 Doris Boeckelmann1†
Doris Boeckelmann1† Mira Wolter1†
Mira Wolter1† Katharina Neubauer1
Katharina Neubauer1 Felix Sobotta1
Felix Sobotta1 Antonia Lenz1
Antonia Lenz1 Hannah Glonnegger1
Hannah Glonnegger1 Barbara Käsmann-Kellner2
Barbara Käsmann-Kellner2 Jasmin Mann3
Jasmin Mann3 Stephan Ehl3
Stephan Ehl3 Barbara Zieger1*
Barbara Zieger1*- 1Department of Pediatrics and Adolescent Medicine, Division of Pediatric Hematology and Oncology, Faculty of Medicine, Medical Center—University of Freiburg, Freiburg, Germany
- 2Department of Ophthalmology, Saarland University Medical Center, Homburg, Germany
- 3Institute for Immunodeficiency, Center for Chronic Immunodeficiency (CCI), Faculty of Medicine, Medical Center—University of Freiburg, Freiburg, Germany
Hermansky-Pudlak syndrome (HPS), a rare heterogeneous autosomal recessive disorder, is characterized by oculocutaneous albinism (OCA) and a bleeding diathesis due to a defect regarding melanosomes and platelet delta (δ)-granule secretion. Interestingly, patients with HPS type 2 (HPS-2) or HPS type 10 (HPS-10) present additionally with an immunological defect. We investigated three patients (IP1, IP2, and IP3) who suffer from a bleeding diathesis. Platelet aggregometry showed impaired platelet function and flow cytometry revealed a severely reduced platelet CD63 expression hinting to either a defect of platelet delta granule secretion or a decreased number of delta granules in these patients. However, only IP3 presents with an apparent OCA. We performed panel sequencing and identified a homozygous deletion of exon 6 in DTNBP1 for IP3. Western analysis confirmed the absence of the encoded protein dysbindin confirming the diagnosis of HPS-7. Interestingly, this patient reported additionally recurrent bacterial infections. Analysis of lymphocyte cytotoxicity showed a slightly reduced NK-degranulation previously documented in a more severe form in patients with HPS-2 or HPS-10. IP1 is carrier of two compound heterozygous variants in the HPS3 gene (c.65C > G and c.1193G > A). A homozygous variant in HPS5 (c.760G > T) was identified in IP2. The novel missense variants were classified as VUS (variant of uncertain significance) according to ACMG guidelines. For IP1 with the compound heterozygous variants in HPS3 a specialized ophthalmological examination showed ocular albinism. HPS3 and HPS5 encode subunits of the BLOC-2 complex and patients with HPS-3 or HPS-5 are known to present with variable/mild hypopigmentation.
Introduction
Hermansky-Pudlak syndrome (HPS) which was first described by Hermansky and Pudlak (Hermansky and Pudlak 1959) has a prevalence of 1–9 per 1,000,000 individuals (Christensen et al., 2017). The key characteristics of HPS include oculocutaneous albinism (OCA) and a bleeding tendency. Typically, the patients present with congenital nystagmus, iris transillumination, decreased visual acuity, and reduced skin/hair pigmentation (Gahl et al., 1998). Bleeding symptoms may manifest as epistaxis, petechiae, extensive bruising or even serious post-traumatic or perioperative complications. To date, 11 types of HPS have been described (HPS-1- HPS-11) explaining some of the clinical variability that relates to the biological functions of the impaired protein complex (Huizing et al., 2020; Pennamen et al., 2020). Malfunctioning of lysosome-related organelles such as melanosomes and platelet δ-granules causes HPS, as they are essential for granule transport (Vincent et al., 2009). Platelet δ-granules secrete serotonin, calcium, ADP, and polyphosphate, therefore, enhancing platelet adhesion and activation (Bowman et al., 2019). Currently, 11 genes associated with HPS (HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1, BLOC1S3, BLOC1S6, AP3D1, and BLOC1S5) have been reported. These genes encode either for the multi-protein complexes BLOC, (biogenesis of lysosome-related organelles complex) or AP-3 (adaptor protein-3).
BLOC-1 comprises the gene products of DTNBP1 (HPS-7), BLOC1S3 (HPS-8), BLOC1S6 (HPS-9), and BLOC1S5 (HPS-11) (Huizing et al., 2020; Pennamen et al., 2020). DTNBP1 (HPS-7) is located on chromosome 6 (6p22.3) and comprises ten exons. The gene codes for 351 amino acid polypeptides (MW 39.5 kD). To our knowledge, only seven patients with four different pathogenic genetic variants in DTNBP1 (HPS-7) have been described (Li et al., 2003; Lowe et al., 2013; Bryan et al., 2017; Lasseaux et al., 2018; Bastida et al., 2019). These patients exhibit a characteristic phenotype of bleeding diathesis and hypopigmentation (OCA). For these patients signs of immunodeficiency or pulmonary fibrosis were not reported, however, the number of patients described with HPS-7 is very low.
