 Yacong Li
Yacong Li Runlan Wan
Runlan Wan Jun Liu
Jun Liu Weichao Liu
Weichao Liu Lei Ma
Lei Ma Henggui Zhang
Henggui Zhang- 1Beijing Academy of Artificial Intelligence, Beijing, China
- 2Department of Oncology, The Affiliated Hospital of Southwest Medical University, Luzhou, China
- 3School of Computer Science and Technology, Harbin Institute of Technology, Harbin, China
- 4Key Laboratory of Medical Electrophysiology, Ministry of Education & Medical Electrophysiological Key Laboratory of Sichuan Province, Institute of Cardiovascular Research, Southwest Medical University, Luzhou, China
- 5National Biomedical Imaging Center, Peking University, Beijing, China
- 6Biological Physics Group, School of Physics and Astronomy, The University of Manchester, Manchester, United Kingdom
It has been found that arsenic trioxide (ATO) is effective in treating acute promyelocytic leukemia (APL). However, long QT syndrome was reported in patients receiving therapy using ATO, which even led to sudden cardiac death. The underlying mechanisms of ATO-induced cardiotoxicity have been investigated in some biological experiments, showing that ATO affects human ether-à-go-go-related gene (hERG) channels, coding rapid delayed rectifier potassium current (IKr), as well as L-type calcium (ICaL) channels. Nevertheless, the mechanism by which these channel reconstitutions induced the arrhythmia in ventricular tissue remains unsolved. In this study, a mathematical model was developed to simulate the effect of ATO on ventricular electrical excitation at cellular and tissue levels by considering ATO’s effects on IKr and ICaL. The ATO-dose-dependent pore block model was incorporated into the IKr model, and the enhanced degree of ATO to ICaL was based on experimental data. Simulation results indicated that ATO extended the action potential duration of three types of ventricular myocytes (VMs), including endocardial cells (ENDO), midmyocardial cells (MCELL), and epicardial cells (EPI), and exacerbated the heterogeneity among them. ATO could also induce alternans in all three kinds of VMs. In a cable model of the intramural ventricular strand, the effects of ATO are reflected in a prolonged QT interval of simulated pseudo-ECG and a wide vulnerable window, thus increasing the possibility of spiral wave formation in ventricular tissue. In addition to showing that ATO prolonged QT, we revealed that the heterogeneity caused by ATO is also an essential hazard factor. Based on this, a pharmacological intervention of ATO toxicity by resveratrol was undertaken. This study provides a further understanding of ATO-induced cardiotoxicity, which may help to improve the treatment for APL patients.
Introduction
Arsenic trioxide (ATO), a traditional Chinese medicine, has been reported to be used to treat acute promyelocytic leukemia (APL) in 1997 (Chen et al., 1997). After that, scientists discovered that a combination of ATO and all-trans retinoic acid (ATRA) almost cured APL (Shen et al., 2004), whose molecular and cellular mechanisms have also been elucidated (Zhang et al., 2010). In recent years, ATO was screened to rescue the p53 folding function (Chen et al., 2021). In oncology, this is a breakthrough, since the p53 mutation is the most common mutation among cancer patients, which has shown great therapeutic potential but had never been rescued before this research. As a result, ATO is a promising drug in oncotherapy and deserves to be further investigated and applied.
In addition to the efficacy, the safety of ATO also needs to be assessed in clinical trials. It has been reported that ATO may generate cardiotoxicity as well as hepatotoxicity (Mathews et al., 2006; Alexandre et al., 2018). Cardiotoxicity is reflected in the prolonged QT interval of electrocardiograms (ECGs) (Soignet et al., 2001), called long QT syndrome (LQT), which may lead to torsade de pointes tachycardia (TdP) (Unnikrishnan et al., 2001; Hai et al., 2015) and even threaten life (Westervelt et al., 2001; Lenihan and Kowey, 2013). There was also a case of ventricular tachycardia with a normal QT interval in ATO therapy (Ducas et al., 2011), which further warned of the potential arrhythmia risk. A study manifested that the combination of ATRA and ATO therapy can reduce side effects and has less toxicity than ATO treatment alone (Hu et al., 2009). Nevertheless, a Position Paper published by the European Society of Cardiology emphasized that ATO was more related to QT prolongation than other reported anticancer drugs, and it also has a higher chance of causing sudden death due to TdP (Zamorano et al., 2016). Therefore, there is a strong need to further investigate the mechanisms of ATO-induced cardiotoxicity.
Because of the clinical observation of ATO-induced cardiotoxicity, a series of animal studies, including subcellular and cellular experiments, were carried out. According to experimental research, ATO acts on cardiomyocytes (CMs) mainly via potassium channels and calcium channels. The human ether-à-go-go-related gene (hERG) codes rapid delayed rectifier potassium current (IKr) in the human heart, which is susceptible to ATO. Exposure to ATO for 20 min can suppress the hERG channel in hERG-transfected CHO cells (Drolet et al., 2004), whereas an experiment in HEK293 cells (Ficker et al., 2004) indicated that short-term application of ATO did not affect the hERG current and could not alter the action potential duration (APD) in guinea pig ventricular myocytes (VMs). This study revealed that long-term ATO suppressed the IKr current in HEK293 cells, and the dosage of ATO directly determined the reduction degree of IKr (Ficker et al., 2004). The same phenomenon can also be observed in other HEK293 experiments (Zhao et al., 2015; Yan et al., 2017), and the inhibiting effect of ATO on the hERG channel has also appeared in rodent animals, such as guinea pig VMs (Ficker et al., 2004; Zhao et al., 2014), neonatal rat VMs (NRVMs) (Zhao et al., 2015), and neonatal mouse cardiomyocytes (Liu et al., 2017). In addition, the underlying RNA regulation mechanisms of ATO-impaired hERG were revealed (Shan et al., 2013; Zhao et al., 2015). The effect of ATO on slow delayed rectifier potassium current (IKs) is controversial. The IKs of CHO cells were susceptible to short-term exposure to ATO (Drolet et al., 2004), but in guinea pig VMs, ATO did not have an apparent influence on IKs density (Ficker et al., 2004). A similar controversy also appeared in the inward rectifier potassium current (IK1). Chronic ATO administration inhibited IK1 significantly by reducing Kir2.1 protein expression levels in guinea pig CMs (Chu et al., 2012; Shan et al., 2013) and neonatal rat CMs (Chen et al., 2010; Chu et al., 2012). However, a guinea pig experiment did not show an obvious change in IK1 after overnight ATO treatment (Ficker et al., 2004). The drug-delivery method and its dosage should be responsible for this difference between the results. Furthermore, calcium channels are sensitive to ATO. Experiments in guinea pig VMs (Sun et al., 2006) and NRVMs (Chen et al., 2010; Yan et al., 2017) reported an increase in L-type calcium current (ICaL) under the action of ATO at different dosages. The peak of intracellular calcium concentration ([Ca2+]i) was also markedly increased in the presence of ATO (Yan et al., 2017), while the diastolic [Ca2+]i level did not change (Chen et al., 2010). Consistently, it has been verified that ATO can prolong APD in different cell types, including guinea pig VMs (Sun et al., 2006), NRVMs (Chen et al., 2010), HEK293 cells (Ficker et al., 2004), and human-induced pluripotent stem cell-derived cardiomyocytes (hiPS-CMs) (Yan et al., 2017). This finding was consistent with the clinical observation that ATO caused LQT in animal studies (Chen et al., 2010). Research on the potential signaling mechanisms of ATO-induced LQT revealed that ATO promoted the secretion of transforming growth factor-β1 (TGF-β1), which led to fibrosis and inhibited hERG and Kir2.1 protein in CMs, thus causing LQT syndrome (Chu et al., 2012). Although the above experimental results greatly helped understand ATO-induced cardiotoxicity, most studies have been conducted on a single ion channel and have not directly examined using human cardiomyocytes.
It is of great significance in clinical practice to find a way to ameliorate the side effects of ATO. To date, several drugs have been attempted to do this, including antiallergic drugs [such as fexofenadine and astemizole (Yan et al., 2017)], hypoglycemic drugs [such as glibenclamide (Drolet et al., 2004)], cardiovascular drugs [such as nisoldipine (Ficker et al., 2004), ranolazine (Yan et al., 2017) and choline (Sun et al., 2006)], antagonists (Chu et al., 2012) and organic compounds [such as resveratrol (Zhao et al., 2014; Yan et al., 2017), eugenol (Binu et al., 2017) and omega-3 fatty acid (Varghese et al., 2017)]. Fexofenadine can increase the IKr of HEK293 cells and shorten APD in both NRVMs and hiPS-CMs treated with 3 μM ATO (Yan et al., 2017). Ranolazine, astemizole and glibenclamide also acted on potassium channel proteins. Ranolazine corrected hERG expression in HEK293 and NRVMs but failed to reverse the damaged hERG channel in hiPS-CMs (Yan et al., 2017). However, astemizole did not have remarkable assuasive effects on the long APD caused by ATO (Yan et al., 2017). Resveratrol (Yan et al., 2017) and choline (Sun et al., 2006) attenuated ATO toxicity by inhibiting the ICaL channel, and resveratrol exerted a better rescue effect than potassium-intervened agents (Yan et al., 2017). Moreover, lead compound optimization was also reported to be a strategy that alleviated ATO toxicity (Zhou et al., 2016). In addition, the regulatory mechanism of TGF-β1 under ATO treatment (Chu et al., 2012; Liu et al., 2017) provided new methods for preventing hERG and Kir2.1 protein damage by treatment with the protein kinase A (PKA) antagonist H89 and the TβR-I inhibitor LY364947 (Chu et al., 2012).
