 Marko Jukič1,2*
Marko Jukič1,2* Urban Bren1,2*
Urban Bren1,2*- 1Laboratory of Physical Chemistry and Chemical Thermodynamics, Faculty of Chemistry and Chemical Engineering, University of Maribor, Maribor, Slovenia
- 2Faculty of Mathematics, Natural Sciences and Information Technologies, University of Primorska, Koper, Slovenia
Advances in computer hardware and the availability of high-performance supercomputing platforms and parallel computing, along with artificial intelligence methods are successfully complementing traditional approaches in medicinal chemistry. In particular, machine learning is gaining importance with the growth of the available data collections. One of the critical areas where this methodology can be successfully applied is in the development of new antibacterial agents. The latter is essential because of the high attrition rates in new drug discovery, both in industry and in academic research programs. Scientific involvement in this area is even more urgent as antibacterial drug resistance becomes a public health concern worldwide and pushes us increasingly into the post-antibiotic era. In this review, we focus on the latest machine learning approaches used in the discovery of new antibacterial agents and targets, covering both small molecules and antibacterial peptides. For the benefit of the reader, we summarize all applied machine learning approaches and available databases useful for the design of new antibacterial agents and address the current shortcomings.
Introduction
Modern antibacterial drug development currently notes a lack of novel antibacterial classes, an observation that is critical in the context of antibacterial drug resistance (Brown and Wright, 2016). Furthermore, not only single-drug resistance but also multiple-drug antibiotic resistance (MDR) has been observed in clinically relevant pathogens worldwide, rendering current established therapies ineffective (Laxminarayan et al., 2020; Vila et al., 2020). The annual number of deaths caused by infections with resistant pathogens alone is currently high and is expected to reach into millions by 2050, making high-quality data collection and reporting and antibacterial research essential (de Kraker et al., 2016; Matamoros-Recio et al., 2021). Recent advances in Computer-aided drug design (CADD) coupled with parallel and high-performance computing (HPC) platforms and new in silico methods represent a new paradigm for antibacterial drug discovery. In particular, machine learning methods have the potential to increase the accuracy of high-throughput virtual screening using ligand-based, structure-based, or consensus-based approaches (Serafim et al., 2020). It should be noted that modern software implementations of machine learning algorithms efficiently utilize computer hardware and are ideal for the bioinformatics or chemoinformatics scenario; however, extreme care should be taken with input data (Bzdok et al., 2017). Most importantly, the increasing availability of data makes machine learning methods even more important, either as a stand-alone method or in a consensus scenario where they can boost traditional medicinal chemistry approaches (He et al., 2021). In this review, we focus on machine learning approaches in CADD that have been reported in recent years and have been used in the development of novel antibacterials. We summarize the relevant databases and consolidate the general workflow along with the methods used in Figure 1.
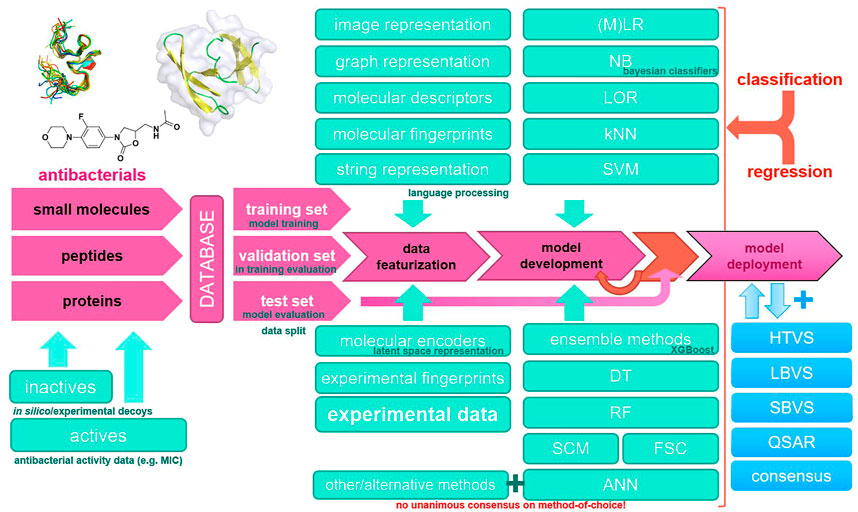
FIGURE 1. Common machine learning methodology in novel antibacterial drug design and a typical modeling workflow. ANN, artificial neural network; DT, decision tree; FSC, feedback system control; HTVS, high-throughput virtual screening; kNN, k-nearest neighbors; LBVS, ligand-based virtual screening; LOR, logistic regression; (M)LR, (multiple) linear regression; NB, naïve Bayes; QSAR, quantitative structure–activity relationship; RF, random forest; SBVS, structure-based virtual screening; SCM, set covering machine; SVM, support vector machines.
Relevant Databases for Antibacterial Drug Design
The currently accessible libraries of antibacterial compounds are enlisted that include small molecules or peptides that can be used for the design of new antibacterial agents and model development (Table 1). The reader should also be aware of tailored or focused libraries and antibacterial libraries offered by commercial compound suppliers and complete online antibacterial drug discovery communities (CO-ADD; of special mention is that the industry also contributes to the CO-ADD community, or previously SPARK-database). The ChEMBL bioinformatics platform is by far the most comprehensive resource (especially considering small molecules), followed by CO-ADD (SPARK) and antimicrobial index. Databases supporting antibacterial peptides are far more common and offer quality data.

TABLE 1. Currently available antibacterial compound and peptide databases suitable for in silico drug design.
Small Molecules
To utilize machine learning approaches in the design of antibacterial small molecules and test different machine learning approaches, Yang et al. computed a simple set of molecular descriptors for small molecules with and without antibacterial properties and evaluated the decision tree, k-nearest neighbor, and support vector machine (SVM) classification models. The authors noted the good accuracy of the SVM approach and the applicability of the methodology for antibacterial drug design. Developed models produced the best prediction accuracies of 96.66 and 98.15% for antibacterial compounds and 99.50 and 98.02% for non-antibacterial compounds (Yang et al., 2009). Ivanenkov et al. (2019) compiled a database of 145,000 small molecules, most of which came from a proprietary high-throughput screening campaign with Escherichia coli (E. coli; 1,786 active and 130,855 inactive compounds; all data points were obtained under the same experimental conditions). 1243 molecular descriptors were calculated using Dragon, ChemoSoft, MOE, and SmartMining software tools. Subsequently, self-organizing maps (Kohonen maps) were used for classification and prediction of antibacterial activity with SmartMining software, and good results were obtained (predictive power of 75.5% on average). The developed models were deployed to identify new agents against E. coli (compound 9, Figure 2). Maltarollo (2019) focused on Staphylococcus aureus (S. aureus), specifically FabI inhibitors. 166 literature compounds were collected and molecular descriptors and fingerprints were calculated using PaDEL software. Decision trees (DTs), random forests (RF), multilayer perceptron (MLP), k-nearest neighbors (kNN), Naive Bayes (NB), and support vector machine (SVM) models were trained for classification. RF models performed best in classifying known connections.
