarize cardiomyocyte resting membrane potentials altering myocyte excitability, slow conduction, shorten APD, and induce spontaneous phase 4 depolarization (Nattel, 2018). Additionally, Ca2+ signaling in fibroblasts employs stretch-sensitive short TRPC3 or melastatin-type TRPM7 transient receptor potential (TRP) channels, both upregulated in AF fibroblasts (Yue et al., 2015).
Other downstream processes may feed forward to modify these changes. Both genetic Nav1.5 (Jeevaratnam et al., 2012; Jeevaratnam et al., 2016) and Pgc1-β-related metabolic deficiencies (Ahmad et al., 2017; Valli et al., 2017b; Ahmad et al., 2018a; Ahmad et al., 2018b) were associated with accentuated TGF-β1-mediated fibrosis, possibly driving observed electrophysiological remodeling underlying age-related SND dysfunction (Hao et al., 2011) and pro-arrhythmic atrial and ventricular fibrosis. The former non-canonical roles for Nav1.5 in cellular biological processes extend to associations with morphological changes in dilated and arrhythmic cardiomyopathies (Remme, 2023).
10 Mechanistic insights translated into novel anti-arrhythmic therapies
Pharmacological intervention underpins much clinical arrhythmia management. Both modern drug innovation and optimizing existing therapeutic strategies require systematically classified mechanisms of action at identified molecular physiological targets and their rational correlation with single cell, tissue, or organ tissue effects and, in turn, clinical indications and therapeutic actions. This requirement had been addressed by successive historic Singh–Vaughan Williams (VW) (Vaughan Williams, 1975) and updated European Society of Cardiology classifications (Task force of the working group on arrhythmias The European Society of Cardiology, 1991). The most recent modernized classification scheme (Lei et al., 2018) responding to the recent cardiac electrophysiological and pharmacological insights outlined here pragmatically retained but extended the original VW classes. It mapped actions on molecular targets within more recently characterized levels of surface membrane ion channel, excitation–contraction coupling, autonomic function, and longer-term energetic and remodeling changes outlined above. The resulting scheme for cardiac arrhythmic mechanisms thereby offered a basis for understanding existing explorations for novel arrhythmic therapies (Lei et al., 2018), and reviewing drug cardiac safety (Saadeh et al., 2022).
Of the introduced novel drug categories, class 0, bearing on cardiac automaticity, includes the use-dependent SAN inhibitor ivabradine. It reduces heart rate by acting on If, sparing myocardial contractility and vascular tone, actions distinct from the existing VW classes II and IV. It is clinically approved for reduced ejection fraction under circumstances of cardiac failure, adjunct therapy improving clinical outcomes by reducing heart rate. It is yet to be established for stable ischemic heart disease. It may also benefit patients with inappropriate sinus tachycardia intolerant of class II or IV agents (Koruth et al., 2017).
Of extended VW classes I–IV, class I now includes class Id drugs acting on INaL this complements the existing Class Ia–c, which reduce early INa thereby reducing AP phase 0 slopes and overshoots, and modify APD and ERP. Class Ia drugs classically bind to the Nav1.5 open state with τ ∼1–10 s dissociation time constants. They concomitantly block IK, slow AV conduction, and increase ERP and APD. Classes Ib and Ic contrastingly bind preferentially to the inactivated state with rapid τ ∼0.1–1.0 s or slow τ > 10 s dissociation. This results in a use-independent and use-dependent channel block and a slowed AV conduction. These insights clarify some of flecainide’s paradoxical actions. Thus, flecainide exerts anti-arrhythmic actions in gain of Na+ channel, particularly INaL, function associated with LQTS3 and experimental murine Scn5a+/Δkpq. In contrast, it shows pro-arrhythmic actions under post-infarct conditions and clinical and experimental Scn5a+/− Brugada syndrome (Martin et al., 2010; Martin et al., 2011a). Of drugs in the new class Id, ranolazine produces a frequency-dependent block of the pro-arrhythmic INaL. This shortens APD and increases refractoriness and repolarization reserve in LQTS3, pathological bradycardic and ischemic conditions, and cardiac failure (Belardinelli et al., 2015). Intracellular Na+ overload, otherwise arising from such INaL, increases reverse-mode NCX, promoting pro-arrhythmic intracellular Ca2+ overload and SR Ca2+ leak. Ranolazine may also reduce atrial peak INa and Kv11.1-mediated IK (Burashnikov et al., 2007; Antzelevitch et al., 2011).
A broadened class II encompassed further modifiers of G-protein-mediated signaling besides the β-adrenergic antagonists originally used to slow down SAN pacing and AVN conduction. These included new selective β1-(atenolol and bisoprolol) and non-selective β-adrenergic antagonists (carvedilol), adenosine A1-activators (adenosine), and cholinergic muscarinic M2 receptor inhibitors (scopolamine) and activators (carbacholine and pilocarpine) (Singh, 2004). Future possibilities could arise from numerous (∼150) further orphan GPCRs (Lei et al., 2018). Finally, centrally acting sympathetic inhibitors, such as monoxidine, have been investigated in relation to AF management (Hu et al., 2023).
An expanded class III encompassed drugs directed at the large number of more recently discovered, sometimes atrial or ventricular-specific, voltage-sensitive and -insensitive K+ channel types. This added to the voltage-gated K+ channel blockers originally employed to delay AP phase 3 repolarization, lengthening ERPs. Exemplars, including investigated agents directed at atrial arrhythmia, included those directed at voltage-sensitive IK such as the atrial IKur, including XEN-D0103 (Ford et al., 2016) and vernakalant, for rapid AF (Wettwer et al., 2013), with the latter producing some INa and INaL, and IKACh in addition to IKur block. New IKr blockers under development include vanoxerine (Lacerda et al., 2010). Investigational agents directed at voltage-insensitive IK include the SK channel inhibitor AP30663 (Bentzen et al., 2020) and the K2P blocker doxapram (Wiedmann et al., 2022).
Major advances in excitation–contraction coupling have yielded significant progress in class IV drugs beyond the L-type Ca2+ channel inhibitors introduced to reduce SAN and AVN rates and conduction (Vaughan Williams, 1975). First, investigational non-selective and phenylalkylamine and benzothiazepine inhibitors supplement dihydropyridine Cav1.2 and Cav1.3 surface membrane ICaL and ICaT inhibitors.
Second, of agents targeting intracellular membrane molecules mediating SR Ca2+ release and reuptake, the experimental drug dantrolene enhances CaM-RyR2 binding and, consequently, N-terminal-central domain interactions, stabilizing RyR2 closed states (Pabel et al., 2020). Recent reports implicate flecainide in class IV RyR2 open-state block reducing RyR2 open probabilities, in addition to its class I actions. Both experimental and clinical reports support its application in catecholaminergic polymorphic ventricular tachycardia (Watanabe et al., 2009; Hilliard et al., 2010; Hwang et al., 2011; Van Der Werf et al., 2011; Salvage et al., 2018). As expected from concentration dependences for the latter effect (Watanabe et al., 2009; Galimberti and Knollmann, 2011; Heath et al., 2011), flecainide (1 µM) was, respectively, pro- and paradoxically anti-arrhythmic in murine WT and homozygotic RyR2-P2328S atria. In WT, it reduced INa, slowing conduction velocity, sparing refractory periods. Contrastingly, in RyR2-P2328S, it restored normal WT INa values, similarly sparing refractory periods. Dantrolene challenge yielded similar results. These findings suggest its rescue of an arrhythmic phenotype in RyR2-P2328S involves relief of Nav1.5 inhibition by reducing RyR2-mediated Ca2+ release in general feedback effects of Ca2+ homeostasis on different Nav subtypes (Salvage et al., 2018; Salvage et al., 2022; Salvage et al., 2023). Higher flecainide concentrations (5 µM) reduced INa in both WT and RyR2-P2328S (Salvage et al., 2015).
Carvedilol has similar dual class IV in addition to β-adrenergic blocking class II actions: its antioxidant actions reduce RyR2 phosphorylation and oxidation, and its open-state channel block decreases RyR2-mediated Ca2+ release (Zhou et al., 2011). Finally, explorations of SERCA activation by istaroxime also have potential translational implications (Ferrandi et al., 2013; Huang, 2013).
Third, continued progress in understanding Ca2+ homeostasis demonstrates novel regulatory novel Ca2+ signaling molecules. In addition to CaMK-II inhibitors, exemplified by RA608 (Mustroph et al., 2020), and investigational PAK1-targeted agents (He et al., 2023) influencing phosphorylation of cytosolic handling proteins, further signaling molecules, including cADP-ribose, nicotinic acid adenine dinucleotide phosphate (NAADP) (Terrar, 2023), and Epac proteins (Li et al., 2017; Valli et al., 2018b), offer potential investigational targets.
The modernized scheme additionally introduced novel classes V–VII. Investigational drugs from class V blockers of mechanically sensitive TRP channels included N-(p-amylcinnamoyl) anthranilic acid, which may also target Cl− channels (Harteneck et al., 2007), the TRPC3 inhibitor pyrazole-3 (Harada et al., 2012), and TRPM7 inhibitor, LBQ657 (Lifei et al., 2020). These may additionally potentially inhibit hypertrophic and fibrotic changes (Harada et al., 2012; Seo et al., 2014). Class VI targeting cell–cell electrotonic coupling includes the gap junction blocker carbenoxalone (De Groot et al., 2003) and Cx43 opening peptide rotigaptide (Hsieh et al., 2016). Class VII targeting upstream signaling and remodeling contains significant representatives for its component processes. Among inflammatory cytokines, these inhibit TNFα decoy receptor [etanercept (Aschar-Sobbi et al., 2015)], IL-1β [colchicine (Wu et al., 2020)], and IL1 [anakinra (De Jesus et al., 2017)]. Microtubule disruption by colchicine may prevent NLRP3 inflammasome assembly (Wu et al., 2020). Profibrotic modeling is targeted by the TGF-β1 inhibitor pirfenidone (Lee et al., 2006) and TGF-β1 receptor inhibitor GW788388 (de Oliveira et al., 2012). Agents targeting further signaling pathways include the ERK-phosphorylation inhibitor mesalazine (Künzel et al., 2021) and the HMG-CoA reductase inhibitors, statins (Adam et al., 2011). PAK1 activators may inhibit maladaptive, pro-arrhythmic, hypertrophic remodeling and progression in cardiac failure (Tsui et al., 2015; Wang et al., 2018; He et al., 2023) in addition to its role regulating ion channel activity, Ca2+ homeostasis, and cardiac contractility (Ke et al., 2009; Wang et al., 2014; Wang et al., 2015). Angiotensin-converting enzyme and angiotensin receptor blockers, aldosterone receptor antagonists, are further exploratory targets (Savelieva and Camm, 2008; Geng et al., 2020; Saljic et al., 2022; Hu et al., 2023).
11 Discussion: future therapeutic prospects
Research in the physiological sciences has long involved work in successive, mutually reinforcing, interacting cycles between laboratory and clinic. Identified clinical or novel physiological phenomena initiate the development of representative experimental physiological models. These insights, in turn, prompt innovative clinical testing, management, and treatment, themselves prompting further iterations of experimental testing to identify novel physiological targets, investigational new drugs, and interventions (Huang et al., 2020; Huang and Lei, 2023). Future such cycles could add immunotherapeutic approaches, including antibody-based agents with further enhanced target specificities even targeting particular channel isoforms (Presta, 2008) and micro-RNAs/shRNAs knocking down protein expression at the mRNA level provided suitable delivery methods become available (Chakraborty et al., 2017). Within a framework of a modernized drug classification, these could themselves add to understanding arrhythmic events and their modification besides facilitating future clinical development.