BLOC-2 subunits are encoded by HPS3, HPS5, and HPS6 (Di Pietro et al., 2004; Gautam et al., 2004). HPS3 is located on chromosome 3 (3q24) and HPS5 on chromosome 22 (22q.12.2), respectively. HPS3 (MW 11.7kD) encompasses 17 exons coding for a 1,004 amino acid polypeptide. In 2001 the first patients with HPS-3 were described (Anikster et al., 2001; Huizing et al., 2001). HPS5 (127.4 kD) codes for a 1,129 amino acid polypeptide and comprises 23 exons. Zhang et al. identified the first pathogenic variant in human HPS5, which is orthologue to ru2, the gene mutated in a HPS mimicking mouse model (Zhang et al., 2003). Individuals with pathogenic variants in BLOC-2 seem to present a milder HPS phenotype causing a moderate bleeding diathesis and an OCA with variable hypopigmentation (Nurden et al., 2020).
BLOC-3 encompasses the gene products of HPS1 and HPS4 (Wei 2006). Deficiencies in these proteins are associated with a more severe bleeding diathesis, OCA, and serious complications such as pulmonary fibrosis and granulomatous colitis (Huizing et al., 2020; Nurden et al., 2020).
HPS-2 and HPS-10 are caused by variants in AP3B1 and AP3D1, respectively, which constitute the adaptor protein-3 (AP-3) complex. Affected patients present with a bleeding diathesis, OCA, and immunodeficiency due to impaired cytotoxic activity of T-lymphocytes and/or natural killer (NK) cells (Fontana et al., 2006; Ammann et al., 2016; Mohammed et al., 2018). HPS-2 patients are at risk to develop pulmonary fibrosis in childhood (Hengst et al., 2018). One patient with HPS-2 developed hemophagocytic lymphohistiocytosis (HLH) (Enders et al., 2006). For patients with HPS-2, the risk to develop HLH is lower than for patients with Griscelli or Chediak-Higashi syndrome (Jessen et al., 2013).
Here, we report novel genetic alterations in HPS3, HPS5, and DTNBP1 (HPS-7) in patients with a platelet delta granule secretion defect and differently pronounced OCA. Interestingly, we also document a mild NK cell degranulation defect in HPS-7, potentially implicating BLOC-1 also in immune functions.
Materials and Methods
Patients
Index Patient 1
The six-year-old girl (ethnic origin: European) presented with frequent epistaxis (once a month) and bruising. Previous surgery had not been performed. Cutaneous albinism was not apparent. After the molecular genetic analysis had identified compound heterozygous variants in the gene HPS3, a specialized ophthalmological examination was initiated and showed ocular albinism (atypical albino-VEP) with normal visual acuity (no nystagmus). The girl’s mother exhibits prolonged menstrual bleeding. Her father and older sister did not show any bleeding symptoms. The parents are not consanguine.
Index Patient 2
The 45-year-old woman (ethnic origin: Arabic) had a history of extensive bruising, epistaxis, menorrhagia, postoperative bleeding (teeth extraction, liposuction), bleeding after deliveries, joint hemorrhages, microhematuria, and impaired wound healing/increased scarring after surgery. She has eight children and suffered from increased bleeding during childbirth. Therefore, she had received red blood cell and platelet concentrates each time. The symptoms of menorrhagia improved significantly after therapy with tranexamic acid and desmopressin. Cutaneous albinism was not apparent. She and her husband are consanguine, however, her husband does not present any bleeding symptoms. Some of their 8 children seem to exhibit only very mild bleeding symptoms, none of them is clinically as severely affected as their mother. We performed panel sequencing for IP2 and two of her children (daughter and son).
Index Patient 3
The 60-year-old woman (ethnic origin: European, Portuguese descent) presented with OCA and frequent gingival bleeding. Furthermore, she had experienced prolonged bleeding after skin excision and adenoma resection. She did not show signs of colitis. She suffers from asthma. She reported that she has always suffered from severe respiratory infections and recurrent skin furuncles. The patient does not have children. Her consanguine parents are deceased. Her brother had OCA and recurring epistaxis. At the age of 54 ears, he died due to liver cirrhosis (caused by a hepatitis B infection) and due to internal bleeding.
Platelet Count and Platelet Aggregometry Analysis
Platelet count was measured using an automated cell counter (Sysmex KX-21 N, Norderstedt, Germany). Platelet-rich plasma (PRP) and platelet-poor plasma (PPP) were obtained by centrifugation of citrate-anticoagulated blood samples. Using the APACT 4004 (LABiTec, Ahrensburg, Germany), platelet aggregometry analyses were performed after stimulation with collagen (2 and 10 μg/ml; Takeda, Linz, Austria), adenosine diphosphate (ADP; 4 and 10 μmol/L; Sigma-Aldrich, St. Luis, MO, United States), epinephrine (8 and 16 μmol/L; Sanofi-Aventis, Frankfurt, Germany) and ristocetin (1.2 mg/ml; American Biochemical and Pharmaceutical LTD., Frankfurt, Germany).