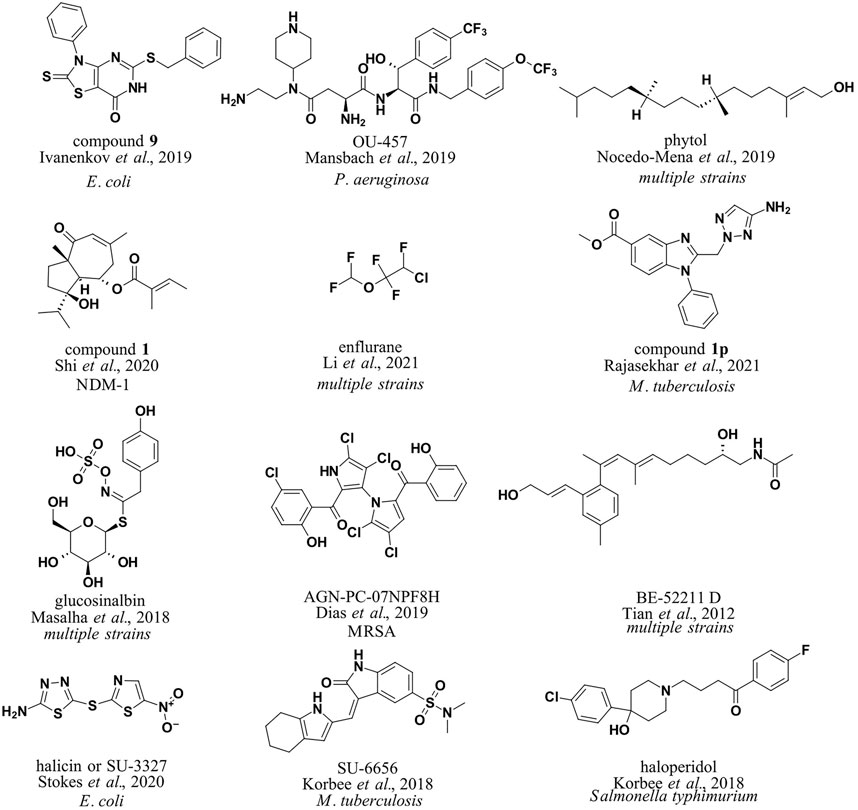
FIGURE 2. Antibacterial compounds identified by machine learning boosted in silico methods in CADD.
Shi et al. collected a database of New Delhi metallo beta-lactamase (NDM-1) inhibitors (511 compounds) from the literature (Shi et al., 2020). This was followed by the calculation of molecular descriptors (34 descriptors, MOE software) and the representation of SMILES strings padded with zeros up to a length of 550. Different methods were tested, such as RF, SVM, and linear discriminant analysis. Finally, it was decided to use the RF model, which performed much better than the classical virtual screening model (90.5 and 69.14%, respectively). The model was used to predict potential NDM-1 inhibitors from a natural product library that contained 2,172 compounds (compound 1, Figure 2). The authors noted that the deep-learning method was not very powerful because of low data availability. Li et al. approached in a more general manner using more data points from the ChEMBL database (Li et al., 2021). The group collected a library of 2708 active antibacterial compounds (IC50 cut-off of 10 μM) and 78,620 inactive compounds and proceeded to calculate fingerprints (FP2, FP3, FP4, DLFP, MACCS, ECFP2, ECFP4, ECFP6, FCFP2, FCFP4, and FCFP6) and vector representations (mol2vec, SMILES2Vec, FP2VEC software; Jaeger et al., 2018; Öztürk et al., 2018; Jeon and Kim, 2019). Several machine learning methods were reviewed, and the FP2 database along with RF, SVM, and MLP methods was selected for screening (scikit-learn library; average accuracy of 0.85). The team then constructed a predictor for antibacterial agents based on all three models and applied it to the FDA-approved small-molecule database (DrugBank, Wishart et al., 2018). Of interest is the observed low FP2 similarity (<0.2) between the predicted and FDA-approved antibacterial agents. The group focused on the nine most different predicted compounds from the FDA antibacterials with the highest screening scores in all three models; however, it did not follow up with biological evaluation. The identified compounds belonged to the classes of anticancer drugs, ocular antihypertensives, and general anesthetics, with enflurane scoring the highest. Enflurane was previously demonstrated to possess antibacterial properties in vitro (enflurane, Figure 2).
The superiority of machine learning–assisted molecular docking was reported by de Avila et al. (2018). The group collected a database of 22 structurally supported 3-dehydroquinate dehydratase (DHQD) inhibitors with measured inhibition constants. They developed a new polynomial scoring function with selected energy terms from classical scoring functions. Using Sandres software (Lasso and Ridge Regression), the newly developed scoring functions performed significantly better in the DHQD system test set supplemented by decoy compounds (the group did not further deploy the model).
Mansbach et al. focused on the permeation of Gram-negative bacteria and developed a fragment-based approach. They collected a database of compounds with MIC values in Pseudomonas aeruginosa (P. aeruginosa) and calculated fragment-based molecular representations for sparse regression and hierarchical clustering to identify the most relevant fragments thought to influence antibacterial activity (Mansbach et al., 2020). The method was used to predict new compounds with antibacterial properties and design “hybrid” molecules from multiple fragments (OU-457, Figure 2). Predicted molecules were experimentally evaluated.
Interestingly, an approach combining both antibacterial small molecules and antimicrobial peptides in a heterogenous library was reported by Nava Lara et al. (2019). To identify compounds with antimicrobial activity in the intestinal flora, 1444 descriptors were calculated (Padel Descriptor software) and 52 different machine learning algorithms were tested (WEKA, AutoWEKA software) to finally select a random committee algorithm classifier with receiver operating characteristic (ROC) area under the curve (AUC) performance of 0.83 for the classification. The model was applied to the FDA-approved antimicrobial agents and found that almost half of them had potential broad-spectrum activity against intestinal bacteria; however, the predictions were not experimentally substantiated. Since antibacterial peptides make up a large proportion of antibacterial chemical substances, they are discussed in more detail in the section Antibacterial Peptides.
Mycobacteria infections are a significant public health problem worldwide. The development of novel antimycobacterial agents remains a challenge, especially in light of the increasing emergence of multidrug-resistant strains of mycobacteria. Several reviews have been published collecting the main therapeutic targets in this field and highlighting the importance of in silico methods, particularly promoted by machine learning approaches and focusing on cell-wall permeability studies (Aleksandrov and Myllykallio, 2019; Pushkaran et al., 2019; Ejalonibu et al., 2021). In this way, classical approaches of virtual screening against the mycobacterial target PrpR (Vina, Glide software), MMGBSA, and molecular dynamics (MD) studies on hit compounds were complemented by the MycoCSM method to identify novel benzimidazole derivatives as potential PrpR inhibitors (compound 1p, Figure 2; Rajasekhar et al., 2021). MycoCSM is a graph-based DT model (scikit-learn library) based on 15,000 unique compounds (featurized with RDkit descriptors) with activity against bacteria of the genus Mycobacterium (MIC cut-off of 1 μM), achieving correlation coefficients of up to 0.89 in predicting bioactivity in terms of minimum inhibitory concentration (Pires and Ascher, 2020).
Korbee et al. used predictive clustering trees (PCTs) to explore host-directed pathways toward antimycobacterial drug design (Clus software; https://sourceforge.net/projects/clus/). The group deployed their models on a library of pharmacologically active compounds in a (LOPAC)-based drug-repurposing screen to identify experimentally validated compounds which target receptor tyrosine kinases (RTKs) and inhibit intracellular mycobacteria (SU-6656, Figure 2) and salmonellae (haloperidol, Figure 2; Korbee et al., 2018).
Natural Compounds
Prediction of antibacterial activity while considering molecular structure and metabolic reaction networks was also attempted by Nocedo-Mena et al. (2019) (dataset: Jeong et al., 2000). The metabolic reaction network data were merged with compounds with MIC properties in ChEMBL, and machine learning modeling with multi-output perturbations was used to build predictive models. The models were deployed to identify natural antibacterial compounds from C. incisa (phytol, Figure 2).
The natural compounds were further explored by Masalha et al., (2018). The group assembled a library of 628 antibacterial compounds (Comprehensive Medicinal Chemistry Database) along with an inactive set of 2892 natural compounds (AnalytiCon Discovery GmbH database) and proceeded to calculate molecular descriptors (MOE software). An iterative indexing model based on stochastic elimination was created for discriminative filtering and antibacterial identification via the calculated molecular bioactivity index (Rayan et al., 2010). The model ROC AUC for antibacterial classification was 0.96, and the model was deployed for screening of the natural product database to identify 10 potential antibacterial hits, two of which were experimentally confirmed as active and others are still under research (glucosinalbin, Figure 2). It is interesting to note that the authors found that comparable performance could not be achieved with either structure-based or ligand-based approaches due to non-efficient scoring or the number of false-positives.
