 Alejandro Ríos-Hoyo
Alejandro Ríos-Hoyo Xavier Monzonís
Xavier Monzonís Joana Vidal
Joana Vidal Jenniffer Linares2
Jenniffer Linares2 Clara Montagut
Clara Montagut- 1Yale Cancer Center, Yale School of Medicine, Yale University, New Haven, CT, United States
- 2Department of Medical Oncology, Hospital del Mar Research Institute, Centro de Investigación Biomédica en Red de Cáncer (CIBERONC), Barcelona, Spain
Emergence of acquired resistance limits the efficacy of the anti-EGFR therapies cetuximab and panitumumab in metastatic colorectal cancer. In the last decade, preclinical and clinical cohort studies have uncovered genomic alterations that confer a selective advantage to tumor cells under EGFR blockade, mainly downstream re-activation of RAS-MEK signaling and mutations in the extracellular domain of EGFR (EGFR-ECD). Liquid biopsies (genotyping of ctDNA) have been established as an excellent tool to easily monitor the dynamics of genomic alterations resistance in the blood of patients and to select patients for rechallenge with anti-EGFR therapies. Accordingly, several clinical trials have shown clinical benefit of rechallenge with anti-EGFR therapy in genomically-selected patients using ctDNA. However, alternative mechanisms underpinning resistance beyond genomics -mainly related to the tumor microenvironment-have been unveiled, specifically relevant in patients receiving chemotherapy-based multi-drug treatment in first line. This review explores the complexity of the multifaceted mechanisms that mediate secondary resistance to anti-EGFR therapies and potential therapeutic strategies to circumvent acquired resistance.
1 Introduction
Colorectal cancer (CRC) represents the third most commonly diagnosed cancer worldwide, and the second leading cause of cancer related deaths (Sung et al., 2021; Siegel et al., 2023). Although lifestyle modifications (e.g., smoking cessation, lowering alcohol intake, increasing dietary fiber, physical activity) can prevent a substantial amount of cases, its estimated incidence is predicted to increase, particularly in countries with a High Development Index (Morgan et al., 2022). Survival rate in metastatic CRC (mCRC) remains poor, with a median overall survival (mOS) of 36 months, and a 5-year OS not exceeding 20% (Cervantes et al., 2022). In the metastatic or unresectable setting, systemic therapy is the treatment of choice, using chemotherapy, targeted therapies, or immunotherapy (Biller and Schrag, 2021; Cervantes et al., 2022). To adequately guide treatment selection in mCRC, biomarker identification is crucial. This involves testing for genomic alterations including KRAS/NRAS/BRAF mutations, and microsatellite instability (MSI)/mismatch repair genes (MMR). In this sense, patients with wild-type (WT) KRAS/NRAS/BRAFV600E benefit from chemotherapy doublets (FOLFOX [5-Fluouracil, folinic acid and oxaliplatin], CAPOX [capecitabine and oxaliplatin] or FOLFIRI [5-Fluouracil, folinic acid and irinotecan]), combined with EGFR-inhibitors such as cetuximab or panitumumab (Benson et al., 2022; Cervantes et al., 2022).
Cetuximab is an anti-EGFR targeted monoclonal antibody (moAb) that consists of a chimeric immunoglobulin G1 (IgG1), which, upon binding to the EGFR receptor, induces the internalization and degradation of the receptor, thus disrupting the downstream pathway. Since cetuximab is an IgG1 moAb, it can elicit immune functions such as antibody-dependent cell-mediated cytotoxicity (ADCC) as an anti-tumoral effect (Mendelsohn et al., 2015). However, panitumumab, a humanized IgG2 moAb with similar anti-proliferative effects to cetuximab, is not able to initiate an ADCC effect (Yarom and Jonker, 2011). Two decades ago, initial trials of anti-EGFR therapy in mCRC showed the efficacy of cetuximab and panitumumab monotherapy as second and subsequent lines of treatment in patients with WT KRAS tumors. These trials reported an objective response rate (ORR) of 12.8% and 17%, a median progression-free survival (mPFS) of 3.7 months and 12.3 weeks, and a median overall survival (mOS) of 9.5 months and 8.1 months, for cetuximab and panitumumab, respectively. Subsequently, the frontline pivotal randomized trials that tested anti-EGFR therapies in combination with chemotherapy doublets (Jonker et al., 2007; Amado et al., 2008; Karapetis et al., 2008), showed a benefit in ORR (46.9%–58%), mPFS (9.9–12 months), and mOS (19.8–24.9 months) (Bokemeyer et al., 2009; 2015; Van Cutsem et al., 2009). In the following years, retrospective analysis of RAS (KRAS, NRAS) and BRAF mutations in tumor tissue samples from patients included in these pivotal trials showed a greater benefit in patients with tumors not harboring RAS/BRAF mutations.
2 Acquired resistance to anti-EGFR moAb in mCRC: heterogeneity and clonal selection
The benefit driven by the addition of anti-EGFR therapy to chemotherapy is undoubtful, however, resistance eventually develops which leads to disease progression. Colorectal tumors are heterogeneous, composed of multiple cellular clones carrying different genetic or epigenetic abnormalities within the same tumor. Understanding this heterogeneity and why tumors are heterogenous is crucial to understand how cancer initiates and evolves, how cancer can be attacked and at the same time how cancer can become resistant to therapy. This heterogeneity may be explained by a branching evolutionary process driven by genetic variation (mainly fostered by genomic instability) and natural selection of the fittest variant driven by microenvironment conditions or external pressures such as therapy (Amirouchene-Angelozzi et al., 2017; Niida et al., 2021). The Darwinian principles of evolution and survival are the basis of tumor heterogeneity and clonal evolution, since the acquisition of different genetic and/or epigenetic alterations endows the tumor with greater survival capabilities, and the capacity to escape drug inhibition (Kreso et al., 2013; Amirouchene-Angelozzi et al., 2017).
Tumor heterogeneity can be studied by sequencing of different regions within a tumor and reconstructing the evolutionary dynamics or the history of a specific cancer, represented in what is called a phylogenetic tree (Siravegna et al., 2018). However, upon metastatic spread and after several lines of drug pressure, heterogeneity becomes greater, and heterogeneity is underrepresented by a single tumor re-biopsy (Gerlinger et al., 2012; Amirouchene-Angelozzi et al., 2017; Dang et al., 2020). A different approach to study tumoral molecular heterogeneity is the use of (serial) liquid biopsies, which are able to detect the genomic landscape shed into the bloodstream by the different subclones (spatial heterogeneity), in a minimally-invasive blood extraction which can be repeated as many times as necessary to track the evolving sub-clonal genetic abnormalities (temporal heterogeneity) (Van Emburgh et al., 2016; Amirouchene-Angelozzi et al., 2017; Dasari et al., 2020; Vidal et al., 2022).
3 Translational models to study acquired resistance to EGFR inhibitors
With the goal of finding the best treatment strategies to circumvent or prevent the emergence of acquired resistance to anti-EGFR therapy in the clinical setting, several preclinical studies have been conducted in the last decades to characterize the molecular drivers of acquired resistance to anti-EGFR therapy in CRC. In vitro and in vivo studies generally include the generation of resistant cells to anti-EGFR therapy by a long-time exposure of cetuximab or panitumumab sensitive cells, followed by molecular characterization of the resistant cells compared to paired initially sensitive cells, and ideally functional studies to confirm causality of the preclinical findings, as well as confirmation of the preclinical findings in tumor samples from patients treated with anti-EGFR therapy. While all studies share these general principles for the generation of drug-resistant cells, each study had its own specificities such as different cancer cell lines (GEO, SW48, DiFi, Lim-1215, CaCo2, NCIH508, OXCO and HCA-46, etc.), or the use of different treatment strategies to generate resistant cells (mainly continuous or increasing exposure of the cells to cetuximab or panitumumab), which may have led to identification of different mechanisms of acquired resistance. These translational models have unveiled a myriad of molecular mechanisms of acquired resistance to anti-EGFR therapy, including c-MET activation, mutations in the extracellular domain of EGFR, mutations in the RAS genes (KRAS or NRAS), KRAS amplification, PIK3CA mutation, ERBB2 amplification and overexpression of EGFR ligands (Ciardiello et al., 2004; Yonesaka et al., 2011; Misale et al., 2012; 2014b; Montagut et al., 2012; Troiani et al., 2013; Hobor et al., 2014; Arena et al., 2015). Table 1 presents different preclinical models of CRC used to study induced resistance to EGFR antibodies.