Flow Cytometry Analyses
Flow cytometry analyses were performed using FACSCalibur (Becton Dickinson, Heidelberg, Germany) (Lahav et al., 2002). Diluted PRP aliquots (5 × 107 platelets/ml) were fixed and stained with FITC-labeled monoclonal surface antibody against CD41 (GPIIb/IIIa-complex), CD42a (GPIb/IX) and CD42b (GPIb) (Coulter, Immunotech, Marseille, France). FITC-labeled anti-VWF (Bio-Rad AbD Serorech, Puchheim, Germany) and Alexa Fluor 488-labeled anti-fibrinogen (Invitrogen, Waltham, MA United States) was used to stain the platelets. In the presence of 1.25 mM Gly-Pro-Arg-Pro (Bachem, Bubendorf, Switzerland) diluted PRP (5 × 107 platelets/ml) was stimulated with a number of concentrations of thrombin (0, 0.05, 0.1, 0.2, 0.5, and 1 U/ml; Siemens Healthineers, Marburg, Germany) to conduct the CD62 and CD63 expression analyses. Additionally, the platelets were stained with monoclonal FITC-labeled anti-CD62 (P-selectin) and anti-CD63 antibodies (lysosomal membrane-associated glycoprotein 3, LAMP-3; Immunotech, Marseille, France). Data of patients and controls (day control and 20 independent measurements from 10 controls as mean ± standard error of the mean, SEM) were analyzed using GraphPad Prism software (version 8, San Diego, CA, United States).
Molecular Genetic Analyses
Informed consent for molecular genetic analysis was obtained for each patient and the investigated family members. To extract genomic DNA from EDTA blood, we used standard procedures and the Blood and Cell Kit by Qiagen (Qiagen GmbH, Hilden, Germany). For index patients panel sequencing (95 genes including all 11 HPS genes; Supplementary Material S1 gene list) was performed using a custom-designed Nextera Rapid Enrichment Kit (Illumina) followed by sequencing on a MiSeq (Illumina). The average sequencing depth overall genes for the 3 patients investigated was 98% for 20x and 91% for 100x, respectively. SeqPilot (JSI medical systems) was used for data analyses. The variants were exported and filtered by allele frequency and serious consequences. We utilized supporting software ALAMUT®(v.2.15), pathogenicity prediction (SIFT, MutTaster, PolyPhen2, and CADD), occurrence in population and disease databases (HGMD public version, Huizing HPS Mutation update (Huizing et al., 2020)) in order to classify the variants. These analyses were performed in accordance with the ACMG (American College of Medical Genetics) guidelines (Richards et al., 2015). For segregation analysis Sanger sequencing was conducted.
cDNA Sequencing Using Reverse Transcripted Platelet-Derived mRNA (for IP3)
Total RNA was isolated from washed human platelets using 2 ml TRIzol® reagent (Thermo Fisher Scientific). Single strand cDNA synthesis was generated with SuperScript® III Reverse Transcriptase (Thermo Fisher Scientific). Amplification was performed using specific primers (fw: TGCAGCAGGATTTCACCTCC; rev: ATCTGCTCCAGCATGTCCAC) covering the coding region from exon 1 to exon 9 of DTNBP1.
Platelet Preparation and Immunoblotting (for IP3)
Platelet-rich plasma (PRP) of the patients and a healthy volunteer was obtained by centrifugation of citrate-anticoagulated whole blood (120 × g for 10 min). Platelets were then isolated from PRP by gel-filtration using a Sepharose CL-2B (GE Healthcare Life Sciences) column, eluted with Tyrode buffer (140 mM NaCl, 2.7 mM KCl, 0.42 mM NaH2PO4, 12 mM NaHCO3, 5.5 mM glucose, and 5 mM HEPES; pH 7.4). Platelets were centrifuged at 1,200 × g for 10 min and lysed in lysis buffer (5 mM Tris-HCl, pH 7,4, 50 mM NaCl, 0,25 mM MgCl2, 0.1% Nonidet P40, 10% glycerol including protease inhibitor cocktail; Roche cOmplete, Merck). Protein content was determined with Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and adjusted to 10 µg. Denatured platelet lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, Invitrogen) and blotted onto Hybond-P polyvinylidene difluoride membrane (PVDF, Amersham, GE Healthcare Life Sciences). Membranes were blocked with 5% milk powder in TBST (20 mM Tris, 140 mM NaCl, 0.1% Tween; pH 7.6) probed with anti-dysbindin antibody (dilution 1:2.000; Abcam) and detected using horseradish peroxidase (HRP)-conjugated goat anti-rabbit (dilution 1:10.000; Cell Signaling Technology) and enhanced chemiluminescence solution (Amersham detection reagent, GE Healthcare Life Sciences). For loading control, the same blot was incubated with anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase; dilution 1:300.000; Abcam) and HRP-coupled goat anti-mouse (dilution 1:10.000; Dianova).
Analysis of NK Cell and CTL Degranulation (for IP3)
NK cell degranulation was analyzed as described (Bryceson et al., 2012). Briefly, fresh NK cell degranulation was assessed by stimulation of isolated peripheral blood mononuclear cells (PBMCs) with K562 target cells followed by flow cytometric analysis of CD107a surface expression. For evaluation of activated NK cell and CTL degranulation, PBMCs were cultured in the presence of Phytohemagglutinin (PHA) and Interleukin-2 (IL-2) for 48 h at 37°C prior to stimulation with K562 target cells and anti-CD3/CD28 beads.
Results
Platelet Count, In-Vivo Bleeding Time, and Platelet Function Analysis
All three index patients presented with normal platelet counts (234 × 109/L, 173 × 109/L, and 189 × 109/L, respectively). The in-vivo bleeding time (Ivy) was severely prolonged (IP1 and IP3 > 15 min, IP2 > 8 min (norm < 6 min)). Platelet aggregation was severely impaired after stimulation with collagen (2 μg/ml) and epinephrine (8 μmol/L) for all index patients. Impaired aggregation after stimulation with low dose ADP (4 μmol/L) was seen in IP1 and IP2. Agglutination after stimulation with ristocetin (1.2 mg/ml) was normal (Table 1).