Author contributions
ML: conceptualization, funding acquisition, project administration, supervision, and writing–review and editing. SCS: investigation, validation, and writing–review and editing. APJ: conceptualization, project administration, validation, and writing–review and editing. CL-HH: conceptualization, data curation, funding acquisition, investigation, project administration, supervision, validation, visualization, writing–original draft, and writing–review and editing.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the British Heart Foundation (BHF), UK (PG/14/79/31102 and PG/19/59/34582: CL-HH, APJ, and SCS; PG/23/11479, PG/22/11217, RE/18/3/34214, PG/21/10512, FS/PhD/20/29053: ML) and the BHF Centres for Research Excellence (CRE) at Cambridge (CH) and Oxford (ML).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adam O., Theobald K., Lavall D., Grube M., Kroemer H. K., Ameling S., et al. (2011). Increased lysyl oxidase expression and collagen cross-linking during atrial fibrillation. J. Mol. Cell. Cardiol. 50, 678–685. doi:10.1016/j.yjmcc.2010.12.019
Adelman J. P., Maylie J., Sah P. (2012). Small-conductance Ca2+-activated K+ channels: form and function. Annu. Rev. Physiol. 74, 245–269. doi:10.1146/ANNUREV-PHYSIOL-020911-153336
Adeniran I., Hancox J., Zhang H. (2013). In silico investigation of the short QT syndrome, using human ventricle models incorporating electromechanical coupling. Card. Electrophysiol. 4, 166. doi:10.3389/fphys.2013.00166
Ahmad F., Jin Y., Grassam-Rowe A., Zhou Y., Yuan M., Fan X., et al. (2023). Generation of cardiomyocytes from human induced pluripotent stem cells resembling atrial cells with ability to respond to adrenoceptor agonists. Philos. Trans. R. Soc. B Biol. Sci. 378, 20220312. doi:10.1098/rstb.2022.0312
Ahmad S., Valli H., Chadda K. R., Cranley J., Jeevaratnam K., Huang C. L.-H. (2018a). Ventricular pro-arrhythmic phenotype, arrhythmic substrate, ageing and mitochondrial dysfunction in peroxisome proliferator activated receptor-γ coactivator-1β deficient (Pgc-1β-/-) murine hearts. Mech. Ageing Dev. 173, 92–103. doi:10.1016/j.mad.2018.05.004
Ahmad S., Valli H., Edling C. E., Grace A. A., Jeevaratnam K., Huang C. L.-H. (2017). Effects of ageing on pro-arrhythmic ventricular phenotypes in incrementally paced murine Pgc-1β -/- hearts. Pflugers Arch. Eur. J. Physiol. 469, 1579–1590. doi:10.1007/s00424-017-2054-3
Ahmad S., Valli H., Salvage S., Grace A., Jeevaratnam K., Huang C. L.-H. (2018b). Age-dependent electrocardiographic changes in Pgc-1β deficient murine hearts. Clin. Exp. Pharmacol. Physiol. 45, 174–186. doi:10.1111/1440-1681.12863
Ahmad S., Valli H., Smyth R., Jiang A. Y., Jeevaratnam K., Matthews H. R., et al. (2019). Reduced cardiomyocyte Na+ current in the age-dependent murine Pgc-1β-/- model of ventricular arrhythmia. J. Cell. Physiol. 234, 3921–3932. doi:10.1002/jcp.27183
Akar F. G., O’Rourke B. (2011). Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol. {and} Ther. 131, 287–294. doi:10.1016/j.pharmthera.2011.04.005
Alrabghi G., Liu Y., Hu W., Hancox J., Zhang H. (2023). Human atrial fibrillation and genetic defects in transient outward currents: mechanistic insights from multi-scale computational models. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220166. doi:10.1098/rstb.2022.0166
Anderson C., Forte G., Boyett M., D’Souza A., D'Souza A. (2023). Non-canonical role of the sympathetic nervous system in the day-night rhythm in heart rate. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220179. doi:10.1098/rstb.2022.0179
Andrade J., Khairy P., Dobrev D., Nattel S. (2014). The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ. Res. 114, 1453–1468. doi:10.1161/CIRCRESAHA.114.303211
Antzelevitch C., Burashnikov A., Sicouri S., Belardinelli L. (2011). Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm 8, 1281–1290. doi:10.1016/j.hrthm.2011.03.045
Arany Z., He H., Lin J., Hoyer K., Handschin C., Toka O., et al. (2005). Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab 1, 259–271. doi:10.1016/j.cmet.2005.03.002
Armoundas A., Osaka M., Mela T., Rosenbaum D., Ruskin J., Garan H., et al. (1998). T-wave alternans and dispersion of the QT interval as risk stratification markers in patients susceptible to sustained ventricular arrhythmias. Am J Cardioll 82, 1127–1129. doi:10.1016/s0002-9149(98)00570-0
Aschar-Sobbi R., Izaddoustdar F., Korogyi A. S., Wang Q., Farman G. P., Yang F., et al. (2015). Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNFα. Nat. Commun. 6, 6018. doi:10.1038/ncomms7018
Ashino S., Watanabe I., Kofune M., Nagashima K., Ohkubo K., Okumura Y., et al. (2010). Abnormal action potential duration restitution property in the right ventricular outflow tract in Brugada syndrome. Circ. J. 74, 664–670. doi:10.1253/circj.cj-09-0872
Bagwe S., Berenfeld O., Vaidya D., Morley G. E., Jalife J. (2005). Altered right atrial excitation and propagation in connexin40 knockout mice. Circulation 112, 2245–2253. doi:10.1161/CIRCULATIONAHA.104.527325
Balasubramaniam R., Grace A., Saumarez R., Vandenberg J., Huang C. L.-H. (2003). Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J. Physiol. 552, 535–546. doi:10.1113/jphysiol.2003.048249
Bardsley O. J., Matthews H. R., Huang C. L.-H. (2021). Finite element analysis predicts Ca2+ microdomains within tubular-sarcoplasmic reticular junctions of amphibian skeletal muscle. Sci. Rep. 11, 14376. doi:10.1038/s41598-021-93083-1
Behr E., Wood D. A., Wright M., Syrris P., Sheppard M. N., Casey A., et al. (2003). Cardiological assessment of first-degree relatives in sudden arrhythmic death syndrome. Lancet 362, 1457–1459. doi:10.1016/S0140-6736(03)14692-2
Belardinelli L., Giles W., Rajamani S., Karagueuzian H., Shryock J. (2015). Cardiac late Na⁺ current: proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Hear. Rhythm 12, 440–448. doi:10.1016/j.hrthm.2014.11.009
Belevych A. E., Terentyev D., Terentyeva R., Ho H. T., Gyorke I., Bonilla I. M., et al. (2012). Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 110, 569–577. doi:10.1161/CIRCRESAHA.111.260455
Benhorin J., Taub R., Goldmit M., Kerem B., Kass R. S., Windman I., et al. (2000). Effects of flecainide in patients with new SCN5A mutation: mutation-specific therapy for long-QT syndrome? Circulation 101, 1698–1706. doi:10.1161/01.CIR.101.14.1698
Bentzen B. H., Bomholtz S. H., Simó-Vicens R., Folkersen L., Abildgaard L., Speerschneider T., et al. (2020). Mechanisms of action of the KCa2-negative modulator AP30663, a novel compound in development for treatment of atrial fibrillation in man. Front. Pharmacol. 11, 610. doi:10.3389/fphar.2020.00610
Bers D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi:10.1038/415198a
Bezzina C. R., Lahrouchi N., Priori S. G. (2015). Genetics of sudden cardiac death. Circ. Res. 116, 1919–1936. doi:10.1161/CIRCRESAHA.116.304030
Black N., D’Souza A., Wang Y., Piggins H., Dobrzynski H., Morris G., et al. (2019). Circadian rhythm of cardiac electrophysiology, arrhythmogenesis, and the underlying mechanisms. Heart Rhythm 16, 298–307. doi:10.1016/j.hrthm.2018.08.026
Bond R. C., Choisy S. C., Bryant S. M., Hancox J. C., James A. F. (2020). Ion currents, action potentials, and noradrenergic responses in rat pulmonary vein and left atrial cardiomyocytes. Physiol. Rep. 8, e14432. doi:10.14814/phy2.14432
Booz G. W., Day J. N. E., Baker K. M. (2002). Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J. Mol. Cell. Cardiol. 34, 1443–1453. doi:10.1006/jmcc.2002.2076
Bovo E., Lipsius S. L., Zima A. V. (2012). Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J. Physiol. 590, 3291–3304. doi:10.1113/jphysiol.2012.230748
Brown D. A., O’Rourke B. (2010). Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 88, 241–249. doi:10.1093/cvr/cvq231
Burashnikov A., Di Diego J. M., Zygmunt A. C., Belardinelli L., Antzelevitch C. (2007). Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 116, 1449–1457. doi:10.1161/CIRCULATIONAHA.107.704890
Carmeliet E. (2019). Conduction in cardiac tissue. Historical reflections. Physiol. Rep. 7, e13860. doi:10.14814/phy2.13860
Carnes C. A., Chung M. K., Nakayama T., Nakayama H., Baliga R. S., Piao S., et al. (2001). Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ. Res. 89, E32–E38. doi:10.1161/hh1801.097644
Chadda K. R., Ajijola O. A., Vaseghi M., Shivkumar K., Huang C. L.-H., Jeevaratnam K. (2018). Ageing, the autonomic nervous system and arrhythmia: from brain to heart. Ageing Res. Rev. 48, 40–50. doi:10.1016/j.arr.2018.09.005
Chadda K. R., Jeevaratnam K., Lei M., Huang C. L.-H. (2017). Sodium channel biophysics, late sodium current and genetic arrhythmic syndromes. Pflugers Arch. Eur. J. Physiol. 469, 629–641. doi:10.1007/s00424-017-1959-1
Chakraborty C., Sharma A. R., Sharma G., Doss C. G. P., Lee S.-S. (2017). Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol. Ther. - Nucleic Acids 8, 132–143. doi:10.1016/j.omtn.2017.06.005
Chaldoupi S. M., Loh P., Hauer R. N. W., De Bakker J. M. T., Van Rijen H. V. M. (2009). The role of connexin40 in atrial fibrillation. Cardiovasc. Res. 84, 15–23. doi:10.1093/cvr/cvp203
Chen C., Zhu X., Xie J., Li X., Wan R., Hong K. (2023). FAT10 regulates the IKs potassium channel by competing for Kv7.1 ubiquitination. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 278, 20220167. doi:10.1098/rtsb.2022.0167
Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. doi:10.1038/nature13909
Clancy C. E., Kass R. S. (2005). Inherited and acquired vulnerability to ventricular arrhythmias: cardiac Na+ and K+ channels. Physiol. Rev. 85, 33–47. doi:10.1152/physrev.00005.2004
Colquitt J. L., Mendes D., Clegg A. J., Harris P., Cooper K., Picot J., et al. (2014). Implantable cardioverter defibrillators for the treatment of arrhythmias and cardiac resynchronisation therapy for the treatment of heart failure: systematic review and economic evaluation. Health Technol. Assess. 18, 1–560. doi:10.3310/hta18560