Another report focused on marine natural sources to identify new compounds with activity against MRSA (Dias et al., 2019). Construction of a database of 6645 small molecules (ChEMBL, PubChem, ZINC; active molecules with MIC <5 μM and inactive molecules with MIC ≥5 μM) was followed by a calculation of a comprehensive list of molecular descriptors and fingerprints (PaDEL and CDK Descriptor Software) to finally build a regression model using RF, SVM, Gaussian processes (GPs), and consensus approaches for pMIC determination against MRSA. The best consensus model (R2 of 0.68) was deployed on the StreptomeDB database and resulted in 150 hits with 12 prioritized compounds, all with confirmed anti-MRSA experimental activity (AGN-PC-07NPF8H, Figure 2). The same group also reported a nuclear magnetic resonance (1H and 13C NMR)–based approach where compounds were featurized using experimental NMR-spectra assignation data. The compound library was a dataset of 155 samples that included 50 crude extracts, 55 fractions, and 50 pure compounds obtained from microbial actinobacteria isolated from marine sediments off the Madeira archipelago. RF, SVN, and convolutional neural network (CNN) models were generated with an accuracy of 0.77 for the test set and were ready for further research and application.
Drug similarity identification was also attempted using molecular descriptors and fingerprints calculated using a database from the Current Medicinal Chemistry Database, MDL Drug Data Repot, World Drug Index (drug-like molecules), and Available Chemicals Directory for non–drug-like molecules (180,000 compounds in total). Naive Bayesian classifiers and recursive partitioning models were developed and used for drug similarity prediction in the Traditional Chinese Medicine Compound Database (TCMD) (Tian et al., 2012). The research found that the classifiers can successfully provide valuable information in the early stages of drug design (drug-like compound identification accuracy of 0.86) and identify important drug-like scaffolds and even classify them by pharmacological activity, for example, label scaffolds of antibacterial compounds (BE-52211D, Figure 2).
Indeed, natural compounds represent an invaluable source of chemical diversity, and their drawbacks (availability, complexity, synergistic pharmacodynamics) in drug development could be mitigated by modern machine learning methods (Rodrigues et al., 2016). To this end, Zhang et al. have collected several machine learning protocols for activity prediction of natural products (Zhang et al., 2021).
Antibacterial Peptides
An important subfield of the discovery of new antibacterials is also the discovery of antibacterial peptides. The latter can serve as active agents, starting points for the design of peptidomimetics, or probes for further studies. The field and in silico tools have been reviewed previously (Lee et al., 2017; Cardoso et al., 2020; Wang et al., 2021), with the emphasis on machine learning–enabled antimicrobial peptide discovery and SVM for the discovery of membrane-active peptides (Lee et al., 2018). However, Frecer reported a successful design of cationic antibacterial peptides derived from protegrin-1 as early as 2006 (Frecer, 2006), and machine learning methodology contributed significantly to the design and discovery of novel peptides, as demonstrated by Fjell et al. To single out just one report, they reinforced the traditional QSAR approach with an artificial neural network model (ANN) that inferred a set of peptides with known antibacterial properties from computed descriptors (MOE software). After deploying the model in a screening scenario (in silico library with random peptides), short cationic peptides with MICs in the range of 0.3–10 μM were identified (Fjell et al., 2009). The extended research group later reported an interesting approach for relational learning algorithms (RelF and WEKA software for regression) to explore patterns from the relational structures of the antibacterial peptides or an approximate attribute-value representation of the peptides (Szaboova et al., 2012). Feature vectors for peptide representation were also usedusing Chou’s pseudo-amino acid composition (PseAAC), and the SVM was successfully used to classify antibacterial peptides (Khosravian et al., 2013).
The later approaches were also extended beyond antibacterial peptide identification to peptide target selectivity or prediction of Gram-positive or Gram-negative activities (Veltri et al., 2015). The group used an evolutionary feature construction and a fast correlation-based filter selection algorithm with logistic regression (WEKA) to successfully identify antibacterial peptides of up to 11 amino acids in length. The same group used APD3 database, converted peptide sequences into zero-padded numerical vectors of length 200, and trained a deep neural network (DNN; Keras, TensorFlow software) model to classify antimicrobial peptides (accuracy of 0.98 on APD3 data). Embedding vector visualization was also performed, and a reduced alphabet learnt from the DNN model was developed. Reduced sequence space retained good classification performance (Veltri et al., 2018). Müller et al. trained a recurrent neural network (RNN) with helical antimicrobial peptides (1554 peptides, APD). The sequences were padded according to the length of the longest sequence, N-terminal token added, and One-hot encoding employed (Müller et al., 2018). The resulting model was developed for de novo sequence generation, where 82% were predicted to be active antimicrobial peptides compared to 65% of randomly sampled sequences with the same amino acid distribution as the training set (CAMP AMP prediction tool; Waghu et al., 2014). Wu et al. used previous amino acid substitution data for antibacterial peptides and developed an amino acid activity contribution matrix (Wu et al., 2014). Using this methodology, the group developed a 12-mer DP7 peptide with antibacterial properties against multiple strains (Zhang et al., 2019). Similarly, Yoshida et al. used a natural antibacterial peptide Temporin-Ali (FFPIVGKLLSGLL-NH2) and PSI BLAST to create a library of distantly related and functionally similar sequences, prepared the peptides, and evaluated their antibacterial activities in vitro on E. coli to construct a fitness matrix. The data were then used to train a model and deploy it to optimize peptide sequences. The group produced a peptide with 163-fold lower activity on E. coli bacteria (Yoshida et al., 2018). Another approach using rough set theory constructed quantitative structure–activity relationship rules for existing antibacterial peptides. New sequence development via a genetic algorithm and further in vitro testing resulted in a peptide being active against Staphylococcus epidermidis (S. epidermidis) (Boone et al., 2021).
Approaches were again extended by considering toxicity data in the development of novel antibacterial peptides intended for human drug development campaigns. Capecchi et al. used the Database of Antimicrobial Activity and Structure of Peptides (DBAASP; 4774 active peptides with an MIC threshold of 32 mg/ml) to train a recurrent neural network (RNN) generative model to develop nonhemolytic antibacterial peptides with activity against P. aeruginosa, Acinetobacter baumannii (A. baumannii), MRSA, and a broader range of MDR strains. To test the performance of machine learning models for antibacterial peptide design, Wani et al. trained models on a database of antibacterials (2638) and inactive peptides (3700) using RF, kNN, SVM, DT, NB, quadratic discriminant analysis (QDA), and ensemble learning. RF models were found to perform best in validation experiments. The group also highlighted three important peptide descriptors as essential for antibacterial activity, namely, charge, polarity, and pseudo-amino acid composition (Wani et al., 2021). The field of in silico tools for designing antibacterial peptides using machine learning is also gaining traction, and targeted tools such as AMPGAN v2 are being developed (Van Oort et al., 2021). AMPGAN v2 is a bidirectional conditional generative adversarial network (BiCGAN) that targets de novo generation of antibacterial peptides. The group used training data by compiling the Database of Antimicrobial Activity and Structure of Peptides (DBAASP), Antiviral Peptide database (AVPdb), and UniProt databases (Apweiler et al., 2004; Gogoladze et al., 2014; Qureshi et al., 2014).