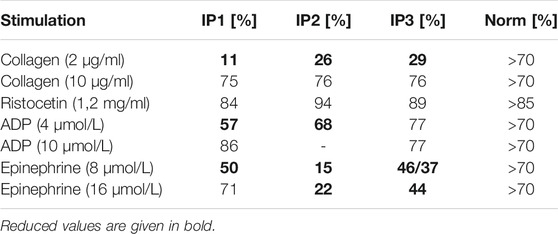
TABLE 1. Platelet aggregometry analyses (LTA). Data are presented as maximal aggregation/agglutination in % compared to normal control levels.
Flow cytometry analysis revealed severely reduced CD63 expression for all index patients (Figure 1). All of them showed normal values for expression of CD62, CD42a, CD42b, CD41, fibrinogen binding, and VWF-binding (data not shown). IP1’s sister presented with only slightly reduced CD63 expression, while her mother`s expression was borderline low. Flow cytometry was not performed for the father. Six of IP2`s children presented with borderline low and two with a slightly reduced CD63 expression.
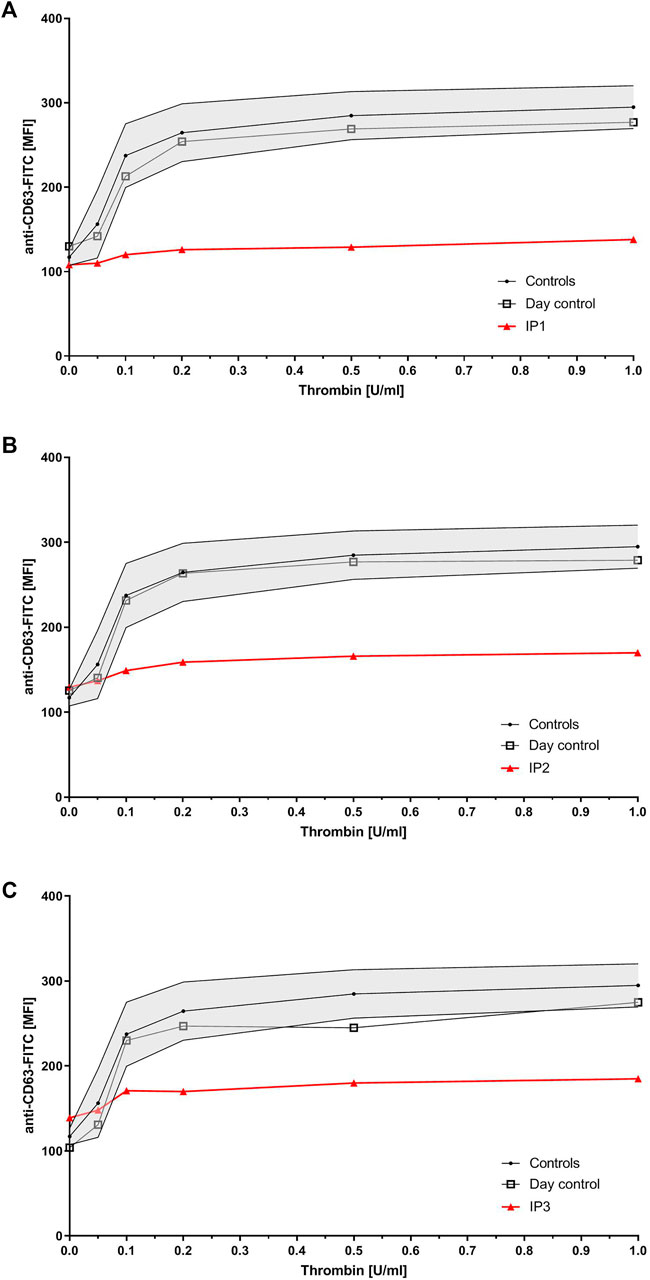
FIGURE 1. Platelet granule secretion stimulated with thrombin (concentrations: 0, 0.05, 0.1, 0.2, 0.5, and 1.0 U/ml) for all three index patients using flow cytometry. Severely impaired δ-granule secretion indicated by reduced platelet CD63 expression in IP1 (A), IP2 (B), and IP3 (C) compared to the healthy controls/day control. Data are expressed as logarithmic arbitrary units (logAU) of anti-CD63-stained unstimulated and thrombin-stimulated platelets.
Molecular Genetic Analysis, cDNA Sequencing, and Immunoblotting
In IP1 two novel heterozygous missense variants in HPS3 (NM_032383.3) were identified (c.65C > G; p.Pro22Arg and c.1193G > A; p.Cys398Tyr). Family genotyping confirmed compound heterozygosity. Father and sister of IP1 are heterozygous carriers of the c.65C > G variant, whereas the mother is a carrier of the c.1193G > A variant. Since the index patient lacked apparent cutaneous albinism we confirmed both variants to be germline by genotyping buccal swab DNA. The c.65C > G variant is absent from GnomAD v2.1 (https://gnomad.broadinstitute.org/) and EVS v.0.0.30 (https://evs.gs.washington.edu/EVS/) databases (ACMG Classification: uncertain significance [PM1, PM2]). The variation shows a moderately conserved nucleotide (phyloP: 4.79 [−19.0, 10.9]. In silico pathogenicity prediction (PP) is predominantly disease causing (SIFT, tolerated; MutationTaster, disease causing; PolyPhen2, probably damaging, CADD score 29.1). The second variant c.1193G > A is reported once in the GnomAD database in a heterozygous state and absent from EVS (ACMG Classification: uncertain significance [PM1, PM2, PP3]). In silico PP is concordant disease causing and CADD score is 29.1.
