 Takudzwa Mugiya
Takudzwa Mugiya Mamosheledi Mothibe1
Mamosheledi Mothibe1 Andile Khathi
Andile Khathi Phikelelani Ngubane
Phikelelani Ngubane Ntethelelo Sibiya
Ntethelelo Sibiya- 1Pharmacology Division, Faculty of Pharmacy, Rhodes University, Makhanda, South Africa
- 2School of Laboratory Medicine and Medical Sciences, University of KwaZulu-Natal, Durban, South Africa
In light of the expected increase in the prevalence of diabetes mellitus due to an aging population, sedentary lifestyles, an increase in obesity, and unhealthy diets, there is a need to identify potential pharmacological agents that can heighten the risk of developing diabetes. Similarly, it is equally important to also identify those agents that show blood glucose-lowering properties. Amongst these agents are tyrosine kinase inhibitors used to treat certain types of cancers. Over the last two decades, there has been an increase in the use of targeted chemotherapy for cancers such as renal cell carcinoma, chronic leukaemia, and gastrointestinal stromal tumours. Small molecule tyrosine kinase inhibitors have been at the forefront of targeted chemotherapy. Studies have shown that small molecule tyrosine kinase inhibitors can alter glycaemic control and glucose metabolism, with some demonstrating hypoglycaemic activities whilst others showing hyperglycaemic properties. The mechanism by which small molecule tyrosine kinase inhibitors cause glycaemic dysregulation is not well understood, therefore, the clinical significance of these chemotherapeutic agents on glucose handling is also poorly documented. In this review, the effort is directed at mapping mechanistic insights into the effect of various small molecule tyrosine kinase inhibitors on glycaemic dysregulation envisaged to provide a deeper understanding of these chemotherapeutic agents on glucose metabolism. Small molecule tyrosine kinase inhibitors may elicit these observed glycaemic effects through preservation of β-cell function, improving insulin sensitivity and insulin secretion. These compounds bind to a spectrum of receptors and proteins implicated in glucose regulation for example, non-receptor tyrosine kinase SRC and ABL. Then receptor tyrosine kinase EGFR, PDGFR, and FGFR.
1 Introduction
Diabetes mellitus (DM) is a chronic metabolic disorder characterised by elevated blood glucose concentration due to insulin resistance, deficiency of insulin, or both (Bastaki, 2005). DM affects the metabolism of fats, carbohydrates, and proteins, resulting in chronic hyperglycaemia (Bastaki, 2005; Saeedi et al., 2019). The global prevalence of DM is on the rise (Bastaki, 2005; Saeedi et al., 2019). The increase in DM prevalence may be attributed to sedentary lifestyles and diets rich in refined carbohydrates (Bastaki, 2005; Saeedi et al., 2019; Sibiya et al., 2022). Diabetes can be classified into type 1, type 2, and gestational diabetes mellitus (Bastaki, 2005; Saeedi et al., 2019; Sibiya et al., 2022). Reports have estimated that 90% of all DM cases are type 2 (Saeedi et al., 2019; Sibiya et al., 2022). An estimated 171 million people were affected by DM before 2010 and this figure is predicted to increase to 300 million by 2025 (Sibiya et al., 2022). An estimated 5.2 million deaths globally per year have been attributed to DM, exerting pressure on health systems worldwide (Glovaci et al., 2019). Many routinely prescribed pharmacological agents are believed to induce glucose intolerance and/or cause DM in non-diabetic individuals or they can negatively impact glycaemic control in diabetic patients (Bressler and DeFronzo, 1994). These effects on glycaemic control by pharmacological agents can be attributed to the induction of insulin resistance, inhibition of insulin secretion, and direct cytotoxic effects on pancreatic cells (Fathallah et al., 2015). Patients with diabetes often have other coexisting medical conditions which often leads to polypharmacy (Alwhaibi et al., 2018). The use of polypharmacy has the potential to worsen glucose tolerance (Dobrică et al., 2019). Several pharmacological agents influence glucose metabolism, including certain antihypertensives, antiretrovirals, atypical antipsychotics, antibiotics, and immunosuppressives (Izzedine et al., 2005). Literature indicates that patients being treated with β-blockers, protease inhibitors, thiazide diuretics, and atypical antipsychotic drugs are at a higher risk of developing DM as the prevalence of hyperglycaemia is higher with these agents (Izzedine et al., 2005). Due to the heightened risks of developing DM to date, there is a need for more antidiabetic options to be available for physicians to utilize in the management of diabetes. Interestingly, anticancer agents known as small molecule kinase inhibitors have shown mixed observations, where some generations have been reported to be associated with blood glucose-lowering effects in patients with cancer, whilst others have been associated with hypoglycaemia. This, therefore, highlights the necessity for more research regarding these agents and their effect on glucose metabolism, thus allowing a deeper insight in terms of their mechanisms of action at both cellular and molecular levels. Where consensus and a solid understanding of these agents on glucose handling is achieved, clear insights on their role in worsening or managing hyperglycaemia can be realised. Considering the burden exerted by DM, together with the time and challenges it takes to discover and develop new antidiabetics, there is merit for studies looking into leveraging on the existing hypoglycaemic effects demonstrated by small molecule tyrosine kinase inhibitors. With a defined mechanism of action, and structure activity relationship, small molecule tyrosine kinase inhibitors can potentially be repurposed for diabetes management, a route that could be time and cost-effective. Small molecule tyrosine kinase inhibitors are already in use and registered for the use of the treatment of cancers; therefore, safety and release profile studies are already available for these drugs. For a better understanding of small molecule tyrosine kinase inhibitors on glucose metabolism, firstly we will start by briefly outlining key processes involved in glucose homeostasis.
2 Glucose metabolism
2.1 Insulin secretion
Insulin secretion is primarily glucose-induced, with other nutrients such as amino acids and fatty acids augmenting the glucose-induced secretion (Rorsman and Braun, 2013). Glucose-induced insulin secretion requires the intracellular uptake of glucose and its metabolic oxidation. The primary transporters of glucose in βcells are glucose transporter 1 (GLUT1) and GLUT3 (McCulloch et al., 2011). Insulin is produced in the rough endoplasmic reticulum as prepro-insulin, a 110 amino acid precursor. It is then packed in the Golgi apparatus as proinsulin after the removal of 24 residue signal sequence from prepro-insulin. Insulin is stored in immature secretory granules (Goodge and Hutton, 2000). Studies of isolated islets with radioactive leucine showed that granules synthesised last are the most likely to be released when β-cells are stimulated (Gold et al., 1982). The breakdown of insulin is by crinophagia and the intracellular destruction of insulin is by lysosomes (Schnell et al., 1988). The secretory granules containing insulin also contain approximately 50 polypeptides and low molecular weight compounds like GABA, glutamate, and serotonin. The granules also contain high concentrations of metal ions like Zn2+ and Ca2+ (Ashcroft and Rorsman, 1989). The secretion of insulin is biphasic, with glucose providing a triggering action and generating triggering and amplifying signals in β-cells (Henquin, 2000). β-cells are electrically excitable and utilize changes in their membrane potential to link changes in plasma glucose concentrations with the stimulation or inhibition of insulin secretion (Rorsman and Braun, 2013). β-cells have ATPsensitive K+ (KATP) channels which at low glucose concentrations maintain a negative membrane potential and close in response to glucose stimulation (Gromada et al., 1998). Insulin secretion also involves ATP production, glucose metabolism, Ca2+-dependent action potential firing, and membrane depolarization (Eliasson et al., 2008). β-cell electrical activity leads to elevation of Ca2+ level which triggers exocytosis of insulin granules. This is referred to as Ca2+-dependent exocytosis of insulin granules (Henquin et al., 2006). Plasma glucose concentration homeostasis is under the feedback loop control of insulin through alterations in KATP channel closure, β-cell metabolism, and electrical activity (Eliasson et al., 2008).
Pharmacological agents that act by depolarization and Ca2+ influx into β-cells lead to desensitization of insulin secretion, which is a reversible state of decreased insulin secretion responsiveness of β-cells due to prolonged exposure to stimuli. Other stimuli include glucose and free fatty acids (Rustenbeck, 2002). Sulphonylureas, namely, glibenclamide and tolbutamide, cause recoverable impairment of insulin secretion (FILIPPONI et al., 1983). Studies have shown that prolonged sulphonylurea exposure of isolated islets reduced glucose-induced insulin secretion by approximately 50% (Anello et al., 1999). Calcineurin inhibitors including tacrolimus and cyclosporine are implicated as diabetogenic drugs. Cyclosporine is believed to alter mitochondrial permeability transition pore function (Chakkera et al., 2017).
2.2 Insulin signaling
Insulin receptors are present on every cell, but they are primarily expressed in skeletal muscle, liver, and fat tissue, which are the predominant insulin target sites. Insulin binds to target cell surface receptors; the insulin receptor is a heterotetramer consisting of β subunits and α subunits linked together through disulphide bonds (Bhattacharya et al., 2007). As illustrated in Figure 1, Insulin binds to cell surface protein receptors, resulting in tyrosine phosphorylation and subsequent phosphorylation of the insulin receptor substrate. This recruits and activates PI3 kinase and its downstream target Akt/PKB (Mora et al., 2004). The activation of Akt/PKB leads to phosphorylation of AS160, resulting in a reduction in Rab-GAP activity. This results in the translocation of GLUT4 and glucose uptake as depicted in Figure 1 (Sakamoto and Holman, 2008). The serine/threonine kinase Akt signaling plays a major role in insulin stimulated glucose uptake in adipose and muscle tissue. Akt signaling also inhibits glucose release from hepatocytes (Mackenzie and Elliott, 2014). The uptake of glucose via Akt signaling occurs through translocation of GLUTs to the cell membrane upon activation of insulin receptors in the presence of glucose (Jiang et al., 2003). Therefore, disruptions in PI3K/Akt/AS160 transduction pathways results in the reduction of glucose uptake in skeletal muscle. Protein tyrosine phosphatases (PTPs) can be classified into several sub-families based on their functional and structural characteristics. The three major sub-families include receptor-type PTPs (RPTPs), non-receptor-type PTPs (NRPTPs), and dual-specificity phosphatases (DSPs) (Li et al., 2016). LAR is a member of the RPTPs family and PTP-1B, PTP-1C, and PTP-1D belong to the NRPTPs family (Tao et al., 2001). From the PTPs mentioned above, only PTP-1B is a target for obesity and diabetes treatment as PTP dephosphorylation activates both leptin and insulin signaling pathways (Goldstein, 2001). The negative regulatory effect of PTP-1B on the insulin signaling pathways is a therapeutic potential in type 2 DM (Zhao et al., 2018). Patients with type 2 DM have disrupted GLUT4 expression and this contributes to insulin resistance (Albrecht et al., 1993). Disrupted intracellular signaling of GLUT4 translocation from intracellular vesicles to the cell membrane is responsible for resistance to insulin-stimulated glucose transport (Bryant et al., 2002). Apart from insulin mediated glucose transport, basal glucose uptake in most cells is facilitated by the presence of GLUT 1 (Medina and Owen, 2002).
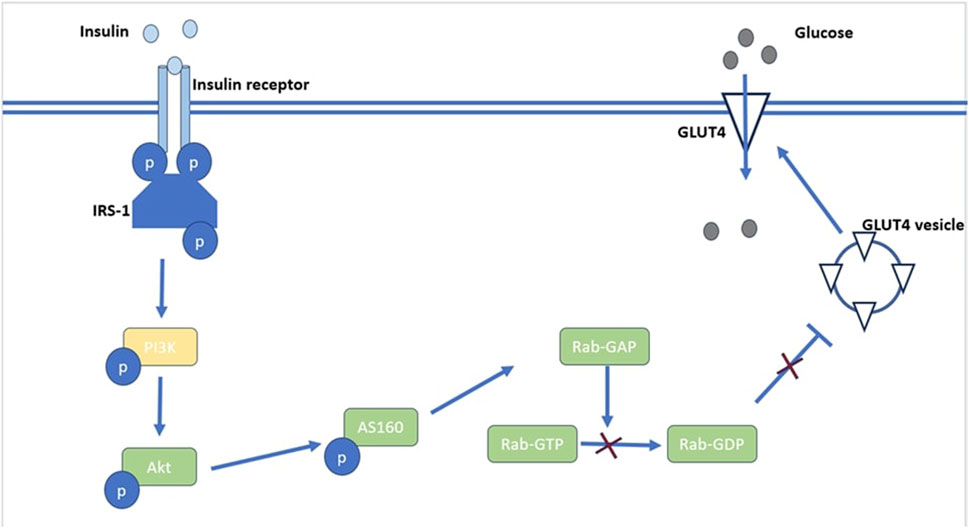
FIGURE 1. An illustration of Akt’s role in the insulin signaling pathway. Akt activation leads to phosphorylation of AS160 which reduces Rab-GAP activity resulting in reduced production of RabGDP which inhibits the translocation of GLUT4 vesicles.