Antibacterial Drug Resistance
Machine learning approaches are also being used to combat antibiotic resistance. Back in 2017, Macesic et al. published a review of antibacterial susceptibility testing using genotype–phenotype prediction, machine learning approaches to identify resistant strains, and the use of machine learning to improve treatment and optimize clinical approaches to MDR infections (Macesic et al., 2017). Interestingly, the authors lamented data abstraction and quality but pointed out that the methodology gains strength with the availability of quality data. A recent review article discusses several bioinformatics approaches involving machine learning that are useful for studying bacterial resistance, such as the use of modern bioinformatics approaches for the interpretation of data from increasing sequencing libraries; study of protein structures; in silico analysis of serovar, serogroup, and antigen markers; the development of in silico plasmid detection methods; in silico identification of resistance genes; antibacterial surveillance; and in turn, the prediction of the evolution of antibacterial drug resistance (Ndagi et al., 2020). In addition, machine learning approaches have been used beyond resistance prediction using genomic data to elucidate resistance mechanisms and for antibacterial stewardship applications. The latter are mainly concerned with patient data analysis, diagnosis, treatment, and prevention of resistance development in a clinical scenario (Anahtar et al., 2021). With the increasing use of antibiotics and the accompanying bacterial resistance, we cannot overemphasize the importance of these new approaches in translational research. Furthermore, the power of reported methods is increasing with the growth of quality data and availability of curated and resistance-focused libraries such as Plasmid ATLAS by Jesus et al., (2019), Ensembl Genomes (Bacteria) by Yates et al., (2022), BacDive by Reimer et al., (2019), Virulence Factor Database VFDB by Chen et al., (2005), Beta-Lactamase Database (BLDB) by Naas et al., (2017), Antibiotic Resistance Genes Database (ARDB, Liu and Pop, 2009), BacMed (Pal et al., 2014), and Comprehensive Antibiotic Resistance Database (CARD, McArthur et al., 2013 and Alcock et al., 2020).
Modern Approaches
As reviewed already by Durrant and Amaro in 2014 (Durrant and Amaro, 2015) up to now, the medicinal chemistry community and pharmaceutical industry are adopting machine learning techniques in medicinal chemistry and drug design in general (Ekins et al., 2019) and antibacterial drug development (Patel et al., 2020; Serafim et al., 2020). Of special mention would be the acknowledgment of enormous data availability, its application toward drug design (Burki, 2020), and utilization of modern artificial intelligence approaches (David et al., 2021). Specifically, the applications of modern deep learning methods in antibacterial drug design are evident from a multitude of published reports in scientific literature, tailored offerings by commercial drug design software developers, and emergence of deep-learning in drug design–focused CROs and start-ups (Schroedl, 2019; Chang et al., 2019; Gupta et al., 2021; da Silva et al., 2021).
Deep-Learning and Artificial Neural Networks
An excellent example of the development and use of deep learning supervised, semi-supervised, or unsupervised models in the area of novel antibacterial drug development and discovery was recently reported (Stokes et al., 2020). The group initially generated the dataset by computing graph representations, Morgan fingerprints, and molecular features computed using RDKit (internal training set of 2560 compounds, 120 positive controls; with a test set: Broad’s Drug Repurposing Hub of 6111 compounds) and used a Directed Message Passing Neural Network (D-MPNN; Chemprop implementation available on Github), a type of graph convolutional neural network for model development. After prioritization by toxicity prediction, the authors identified one promising new antibiotic, halicin (SU -3327, Figure 2), and eight (ZINC000098210492, ZINC000001735150, ZINC000225434673, ZINC000019771150, ZINC000004481415, ZINC000004623615, ZINC000238901709, and ZINC000100032716) other potential antibiotic candidates and experimentally validated the obtained hits to have an antibiotic activity on E. coli.
K-Nearest Neighbor
kNN is a supervised learning method that can be applied for classification and regression tasks and is effectively utilized in medicinal chemistry for novel antibacterial drug design. A classification application of kNN was reported by Karakoc et al. for classification of small molecules based on selecting the most relevant set of chemical descriptors used for ultimate discrimination between active and inactive compounds on various biological systems (Karakoc et al., 2007). A comprehensive list of kNN applications in classification and regression tasks all applied toward drug delivery for infectious disease treatment, treatment regimen optimization, drug delivery system and administration route design, and drug delivery outcome prediction was reported by He et al. (2021).
Support Vector Machines
SVM supervised learning models are also widely applied for classification, regression, and ranking/virtual screening tasks in medicinal chemistry in a range of fields such as novel anticancer research, design of antivirals, protein–protein interaction research etc. (Romero-Molina et al., 2019). Focusing on antibacterial drug design, Li et al. reported SVM model development from the fingerprint-featurized ChEMBL database in order to identify novel antibacterial compounds (Li et al., 2021). SVM model applications in antibacterial design and antibacterial drug resistance research were reviewed by Serafim et al. (2020). In a broader scope, recent advances in SVMs and their numerous drug discovery applications are summarized by Maltarollo et al. (2019).
Random Forest and Decision Trees
RF is a supervised ensemble learning method that consists of a multitude of decision trees, constructed at a training phase. Upon reviewing literature on novel antibacterial design supported by machine learning, RF models were found to be one of the most commonly applied for classification, regression, and other tasks and represent a performance and computationally lean approach. In this review, a number of RF applications are presented, for small molecules, peptides (Bhadra et al., 2018), natural product–based antibacterial design, and studying antibacterial drug resistance (Dias et al., 2019; Maltarollo et al., 2019; Shi et al., 2020; Li et al., 2021; Wani et al., 2021). A good example of underlying supervised learning DT method was reported by Suay-Garcia et al. (2020). The authors created a QSAR model to predict antibacterial activity against E. coli. The compounds were classified using a tree-based method and linear discriminant analysis. A comprehensive review on other DT applications is also provided by Serafim et al., (2020).
Coupling to Big Data
Needless to say, we must emphasize the coupling of modern machine learning approaches to valuable data sources. Sripriya Akondi et al., (2022) emphasize the use of compound and protein conformational data which in its abundance classifies as big data in all respects. However, common problems with big data sources such as data quality, over-fitting, and difficult or lengthy protocols should be taken in consideration (Motamedi et al., 2022). Taken together, the big data era will walk hand-in-hand with future drug design and will have a significant impact on how to approach a drug discovery campaign (Zhu, 2020; Bhattarai, et al., 2022; Lee et al., 2022). Zhao et al. point out in a wonderful report “10 Vs.” or characteristics that are intrinsic in drug discovery big data that we should be aware of and utilize, namely: volume (size of data), velocity (data growth), variety (lots of data sources), veracity (variable data quality), validity (authenticity of data), vocabulary (aware of the terminology), venue (numerous data platforms), visualization (presentation and patterns in data), volatility (time domain of the data and usefulness time window), and value (associated economic and added value, Zhao et al., 2020).
Conclusion
In conjunction with antibacterial compound databases (Table 1) and general (big) data sources such as ChEMBL and CO-ADD (SPARK), efficient research in the area of new antibacterial drug design and target identification is possible (Gaulton et al., 2017; Wishart et al., 2018). Incorporating novel machine learning methods can successfully boost the traditional medicinal chemistry approaches, and this review highlights a host of applications and machine learning model deployments. The examples include synthetic and natural small molecules, as well as peptides, ranging from a narrow spectrum of Gram-positive or Gram-negative bacteria to a broad spectrum of compounds acting on mycobacteria and eventually even MDR bacteria. However, in reviewing the literature, it is immediately apparent that medicinal chemistry is currently still in the introductory phase of exploring modern (and also established) machine learning methods and adapting them to the field. Most of the reports are proof-of-concept works where the models are only deployed to test the data and no experimental biological evaluation is performed. However, the analysis of the best performing featurization approaches and the methods themselves may be even more important takeaways.
Input data is of critical importance, and the available tailored or focused antibacterial data libraries, especially public resources, leave much to be desired. The good availability of antimicrobial peptide data and general relational databases, such as the ones mentioned above, improves the situation. In conclusion, the immense value of modern machine learning methods is obvious—coupled with classical and experimental approaches in medicinal chemistry— and new advances in antibacterial drug design and mode of action research are possible.
Author Contributions
MJ and UB interpreted the data from the literature. MJ and UB wrote the original draft. MJ and UB reviewed, edited, and drafted the manuscript and approved the final version.
Funding
This work was supported by the Slovenian Ministry of Science and Education infrastructure project grants HPC-RIVR and RI-SI-ELIXIR and by the Slovenian Research Agency (ARRS) program and project grants P2-0046, J1-2471, J1-1715, N1-0209, P1-0403, L2-3175 and J1-9186.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AI, artificial intelligence; ANN, artificial neural network; CADD, computer-assisted drug design; DT, decision tree; FSC, feedback system control; kNN, k-nearest neighbors; LOR, logistic regression; (M)LR, (multiple) linear regression; MDR, multidrug resistant; MIC, minimum inhibitory concentration; MRSA, methicillin-resistant Staphylococcus aureus; NB, naïve Bayes; RF, random forest; RiPPS, ribosomally synthesized and posttranslationally modified peptides; SCM, set covering machine; SVM, support vector machines.