ATO is a vital agent in the field of clinical oncotherapy. Research is being conducted to unravel and alleviate ATO-induced cardiotoxicity. However, the mechanism of ATO-induced cardiotoxicity is not well understood, especially at the myocardial tissue level. In the present study, we constructed a multiscale mathematical model to simulate cardiac electrical activity in the presence of ATO, by which the generating process of arrhythmia induced by ATO treatment can be delineated from ionic channels to cardiac tissue. In this way, the effect of ATO on single CMs can be extended to a macroscopic level to further predict and analyze its underlying risks. Using the results from this study, we gained a new perspective on ATO-induced cardiotoxicity, such as tissue electrical heterogeneity, vulnerability to arrhythmogenesis and electrical alternans, and provided a method for finding the right dose and a pharmacological rescue scheme for ATO treatment.
Methods
Modeling single VMs and the binding interaction between ATO and the IKr/ICaL channel
The human VM models, including endocardial cells (ENDO), midmyocardial cells (MCELL) and epicardial cells (EPI), followed ten Tusscher’s model (TNNP06) because of its application in alternans and reentry (ten Tusscher and Panfilov, 2006). The membrane potential of a single VM can be described by the following ordinary differential equation:
where V is the membrane potential; t is time; Istim is the stimulation current; Cm is the cell capacitance. Iion is the sum of transmembrane ionic currents, including:
Here, we simulated the electrophysiological activities of VMs incubated with ATO by modifying a potassium channel, IKr, and a calcium current, ICaL. The formulations of all ionic channel currents can be referenced in the TNNP06 model (ten Tusscher and Panfilov, 2006).
According to a simple pore block theory (Yuan et al., 2014), the binding interaction between ATO and IKr can be modeled by a blocking factor λ that denoted the blocking degree of ATO to the maximum conductance of the targeted ion channel. This blocking factor λ can be described by a Hill equation as follows:
where D is the concentration of ATO, IC50 is the ATO concentration at which 50% blockade of the binding site occurs and nH is the Hill coefficient. As a result, the remaining maximum conductance index of IKr is:
Consequently, the formulation of IKr can be described as follows:
where GKr is the conductance of IKr, Ko is the extracellular K+ concentration, xr1 is an activation gate, xr2 is an inactivation gate, and EK is the reversal potential.
According to the experimental data (Ficker et al., 2004), IC50 was 1.5 μM and nH was fitted at −1.2, so the interaction between ATO concentration and k is demonstrated in Figure 1A. The tail currents of IKr under control and 3 μM ATO in the present simulation model and experimental measurement (Yan et al., 2017) are shown in Figure 1B. Our model could well fit the experimental data, which indicated the accuracy of the present model.
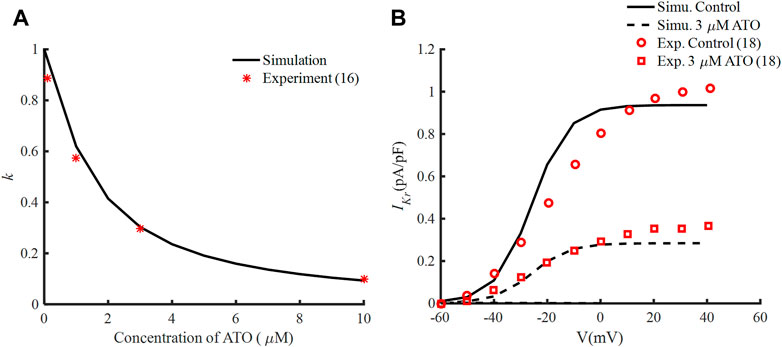
FIGURE 1. Effects of ATO and the hERG channel current. (A) The normalized remaining maximum conductance of IKr blocked by ATO. (B) Tail current of IKr under the conditions of control and 3 μM ATO in the simulation model (black lines) and an experiment (red circle and square), respectively.
Experiments indicated that exposure to 3 μM ATO for 24 h tripled the density of ICaL in guinea pig VMs (Ficker et al., 2004) or doubled it in NRVMs (Yan et al., 2017). Another animal experiment showed that when ATO administration led to a double ICaL density, the activation curve of ICaL was shifted negatively (Chen et al., 2010).
In the present model, the formulation of ICaL was as follows:
where d is a voltage-dependent activation gate:
Two parameters that were modified to simulate the effect of ATO on ICaL were θ and V1/2, which represented the change in the conductance of ICaL (GCaL) and the half activation voltage (V1/2) of the activation gate d) respectively. They were estimated according to the above biological experimental results, and the corresponding values are listed in Table 1. The meanings of the other parameters in Eq. (6) can be seen in Ref. (ten Tusscher and Panfilov, 2006).
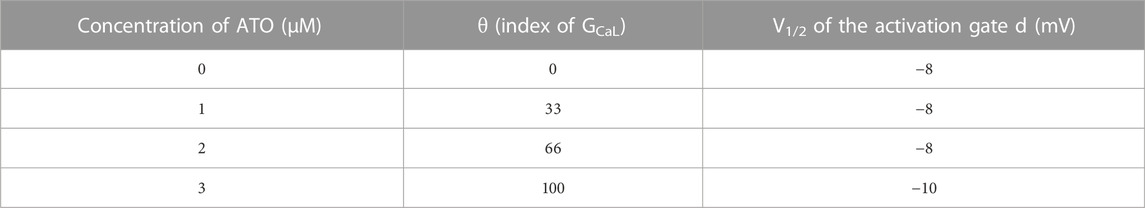
TABLE 1. The effect of ATO on ICaL.
Experiments observed that resveratrol can suppress the ICaL channel current, thus shortening APD (Zhang et al., 2006), and it was shown to increase the IKr channel current (Zhao et al., 2014). Consequently, it was suitable to alleviate cardiotoxicity caused by ATO (Zhao et al., 2014; Yan et al., 2017). The effect of resveratrol on the VMs treated with 3 µM ATO was simulated by manipulating the conductance of ICaL and IKr. According to experimental data, under the condition of 3 µM ATO, 10 µM resveratrol decreased the ICaL density from twice the original value to approximately 1.3 times (Yan et al., 2017). And 10 µM resveratrol recovered IKr by approximately 33% of its density under the impact of 3 µM ATO (Zhao et al., 2014). The same rescue ratio of resveratrol was applied in the present model; thus, the formulation of IKr and ICaL was changed as follows:
where nRes and mRes are the coefficients of resveratrol’s effect on IKr and ICaL, whose values were 1.33 and 1.3, respectively, under 10 µM resveratrol.
To assess the degree of pharmacological rescue, we defined the rescue ratio as follows:
In which APD90 means time duration from depolarization to 90% repolarization,
Eq. (1) was solved by the forward Euler method with a time step of 0.02 m. The single VM model was pulsed under stimulus currents of −52 pA/pF with a basic cycle length (BCL) of 800 m. The S1-S2 standard protocol was used to depict the restitution curve of a single cell. Ten S1 stimulation currents were applied under a BCL of 800 m, following an S2 stimulation current after a dynamic shortening period. This period was called the S1-S2 interval, and the corresponding APD90 of the last cycle was calculated. A dynamic protocol was used to estimate the risk of alternans of VMs, which was conducted by a series of S1 with tapering BCL and corresponding APD90 being calculated.
Modeling ventricular tissue
Non-linear cable theory was applied to build a monodomain ventricular tissue model. As such, the electrical activity of ventricular tissue can be described by a partial differential equation as follows:
where
Eq. (11) was solved by the finite difference method with a time step of 0.02 m and a space step of 0.25 mm. The D was set at 0.08 mm2/ms (Luo et al., 2017) so that the conductivity velocity of electrical waves in ventricular tissue was 0.7 m/s, which was consistent with experimental observations (Taggart et al., 2000).
ECG is used to describe the body surface potential in the clinic. It can be estimated according to cellular electrophysiological processes (Gima and Rudy, 2002). Here, we calculated a pseudo-ECG by the following equation:
where k is a constant; V is the membrane potential;
S1-S2 stimuli were used to evaluate the vulnerability of ventricular cables and tissues via the genesis of unidirectional conduction block. In the ventricular cable, S1 stimuli were applied at first five ENDO cells, and S2 stimuli were applied at five EPI cells. Different locations of EPI that applied S1 stimuli were from the site that neighboring MCELLs to the site that was far away from 5 MCELLs, i.e., the 61–65th cells to 65–69th cells. In ventricular tissue, S1 stimuli were also located at ENDO cells, while S2 stimuli were applied at a block of epicardial tissue whose width was less than the whole cardiac width so that spiral waves could be motivated.
The dynamic protocol was conducted in a heterogeneous ventricular cable, in which the S1 stimuli were applied at the first five ENDO cells with a variable cycle length from 300–500 m.