3 Tyrosine kinases and cancer
Tyrosine kinases are enzymes that selectively phosphorylate tyrosine residues in specific proteins by transferring a phosphate group from ATP to the tyrosine residue. They alter the signaling cascade which modifies cell growth, differentiation, migration, death, and apoptosis, as illustrated in Figure 2 (Thomson et al., 2022). Tyrosine kinase signaling pathways are typically responsible for preventing dysregulated cell proliferation and play a role in sensitising cells to apoptotic stimuli. In cancer cells, tyrosine kinase signaling pathways are often epigenetically or genetically altered, which gives cancer cells a selection advantage (Paul and Mukhopadhyay, 2004). Tyrosine kinases comprise the major group of all oncoproteins (Rodrigues and Park, 1994). Out of the 518 kinases found in the human kinome, approximately 90 are tyrosine kinases (Bardelli et al., 2003). Therapeutic agents that are utilized in diseases linked to unusual activation of tyrosine kinase resulting in abnormal downstream oncogenic signaling are at the forefront as targets for chemotherapy (Baselga, 2006). Tyrosine kinases are primarily classified as receptor tyrosine kinases (RTKs), e.g., fibroblast growth factor receptors (FGFRs), epidermal growth factor receptors (EGFRs), platelet-derived growth factor receptors (PDGFRs), and insulin receptors (IRs) and non-receptor tyrosine kinases (NRTKs), e.g., Janus kinases (JAKs) and proto-oncogene tyrosine-protein kinase SRC (Hunter, 1995). Receptor tyrosine kinases are more than cell surface transmembrane receptors, as they are also enzymes that have intracellular kinase activity (Hunter, 1995). Non-receptor tyrosine kinases are cytoplasmic proteins varying in structure (Schenk and Snaar-Jagalska, 1999).
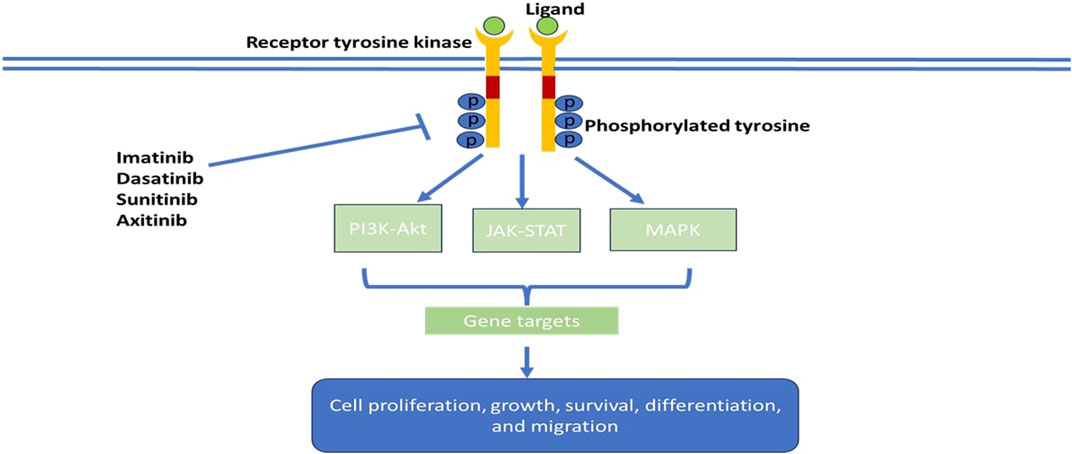
FIGURE 2. Schematic model of tumorigenic signaling pathways and their inhibition by anti-cancer- tyrosine kinase inhibitors.
3.1 Tyrosine kinase mechanism of action
Similar to an insulin receptor activation, ligands bind to the receptor tyrosine kinase resulting in its conformational change; which enables the autophosphorylation of tyrosine residues (Figure 1). Activated receptors attract various signaling molecules which leads to the activation of kinases like the Serine/threonine kinase which leads to the phosphorylation and activation of mitogen-activated protein kinase thereby activating extracellular signal-regulated kinases 1 and 2 (Banning et al., 2014). Ligands such as epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) are extracellular signaling molecules that induce receptor dimerization (Paul and Mukhopadhyay, 2004). Two types of ligand-receptor complexes can be formed, viz., one ligand may bind to two receptors resulting in a 1:2 ligand: receptor complex or two ligands can simultaneously bind to two receptor molecules resulting in a 2:2 receptor complex, for example, EGF: EGFR (epidermal growth factor: epidermal growth factor receptor) and VEGF: VEGFR (vascular endothelial growth factor: vascular endothelial growth factor receptor) (Paul and Mukhopadhyay, 2004; Hubbard and Miller, 2007). The activation loop has an open conformation after tyrosine phosphorylation and this gives access to ATP and substrates; ATP from Mg-ATP is transferred to the tyrosine residue (Schlessinger, 2000). The ATP binding sites function as a docking site for numerous downstream signaling proteins containing SRC homology-2 (SH2) or protein tyrosine binding (PTB) domain (Du and Lovly, 2018). These docking proteins recruit other effector molecules which contain SH2, SH3, PTB, and Pleckstrin homology domain (Paul and Mukhopadhyay, 2004; Du and Lovly, 2018). As a result, signaling complexes are formed to an activated receptor and membrane which leads to activating a cascade of intracellular biochemical signals resulting in activation or regression of genes and this is what defines biological responses to signals (Östman and Böhmer, 2001). Receptor tyrosine kinases transfer complex information relating to cell growth from the cell’s external environment to the cell nucleus thus activating transcriptional pathways which control cellular functions (Du and Lovly, 2018).
3.2 Tyrosine kinase inhibitors
Cancer growth is usually promoted by the activation of mutations or gene amplifications of specific serine kinase or tyrosine kinases (Cheng and Force, 2010). The non-specificity of traditional chemotherapy drugs, impacting both cancerous and healthy cells, lead to a range of side effects and pose a narrow therapeutic index and potential drug resistance. Tyrosine kinase inhibitors in cancer therapy offer the advantage of selectively targeting and disrupting the upregulated activities of enzymes involved in cancer cell signaling, providing a more precise and less toxic treatment approach (Arora and Scholar, 2005). The first tyrosine kinase inhibitor approved by the FDA was imatinib in 2001. By 2015, 28 small molecule tyrosine kinase inhibitors had been approved by the FDA, and by 2021 the FDA had approved 73 small molecule kinase inhibitors (Wu et al., 2015; Ayala-Aguilera et al., 2022). Tyrosine kinase inhibitors have two classes, small molecules that target specific kinases and monoclonal antibodies that target receptor kinases or their ligands (Cheng and Force, 2010). Tyrosine kinases play an important role in modulating growth factor signaling (Arora and Scholar, 2005). Tyrosine kinase inhibitors are orally active and work by competitive ATP inhibition at catalytic binding sites of oncogenic tyrosine kinases, as illustrated in Figure 2. Tyrosine kinase inhibitors differ from each other through targeting different kinases, differing pharmacokinetics, and adverse effects. Skin toxicity is an adverse effect observed in more than 50% of the patients on tyrosine kinase inhibitors (Hartmann et al., 2009). The use of tyrosine kinase inhibitors is limited by the development of resistance and lack of tumor response in the general population (Arora and Scholar, 2005).
3.3 Tyrosine kinase inhibitors mechanism
Tyrosine kinase inhibitors are categorized by their binding modes into two classes: irreversible and reversible. Irreversible kinase inhibitors form covalent bonds with reactive nucleophilic cysteine residue proximal to the ATP binding site, blocking the ATP site resulting in irreversible inhibition (Wu et al., 2015). Reversible kinase inhibitors can be categorised into five types based on the conformation of the binding pockets and the Asp-Phe-Gly DFG motif which is a conserved amino acid sequence from the activation loop (Noble et al., 2004). Type I inhibitors competitively bind to ATP binding sites on active kinases that have aspartate residue of the DFG motif facing the catalytic site of the kinase. Type I½ inhibitors bind to a DFG-Asp in inactive conformation (Roskoski, 2016). Type II inhibitors bind to inactive kinase with the aspartate residue protruding outward from the ATP binding site (Noble et al., 2004; Wu et al., 2015). Type I, I½, and II inhibitors reside in a part of the adenine binding pocket (Roskoski, 2016). Type III inhibitors only bind to allosteric pockets next to the ATP binding site as they do not interact with or alter the ATP binding pocket (Noble et al., 2004). Type IV inhibitors bind to allosteric sites that are not on the ATP binding pocket (Cox et al., 2010). Type V includes kinase Inhibitors like bisubstrate and bivalent inhibitors which display multiple binding modes (Cox et al., 2011). Irreversible kinase inhibitors are known as type VI inhibitors. Tyrosine inhibitors can inhibit either receptor or cytosolic tyrosine kinases. They compete with ATP for binding sites on tyrosine kinase (e.g., imatinib, gefitinib, PP1-AG1872) (Table 1). Other tyrosine kinase inhibitors such as lavendustin A exert their effect through allosteric inhibition on tyrosine kinase. Some tyrosine kinase inhibitors inhibit tyrosine kinase interaction with other proteins, including UCS15A. Tyrosine kinase inhibitors such as cetuximab inhibit ligands from binding to receptor tyrosine kinases (Cismowski et al., 2007).
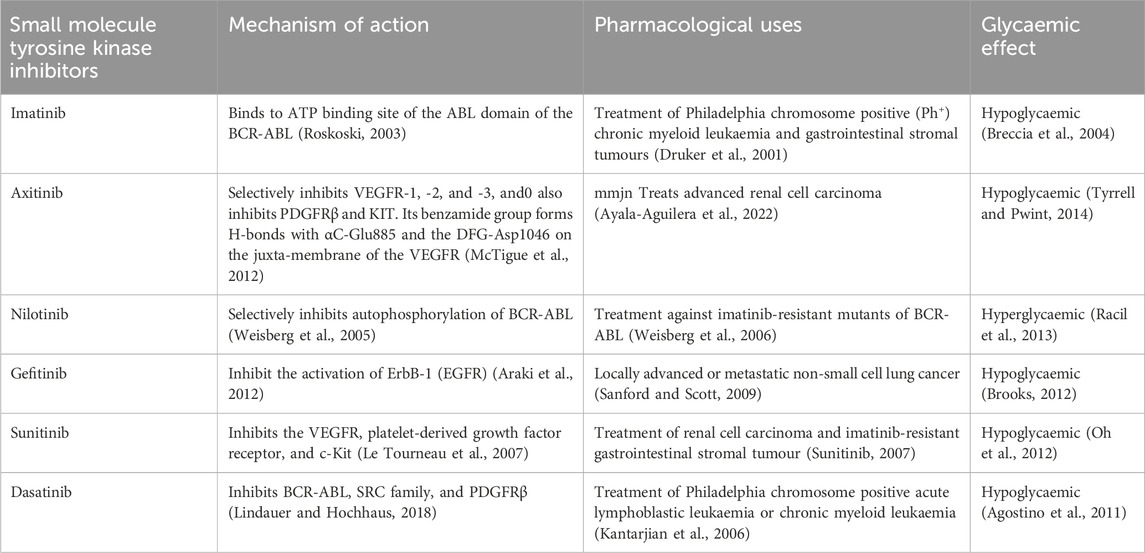
TABLE 1. Summary of small molecule tyrosine kinase inhibitors investigated for their antidiabetic activitiy.