References
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 48 (D1), D517–D525. doi:10.1093/nar/gkz935
Aleksandrov, A., and Myllykallio, H. (2019). Advances and Challenges in Drug Design against Tuberculosis: Application of In Silico Approaches. Expert Opin. Drug Discov. 14 (1), 35–46. doi:10.1080/17460441.2019.1550482
Amirkia, V. D., and Qiubao, P. (2011). The Antimicrobial Index: a Comprehensive Literature-Based Antimicrobial Database and Reference Work. Bioinformation 5 (8), 365–366. doi:10.6026/97320630005365
Anahtar, M. N., Yang, J. H., and Kanjilal, S. (2021). Applications of Machine Learning to the Problem of Antimicrobial Resistance: An Emerging Model for Translational Research. J. Clin. Microbiol. 59 (7), e0126020. doi:10.1128/jcm.01260-20
Apweiler, R., Bairoch, A., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., et al. (2004). UniProt: the Universal Protein Knowledgebase. Nucleic Acids Res. 32 (Suppl. l_1), D115–D119. doi:10.1093/nar/gkh131
Bhadra, P., Yan, J., Li, J., Fong, S., and Siu, S. W. I. (2018). AmPEP: Sequence-Based Prediction of Antimicrobial Peptides Using Distribution Patterns of Amino Acid Properties and Random forest. Sci. Rep. 8 (1), 1697–1710. doi:10.1038/s41598-018-19752-w
Bhattarai, S., Kumar, R., Nag, S., and Namasivayam, V. (2022). “Big Data in Drug Discovery,” in Machine Learning and Systems Biology in Genomics and Health (Singapore: Springer), 17–48. doi:10.1007/978-981-16-5993-5_2
Boone, K., Wisdom, C., Camarda, K., Spencer, P., and Tamerler, C. (2021). Combining Genetic Algorithm with Machine Learning Strategies for Designing Potent Antimicrobial Peptides. BMC bioinformatics 22 (1), 239–317. doi:10.1186/s12859-021-04156-x
Brown, E. D., and Wright, G. D. (2016). Antibacterial Drug Discovery in the Resistance Era. Nature 529 (7586), 336–343. doi:10.1038/nature17042
Burki, T. (2020). A New Paradigm for Drug Development. The Lancet Digital Health 2 (5), e226–e227. doi:10.1016/S2589-7500(20)30088-1
Bzdok, D., Krzywinski, M., and Altman, N. (2017). Points of Significance: Machine Learning: a Primer. Nat. Methods 14 (12), 1119–1120. doi:10.1038/nmeth.4526
Cardoso, M. H., Orozco, R. Q., Rezende, S. B., Rodrigues, G., Oshiro, K. G. N., Cândido, E. S., et al. (2020). Computer-aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front Microbiol. 10, 3097. doi:10.3389/fmicb.2019.03097
Chang, C. H., Hung, C. L., and Tang, C. Y. (2019). “November). A Review of Deep Learning in Computer-Aided Drug Design,” in 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) (IEEE), 1856–1861.
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a Reference Database for Bacterial Virulence Factors. Nucleic Acids Res. 33 (Suppl. l_1), D325–D328. doi:10.1093/nar/gki008
Cooper, M. A. (2015). A Community-Based Approach to New Antibiotic Discovery. Nat. Rev. Drug Discov. 14 (9), 587–588. doi:10.1038/nrd4706
Corsello, S. M., Bittker, J. A., Liu, Z., Gould, J., McCarren, P., Hirschman, J. E., Johnston, S. E., Vrcic, A., Wong, B., Khan, M., Asiedu, J., Narayan, R., Mader, C. C., Subramanian, A., and Golub, T. R. (2017). The Drug Repurposing Hub: a Next-Generation Drug Library and Information Resource. Nat. Med. 23 (4), 405–408. doi:10.1038/nm.4306
da Silva, T. H., Hachigian, T. Z., Lee, J., and King, M. D. (2022). Using Computers to ESKAPE the Antibiotic Resistance Crisis. Drug Discov. Today 27 (2), 456–470. doi:10.1016/j.drudis.2021.10.005
David, L., Brata, A. M., Mogosan, C., Pop, C., Czako, Z., Muresan, L., Ismaiel, A., Dumitrascu, D. I., Leucuta, D. C., Stanculete, M. F., Iaru, I., and Popa, S. L. (2021). Artificial Intelligence and Antibiotic Discovery. Antibiotics (Basel) 10 (11), 1376. doi:10.3390/antibiotics10111376
de Avila, M. B., and de Azevedo, W. F. (2018). Development of Machine Learning Models to Predict Inhibition of 3‐dehydroquinate Dehydratase. Chem. Biol. Drug Des. 92 (2), 1468–1474.
de Kraker, M. E., Stewardson, A. J., and Harbarth, S. (2016). Will 10 million people die a year due to antimicrobial resistance by 2050? Plos Med. 13 (11), e1002184. doi:10.1371/journal.pmed.1002184
Di Luca, M., Maccari, G., Maisetta, G., and Batoni, G. (2015). BaAMPs: the Database of Biofilm-Active Antimicrobial Peptides. Biofouling 31 (2), 193–199. doi:10.1080/08927014.2015.1021340
Dias, T., Gaudêncio, S. P., and Pereira, F. (2019). A Computer-Driven Approach to Discover Natural Product Leads for Methicillin-Resistant Staphylococcus aureus Infection Therapy. Mar. Drugs 17 (1), 16. doi:10.3390/md17010016
Doster, E., Lakin, S. M., Dean, C. J., Wolfe, C., Young, J. G., Boucher, C., et al. (2020). MEGARes 2.0: a Database for Classification of Antimicrobial Drug, Biocide and Metal Resistance Determinants in Metagenomic Sequence Data. Nucleic Acids Res. 48 (D1), D561–D569. doi:10.1093/nar/gkz1010
Durrant, J. D., and Amaro, R. E. (2015). Machine-learning Techniques Applied to Antibacterial Drug Discovery. Chem. Biol. Drug Des. 85 (1), 14–21. doi:10.1111/cbdd.12423
Ejalonibu, M. A., Ogundare, S. A., Elrashedy, A. A., Ejalonibu, M. A., Lawal, M. M., Mhlongo, N. N., et al. (2021). Drug Discovery for Mycobacterium tuberculosis Using Structure-Based Computer-Aided Drug Design Approach. Int. J. Mol. Sci. 22 (24), 13259. doi:10.3390/ijms222413259
Ekins, S., Puhl, A. C., Zorn, K. M., Lane, T. R., Russo, D. P., Klein, J. J., et al. (2019). Exploiting Machine Learning for End-To-End Drug Discovery and Development. Nat. Mater. 18 (5), 435–441. doi:10.1038/s41563-019-0338-z
Farrell, L. J., Lo, R., Wanford, J. J., Jenkins, A., Maxwell, A., and Piddock, L. J. V. (2018). Revitalizing the Drug Pipeline: AntibioticDB, an Open Access Database to Aid Antibacterial Research and Development. J. Antimicrob. Chemother. 73 (9), 2284–2297. doi:10.1093/jac/dky208