Results
Effects of ATO on the action potential of VMs
There were experimental data of VMs incubated with 3 µM ATO (Ficker et al., 2004; Yan et al., 2017), in which the subcellular effects of ATO on IKr and ICaL were provided. As a result, we simulated the electrophysiology of three types of VMs in the presence of 3 μM ATO and exerted different stimulation protocols to investigate the change in single-cell membrane potential under ATO intervention. First, a series of periodic stimulation protocol with a BCL of 800 m was conducted, whose corresponding heartbeat was 75 times per minute. According to Eq. (3), 3 μM ATO inhibited the conductance of IKr by 70%, thus suppressing IKr density in ENDO, MCELL and EPI (Figure 2A(ii)–C(ii)). The administration of 3 μM ATO also doubled the conductance of ICaL and shifted the activation curve, thus increasing ICaL density (Figure 2A(iii)–C(iii)). The increased ICaL also accumulated the intracellular calcium concentration ([Ca2+]i) via the calcium dynamics in VMs, thus increasing the Na+/Ca2+ exchanger current (INaCa) as shown in Supplementary Figure S1. As expected, the decrease in IKr and increase in ICaL and INaCa prolonged the APD of VMs to different degrees. ATO increased the APD90 of ENDO from 306 m to 391 m, that of MCELL from 410 m to 602 m, and that of EPI from 307 m to 394 m. The increase ratio of APD90 were approximately 27.8%, 46.8%, and 28.3%, respectively, in the three types of VMs. It was noteworthy that the APD90 increase ratio showed a great difference among different VMs, which may increase the risk of arrhythmia in ventricular tissue. Specifically, the difference in the action potential properties between two adjacent VMs can extend the time window that one kind of cell was in the resting state and the other was in the refractory period, which may lead to unidirectional conduction block, thus producing reentry in cardiac tissue. This will be further analyzed in the following section.
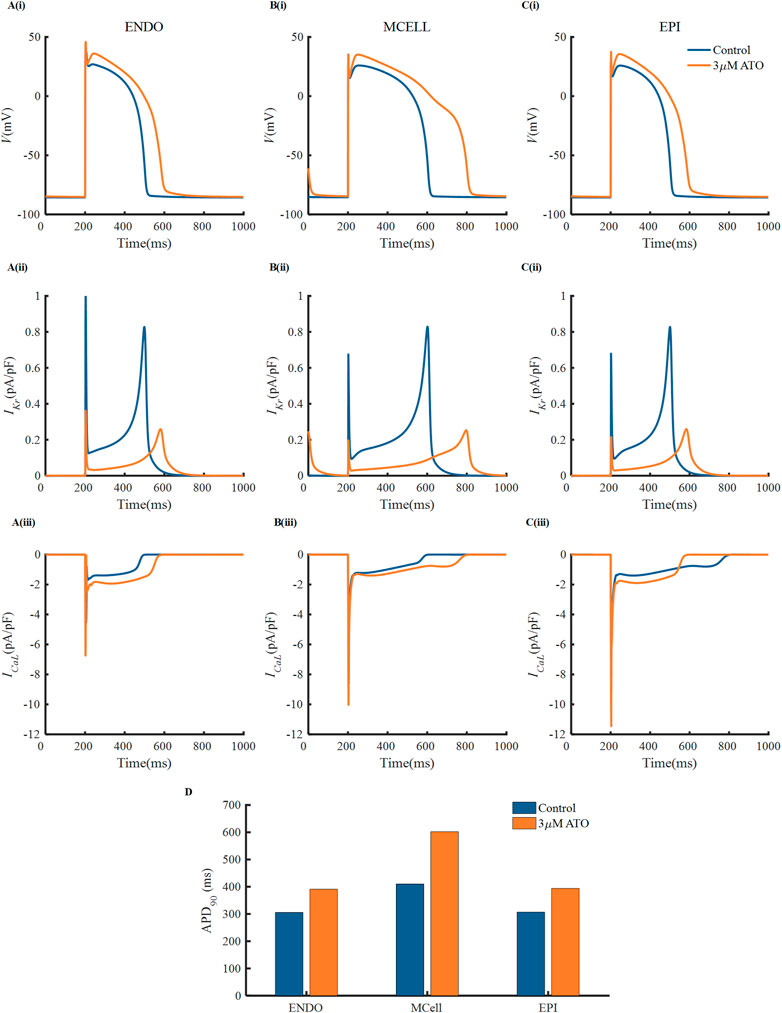
FIGURE 2. The effect of ATO on the action potentials of different ventricular myocytes. (A–C) The membrane potential, (i) (V), IKr (ii) and ICaL (iii) of endocardial cells (ENDO), middle cells (MCELL) and epicardial cells (EPI) on the condition of control and 3 μM ATO. (D) The APD90 of the action potentials in Figure A.
The APD restitution curve of a single cell was deemed to relate to the dynamical behavious of spiral waves in cardiac tissue. Here, we drew an APD restitution curve via the S1-S2 stimulation protocol, as shown in Figure 3A. Ten uniform S1 stimuli with a BCL of 800 m were applied before the S2 stimulus (only five S1 stimuli are shown in Figure 3A). With the reduction of the S1-S2 interval, the APD90 of the action potential triggered by the S2 stimulus declined until the S2 stimulus could not ignite depolarizing activity. The relationship between the S1-S2 interval and the corresponding APD90 formed the restitution curve as shown in Figure 3B. The restitution curve was shifted rightward slightly in ENDO and EPI and dramatically in MCELL, implying that the ATO-incubated VMs cannot support high-frequency pacing activity. The slope of the restitution curve reflects the stability of spiral waves. Results showed that ATO steepened the restitution curves of all kinds of VMs to varying degrees, whose slope is shown in Figure 3C, indicating unstable electrical activities.
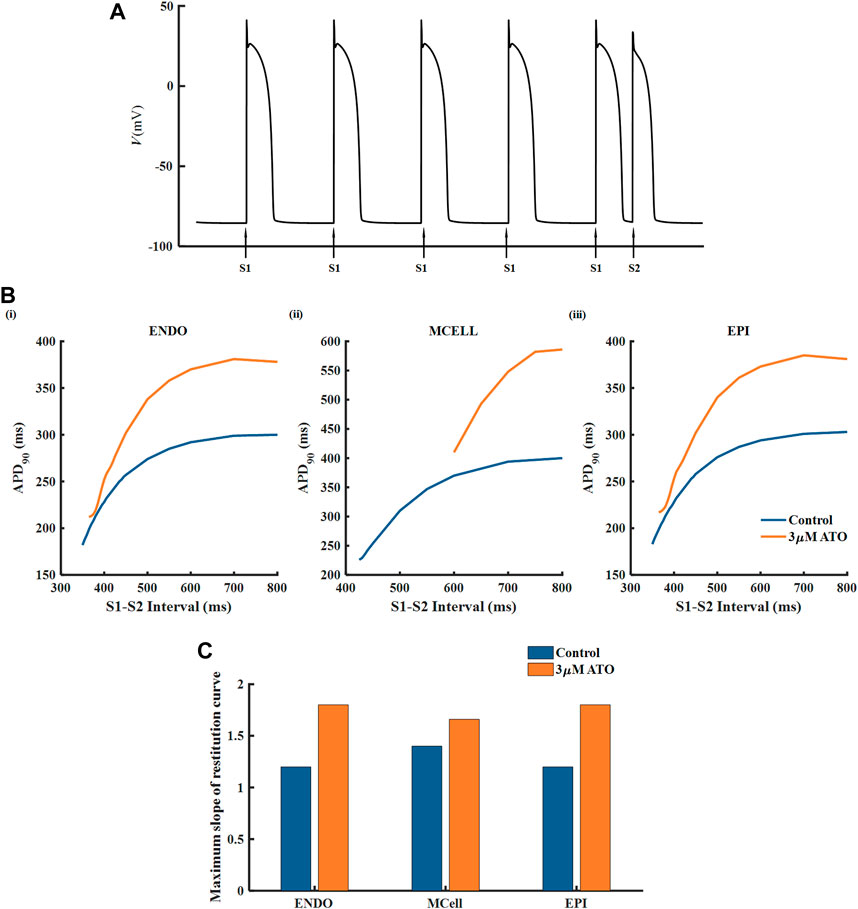
FIGURE 3. The restitution curves of ventricular myocytes. (A) The S1-S2 stimulation protocol. (B) The restitution curves of endocardial cells (ENDO), middle cells (MCELL) and epicardial cells (EPI) on the condition of control and 3 μM ATO. (C) The maximum slope of restitution curves in Figure (B).
To inspect the electrical activity of ATO-induced VMs under the high-frequency stimulus, a dynamic stimulation protocol was executed by gradually shortening the BCL. In the normal VM model, APD90 was unchanged under a specific BCL no matter how small the BCL was (results not shown). In the presence of 3 μM ATO, the APD90 in two consecutive beats may be different when BCL was reduced to a threshold. For example, when the BCL was 360 m, the membrane potential of ENDO had two alternans APD90 with a long APD90 at 334 m and a short APD90 at 238 m (Figure 4A(ii)). This kind of periodic APD90 change in a fixed BCL is called alternans. Figure 4 indicates that the alternans occurred during a BCL of 350–375 m in ENDO, that of 550–600 m in MCELL and that of 350–380 m in EPI, whose corresponding representative membrane potentials are given in Figure 4A(ii)–C(ii).