3.4 Tyrosine kinase inhibitors and their glycaemic effects
Studies have shown that some small molecule tyrosine kinase inhibitors such as imatinib, erlotinib, and sunitinib exhibit antihyperglycemic effects, the mechanisms of which are not yet fully understood (Prada and Saad, 2013). However, certain tyrosine kinase inhibitors like nilotinib have been observed to cause hyperglycaemia (Breccia et al., 2007). Understanding how these drugs cause antihyperglycemic effects could provide further insight on glucose handling of pharmacological targets and shed light on whether tyrosine inhibitors could be useful as pharmacophores to develop novel agents towards the management of DM. Small molecule tyrosine kinase inhibitors have been shown to reverse or even prevent type 1 and type 2 DM by reducing insulin resistance and improving β cell dysfunction, thus reversing hyperglycaemia (Fountas et al., 2015). Literature indicates that tyrosine kinase inhibitors can cause hypoglycaemia in both type 1 and type 2 DM (Hadova et al., 2021). Improvement in HbA1c and glycaemia has been noted in several cases, leading to either termination or reduction of insulin therapy when patients are being treated with imatinib and sunitinib (Buffier et al., 2018). A retrospective study investigated the blood glucose concentration in 17 diabetic and 61 non-diabetic patients being treated with imatinib, sunitinib, dasatinib, and sorafenib. A mean decline in blood glucose concentration was observed with the use of all four drugs; imatinib had a mean decline of blood glucose concentration of 9 mg/dL, sunitinib 14 mg/dL, dasatinib 53 mg/dL, and sorafenib 12 mg/dL. The decrease in blood glucose concentration observed in this retrospective study was statistically significant and it was noted that 47% of patients with diabetes were able to successfully discontinue their diabetic treatment including insulin therapy (Agostino et al., 2011).
This literature is envisaged to provide and consolidate evidence of glycaemic aberrations associated with the small molecule tyrosine kinase inhibitors. We envisage that corroborating retrospective evidence together in vivo and in vitro observation may assist in shedding deeper mechanistic insight on how small molecule tyrosine kinase inhibitors modulate glucose handling. As mentioned in the introduction, some small kinase inhibitors exhibit hyperglycaemic effects, hence understanding and differentiating the mechanisms of actions of small molecule tyrosine kinase inhibitors is crucial. This may pave the way for the repurposing of these drugs in diabetes management. We envisage discussion in the review will provide a motive for further research and development to explore the worthiness of repurposing small molecule tyrosine kinase inhibitors for the management of DM. In the next section, we present a discussion of the small molecule tyrosine kinase inhibitors outlining the indications and highlighting the known glycaemic effects to date, of each one. Table 1 provides a summary of some small molecule tyrosine kinase inhibitors investigated for their antidiabetic activity.
3.4.1 Imatinib
Imatinib was approved in 2001 by the FDA after successful clinical trials for the treatment of all phases of Philadelphia chromosome positive (Ph+) chronic myeloid leukaemia. It was then approved for the treatment of gastrointestinal stromal tumours (GIST) in 2002 (Druker et al., 2001; Ayala-Aguilera et al., 2022). Imatinib is a selective and potent inhibitor of the protein tyrosine kinase BCR-ABL, KIT and platelet derived growth factor receptors (PDGFR-α and PDGFR-β) (Peng et al., 2005). The protein tyrosine kinase BCR-ABL is a ligand independent (constitutively) activated tyrosine kinase that causes myeloid leukaemia (CML) (Druker et al., 2001). Imatinib is a type II tyrosine kinase inhibitor and a phenylamino-pyrimidine derivative (Roskoski, 2016). After binding to the ATP binding site of the ABL domain of the BCR-ABL tyrosine kinase protein, imatinib traps the chimeric protein in an inactive conformation thus inhibiting it with nanomolar potency (Roskoski, 2003). The BCR-ABL is constitutively activated and is only present in the leukemic clone, hence imatinib’s selectivity. Imatinib inhibition of KIT and tyrosine kinase PDGFR is the basis of its selective treatment in GIST. This selectivity results in reduced side effects. Inhibition of KIT may lead to myelosuppression, dermatologic toxicity, while ABL inhibition may lead to rare cardiac events and inhibition of PDGFR may lead to altered bone metabolism and fluid retentions (Giles et al., 2009).
BCR-ABL positive cells express high affinity for the GLUT-1 glucose transporter and increased glucose uptake. This supports the theory that the control of glucose substrate flux is a vital mechanism of the antiproliferative action demonstrated by imatinib (Breccia and Alimena, 2009). The potential therapeutic benefit of imatinib in GIST cells is via decreased glucose uptake (Stroobants et al., 2003; Verweij et al., 2004). Treatment of BCR-ABL positive cells with imatinib resulted in approximately 90% of the cell surface transporter GLUT-1 being internalized (Barnes et al., 2005). Furthermore, imatinib treatment has been found to significantly reduce glucose uptake by reduced plasma membrane-bound GLUT-4 in GIST cells (Tarn et al., 2006). Boren et al. (Boren et al., 2001) have shown that imatinib treatment might reduce de novo fatty acid and nucleic acid synthesis through restriction of glucose-6phosphate 1- dehydrogenase activities and hexokinase in myeloid tumour cells. In a study by Gottschalk et al. (Gottschalk et al., 2004), human BCR-ABL positive cell lines treated with imatinib 0.1–1.0 μmol/L demonstrated a decrease in glucose uptake from the media via the suppression of glycolytic cell activity and increased mitochondrial Krebs cycle activity. However, we should note that these observations are reported in cancerous cells, which could suggest that a poor-glucose uptake as a cytotoxic tool to kill cancerous cells.
In a clinical observation of seven diabetic chronic myeloid leukaemia patients being treated with imatinib, six patients showed improvement in fasting blood glucose concentration which led to a reduction of their insulin dosage or oral antidiabetic drugs (Breccia et al., 2004). Imatinib has been shown to increase insulin sensitivity, as studies have shown that imatinib treatment led to stable insulin plasma levels and progressive reduction of HbA1c fraction (Breccia et al., 2005). The exact molecular mechanisms in which imatinib confers its antidiabetic properties is not clearly understood and this is a strong motive of this paper. So far, the proposed hypothesis of how imatinib improves glycaemia include preservation of beta cells viability and mass. Imatinib is thought to inhibit beta cell apoptosis through increased activation of NF-KB, an antiapoptotic factor and via decreased activation of proapoptotic MAPK JNK. Insulin sensitivity is improved via inhibition of tumour necrosis factor-α production (Wolf et al., 2005). This is believed to occur via imatinib’s inhibition of the non-receptor tyrosine kinase c-Abl (Breccia et al., 2004). Studies show that moderate dose of imatinib efficiently neutralize high-fat induced peripheral insulin resistance (Hägerkvist et al., 2007). Increased insulin sensitivity may also be due to imatinib’s slimming effect (Breccia et al., 2005; Hägerkvist et al., 2007). Imatinib inhibits phosphorylation of several kinases such as extracellular regulated kinase 1 and 2 (ERK1 and ERK2), AKT and PDGFR. This inhibition of phosphorylation leads to better signaling and functioning of effectors with improvements in insulin sensitivity (Veneri et al., 2005).
3.4.2 Axitinib
Axitinib is a small molecule tyrosine kinase inhibitors derived from indazole. It is an oral multitargeted potent tyrosine kinase receptor inhibitor. Axitinib selectively inhibits VEGFR-1, -2 and -3 at sub nanomolar concentrations in vitro (Escudier and Gore, 2011). Axitinib was approved by the FDA for the treatment of advanced renal cell carcinoma in 2012 (Ayala-Aguilera et al., 2022). It is a second generation type IIA receptor inhibitor of VEGFR with sub nanomolar potency (Roskoski, 2016). The VEGFR is a component of the VEGF/VEGFR signaling pathways which play a role in vascular development during embryogenesis and angiogenesis during adulthood. These pathways are also implicated in growth metastasis and survival of tumours (Chen et al., 2013). Axitinib binds to intracellular tyrosine kinase domain of VEGFR and inhibits autophosphorylation of VEGFRs thus blocking downstream signaling pathways, cell survival and tube formation at picomolar concentration (Hu-Lowe et al., 2008). Axitinib is more selective against VEGFR tyrosine kinase than the tyrosine kinase inhibitors such as soraferinib, sunitinib and pazopib, as they are multitargeting (Bhargava and Robinson, 2011). Tyrrell and Pwint (Tyrrell and Pwint, 2014) observed a decrease in blood glucose concentrations and HbA1c level in a patient receiving axitinib, 5 mg twice daily. In vitro studies by Hudson et al. (2014) showed that axitinib increases [C-14] deoxy glucose uptake and translocation of GLUT-1 transporters from cytosolic pools to the cell surface membrane in pancreatic adenocarcinoma (PDAC) cells.
3.4.3 Sunitinib
Sunitinib is a potent orally bioavailable ATP-competitive type I1/2B inhibitor. It is a multitargeted receptor tyrosine kinase inhibitor of PDGFR-α and β, FMS-like tyrosine kinase 3, VEGFR-1, -2 and 3, colony stimulating factor 1 receptor and glial cell-line derived neurotrophic factor receptor (Blay, 2010). These receptors are associated with tumour growth and angiogenesis. Sunitinib is used in the treatment of renal cell carcinoma and gastrointestinal stromal tumours (Gan et al., 2009). The development of sunitinib was brought by the development of resistance to imatinib in gastrointestinal stromal tumour potent caused by mutation of KIT (Atkins et al., 2006). Sunitinib forms polar bonds with Asp86, Glu81 and Lue83 in the CDK2 hinge region (Roskoski, 2016).
In a study that was done at Seoul National University Bundang Hospital in the Republic of Korea, 10 out of 48 diabetic patients with metastatic renal cell carcinoma, were reported to have a significant decrease in blood glucose levels after treatment with sunitinib dose for 4 weeks. However, their blood glucose concentrations reverted to normal 2 weeks after treatment cessation. Sunitinib doses were not disclosed by the authors, however since no special note was highlighted, we propose that the doses were within the global recommendations of 50 mg daily for an adult patient. One of the 10 patients was able to discontinue their diabetic medication and 3 patients had dose reductions of their oral antidiabetics during the 4-week sunitinib treatment. Conversely, 38 non-diabetic patients showed no significant changes in blood glucose levels during the 4 weeks of sunitinib treatment (Oh et al., 2012). In a retrospective review, all 19 types 2 diabetes mellitus patients showed a reduction in blood glucose concentrations after 4 weeks sunitinib treatment. Some studies reported discontinuation of diabetic medications as a results of sunitinib treatments, whilst others reported patients having normalized blood glucose (Billemont et al., 2008).
The exact blood glucose lowering mechanism of tyrosine kinase inhibitors is unknown. It has been suggested that tyrosine kinase inhibitors’ effect on c-KIT tyrosine kinase is in part responsible for the alteration in blood glucose levels. C-KIT is associated with β-cell survival (Rachdi et al., 2001). Sunitinib is believed to have a direct effect on β-cells resulting in glucose-induced insulin secretion which leads to lowered glucose in patients. Sunitinib stimulates insulin secretion in a concentration and dose dependent manner (Lutz et al., 2017). Billemont et al. (2008) suggested that sunitinib affects insulin resistance by interfering with insulin-like growth factor-1 pathway. Louvet et al. (2008) using data from mouse models suggested that inhibition of PDGF downstream-mediated inflammatory response improves pancreatic β-cell survival and insulin resistance.