Fjell, C. D., Jenssen, H., Hilpert, K., Cheung, W. A., Panté, N., Hancock, R. E., et al. (2009). Identification of Novel Antibacterial Peptides by Chemoinformatics and Machine Learning. J. Med. Chem. 52 (7), 2006–2015. doi:10.1021/jm8015365
Frecer, V. (2006). QSAR Analysis of Antimicrobial and Haemolytic Effects of Cyclic Cationic Antimicrobial Peptides Derived from Protegrin-1. Bioorg. Med. Chem. 14 (17), 6065–6074. doi:10.1016/j.bmc.2006.05.005
Gaulton, A., Hersey, A., Nowotka, M., Bento, A. P., Chambers, J., Mendez, D., et al. (2017). The ChEMBL Database in 2017. Nucleic Acids Res. 45 (D1), D945–D954. doi:10.1093/nar/gkw1074
Gogoladze, G., Grigolava, M., Vishnepolsky, B., Chubinidze, M., Duroux, P., Lefranc, M. P., et al. (2014). DBAASP: Database of Antimicrobial Activity and Structure of Peptides. FEMS Microbiol. Lett. 357 (1), 63–68. doi:10.1111/1574-6968.12489
Gupta, R., Srivastava, D., Sahu, M., Tiwari, S., Ambasta, R. K., and Kumar, P. (2021). Artificial Intelligence to Deep Learning: Machine Intelligence Approach for Drug Discovery. Mol. Divers. 25 (3), 1315–1360. doi:10.1007/s11030-021-10217-3
He, S., Leanse, L. G., and Feng, Y. (2021). Artificial Intelligence and Machine Learning Assisted Drug Delivery for Effective Treatment of Infectious Diseases. Adv. Drug Deliv. Rev. 178, 113922. doi:10.1016/j.addr.2021.113922
Ivanenkov, Y. A., Zhavoronkov, A., Yamidanov, R. S., Osterman, I. A., Sergiev, P. V., Aladinskiy, V. A., Aladinskaya, A. V., Terentiev, V. A., Veselov, M. S., Ayginin, A. A., Kartsev, V. G., Skvortsov, D. A., Chemeris, A. V., Baimiev, A. K., Sofronova, A. A., Malyshev, A. S., Filkov, G. I., Bezrukov, D. S., Zagribelnyy, B. A., Putin, E. O., Puchinina, M. M., and Dontsova, O. A. (2019). Identification of Novel Antibacterials Using Machine Learning Techniques. Front Pharmacol. 10, 913. doi:10.3389/fphar.2019.00913
Jaeger, S., Fulle, S., and Turk, S. (2018). Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition. J. Chem. Inf. Model. 58 (1), 27–35. doi:10.1021/acs.jcim.7b00616
Jeon, W., and Kim, D. (2019). FP2VEC: a New Molecular Featurizer for Learning Molecular Properties. Bioinformatics 35 (23), 4979–4985. doi:10.1093/bioinformatics/btz307
Jeong, H., Tombor, B., Albert, R., Oltvai, Z. N., and Barabási, A. L. (2000). The Large-Scale Organization of Metabolic Networks. Nature 407 (6804), 651–654. doi:10.1038/35036627
Jesus, T. F., Ribeiro-Gonçalves, B., Silva, D. N., Bortolaia, V., Ramirez, M., and Carriço, J. A. (2019). Plasmid ATLAS: Plasmid Visual Analytics and Identification in High-Throughput Sequencing Data. Nucleic Acids Res. 47 (D1), D188–D194. doi:10.1093/nar/gky1073
Jhong, J. H., Yao, L., Pang, Y., Li, Z., Chung, C. R., Wang, R., et al. (2022). dbAMP 2.0: Updated Resource for Antimicrobial Peptides with an Enhanced Scanning Method for Genomic and Proteomic Data. Nucleic Acids Res. 50 (D1), D460–D470. doi:10.1093/nar/gkab1080
Jorge, P., Alves, D., and Pereira, M. O. (2019). Catalysing the Way towards Antimicrobial Effectiveness: A Systematic Analysis and a New Online Resource for Antimicrobial-Enzyme Combinations against Pseudomonas aeruginosa and Staphylococcus aureus. Int. J. Antimicrob. Agents 53 (5), 598–605. doi:10.1016/j.ijantimicag.2019.01.001
Jorge, P., Pérez-Pérez, M., Pérez Rodríguez, G., Fdez-Riverola, F., Olivia Pereira, M., and Lourenço, A. (2016). Reconstruction of the Network of Experimentally Validated AMP-Drug Combinations against Pseudomonas aeruginosa Infections. Cbio 11 (5), 523–530. doi:10.2174/1574893611666160617093955
Kang, X., Dong, F., Shi, C., Liu, S., Sun, J., Chen, J., et al. (2019). DRAMP 2.0, an Updated Data Repository of Antimicrobial Peptides. Sci. Data 6 (148), 1–10. doi:10.1038/s41597-019-0154-y
Karakoc, E., Cherkasov, A., and Sahinalp, S. C. (2007). Novel Approaches for Small Biomolecule Classification and Structural Similarity Search. SIGKDD Explor. Newsl. 9 (1), 14–21. doi:10.1145/1294301.1294307
Khosravian, M., Faramarzi, F. K., Beigi, M. M., Behbahani, M., and Mohabatkar, H. (2013). Predicting Antibacterial Peptides by the Concept of Chou's Pseudo-amino Acid Composition and Machine Learning Methods. Protein Pept. Lett. 20 (2), 180–186. doi:10.2174/092986613804725307
Korbee, C. J., Heemskerk, M. T., Kocev, D., van Strijen, E., Rabiee, O., Franken, K. L. M. C., et al. (2018). Combined Chemical Genetics and Data-Driven Bioinformatics Approach Identifies Receptor Tyrosine Kinase Inhibitors as Host-Directed Antimicrobials. Nat. Commun. 9 (358), 1–14. doi:10.1038/s41467-017-02777-6
Laxminarayan, R., Van Boeckel, T., Frost, I., Kariuki, S., Khan, E. A., Limmathurotsakul, D., et al. (2020). The Lancet Infectious Diseases Commission on Antimicrobial Resistance: 6 Years Later. Lancet Infect. Dis. 20 (4), e51–e60. doi:10.1016/S1473-3099(20)30003-7
Lee, E. Y., Lee, M. W., Fulan, B. M., Ferguson, A. L., and Wong, G. C. L. (2017). What Can Machine Learning Do for Antimicrobial Peptides, and what Can Antimicrobial Peptides Do for Machine Learning? Interf. Focus 7 (6), 20160153. doi:10.1098/rsfs.2016.0153
Lee, E. Y., Wong, G. C. L., and Ferguson, A. L. (2018). Machine Learning-Enabled Discovery and Design of Membrane-Active Peptides. Bioorg. Med. Chem. 26 (10), 2708–2718. doi:10.1016/j.bmc.2017.07.012
Lee, J. W., Maria-Solano, M. A., Vu, T. N. L., Yoon, S., and Choi, S. (2022). Big Data and Artificial Intelligence (AI) Methodologies for Computer-Aided Drug Design (CADD). Biochem. Soc. Trans. 50 (1), 241–252. doi:10.1042/bst20211240
Li, W. X., Tong, X., Yang, P. P., Zheng, Y., Liang, J. H., Li, G. H., et al. (2021). Screening of Antibacterial Compounds with Novel Structure from the FDA Approved Drugs Using Machine Learning Methods.