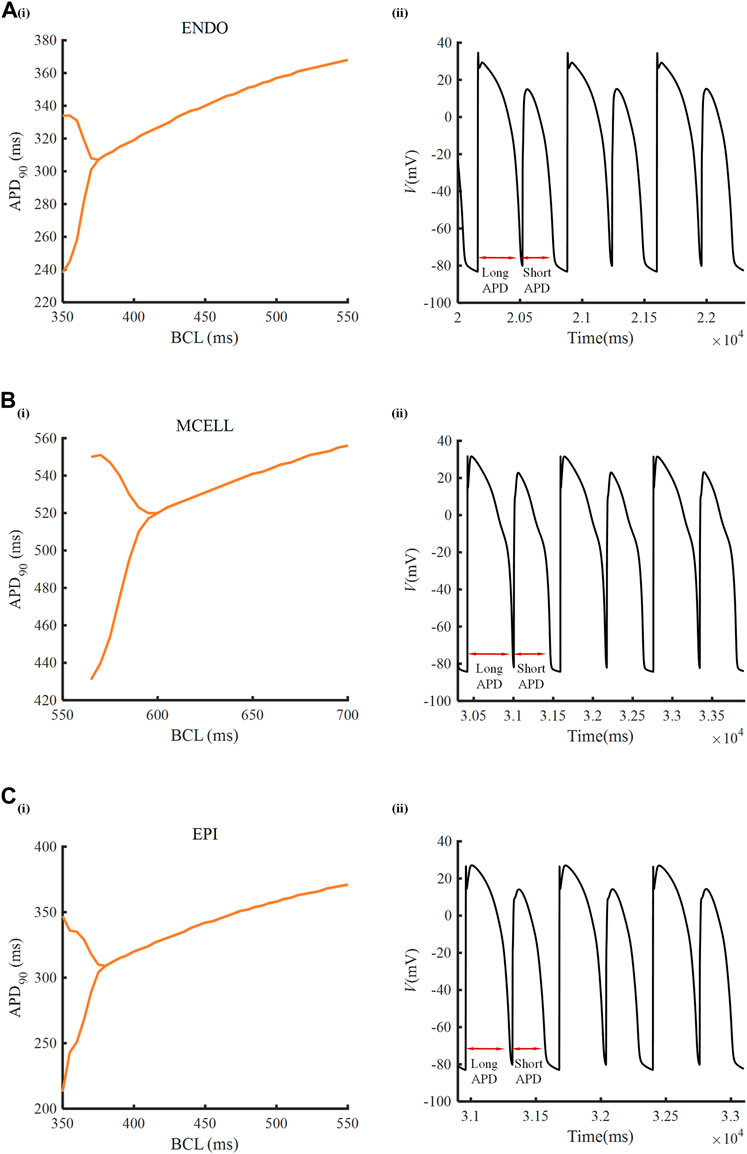
FIGURE 4. Alternans induced by ATO. [A(i)–C(i)] APD90 rate-dependent curves of endocardial cells (ENDO), middle cells (MCELL) and epicardial cells (EPI) incubated with 3 μM ATO. [A(ii)–C(ii)] The representative membrane potentials of Figure [A(i)–C(i)] with the basic cycle lengths at 360, 585, and 360 ms, respectively.
Effects of ATO on the vulnerability of heterogeneous ventricular cables
A heterogeneous ventricular cable including ENDO, MCELL and EPI with a ratio of 25:35:40 (Luo et al., 2017) was designed, and its electrical activities were simulated by solving Eq. (11). According to the membrane potential of the ventricular cable, the pseudo ECG, whose virtual electrode was placed 2.0 cm away from the last EPI, was calculated by Eq. (12). The ECG under different ATO concentrations is shown in Figure 5A. With the increase in ATO concentration, the QT interval rose from 362 m at 0 μM ATO to 477 m at 3 μM ATO, and the amplitude of the T wave slightly increased.
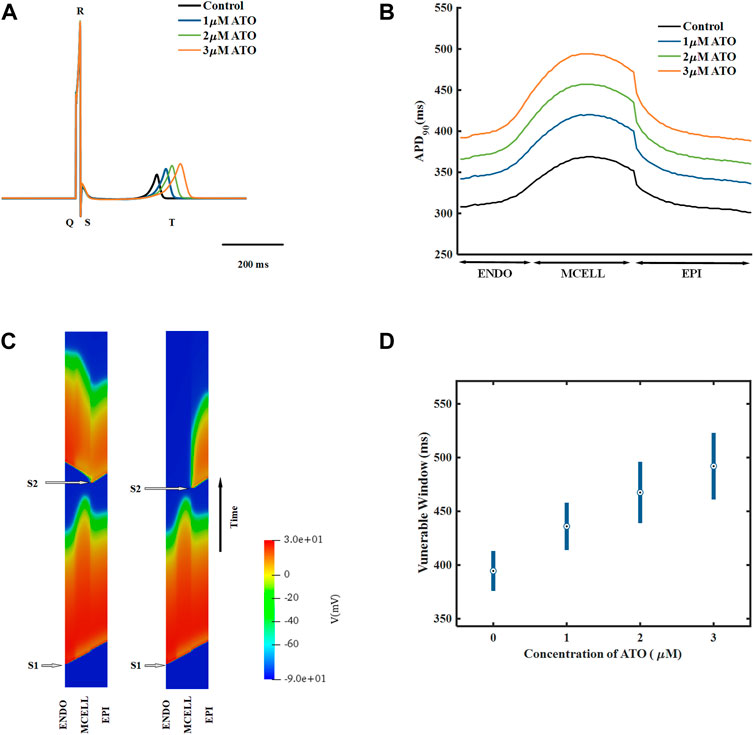
FIGURE 5. The effect of ATO on the heterogeneous ventricular cable. (A) ECG of ventricular cables with varying ATO concentrations. (B) Spatial distribution of APD90 across the ventricular cable with varying ATO concentrations. (C) Space-time plot of normal and unidirectional conduction. (D) Vulnerable windows of ventricular cables under varying ATO concentrations.
The dispersion of APD in the ventricular cable directly influenced the vulnerability of the cable. Compared with the isolated single cell, the gap junction between VMs decreased the difference in membrane potential duration between cells. Nevertheless, there was still an obvious difference in APD90 in space, particularly between MCELL and EPI, as shown in Figure 5B. The incorporation of ATO exacerbated the dispersion. The maximum gap of APD90 between two adjacent cells was 17 m in the control and 26 m on the condition of 3 μM ATO (Figure 5B). An S1-S2 stimulation protocol was applied to heterogeneous ventricular cables to detect vulnerability under varying ATO concentrations. Under the condition of long S1-S2 intervals, S2 inspired an electrical wave that can propagate into both MCELL and EPI tissue, as shown in the left panel in Figure 5C. However, in a short S1-S2 interval, the S2-inspired wave can only propagate into EPI tissue because the MCELL was in the refractory period and could not depolarize, as shown in the right panel of Figure 5C. This kind of unidirectional conduction may lead to the formation of reentry in ventricular tissue. As a result, the S1-S2 interval that led to unidirectional conduction was measured to evaluate the underlying arrhythmia risk, which was called the vulnerable window. Figure 5D presents the results. With the increment of ATO dosage, the vulnerable window expanded from 37 m at 0 μM ATO to 62 m at 3 μM ATO, demonstrating an increasing possibility of reentry at the tissue level. The value of the vulnerable window also increased from 376–413 m at 0 μM ATO to 461–532 m at 3 μM ATO. This was because extra ATO prolonged the APD in single VM cells; thus, the refractory period extended. As a result, the S1-S2 interval with a unidirectional conduction block was greater, which was reflected in the increase in the value of the vulnerable window. In addition, S2 was exerted at more locations as described in the Method section. The results at all locations had a coincident density of VM as shown in Table 2.
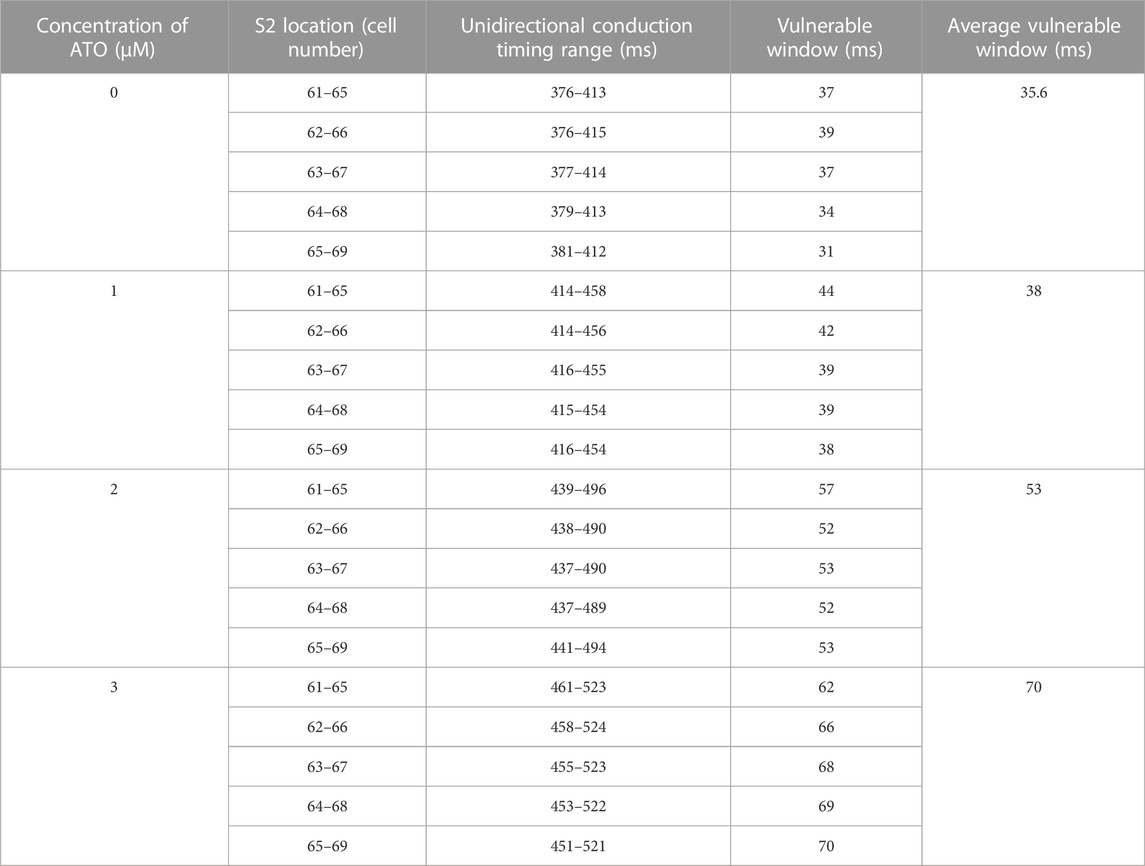
TABLE 2. Effect of ATO on VM in the ventricular cable model.