3.4.4 Nilotinib
Nilotinib is an orally administered type IIA BCR-ABL inhibitor, second generation tyrosine kinase inhibitor developed by Novartis for treatment against imatinib-resistant mutants of BCR-ABL. Nilotinib binds to the inactive DFG-out conformation of the BCR-ABL (Weisberg et al., 2006). Nilotinib is a high-affinity aminopyrimidine-based ATP competitive inhibitor that reduces cell proliferation and viability through selective inhibition of BCR-ABL autophosphorylation (Weisberg et al., 2005). To maximize efficacy, nilotinib was designed to bind more tightly to the BCR-ABL than imatinib, by incorporating a N-methypiperamine moiety while keeping the amide pharmacophore to preserve the H-bond interaction to Glu286 and Asp381. Hence nilotinib is able to maintain binding to the inactive conformation of the ABL kinase domain (Golemovic et al., 2005). Out of the 33 imatinib-resistant BCR-ABL mutants, nilotinib is active against 32 except for the T315I mutant. Nilotinib has more lipophilic interactions than imatinib; it has 4 hydrogen-bond interactions with the ABL kinase domain, which results in nilotinib expressing higher affinity for binding than imatinib (Manley et al., 2005).
Nilotinib and imatinib are similar in structure but reports have shown them to have opposite effects on glucose metabolism (Breccia et al., 2007). Iurlo et al. (2015) observed that patients on nilotinib had significantly higher levels of insulin, C-peptide, fasting plasma glucose, insulin resistance and LDL cholesterol compared to patients that were being treated with imatinib and dasatinib. It was however concluded that nilotinib treatment does not induce diabetes mellitus or impair fasting glucose to a significantly higher extent than imatinib and dasatinib, but nilotinib causes a worse glucometabolic profile. Hence it has been advised that patients on nilotinib require dose monitoring of glucose and lipid metabolism. A large phase III trial comparing the efficacy of nilotinib and imatinib showed that 53% of the patients being treated with nilotinib 400 mg b.i.d were observed to have hyperglycaemia, 50% of patients treated with 300 mg b.i.d and only 30% of patients treated with imatinib 400 mg/day were observed to have hyperglycaemia (Larson et al., 2014). Nine out of thirteen chronic myeloid leukaemia patients being treated with nilotinib showed increased fasting glucose levels as compared to baseline pre-nilotinib therapy (Breccia et al., 2007). In a study by Philipp le Courte an increase in grade ¾ hyperglycaemia was observed during nilotinib therapy (Larson et al., 2014). Fasting 1-h and 2-h plasma glucose concentrations increased after 3 months of nilotinib treatment (Racil et al., 2013). Based of fasting glucose (8.1 and 7.3 mmol/L), 2 patients fulfilled the criteria of diabetes mellitus while on nilotinib therapy and 2 patients had impaired glucose tolerance. It is worth noting that most studies have concluded that the hyperglycaemia caused by nilotinib is clinically insignificant as it is usually mild, transient and manageable (Saglio et al., 2010). However, understanding the mechanism by which the hyperglycaemia is brought about is crucial, especially for patients with both chronic myeloid leukaemia and diabetes mellitus.
The mechanism in which nilotinib impairs glucose metabolism is not clearly known. In one case of pre-existing severe type 2 diabetes mellitus, nilotinib was shown to directly affect glucose metabolism via impairment of endogenous insulin secretion. When nilotinib was discontinued the impairment to endogenous insulin secretion was reserved (Ito et al., 2013). Insulin resistance which was calculated by the mathematical model HOMA2-IR increased significantly during nilotinib therapy (Racil et al., 2013). Insulin sensitivity index HOMA2-%S significantly decreased as well as ISI0,120. HOMA2-%S and HOMA2IR represent hepatic insulin resistance while ISI0,120 represents peripheral insulin resistance. The cABL domain is associated with the insulin signaling pathway. It has been hypothesized that by blocking the c-ABL, tyrosine kinase therapy affects the insulin receptor pathway (Genua et al., 2009). Plasma concentration of adiponectin has been observed to have decreased during nilotinib therapy. Adiponectin exerts a potent insulin-sensitizing effect (Shehzad et al., 2012; Racil et al., 2013).
3.4.5 Gefitinib
Gefitinib is a 4-anilinoquinazoline derivative, a potent and selective ATP competitive inhibitor of EGFR and HER-2 kinases (Rahman et al., 2014). Gefitinib binds to the ATP binding pockets of EGFR’s active form, hence it is classified as a Type-I kinase inhibitor. Gefitinib forms H-bonds with Met793 in the hinge region (Ayala-Aguilera et al., 2022). Mutations in the extracellular segment of the EGFR such as deletion have been shown to result in constitutive growth factor independent EGFR activation leading to the overexpression of the EGFR, which causes the progression of numerous malignancies (Ekstrand et al., 1992). A common mutation of the EGFR is the L858R which results from exon 19 substitution of leucine with arginine, and this accounts for 40% of mutations in non-small cell lung cancer. The other mutation is exon 19 deletion. These mutations cluster around the active site cleft of the tyrosine kinase domain of the EGFR (Chan et al., 2006). Studies of direct binding measurements have shown that gefitinib binds 20-fold more tightly to the L858R mutant form of the EGFR as compared to the wild type receptors. Cells with mutant EGFR tend to be more sensitive to tyrosine kinase inhibitors than cells that express the wild type kinase (Yun et al., 2007). Gefitinib is considered to be relatively selective, however it demonstrates moderate activity in other receptor tyrosine kinase resulting in common adverse events such as diarrhoea, rash, nausea and dry skin (Culy and Faulds, 2002). In 2004 gefitinib became the first epidermal growth factor receptor tyrosine kinase inhibitor for molecular targeted chemotherapy of unresectable non-small cell lung cancer in Japan. The antineoplastic effect of gefitinib comes from inhibiting the activation of ErbB-1 (EGFR) (Araki et al., 2012). Clinically gefitinib is used for treatment of chemo resistant non-small cell lung cancer (Rahman et al., 2014).
Gefitinib has been shown to have a dramatic effect on a limited number of patients. Studies have suggested that it is ineffective in 70%–80% of patients with non-small cell lung cancer (Fukuoka et al., 2023). EGFR inhibitors, erlotinib and gefitinib seem to have secondary pharmacological mechanisms of action, as they appear to be inhibitors of Tumour Necrosis Factor Alpha (TNF-α). They may prevent progression of type 1 diabetes mellitus as they appear to be effective in the treatment of related TNF-α mediated inflammatory diseases. This is achieved through the inhibition of the TNF-α (Brooks, 2012). Inflammation associated with TNF-α has been linked to the development of insulin resistance (Borst, 2004). Gefitinib may be of value in the treatment of type I diabetes mellitus by inhibition of TNF-α, leading to reduction in inflammation, decreased insulin resistance and slowed progression of Beta-cell destruction (Brooks, 2012).
In a rat model study, gefitinib improved insulin sensitivity in rats with type 2 diabetes. This is hypothesised to occur through the increase in PI3K/AKT pathway of insulin signaling (Sun et al., 2009). Gefitinib has inhibitory action on receptor-interacting protein kinase 2 (RIPK2). RIPK2 propagate immune responses that are associated with dysglycemia during obesity and insulin resistance (Tigno-Aranjuez et al., 2014; Denou et al., 2015). In a mice model study, gefitinib improved glucose control independently of RIPK2. In the absence of RIPK2 gefitinib improved insulin sensitivity and lowered insulin secretion (Duggan et al., 2020). One study showed that there was a significant decrease in fluorodeoxyglucose (FDG) uptake in gefitinib sensitive cell lines (Su et al., 2006). FDG uptake is a marker for tissue uptake of glucose (Ashraf and Goyal, 2023). In the same study immunoblots showed the translocation of GLUT3 from the plasma to the cytosol (Su et al., 2006).
3.4.6 Dasatinib
Dasatinib is a type 1 inhibitor of ABL tyrosine kinase and a type I1/2A inhibitor of LYN. It is an orally available ABL inhibitor which is different to imatinib such that it binds to both the active and inactive conformations of the ABL kinase domain (Talpaz et al., 2006). In 2006, the FDA granted accelerated approval for the treatment of chronic myeloid leukaemia and Ph+ acute lymphoblastic leukaemia (Ph+ ALL) with dasatinib in patients that had demonstrated resistance or intolerance to prior therapy with imatinib (Kantarjian et al., 2006; Jabbour and Lipton, 2013). Resistance in the large proportion of patients on imatinib therapy is a result of point mutations of the BCR-ABL gene and activation of alternative signaling pathways the likes of SRC family kinase (Kantarjian et al., 2006). Dasatinib is a potent ATP competitive inhibitor, it has less stringent binding requirements as compared to imatinib; hence it shows inhibition activity against many imatinib resistant BCR-ABL mutations. Dasatinib is active against 14 out of 15 imatinib resistant BCR-ABL mutants (Shah et al., 2004). The one mutant not inhibited by dasatinib is the T315I mutant, which is a single mutation deep within the ATP-biding pockets of the ABL tyrosine kinase (Weisberg et al., 2005). Dasatinib is a multikinase inhibitor that shows activity against PDGFR- β, KIT, and SRC family kinase, and because of this range of inhibition, dasatinib has many side effects including haemorrhage, teratogenicity, fluid retention and myelosuppression (Steegmann et al., 2012).
In a retrospective study on blood glucose concentrations in eight diabetic and non-diabetic patients treated with dasatinib, a mean decrease in blood glucose of 53 mg/dL was observed. Some patients on dasatinib treatment were able to discontinue their antidiabetic medication (Agostino et al., 2011). A case report of a 57-year-old diabetic chronic myeloid leukaemia patient had their insulin requirements declined upon the introduction of dasatinib. Within 2 weeks of dasatinib treatment, insulin treatment was discontinued. The patient’s fasting C-peptide immunoreactivity increased two-fold, suggesting that dasatinib allowed for the recovery of insulin production (Ono et al., 2012). In another case report, a 65-year-old woman with type 2 diabetes mellitus, had her glycaemic index improved to <6.0% upon dasatinib treatment initiation. Quantitative insulin sensitivity check index rapidly improved followed by an improvement in insulin sensitivity. This case report demonstrated that dasatinib might have negative as well as positive effects on glucose metabolism (Iizuka et al., 2016). In a retrospective study, patients that were treated with dasatinib had a mean decrease in HbA1c of 0.80 absolute point and these patients required 8.2 less total daily insulin units on average. This led to a conclusion that dasatinib demonstrates antidiabetic effects and may be considered for the use as a novel diabetic therapy. The mechanism by which dasatinib instigates these antidiabetic effects have not been stated and remains unknown (Salaami et al., 2021).
Evidence suggests that tyrosine kinase play a role in regulating insulin secretion. Various mechanisms underpin the involvement of SRC kinase, including their potential regulation through myristylation, dynamic palmitoylation, and acylation. All which contribute to the inhibition of insulin secretion (Breccia et al., 2008). SRC kinase pathways are integral in the extracellular calcium-dependent secretion of pancreatic enzymes and insulin (Cheng et al., 2007). Dasatinib’s inhibition of SRC kinase pathways leads to the reduction of fasting glucose observed in the studies mentioned.
SRC-family kinase are proto-oncogenes that play a role in cell processes, including cell differentiation, cell growth, cell morphology, mortality and survival (Parsons and Parsons, 2004). Insulin secretion and neurotransmitter release both occur via exocytosis (Cheng et al., 2003; Baldwin et al., 2006). Exocytosis is an ubiquitously occurring mechanism responsible for cell to cell communication (SIM et al., 2003). It has been suggested that SRC-family tyrosine kinase inhibit neurotransmitter release and it is believed that insulin secretion and neurotransmitter release have similar regulatory mechanisms. Cheng et al. (2007) observed that SU-6656 and PP2, structurally different SRC-family tyrosine kinase inhibitors, enhanced Ca2+-induced insulin secretion in INS-1 cells and rat pancreatic islets in both a time and dose dependent manner. This study illustrates that one or more SRC-family tyrosine kinase exert a tonic inhibitory role on Ca2+-dependent insulin secretion. The role of SRC-family tyrosine kinase in Ca2+-induced insulin secretion is illustrated in Figure 3.