Dascal N., Kahanovitch U. (2015). The roles of Gβγ and Gα in gating and regulation of GIRK channels. Int. Rev. Neurobiol. 123, 27–85. doi:10.1016/bs.irn.2015.06.001
Davidenko J. M., Salomonsz R., Pertsov A. M., Baxter W. T., Jalife J. (1995). Effects of pacing on stationary reentrant activity. Theoretical and experimental study. Circ. Res. 77, 1166–1179. doi:10.1161/01.RES.77.6.1166
Davies L., Jin J., Shen W., Tsui H., Shi Y., Wang Y., et al. (2014). Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J. Am. Heart Assoc. 3, e000340. doi:10.1161/JAHA.113.000340
De Groot J. R., Veenstra T., Verkerk A. O., Wilders R., Smits J. P. P., Wilms-Schopman F. J. G., et al. (2003). Conduction slowing by the gap junctional uncoupler carbenoxolone. Cardiovasc. Res. 60, 288–297. doi:10.1016/j.cardiores.2003.07.004
De Jesus N. M., Wang L., Lai J., Rigor R. R., Francis Stuart S. D., Bers D. M., et al. (2017). Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Hear. Rhythm 14, 727–736. doi:10.1016/j.hrthm.2017.01.027
De Marneffe M., Gregoire J. M., Waterschoot P., Kestemont M. P. (1993). The sinus node function: normal and pathological. Eur. Heart J. 14, 649–654. doi:10.1093/eurheartj/14.5.649
De Oliveira F. L., Araújo-Jorge T. C., de Souza E. M., de Oliveira G. M., Degrave W. M., Feige J. J., et al. (2012). Oral administration of gw788388, an inhibitor of transforming growth factor beta signaling, prevents heart fibrosis in chagas disease. PLoS Negl. Trop. Dis. 6, e1696. doi:10.1371/journal.pntd.0001696
Desmouliere A., Geinoz A., Gabbiani F., Gabbiani G. (1993). Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111. doi:10.1083/jcb.122.1.103
Despa S., Shui B., Bossuyt J., Lang D., Kotlikoff M. I., Bers D. M. (2014). Junctional cleft [Ca²⁺]i measurements using novel cleft-targeted Ca²⁺ sensors. Circ. Res. 115, 339–347. doi:10.1161/CIRCRESAHA.115.303582
DiFrancesco D. (1993). Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 55, 455–472. doi:10.1146/annurev.ph.55.030193.002323
Dillon L. M., Rebelo A. P., Moraes C. T. (2012). The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 64, 231–241. doi:10.1002/iub.608
Dobaczewski M., Chen W., Frangogiannis N. G. (2011). Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 51, 600–606. doi:10.1016/j.yjmcc.2010.10.033
Dobrev D. (2009). Editorial: ion channel portrait of the human sinus node: useful for a better understanding of sinus node function and dysfunction in humans? Circulation 119, 1556–1558. doi:10.1161/CIRCULATIONAHA.108.836866
Dobrev D., Nattel S. (2010). New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet 375, 1212–1223. doi:10.1016/S0140-6736(10)60096-7
Dobrzynski H., Boyett M. R., Anderson R. H. (2007). New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115, 1921–1932. doi:10.1161/CIRCULATIONAHA.106.616011
Donald L., Lakatta E. (2023). What makes the sinoatrial node tick? A question not for the faint of heart. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220180. doi:10.1098/rstb.2022.0180
Draper M. H., Weidmann S. (1951). Cardiac resting and action potentials recorded with an intracellular electrode. J. Physiol. 115, 74–94. doi:10.1113/jphysiol.1951.sp004653
D’Souza A., Wang Y., Anderson C., Bucchi A., Baruscotti M., Olieslagers S., et al. (2021). A circadian clock in the sinus node mediates day-night rhythms in Hcn4 and heart rate. Heart Rhythm 18, 801–810. doi:10.1016/j.hrthm.2020.11.026
Dudley S. C., Hoch N. E., McCann L. A., Honeycutt C., Diamandopoulos L., Fukai T., et al. (2005). Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation 112, 1266–1273. doi:10.1161/CIRCULATIONAHA.105.538108
Dulhunty A. F. (2006). Excitation-contraction coupling from the 1950s into the new millennium. Clin. Exp. Pharmacol. Physiol. 33, 763–772. doi:10.1111/j.1440-1681.2006.04441.x
Dun W., Boyden P. A. (2009). Aged atria: electrical remodeling conducive to atrial fibrillation. J. Interv. Card. Electrophysiol. 25, 9–18. doi:10.1007/s10840-008-9358-3
Dzeshka M. S., Lip G. Y. H., Snezhitskiy V., Shantsila E. (2015). Cardiac fibrosis in patients with atrial fibrillation: mechanisms and clinical implications. J. Am. Coll. Cardiol. 66, 943–959. doi:10.1016/j.jacc.2015.06.1313
Eager K. R., Dulhunty A. F. (1998). Activation of the cardiac ryanodine receptor by sulfhydryl oxidation is modified by Mg2+ and ATP. J. Membr. Biol. 163, 9–18. doi:10.1007/s002329900365
Eloff B. C., Lerner D. L., Yamada K. a., Schuessler R. B., Saffitz J. E., Rosenbaum D. S. (2001). High resolution optical mapping reveals conduction slowing in connexin43 deficient mice. Cardiovasc. Res. 51, 681–690. doi:10.1016/s0008-6363(01)00341-8
Emelyanova L., Ashary Z., Cosic M., Negmadjanov U., Ross G., Rizvi F., et al. (2016). Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am. J. Physiol. - Hear. Circ. Physiol. 311, H54–H63. doi:10.1152/ajpheart.00699.2015
Endo M. (2009). Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 89, 1153–1176. doi:10.1152/physrev.00040.2008
Enyedi P., Czirjak G. (2010). Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev. 90, 559–605. doi:10.1152/physrev.00029.2009
Erickson J. R., Joiner M. ling A., Guan X., Kutschke W., Yang J., Oddis C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi:10.1016/j.cell.2008.02.048
Faivre J. F., Findlay I. (1990). Action potential duration and activation of ATP-sensitive potassium current in isolated Guinea-pig ventricular myocytes. Biochim. Biophys. Acta 1029, 167–172. doi:10.1016/0005-2736(90)90450-3
Ferrandi M., Barassi P., Tadini-Buoninsegni F., Bartolommei G., Molinari I., Tripodi M. G., et al. (2013). Istaroxime stimulates SERCA2a and accelerates calcium cycling in heart failure by relieving phospholamban inhibition. Br. J. Pharmacol. 169, 1849–1861. doi:10.1111/bph.12278
Ford J., Milnes J., El Haou S., Wettwer E., Loose S., Matschke K., et al. (2016). The positive frequency-dependent electrophysiological effects of the IKur inhibitor XEN-D0103 are desirable for the treatment of atrial fibrillation. Hear. Rhythm 13, 555–564. doi:10.1016/j.hrthm.2015.10.003
Fosset M., De Weille J. R., Green R. D., Schmid-Antomarchi H., Lazdunski M. (1988). Antidiabetic sulfonylureas control action potential properties in heart cells via high affinity receptors that are linked to ATP-dependent K+ channels. J. Biol. Chem. 263, 7933–7936. doi:10.1016/s0021-9258(18)68422-4
Foster M. N., Coetzee W. A. (2016). KATP Channels in the cardiovascular system. Physiol. Rev. 96, 177–252. doi:10.1152/physrev.00003.2015
Friedrichs K., Baldus S., Klinke A. (2012). Fibrosis in atrial fibrillation - role of reactive species and MPO. Front. Physiol. 3, 214. doi:10.3389/fphys.2012.00214
Galimberti E. S., Knollmann B. C. (2011). Efficacy and potency of class I antiarrhythmic drugs for suppression of Ca2+ waves in permeabilized myocytes lacking calsequestrin. J. Mol. Cell. Cardiol. 51, 760–768. doi:10.1016/j.yjmcc.2011.07.002
Garesse R., Vallejo C. G. (2001). Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes. Gene 263, 1–16. doi:10.1016/S0378-1119(00)00582-5
Geng M., Lin A., Nguyen T. P. (2020). Revisiting antiarrhythmic drug therapy for atrial fibrillation: reviewing lessons learned and redefining therapeutic paradigms. Front. Pharmacol. 11, 581837. doi:10.3389/fphar.2020.581837
Grivennikova V. G., Kareyeva A. V., Vinogradov A. D. (2010). What are the sources of hydrogen peroxide production by heart mitochondria? Biochim. Biophys. Acta 1797, 939–944. doi:10.1016/j.bbabio.2010.02.013
Gu M., Zhu Y., Yin X., Zhang D. M. (2018). Small-conductance Ca2+-activated K+ channels: insights into their roles in cardiovascular disease. Exp. Mol. Med. 50, 23–27. doi:10.1038/S12276-018-0043-Z
Gurung I., Medina-Gomez G., Kis A., Baker M., Velagapudi V., Neogi S. G., et al. (2011). Deletion of the metabolic transcriptional coactivator PGC1β induces cardiac arrhythmia. Cardiovasc. Res. 92, 29–38. doi:10.1093/cvr/cvr155
Gutstein D. E., Morley G. E., Tamaddon H., Vaidya D., Schneider M. D., Chen J., et al. (2001). Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 88, 333–339. doi:10.1161/01.res.88.3.333
Hagendorff A., Schumacher B., Kirchhoff S., Lüderitz B., Willecke K. (1999). Conduction disturbances and increased atrial vulnerability in connexin40-deficient mice analyzed by transesophageal stimulation. Circulation 99, 1508–1515. doi:10.1161/01.CIR.99.11.1508
Hakim P., Brice N., Thresher R., Lawrence J., Zhang Y., Jackson A. P., et al. (2010). Scn3b knockout mice exhibit abnormal sino-atrial and cardiac conduction properties. Acta Physiol. (Oxf). 198, 47–59. doi:10.1111/j.1748-1716.2009.02048.x
Hakim P., Gurung I. S., Pedersen T. H., Thresher R., Brice N., Lawrence J., et al. (2008). Scn3b knockout mice exhibit abnormal ventricular electrophysiological properties. Prog. Biophys. Mol. Biol. 98, 251–266. doi:10.1016/j.pbiomolbio.2009.01.005
Hancox J., Du C., Butler A., Zhang Y., Dempsey C., Harmer S., et al. (2023). Pro-arrhythmic effects of gain-of-function potassium channel mutations in the short QT syndrome. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220165. doi:10.1098/rstb.2022.0165
Hao X., Zhang Y., Zhang X., Nirmalan M., Davies L., Konstantinou D., et al. (2011). TGF-β1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ. Arrhythmia Electrophysiol. 4, 397–406. doi:10.1161/CIRCEP.110.960807
Harada M., Luo X., Qi X. Y., Tadevosyan A., Maguy A., Ordog B., et al. (2012). Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 126, 2051–2064. doi:10.1161/CIRCULATIONAHA.112.121830
Harteneck C., Frenzel H., Kraft R. (2007). N-(p-amylcinnamoyl)anthranilic acid (ACA): a phospholipase A2 inhibitor and TRP channel blocker. Cardiovasc. Drug Rev. 25, 61–75. doi:10.1111/j.1527-3466.2007.00005.x
Hayashi H., Shiferaw Y., Sato D., Nihei M., Lin S.-F., Chen P.-S., et al. (2007). Dynamic origin of spatially discordant alternans in cardiac tissue. Biophys. J. 92, 448–460. doi:10.1529/biophysj.106.091009
He Y., Grassam-Rowe A., Lei M., Bae J. (2023). Targeting p21-activated kinase 1 for development of a novel anti-arrhythmic drug class. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220285. doi:10.1098/rstb.2022.0285