The results of the dynamic protocol indicated that discordant alternans can be induced in a heterogeneous ventricular cable. A representative result with a BCL of 410 m is shown in Supplementary Figure S2. The ENDO cells presented alternans APD, while some of the short APD was blocked by the MCELL because of its long refractory period.
Effects of ATO on the vulnerability of heterogeneous ventricular tissue
We designed a heterogeneous ventricular tissue with a size of 100 × 400 cells (Figure 6A). The length of the tissue included 100 heterogeneous VMs with an ENDO:MCELL:EPI ratio of 25:35:40. Electrophysiology activity with time can be solved by Eq. (11). An essential evaluation index in the two-dimensional (2D) ventricular tissue was reentry, i.e., spiral wave. It was a curved wavefront generated due to unidirectional conduction in tissue. We induced spiral waves through an S1-S2 stimulation protocol in ventricular tissue. A case in the control condition is shown in Figure 6C and the complete videos were attached in the supplementary materials. A case of reentry under the 3 μM ATO condition is also shown in the supplementary materials. An S1 stimulus was applied to the peripheral ENDO tissue with a length of five cells and a width of 400 cells (whole tissue width) to induce a plane wave (the first panel in Figure 6C(i)). During the refractory period of MCELL, an S2 stimulus was applied to the EPI cells that neighbored MCELL, whose width was less than that of the whole tissue (the second panel in Figure 6C(i)). With a specific S1-S2 interval and sufficient S2 stimulus width, a spiral wave can be provoked. The S1-S2 interval in Figure 6C(i) were 378 m. The membrane potentials of horizontal cells in 2D tissue are shown in Figure 6C(ii)), in which a single S2 stimulus could provoke more than one depolarizing potential. In the control condition (the ATO concentration was 0 μM), the S1-S2 interval that can induce spiral waves was from 376 m to 414 m, with a time window of 38 m. When the ATO concentration increased to 3 μM, reentry occurred during the S1-S2 interval of 461–507 m, whose vulnerable window rose to 46 m (Figure 6B).
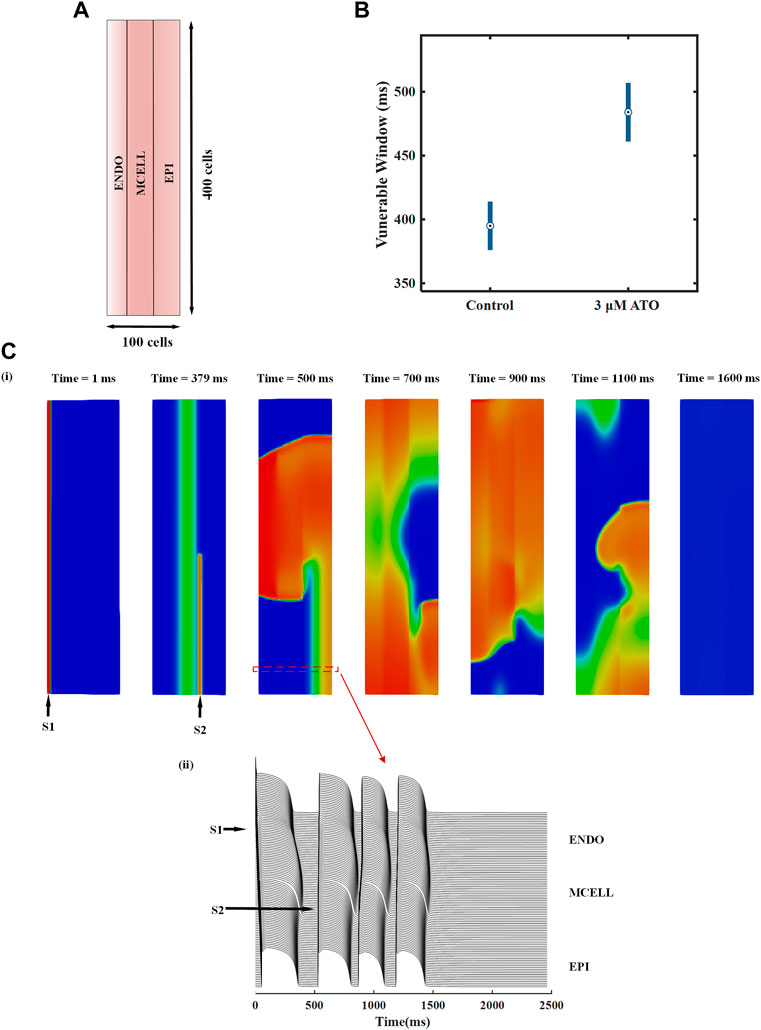
FIGURE 6. The effect of ATO on heterogeneous ventricular tissue. (A) The structure of heterogeneous ventricular tissue. (B) The vulnerable window of ventricular tissue in control and 3 μM ATO conditions. (C) Snapshots of reentrant excitation waves in the control condition with S1-S2 intervals at 378 m and sequence diagram of the membrane potential of horizontal continuous ventricular myocytes.
Effects of resveratrol on ATO-induced cardiotoxicity
When treated with 3 μM ATO in VMs, 10 μM resveratrol can surpress the excessive ICaL conductance from twice to 1.3 times (Yan et al., 2017) and increase the remaining maximum conductance index k of IKr (Eq. (4)) to 1.3 times (Zhao et al., 2014). The membrane potential of three types of VMs in the presence of 3 μM ATO and 10 μM resveratrol were simulated. The results showed that resveratrol shortened the APD90 from 391 to 357 m in ENDO cells, from 602 to 511 m in MCELL cells and from 394 to 360 m in EPI cells. Resveratrol also narrowed the maximum gap of APD90 between the three types of VMs from 211 to 154 m. The action potentials of different types of VMs treated with ATO and resveratrol are shown in Supplementary Figure S3.
The effect of resveratrol was further predicted in a ventricular cable model. The S1-S2 stimulation protocol was conducted with 3 μM ATO and 10 μM resveratrol. Simulation results showed that the time window that produced unidirectional conduction was narrowed from 62 m to 45 m under the action of resveratrol. The vulnerable window in different conditions is presented in Table 3.
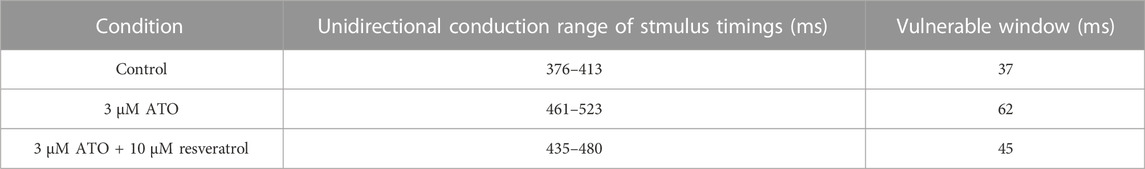
TABLE 3. Effect of resveratrol on the ATO-induced ventricular cable model.
Discussion
Summary of major findings
As has been widely reported, ATO may cause severe cardiotoxicity when applied to treat APL (Haybar et al., 2019) by interfering with hERG channels (Zhao et al., 2015) as well as ICaL channels (Chen et al., 2010). Even so, ATO remains one of the most effective drugs for rescuing cancer patients since it has a high long-term survival rate in newly diagnosed APL patients (Hu et al., 2009). As a result, the mechanisms of ATO-induced cardiotoxicity need to be elucidated, based on which more methods can be developed to improve the safety of ATO therapy. The in silico method provided an efficient approach to do this. Some mature models have been widely used in drug screening, such as the CiPA model (Park et al., 2019; Han et al., 2020; Ridder et al., 2020; Strauss et al., 2021) and a virtual heart model (Yuan et al., 2014). In this study, a mathematical model was built to delineate the cardiotoxicity of the human ventricle implicated in ATO by modulating the IKr and ICaL channels according to corresponding patch clamp data (Ficker et al., 2004; Chen et al., 2010; Yan et al., 2017). Based on the constructed model, we explored the process of ATO-induced arrhythmia from the subcellular level to the tissue level. The long APD in cells and LQT in cables were triggered by ATO-induced inhibition of IKr and facilitation of the ICaL channel, coinciding with clinical ECG diagnosis (Soignet et al., 2001). Beyond ATO’s explicit side-effect on LQT, it was first uncovered that ATO could augment the heterogeneity between different types of ventricular tissue, which was also an essential predisposing factor of tachycardia. In addition, ATO may induce alternans in all three types of VMs, which indicated an increase in the arrhythmia risk. Moreover, the vulnerability of ventricular tissue increased under the action of ATO, which was a direct factor of cardiac arrhythmia. Despite the underlying detrimental effects of ATO on the human heart, this study demonstrated a potential pharmacological remedy by resveratrol, which is expected to be beneficial for the safety of ATO therapy and provided better prognosis for newly diagnosed APL patients.