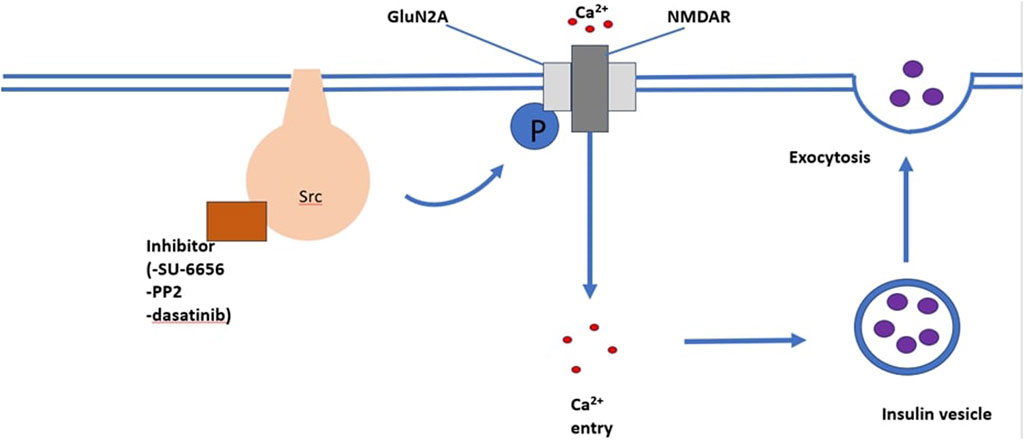
FIGURE 3. An illustration of the mechanism through which the SRC-family kinase pathway enhances insulin secretion. The binding of inhibitors such as SU-6656, PP2, and dasatinib enhances Ca2+ induced insulin secretion.
SRC-family tyrosine kinase are non-receptor tyrosine kinase. They are expressed in high levels on secondary vesicles and plasma membranes of cells that are specialised for exocytosis. Cells which fall under this category include neuronal and endocrine cells (Ely et al., 1994). Evidence from studies suggests that protein tyrosine kinase are associated with the regulation of insulin secretion (Sorenson et al., 1994). Small molecule tyrosine kinase inhibitors have been observed to induce increases in insulin secretion in INS-1 cells (Verspohl et al., 1995). On the other hand, some studies have reported decreased insulin secretion due to small molecule tyrosine kinase inhibitors in adult rat islets (Persaud et al., 1999). These varying observations highlight that different protein tyrosine kinases have different or opposing outcomes on insulin secretion. The exact function of protein tyrosine kinase in insulin secretion is unknown.
4 Authors perspective and recommendations
The development of small molecule tyrosine kinase inhibitors has revolutionized the cancer treatment landscape. Small molecule kinase inhibitors have been developed for targeted cancer therapy, hence the increase in their popularity and use. Despite their great potential, there aremajor challenges pertaining to toxicity and resistance. Small molecule tyrosine kinase inhibitors have been linked to altered glucose metabolism in numerous studies, with some small molecule tyrosine kinase inhibitors inducing hyperglycaemia including nilotinib, ceritinib and rociletinib. The hyperglycaemia induced have been noted as one of their dose-limiting side effects which has been attributed to impaired pancreatic β-cell insulin secretion or development of insulin resistance and inhibition of the insulin receptor. However, studies have shown diabetic cancer patients on small molecule tyrosine kinase inhibitors demonstrate improved glycaemic control and require a reduction in their daily insulin requirements or diabetic medication in both type 1 and type 2 diabetes. Examples of such kinase inhibitors include sunitinib, imatinib, and erlotinib. The improved blood glucose concentration is achieved potentially in part through preserving functional βcell mass and increased insulin sensitivity or insulin secretion.
The exact mechanism by which small molecule tyrosine kinase inhibitors elicit an increase or a decrease in patients largely remains elusive and unknown. Given their role and increase in use, this is a cause for concern, particularly in hospitalized comorbid patients, as slight alterations in blood glucose may result in complications or mortality. Research into the mechanism needs to be prioritized, with the aim to shed light and assist prescribers with the administration of treatment which will offer the patients the best possible prognosis.
Some authors have highlighted small molecule tyrosine kinase inhibitors potential as novel therapies for diabetes (Prada and Saad, 2013; Fountas et al., 2015). Considering the increasing incidence and prevalence of diabetes, the novel therapies school of thought is a well-grounded approach. However, before we explore their potential use as antidiabetics, there is a necessity to fully elucidate their glycaemic altering mechanism at both cellular and molecular level. There is also a necessity to establish discrepancies observed amongst small molecule tyrosine kinase inhibitors in the context of glycaemic control. Perhaps the answer lays in the chemical structural differences. This includes uncovering whether the alteration is dependent on the type of the small molecule tyrosine kinase inhibitors. In the short term, this will be a valuable tool for physicians, prescribers, and other healthcare professionals. For example, diabetic patients with cancer being treated by small molecule tyrosine kinase inhibitors may need to be assigned to those that are known to improve blood glucose concentration. This may have potential benefits including a reduction in polypharmacy which can prove to have precipitate adverse effects. Our line of thought can be backed by studies which indicated that some patients on small molecule tyrosine kinase inhibitors required dose reductions in their diabetic medication with some case patients discontinuing diabetes treatment while under a small molecule kinase inhibitor therapy. With the opposite being true as well, patients at risk of hyperglycaemia complications including neuropathy or kidney damage may need to be kept away from small molecule tyrosine kinase inhibitors known to induce hyperglycaemia.
These strategies are only possible if the glycaemic-altering mechanisms are known, hence further research into the inner workings of small molecule tyrosine kinase inhibitors is a necessity for the healthcare system. Uncovering how glucose metabolism is altered by small molecule tyrosine kinase inhibitors will provide more insight into glycaemic control in cancer patients. This will not only open the possibility of potential alternative methods to manage glycaemic dysregulation, but it may shed light on what is currently known about the pathophysiology of diabetes and cancer. This is of paramount importance given the global demography of an aging population. It is well documented that the incidence of morbidities such as cancer and diabetes is higher in the geriatric population as compared to the others.
The long-term effects of the altered glucose metabolism by small molecule tyrosine kinase inhibitors are poorly documented. There is a necessity to elucidate whether the cases of glycaemic dysregulation in patients are isolated events that are due to particular conditions. For instance, certain morbidities render patients more susceptible to phenomena which individuals where these morbidities are absent are not as susceptible. This calls for large scale follow up studies and not just limit our understanding to retrospective case studies. This would help uncover if the effects elicited by the small molecule tyrosine kinase inhibitors are limited to a particular demography. In the event that the effects can be expected in the general population, the relevant healthcare professionals, especially oncologists need to be informed accordingly for the safe and optimum use of these compounds.
Another vital area of research is to uncover the factors that may sensitize a patient to glycaemic dysregulation observed in small molecule tyrosine kinase inhibitors. Highlighting these factors before commencing therapy has the potential to improve patient health outcomes and possibly reduce the number of hospital days.
Existing evidence suggests that small molecule tyrosine kinase inhibitors have a lot of potential as novel therapy for DM. This is not the case with all but small molecule tyrosine kinase inhibitors by way of example imatinib. The chronic nature of DM costs and side effects are major concerns when proposing the use of small molecule tyrosine kinase inhibitors in DM treatment. The estimated cost of small molecule tyrosine kinase inhibitors for chronic myeloid leukaemia ranges between $3 632 and $8 429 USD (Talon et al., 2021). While according to drugs.com the price of a pack of 30 metformin 500 mg oral tablets is approximately $13,15 (Drugs.com, 2024a). According to drugs.com 30 tablets of 100 mg oral imatinib tablets and 28 oral tablets of 12,5 mg sunitinib have an approximate cost of $219 and $5 198–$5 492 respectively (Drugs.com, 2024b). For chronic use in the management of DM the cost-benefit ratio is not in favour of small molecule kinase inhibitors when compared to current antidiabetic agents which already have healthcare budgets across the globe stretched. The cost of small molecule tyrosine kinase inhibitors is a major obstacle which would limit their use in DM. Another concern to highlight is that the current price of small molecule tyrosine kinase inhibitors is a direct contributing factor of non-adherence mortality from certain cancers.
It has been reported that nearly all patients experience treatment related adverse events while on small molecule kinase inhibitor treatment. One study stated that up to 70% of the patients undergoing small molecule tyrosine kinase inhibitors treatment experienced adverse events. Caldemeyer et al. (2016) reported that 60% of the patients on imatinib treatment experienced oedema, 50% nausea, 49% muscle cramps, and 47% musculoskeletal pain. However overall small molecule tyrosine kinase inhibitors appear to be well tolerated with patients experiencing mild or moderate adverse events. Some common adverse events include skin toxicity, diarrhoea, and fatigue (Hartmann et al., 2009; Caldemeyer et al., 2016; Płużański and Piórek, 2016). Adverse events led to discontinuation of imatinib in only 4% of patients in some studies (Druker et al., 2006). Small molecule tyrosine kinase inhibitors share the same mechanism of action of competitive inhibition of ATP at the catalytic site of tyrosine kinase. However, they differ in the specific tyrosine kinase they target, hence they elicit the varied pharmacological responses and side effects. This difference opens the possibility for targeted therapy and minimal side effects with regards to their use in DM management.
Indeed, understanding the mechanism of action at the molecular level is a starting point, which should then be followed by elucidating the structure activity relationships. Deeper understanding of structure activity relationships will allow these compounds to be modified accordingly. As mentioned earlier, the mechanism of action of small molecule tyrosine kinase inhibitors is largely the same (ATP competitive inhibition), while the differences are conferred by the spectrum of targeted tyrosine kinases. Elucidating why certain small molecule tyrosine kinase inhibitors have higher affinity for specific tyrosine kinase as compared to others is key. Small molecule tyrosine kinase inhibitors effect on glucose metabolism is strongly linked to the tyrosine kinase they interact with. Structural modification of small molecule tyrosine kinase inhibitors paves the way for specificity in terms of the tyrosine kinase they will interact strongly with. This in turn may be utilized to optimise their role in glucose metabolism.
5 Conclusion
In conclusion, the development of small molecule tyrosine kinase inhibitors has undoubtedly transformed the landscape of cancer treatment, offering new hope for targeted therapy. Small molecule tyrosine kinase inhibitors have however been associated with glycaemic dysregulation in patients. With some small molecule tyrosine kinase inhibitors improving glycaemic regulation in diabetic patients while others induce clinically insignificant hyperglycaemia. The focus of this paper aimed to understand small molecule tyrosine kinase inhibitors on glycaemic control and their mechanisms with the goal of improving patient health outcomes and prognosis. This is vital given the incidence and prevalence of diabetes and cancer. As the global population ages and the incidence of cancer and diabetes continues to rise, understanding the complex interplay between kinase inhibitors and glucose metabolism is not just a medical imperative but a fundamental step toward advancing our knowledge of the pathophysiology of these diseases. Research in this field holds the promise of improving patient care, optimizing treatment strategies, and enhancing the overall quality of healthcare for a growing demographic in need of effective therapeutic solutions. Furthermore, considering the prevalence of diabetes, it would be worthwhile for researchers to explore repurposing strategies of small molecule tyrosine kinase inhibitors towards diabetes mellitus.