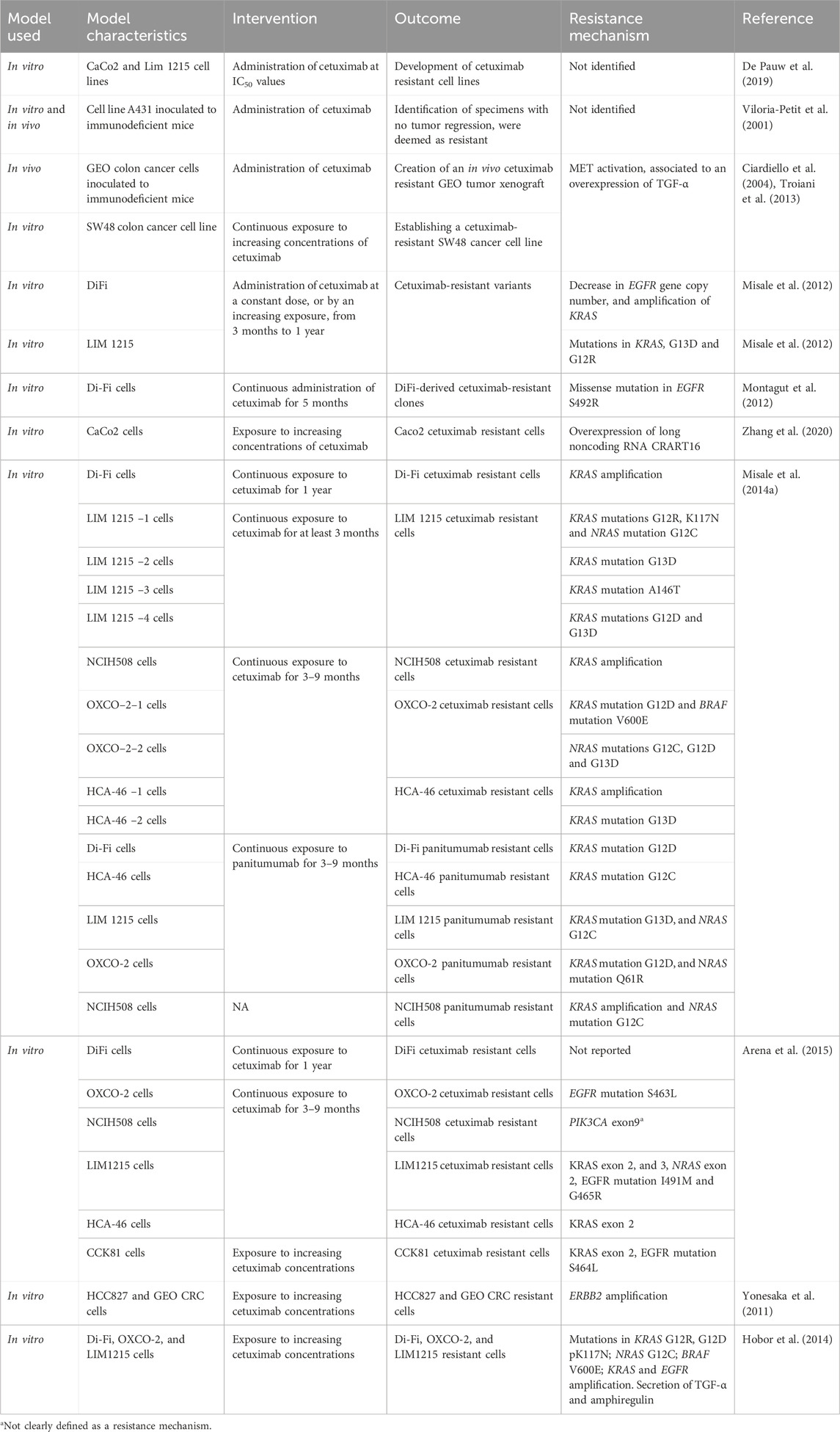
Table 1. Preclinical models to induce resistance to anti EGFR antibodies, and the resistance mechanism identified.
4 Genetic mechanisms of acquired resistance to anti-EGFR therapies
Overall, colorectal cancer cells evade EGFR blockade through two main strategies: (a) reactivation of the MAPK-ERK signaling pathway either by mutations in the pathway genes, alterations in alternative tyrosine-kinase receptors, or ligands overexpression (b) lack of binding of cetuximab/panitumumab to the receptor by mutations in the binding epitope located in the extracellular domain of EGFR (Misale et al., 2015; Siravegna et al., 2015). Notably, these mechanisms of resistance can coexist within one same tumor (Figure 1).

Figure 1. Molecular mechanisms of acquired resistance to anti-EGFR therapies.
4.1 RAS alterations
KRAS and NRAS belong to the RAS membrane-bound family proteins, they possess an inherent GTPase activity, and can activate different effector targets, such as the RAF-MAPK, and PI3K-ATK-mTOR pathways (Uprety and Adjei, 2020). Both KRAS mutations and amplifications, as well as NRAS mutations have been identified as mechanisms of resistance to cetuximab and panitumumab both in liquid biopsy and tumor biopsy specimens (Table 2). The development of these mutations following anti-EGFR targeted treatment, can be a consequence of alterations rising from pre-existent KRAS altered clones, or due to new mutations derived from stress conditions induced by targeted therapy to the tumor and tumor microenvironment. Furthermore, it has been reported that several alterations can coexist (Diaz Jr et al., 2012; Misale et al., 2012; Misale et al., 2014a).
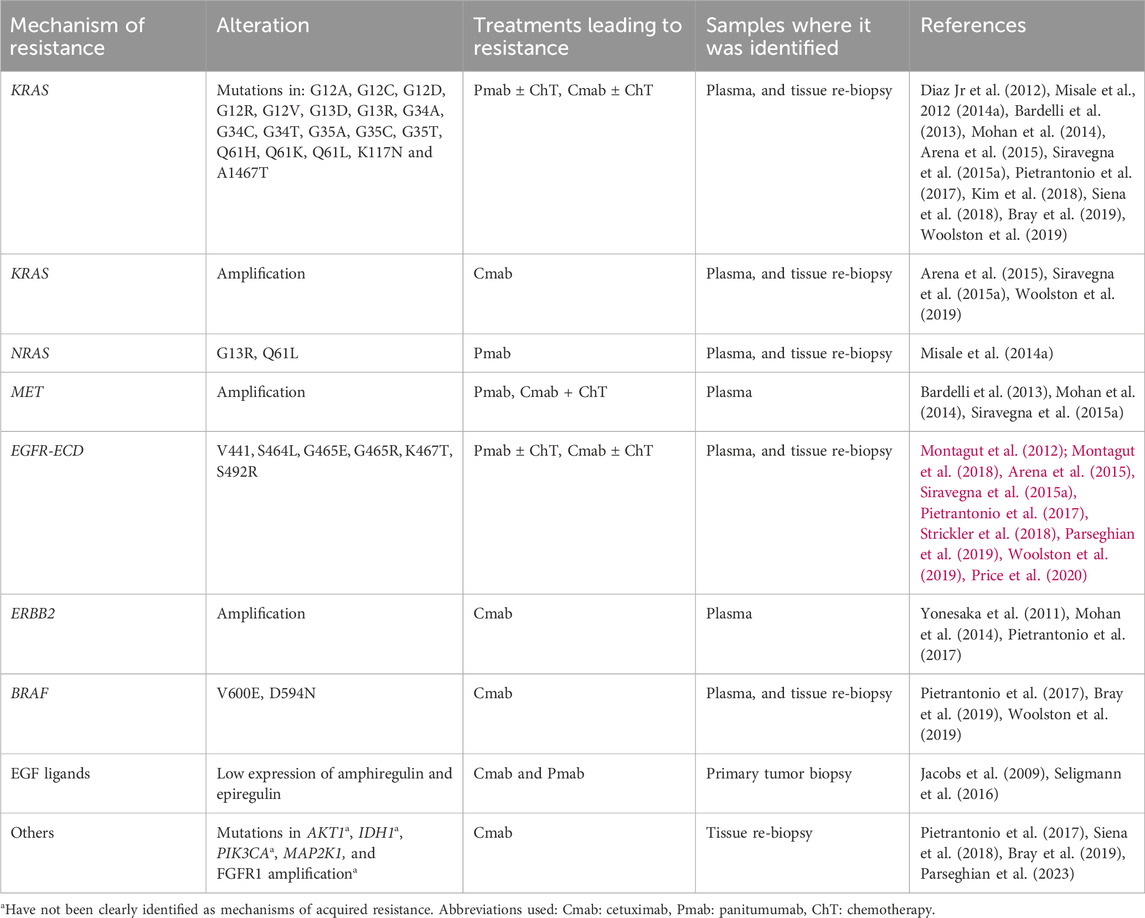
Table 2. Selected mechanisms of acquired resistance to anti-EGFR therapies.