ATO-induced cardiotoxicity
In clinical trials, APL patients who received ATO had a higher risk of suffering from LQT syndrome and even sudden cardiac death (Westervelt et al., 2001). Biologists tried to explain the underlying ionic mechanisms of ATO-induced arrhythmia and found that K+ channels (including IKr, IKs and IK1) (Ficker et al., 2004) and Ca2+ dynamics (including ICaL and [Ca2+]i) (Chen et al., 2010) might be responsible for this. The effect of ATO on IK1 and IKs is controversial. An experiment in CHO cells claimed that ATO could inhibit the IKs channel (Drolet et al., 2004). ATO can also impair the expression of Kir2.1 in guinea pigs (Shan et al., 2013) and reduce the IK1 density in guinea pig VMs (Shan et al., 2013) and rat VMs (Chen et al., 2010). However, another study announced that the IKs and IK1 of guinea pig VMs had no obvious changes with overnight ATO incubation (Ficker et al., 2004). There were unified results of the inhibitory effect of ATO on IKr and its promotive effect on ICaL, whose experimental data were more abundant. As a result, we mainly focused on ATO’s effect on the ventricle via IKr and ICaL channels.
The relationship between several ATO dosages and the degree of inhibition of IKr was given in an experiment (Ficker et al., 2004), which provided basic data to depict the binding interaction between ATO and IKr in the present model. The I-V curve data of ICaL were usually measured in the condition of 3 µM ATO. Experiments displayed that long-term exposure to ATO facilitated ICaL density to approximately 2–3 times and negatively shifted the V1/2 of the activation curve (Ficker et al., 2004; Chen et al., 2010; Yan et al., 2017). According to this, we estimated the subcellular effect of ATO on human VMs and simulated the electrical activity of ATO-disrupted ventricular single cells, cables, and tissue. The simulation result implied a high risk of arrhythmia due to the steep restitution curve and electrical alternans in single VMs. Additionally, beyond the biological experiments, we found that ATO-reconstructed ionic channels not only prolonged the action potential of ventricular cells, but also aggravated the heterogeneity between the three types of VMs. On the one hand, in the present simulation, the QT interval of heterogeneous ventricular cable was 362 m in control and treated with 3 μM ATO prolonged QT interval to 477 m. Clinically, the normal QT interval should range from 350–420 m, over 460 m in women and 440 m in men can be diagnosed with LQT (Vadavanath et al., 2019). Particularly, when QT is more than 500 m, the risk of TdP increases dramatically (Zamorano et al., 2016). This means that the present simulation result with 3 μM ATO was in a hazardous range of QT intervals, which was coincident with clinical manifestations and thus reliable. In addition, simulation studies also reported the underlying risk of long APD. A study indicated that the prolonged APD impaired the repolarization of action potential, so in a short cycle length, the early afterdepolarizations may be evoked (Bai et al., 2017). Another study also proved that the adaptability of ventricular tissue was impaired because of the prolonged APD (Bi et al., 2022). On the other hand, increased ventricular heterogeneity among single VM cells can result in a further detriment in the ventricle, raising the possibility of reentry within the heart. Both factors led to a wide vulnerable window in ventricular tissue, indicating a higher risk of arrhythmia in the heart.
An uncharacteristic discordant alternans can be observed under the action of ATO (Supplementary Figure S2). The tissue alternans in this study were not as obvious as those in heart failure-associated atrial alternans research (Zhao et al., 2020) because except for single cell characters, the decreased CV in atrial tissue was also an essential factor in inducing tissue alternans. There is no evidence that ATO reduced the CV in ventricular tissue, so the alternans did not easily occur in this present simulation.
The ATO concentration in plasma reached 0.34–2 µM with intravenous treatment for APL and acute myeloid leukemia (AML) patients at a dosage of 10 mg/day (Siu et al., 2006). As a result, although there were animal experimental data with ATO concentrations of 0.1–50 µM (Ficker et al., 2004; Sun et al., 2006), we mainly adopted and modeled the data within 0.1–3 µM ATO. As a result, the present model was clinically valuable, based on which ATO-induced arrhythmogenesis can be investigated.
Pharmacological rescue of ATO toxicity
Despite the possibility of cardiac arrhythmia after administering ATO in both experiments and the simulation, it was still crucial for APL patients. Consequently, drug combinations have been proposed as a means of reducing cardiotoxicity resulting from ATO. Various kinds of drugs have been investigated to attenuate ATO-induced toxicity, but only a few have shown remarkable rescue properties. Fexofenadine is an antiallergic agent. Experiments have shown that 1 μM fexofenadine can increase IKr from 30% of the original density to approximately 60% in HEK293 cells incubated with 3 µM ATO (Yan et al., 2017). It was verified to shorten the APD90 from 396.43 ± 25.33 (with 3 µM ATO) to 233.30 ± 18.75 m in NRVMs and from 1164.71 ± 40.25 to 942.86 ± 103.11 m in hiPS-CMs (Yan et al., 2017). Nisoldipine, a hypotensive drug, could shorten the APD90 of guinea pig VMs with 3 µM ATO treatment from 880 ± 61 to 686 ± 36 m (Ficker et al., 2004). It is known to be the ICaL blocker, but the quantitative relation between Nisoldipine concentration and ICaL properties was not given on the condition of ATO. Resveratrol is a natural antioxidation ingredient that can protect the cardiovascular system (Sulaiman et al., 2010; Dudka et al., 2012) by ameliorating structural abnormalities and oxidative damage (Zhao et al., 2008). It can act on both the IKr (Zhao et al., 2014) and ICaL (Yan et al., 2017) channels and shorten the APD90 of ATO-incubated cells from 948.3 ± 63.7 m to 522.6 ± 26.3 m in guinea pig VMs (Zhao et al., 2014) and from 1164.71 ± 40.25 to 942.86 ± 103.11 m in hiPS-CMs (Yan et al., 2017). It can be found that resveratrol performed the best rescue effect among all three drugs that had significant protective effects on ATO-incubated CMs, and the rescue ratio was less in hiPS-CMs than in rodent VMs. The data on drug rescue in ATO-incubated CMs are summarized in Table 4.
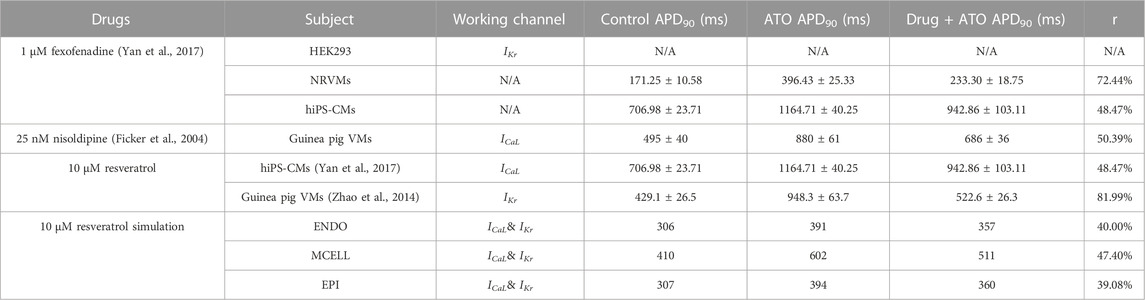
TABLE 4. Effect of drugs on the APD of cardiomyocytes incubated with 3 μM ATO.
To further predict the effect of resveratrol on human VMs incubated with ATO, we simulated ATO/resveratrol-incorporated ventricular models by modifying the IKr and ICaL channels. Although the rescue ratio of resveratrol declined in human VMs, it still remarkably ameliorated ATO cardiotoxicity (Table 4). In the present model, resveratrol not only shortened the APD90 of ENDO, MCELL and EPI but also narrowed the difference in APD90 between VMs, thus decreasing the vulnerable window of the ventricular cable (Table 3). This study verified that resveratrol has the potential to be applied in clinics to protect the cardiovascular system in ATO-treated patients.
Limitations
Except for the intrinsic limitations of the basic TNNP06 model (ten Tusscher and Panfilov, 2006), the pharmacological model in this study was not completely accurate due to a lack of abundant experimental data. The patch clamp experiment was performed on HEK293 (Yan et al., 2017) or rodent VMs (Chen et al., 2010), so it was only a prediction of the binding interaction between ATO and human VMs. In another human VM model (O'Hara et al., 2011), the IKr channel model was built by the Markov chain model, and the corresponding drug model was extended by incorporating two drug states (Whittaker et al., 2017). In this study, the simple pore block theory was used to depict the effect of ATO on the IKr channel because it was suitable for the IKr model of TNNP06.
Experiments reported that ATO had an underlying effect on calcium homeostasis by upregulating the expression of CaMKII, which finally caused abnormal CM contraction (Zhang et al., 2016; Zhang et al., 2017). The change in calcium dynamics caused by extra ATO is worth discussing in silico after more specific ionic data are obtained.