In IP2 a novel homozygous missense variant (c.760G > T; p.Val254Phe) in HPS5 (NM_181507.1) was identified. The patient’s husband showed wild type sequence at this position. NGS and Sanger sequencing revealed that all of her eight children were heterozygous carriers for this HPS5 variant as expected. The variant c.760G > T is reported six times in the GnomAD database in a heterozygous state (all counts in South Asian population) and absent from EVS (ACMG Classification: uncertain significance [PM1, PM2, PP3]). In silico PP is concordant disease causing and CADD score is 28.1. Additionally we identified a variant (NM_018668.5:c.1780A > G) in the gene VPS33B in a heterozygous state. This variant was also detected in the NGS analysis of her daughter. According to OMIM alterations in the gene VPS33P are autosomal recessive associated with ARC (Arthrogryposis, renal dysfunction, and cholestasis) and α-granule deficiency.
IP3 showed a novel homozygous deletion of exon 6 in DTNBP1 (Figure 2B). Exon 6 could not be amplified from genomic DNA using PCR analysis. We performed cDNA sequencing out of platelet derived mRNA and confirmed the absence of exon 6. Interestingly, the small exon 7 was spliced out. It seems that the spliceosome used the acceptor splice site of exon 8. This would result in an in-frame loss of 52 amino acids in the middle of the expected protein (p.H120_A171del). (Figure 2C). To investigate if the aberrant mRNA is leading to detectable dysbindin we performed Western blot analysis. Dysbindin, which is encoded by DTNBP1, was not detectable in platelets (Figure 2D). The antibody used recognizes the C-terminal sequence within human dysbindin (aa 251-301). However, it is possible that the protein will fold incorrectly and in addition protein degradation will occur. Genetic alterations of all patients are summarized in Table 2.
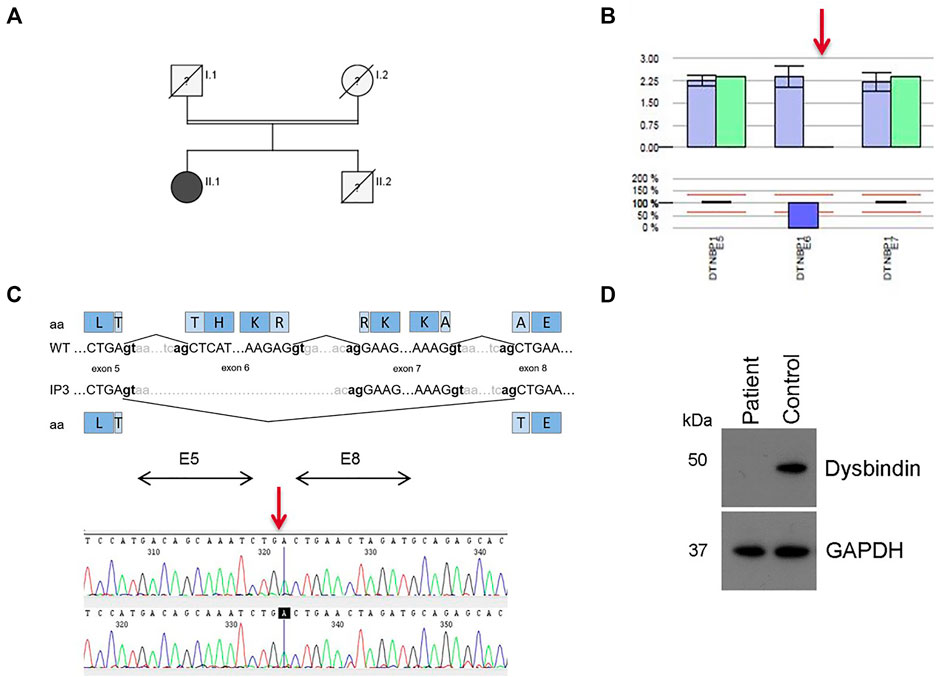
FIGURE 2. Results for patient IP3 (HPS-7). (A) Pedigree for IP3 (II.1). Her consanguine parents and her brother are already deceased, her brother presented with OCA and bleeding disorder. (B) Bioinformatics software SeqPilot CNV analysis indicates a homozygous deletion of exon 6 in DTNBP1. (C) Upper Panel: Schematic representation of the normally spliced mRNA (WT) and the aberrantly spliced mRNA of IP3. Lower Panel: cDNA sequencing of platelet derived DTNBP1 mRNA shows deletion of exon 6 and 7 in IP3. (D) Dysbindin expression in gel-filtrated platelets of the patient (IP3) and a healthy volunteer performed by Western analysis: Dysbindin is not expressed in patient’s platelets. GAPDH (37 kDa) was used as loading control (lower bands).