4.2 BRAF mutations
BRAF belongs to the serine/threonine kinases RAF family, its downstream signaling consists of MEK one and two and ERK, leading to further phosphorylation of multiple molecules (Subbiah et al., 2020). Different mutations in the BRAF gene have been identified as acquired mechanisms of resistance to anti-EGFR inhibitors, such as V600E and D594N, which lead to a persistent activation on the downstream pathway of RAF-ERK (Pietrantonio et al., 2017; Bray et al., 2019; Woolston et al., 2019).
4.3 ERBB2 amplifications
HER2 belongs to the EGFR tyrosine kinase family, it presents the most potent catalytic kinase activity, and its phosphorylation leads to a downstream activation of the PI3K-AKT-mTOR, and MAPK pathways (Yan et al., 2015; Ríos-Hoyo et al., 2022). ERBB2 amplifications have been described as resistance mechanisms in plasma samples from patients with acquired resistance to cetuximab, detection in serum of the HER2/HER2 ECD was correlated to resistance to cetuximab at progression. Abnormal activation of HER2 signaling led to persistent ERK 1/2 signaling, induced by treatment with cetuximab (Yonesaka et al., 2011; Mohan et al., 2014).
4.4 Mutations in the EGFR-extracellular domain (ECD)
Our group identified mutations in the EGFR-ECD as a mechanism of resistance to anti-EGFR therapies, these mutations are located in domain III of EGFR, in the binding sites of cetuximab, thus impairing the drug-receptor interaction. The most frequent described EGFR-ECD mutations emerging during anti-EGFR therapy are V441, S464, G465, and S492 mutations (Montagut et al., 2018; Strickler et al., 2018). It is worth noting that because the binding epitopes of cetuximab and panitumumab do not fully overlap, some mutations confer resistance to cetuximab but not to panitumumab. This is the case of S492 mutation which does not affect the binding activity of panitumumab, whereas S464L, G465R and 1491M mutations do not allow the binding of neither cetuximab nor panitumumab to the receptor (Montagut et al., 2012; Arena et al., 2015; Price et al., 2020). In this sense, one patient with an S492 mutation after cetuximab treatment responded to treatment with panitumumab monotherapy (Montagut et al., 2012). Importantly, EGFR-ECD mutations have not been detected in untreated samples and therefore are thought to drive acquired resistance but not primary resistance. Interestingly, patients who develop mutations in the EGFR-ECD experience greater and more lasting tumor responses con anti-EGFR treatment, compare to patients who develop other mechanisms of resistance, such as RAS mutations (Van Emburgh et al., 2016). This data highlights that the absence of EGFR-ECD mutant clones in treatment naïve tumors confers an advantage in terms of the response to anti-EGFR treatment.
4.5 MET amplifications
The mesenchymal-epithelial transition factor (MET) serves as a transmembrane receptor tyrosine kinase, and it is usually activated by the binding of the hepatocyte growth factor ligands. MET activation further activates other signaling pathways including the RAS-ERK-MAPK, PI3K-AKT-mTOR, Wnt/β-catenin, and STAT pathways (Singh Raghav et al., 2012; Drilon et al., 2017). MET amplifications have been detected in plasma samples from patients with acquired resistance to anti-EGFR therapies, methods such as BEAMing and FISH have been used to confirm this finding. It has been suggested that anti-EGFR treatment elicits a selective pressure, and therefore an expansion of preexisting subclones with MET amplification. MET initiated signaling has been proposed as a mechanism to bypass the EGFR blockade (Bardelli et al., 2013).
5 Liquid biopsy to monitor clonal dynamics and track mechanisms of acquired resistance
The term liquid biopsy applied to oncology encompasses the isolation and analysis of tumor derived material in corporal fluids, such as circulating tumor cells, circulating tumor DNA (ctDNA), extracellular vesicles, miRNA, among others. ctDNA is released from tumors into bodily fluids, including blood, cerebrospinal fluid, saliva, pleural fluid, ascites and urine (Wan et al., 2017; Corcoran and Chabner, 2018; Heitzer et al., 2019). Liquid biopsy has been proposed as an exquisite tool to assess intratumor molecular heterogeneity, track clonal dynamics and detect emergent resistant subclones (Wan et al., 2017; Corcoran and Chabner, 2018; Heitzer et al., 2019) (Figure 2). ctDNA is able to comprehensively capture heterogeneity with a high sensitivity for subclones arising under drug pressure. Moreover, the ease-of-use and minimally-invasive procedure allow serial assessment of the genomic landscape to closely track emerging subclones of resistance. In mCRC, several cohort studies and retrospective analysis from clinical trials have shown the utility of liquid biopsy to monitor the genomic landscape and track the emergence of resistant clones in patients treated with anti-EGFR therapies (Diaz Jr et al., 2012; Misale et al., 2012; Siravegna et al., 2015a; Montagut et al., 2018; Vitiello et al., 2019; Dasari et al., 2020; Vidal et al., 2022).
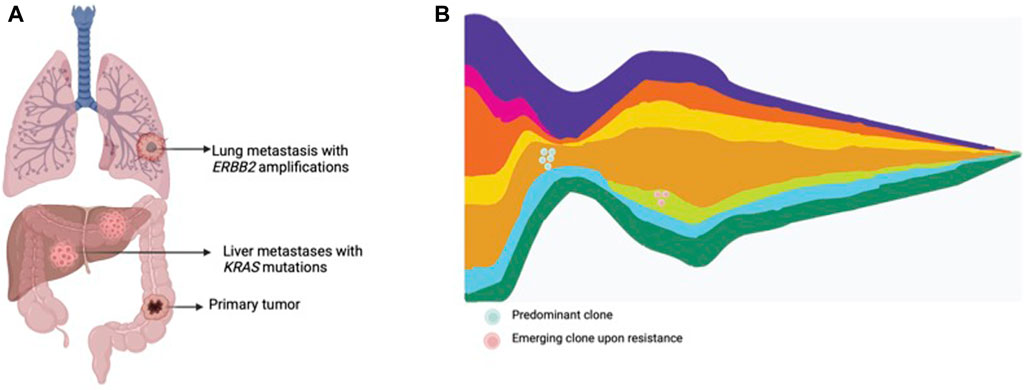
Figure 2. Tumor heterogeneity and clonal dynamics in metastatic CRC. (A) Multiple metastatic lesions showing the presence of various subclones within the tumor, each with different genomic alterations. (B) Clonal dynamics representing an original dominant clone, responding to treatment, and the rise of other treatment resistant clones.
In 2012, the first two studies to show the utility of liquid biopsy in detecting the emergence of RAS mutations during treatment with anti-EGFR therapy were concomitantly published (Diaz et al., 2012; Misale et al., 2012). That same year, our group identified the emergence of mutations of acquired resistance in the EGFR extracellular domain during anti-EGFR therapy, which later were also detected in ctDNA. In the following years, other mutations of resistance were detected in ctDNA, including mutations in BRAF and MAP2K1 (Misale et al., 2014b; Siravegna et al., 2015a). Interestingly, using liquid biopsy, our group in collaboration with Bardelli’s group was able to show that not all mutations are the same in regard to treatment response and duration of response. In 27 patients with mCRC, RAS, EGFR-ECD and co-occurrence of both mutations were detected in 20, 14 and 7 cases respectively at the time of progression to anti-EGFR therapy. Interestingly, RAS mutations were mostly detected in patients who presented stable disease as best response with a shorter duration of response (mPFS of 25.6 weeks), compared to EGFR-ECD mutations which were more frequently detected in patients achieving a higher decrease in tumor size (partial response) and a longer duration of response (mPFS of 44.6 weeks). Moreover, in vitro studies supported the same concept that RAS mutations emerge earlier during anti-EGFR therapy than EGFR-ECD mutations (Van Emburgh et al., 2016). It is important to highlight, that multiple mutations of acquired resistance usually co-exist within one same patient after treatment with anti-EGFR therapy, as a consequence of the selection of several clones of resistance (Pietrantonio et al., 2017; Montagut et al., 2018; Strickler et al., 2018) Interestingly, mutation upsurge/emergence of multiple subclones anticipates a remarkable clinical deterioration, especially when EGFR-ECD mutations emerge (Toledo et al., 2017; Montagut et al., 2018). Therefore, it may be extremely challenging to pharmacologically target the complex molecular heterogeneity associated with emergence of resistance to cetuximab/panitumumab in mCRC patients.