ATO may also have an impact on other ionic currents, such as IK1 (Chen et al., 2010), IKs (Drolet et al., 2004) and INa (Ficker et al., 2004), but the literature shows that their effect is not obvious. So did the present model study. We did not perform further study in this paper because of insufficient biological evidence as well as the minor effect. A related study can be conducted if more data are provided.
This simulation study revealed the acute effect of ATO on ionic channel currents rather than on protein expression. In the future, a Markov pharmacological model of IKr can be built to simulate the effect of ATO on hERG protein, which can refer to the previous short-QT simulation work (Whittaker et al., 2017; Zhang et al., 2022).
Many drugs or chemical compounds have been explored to relieve ATO toxicity (Haybar et al., 2019), but most of their ionic reactions were not clear, so only two drugs were modeled in this study. Nevertheless, this study provided an approach for simulating and evaluating the effectiveness of new drugs that can ameliorate ATO toxicity. Drugs that can protect the hearts treated with ATO should be compared in the future so that the optimal medication regimen can be screened.
Conclusion
This study provided a computational method for investigating the cardiotoxicity induced by ATO. The mechanisms of arrhythmia attributed to ATO were investigated from the ionic level to the tissue level. Simulation results showed that ATO not only extended the QT interval of ECG but also aggravated the heterogeneity of VM cells and led to alternans, thus raising the possibility of reentry in the human ventricle. Under the actions of ATO, resveratrol was incorporated into the ventricular model by intervening in the ionic channel, by which the side effects of ATO were ameliorated. The method of this study can also be used to screen drugs that may ameliorate ATO toxicity. This study elucidated ATO cardiotoxicity pathogenesis and its attenuation mechanisms, which is expected to improve ATO treatment in its clinical use.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
YL and JL conceived the study, designed most of the study, performed the simulations and analyses, and wrote most of the manuscript. RW, WL, and HZ contributed to the design of simulations, figure design, and manuscript writing. LM and HZ supervised the project. All authors contributed to the article and approved the submitted version.
Funding
The work is supported by the National Key R&D Program of China (2020AAA0105200), the Open Fund of the Key Laboratory of Medical Electrophysiology of Ministry of Education and Sichuan Province, China (KeyME-2020-001), the Science & Technology Program from Administration for Market Regulation of Sichuan Province (SCSJ2021009), the Science and Technology Project of Sichuan Province (2021YFS0089), and the Science and Technology Project of Luzhou City (2020-SYF-23).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.1004605/full#supplementary-material
Supplementary Figure S1 | The calcium dynamic in VMs under the action of ATO. (A–D) The membrane potential (V), ICaL, [Ca2+]i, and INaCa of endocardial cells (ENDO) on the condition of control and 3 μM ATO.
Supplementary Figure S2 | The action potential of heterogeneous ventricular cable with a BCL of 410 ms.
Supplementary Figure S3 | The action potential of endocardial cells (ENDO), middle cells (MCELL) and epicardial cells (EPI) incubated with 3 µM ATO on the condition of pharmacological rescue.
Supplementary Video S1 | Control reentry in heterogeneous ventricular tissue. This video shows the generation and elimination of a reentrant excitation wave in a control condition. At the beginning of the simulation, an S1 stimulus was conducted at the peripheral endocardial cells so that a planar wave was initiated. At 378 m, a S2 stimulus was applied at a piece of epicardial cells neighboring the middle cells so that a series of spiral waves were provoked.
Supplementary Video S2 | ATO reentry in a heterogeneous ventricular tissue. This video shows the generation and elimination of a reentrant excitation wave in 3 µM ATO. At the beginning of the simulation, an S1 stimulus was conducted at the peripheral endocardial cells so that a planar wave was initiated. At 475 ms, an S2 stimulus was applied at a piece of epicardial cells neighboring the middle cells so that a series of spiral waves were provoked.
References
Alexandre J., Moslehi J. J., Bersell K. R., Funck-Brentano C., Roden D. M., Salem J. E. (2018). Anticancer drug-induced cardiac rhythm disorders: Current knowledge and basic underlying mechanisms. Pharmacol. Ther. 189, 89–103. doi:10.1016/j.pharmthera.2018.04.009
Bai J., Wang K., Liu Y., Li Y., Liang C., Luo G., et al. (2017). Computational cardiac modeling reveals mechanisms of ventricular arrhythmogenesis in long QT syndrome type 8: CACNA1C R858H mutation linked to ventricular fibrillation. Front. Physiol. 8, 771. doi:10.3389/fphys.2017.00771
Bi X., Zhang S., Jiang H., Ma W., Li Y., Lu W., et al. (2022). Mechanistic insights into inflammation-induced arrhythmias: A simulation study. Front. Physiol. 13, 843292. doi:10.3389/fphys.2022.843292
Binu P., Priya N., Abhilash S., Vineetha R. C., Nair R. H. (2017). Studies on curative efficacy of monoterpene eugenol on anti- leukemic drug arsenic trioxide induced cardiotoxicity. Biomed. Pharmacother. 91, 559–566. doi:10.1016/j.biopha.2017.04.087
Chen G. Q., Shi X. G., Tang W., Xiong S. M., Zhu J., Cai X., et al. (1997). Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood 89 (9), 3345–3353.
Chen S., Wu J. L., Liang Y., Tang Y. G., Song H. X., Wu L. L., et al. (2021). Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell 39 (2), 225–239.e8. doi:10.1016/j.ccell.2020.11.013
Chen X., Shan H., Zhao J., Hong Y., Du Z., Sun I., et al. (2010). L-type calcium current (ICa, L) and inward rectifier potassium current (Ik1) are involved in QT prolongation induced by arsenic trioxide in rat. Cell. Physiol. biochem. 26 (6), 967–974. doi:10.1159/000324005
Chu W., Li C., Qu X., Zhao D., Wang X., Yu X., et al. (2012). Arsenic-induced interstitial myocardial fibrosis reveals a new insight into drug-induced long QT syndrome. Cardiovasc. Res. 96 (1), 90–98. doi:10.1093/cvr/cvs230
Drolet B., Simard C., Roden D. M. (2004). Unusual effects of a QT-prolonging drug, arsenic trioxide, on cardiac potassium currents. Circulation 109 (1), 26–29. doi:10.1161/01.CIR.0000109484.00668.CE
Ducas R. A., Seftel M. D., Ducas J., Seifer C. (2011). Monomorphic ventricular tachycardia caused by arsenic trioxide therapy for acute promyelocytic leukaemia. J. R. Coll. Physicians Edinb. 41 (2), 117–118. doi:10.4997/JRCPE.2011.204
Dudka J., Gieroba R., Korga A., Burdan F., Matysiak W., Jodlowska-Jedrych B., et al. (2012). Different effects of resveratrol on dose-related Doxorubicin-induced heart and liver toxicity. Evid. Based. Complement. Altern. Med. 2012, 606183. doi:10.1155/2012/606183
Ficker E., Kuryshev Y. A., Dennis A. T., Obejero-Paz C., Wang L., Hawryluk P., et al. (2004). Mechanisms of arsenic-induced prolongation of cardiac repolarization. Mol. Pharmacol. 66 (1), 33–44. doi:10.1124/mol.66.1.33
Gima K., Rudy Y. (2002). Ionic current basis of electrocardiographic waveforms: A model study. Circ. Res. 90 (8), 889–896. doi:10.1161/01.res.0000016960.61087.86
Hai J. J., Gill H., Tse H. F., Kumana C. R., Kwong Y. L., Siu C. W. (2015). Torsade de Pointes during oral arsenic trioxide therapy for acute promyelocytic leukemia in a patient with heart failure. Ann. Hematol. 94 (3), 501–503. doi:10.1007/s00277-014-2174-1
Han X., Samieegohar M., Ridder B. J., Wu W. W., Randolph A., Tran P., et al. (2020). A general procedure to select calibration drugs for lab-specific validation and calibration of proarrhythmia risk prediction models: An illustrative example using the CiPA model. J. Pharmacol. Toxicol. Methods 105, 106890. doi:10.1016/j.vascn.2020.106890
Haybar H., Shahrabi S., Rezaeeyan H., Jodat H., Saki N. (2019). Strategies to inhibit arsenic trioxide-induced cardiotoxicity in acute promyelocytic leukemia. J. Cell. Physiol. 234, 14500–14506. doi:10.1002/jcp.28292
Sun H. L., Chu W. F., Dong D. L., Liu Y., Bai Y. L., Wang X. H., et al. (2006). Choline-modulated arsenic trioxide-induced prolongation of cardiac repolarization in Guinea pig. Basic Clin. Pharmacol. Toxicol. 98 (4), 381–388. doi:10.1111/j.1742-7843.2006.pto_319.x