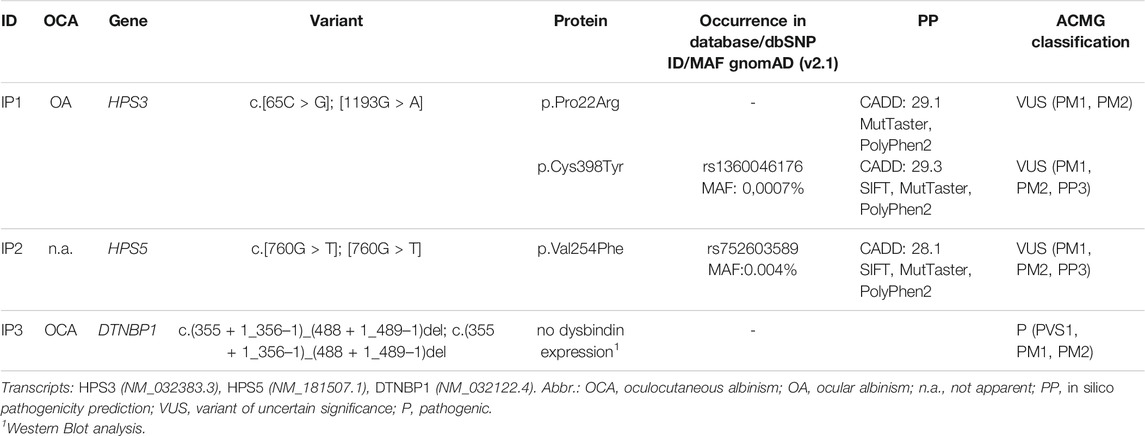
TABLE 2. OCA phenotype and genetic variants identified using NGS for each patient.
Analysis of Lymphocyte Cytotoxicity
We analyzed the degranulation capacity of NK cells and cytotoxic T lymphocytes (CTL) from IP3 using CD107 degranulation assays. “Fresh” NK cell degranulation in response to K562 target cell stimulation was slightly impaired compared to more than 30 historical controls and additional day controls in two independent experiments (Figure 3A). Following IL-2 stimulation for 48h, the degranulation of patient NK cells increased to normal values. Moreover, degranulation of PHA/IL-2 blasts following stimulation with anti-CD3/28 was in the normal range (Figure 3B).
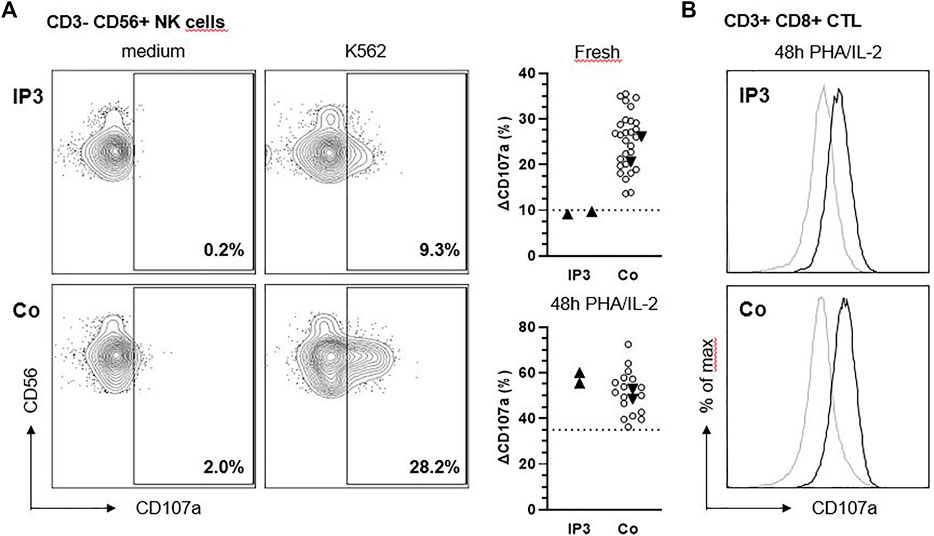
FIGURE 3. NK cell degranulation of IP3 is slightly reduced in response to K562. (A) Ex vivo degranulation of CD3− CD56+ NK cells from IP3 and a healthy day control (Co.) after incubation with medium (left panel) or NK-sensitive K562 target cells (middle panel) assessed by flow cytometric analysis of CD107a surface expression. Graphs in the right panel show ∆CD107a for the patient and healthy day controls (solid triangles) pooled from two independent experiments assessing fresh and activated (PHA/IL-2 pre-culture for 48 h) NK cell degranulation. ∆CD107a was calculated as the percentage of NK cells expressing CD107a after stimulation with K562 minus the percentage of NK cells expressing CD107a after incubation with medium. Blank circles represent historical controls (n = 28,17). The dashed line indicates the diagnostic cutoff below which NK cell degranulation is considered abnormal and equals the 10th percentile of a reference range. (B) Degranulation of CD3+ CD8+ CTL after incubation with medium (grey line) or anti-CD3/CD28 beads (black line).
Discussion
We report three patients with a severely impaired δ-granule secretion. NGS identified genetic alterations in three genes associated with different types of HPS. For IP3 the diagnosis of the rare HPS-7 was made after extensive investigations (cDNA sequencing, Western blot). The patient presented with an apparent OCA. Missense variants identified in HPS3 (IP1) and HPS5 (IP2) have to be classified as variant of uncertain significance (VUS), however we think it is valuable to report the findings of these variants in our patients.