The use of serial liquid biopsies to track mutations of resistance has showed a decrease in RAS and EGFR-ECD mutations upon withdrawal of anti-EGFR therapy. Siravegna et al. reported the first study to prove this concept in mCRC patients, in whom KRAS mutant alleles, EGFR-ECD mutations, and MET amplifications detected in ctDNA upon progression to anti-EGFR drugs, diminished and were undetectable several months after finishing anti-EGFR therapy. The intermittent detection of KRAS mutant clones in blood of patients treated with anti-EGFR therapies, supports the concept that CRC cells possess an outstanding plasticity (Siravegna et al., 2015a). A similar study showed that the exponential decay of RAS and EGFR-ECD mutant allele frequency presented a median of 3.4 and 6.9 months, respectively. (Parseghian et al., 2019). Altogether, decay and absence of detection of subclones of resistance few months upon withdrawal of EGFR blockade sets a strong biological rationale for testing clinical strategies of rechallenge with anti-EGFR therapy in mCRC patients.
6 Mechanisms of acquired resistance beyond genomics
So far, most studies have focused on genomic alterations as the drivers of acquired resistance to anti-EGFR therapy. However, genomic alterations in EGFR and the MAPKs pathway occur in less than 50% of tumors progressing to anti-EGFR therapy, and recent data show that this percentage is even lower in mCRC patients treated with anti-EGFR therapy plus chemotherapy in the first-line setting. This has led to explore alternative mechanisms of acquired resistance. In an effort to identify novel biomarkers of resistance, transcriptomic profiles from three clinical and two preclinical cohorts treated with cetuximab were used to assign consensus molecular subtypes (CMS) and found excellent responses to cetuximab in CMS2 tumors, independently of primary tumor laterality (Parseghian et al., 2023). Conversely, resistance to anti-EGFR therapy was associated with a transition from CMS2 tumors to CMS4 tumors, characterized by mesenchymal infiltration (Woolston et al., 2019). In this sense, preclinical modeling demonstrated that acquired resistance to either cetuximab or chemotherapy was a result of cross-resistant transcriptomic profiles consistent with epithelial-to-mesenchymal transition. In addition, recent preclinical studies have suggested that anti-EGFR resistance may be driven by cancer associated fibroblasts populating the tumor microenvironment, and their secreted factors (Woolston et al., 2019; Garvey et al., 2020). More recently, data on patients treated in first line and in combination with chemotherapy has revealed novel data and a vastly different profile of mechanisms of resistance to anti-EGFR therapy. Biomarker analysis of the CALBG/SWOG-80405 trial evaluated the development of acquired mechanisms of resistance to anti-EGFR inhibitors using liquid biopsy in patients with metastatic CRC who received a first line treatment with chemotherapy (FOLFOX or FOLFIRI) and cetuximab (n = 61), or chemotherapy (FOLFOX or FOLFIRI) and bevacizumab (n = 69). The authors reported emergence of 6.6% and 10.1% genomic alterations of anti-EGFR resistance in ctDNA at the time of progression to cetuximab and bevacizumab, respectively. Among the reported genomic alterations, mutations in KRAS, NRAS, BRAF, EGFR-ECD and amplifications in ERBB2 and MET were reported (Raghav et al., 2023). Parseghian et al. retrospectively analyzed paired ctDNA samples before and after anti-EGFR therapy from three different trials and also demonstrated unique molecular patterns of resistance between first-line and later-line anti-EGFR therapies (Parseghian et al., 2023). Similarly, our group analyzed serial ctDNA samples of patients treated with cetuximab plus chemotherapy in first-line within the PLATFORM-B study, and found that in five out of nine patients with RAS/BRAF subclones emerging early (cycle 2) during anti-EGFR plus chemotherapy did not expanded (Vidal et al., 2023). Altogether, these studies suggest that chemotherapy-based multi-drug treatment may favor a specific resistance profile that may include additional mechanisms of resistance (transcriptomic, epigenetic, tumor-microenvironment-derived factors) rather than genomic-driven resistance to the anti-EGFR component of the regimen. Therefore, the use of liquid biopsy to also detect non-genomic alterations of the tumor could provide a comprehensive understanding of tumor evolution during the course of treatment. In addition, understanding the complexity of mechanisms of resistance beyond point mutations of driver genes in cancer cells is crucial to design future successful combination regimens.
7 Clinical strategies to overcome resistance
Different clinical strategies have been proposed to prevent or circumvent acquired resistance to anti-EGFR therapies. Targeting mutations of resistance is potentially limited by the complex heterogeneity of coexisting subclones of resistance. Another treatment strategy is to take advantage of clonal dynamics and rechallenge with anti-EGFR drugs after a wash-off period and decay of mutations of resistance in ctDNA. Rechallenge refers to the concept of re-treating with anti-EGFR therapy in patients who previously derived a benefit from this drug (Mauri et al., 2019; Martinelli et al., 2020; Mauri et al., 2022) (Figure 3).
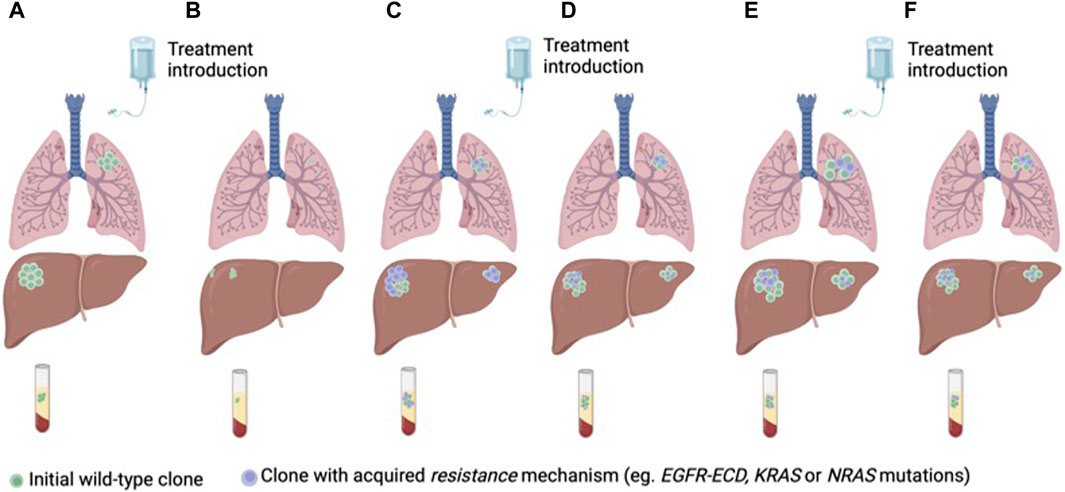
Figure 3. Anti-EGFR treatment of RAS/BRAF wt mCRC according to clonal dynamics assessed in ctDNA (A) Treatment naïve CRC with a predominant clone of anti-EGFR sensitive cancer cells (B) Tumor response to first-line treatment with anti-EGFR therapy. (C) Tumor progression and emergence of sub-clones of resistance to anti-EGFR therapy. (D) Decay of the anti-EGFR resistant clones with second-line treatment without anti-EGFR therapy. (E) Tumor progression. (F) Tumor response to third-line anti-EGFR rechallenge.