Healey J. S., Morillo C. A., Connolly S. J. (2005). Role of the renin-angiotensin-aldosterone system in atrial fibrillation and cardiac remodeling. Curr. Opin. Cardiol. 20, 31–37. doi:10.1097/01.hco.0000147883.32107.1d
Heath B. M., Cui Y., Worton S., Lawton B., Ward G., Ballini E., et al. (2011). Translation of flecainide- and mexiletine-induced cardiac sodium channel inhibition and ventricular conduction slowing from nonclinical models to clinical. J Pharmacol Toxicol Methods 63, 258–268. doi:10.1016/j.vascn.2010.12.004
Heijman J., Muna A. P., Veleva T., Molina C. E., Sutanto H., Tekook M., et al. (2020). Atrial myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation. Circ. Res. 127, 1036–1055. doi:10.1161/CIRCRESAHA.120.316710
Hichri E., Abriel H., Kucera J. P. (2018). Distribution of cardiac sodium channels in clusters potentiates ephaptic interactions in the intercalated disc. J. Physiol. 596, 563–589. doi:10.1113/JP275351
Hilliard F. A., Steele D. S., Laver D., Yang Z., Le Marchand S. J., Chopra N., et al. (2010). Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J. Mol. Cell. Cardiol. 48, 293–301. doi:10.1016/j.yjmcc.2009.10.005
Hothi S. S., Gurung I. S., Heathcote J. C., Zhang Y., Booth S. W., Skepper J. N., et al. (2008). Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflugers Arch 457, 253–270. doi:10.1007/s00424-008-0508-3
Hothi S. S., Thomas G., Killeen M. J., Grace A. A., Huang C. L.-H. (2009). Empirical correlation of triggered activity and spatial and temporal re-entrant substrates with arrhythmogenicity in a murine model for Jervell and Lange-Nielsen syndrome. Pflugers Arch 458, 819–835. doi:10.1007/s00424-009-0671-1
Hsieh Y. C., Lin J. C., Hung C. Y., Li C. H., Lin S. F., Yeh H. I., et al. (2016). Gap junction modifier rotigaptide decreases the susceptibility to ventricular arrhythmia by enhancing conduction velocity and suppressing discordant alternans during therapeutic hypothermia in isolated rabbit hearts. Heart Rhythm 13, 251–261. doi:10.1016/j.hrthm.2015.07.023
Hu D., Barajas-Martinez H., Zhang Z., Duan H., Zhao Q., Bao M., et al. (2023). Advances in basic and translational research in atrial fibrillation. Philos. Trans. R. Soc. B Biol. Sci. 378. doi:10.1098/rstb.2022.0174
Hu Y. F., Chen Y. J., Lin Y. J., Chen S. A. (2015). Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 12, 230–243. doi:10.1038/nrcardio.2015.2
Huang C.L.-H. (2021). Keynes and aidley’s nerve and muscle. 5th ed. Cambridge: Cambridge University Press.
Huang C. L.-H., Lei M. (2023). Cardiomyocyte electrophysiology and its modulation: current views and future prospects. Philos. Trans. R. Soc. B Biol. Sci. 378, 20220160. doi:10.1098/rstb.2022.0160
Huang C. L.-H., Solaro R., Ke Y., Lei M. (2016). Editorial: Ca(2+) signaling and heart rhythm. Front. Physiol. 6, 423. doi:10.3389/fphys.2015.00423
Huang C. L.-H. (2013). SERCA2a stimulation by istaroxime: a novel mechanism of action with translational implications. Br. J. Pharmacol. 170, 486–488. doi:10.1111/bph.12288
Huang C. L.-H. (2015). Computational analysis of the electromechanical consequences of short QT syndrome. Front Physiol 6, 44. doi:10.3389/fphys.2015.00044
Huang C. L.-H. (2017). Murine electrophysiological models of cardiac arrhythmogenesis. Physiol. Rev. 97, 283–409. doi:10.1152/physrev.00007.2016
Huang C. L.-H., Wu L., Jeevaratnam K., Lei M. (2020). Update on antiarrhythmic drug pharmacology. J. Cardiovasc. Electrophysiol. 31, 579–592. doi:10.1111/jce.14347
Huss J. M., Torra I. P., Staels B., Giguère V., Kelly D. P. (2004). Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 24, 9079–9091. doi:10.1128/MCB.24.20.9079-9091.2004
Hwang H. S., Hasdemir C., Laver D., Mehra D., Turhan K., Faggioni M., et al. (2011). Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ. Arrhythm. Electrophysiol. 4, 128–135. doi:10.1161/CIRCEP.110.959916
Iwasaki Y., Nishida K., Kato T., Nattel S. (2011). Atrial fibrillation pathophysiology: implications for management. Circulation 124, 2264–2274. doi:10.1161/CIRCULATIONAHA.111.019893
January C. T., Riddle J. M. (1989). Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ. Res. 64, 977–990. doi:10.1161/01.RES.64.5.977
Jeevaratnam K., Chadda K., Huang C. L.-H., Camm A. (2017). Cardiac potassium channels: physiological insights for targeted therapy. J. Cardiovasc. Pharmacol. Ther. 23, 119–129. doi:10.1177/1074248417729880
Jeevaratnam K., Guzadhur L., Goh Y. M., Grace A. A., Huang C. L.-H. (2016). Sodium channel haploinsufficiency and structural change in ventricular arrhythmogenesis. Acta Physiol 216, 186–202. doi:10.1111/apha.12577
Jeevaratnam K., Poh Tee S., Zhang Y., Rewbury R., Guzadhur L., Duehmke R., et al. (2011). Delayed conduction and its implications in murine Scn5a+/- hearts: independent and interacting effects of genotype, age, and sex. Pflugers Arch. Eur. J. Physiol. 461, 29–44. doi:10.1007/s00424-010-0906-1
Jeevaratnam K., Rewbury R., Zhang Y., Guzadhur L., Grace A. A., Lei M., et al. (2012). Frequency distribution analysis of activation times and regional fibrosis in murine Scn5a+/- hearts: the effects of ageing and sex. Mech. Ageing Dev. 133, 591–599. doi:10.1016/j.mad.2012.07.006
Jesus I. C. G., Araújo F. M., Mesquita T., Júnior N. N. S., Silva M. M., Morgan H. J. N., et al. (2021). Molecular basis of Period 1 regulation by adrenergic signaling in the heart. FASEB J 35, e21886. doi:10.1096/fj.202100441R
Jung E., Capel R., Jiang C., Venturi E., Neagu G., Pearcey S., et al. (2023). Cardiac deficiency of P21-activated kinase 1 promotes atrial arrhythmogenesis in mice following adrenergic challenge. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220168. doi:10.1098/rstb.2022.0168
Kannel W. B., Kannel C., Paffenbarger R. S., Cupples L. A. (1987). Heart rate and cardiovascular mortality: the Framingham Study. Am. Heart J. 113, 1489–1494. doi:10.1016/0002-8703(87)90666-1
Ke Y., Lei M., Solaro R. J. (2009). Regulation of cardiac excitation and contraction by p21 activated kinase-1. Prog. Biophys. Mol. Biol. 98, 238–250. doi:10.1016/j.pbiomolbio.2009.01.007
Killeen M. J. (2009). Drug-induced arrhythmias and sudden cardiac death: implications for the pharmaceutical industry. Drug Discov 14, 589–597. doi:10.1016/j.drudis.2009.03.004
Killeen M. J., Sabir I. N., Grace A. A., Huang C. L.-H. (2008a). Dispersions of repolarization and ventricular arrhythmogenesis: lessons from animal models. Prog. Biophys. Mol. Biol. 98, 219–229. doi:10.1016/j.pbiomolbio.2008.10.008
Killeen M. J., Thomas G., Gurung I. S., Goddard C. A., Fraser J. A., Mahaut-Smith M. P., et al. (2007). Arrhythmogenic mechanisms in the isolated perfused hypokalaemic murine heart. Acta Physiol. (Oxf). 189, 33–46. doi:10.1111/j.1748-1716.2006.01643.x
Killeen M. J., Thomas G., Sabir I. N., Grace A. A., Huang C. L.-H. (2008b). Mouse models of human arrhythmia syndromes. Acta Physiol. (Oxf). 192, 455–469. doi:10.1111/j.1748-1716.2007.01822.x
King J., Huang C. L.-H., Fraser J. A. (2013a). Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front. Physiol. 4, 154. doi:10.3389/fphys.2013.00154
King J., Wickramarachchi C., Kua K., Du Y., Jeevaratnam K., Matthews H. R., et al. (2013b). Loss of Nav1.5 expression and function in murine atria containing the RyR2-P2328S gain-of-function mutation. Cardiovasc. Res. 99, 751–759. doi:10.1093/cvr/cvt141
King J., Zhang Y., Lei M., Grace A. A., Huang C. L.-H., Fraser J. A. (2013c). Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol 207, 308–323. doi:10.1111/apha.12006
Kléber A. G., Rudy Y. (2004). Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 84, 431–488. doi:10.1152/physrev.00025.2003
Koruth J. S., Lala A., Pinney S., Reddy V. Y., Dukkipati S. R. (2017). The clinical use of ivabradine. J. Am. Coll. Cardiol. 70, 1777–1784. doi:10.1016/j.jacc.2017.08.038
Krogh-Madsen T., Abbott G. W., Christini D. J. (2012). Effects of electrical and structural remodeling on atrial fibrillation maintenance: a simulation study. PLoS Comput. Biol 8, e1002390. doi:10.1371/journal.pcbi.1002390
Kucera J. P., Kléber A. G., Rohr S. (1998). Slow conduction in cardiac tissue, II: effects of branching tissue geometry. Circ. Res. 83, 795–805. doi:10.1161/01.res.83.8.795
Kucharska-Newton A. M., Couper D. J., Pankow J. S., Prineas R. J., Rea T. D., Sotoodehnia N., et al. (2010). Diabetes and the risk of sudden cardiac death, the Atherosclerosis Risk in Communities study. Acta Diabetol 47 (1), 161–168. doi:10.1007/s00592-009-0157-9
Kukreja R. C., Okabe E., Schrier G. M., Hess M. L. (1988). Oxygen radical-mediated lipid peroxidation and inhibition of Ca2+-ATPase activity of cardiac sarcoplasmic reticulum. Arch. Biochem. Biophys. 261, 447–457. doi:10.1016/0003-9861(88)90361-X
Künzel S. R., Hoffmann M., Weber S., Künzel K., Kämmerer S., Günscht M., et al. (2021). Diminished PLK2 induces cardiac fibrosis and promotes atrial fibrillation. Circ. Res. 129, 804–820. doi:10.1161/CIRCRESAHA.121.319425
Lacerda A. E., Kuryshev Y. A., Yan G. X., Waldo A. L., Brown A. M. (2010). Vanoxerine: cellular mechanism of a new antiarrhythmic. J. Cardiovasc. Electrophysiol. 21, 301–310. doi:10.1111/j.1540-8167.2009.01623.x
Lakatta E. G., Maltsev V. A., Vinogradova T. M. (2010). A coupled system of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 106, 659–673. doi:10.1161/CIRCRESAHA.109.206078
Langa P., Shafaattalab S., Goldspink P., Wolska B., Fernandes A., Tibbits G., et al. (2023). A perspective on Notch signalling in progression and arrhythmogenesis in familial hypertrophic and dilated cardiomyopathies. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220176. doi:10.1098/rstb.2022.0176
Laver D. R., Curtis B. A. (1996). Response of ryanodine receptor channels to Ca2+ steps produced by rapid solution exchange. Biophys. J. 71, 732–741. doi:10.1016/S0006-3495(96)79272-X
Lee K. W., Everett IV T. H., Rahmutula D., Guerra J. M., Wilson E., Ding C., et al. (2006). Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation 114, 1703–1712. doi:10.1161/CIRCULATIONAHA.106.624320