Liu, B., and Pop, M. (2009). ARDB--Antibiotic Resistance Genes Database. Nucleic Acids Res. 37 (Suppl. l_1), D443–D447. doi:10.1093/nar/gkn656
Macesic, N., Polubriaginof, F., and Tatonetti, N. P. (2017). Machine Learning: Novel Bioinformatics Approaches for Combating Antimicrobial Resistance. Curr. Opin. Infect. Dis. 30 (6), 511–517. doi:10.1097/QCO.0000000000000406
Maltarollo, V. G., Kronenberger, T., Espinoza, G. Z., Oliveira, P. R., and Honorio, K. M. (2019). Advances with Support Vector Machines for Novel Drug Discovery. Expert Opin. Drug Discov. 14 (1), 23–33. doi:10.1080/17460441.2019.1549033
Maltarollo, V. G. (2019). Classification of Staphylococcus aureus FabI Inhibitors by Machine Learning Techniques. Int. J. Quantitative Structure-Property Relationships (Ijqspr) 4 (4), 1–14. doi:10.4018/ijqspr.2019100101
Mansbach, R. A., Leus, I. V., Mehla, J., Lopez, C. A., Walker, J. K., Rybenkov, V. V., Hengartner, N. W., Zgurskaya, H. I., and Gnanakaran, S. (2020). Machine Learning Algorithm Identifies an Antibiotic Vocabulary for Permeating Gram-Negative Bacteria. J. Chem. Inf. Model. 60 (6), 2838–2847. doi:10.1021/acs.jcim.0c00352
Masalha, M., Rayan, M., Adawi, A., Abdallah, Z., and Rayan, A. (2018). Capturing Antibacterial Natural Products with In Silico Techniques. Mol. Med. Rep. 18 (1), 763–770. doi:10.3892/mmr.2018.9027
Matamoros-Recio, A., Franco-Gonzalez, J. F., Forgione, R. E., Torres-Mozas, A., Silipo, A., and Martín-Santamaría, S. (2021). Understanding the Antibacterial Resistance: Computational Explorations in Bacterial Membranes. ACS omega 6 (9), 6041–6054. doi:10.1021/acsomega.0c05590
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The Comprehensive Antibiotic Resistance Database. Antimicrob. Agents Chemother. 57 (7), 3348–3357. doi:10.1128/AAC.00419-13
Mendez, D., Gaulton, A., Bento, A. P., Chambers, J., De Veij, M., Félix, E., et al. (2019). ChEMBL: towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 47 (D1), D930–D940. doi:10.1093/nar/gky1075
Motamedi, F., Pérez-Sánchez, H., Mehridehnavi, A., Fassihi, A., and Ghasemi, F. (2022). Accelerating Big Data Analysis through LASSO-Random Forest Algorithm in QSAR Studies. Bioinformatics 38 (2), 469–475. doi:10.1093/bioinformatics/btab659
Müller, A. T., Hiss, J. A., and Schneider, G. (2018). Recurrent Neural Network Model for Constructive Peptide Design. J. Chem. Inf. Model. 58 (2), 472–479.
Naas, T., Oueslati, S., Bonnin, R. A., Dabos, M. L., Zavala, A., Dortet, L., et al. (2017). Beta-lactamase Database (BLDB) - Structure and Function. J. Enzyme Inhib. Med. Chem. 32 (1), 917–919. doi:10.1080/14756366.2017.1344235
Nava Lara, R. A., Aguilera-Mendoza, L., Brizuela, C. A., Peña, A., and Del Rio, G. (2019). Heterologous Machine Learning for the Identification of Antimicrobial Activity in Human-Targeted Drugs. Molecules 24 (7), 1258. doi:10.3390/molecules24071258
Ndagi, U., Falaki, A. A., Abdullahi, M., Lawal, M. M., and Soliman, M. E. (2020). Antibiotic Resistance: Bioinformatics-Based Understanding as a Functional Strategy for Drug Design. RSC Adv. 10 (31), 18451–18468. doi:10.1039/d0ra01484b
Nocedo-Mena, D., Cornelio, C., Camacho-Corona, M. D. R., Garza-González, E., Waksman de Torres, N., Arrasate, S., et al. (2019). Modeling Antibacterial Activity with Machine Learning and Fusion of Chemical Structure Information with Microorganism Metabolic Networks. J. Chem. Inf. Model. 59 (3), 1109–1120. doi:10.1021/acs.jcim.9b00034
Öztürk, H., Ozkirimli, E., and Özgür, A. (2018). A Novel Methodology on Distributed Representations of Proteins Using Their Interacting Ligands. Bioinformatics 34 (13), i295–i303.
Pal, C., Bengtsson-Palme, J., Rensing, C., Kristiansson, E., and Larsson, D. G. (2014). BacMet: Antibacterial Biocide and Metal Resistance Genes Database. Nucleic Acids Res. 42 (D1), D737–D743. doi:10.1093/nar/gkt1252
Patel, L., Shukla, T., Huang, X., Ussery, D. W., and Wang, S. (2020). Machine Learning Methods in Drug Discovery. Molecules 25 (22), 5277. doi:10.3390/molecules25225277
Pires, D. E. V., and Ascher, D. B. (2020). mycoCSM: Using Graph-Based Signatures to Identify Safe Potent Hits against Mycobacteria. J. Chem. Inf. Model. 60 (7), 3450–3456. doi:10.1021/acs.jcim.0c00362
Pirtskhalava, M., Gabrielian, A., Cruz, P., Griggs, H. L., Squires, R. B., Hurt, D. E., et al. (2016). DBAASP v.2: an Enhanced Database of Structure and Antimicrobial/cytotoxic Activity of Natural and Synthetic Peptides. Nucleic Acids Res. 44 (D1), 6503–D1112. doi:10.1093/nar/gkw243
Pushkaran, A. C., Biswas, R., and Mohan, C. G. (2019). “Impact of Target-Based Drug Design in Anti-bacterial Drug Discovery for the Treatment of Tuberculosis,” in Structural Bioinformatics: Applications in Preclinical Drug Discovery Process (Cham: Springer), 307–346. doi:10.1007/978-3-030-05282-9_10
Qureshi, A., Thakur, N., Tandon, H., and Kumar, M. (2014). AVPdb: a Database of Experimentally Validated Antiviral Peptides Targeting Medically Important Viruses. Nucleic Acids Res. 42 (D1), D1147–D1153. doi:10.1093/nar/gkt1191
Rajasekhar, S., Karuppasamy, R., and Chanda, K. (2021). Exploration of Potential Inhibitors for Tuberculosis via Structure-Based Drug Design, Molecular Docking, and Molecular Dynamics Simulation Studies. J. Comput. Chem. 42 (24), 1736–1749. doi:10.1002/jcc.26712
Rayan, A., Marcus, D., and Goldblum, A. (2010). Predicting Oral Druglikeness by Iterative Stochastic Elimination. J. Chem. Inf. Model. 50 (3), 437–445. doi:10.1021/ci9004354
Reimer, L. C., Vetcininova, A., Carbasse, J. S., Söhngen, C., Gleim, D., Ebeling, C., et al. (2019). BacDive in 2019: Bacterial Phenotypic Data for High-Throughput Biodiversity Analysis. Nucleic Acids Res. 47 (D1), D631–D636. doi:10.1093/nar/gky879
Rodrigues, T., Reker, D., Schneider, P., and Schneider, G. (2016). Counting on Natural Products for Drug Design. Nat. Chem. 8 (6), 531–541. doi:10.1038/nchem.2479
Romero‐Molina, S., Ruiz‐Blanco, Y. B., Harms, M., Münch, J., and Sanchez‐Garcia, E. (2019). PPI‐detect: A Support Vector Machine Model for Sequence‐based Prediction of Protein–Protein Interactions. J. Comput. Chem. 40 (11), 1233–1242.