Several small phase II clinical trials assessing the efficacy of rechallenge with anti-EGFR therapies have been conducted. In common, all trials include patients that previously responded to anti-EGFR therapy, followed by a subsequent treatment with no anti-EGFR treatment. The first study assessing re-challenge was conducted more than a decade ago and included 39 patients with KRAS WT (codons 12 and 13) metastatic CRC re-treated with cetuximab plus irinotecan in third-line. The ORR was 53.8%, and the mPFS 6.6 months (Santini et al., 2012). Following this study, the CRICKET trial was a single-arm phase II study that included 28 patients with metastatic tissue RAS/BRAF WT mCRC who previously benefited for at least 6 months of irinotecan-based chemotherapy and cetuximab. The trial aimed to evaluate the activity of these compounds in the third-line setting, and achieved an ORR of 21%, mPFS 3.4 months, and mOS 9.8 months. Importantly, for the first time, the use of ctDNA to select for anti-EGFR rechallenge was retrospectively analyzed. Patients with baseline (before rechallenge) ctDNA RAS WT achieved a partial response in 57% of the cases, and had a longer mPFS compared to patients with mutations in RAS detected in ctDNA (4 vs. 1.9 months, respectively) (Cremolini et al., 2019). The JACCRO CC-08 trial also evaluated the efficacy of irinotecan plus cetuximab rechallenge in the third-line setting in 34 patients with KRAS WT mCRC. One patient achieved a partial response, the disease control rate (DCR) was 55.9%, the mPFS was 2.4 months, and the mOS was 8.2 months. In an attempt to find clinical surrogate markers of clinical benefit, the authors identified that patients with a longer cetuximab free interval (CFI), derived a greater benefit from the rechallenge strategy, as opposed to patients with a short CFI (DCR of 82% vs. 44%, mPFS of 4.6 vs. 2.1 months, and mOS of 14.1 and 6.3 months, respectively for the long and short CFI) (Masuishi et al., 2020). The VELO clinical trial was a randomized trial that evaluated rechallenge with panitumumab plus trifluridine-tipiracil (also known as TAS102) vs. trifluridine-tipiracil alone as control arm. The study included 62 patients with chemo-refractory tissue RAS WT mCRC and was positive in favor of the rechallenge strategy (mPFS4 and 2.5 months in panitumumab + trifluridine-tipiracil vs. trifluridine-tipiracil, respectively). Retrospective analysis of baseline ctDNA RAS/BRAF WT identified patients obtaining prolonged clinical benefit with panitumumab plus trifluridine-tipiracil compared with trifluridine-tipiracil (PFS rates at 6months 38.5% vs. 13.0% and at 12 months 15.4% vs. 0%). Interestingly, extended ctDNA hyperselection (WT for KRAS, NRAS, BRAFV600E, EGFR ECD, ERBB2, MAP2K1, andPIK3CA) selected patients with a mPFS of 6.4 months, partial response of 13.3% and stable disease of 73.3% (Napolitano et al., 2023). A chemotherapy-free treatment strategy was evaluated in the CAVE trial, a single arm phase II trial that included 77 patients with tissue RAS WT mCRC patients rechallenged with cetuximab plus the anti-PD-L1 drug avelumab. The ORR was 7.8%, DCR was 65%, mPFS was 3.6 months, and mOS was 11.6 months. Post-hoc analysis of baseline ctDNA revealed, that patients with RAS/BRAF WT ctDNA had a better survival than patients with mutated RAS/BRAF (mPFS of 4.1 vs. 3 months, and a mOS of 17.3 vs. 10.4 months, respectively). At progression to rechallenge, ctDNA detected KRAS/BRAF and EGFR-ECD S292R mutations as mechanisms of resistance (Martinelli et al., 2021)
The CHRONOS clinical trial was the first trial to include genomic selection by ctDNA as an inclusion criterion. Moreover, the trial used a clean design in which panitumumab rechallenge was administered alone to evaluate the effect of anti-EGFR treatment without the effect of concomitant chemotherapy. Panitumumab was administered in 27 patients with RAS/BRAF and EGFR ECD WT in ctDNA (mutation zero). The study achieved it primary endpoint, with an ORR of 30%, a DCR of 63%, with a median duration of response of 17 weeks, a mPFS of 16 weeks, and a mOS of 55 weeks. Following panitumumab rechallenge, ctDNA identified different resistance mechanisms including mutations or amplifications in KRAS, NRAS, EGFR, PTEN, and MET, 48% of the patients had at least two co-occurring mechanisms of resistance (Sartore-Bianchi et al., 2021; Sartore-Bianchi et al., 2022). The ongoing CITRIC trial (EudraCT 2020-000443-31) is the first randomized clinical trial aimed to evaluate the efficacy of cetuximab plus irinotecan rechallenge in the third-line setting in comparison to standard treatment at investigator’s choice in patients genomically selected with no detection of mutations of acquired resistance (RAS, BRAF and EGFR-ECD wild-type) in the blood of patients before rechallenge. Recruitment was recently completed. Table 3 presents different completed and ongoing rechallenge strategies.
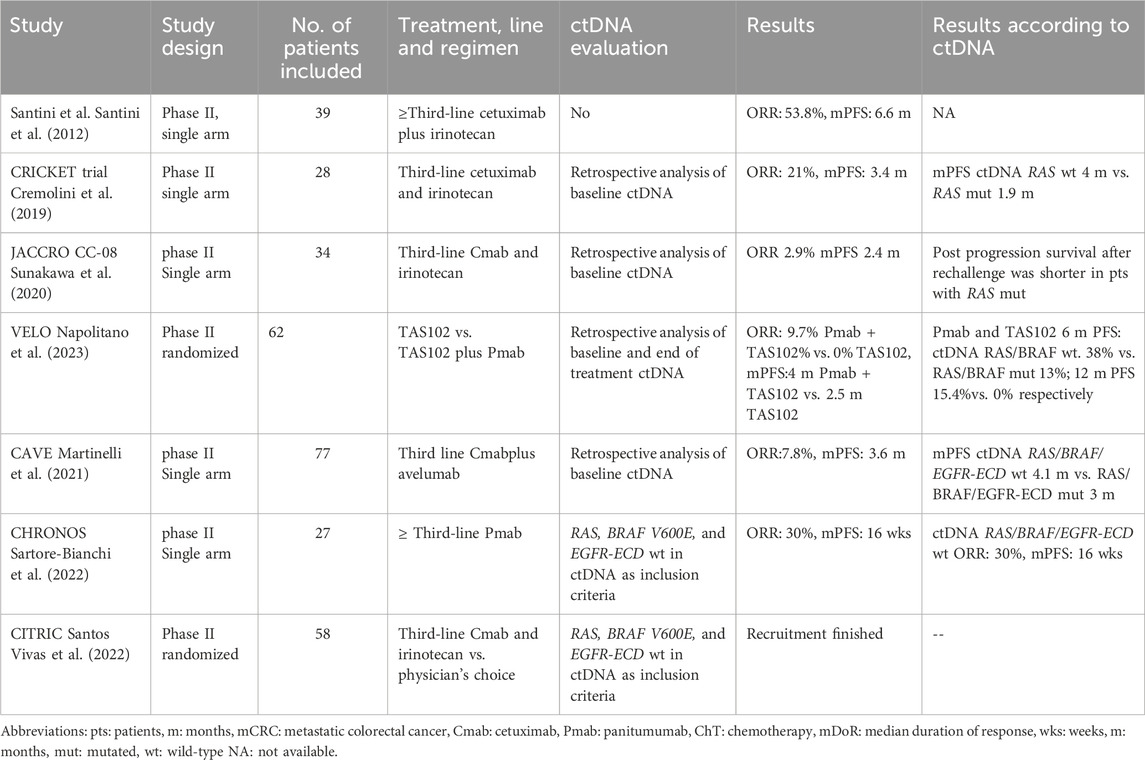
Table 3. Clinical trials using rechallenge strategies with anti-EGFR therapies in patients with colorectal cancer and ctDNA evaluation.
In a different approach, the Sym004-005 clinical trial evaluated the use of Sym004, a mixture of two synergistic antibodies, futuximab and modotuximab, directed against nonoverlapping epitopes in EGFR, leading to internalization and degradation of the receptor (Sánchez-Martín et al., 2016). A phase II clinical trial evaluated the use of two regimens of Sym004 (higher dose: arm A, lower dose: arm B), compared to chemotherapy (arm C). The study included 254 patients with KRAS exon 2 WT mCRC who were refractory to standard chemotherapy and had acquired resistance to anti-EGFR therapies. The mOS was 7.9, 10.3 and 9.6 months for arms A, B and C, respectively. A preplanned retrospective analysis of patients with no detection of mutations in RAS, BRAF and EGFR ECD in ctDNA, showed a dramatic statistically significant improvement in mOS for treatment with low-dose Sym004 (12.8 vs. 7.3 for the control arm). Again, this study shows the necessity of ctDNA genomic analysis to select patients that benefit from anti-EGFR therapy (Montagut et al., 2018).