Author contributions
TM: Conceptualization, Investigation, Writing–original draft, Writing–review and editing. MM: Writing–review and editing. AK: Writing–review and editing. PN: Writing–review and editing. NS: Conceptualization, Funding acquisition, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The study was supported by the National Research Foundation South Africa: TTK220318196.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agostino, N. M., Chinchilli, V. M., Lynch, C. J., Koszyk-Szewczyk, A., Gingrich, R., Sivik, J., et al. (2011). Effect of the tyrosine kinase inhibitors (sunitinib, sorafenib, dasatinib, and imatinib) on blood glucose levels in diabetic and nondiabetic patients in general clinical practice. J. Oncol. Pharm. Pract. Off. Publ. Int. Soc. Oncol. Pharm. Pract. 17, 197–202. doi:10.1177/1078155210378913
Albrecht, H., Stellbrink, H. J., and Arasth, K. (1993). Didanosine-induced disorders of glucose tolerance. Ann. Intern Med. 119, 1050. doi:10.7326/0003-4819-119-10-199311150-00017
Alwhaibi, M., Balkhi, B., Alhawassi, T. M., Alkofide, H., Alduhaim, N., Alabdulali, R., et al. (2018). Polypharmacy among patients with diabetes: a cross-sectional retrospective study in a tertiary hospital in Saudi Arabia. BMJ Open 8, 020852. doi:10.1136/bmjopen-2017-020852
Anello, M., Gilon, P., and Henquin, J. C. (1999). Alterations of insulin secretion from mouse islets treated with sulphonylureas: perturbations of Ca2+ regulation prevail over changes in insulin content. Br. J. Pharmacol. 127, 1883–1891. doi:10.1038/sj.bjp.0702731
Araki, T., Yashima, H., Shimizu, K., Aomori, T., Hashita, T., Kaira, K., et al. (2012). Review of the treatment of non-small cell lung cancer with gefitinib. Clin. Med. Insights Oncol. 6, 407–421. CMO.S7340. doi:10.4137/CMO.S7340
Arora, A., and Scholar, E. M. (2005). Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 315, 971–979. doi:10.1124/jpet.105.084145
Ashcroft, F. M., and Rorsman, P. (1989). Electrophysiology of the pancreatic beta-cell. Prog. Biophys. Mol. Biol. 54, 87–143. doi:10.1016/0079-6107(89)90013-8
Ashraf, M. A., and Goyal, A. (2023). 2 fludeoxyglucose (18F). St. Petersburg, FL: StatPearls. Treasure Island (FL): StatPearls Publishing.
Atkins, M., Jones, C. A., and Kirkpatrick, P. (2006). Sunitinib maleate. Nat. Rev. Drug Discov. 5, 279–280. doi:10.1038/nrd2012
Ayala-Aguilera, C. C., Valero, T., Lorente-Macías, Á, Baillache, D. J., Croke, S., and Unciti-Broceta, A. (2022). Small molecule kinase inhibitor drugs (1995–2021): medical indication, Pharmacology, and synthesis. J. Med. Chem. 65, 1047–1131. doi:10.1021/acs.jmedchem.1c00963
Baldwin, M. L., Cammarota, M., Sim, A. T. R., and Rostas, J. A. P. (2006). Src family tyrosine kinases differentially modulate exocytosis from rat brain nerve terminals. Neurochem. Int. 49, 80–86. doi:10.1016/j.neuint.2006.01.002
Banning, A., Kurrle, N., Meister, M., and Tikkanen, R. (2014). Flotillins in receptor tyrosine kinase signaling and cancer. Cells 3, 129–149. doi:10.3390/cells3010129
Bardelli, A., Parsons, D. W., Silliman, N., Ptak, J., Szabo, S., Saha, S., et al. (2003). Mutational analysis of the tyrosine kinome in colorectal cancers. Science 300, 949. doi:10.1126/science.1082596
Barnes, K., McIntosh, E., Whetton, A. D., Daley, G. Q., Bentley, J., and Baldwin, S. A. (2005). Chronic myeloid leukaemia: an investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene 24, 3257–3267. doi:10.1038/sj.onc.1208461
Baselga, J. (2006). Targeting tyrosine kinases in cancer: the second wave. Science 312, 1175–1178. doi:10.1126/science.1125951
Bastaki, S. S. (2005). Diabetes mellitus and its treatment. Int. J. Diabetes Metab. 13, 111–134. doi:10.1159/000497580
Bhargava, P., and Robinson, M. O. (2011). Development of second-generation VEGFR tyrosine kinase inhibitors: current status. Curr. Oncol. Rep. 13, 103–111. doi:10.1007/s11912-011-0154-3
Bhattacharya, S., Dey, D., and Roy, S. S. (2007). Molecular mechanism of insulin resistance. J. Biosci. 32, 405–413. doi:10.1007/s12038-007-0038-8
Billemont, B., Medioni, J., Taillade, L., Helley, D., Meric, J. B., Rixe, O., et al. (2008). Blood glucose levels in patients with metastatic renal cell carcinoma treated with sunitinib. Br. J. Cancer 99, 1380–1382. doi:10.1038/sj.bjc.6604709
Blay, J. Y. (2010). Pharmacological management of gastrointestinal stromal tumours: an update on the role of sunitinib. Ann. Oncol. 21, 208–215. doi:10.1093/annonc/mdp291
Boren, J., Cascante, M., Marin, S., Comin-Anduix, B., Centelles, J. J., Lim, S., et al. (2001). Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J. Biol. Chem. 276, 37747–37753. doi:10.1074/jbc.M105796200
Borst, S. E. (2004). The role of TNF-alpha in insulin resistance. Endocrine 23, 177–182. doi:10.1385/ENDO:23:2-3:177
Breccia, M., and Alimena, G. (2009). The metabolic consequences of imatinib mesylate: changes on glucose, lypidic and bone metabolism. Leuk. Res. 33, 871–875. doi:10.1016/j.leukres.2009.01.040
Breccia, M., Muscaritoli, M., and Alimena, G. (2005). Reduction of glycosylated hemoglobin with stable insulin levels in a diabetic patient with chronic myeloid leukemia responsive to imatinib. Haematologica 90, ECR21. doi:10.3324/%x
Breccia, M., Muscaritoli, M., Aversa, Z., Mandelli, F., and Alimena, G. (2004). Imatinib mesylate may improve fasting blood glucose in diabetic Ph+ chronic myelogenous leukemia patients responsive to treatment. J. Clin. Oncol. 22, 4653–4655. doi:10.1200/JCO.2004.04.217
Breccia, M., Muscaritoli, M., Cannella, L., Stefanizzi, C., Frustaci, A., and Alimena, G. (2008). Fasting glucose improvement under dasatinib treatment in an accelerated phase chronic myeloid leukemia patient unresponsive to imatinib and nilotinib. Leuk. Res. 32, 1626–1628. doi:10.1016/j.leukres.2008.01.015
Breccia, M., Muscaritoli, M., Gentilini, F., Latagliata, R., Carmosino, I., Rossi Fanelli, F., et al. (2007). Impaired fasting glucose level as metabolic side effect of nilotinib in non-diabetic chronic myeloid leukemia patients resistant to imatinib. Leuk. Res. 31, 1770–1772. doi:10.1016/j.leukres.2007.01.024
Brooks, M. B. (2012). Erlotinib and gefitinib, small-molecule EGFR inhibitors. New uses for old drugs? Br. J. Diabetes Vasc. Dis. 12, 195–196. doi:10.1177/1474651412458460
Bryant, N. J., Govers, R., and James, D. E. (2002). Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 3, 267–277. doi:10.1038/nrm782
Buffier, P., Bouillet, B., Smati, S., Archambeaud, F., Cariou, B., and Verges, B. (2018). Expert opinion on the metabolic complications of new anticancer therapies: tyrosine kinase inhibitors. Ann. Endocrinol. 79, 574–582. doi:10.1016/j.ando.2018.07.011
Caldemeyer, L., Dugan, M., Edwards, J., and Akard, L. (2016). Long-term side effects of tyrosine kinase inhibitors in chronic myeloid leukemia. Curr. Hematol. Malig. Rep. 11, 71–79. doi:10.1007/s11899-016-0309-2
Chakkera, H., Kudva, Y., and Kaplan, B. (2017). Calcineurin inhibitors: pharmacologic mechanisms impacting both insulin resistance and insulin secretion leading to glucose dysregulation and diabetes mellitus. Clin. Pharmacol. Ther. 101, 114–120. doi:10.1002/cpt.546
Chan, S. K., Gullick, W. J., and Hill, M. E. (2006). Mutations of the epidermal growth factor receptor in non-small cell lung cancer – search and destroy. Eur. J. Cancer 42, 17–23. doi:10.1016/j.ejca.2005.07.031
Chen, Y., Tortorici, M. A., Garrett, M., Hee, B., Klamerus, K. J., and Pithavala, Y. K. (2013). Clinical Pharmacology of axitinib. Clin. Pharmacokinet. 52, 713–725. doi:10.1007/s40262-013-0068-3
Cheng, H., and Force, T. (2010). Why do kinase inhibitors cause cardiotoxicity and what can be done about it? Prog. Cardiovasc Dis. 53, 114–120. doi:10.1016/j.pcad.2010.06.006
Cheng, H., Straub, S. G., and Sharp, G. W. G. (2003). Protein acylation in the inhibition of insulin secretion by norepinephrine, somatostatin, galanin, and PGE2. Am. J. Physiol-Endocrinol Metab. 285, 287–294. doi:10.1152/ajpendo.00535.2002
Cheng, H., Straub, S. G., and Sharp, G. W. G. (2007). Inhibitory role of Src family tyrosine kinases on Ca2+-dependent insulin release. Am. J. Physiol-Endocrinol Metab. 292, 845–852. doi:10.1152/ajpendo.00103.2006
Cismowski, M. J. (2007). “Tyrosine kinase inhibitors,” in xPharm: the comprehensive Pharmacology reference. Editors S. J. Enna, and D. B. Bylund (New York: Elsevier), 1–4.
Cox, D. S., Cox, A. D., Sturm, L., and Zimet, G. (2010). Behavioral interventions to increase HPV vaccination acceptability among mothers of young girls. Health Psychol. Off. J. Div. Health Psychol. Am. Psychol. Assoc. 29, 29–39. doi:10.1037/a0016942
Cox, K. J., Shomin, C. D., and Ghosh, I. (2011). Tinkering outside the kinase ATP box: allosteric (type IV) and bivalent (type V) inhibitors of protein kinases. Future Med. Chem. 3, 29–43. doi:10.4155/fmc.10.272
Culy, C. R., and Faulds, D. (2002). Gefitinib. Drugs. 62, 2237–2250. doi:10.2165/00003495-200262150-00008
Denou, E., Lolmède, K., Garidou, L., Pomie, C., Chabo, C., Lau, T. C., et al. (2015). Defective NOD2 peptidoglycan sensing promotes diet-induced inflammation, dysbiosis, and insulin resistance. EMBO Mol. Med. 7, 259–274. doi:10.15252/emmm.201404169
Dobrică, E. C., Găman, M. A., Cozma, M. A., Bratu, O. G., Pantea Stoian, A., and Diaconu, C. C. (2019). Polypharmacy in type 2 diabetes mellitus: insights from an internal medicine department. Med. Mex. 55, 436. doi:10.3390/medicina55080436
Drugs.com (2024a). Metformin prices, coupons, copay & patient assistance. Available at: https://www.drugs.com/price-guide/metformin (Accessed January 3, 2024).
Drugs.com (2024b). Sunitinib prices, coupons, copay & patient assistance. Available at: https://www.drugs.com/price-guide/sunitinib (Accessed January 3, 2024).