Schroedl, S. (2019). Current Methods and Challenges for Deep Learning in Drug Discovery. Drug Discov. Today Technol. 32-33, 9–17. doi:10.1016/j.ddtec.2020.07.003
Seebah, S., Suresh, A., Zhuo, S., Choong, Y. H., Chua, H., Chuon, D., et al. (2007). Defensins Knowledgebase: a Manually Curated Database and Information Source Focused on the Defensins Family of Antimicrobial Peptides. Nucleic Acids Res. 35 (Suppl. l_1), D265–D268. doi:10.1093/nar/gkl866
Serafim, M. S. M., Kronenberger, T., Oliveira, P. R., Poso, A., Honório, K. M., Mota, B. E. F., et al. (2020). The Application of Machine Learning Techniques to Innovative Antibacterial Discovery and Development. Expert Opin. Drug Discov. 15 (10), 1165–1180. doi:10.1080/17460441.2020.1776696
Shi, C., Dong, F., Zhao, G., Zhu, N., Lao, X., and Zheng, H. (2020). Applications of Machine-Learning Methods for the Discovery of NDM-1 Inhibitors. Chem. Biol. Drug Des. 96 (5), 1232–1243. doi:10.1111/cbdd.13708
Sripriya Akondi, V., Menon, V., Baudry, J., and Whittle, J. (2022). Novel Big Data-Driven Machine Learning Models for Drug Discovery Application. Molecules 27 (3), 594. doi:10.3390/molecules27030594
Stokes, J. M., Yang, K., Swanson, K., Jin, W., Cubillos-Ruiz, A., Donghia, N. M., MacNair, C. R., French, S., Carfrae, L. A., Bloom-Ackermann, Z., Tran, V. M., Chiappino-Pepe, A., Badran, A. H., Andrews, I. W., Chory, E. J., Church, G. M., Brown, E. D., Jaakkola, T. S., Barzilay, R., and Collins, J. J. (2020). A Deep Learning Approach to Antibiotic Discovery. Cell 180 (4), 688–e13. doi:10.1016/j.cell.2020.01.021
Suay-Garcia, B., Falcó, A., Bueso-Bordils, J. I., Anton-Fos, G. M., Pérez-Gracia, M. T., and Alemán-López, P. A. (2020). Tree-based QSAR Model for Drug Repurposing in the Discovery of New Antibacterial Compounds against Escherichia coli. Pharmaceuticals 13 (12), 431. doi:10.3390/ph13120431
Szaboova, A., Kuželka, O., and Železný, F. (2012). Prediction of Antimicrobial Activity of Peptides Using Relational Machine Learning In IEEE International Conference on Bioinformatics and Biomedicine Workshops. IEEE, 575–580.
Thomas, J., Navre, M., Rubio, A., and Coukell, A. (2018). Shared Platform for Antibiotic Research and Knowledge: a Collaborative Tool to SPARK Antibiotic Discovery. ACS Infect. Dis. 4 (11), 1536–1539. doi:10.1021/acsinfecdis.8b00193
Tian, S., Wang, J., Li, Y., Xu, X., and Hou, T. (2012). Drug-likeness Analysis of Traditional Chinese Medicines: Prediction of Drug-Likeness Using Machine Learning Approaches. Mol. Pharm. 9 (10), 2875–2886. doi:10.1021/mp300198d
van Heel, A. J., de Jong, A., Song, C., Viel, J. H., Kok, J., and Kuipers, O. P. (2018). BAGEL4: a User-Friendly Web Server to Thoroughly Mine RiPPs and Bacteriocins. Nucleic Acids Res. 46 (W1), W278–W281. doi:10.1093/nar/gky383
Van Oort, C. M., Ferrell, J. B., Remington, J. M., Wshah, S., and Li, J. (2021). AMPGAN V2: Machine Learning-Guided Design of Antimicrobial Peptides. J. Chem. Inf. Model. 61 (5), 2198–2207. doi:10.1021/acs.jcim.0c01441
Veltri, D., Kamath, U., and Shehu, A. (2018). Deep Learning Improves Antimicrobial Peptide Recognition. Bioinformatics 34 (16), 2740–2747. doi:10.1093/bioinformatics/bty179
Veltri, D., Kamath, U., and Shehu, A. (2015). Improving Recognition of Antimicrobial Peptides and Target Selectivity through Machine Learning and Genetic Programming. Ieee/acm Trans. Comput. Biol. Bioinform 14 (2), 300–313. doi:10.1109/TCBB.2015.2462364
Vila, J., Moreno-Morales, J., and Ballesté-Delpierre, C. (2020). Current Landscape in the Discovery of Novel Antibacterial Agents. Clin. Microbiol. Infect. 26 (5), 596–603. doi:10.1016/j.cmi.2019.09.015
Waghu, F. H., Barai, R. S., Gurung, P., and Idicula-Thomas, S. (2016). CAMPR3: a Database on Sequences, Structures and Signatures of Antimicrobial Peptides. Nucleic Acids Res. 44 (D1), D1094–D1097. doi:10.1093/nar/gkv1051
Waghu, F. H., Gopi, L., Barai, R. S., Ramteke, P., Nizami, B., and Idicula-Thomas, S. (2014). CAMP: Collection of Sequences and Structures of Antimicrobial Peptides. Nucleic Acids Res. 42 (D1), D1154–D1158. doi:10.1093/nar/gkt1157
Wang, C., Garlick, S., and Zloh, M. (2021). Deep Learning for Novel Antimicrobial Peptide Design. Biomolecules 11 (3), 471. doi:10.3390/biom11030471
Wang, G., Li, X., and Wang, Z. (2016). APD3: the Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 44 (D1), D1087–D1093. doi:10.1093/nar/gkv1278
Wani, M. A., Garg, P., and Roy, K. K. (2021). Machine Learning-Enabled Predictive Modeling to Precisely Identify the Antimicrobial Peptides. Med. Biol. Eng. Comput. 59 (11), 2397–2408. doi:10.1007/s11517-021-02443-6
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2018). DrugBank 5.0: a Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 46 (D1), D1074–D1082. doi:10.1093/nar/gkx1037
Wu, X., Wang, Z., Li, X., Fan, Y., He, G., Wan, Y., Yu, C., Tang, J., Li, M., Zhang, X., Zhang, H., Xiang, R., Pan, Y., Liu, Y., Lu, L., and Yang, L. (2014). In Vitro and In Vivo Activities of Antimicrobial Peptides Developed Using an Amino Acid-Based Activity Prediction Method. Antimicrob. Agents Chemother. 58 (9), 5342–5349. doi:10.1128/AAC.02823-14
Yang, X. G., Chen, D., Wang, M., Xue, Y., and Chen, Y. Z. (2009). Prediction of Antibacterial Compounds by Machine Learning Approaches. J. Comput. Chem. 30 (8), 1202–1211. doi:10.1002/jcc.21148
Yates, A. D., Allen, J., Amode, R. M., Azov, A. G., Barba, M., Becerra, A., et al. (2022). Ensembl Genomes 2022: an Expanding Genome Resource for Non-vertebrates. Nucleic Acids Res. 50 (D1), D996–D1003. doi:10.1093/nar/gkab1007
Yoshida, M., Hinkley, T., Tsuda, S., Abul-Haija, Y. M., McBurney, R. T., Kulikov, V., et al. (2018). Using Evolutionary Algorithms and Machine Learning to Explore Sequence Space for the Discovery of Antimicrobial Peptides. Chem 4 (3), 533–543. doi:10.1016/j.chempr.2018.01.005
Zhang, R., Li, X., Zhang, X., Qin, H., and Xiao, W. (2021). Machine Learning Approaches for Elucidating the Biological Effects of Natural Products. Nat. Prod. Rep. 38 (2), 346–361. doi:10.1039/d0np00043d
Zhang, R., Wang, Z., Tian, Y., Yin, Q., Cheng, X., Lian, M., Zhou, B., Zhang, X., and Yang, L. (2019). Efficacy of Antimicrobial Peptide DP7, Designed by Machine-Learning Method, against Methicillin-Resistant Staphylococcus aureus. Front Microbiol. 10, 1175. doi:10.3389/fmicb.2019.01175
Zhao, L., Ciallella, H. L., Aleksunes, L. M., and Zhu, H. (2020). Advancing Computer-Aided Drug Discovery (CADD) by Big Data and Data-Driven Machine Learning Modeling. Drug Discov. Today 25 (9), 1624–1638. doi:10.1016/j.drudis.2020.07.005
Keywords: artificial intelligence, machine learning, computer-aided drug design (CADD), infectious diseases, antibacterial drug design, antibacterial, antibacterial target discovery, antibacterial drug resistance
Citation: Jukič M and Bren U (2022) Machine Learning in Antibacterial Drug Design. Front. Pharmacol. 13:864412. doi: 10.3389/fphar.2022.864412
Received: 28 January 2022; Accepted: 28 March 2022;
Published: 03 May 2022.
Edited by:
Leonardo L. G. Ferreira, University of São Paulo, BrazilReviewed by:
Tihomir Tomašič, University of Ljubljana, SloveniaAmit Kumar Banerjee, Indian Institute of Chemical Technology (CSIR), India
Copyright © 2022 Jukič and Bren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marko Jukič, marko.jukic@um.si; Urban Bren, urban.bren@um.si