Lee P., Klos M., Bollensdorff C., Hou L., Ewart P., Kamp T. J., et al. (2012). Simultaneous voltage and calcium mapping of genetically purified human induced pluripotent stem cell-derived cardiac myocyte monolayers. Circ. Res. 110, 1556–1563. doi:10.1161/CIRCRESAHA.111.262535
Lee S. H., Chen Y. C., Chen Y. J., Chang S. L., Tai C. T., Wongcharoen W., et al. (2007). Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci 80, 1806–1815. doi:10.1016/j.lfs.2007.02.029
Lehman J. J., Barger P. M., Kovacs A., Saffitz J. E., Medeiros D. M., Kelly D. P. (2000). Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 106, 847–856. doi:10.1172/JCI10268
Lei M., Goddard C., Liu J., Léoni A.-L., Royer A., Fung S. S.-M., et al. (2005). Sinus node dysfunction following targeted disruption of the murine cardiac sodium channel gene Scn5a. J. Physiol. 567, 387–400. doi:10.1113/jphysiol.2005.083188
Lei M., Wu L., Terrar D. A., Huang C. L.-H. (2018). Modernized classification of cardiac antiarrhythmic drugs. Circulation 138, 1879–1896. doi:10.1161/CIRCULATIONAHA.118.035455
Lei M., Zhang H., Grace A. A., Huang C. L.-H. (2007). SCN5A and sinoatrial node pacemaker function. Cardiovasc. Res. 74, 356–365. doi:10.1016/j.cardiores.2007.01.009
Lelliott C. J., Medina-Gomez G., Petrovic N., Kis A., Feldmann H. M., Bjursell M., et al. (2006). Ablation of PGC-1β results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol 4, e369–e2056. doi:10.1371/journal.pbio.0040369
Leone T. C., Kelly D. P. (2011). Transcriptional control of cardiac fuel metabolism and mitochondrial function. Cold Spring Harb. Symp. Quant. Biol. 76, 175–182. doi:10.1101/sqb.2011.76.011965
Lerman B. B. (2015). Ventricular tachycardia: mechanistic insights derived from adenosine. Circ. Arrhythmia Electrophysiol 8, 483–491. doi:10.1161/CIRCEP.115.001693
Li L., fei Y., Lou Q., Liu G., Yun F. X., Li T. K., et al. (2020). Sacubitril/valsartan attenuates atrial electrical and structural remodelling in a rabbit model of atrial fibrillation. Eur. J. Pharmacol. 881, 173120. doi:10.1016/j.ejphar.2020.173120
Li M., Hothi S., Salvage S., Jeevaratnam K., Grace A., Huang C. L.-H. (2017). Arrhythmic effects of Epac-mediated ryanodine receptor activation in Langendorff-perfused murine hearts are associated with reduced conduction velocity. Clin Exp Pharmacol Physiol 44, 686–692. doi:10.1111/1440-1681.12751
Li N., Edel M., Liu K., Denning C., Betts J., Neely O., et al. (2023a). Human induced pluripotent stem cell-derived cardiac myocytes and sympathetic neurons in disease modelling. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220173. doi:10.1098/rstb.2022.0173
Li T., Liu T., Yang B., Jiang Q., Zhou Y., Wang J., et al. (2023b). Small-conductance calcium-activated potassium channels in the heart: expression, regulation and pathological implications. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220171. doi:10.1098/rstb.2022.0171
Liu M., Galli G., Wang Y., Fan Q., Wang Z., Wang X., et al. (2020). Novel therapeutic targets for hypoxia-related cardiovascular diseases: the role of HIF-1. Front. Physiol. 11, 774. doi:10.3389/fphys.2020.00774
Liu M., Liu H., Dudley S. C. (2010). Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 107, 967–974. doi:10.1161/CIRCRESAHA.110.220673
Liu S. X., Matthews H. R., Huang C. L.-H. (2021). Sarcoplasmic reticular Ca2+-ATPase inhibition paradoxically upregulates murine skeletal muscle Nav1.4 function. Sci. Rep. 11, 2846. doi:10.1038/s41598-021-82493-w
Liu W., Zi M., Naumann R., Ulm S., Jin J., Taglieri D. M., et al. (2011). Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation 124, 2702–2715. doi:10.1161/CIRCULATIONAHA.111.048785
Liu X., Ren L., Yu S., Li G., He P., Yang Q., et al. (2023). Late sodium current in synergism with Ca2+/calmodulin-dependent protein kinase II contributes to β-adrenergic activation-induced atrial fibrillation. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220163. doi:10.1098/rstb.2022.0163
Lloyd-Jones D. M., Wang T. J., Leip E. P., Larson M. G., Levy D., Vasan R. S., et al. (2004). Lifetime risk for development of atrial fibrillation: the Framingham heart study. Circulation 110, 1042–1046. doi:10.1161/01.CIR.0000140263.20897.42
Logantha S., Yanni J., Yamanushi T., Absi M., Temple I., Kabuto H., et al. (2023). Remodelling and dysfunction of the sinus node in pulmonary arterial hypertension. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220178. doi:10.1098/rstb.2022.0178
Lu Y., Mahaut-Smith M. P., Huang C. L.-H., Vandenberg J. I. (2003). Mutant MiRP1 subunits modulate HERG K+ channel gating: a mechanism for pro-arrhythmia in long QT syndrome type 6. J. Physiol. 551, 253–262. doi:10.1113/jphysiol.2003.046045
Lu Y., Mahaut-Smith M. P., Varghese A., Huang C. L.-H., Kemp P. R., Vandenberg J. I. (2001). Effects of premature stimulation on HERG K+ channels. J. Physiol. 537, 843–851. doi:10.1111/j.1469-7793.2001.00843.x
Martin C. A., Grace A. A., Huang C. L.-H. (2011a). Refractory dispersion promotes conduction disturbance and arrhythmias in a Scn5a+/− mouse model. Pflugers Arch 462, 495–504. doi:10.1007/s00424-011-0989-3
Martin C. A., Grace A. A., Huang C. L.-H. (2011b). Spatial and temporal heterogeneities are localized to the right ventricular outflow tract in a heterozygotic Scn5a mouse model. Am. J. Physiol. Heart Circ. Physiol. 300, H605–H616. doi:10.1152/ajpheart.00824.2010
Martin C. A., Guzadhur L., Grace A. A., Lei M., Huang C. L.-H. (2011c). Mapping of reentrant spontaneous polymorphic ventricular tachycardia in a Scn5a+/- mouse model. Am. J. Physiol. Heart Circ. Physiol. 300, H1853–H1862. doi:10.1152/ajpheart.00034.2011
Martin C. A., Huang C. L.-H., Matthews G. D. (2011d). Recent developments in the management of patients at risk for sudden cardiac death. Postgrad. Med. 123, 84–94. doi:10.3810/pgm.2011.03.2266
Martin C. A., Huang C. L.-H., Matthews G. D. K. (2013). The role of ion channelopathies in sudden cardiac death: implications for clinical practice. Ann. Med. 45, 364–374. doi:10.3109/07853890.2013.783994
Martin C. A., Matthews G. D. K., Huang C. L.-H. (2012). Sudden cardiac death and inherited channelopathy: the basic electrophysiology of the myocyte and myocardium in ion channel disease. Heart 98, 536–543. doi:10.1136/heartjnl-2011-300953
Martin C. A., Petousi N., Chawla S., Hockaday A. R., Burgess A. J., Fraser J. A., et al. (2003). The effect of extracellular tonicity on the anatomy of triad complexes in amphibian skeletal muscle. J. Muscle Res. Cell Motil. 24, 407–415. doi:10.1023/A:1027356410698
Martin C. A., Zhang Y., Grace A. A., Huang C. L.-H. (2010). Increased right ventricular repolarization gradients promote arrhythmogenesis in a murine model of Brugada syndrome. J. Cardiovasc. Electrophysiol. 21, 1153–1159. doi:10.1111/j.1540-8167.2010.01767.x
Matthews G., Guzadhur L., Grace A., Huang C. L.-H. (2012). Nonlinearity between action potential alternans and restitution, which both predict ventricular arrhythmic properties in Scn5a+/- and wild-type murine hearts. J. Appl. Physiol. 112, 1847–1863. doi:10.1152/japplphysiol.00039.2012
Matthews G., Guzadhur L., Sabir I., Grace A., Huang C. L.-H. (2013). Action potential wavelength restitution predicts alternans and arrhythmia in murine Scn5a+/- hearts. J. Physiol. 591, 4167–4188. doi:10.1113/jphysiol.2013.254938
Matthews G. D. K., Martin C. A., Grace A. A., Zhang Y., Huang C. L.-H. (2010). Regional variations in action potential alternans in isolated murine Scn5a (+/-) hearts during dynamic pacing. Acta Physiol. (Oxf). 200, 129–146. doi:10.1111/j.1748-1716.2010.02138.x
Matthews H. R., Tan S. R. X., Shoesmith J. A., Ahmad S., Valli H., Jeevaratnam K., et al. (2019). Sodium current inhibition following stimulation of exchange protein directly activated by cyclic-3′,5′-adenosine monophosphate (Epac) in murine skeletal muscle. Sci. Rep. 9, 1927. doi:10.1038/s41598-018-36386-0
Mauviel A. (2005). Transforming growth factor-beta: a key mediator of fibrosis. Methods Mol. Med. 117, 69–80. doi:10.1385/1-59259-940-0:069
Mehta P. K., Griendling K. K. (2007). Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. - Cell Physiol. 292, C82–C97. doi:10.1152/ajpcell.00287.2006
Meredith A. L., Wu Y., Gao Z., Anderson M. E., Dalziel J. E., Lai M. H. (2014). BK channels regulate sinoatrial node firing rate and cardiac pacing in vivo. Am. J. Physiol. - Hear. Circ. Physiol. 307, H1327–H1338. doi:10.1152/ajpheart.00354.2014
Moreno A. P., Sáez J. C., Fishman G. I., Spray D. C. (1994). Human connexin43 gap junction channels: regulation of unitary conductances by phosphorylation. Circ. Res. 74, 1050–1057. doi:10.1161/01.RES.74.6.1050
Mori Y., Fishman G. I., Peskin C. S. (2008). Ephaptic conduction in a cardiac strand model with 3D electrodiffusion. Proc. Natl. Acad. Sci. U. S. A. 105, 6463–6468. doi:10.1073/pnas.0801089105
Morris J., Bardsley O., Salvage S., Jackson A., Matthews H., Huang C. L.-H. (2024). Nernst-Planck-Gaussian modelling of electrodiffusional recovery from ephaptic excitation between mammalian cardiomyocytes. Front. Physiol. Sect. Card. Electrophysiol. 14, 1280151. doi:10.3389/fphys.2023.1280151
Murphy A. M., Wong A. L., Bezuhly M. (2015). Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenes. Tissue Repair 8, 7. doi:10.1186/s13069-015-0023-z
Mustroph J., Drzymalski M., Baier M., Pabel S., Biedermann A., Memmel B., et al. (2020). The oral Ca/calmodulin-dependent kinase II inhibitor RA608 improves contractile function and prevents arrhythmias in heart failure. ESC Hear. Fail. 7, 2871–2883. doi:10.1002/ehf2.12895
Nakamura K., Fushimi K., Kouchi H., Mihara K., Miyazaki M., Ohe T., et al. (1998). Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation 98, 794–799. doi:10.1161/01.CIR.98.8.794
Namadurai S., Yereddi N. R., Cusdin F. S., Huang C. L.-H., Chirgadze D. Y., Jackson A. P. (2015). A new look at sodium channel β subunits. Open Biol 5, 140192. doi:10.1098/rsob.140192