8 Conclusion
Therapeutic anti-EGFR moAbs (cetuximab and panitumumab) remain the mainstay of targeted therapy in RAS/BRAF wild-type metastatic colorectal cancer. However, resistance eventually develops leading to cancer progression. In the last decade, preclinical and translational models have identified two main strategies for colorectal cancer cells to evade EGFR inhibition: reactivation of the MAPK pathway and mutations in the extracellular domain of EGFR (EGFR ECD). These genomic alterations arise as a consequence of heterogeneity and clonal selection under drug pressure. Interestingly, liquid biopsy (i.e., genotyping of ctDNA) is a minimally invasive method to track genomic alterations of resistance in the blood of patients treated with cetuximab/panitumumab. Treatment of resistance to anti-EGFR therapies remains a challenge, since genomic alterations of resistance are multiple and coexist within one same tumor. Because mutations of acquired resistance decline over time following anti-EGFR withdrawal, an alternative strategy that is showing promising results in several phase II clinical trials is to rechallenge with anti-EGFR therapy in patients selected by no detection of mutations of acquired resistance in liquid biopsy. More recently, alternative mechanisms of resistance beyond genomics, mainly related to the tumor microenvironment, have been identified, specifically in patients treated with chemotherapy-based multi-drug treatment in first line of treatment (vs. anti-EGFR single treatment in heavily pretreated patients). In the era of personalized medicine, it is of the utmost importance to better understand the complexity of the mechanisms of acquired resistance to anti-EGFR therapy to be able to design appropriate clinical trials and ultimately improve treatment and care of mCRC patients.
Author contributions
AR-H: Conceptualization, Data curation, Formal Analysis, Investigation, Project administration, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. XM: Formal Analysis, Methodology, Visualization, Writing–review and editing. JV: Formal Analysis, Methodology, Visualization, Writing–review and editing. JL: Formal Analysis, Methodology, Visualization, Writing–review and editing. CM: Conceptualization, Data curation, Formal Analysis, Supervision, Validation, Visualization, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. AR-H is supported by a grant from the Spanish Society of Medical Oncology (SEOM). XM is supported by a Río Hortega contract, ISCIII (Instituto de Salut Carlos III). JL is the recipient of a Junior Clinician fellowship from Spanish Association Against Cancer (AECC) (CLJUN19004LINA). CM is supported by grants by CRIS EXCELLENCE 19-30, funded by CRIS Contra el Cáncer.
Acknowledgments
Figures created using BioRender.com
Conflict of interest
AR-H reports traveling accommodations from Novartis and Roche. CM reports scientific consultancy role, travel grants or research grants past 5 years from Amgen, Biocartis, BMS, Guardant-Health, Merck-Serono, Roche, Sanofi Aventis, and SeaGen. JV reports personal fees and other support from Merck and personal fees from Amgen, Sanofi, and BMS outside the submitted work.
The remaining authors declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amado, R. G., Wolf, M., Peeters, M., Van Cutsem, E., Siena, S., Freeman, D. J., et al. (2008). Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 1626–1634. doi:10.1200/JCO.2007.14.7116
Amirouchene-Angelozzi, N., Swanton, C., and Bardelli, A. (2017). Tumor evolution as a therapeutic target. Cancer Discov. 7, 805–817. doi:10.1158/2159-8290.CD-17-0343
Arena, S., Bellosillo, B., Siravegna, G., Martínez, A., Cañadas, I., Lazzari, L., et al. (2015). Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin. Cancer Res. 21, 2157–2166. doi:10.1158/1078-0432.CCR-14-2821
Bardelli, A., Corso, S., Bertotti, A., Hobor, S., Valtorta, E., Siravegna, G., et al. (2013). Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 3, 658–673. doi:10.1158/2159-8290.CD-12-0558
Benson, A. B., Venook, A. P., Al-Hawary, M. M., Azad, N., Chen, Y.-J., K, C. K., et al. (2022). NCCN clinical practice guidelines in oncology (NCCN Guidelines®). Rectal Cancer. Version 3.2022.
Biller, L. H., and Schrag, D. (2021). Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA 325, 669–685. doi:10.1001/jama.2021.0106
Bokemeyer, C., Bondarenko, I., Makhson, A., Hartmann, J. T., Aparicio, J., de Braud, F., et al. (2009). Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 27, 663–671. doi:10.1200/JCO.2008.20.8397
Bokemeyer, C., Köhne, C.-H., Ciardiello, F., Lenz, H.-J., Heinemann, V., Klinkhardt, U., et al. (2015). FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 51, 1243–1252. doi:10.1016/j.ejca.2015.04.007
Bray, S. M., Lee, J., Kim, S. T., Hur, J. Y., Ebert, P. J., Calley, J. N., et al. (2019). Genomic characterization of intrinsic and acquired resistance to cetuximab in colorectal cancer patients. Sci. Rep. 9, 15365. doi:10.1038/s41598-019-51981-5
Cervantes, A., Adam, R., Roselló, S., Arnold, D., Normanno, N., Taïeb, J., et al. (2022). Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 141604, 10–32. doi:10.1016/j.annonc.2022.10.003
Ciardiello, F., Bianco, R., Caputo, R., Caputo, R., Damiano, V., Troiani, T., et al. (2004). Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin. Cancer Res. 10, 784–793. doi:10.1158/1078-0432.CCR-1100-03
Corcoran, R. B., and Chabner, B. A. (2018). Application of cell-free DNA analysis to cancer treatment. N. Engl. J. Med. 379, 1754–1765. doi:10.1056/NEJMra1706174
Cremolini, C., Rossini, D., Dell’Aquila, E., Lonardi, S., Conca, E., Del Re, M., et al. (2019). Rechallenge for patients with RAS and BRAF wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: a phase 2 single-arm clinical trial. JAMA Oncol. 5, 343–350. doi:10.1001/jamaoncol.2018.5080
Dang, H. X., Krasnick, B. A., White, B. S., Grossman, J. G., Strand, M. S., Zhang, J., et al. (2020). The clonal evolution of metastatic colorectal cancer. Sci. Adv. 6, eaay9691. doi:10.1126/sciadv.aay9691
Dasari, A., Morris, V. K., Allegra, C. J., Atreya, C., Benson, A. B., Boland, P., et al. (2020). ctDNA applications and integration in colorectal cancer: an NCI Colon and Rectal–Anal Task Forces whitepaper. Nat. Rev. Clin. Oncol. 17, 757–770. doi:10.1038/s41571-020-0392-0
De Pauw, I., Lardon, F., Van den Bossche, J., Baysal, H., Pauwels, P., Peeters, M., et al. (2019). Overcoming intrinsic and acquired cetuximab resistance in RAS wild-type colorectal cancer: an in vitro study on the expression of HER receptors and the potential of afatinib. Cancers (Basel) 11, 98. doi:10.3390/cancers11010098
Diaz, L. A., Williams, R. T., Wu, J., Kinde, I., Hecht, J. R., Berlin, J., et al. (2012). The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486, 537–540. doi:10.1038/nature11219
Drilon, A., Cappuzzo, F., Ou, S.-H. I., and Camidge, D. R. (2017). Targeting MET in lung cancer: will expectations finally Be MET? J. Thorac. Oncol. 12, 15–26. doi:10.1016/j.jtho.2016.10.014
Garvey, C. M., Lau, R., Sanchez, A., Sun, R. X., Fong, E. J., Doche, M. E., et al. (2020). Anti-EGFR therapy induces EGF secretion by cancer-associated fibroblasts to confer colorectal cancer chemoresistance. Cancers (Basel) 12, 1393. doi:10.3390/cancers12061393
Gerlinger, M., Rowan, A. J., Horswell, S., Larkin, J., Endesfelder, D., Gronroos, E., et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892. doi:10.1056/NEJMoa1113205