Druker, B. J., Guilhot, F., O’Brien, S. G., Gathmann, I., Kantarjian, H., Gattermann, N., et al. (2006). Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 355, 2408–2417. doi:10.1056/NEJMoa062867
Druker, B. J., Talpaz, M., Resta, D. J., Peng, B., Buchdunger, E., Ford, J. M., et al. (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031–1037. doi:10.1056/NEJM200104053441401
Du, Z., and Lovly, C. M. (2018). Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 17, 58. doi:10.1186/s12943-018-0782-4
Duggan, B. M., Cavallari, J. F., Foley, K. P., Barra, N. G., and Schertzer, J. D. (2020). RIPK2 dictates insulin responses to tyrosine kinase inhibitors in obese mice. bioRxiv. doi:10.1210/endocr/bqaa086
Ekstrand, A. J., Sugawa, N., James, C. D., and Collins, V. P. (1992). Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. 89, 4309–4313. doi:10.1073/pnas.89.10.4309
Eliasson, L., Abdulkader, F., Braun, M., Galvanovskis, J., Hoppa, M. B., and Rorsman, P. (2008). Novel aspects of the molecular mechanisms controlling insulin secretion. J. Physiol. 586, 3313–3324. doi:10.1113/jphysiol.2008.155317
Ely, C. M., Tomiak, W. M., Allen, C. M., Thomas, L., Thomas, G., and Parsons, S. J. (1994). pp60c-src enhances the acetylcholine receptor-dependent catecholamine release in vaccinia virus-infected bovine adrenal chromaffin cells. J. Neurochem. 62, 923–933. doi:10.1046/j.1471-4159.1994.62030923.x
Escudier, B., and Gore, M. (2011). Axitinib for the management of metastatic renal cell carcinoma. Drugs rd. 11, 113–126. doi:10.2165/11591240-000000000-00000
Fathallah, N., Slim, R., Larif, S., Hmouda, H., and Ben Salem, C. (2015). Drug-induced hyperglycaemia and diabetes. Drug Saf. 38, 1153–1168. doi:10.1007/s40264-015-0339-z
Filipponi, P., Marcelli, M., Nicoletti, I., Pacifici, R., Santeusanio, F., and Brunetti, P. (1983). Suppressive effect of long term sulfonylurea treatment on A, B, and D cells of normal rat pancreas. Endocrinology 113, 1972–1979. doi:10.1210/endo-113-6-1972
Fountas, A., Diamantopoulos, L. N., and Tsatsoulis, A. (2015). Tyrosine kinase inhibitors and diabetes: a novel treatment paradigm? Trends Endocrinol. Metab. 26, 643–656. doi:10.1016/j.tem.2015.09.003
Fukuoka, M., Yano, S., Giaccone, G., Tamura, T., Nakagawa, K., Douillard, J. Y., et al. (2023). Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non–small-cell lung cancer. J. Clin. Oncol. 41, 1162–1171. doi:10.1200/JCO.22.02499
Gan, H. K., Seruga, B., and Knox, J. J. (2009). Sunitinib in solid tumors. Expert Opin. Investig. Drugs 18, 821–834. doi:10.1517/13543780902980171
Genua, M., Pandini, G., Cassarino, M. F., Messina, R. L., and Frasca, F. (2009). “Chapter 4 c-abl and insulin receptor signalling,” in Vitamins & hormones (Academic Press), 77–105.
Giles, F. J., O’Dwyer, M., and Swords, R. (2009). Class effects of tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia. Leukemia 23, 1698–1707. doi:10.1038/leu.2009.111
Glovaci, D., Fan, W., and Wong, N. D. (2019). Epidemiology of diabetes mellitus and cardiovascular disease. Curr. Cardiol. Rep. 21, 21. doi:10.1007/s11886-019-1107-y
Gold, G., Gishizky, M. L., and Grodsky, G. M. (1982). Evidence that glucose "marks" beta cells resulting in preferential release of newly synthesized insulin. Science 218, 56–58. doi:10.1126/science.6181562
Goldstein, B. J. (2001). Protein-tyrosine phosphatase 1B (PTP1B): a novel therapeutic target for type 2 diabetes mellitus, obesity and related states of insulin resistance. Curr. Drug Targets - Immune Endocr. Metab. Disord. 1, 265–275. doi:10.2174/1568008013341163
Golemovic, M., Verstovsek, S., Giles, F., Cortes, J., Manshouri, T., Manley, P. W., et al. (2005). AMN107, a novel aminopyrimidine inhibitor of bcr-abl, has in vitro activity against imatinib-resistant chronic myeloid leukemia. Clin. Cancer Res. 11, 4941–4947. doi:10.1158/1078-0432.CCR-04-2601
Goodge, K. A., and Hutton, J. C. (2000). Translational regulation of proinsulin biosynthesis and proinsulin conversion in the pancreatic beta-cell. Semin. Cell Dev. Biol. 11, 235–242. doi:10.1006/scdb.2000.0172
Gottschalk, S., Anderson, N., Hainz, C., Eckhardt, S. G., and Serkova, N. J. (2004). Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin. Cancer Res. 10, 6661–6668. doi:10.1158/1078-0432.CCR-04-0039
Gromada, J., Bokvist, K., Ding, W. G., Holst, J. J., Nielsen, J. H., and Rorsman, P. (1998). Glucagon-like peptide 1 (7-36) amide stimulates exocytosis in human pancreatic beta-cells by both proximal and distal regulatory steps in stimulus-secretion coupling. Diabetes 47, 57–65. doi:10.2337/diab.47.1.57
Hadova, K., Mesarosova, L., Kralova, E., Doka, G., Krenek, P., and Klimas, J. (2021). The tyrosine kinase inhibitor crizotinib influences blood glucose and mRNA expression of GLUT4 and PPARs in the heart of rats with experimental diabetes. Can. J. Physiol. Pharmacol. 99, 635–643. doi:10.1139/cjpp-2020-0572
Hägerkvist, R., Jansson, L., and Welsh, N. (2007). Imatinib mesylate improves insulin sensitivity and glucose disposal rates in rats fed a high-fat diet. Clin. Sci. 114, 65–71. doi:10.1042/CS20070122
Hartmann, J. T., Haap, M., Kopp, H. G., and Lipp, H. P. (2009). Tyrosine kinase inhibitors - a review on Pharmacology, metabolism and side effects. Curr. Drug Metab. 10, 470–481. doi:10.2174/138920009788897975
Henquin, J. C. (2000). Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49, 1751–1760. doi:10.2337/diabetes.49.11.1751
Henquin, J. C., Dufrane, D., and Nenquin, M. (2006). Nutrient control of insulin secretion in isolated normal human islets. Diabetes 55, 3470–3477. doi:10.2337/db06-0868
Hubbard, S. R., and Miller, W. T. (2007). Receptor tyrosine kinases: mechanisms of activation and signaling. Curr. Opin. Cell Biol. 19, 117–123. doi:10.1016/j.ceb.2007.02.010
Hudson, C. D., Hagemann, T., Mather, S. J., and Avril, N. (2014). Resistance to the tyrosine kinase inhibitor axitinib is associated with increased glucose metabolism in pancreatic adenocarcinoma. Cell Death Dis. 5, 1160. doi:10.1038/cddis.2014.125
Hu-Lowe, D. D., Zou, H. Y., Grazzini, M. L., Hallin, M. E., Wickman, G. R., Amundson, K., et al. (2008). Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 14, 7272–7283. doi:10.1158/1078-0432.CCR-08-0652
Hunter, T. (1995). Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80, 225–236. doi:10.1016/0092-8674(95)90405-0
Iizuka, K., Niwa, H., Kato, T., and Takeda, J. (2016). Dasatinib improves insulin sensitivity and affects lipid metabolism in a patient with chronic myeloid leukaemia. Case Rep. 2016, bcr2015214284. doi:10.1136/bcr-2015-214284
Ito, Y., Miyamoto, T., Chong, Y., Maki, T., Akashi, K., and Kamimura, T. (2013). Nilotinib exacerbates diabetes mellitus by decreasing secretion of endogenous insulin. Int. J. Hematol. 97, 135–138. doi:10.1007/s12185-012-1222-7
Iurlo, A., Orsi, E., Cattaneo, D., Resi, V., Bucelli, C., Orofino, N., et al. (2015). Effects of first- and second-generation tyrosine kinase inhibitor therapy on glucose and lipid metabolism in chronic myeloid leukemia patients: a real clinical problem? Oncotarget 6, 33944–33951. doi:10.18632/oncotarget.5580
Izzedine, H., Launay-Vacher, V., Deybach, C., Bourry, E., Barrou, B., and Deray, G. (2005). Drug-induced diabetes mellitus. Expert Opin. Drug Saf. 4, 1097–1109. doi:10.1517/14740338.4.6.1097
Jabbour, E., and Lipton, J. H. (2013). A critical review of trials of first-line BCR-ABL inhibitor treatment in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Clin. Lymphoma Myeloma Leuk. 13, 646–656. doi:10.1016/j.clml.2013.05.012
Jiang, Z. Y., Zhou, Q. L., Coleman, K. A., Chouinard, M., Boese, Q., and Czech, M. P. (2003). Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc. Natl. Acad. Sci. 100, 7569–7574. doi:10.1073/pnas.1332633100
Kantarjian, H., Jabbour, E., Grimley, J., and Kirkpatrick, P. (2006). Dasatinib. Nat. Rev. Drug Discov. 5, 717–719. doi:10.1038/nrd2135
Larson, R. A., Kim, D. W., Issaragrilsil, S., le Coutre, P., Dorlhiac Llacer, P. E., Etienne, G., et al. (2014). Efficacy and safety of nilotinib (NIL) vs imatinib (IM) in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): long-term follow-up (f/u) of ENESTnd. . Blood. 124, 4541. doi:10.1182/blood.v124.21.4541.4541
Le Tourneau, C., Raymond, E., and Faivre, S. (2007). Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST). Ther. Clin. Risk Manag. 3, 341–348. doi:10.2147/tcrm.2007.3.2.341
Li, X., Tran, K. M., Aziz, K. E., Sorokin, A. V., Chen, J., and Wang, W. (2016). Defining the protein-protein interaction network of the human protein tyrosine phosphatase family. Mol. Cell Proteomics 15, 3030–3044. doi:10.1074/mcp.M116.060277
Lindauer, M., and Hochhaus, A. (2018). Dasatinib. Recent Results Cancer Res. 212, 29–68. doi:10.1007/978-3-319-91439-8_2
Louvet, C., Szot, G. L., Lang, J., Lee, M. R., Martinier, N., Bollag, G., et al. (2008). Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. 105, 18895–18900. doi:10.1073/pnas.0810246105
Lutz, S. Z., Ullrich, A., Häring, H. U., Ullrich, S., and Gerst, F. (2017). Sunitinib specifically augments glucose-induced insulin secretion. Cell Signal 36, 91–97. doi:10.1016/j.cellsig.2017.04.018
Mackenzie, R. W., and Elliott, B. T. (2014). Akt/PKB activation and insulin signaling: a novel insulin signaling pathway in the treatment of type 2 diabetes. Diabetes Metab. Syndr. Obes. 7, 55–64. doi:10.2147/DMSO.S48260
Manley, P. W., Cowan-Jacob, S. W., Fendrich, G., and Mestan, J. (2005). Molecular interactions between the highly selective pan-bcr-abl inhibitor, AMN107, and the tyrosine kinase domain of abl. Blood 106, 3365. doi:10.1182/blood.v106.11.3365.3365
McCulloch, L. J., van de Bunt, M., Braun, M., Frayn, K. N., Clark, A., and Gloyn, A. L. (2011). GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 104, 648–653. doi:10.1016/j.ymgme.2011.08.026
McTigue, M., Murray, B. W., Chen, J. H., Deng, Y. L., Solowiej, J., and Kania, R. S. (2012). Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. 109, 18281–18289. doi:10.1073/pnas.1207759109
Medina, R. A., and Owen, G. I. (2002). Glucose transporters: expression, regulation and cancer. Biol. Res. 35, 9–26. doi:10.4067/s0716-97602002000100004
Mora, A., Komander, D., van Aalten, D. M. F., and Alessi, D. R. (2004). PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 15, 161–170. doi:10.1016/j.semcdb.2003.12.022
Noble, M. E. M., Endicott, J. A., and Johnson, L. N. (2004). Protein kinase inhibitors: insights into drug design from structure. Science 303, 1800–1805. doi:10.1126/science.1095920
Oh, J. J., Hong, S. K., Joo, Y. M., Lee, B. K., Min, S. H., Lee, S., et al. (2012). Impact of sunitinib treatment on blood glucose levels in patients with metastatic renal cell carcinoma. Jpn. J. Clin. Oncol. 42, 314–317. doi:10.1093/jjco/hys002
Ono, K., Suzushima, H., Watanabe, Y., Kikukawa, Y., Shimomura, T., Furukawa, N., et al. (2012). Rapid amelioration of hyperglycemia facilitated by dasatinib in a chronic myeloid leukemia patient with type 2 diabetes mellitus. Intern Med. 51, 2763–2766. doi:10.2169/internalmedicine.51.8314
Östman, A., and Böhmer, F. D. (2001). Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. 11, 258–266. doi:10.1016/s0962-8924(01)01990-0
Parsons, S. J., and Parsons, J. T. (2004). Src family kinases, key regulators of signal transduction. Oncogene 23, 7906–7909. doi:10.1038/sj.onc.1208160
Paul, M. K., and Mukhopadhyay, A. K. (2004). Tyrosine kinase – role and significance in cancer. Int. J. Med. Sci. 1, 101–115. doi:10.7150/ijms.1.101
Peng, B., Lloyd, P., and Schran, H. (2005). Clinical pharmacokinetics of imatinib. Clin. Pharmacokinet. 44, 879–894. doi:10.2165/00003088-200544090-00001
Persaud, S. J., Harris, T. E., Burns, C. J., and Jones, P. M. (1999). Tyrosine kinases play a permissive role in glucose-induced insulin secretion from adult rat islets. J. Mol. Endocrinol. 22, 19–28. doi:10.1677/jme.0.0220019
Płużański, A., and Piórek, A. (2016). Side effects of tyrosine kinase inhibitors — management guidelines. Oncol. Clin. Pract. 12, 113–118. doi:10.5603/OCP.2016.0004
Prada, P. O., and Saad, M. J. (2013). Tyrosine kinase inhibitors as novel drugs for the treatment of diabetes. Expert Opin. Investig. Drugs 22, 751–763. doi:10.1517/13543784.2013.802768
Rachdi, L., El Ghazi, L., Bernex, F., Panthier, J. J., Czernichow, P., and Scharfmann, R. (2001). Expression of the receptor tyrosine kinase KIT in mature beta-cells and in the pancreas in development. Diabetes 50, 2021–2028. doi:10.2337/diabetes.50.9.2021
Racil, Z., Razga, F., Drapalova, J., Buresova, L., Zackova, D., Palackova, M., et al. (2013). Mechanism of impaired glucose metabolism during nilotinib therapy in patients with chronic myelogenous leukemia. Haematologica 98, 124–126. doi:10.3324/haematol.2013.086355
Rahman, AFMM, Korashy, H. M., and Kassem, M. G. (2014). “Chapter five - gefitinib,” in Profiles of drug substances, excipients and related methodology. Editor H. G. Brittain (Academic Press), 239–264.