Nattel S. (2018). Electrical coupling between cardiomyocytes and fibroblasts: experimental testing of a challenging and important concept. Cardiovasc. Res. 114, 349–352. doi:10.1093/cvr/cvy003
Nattel S., Maguy A., Le Bouter S., Yeh Y.-H. (2007). Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 87, 425–456. doi:10.1152/physrev.00014.2006
Nearing B. D., Huang A. H., Verrier R. L. (1991). Dynamic tracking of cardiac vulnerability by complex demodulation of the T wave. Science 252, 437–440. doi:10.1126/science.2017682
Neelands T. R., Herson P. S., Jacobson D., Adelman J. P., Maylie J. (2001). Small-conductance calcium-activated potassium currents in mouse hyperexcitable denervated skeletal muscle. J. Physiol. 536, 397–407. doi:10.1111/j.1469-7793.2001.0397c.xd
Nerbonne J. M., Kass R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. doi:10.1152/physrev.00002.2005
Nguyen B. Y., Ruiz-Velasco A., Bui T., Collins L., Wang X., Liu W. (2019). Mitochondrial function in the heart: the insight into mechanisms and therapeutic potentials. Br. J. Pharmacol. 176, 4302–4318. doi:10.1111/bph.14431
Nolasco J. B., Dahlen R. W. (1968). A graphic method for the study of alternation in cardiac action potentials. J. Appl. Physiol. 25, 191–196. doi:10.1152/jappl.1968.25.2.191
Pabel S., Mustroph J., Stehle T., Lebek S., Dybkova N., Keyser A., et al. (2020). Dantrolene reduces CaMKIIδC-mediated atrial arrhythmias. Europace 22, 1111–1118. doi:10.1093/europace/euaa079
Pandit S. V., Jalife J. (2013). Rotors and the dynamics of cardiac fibrillation. Circ. Res. 112, 849–862. doi:10.1161/CIRCRESAHA.111.300158
Pastore J. M., Girouard S. D., Laurita K. R., Akar F. G., Rosenbaum D. S. (1999). Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 99, 1385–1394. doi:10.1161/01.CIR.99.10.1385
Pathmanathan P., Whiteley J. P. (2009). A numerical method for cardiac mechanoelectric simulations. Ann. Biomed. Eng. 37, 860–873. doi:10.1007/s10439-009-9663-8
Pineda S., Nikolova-Krstevski V., Leimena C., Atkinson A. J., Altekoester A. K., Cox C. D., et al. (2021). Conserved role of the large conductance calcium-activated potassium channel, KCa1.1, in sinus node function and arrhythmia risk. Circ. Genomic Precis. Med. 14, e003144. doi:10.1161/CIRCGEN.120.003144
Pitzalis M. V., Mastropasqua F., Massari F., Totaro P., Scrutinio D., Rizzon P. (1996). Sleep suppression of ventricular arrhythmias: a predictor of beta-blocker efficacy. Eur. Heart J. 17, 917–925. doi:10.1093/oxfordjournals.eurheartj.a014974
Presta L. G. (2008). Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol 20, 460–470. doi:10.1016/j.coi.2008.06.012
Remme C. (2023). SCN5A channelopathy: arrhythmia, cardiomyopathy, epilepsy and beyond. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220164. doi:10.1098/rstb.2022.0164
Rhett J. M., Gourdie R. G. (2012). The perinexus: a new feature of Cx43 gap junction organization. Hear. Rhythm 9, 619–623. doi:10.1016/j.hrthm.2011.10.003
Rhett J. M., Veeraraghavan R., Poelzing S., Gourdie R. G. (2013). The perinexus: sign-post on the path to a new model of cardiac conduction? Trends Cardiovasc. Med. 23, 222–228. doi:10.1016/j.tcm.2012.12.005
Rice J. J., Wang F., Bers D. M., De Tombe P. P. (2008). Approximate model of cooperative activation and crossbridge cycling in cardiac muscle using ordinary differential equations. Biophys. J. 95, 2368–2390. doi:10.1529/biophysj.107.119487
Rohr S. (2004). Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc. Res. 62, 309–322. doi:10.1016/j.cardiores.2003.11.035
Rosenbaum D. S., Jackson L. E., Smith J. M., Garan H., Ruskin J. N., Cohen R. J. (1994). Electrical alternans and vulnerability to ventricular arrhythmias. N. Engl. J. Med. 330, 235–241. doi:10.1056/NEJM199401273300402
Rubart M., Zipes D. P. (2005). Mechanisms of sudden cardiac death. J. Cllinical Investig. 115, 2305–2315. doi:10.1172/JCI26381
Rusciano M. R., Sommariva E., Douin-Echinard V., Ciccarelli M., Poggio P., Maione A. S. (2019). CaMKII activity in the inflammatory response of cardiac diseases. Int. J. Mol. Sci. 20, 4374. doi:10.3390/ijms20184374
Saadeh K., Nantha Kumar N., Fazmin I. T., Edling C. E., Jeevaratnam K. (2022). Anti-malarial drugs: mechanisms underlying their proarrhythmic effects. Br. J. Pharmacol. 179, 5237–5258. doi:10.1111/bph.15959
Sabir I. N., Fraser J. A., Killeen M. J., Grace A. A., Huang C. L.-H. (2007). The contribution of refractoriness to arrhythmic substrate in hypokalemic Langendorff-perfused murine hearts. Pflugers Arch 454, 209–222. doi:10.1007/s00424-007-0217-3
Sabir I. N., Killeen M. J., Grace A. A., Huang C. L.-H. (2008a). Ventricular arrhythmogenesis: insights from murine models. Prog. Biophys. Mol. Biol. 98, 208–218. doi:10.1016/j.pbiomolbio.2008.10.011
Sabir I. N., Li L. M., Grace A. A., Huang C. L.-H. (2008b). Restitution analysis of alternans and its relationship to arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch. Eur. J. Physiol. 455, 653–666. doi:10.1007/s00424-007-0327-y
Sabir I. N., Matthews G. D. K., Huang C. L.-H. (2013). Sudden arrhythmic death: from basic science to clinical practice. Front. Physiol. 4, 339. doi:10.3389/fphys.2013.00339
Saljic A., Heijman J., Dobrev D. (2022). Emerging antiarrhythmic drugs for atrial fibrillation. Int. J. Mol. Sci. 23, 4096. doi:10.3390/ijms23084096
Salvage S., Chandrasekharan K. H., Jeevaratnam K., Dulhunty A., Thompson A., Jackson A., et al. (2018). Multiple targets for flecainide action: implications for cardiac arrhythmogenesis. Br J Pharmacol 175, 1260–1278. doi:10.1111/bph.13807
Salvage S., Dulhunty A., Jeevaratnan K., Jackson A., Huang C. (2023b). Feedback contributions to excitation-contraction coupling in native functioning striated muscle. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220162. doi:10.1098/rstb.2022.0162
Salvage S., Huang C., Fraser J., Dulhunty A. (2022). How does flecainide impact RyR2 channel function? J Gen Physiol 154, e202213089. doi:10.1085/jgp.202213089
Salvage S., Rees J., McStea A., Hirsch M., Wang L., Tynan C., et al. (2020c). Supramolecular clustering of the cardiac sodium channel Nav1.5 in HEK293F cells, with and without the auxiliary β3-subunit. FASEB J 34, 3537–3553. doi:10.1096/fj.201701473RR
Salvage S. C., Habib Z. F., Matthews H. R., Jackson A. P., Huang C. L.-H. (2021). Ca2+-dependent modulation of voltage-gated myocyte sodium channels. Biochem. Soc. Trans. 49, 1941–1961. doi:10.1042/bst20200604
Salvage S. C., Huang C. L.-H., Jackson A. P. (2020a). Cell-adhesion properties of β-subunits in the regulation of cardiomyocyte sodium channels. Biomolecules 10, 989. doi:10.3390/biom10070989
Salvage S. C., Jeevaratnam K., Huang C. L.-H., Jackson A. P. (2023a). Cardiac sodium channel complexes and arrhythmia: structural and functional roles of the β1 and β3 subunits. J. Physiol. 601, 923–940. doi:10.1113/JP283085
Salvage S. C., King J. H., Chandrasekharan K. H., Jafferji D. I. G., Guzadhur L., Matthews H. R., et al. (2015). Flecainide exerts paradoxical effects on sodium currents and atrial arrhythmia in murine RyR2-P2328S hearts. Acta Physiol. (Oxf). 214, 361–375. doi:10.1111/apha.12505
Salvage S. C., Rees J. S., McStea A., Hirsch M., Wang L., Tynan C. J., et al. (2020b). Supramolecular clustering of the cardiac sodium channel Nav1.5 in HEK293F cells, with and without the auxiliary β3-subunit. FASEB J 34, 3537–3553. doi:10.1096/fj.201701473RR
Sanchez C., Berthier C., Tourneur Y., Monteiro L., Allard B., Csernoch L., et al. (2021). Detection of Ca2+transients near ryanodine receptors by targeting fluorescent Ca2+sensors to the triad. J. Gen. Physiol. 153, e202012592. doi:10.1085/JGP.202012592
Sancho M., Kyle B. D. (2021). The large-conductance, calcium-activated potassium channel: a big key regulator of cell physiology. Front. Physiol. 12, 750615. doi:10.3389/fphys.2021.750615
Sanguinetti M. C., Tristani-Firouzi M. (2006). hERG potassium channels and cardiac arrhythmia. Nature 440, 463–469. doi:10.1038/nature04710
Sarbjit-Singh S. S., Matthews H. R., Huang C. L.-H. (2020). Ryanodine receptor modulation by caffeine challenge modifies Na+ current properties in intact murine skeletal muscle fibres. Sci. Rep. 10, 2199. doi:10.1038/s41598-020-59196-9
Sato D., Xie L. H., Sovari A., Tran D. X., Morita N., Xie F., et al. (2009). Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc. Natl. Acad. Sci. U. S. A. 106, 2983–2988. doi:10.1073/pnas.0809148106
Saucerman J. J., Bers D. M. (2012). Calmodulin binding proteins provide domains of local Ca2+ signaling in cardiac myocytes. J. Mol. Cell. Cardiol. 52, 312–316. doi:10.1016/j.yjmcc.2011.06.005
Savelieva I., Camm J. (2008). Anti-arrhythmic drug therapy for atrial fibrillation: current anti-arrhythmic drugs, investigational agents, and innovative approaches. Europace 10, 647–665. doi:10.1093/europace/eun130
Schmitt N., Grunnet M., Olesen S. (2014). Cardiac potassium channel subtypes: new roles in repolarization and arrhythmia. Physiol. Rev. 94, 609–653. doi:10.1152/physrev.00022.2013
Schnee J. M., Hsueh W. A. (2000). Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 46, 264–268. doi:10.1016/S0008-6363(00)00044-4
Schultz J. E. J., Witt S. A., Glascock B. J., Nieman M. L., Reiser P. J., Nix S. L., et al. (2002). TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Invest. 109, 787–796. doi:10.1172/JCI14190
Seo K., Rainer P. P., Hahn V. S., Lee D. I., Jo S. H., Andersen A., et al. (2014). Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 111, 1551–1556. doi:10.1073/pnas.1308963111
Shattock M. J., Bers D. M. (1989). Rat vs. rabbit ventricle: Ca flux and intracellular Na assessed by ion-selective microelectrodes. Am. J. Physiol. 256, C813–C822. doi:10.1152/ajpcell.1989.256.4.C813
Shy D., Gillet L., Abriel H. (2013). Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim. Biophys. Acta - Mol. Cell Res. 1833, 886–894. doi:10.1016/j.bbamcr.2012.10.026
Singh B. (2004). “Beta-blockers and calcium channel blockers as anti-arrhythmic drugs,” in Cardiac Electrophysiology from cell to bedside. Editors D. Zipes, and J. Jalife (Philadelphia: Saunders), 918–931.