Heitzer, E., Haque, I. S., Roberts, C. E. S., and Speicher, M. R. (2019). Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 20, 71–88. doi:10.1038/s41576-018-0071-5
Hobor, S., Van Emburgh, B. O., Crowley, E., Misale, S., Di Nicolantonio, F., and Bardelli, A. (2014). TGFα and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin. Cancer Res. 20, 6429–6438. doi:10.1158/1078-0432.CCR-14-0774
Jacobs, B., De Roock, W., Piessevaux, H., Van Oirbeek, R., Biesmans, B., De Schutter, J., et al. (2009). Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J. Clin. Oncol. 27, 5068–5074. doi:10.1200/JCO.2008.21.3744
Jonker, D. J., O’Callaghan, C. J., Karapetis, C., Zalcberg, J. R., Tu, D., Au, H. J., et al. (2007). Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 357, 2040–2048. doi:10.1056/NEJMoa071834
Karapetis, C. S., Khambata-Ford, S., Jonker, D. J., O’Callaghan, C. J., Tu, D., Tebbutt, N. C., et al. (2008). K-Ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 359, 1757–1765. doi:10.1056/nejmoa0804385
Kim, T. W., Peeters, M., Thomas, A., Gibbs, P., Hool, K., Zhang, J., et al. (2018). Impact of emergent circulating tumor DNA RAS mutation in panitumumab-treated chemoresistant metastatic colorectal cancer. Clin. Cancer Res. 24, 5602–5609. doi:10.1158/1078-0432.CCR-17-3377
Kreso, A., O’Brien, C. A., van Galen, P., Gan, O. I., Notta, F., Brown, A. M. K., et al. (2013). Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 339, 543–548. doi:10.1126/science.1227670
Martinelli, E., Ciardiello, D., Martini, G., Troiani, T., Cardone, C., Vitiello, P. P., et al. (2020). Implementing anti-epidermal growth factor receptor (EGFR) therapy in metastatic colorectal cancer: challenges and future perspectives. Ann. Oncol. 31, 30–40. doi:10.1016/j.annonc.2019.10.007
Martinelli, E., Martini, G., Famiglietti, V., Troiani, T., Napolitano, S., Pietrantonio, F., et al. (2021). Cetuximab rechallenge plus avelumab in pretreated patients with RAS wild-type metastatic colorectal cancer: the phase 2 single-arm clinical CAVE trial. JAMA Oncol. 7, 1529–1535. doi:10.1001/jamaoncol.2021.2915
Masuishi, T., Tsuji, A., Kotaka, M., Nakamura, M., Kochi, M., Takagane, A., et al. (2020). Phase 2 study of irinotecan plus cetuximab rechallenge as third-line treatment in KRAS wild-type metastatic colorectal cancer: JACCRO CC-08. Br. J. Cancer 123, 1490–1495. doi:10.1038/s41416-020-01042-w
Mauri, G., Pizzutilo, E. G., Amatu, A., Bencardino, K., Palmeri, L., Bonazzina, E. F., et al. (2019). Retreatment with anti-EGFR monoclonal antibodies in metastatic colorectal cancer: systematic review of different strategies. Cancer Treat. Rev. 73, 41–53. doi:10.1016/j.ctrv.2018.12.006
Mauri, G., Vitiello, P. P., Sogari, A., Crisafulli, G., Sartore-Bianchi, A., Marsoni, S., et al. (2022). Liquid biopsies to monitor and direct cancer treatment in colorectal cancer. Br. J. Cancer 127, 394–407. doi:10.1038/s41416-022-01769-8
Mendelsohn, J., Prewett, M., Rockwell, P., and Goldstein, N. I. (2015). CCR 20th anniversary commentary: a chimeric antibody, C225, inhibits EGFR activation and tumor growth. Clin. Cancer Res. 21, 227–229. doi:10.1158/1078-0432.CCR-14-2491
Misale, S., Arena, S., Lamba, S., Siravegna, G., Lallo, A., Hobor, S., et al. (2014a). Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to Anti-EGFR therapies in colorectal cancer. Sci. Transl. Med. 6, 224ra26–11. doi:10.1126/scitranslmed.3007947
Misale, S., Bozic, I., Tong, J., Peraza-Penton, A., Lallo, A., Baldi, F., et al. (2015). Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat. Commun. 6, 8305. doi:10.1038/ncomms9305
Misale, S., Di Nicolantonio, F., Sartore-Bianchi, A., Siena, S., and Bardelli, A. (2014b). Resistance to Anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov. 4, 1269–1280. doi:10.1158/2159-8290.CD-14-0462
Misale, S., Yaeger, R., Hobor, S., Scala, E., Janakiraman, M., Liska, D., et al. (2012). Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486, 532–536. doi:10.1038/nature11156
Mohan, S., Heitzer, E., Ulz, P., Lafer, I., Lax, S., Auer, M., et al. (2014). Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet. 10, e1004271. doi:10.1371/journal.pgen.1004271
Montagut, C., Argilés, G., Ciardiello, F., Poulsen, T. T., Dienstmann, R., Kragh, M., et al. (2018). Efficacy of Sym004 in patients with metastatic colorectal cancer with acquired resistance to anti-EGFR therapy and molecularly selected by circulating tumor DNA analyses a phase 2 randomized clinical trial. JAMA Oncol. 4, e175245–e175249. doi:10.1001/jamaoncol.2017.5245
Montagut, C., Dalmases, A., Bellosillo, B., Crespo, M., Pairet, S., Iglesias, M., et al. (2012). Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 18, 221–223. doi:10.1038/nm.2609
Morgan, E., Arnold, M., Gini, A., Lorenzoni, V., Cabasag, C. J., Laversanne, M., et al. (2022). Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut 72, 338–344. gutjnl-2022-327736. doi:10.1136/gutjnl-2022-327736
Napolitano, S., De Falco, V., Martini, G., Esposito, L., Famiglietti, V., Martinelli, E., et al. (2023). Panitumumab plus trifluridine/tipiracil as third-line anti-EGFR rechallenge therapy in chemo-refractory RAS WT metastatic colorectal cancer: the VELO randomized phase II clinical trial. J. Clin. Oncol. 41, 129. doi:10.1200/JCO.2023.41.4_suppl.129
Niida, A., Mimori, K., Shibata, T., and Miyano, S. (2021). Modeling colorectal cancer evolution. J. Hum. Genet. 66, 869–878. doi:10.1038/s10038-021-00930-0
Parseghian, C. M., Loree, J. M., Morris, V. K., Liu, X., Clifton, K. K., Napolitano, S., et al. (2019). Anti-EGFR-resistant clones decay exponentially after progression: implications for anti-EGFR re-challenge. Ann. Oncol. 30, 243–249. doi:10.1093/annonc/mdy509
Parseghian, C. M., Sun, R., Woods, M., Napolitano, S., Lee, H. M., Alshenaifi, J., et al. (2023). Resistance mechanisms to anti–epidermal growth factor receptor therapy in RAS/RAF wild-type colorectal cancer vary by regimen and line of therapy. J. Clin. Oncol. 41, 460–471. doi:10.1200/JCO.22.01423
Pietrantonio, F., Vernieri, C., Siravegna, G., Mennitto, A., Berenato, R., Perrone, F., et al. (2017). Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin. Cancer Res. 23, 2414–2422. doi:10.1158/1078-0432.CCR-16-1863
Price, T., Ang, A., Boedigheimer, M., Kim, T. W., Li, J., Cascinu, S., et al. (2020). Frequency of S492R mutations in the epidermal growth factor receptor: analysis of plasma DNA from patients with metastatic colorectal cancer treated with panitumumab or cetuximab monotherapy. Cancer Biol. Ther. 21, 891–898. doi:10.1080/15384047.2020.1798695
Raghav, K., Ou, F.-S., Venook, A. P., Innocenti, F., Sun, R., Lenz, H.-J., et al. (2023). Acquired genomic alterations on first-line chemotherapy with cetuximab in advanced colorectal cancer: circulating tumor DNA analysis of the CALGB/SWOG-80405 trial (alliance). J. Clin. Oncol. 41, 472–478. doi:10.1200/JCO.22.00365
Ríos-Hoyo, A., Moliner, L., and Arriola, E. (2022). Acquired mechanisms of resistance to osimertinib—the next challenge. Cancers (Basel) 14, 1931. doi:10.3390/cancers14081931
Sánchez-Martín, F. J., Bellosillo, B., Gelabert-Baldrich, M., Dalmases, A., Cañadas, I., Vidal, J., et al. (2016). The first-in-class anti-EGFR antibody mixture Sym004 overcomes cetuximab resistance mediated by EGFR extracellular domain mutations in colorectal cancer. Clin. Cancer Res. 22, 3260–3267. doi:10.1158/1078-0432.CCR-15-2400