Mild Phenotype in BLOC-2 Deficiencies
For IP1 (HPS-3) two compound heterozygous missense variants in HPS3 with predominantly and concordant disease-causing prediction were identified. Both variants are classified as VUS according to ACMG guidelines. The patient presented without cutaneous albinism, however, significantly hypopigmentation of the retina was detected in a specialized ophthalmological investigation. So far, the young girl exhibits mild bleeding symptoms. Taking these findings together these compound heterozygous variants most probably cause the patient’s mild phenotype. Such a mild phenotype has been described also in other patients with HPS-3 (Liu et al., 2021).
IP2 (HPS-5) carries a homozygous missense variant in HPS5 (c.760G > T). The PP is concordant disease-causing. According to the ACMG criteria the variant classifies VUS. The patient does not exhibit apparent albinism, however, a life-long history of bleeding diathesis. None of her 8 children (all heterozygous carriers) are clinically affected as severely as their mother (IP2), although some show very minor bleeding symptoms, such as gingival bleeding and easy bruising. Because the patient lives outside of Germany, a specialized opthalmological investigation has not been performed to date. IP2 suffered from a cystic lesion of her gall bladder, therefore the gall bladder was removed. Clinically, she did not show any symptoms of colitis and the ultrasound investigations did not reveal any signs of colitis. Besides the HPS5 variant, which we detected homozygous in the severely affected mother (IP2), we identified a heterozygous VUS in the gene VPS33B. According to OMIM alterations in the gene VPS33B are autosomal recessive associated with ARC (Arthrogryposis, renal dysfunction, and cholestasis). IP2 and the daughter who we investigated with NGS are carrier of the VPS33B variant; however, they do not show any symptoms of ARC. The son investigated with NGS presented wild type at the position. We did not investigate the other siblings for occurrence of the VPS33B variant. VPS33B is a protein essential for alpha granule biogenesis (Lo et al., 2005). Thrombin induced alpha-granule specific membrane protein P-selectin (CD62) exposure measured by flow cytometry was normal compared to healthy controls. However, a synergistic effect along with the homozygous HPS5 variant, which is also classified as VUS is conceivable. We excluded Chediak-Higashi syndrome an autosomal recessive disease associated with alterations in the gene LYST. One of the features is a partial or severe reduction of dense-granules. LYST is included in our NGS gene panel and the analysis did not show any pathological findings. Inclusions in polymorph nuclear leukocytes have typically been reported in the blood smear of patients with Chediak-Higashi syndrome. IP2 did not show any inclusions in polymorph nuclear leukocytes and no signs of immunodeficiency.
Both HPS3 and HPS5 gene products are part of the BLOC-2 complex and patients primarily present with mild bleeding symptoms (Huizing et al., 2020; Nurden et al., 2020) and not necessarily with noticeable hypopigmentation (Liu et al., 2021). Complications usually did not include pulmonary fibrosis and immunodeficiency. Granulomatous colitis has been reported in about 10–20% of patients (Hussain et al., 2006). Most of the reported variants in HPS3 and HPS5 are variants with serious consequences like deletions, duplications, and variants affecting splicing (Huizing et al., 2020; Liu et al., 2021), only a few missense variants have been described (Huizing et al., 2004; Michaud et al., 2017; Lasseaux et al., 2018). It has also been described that two different nonsense variants in HPS3 lead to different degrees of severities of OCA, suggesting a wide spectrum. One patient presented with a mild and two brothers exhibited a clear OCA (Sandrock-Lang et al., 2017; Lecchi et al., 2020). Both IP1 and IP2 show a mild phenotype, especially concerning the albinism. No further complications, like pulmonary fibrosis or immunodeficiency, were clinically observed in IP1 and IP2, so far.
BLOC-1 Deficiency
The homozygous deletion of exon 6 in DTNBP1, found in IP3, has not been previously described. This deletion is pathogenic according to the ACMG guidelines due to the serious consequence. In addition, we showed the absence of dysbindin in platelet lysate. Although this patient has exhibited the characteristic HPS symptoms since birth, the genetic defect was detected rather late in life. Even if the symptoms of OCA are obvious, the diagnosis of HPS can be delayed if the bleeding diathesis is not recognized as part of the disease: the patient was 60 years old at the time when HPS-7 was diagnosed. To our knowledge only seven patients with HPS-7 have been reported worldwide comprising 4 nonsense or frameshift variants in DTNBP1 (Li et al., 2003; Lowe et al., 2013; Bryan et al., 2017; Bastida et al., 2019). Due to the small number of patients, a definite statement regarding additional complications cannot be made. So far, only one patient has been reported developing granulomatous colitis (Lowe et al., 2013). The DTNBP1 encoded Dysbindin-1 is involved in neurotransmission regulation and neurodevelopment (Tang et al., 2009; Ghiani et al., 2010) and seems to be involved in the etiology of Schizophrenia (Cheah et al., 2015; Wang et al., 2017). IP3 showed no symptoms of schizophrenia or other psychiatric disorders, however mild deficits in social interaction or depressive-like emotion are difficult to identify.