Rodrigues, G. A., and Park, M. (1994). Oncogenic activation of tyrosine kinases. Curr. Opin. Genet. Dev. 4, 15–24. doi:10.1016/0959-437x(94)90086-8
Rorsman, P., and Braun, M. (2013). Regulation of insulin secretion in human pancreatic islets. Annu. Rev. Physiol. 75, 155–179. doi:10.1146/annurev-physiol-030212-183754
Roskoski, R. (2003). STI-571: an anticancer protein-tyrosine kinase inhibitor. Biochem. Biophys. Res. Commun. 309, 709–717. doi:10.1016/j.bbrc.2003.08.055
Roskoski, R. (2016). Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 103, 26–48. doi:10.1016/j.phrs.2015.10.021
Rustenbeck, I. (2002). Desensitization of insulin secretion. Biochem. Pharmacol. 63, 1921–1935. doi:10.1016/s0006-2952(02)00996-6
Saeedi, P., Petersohn, I., Salpea, P., Malanda, B., Karuranga, S., Unwin, N., et al. (2019). Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas, 9th edition. Diabetes Res. Clin. Pract. 157, 107843. doi:10.1016/j.diabres.2019.107843.157
Saglio, G., Larson, R. A., Hughes, T. P., Issaragrisil, S., Turkina, A. G., Marin, D., et al. (2010). Efficacy and safety of nilotinib in chronic phase (CP) chronic myeloid leukemia (CML) patients (pts) with type 2 diabetes in the ENESTnd trial. Blood 116, 3430. doi:10.1182/blood.v116.21.3430.3430
Sakamoto, K., and Holman, G. D. (2008). Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol-Endocrinol Metab. 295, 29–37. doi:10.1152/ajpendo.90331.2008
Salaami, O., Kuo, C. L., Drake, M. T., Kuchel, G. A., Kirkland, J. L., and Pignolo, R. J. (2021). Antidiabetic effects of the senolytic agent dasatinib. Mayo Clin. Proc. 96, 3021–3029. doi:10.1016/j.mayocp.2021.06.025
Sanford, M., and Scott, L. J. (2009). Gefitinib: a review of its use in the treatment of locally advanced/metastatic non-small cell lung cancer. Gefitinib. Drugs. 69, 2303–2328. doi:10.2165/10489100-000000000-00000
Schenk, P. W., and Snaar-Jagalska, B. E. (1999). Signal perception and transduction: the role of protein kinases. Biochim. Biophys. Acta 1449, 1–24. doi:10.1016/s0167-4889(98)00178-5
Schlessinger, J. (2000). Cell signaling by receptor tyrosine kinases. Cell 103, 211–225. doi:10.1016/s0092-8674(00)00114-8
Schnell, A. H., Swenne, I., and Borg, L. A. H. (1988). Lysosomes and pancreatic islet function. A quantitative estimation of crinophagy in the mouse pancreatic B-cell. Cell Tissue Res. 252, 9–15. doi:10.1007/BF00213820
Shah, N. P., Tran, C., Lee, F. Y., Chen, P., Norris, D., and Sawyers, C. L. (2004). Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 305, 399–401. doi:10.1126/science.1099480
Shehzad, A., Iqbal, W., Shehzad, O., and Lee, Y. S. (2012). Adiponectin: regulation of its production and its role in human diseases. Horm. Athens Greece 11, 8–20. doi:10.1007/BF03401534
Sibiya, N., Mzimela, N., Mbatha, B., Ngubane, P., and Khathi, A. (2022). The insights on why diabetes prevalence may increase amid or post COVID 19 pandemic. Curr. Diabetes Rev. 19, e110422203401. doi:10.2174/1573399818666220411122345
Sim, A. T. R., Baldwin, M. L., Rostas, J. A. P., Holst, J., and Ludowyke, R. I. (2003). The role of serine/threonine protein phosphatases in exocytosis. Biochem. J. 373, 641–659. doi:10.1042/BJ20030484
Sorenson, R. L., Brelje, T. C., and Roth, C. (1994). Effect of tyrosine kinase inhibitors on islets of Langerhans: evidence for tyrosine kinases in the regulation of insulin secretion. Endocrinology 134, 1975–1978. doi:10.1210/endo.134.4.8137766
Steegmann, J. L., Cervantes, F., le Coutre, P., Porkka, K., and Saglio, G. (2012). Off-target effects of BCR–ABL1 inhibitors and their potential long-term implications in patients with chronic myeloid leukemia. Leuk. Lymphoma 53, 2351–2361. doi:10.3109/10428194.2012.695779
Stroobants, S., Goeminne, J., Seegers, M., Dimitrijevic, S., Dupont, P., Nuyts, J., et al. (2003). 18FDG-Positron emission tomography for the early prediction of response in advanced soft tissue sarcoma treated with imatinib mesylate (Glivec). Eur. J. Cancer 39, 2012–2020. doi:10.1016/s0959-8049(03)00073-x
Su, H., Bodenstein, C., Dumont, R. A., Seimbille, Y., Dubinett, S., Phelps, M. E., et al. (2006). Monitoring tumor glucose utilization by positron emission tomography for the prediction of treatment response to epidermal growth factor receptor kinase inhibitors. Clin. Cancer Res. 12, 5659–5667. doi:10.1158/1078-0432.CCR-06-0368
Sun, Q., Wangnan, S., Lixiu, Y., and Wang, H. (2009). Gefitinib improves insulin sensitivity in Wistar diabetes rats models. Zhonghua Yi Xue Za Zhi 89, 3441–3443. doi:10.3760/cma.j.issn.0376-2491.2009.48.015
Talon, B., Calip, G. S., Lee, T. A., Sharp, L. K., Patel, P., and Touchette, D. R. (2021). Trend in tyrosine kinase inhibitor utilization, price, and out-of-pocket costs in patients with chronic myelogenous leukemia. JCO Oncol. Pract. 17, 1811–1820. doi:10.1200/OP.20.00967
Talpaz, M., Shah, N. P., Kantarjian, H., Donato, N., Nicoll, J., Paquette, R., et al. (2006). Dasatinib in imatinib-resistant Philadelphia chromosome–positive leukemias. N. Engl. J. Med. 354, 2531–2541. doi:10.1056/NEJMoa055229
Tao, J., Malbon, C. C., and Wangyu, H. (2001). Insulin stimulates tyrosine phosphorylation and inactivation of protein-tyrosine phosphatase 1B in vivo. J. Biol. Chem. 276, 29520–29525. doi:10.1074/jbc.M103721200
Tarn, C., Skorobogatko, Y. V., Taguchi, T., Eisenberg, B., Von Mehren, M., and Godwin, A. K. (2006). Therapeutic effect of imatinib in gastrointestinal stromal tumors: AKT signaling dependent and independent mechanisms. Cancer Res. 66, 5477–5486. doi:10.1158/0008-5472.CAN-05-3906
Thomson, R. J., Moshirfar, M., and Ronquillo, Y. (2022). Tyrosine kinase inhibitors. St. Petersburg, FL: StatPearls Publishing. StatPearls. Treasure Island (FL). Available at: http://www.ncbi.nlm.nih.gov/books/NBK563322/ (Accessed March 16, 2023).
Tigno-Aranjuez, J. T., Benderitter, P., Rombouts, F., Deroose, F., Bai, X., Mattioli, B., et al. (2014). In vivo inhibition of RIPK2 kinase alleviates inflammatory disease. J. Biol. Chem. 289, 29651–29664. doi:10.1074/jbc.M114.591388
Tyrrell, H. E. J., and Pwint, T. (2014). Sunitinib and improved diabetes control. BMJ Case Rep. 2014, bcr2014207521. doi:10.1136/bcr-2014-207521
Veneri, D., Franchini, M., and Bonora, E. (2005). Imatinib and regression of type 2 diabetes. N. Engl. J. Med. 352, 1049–1050. doi:10.1056/NEJM200503103521023
Verspohl, E. J., Tollkühn, B., and Kloss, H. (1995). Role of tyrosine kinase in insulin release in an insulin secreting cell line (INS-1). Cell Signal 7, 505–512. doi:10.1016/0898-6568(95)00020-p
Verweij, J., Casali, P. G., Zalcberg, J., LeCesne, A., Reichardt, P., Blay, J. Y., et al. (2004). Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 364, 1127–1134. doi:10.1016/S0140-6736(04)17098-0
Weisberg, E., Manley, P., Mestan, J., Cowan-Jacob, S., Ray, A., and Griffin, J. D. (2006). AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br. J. Cancer 94, 1765–1769. doi:10.1038/sj.bjc.6603170
Weisberg, E., Manley, P. W., Breitenstein, W., Brüggen, J., Cowan-Jacob, S. W., Ray, A., et al. (2005). Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7, 129–141. doi:10.1016/j.ccr.2005.01.007
Wolf, A. M., Wolf, D., Rumpold, H., Ludwiczek, S., Enrich, B., Gastl, G., et al. (2005). The kinase inhibitor imatinib mesylate inhibits TNF-{alpha} production in vitro and prevents TNF-dependent acute hepatic inflammation. Proc. Natl. Acad. Sci. 102, 13622–13627. doi:10.1073/pnas.0501758102
Wu, P., Nielsen, T. E., and Clausen, M. H. (2015). FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 36, 422–439. doi:10.1016/j.tips.2015.04.005
Yun, C. H., Boggon, T. J., Li, Y., Woo, M. S., Greulich, H., Meyerson, M., et al. (2007). Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 11, 217–227. doi:10.1016/j.ccr.2006.12.017
Keywords: diabetes mellitus, small molecule tyrosine kinase inhibitors, insulin resistance, blood glucose, cancer
Citation: Mugiya T, Mothibe M, Khathi A, Ngubane P and Sibiya N (2024) Glycaemic abnormalities induced by small molecule tryosine kinase inhibitors: a review. Front. Pharmacol. 15:1355171. doi: 10.3389/fphar.2024.1355171
Received: 13 December 2023; Accepted: 12 January 2024;
Published: 01 February 2024.
Edited by:
Virginia Flores-Morales, Universidad Autonoma de Zacatecas, MexicoReviewed by:
Zhixing Wu, University of Florida, United StatesLeandro Fernández-Pérez, University of Las Palmas de Gran Canaria, Spain
Copyright © 2024 Mugiya, Mothibe, Khathi, Ngubane and Sibiya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ntethelelo Sibiya, n.sibiya@ru.ac.za