Hu J., Liu Y. F., Wu C. F., Xu F., Shen Z. X., Zhu Y. M., et al. (2009). Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 106 (9), 3342–3347. doi:10.1073/pnas.0813280106
Lenihan D. J., Kowey P. R. (2013). Overview and management of cardiac adverse events associated with tyrosine kinase inhibitors. Oncologist 18 (8), 900–908. doi:10.1634/theoncologist.2012-0466
Liu Y., Li D., Nie D., Liu S. K., Yang B. F., Liu M. T., et al. (2017). Arsenic trioxide and angiotensin II have inhibitory effects on HERG protein expression: Evidence for the role of PML SUMOylation. Oncotarget 8 (28), 45447–45458. doi:10.18632/oncotarget.17563
Luo C., Wang K., Zhang H. (2017). In silico assessment of the effects of quinidine, disopyramide and E-4031 on short QT syndrome variant 1 in the human ventricles. PLoS One 12 (6), e0179515. doi:10.1371/journal.pone.0179515
Mathews V., George B., Lakshmi K. M., Viswabandya A., Bajel A., Balasubramanian P., et al. (2006). Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: Durable remissions with minimal toxicity. Blood 107 (7), 2627–2632. doi:10.1182/blood-2005-08-3532
O'Hara T., Virag L., Varro A., Rudy Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Comput. Biol. 7 (5), e1002061. doi:10.1371/journal.pcbi.1002061
Park J. S., Jeon J. Y., Yang J. H., Kim M. G. (2019). Introduction to in silico model for proarrhythmic risk assessment under the CiPA initiative. Transl. Clin. Pharmacol. 27 (1), 12–18. doi:10.12793/tcp.2019.27.1.12
Ridder B. J., Leishman D. J., Bridgland-Taylor M., Samieegohar M., Han X., Wu W. W., et al. (2020). A systematic strategy for estimating hERG block potency and its implications in a new cardiac safety paradigm. Toxicol. Appl. Pharmacol. 394, 114961. doi:10.1016/j.taap.2020.114961
Shan H., Zhang Y., Cai B., Chen X., Fan Y., Yang L., et al. (2013). Upregulation of microRNA-1 and microRNA-133 contributes to arsenic-induced cardiac electrical remodeling. Int. J. Cardiol. 167 (6), 2798–2805. doi:10.1016/j.ijcard.2012.07.009
Shen Z. X., Shi Z. Z., Fang J., Gu B. W., Li J. M., Zhu Y. M., et al. (2004). All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 101 (15), 5328–5335. doi:10.1073/pnas.0400053101
Siu C. W., Au W. Y., Yung C., Kumana C. R., Lau C. P., Kwong Y. L., et al. (2006). Effects of oral arsenic trioxide therapy on QT intervals in patients with acute promyelocytic leukemia: Implications for long-term cardiac safety. Blood 108 (1), 103–106. doi:10.1182/blood-2006-01-0054
Soignet S. L., Frankel S. R., Douer D., Tallman M. S., Kantarjian H., Calleja E., et al. (2001). United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J. Clin. Oncol. 19 (18), 3852–3860. doi:10.1200/JCO.2001.19.18.3852
Strauss D. G., Wu W. W., Li Z., Koerner J., Garnett C. (2021). Translational models and tools to reduce clinical trials and improve regulatory decision making for QTc and proarrhythmia risk (ICH E14/S7B updates). Clin. Pharmacol. Ther. 109 (2), 319–333. doi:10.1002/cpt.2137
Sulaiman M., Matta M. J., Sunderesan N. R., Gupta M. P., Periasamy M., Gupta M. (2010). Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 298 (3), H833–H843. doi:10.1152/ajpheart.00418.2009
Taggart P., Sutton P. M., Opthof T., Coronel R., Trimlett R., Pugsley W., et al. (2000). Inhomogeneous transmural conduction during early ischaemia in patients with coronary artery disease. J. Mol. Cell. Cardiol. 32 (4), 621–630. doi:10.1006/jmcc.2000.1105
ten Tusscher K. H., Panfilov A. V. (2006). Alternans and spiral breakup in a human ventricular tissue model. Am. J. Physiol. Heart Circ. Physiol. 291 (3), H1088–H1100. doi:10.1152/ajpheart.00109.2006
Unnikrishnan D., Dutcher J. P., Varshneya N., Lucariello R., Api M., Garl S., et al. (2001). Torsades de pointes in 3 patients with leukemia treated with arsenic trioxide. Blood 97 (5), 1514–1516. doi:10.1182/blood.v97.5.1514
Vadavanath P., Vineetha K., Gopalan R. (2019). An overview on arsenic trioxide-induced cardiotoxicity. Cardiovasc. Toxicol. 19, 105–119. doi:10.1007/s12012-018-09504-7
Varghese M. V., Abhilash M., Paul M. V., Alex M., Nair R. H. (2017). Omega-3 fatty acid protects against arsenic trioxide-induced cardiotoxicity in vitro and in vivo. Cardiovasc. Toxicol. 17 (2), 109–119. doi:10.1007/s12012-016-9361-3
Westervelt P., Brown R. A., Adkins D. R., Khoury H., Dipersio J. F., HurD D., et al. (2001). Sudden death among patients with acute promyelocytic leukemia treated with arsenic trioxide. Blood 98 (2), 266–271. doi:10.1182/blood.v98.2.266
Whittaker D. G., Ni H., Benson A. P., Hancox J. C., Zhang H. (2017). Computational analysis of the mode of action of disopyramide and quinidine on hERG-linked short QT syndrome in human ventricles. Front. Physiol. 8, 759. doi:10.3389/fphys.2017.00759
Yan M., Feng L., Shi Y., Wang J., Liu Y., Li F., et al. (2017). Mechanism of As2O3-induced action potential prolongation and using hiPS-CMs to evaluate the rescue efficacy of drugs with different rescue mechanism. Toxicol. Sci. 158 (2), 379–390. doi:10.1093/toxsci/kfx098
Yuan Y., Bai X., Luo C., Wang K., Zhang H. (2014). The virtual heart as a platform for screening drug cardiotoxicity. Br. J. Pharmacol. 172 (23), 5531–5547. doi:10.1111/bph.12996
Zamorano J. L., Lancellotti P., Rodriguez Munoz D., Aboyans V., Asteggiano R., Galderisi M., et al. (2016). 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC committee for practice guidelines: The task force for cancer treatments and cardiovascular toxicity of the European society of Cardiology (ESC). Eur. Heart J. 37 (36), 2768–2801. doi:10.1093/eurheartj/ehw211
Zhang J. Y., Sun G. B., Wang M., Liao P., Du Y. Y., Yang K., et al. (2016). Arsenic trioxide triggered calcium homeostasis imbalance and induced endoplasmic reticulum stress-mediated apoptosis in adult rat ventricular myocytes. Toxicol. Res. 5 (2), 682–688. doi:10.1039/c5tx00463b
Zhang J. Y., Zhang B., Wang M., Wang W., Liao P., Sun G. B., et al. (2017). Calcium homeostasis and endoplasmic reticulum stress are involved in Salvianolic acid B-offered protection against cardiac toxicity of arsenic trioxide. Oncotarget 8 (57), 97384–97393. doi:10.18632/oncotarget.22127
Zhang S., Lu W., Yang F., Li Z., Wang S., Jiang M., et al. (2022). Computational analysis of arrhythmogenesis in KCNH2 T618I mutation-associated short QT syndrome and the pharmacological effects of quinidine and sotalol. NPJ Syst. Biol. Appl. 8 (1), 43. doi:10.1038/s41540-022-00254-5
Zhang X. W., Yan X. J., Zhou Z. R., Yang F. F., Wu Z. Y., Sun H. B., et al. (2010). Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 328 (5975), 240–243. doi:10.1126/science.1183424
Zhang Y., Liu Y., Wang T., Li B., Li H., Wang Z., et al. (2006). Resveratrol, a natural ingredient of grape skin: Antiarrhythmic efficacy and ionic mechanisms. Biochem. Biophys. Res. Commun. 340 (4), 1192–1199. doi:10.1016/j.bbrc.2005.12.124
Zhao N., Li Q., Zhang K., Wang K., He R., Yuan Y., et al. (2020). Heart failure-induced atrial remodelling promotes electrical and conduction alternans. PLoS Comput. Biol. 16 (7), e1008048. doi:10.1371/journal.pcbi.1008048
Zhao X., Shi Y. Q., Yan C. C., Feng P. F., Wang X., Zhang R., et al. (2015). Up-regulation of miR-21 and miR-23a Contributes to As2O3-induced hERG Channel Deficiency. Basic Clin. Pharmacol. Toxicol. 116 (6), 516–523. doi:10.1111/bcpt.12348
Zhao X., Zhang K. P., Huang T., Yan C. C., Liu L. R., Zhu Q. L., et al. (2014). The rescuable function and mechanism of resveratrol on As₂O₃-induced hERG K⁺ channel deficiency. Naunyn. Schmiedeb. Arch. Pharmacol. 387 (11), 1079–1089. doi:10.1007/s00210-014-1019-8
Zhao X. Y., Li G. Y., Liu Y., Chai L. M., Chen J. X., Zhang Y., et al. (2008). Resveratrol protects against arsenic trioxide-induced cardiotoxicity in vitro and in vivo. Br. J. Pharmacol. 154 (1), 105–113. doi:10.1038/bjp.2008.81
Keywords: arsenic trioxide, drug cardiotoxicity, ionic channel, cardiac modeling, long QT
Citation: Li Y, Wan R, Liu J, Liu W, Ma L and Zhang H (2022) In silico mechanisms of arsenic trioxide-induced cardiotoxicity. Front. Physiol. 13:1004605. doi: 10.3389/fphys.2022.1004605
Received: 27 July 2022; Accepted: 28 November 2022;
Published: 15 December 2022.
Edited by:
Jieyun Bai, Jinan University, ChinaReviewed by:
Biyi Chen, The University of Iowa, United StatesShugang Zhang, Ocean University of China, China
Copyright © 2022 Li, Wan, Liu, Liu, Ma and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Liu, liujun665@hotmail.com; Weichao Liu, w_liu2019@swmu.edu.cn; Henggui Zhang, h.zhang-3@manchester.ac.uk