Santini, D., Vincenzi, B., Addeo, R., Garufi, C., Masi, G., Scartozzi, M., et al. (2012). Cetuximab rechallenge in metastatic colorectal cancer patients: how to come away from acquired resistance? Ann. Oncol. 23, 2313–2318. doi:10.1093/annonc/mdr623
Santos Vivas, C., Salva, F., Fernández-Rodríguez, C., Alonso Orduña, V., Losa, F., Paez, D., et al. (2022). P-118 Cetuximab rechallenge in RAS, BRAF, EGFR-ECD wild type metastatic colorectal cancer (mCRC) patients treated with anti-EGFR therapies in first line: the CITRIC study. Ann. Oncol. 33, S291. doi:10.1016/j.annonc.2022.04.208
Sartore-Bianchi, A., Pietrantonio, F., Lonardi, S., Mussolin, B., Rua, F., Crisafulli, G., et al. (2022). Circulating tumor DNA to guide rechallenge with panitumumab in metastatic colorectal cancer: the phase 2 CHRONOS trial. Nat. Med. 28, 1612–1618. doi:10.1038/s41591-022-01886-0
Sartore-Bianchi, A., Pietrantonio, F., Lonardi, S., Mussolin, B., Rua, F., Fenocchio, E., et al. (2021). Phase II study of anti-EGFR rechallenge therapy with panitumumab driven by circulating tumor DNA molecular selection in metastatic colorectal cancer: the CHRONOS trial. J. Clin. Oncol. 39, 3506. doi:10.1200/JCO.2021.39.15_suppl.3506
Seligmann, J. F., Elliott, F., Richman, S. D., Jacobs, B., Hemmings, G., Brown, S., et al. (2016). Combined epiregulin and amphiregulin expression levels as a predictive biomarker for panitumumab therapy benefit or lack of benefit in patients with RAS wild-type advanced colorectal cancer. JAMA Oncol. 2, 633–642. doi:10.1001/jamaoncol.2015.6065
Siegel, R. L., Wagle, N. S., Cercek, A., Smith, R. A., and Jemal, A. (2023). Colorectal cancer statistics, 2023. Ca. Cancer J. Clin. 73, 233–254. doi:10.3322/caac.21772
Siena, S., Sartore-Bianchi, A., Garcia-Carbonero, R., Karthaus, M., Smith, D., Tabernero, J., et al. (2018). Dynamic molecular analysis and clinical correlates of tumor evolution within a phase II trial of panitumumab-based therapy in metastatic colorectal cancer. Ann. Oncol. 29, 119–126. doi:10.1093/annonc/mdx504
Singh Raghav, K. P., Gonzalez-Angulo, A. M., and Blumenschein, G. R. (2012). Role of HGF/MET axis in resistance of lung cancer to contemporary management. Transl. Lung Cancer Res. 1, 179–193. doi:10.3978/j.issn.2218-6751.2012.09.04
Siravegna, G., Lazzari, L., Crisafulli, G., Sartore-Bianchi, A., Mussolin, B., Cassingena, A., et al. (2018). Radiologic and genomic evolution of individual metastases during HER2 blockade in colorectal cancer. Cancer Cell 9 (1), 148–162. doi:10.1016/j.ccell.2018.06.004
Siravegna, G., Mussolin, B., Buscarino, M., Corti, G., Cassingena, A., Crisafulli, G., et al. (2015a). Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 21, 795–801. doi:10.1038/nm.3870
Strickler, J. H., Loree, J. M., Ahronian, L. G., Parikh, A. R., Niedzwiecki, D., Pereira, A. A. L., et al. (2018). Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov. 8, 164–173. doi:10.1158/2159-8290.CD-17-1009
Subbiah, V., Baik, C., and Kirkwood, J. M. (2020). Clinical development of BRAF plus MEK inhibitor combinations. Trends Cancer 6, 797–810. doi:10.1016/j.trecan.2020.05.009
Sunakawa, Y., Nakamura, M., Ishizaki, M., Kataoka, M., Satake, H., Kitazono, M., et al. (2020). RAS mutations in circulating tumor DNA and clinical outcomes of rechallenge treatment with anti-EGFR antibodies in patients with metastatic colorectal cancer. JCO Precis. Oncol. 4, 898–911. doi:10.1200/PO.20.00109
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer J. Clin. 71, 209–249. doi:10.3322/caac.21660
Toledo, R. A., Cubillo, A., Vega, E., Garralda, E., Alvarez, R., de la Varga, L. U., et al. (2017). Clinical validation of prospective liquid biopsy monitoring in patients with wild-type RAS metastatic colorectal cancer treated with FOLFIRI-cetuximab. Oncotarget 8, 35289–35300. doi:10.18632/oncotarget.13311
Troiani, T., Martinelli, E., Napolitano, S., Vitagliano, D., Ciuffreda, L. P., Costantino, S., et al. (2013). Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR–MET interaction and activation of MET signaling in colon cancer cells. Clin. Cancer Res. 19, 6751–6765. doi:10.1158/1078-0432.CCR-13-0423
Uprety, D., and Adjei, A. A. (2020). KRAS: from undruggable to a druggable Cancer Target. Cancer Treat. Rev. 89, 102070. doi:10.1016/j.ctrv.2020.102070
Van Cutsem, E., Köhne, C.-H., Hitre, E., Zaluski, J., Chang Chien, C.-R., Makhson, A., et al. (2009). Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 360, 1408–1417. doi:10.1056/NEJMoa0805019
Van Emburgh, B. O., Arena, S., Siravegna, G., Lazzari, L., Crisafulli, G., Corti, G., et al. (2016). Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 7, 13665. doi:10.1038/ncomms13665
Vidal, J., Fernández-Rodríguez, M. C., Casadevall, D., García-Alfonso, P., Páez, D., Guix, M., et al. (2022). Liquid biopsy detects early molecular response and predicts benefit to first-line chemotherapy plus cetuximab in metastatic colorectal cancer: PLATFORM-B study. Clin. Cancer Res. 29, 379–388. OF1–OF10. doi:10.1158/1078-0432.CCR-22-1696
Viloria-Petit, A., Crombet, T., Jothy, S., Hicklin, D., Bohlen, P., Schlaeppi, J. M., et al. (2001). Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 61, 5090–5101.
Vitiello, De F., Giunta, C., Cardone, V., Ciardiello, D., Cardone, C., Vitale, P., et al. (2019). Clinical practice use of liquid biopsy to identify RAS/BRAF mutations in patients with metastatic colorectal cancer (mCRC): a single institution experience. Cancers (Basel) 11, 1504. doi:10.3390/cancers11101504
Wan, J. C. M., Massie, C., Garcia-Corbacho, J., Mouliere, F., Brenton, J. D., Caldas, C., et al. (2017). Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat. Rev. Cancer 17, 223–238. doi:10.1038/nrc.2017.7
Woolston, A., Khan, K., Spain, G., Barber, L. J., Griffiths, B., Gonzalez-Exposito, R., et al. (2019). Genomic and transcriptomic determinants of therapy resistance and immune landscape evolution during anti-EGFR treatment in colorectal cancer. Cancer Cell 36, 35–50. doi:10.1016/j.ccell.2019.05.013
Yan, M., Schwaederle, M., Arguello, D., Millis, S. Z., Gatalica, Z., and Kurzrock, R. (2015). HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 34, 157–164. doi:10.1007/s10555-015-9552-6
Yarom, N., and Jonker, D. J. (2011). The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov. Med. 11, 95–105.
Yonesaka, K., Zejnullahu, K., Okamoto, I., Satoh, T., Cappuzzo, F., Souglakos, J., et al. (2011). Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 3, 99ra86–12. doi:10.1126/scitranslmed.3002442
Keywords: colorectal cancer, acquired resistance, anti-EGFR, liquid biopsy, CtDNA, clonal dynamics, tumor heterogeneity, anti-EGFR rechallenge
Citation: Ríos-Hoyo A, Monzonís X, Vidal J, Linares J and Montagut C (2024) Unveiling acquired resistance to anti-EGFR therapies in colorectal cancer: a long and winding road. Front. Pharmacol. 15:1398419. doi: 10.3389/fphar.2024.1398419
Received: 09 March 2024; Accepted: 03 April 2024;
Published: 22 April 2024.
Edited by:
Edward Van Der Horst, Sensei Biotherapeutics, United StatesReviewed by:
Andrea Sartore-Bianchi, University of Milan, ItalyFrancisco Javier Sánchez Martín, Other, United Kingdom
Copyright © 2024 Ríos-Hoyo, Monzonís, Vidal, Linares and Montagut. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clara Montagut, cmontagut@psmar.cat