Smith T., Cain M. (2004). “Class III antiarrhythmic drugs: amiodarone, ibutilide, and sotalol,” in Cardiac Electrophysiology from cell to bedside. Editors D. Zipes, and J. Jalife (United States: Saunders Philadephia), 933–958.
Smyth J. W., Hong T. T., Gao D., Vogan J. M., Jensen B. C., Fong T. S., et al. (2010). Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Invest. 120, 266–279. doi:10.1172/JCI39740
Sonoda J., Mehl I. R., Chong L.-W., Nofsinger R. R., Evans R. M. (2007). PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl. Acad. Sci. U. S. A. 104, 5223–5228. doi:10.1073/pnas.0611623104
Spector P. (2013). Principles of cardiac electric propagation and their implications for re-entrant arrhythmias. Circ. Arrhythm. Electrophysiol. 6, 655–661. doi:10.1161/CIRCEP.113.000311
Sperelakis N. (2002). An electric field mechanism for transmission of excitation between myocardial cells. Circ. Res. 91, 985–987. doi:10.1161/01.RES.0000045656.34731.6D
Sperelakis N., Mann J. E. (1977). Evaluation of electric field changes in the cleft between excitable cells. J. Theor. Biol. 64, 71–96. doi:10.1016/0022-5193(77)90114-x
Stewart S., Hart C. L., Hole D. J., McMurray J. J. V. (2002). A population-based study of the long-term risks associated with atrial fibrillation: 20-Year follow-up of the Renfrew/Paisley study. Am. J. Med. 113, 359–364. doi:10.1016/S0002-9343(02)01236-6
Stokoe K. S., Balasubramaniam R., Goddard C. A., Colledge W. H., Grace A. A., Huang C. L.-H. (2007). Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a +/- murine hearts modelling the Brugada syndrome. J. Physiol. 581, 255–275. doi:10.1113/jphysiol.2007.128785
Struckman H., Moise N., Dunlap I., Chen Z., Weinberg S., Veeraraghavan R. (2021). Abstract 12293: intercalated disk nanostructure and molecular organization underlie conduction differences between atria and ventricles. Circulation 144, A12293. doi:10.1161/circ.144.suppl_1.12293
Sugden P. H., Clerk A. (1998). Cellular mechanisms of cardiac hypertrophy. J. Mol. Med. 76, 725–746. doi:10.1007/s001090050275
Swaminathan P. D., Purohit A., Hund T. J., Anderson M. E. (2012). Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ. Res. 110, 1661–1677. doi:10.1161/CIRCRESAHA.111.243956
Taggart P., Nash M., Lambiase P. (2022). Ventricular fibrillation: combined myocardial substrate and Purkinje ablation. Eur. Heart J. 43, 1248–1250. doi:10.1093/eurheartj/ehab912
Takla M., Huang C. L.-H., Jeevaratnam K. (2020). The cardiac CaMKII-Nav1.5 relationship: from physiology to pathology. J. Mol. Cell. Cardiol. 139, 190–200. doi:10.1016/j.yjmcc.2019.12.014
Takla M., Saadeh K., Tse G., Huang C. L.-H., Jeevaratnam K. (2023). Ageing and the autonomic nervous system. Subcellular Biochemistry 103, 201–252. doi:10.1007/978-3-031-26576-1_10
Tamargo J., Tamargo J., Caballero R., Caballero R., Gómez R., Gómez R., et al. (2004). Pharmacology of cardiac potassium channels. Cardiovasc. Res. 62, 9–33. doi:10.1016/j.cardiores.2003.12.026
Task force of the working group on arrhythmias The European Society of Cardiology (1991). The Sicilian gambit. A new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. Task Force of the Working Group on Arrhythmias of the European Society of Cardiology. Circulation 84, 1831–1851. doi:10.1161/01.cir.84.4.1831
Ten Tusscher K. H., Noble D., Noble P. J., Panfilov A. V. (2004). A model for human ventricular tissue. Am J Physiol Hear. Circ Physiol 286, H1573–H1589. doi:10.1152/ajpheart.00794.2003
Terentyev D., Gyorke I., Belevych A. E., Terentyeva R., Sridhar A., Nishijima Y., et al. (2008). Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 103, 1466–1472. doi:10.1161/CIRCRESAHA.108.184457
Terentyev D., Rochira J. A., Terentyeva R., Roder K., Koren G., Li W. (2014). Sarcoplasmic reticulum Ca2+ release is both necessary and sufficient for SK channel activation in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 306, H738–H746. doi:10.1152/AJPHEART.00621.2013
Terrar D. (2023). Timing mechanisms to control heart rhythm and initiate arrhythmias: roles for intracellular organelles, signalling pathways and subsarcolemmal Ca2. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220170. doi:10.1098/rstb.2022.0170
Tester D. J., Ackerman M. J. (2007). Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J. Am. Coll. Cardiol. 49, 240–246. doi:10.1016/j.jacc.2006.10.010
Thomas G., Killeen M. J., Gurung I. S., Hakim P., Balasubramaniam R., Goddard C. A., et al. (2007). Mechanisms of ventricular arrhythmogenesis in mice following targeted disruption of KCNE1 modelling long QT syndrome 5. J. Physiol. 578, 99–114. doi:10.1113/jphysiol.2006.118133
Traub O., Eckert R., Lichtenberg-Frate H., Elfgang C., Bastide B., Scheidtmann K. H., et al. (1994). Immunochemical and electrophysiological characterization of murine connexin40 and -43 in mouse tissues and transfected human cells. Eur. J. Cell Biol. 64, 101–112.
Travnickova-Bendova Z., Cermakian N., Reppert S. M., Sassone-Corsi P. (2002). Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc. Natl. Acad. Sci. U. S. A. 99, 7728–7733. doi:10.1073/pnas.102075599
Tsui H., Zi M., Wang S., Chowdhury S. K., Prehar S., Liang Q., et al. (2015). Smad3 couples Pak1 with the antihypertrophic pathway through the E3 ubiquitin ligase, Fbxo32. Hypertension 66, 1176–1183. doi:10.1161/HYPERTENSIONAHA.115.06068
Tung R. T., Shen W. K., Hammill S. C., Gersh B. J. (1994). Idiopathic ventricular fibrillation in out-of-hospital cardiac arrest survivors. Pacing Clin. Electrophysiol. 17, 1405–1412. doi:10.1111/j.1540-8159.1994.tb02460.x
Turakhia M., Tseng Z. H. (2007). Sudden cardiac death: epidemiology, mechanisms, and therapy. Curr. Probl. Cardiol. 32, 501–546. doi:10.1016/j.cpcardiol.2007.05.002
Valli H., Ahmad S., Chadda K., Al-Hadithi A., Grace A., Jeevaratnam K., et al. (2017a). Age-dependent atrial arrhythmic phenotype secondary to mitochondrial dysfunction in Pgc-1β deficient murine hearts. Mech. Ageing Dev. 167, 30–45. doi:10.1016/j.mad.2017.09.002
Valli H., Ahmad S., Chadda K. R., Al-Hadithi A. B. A. K., Grace A. A., Jeevaratnam K., et al. (2017b). Age-dependent atrial arrhythmic phenotype secondary to mitochondrial dysfunction in Pgc-1β deficient murine hearts. Mech. Ageing Dev. 167, 30–45. doi:10.1016/j.mad.2017.09.002
Valli H., Ahmad S., Fraser J. A., Jeevaratnam K., Huang C. L. H. (2017c). Pro-arrhythmic atrial phenotypes in incrementally paced murine Pgc1β-/- hearts: effects of age. Exp. Physiol. 102, 1619–1634. doi:10.1113/EP086589
Valli H., Ahmad S., Jiang A. Y., Smyth R., Jeevaratnam K., Matthews H. R., et al. (2018a). Cardiomyocyte ionic currents in intact young and aged murine Pgc-1β-/- atrial preparations. Mech. Ageing Dev. 169, 1–9. doi:10.1016/j.mad.2017.11.016
Valli H., Ahmad S., Sriharan S., Dean L. D., Grace A. A., Jeevaratnam K., et al. (2018b). Epac-induced ryanodine receptor type 2 activation inhibits sodium currents in atrial and ventricular murine cardiomyocytes. Clin. Exp. Pharmacol. Physiol. 45, 278–292. doi:10.1111/1440-1681.12870
Vandenberg J. I., Perry M. D., Perrin M. J., Mann S. A., Ke Y., Hill A. P. (2012). hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478. doi:10.1152/physrev.00036.2011
Vandenberg J. I., Varghese A., Lu Y., Bursill J. A., Mahaut-Smith M. P., Huang C. L.-H. (2006). Temperature dependence of human ether-a-go-go-related gene K(+) currents. Am. J. Physiol. Cell Physiol. 291, C165–C175. doi:10.1152/ajpcell.00596.2005
Van Der Werf C., Kannankeril P. J., Sacher F., Krahn A. D., Viskin S., Leenhardt A., et al. (2011). Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J. Am. Coll. Cardiol. 57, 2244–2254. doi:10.1016/j.jacc.2011.01.026
Van Der Werf C., Lambiase P. D. (2021). Initiation and management of polymorphic ventricular tachycardia: history gone full circle. Eur. Heart J. 42, 3976–3978. doi:10.1093/eurheartj/ehab428
Vaughan Williams E. (1975). Classification of antidysrhythmic drugs. Pharmacol Ther B 1, 115–138. doi:10.1016/0306-039x(75)90019-7
Veeraraghavan R., Lin J., Hoeker G. S., Keener J. P., Gourdie R. G., Poelzing S. (2015). Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflügers Arch. - Eur. J. Physiol. 467, 2093–2105. doi:10.1007/s00424-014-1675-z
Veeraraghavan R., Poelzing S., Gourdie R. G. (2014). Intercellular electrical communication in the heart: a new, active role for the intercalated disk. Cell Commun. Adhes. 21, 161–167. doi:10.3109/15419061.2014.905932
Vega R. B., Huss J. M., Kelly D. P. (2000). The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 20, 1868–1876. doi:10.1128/mcb.20.5.1868-1876.2000
Verheule S., van Kempen M. J., te Welscher P. H., Kwak B. R., Jongsma H. J. (1997). Characterization of gap junction channels in adult rabbit atrial and ventricular myocardium. Circ. Res. 80, 673–681. doi:10.1161/01.res.80.5.673
Vianna C. R., Huntgeburth M., Coppari R., Choi C. S., Lin J., Krauss S., et al. (2006). Hypomorphic mutation of PGC-1beta causes mitochondrial dysfunction and liver insulin resistance. Cell Metab 4, 453–464. doi:10.1016/j.cmet.2006.11.003
Wakili R., Voigt N., Kääb S., Dobrev D., Nattel S. (2011). Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 121, 2955–2968. doi:10.1172/JCI46315
Wang J., Wang H., Zhang Y., Gao H., Nattel S., Wang Z. (2004). Impairment of HERG K+ channel function by tumor necrosis factor-alpha: role of reactive oxygen species as a mediator. J Biol.Chem. 279, 13289–13292. doi:10.1074/jbc.C400025200
Wang R., Wang Y., Lin W. K., Zhang Y., Liu W., Huang K., et al. (2014a). Inhibition of angiotensin II-induced cardiac hypertrophy and associated ventricular arrhythmias by a p21 activated kinase 1 bioactive peptide. PLoS One 9, e101974. doi:10.1371/journal.pone.0101974
Wang X., Galli G., Campanella M. (2019). Special issue: themed section: mitochondrial pharmacology: featured mechanisms and approaches for therapy translation. Br. J. Pharmacol. 176, 4245–4246. doi:10.1111/bph.14820
Wang Y., Anderson C., Dobrzynski H., Hart G., D’Souza A., Boyett M. R. (2021). RNAseq shows an all-pervasive day-night rhythm in the transcriptome of the pacemaker of the heart. Sci. Rep. 11, 3565. doi:10.1038/s41598-021-82202-7
Wang Y., Lin W. K., Crawford W., Ni H., Bolton E. L., Khan H., et al. (2017). Optogenetic control of heart rhythm by selective stimulation of cardiomyocytes derived from pnmt + cells in murine heart. Sci. Rep. 7, 40687. doi:10.1038/srep40687
Wang Y., Tsui H., Bolton E. L., Wang X., Huang C. L.-H., Solaro R. J., et al. (2015). Novel insights into mechanisms for Pak1-mediated regulation of cardiac Ca2+ homeostasis. Front. Physiol. 6, 76. doi:10.3389/fphys.2015.00076
Wang Y., Tsui H., Ke Y., Shi Y., Li Y., Davies L., et al. (2014b). Pak1 is required to maintain ventricular Ca²⁺ homeostasis and electrophysiological stability through SERCA2a regulation in mice. Circ. Arrhythm. Electrophysiol. 7, 938–948. doi:10.1161/CIRCEP.113.001198
Wang Y., Wang S., Lei M., Boyett M., Tsui H., Liu W., et al. (2018). The p21-activated kinase 1 (Pak1) signalling pathway in cardiac disease: from mechanistic study to therapeutic exploration. Br. J. Pharmacol. 175, 1362–1374. doi:10.1111/bph.13872
Watanabe H., Chopra N., Laver D., Hwang H. S., Davies S. S., Roach D. E., et al. (2009). Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med. 15, 380–383. doi:10.1038/nm.1942
Weidmann S. (1951). Effect of current flow on the membrane potential of cardiac muscle. J. Physiol. 115, 227–236. doi:10.1113/jphysiol.1951.sp004667
Weisbrod D. (2020). Small and intermediate calcium activated potassium channels in the heart: role and strategies in the treatment of cardiovascular diseases. Front. Physiol. 11, 590534. doi:10.3389/FPHYS.2020.590534
Weiss J., Lamp S., Shine K. (1989). Cellular K(+) loss and anion efflux during myocardial ischemia and metabolic inhibition. Am J Physiol 256, H1165–H1175. doi:10.1152/ajpheart.1989.256.4.H1165
Weiss J. N., Qu Z., Chen P. S., Lin S. F., Karagueuzian H. S., Hayashi H., et al. (2005). The dynamics of cardiac fibrillation. Circulation 112, 1232–1240. doi:10.1161/CIRCULATIONAHA.104.529545
Wettwer E., Christ T., Endig S., Rozmaritsa N., Matschke K., Lynch J. J., et al. (2013). The new antiarrhythmic drug vernakalant: ex vivo study of human atrial tissue from sinus rhythm and chronic atrial fibrillation. Cardiovasc. Res. 98, 145–154. doi:10.1093/cvr/cvt006
Wiedmann F., Beyersdorf C., Zhou X. B., Kraft M., Paasche A., Jávorszky N., et al. (2022). Treatment of atrial fibrillation with doxapram: TASK-1 potassium channel inhibition as a novel pharmacological strategy. Cardiovasc. Res. 118, 1728–1741. doi:10.1093/cvr/cvab177
Wolf P. A., Abbott R. D., Kannel W. B. (1991). Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke 22, 983–988. doi:10.1161/01.STR.22.8.983
Wollert K. C., Drexler H. (1999). The renin-angiotensin system and experimental heart failure. Cardiovasc. Res. 43, 838–849. doi:10.1016/S0008-6363(99)00145-5
Wu Q., Liu H., Liao J., Zhao N., Tse G., Han B., et al. (2020). Colchicine prevents atrial fibrillation promotion by inhibiting IL-1β-induced IL-6 release and atrial fibrosis in the rat sterile pericarditis model. Biomed. Pharmacother. 129, 110384. doi:10.1016/j.biopha.2020.110384
Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. doi:10.1016/S0092-8674(00)80611-X
Yan J., Zhao W., Thomson J. K., Gao X., DeMarco D. M., Carrillo E., et al. (2018). Stress signaling JNK2 crosstalk with CaMKII underlies enhanced atrial arrhythmogenesis. Circ. Res. 122, 821–835. doi:10.1161/CIRCRESAHA.117.312536
Yang B., Jiang Q., He S., Li T., Ou X., Chen T., et al. (2021). Ventricular SK2 upregulation following angiotensin II challenge: modulation by p21-activated kinase-1. J. Mol. Cell. Cardiol. 164, 110–125. doi:10.1016/J.YJMCC.2021.11.001
Yang H., Zhang G., Cui J. (2015a). BK channels: multiple sensors, one activation gate. Front. Physiol. 6, 29. doi:10.3389/fphys.2015.00029
Yang K.-C., Bonini M. G., Dudley S. C. (2014). Mitochondria and arrhythmias. Free Radic. Biol. Med. 71, 351–361. doi:10.1016/j.freeradbiomed.2014.03.033
Yang K.-C., Kyle J. W., Makielski J. C., Dudley S. C. (2015b). Mechanisms of sudden cardiac death: oxidants and metabolism. Circ. Res. 116, 1937–1955. doi:10.1161/CIRCRESAHA.116.304691
Yang L., Pang Y., Moses H. L. (2010). TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol 31, 220–227. doi:10.1016/j.it.2010.04.002
Yang T., Colecraft H. M. (2016). Calmodulin regulation of TMEM16A and 16B Ca2+-activated chloride channels. Channels (Austin) 10, 38–44. doi:10.1080/19336950.2015.1058455
Yao C., Veleva T., Scott L., Cao S., Li L., Chen G., et al. (2018). Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 138, 2227–2242. doi:10.1161/CIRCULATIONAHA.118.035202
Yu S., Li G., Huang C. L.-H., Lei M., Wu L. (2018). Late sodium current associated cardiac electrophysiological and mechanical dysfunction. Pflugers Arch. Eur. J. Physiol. 470, 461–469. doi:10.1007/s00424-017-2079-7
Yue Z., Xie J., Yu A. S., Stock J., Du J., Yue L. (2015). Role of trp channels in the cardiovascular system. Am. J. Physiol. - Hear. Circ. Physiol. 308, H157–H182. doi:10.1152/ajpheart.00457.2014
Zaccolo M. (2023). Compartmentalized cAMP signalling and control of cardiac rhythm. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220172. doi:10.1098/rstb.2022.0172
Zaitsev A. V., Berenfeld O., Mironov S. F., Jalife J., Pertsov A. M. (2000). Distribution of excitation frequencies on the epicardial and endocardial surfaces of fibrillating ventricular wall of the sheep heart. Circ. Res. 86, 408–417. doi:10.1161/01.RES.86.4.408
Zeng B., Zhang X., Schimpf R., Powers A., Glikson M., Antzelevitch C., et al. (2023). Functional identification of hot-spot mutations in cardiac calcium channel genes associated with the J wave syndromes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220286. doi:10.1098/rstb.2022.0286
Zeng X. H., Xia X. M., Lingle C. J. (2005). Divalent cation sensitivity of BK channel activation supports the existence of three distinct binding sites. J. Gen. Physiol. 125, 273–286. doi:10.1085/jgp.200409239
Zhang Y., Wu J., Jeevaratnam K., King J. H., Guzadhur L., Ren X., et al. (2013). Conduction slowing contributes to spontaneous ventricular arrhythmias in intrinsically active murine RyR2-P2328S hearts. J. Cardiovasc. Electrophysiol. 24, 210–218. doi:10.1111/jce.12015
Zhou Q., Xiao J., Jiang D., Wang R., Vembaiyan K., Wang A., et al. (2011). Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat. Med. 17, 1003–1009. doi:10.1038/nm.2406
Zhou Y., Huang W., Liu L., Li A., Jiang C., Zhou R., et al. (2023). Patient-specific induced pluripotent stem cell properties implicate Ca2+-homeostasis in clinical arrhythmia associated with combined heterozygous RYR2 and SCN10A variants. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 378, 20220175. doi:10.1098/rstb.2022.0175
Keywords: cardiac arrhythmias, ion channels, excitation–contraction coupling, drug discovery, cardiac remodeling
Citation: Lei M, Salvage SC, Jackson AP and Huang CL-H (2024) Cardiac arrhythmogenesis: roles of ion channels and their functional modification. Front. Physiol. 15:1342761. doi: 10.3389/fphys.2024.1342761
Received: 22 November 2023; Accepted: 22 January 2024;
Published: 04 March 2024.
Edited by:
Mona El Refaey, The Ohio State University, United StatesReviewed by:
Richard Ang, University College London, United KingdomHyun Seok Hwang, Florida State University, United States
Copyright © 2024 Lei, Salvage, Jackson and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher L.-H. Huang, clh11@cam.ac.uk