IP3 shows a history of severe and recurring airway infections which may possibly hint to immunodeficiency. The most obvious immune defect in lysosomal trafficking disorders is impaired NK cell and CTL cytotoxicity (Fischer et al., 2007). Strong degranulation defects predispose to hemophagocytic lymphohistiocytosis (HLH), a severe disorder of hyperinflammation, while the clinical consequences of milder defects are poorly characterized. A previous report has concluded that CTL cytotoxicity is normal in “sandy” mice (a model of HPS-7) (Bossi et al., 2005), which we could confirm in our patient. However, as previously documented in patients and mice with Chediak-Higashi-Syndrome (Jessen et al., 2011) the fresh NK cell degranulation assay is more sensitive than the CTL assay in detecting subtle impairments of the lytic machinery. Moreover, the degranulation defect could also affect other immune cells such as neutrophils or mast cells (Ramadass and Catz 2016), which could make an additional contribution to the observed infection susceptibility.
Two patients with HPS-9 (subunit of BLOC-1) have been reported presenting immunodeficiency (Badolato et al., 2012; Okamura et al., 2019). This might point to a possible complication in patients with BLOC-1 variants. However, only a few patients with BLOC-1 deficiencies have been described at all. More data are required to understand the role of BLOC-1 proteins in the immune system.
Conclusion
Patients with variants of the BLOC-2 complex may not show the characteristic phenotype of a severe bleeding diathesis and oculocutaneous albinism; therefore, molecular genetic analysis including all HPS genes is needed for patients with a platelet delta granule secretion defect. Only a few patients with BLOC-1 deficiencies have been described so far and more data are needed to predict the phenotype and possible additional consequences. NGS can facilitate and accelerate the process of identifying the genetic defects regarding the different HPS-subtypes, especially if the patient presents with only a mild phenotype. Identification of a larger cohort for each subtype of HPS is important for the precise prediction of possible complications. This also offers new opportunities for further understanding the pathophysiology of HPS.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Albert Ludwig University of Freiburg. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
DB and BZ contributed to the design of the study. BZ, HG, and BK-K took care of the patients. DB, MW, FS, AL, KN, and JM conducted all the experiments. DB, MW, and JM wrote the manuscript. BZ and SE participated in the Writing-review and editing.
Funding
This work was supported by the DFG (SFB110 TP1 to SE).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Anja Kahle and Eileen Lerner for excellent laboratory work. The article processing charge was funded by the Baden-Wuerttemberg Ministry of Science, Research and Art and the University of Freiburg in the funding programme Open Access Publishing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.786937/full#supplementary-material
References
Ammann, S., Schulz, A., Krägeloh-Mann, I., Dieckmann, N. M., Niethammer, K., Fuchs, S., et al. (2016). Mutations in AP3D1 Associated with Immunodeficiency and Seizures Define a New Type of Hermansky-Pudlak Syndrome. Blood 127 (8), 997–1006. doi:10.1182/blood-2015-09-671636
Anikster, Y., Huizing, M., White, J., Shevchenko, Y. O., Fitzpatrick, D. L., Touchman, J. W., et al. (2001). Mutation of a New Gene Causes a Unique Form of Hermansky-Pudlak Syndrome in a Genetic Isolate of central Puerto Rico. Nat. Genet. 28 (4), 376–380. doi:10.1038/ng576
Badolato, R., Prandini, A., Caracciolo, S., Colombo, F., Tabellini, G., Giacomelli, M., et al. (2012). Exome Sequencing Reveals a Pallidin Mutation in a Hermansky-Pudlak-like Primary Immunodeficiency Syndrome. Blood 119 (13), 3185–3187. doi:10.1182/blood-2012-01-404350
Bastida, J. M., Morais, S., Palma-Barqueros, V., Benito, R., Bermejo, N., Karkucak, M., et al. (2019). Identification of Novel Variants in Ten Patients with Hermansky-Pudlak Syndrome by High-Throughput Sequencing. Ann. Med. 51 (2), 141–148. doi:10.1080/07853890.2019.1587498
Bossi, G., Booth, S., Clark, R., Davis, E. G., Liesner, R., Richards, K., et al. (2005). Normal Lytic Granule Secretion by Cytotoxic T Lymphocytes Deficient in BLOC-1, -2 and -3 and Myosins Va, VIIa and XV. Traffic 6 (3), 243–251. doi:10.1111/j.1600-0854.2005.00264.x
Bowman, S. L., Bi-Karchin, J., Le, L., and Marks, M. S. (2019). The Road to Lysosome-Related Organelles: Insights from Hermansky-Pudlak Syndrome and Other Rare Diseases. Traffic 20 (6), 404–435. doi:10.1111/tra.12646
Bryan, M. M., Tolman, N. J., Simon, K. L., Huizing, M., Hufnagel, R. B., Brooks, B. P., et al. (2017). Clinical and Molecular Phenotyping of a Child with Hermansky-Pudlak Syndrome-7, an Uncommon Genetic Type of HPS. Mol. Genet. Metab. 120 (4), 378–383. doi:10.1016/j.ymgme.2017.02.007
Bryceson, Y. T., Pende, D., Maul-Pavicic, A., Gilmour, K. C., Ufheil, H., Vraetz, T., et al. (2012). A Prospective Evaluation of Degranulation Assays in the Rapid Diagnosis of Familial Hemophagocytic Syndromes. Blood 119 (12), 2754–2763. doi:10.1182/blood-2011-08-374199
Cheah, S. Y., Lawford, B. R., Young, R. M., Morris, C. P., and Voisey, J. (2015). Dysbindin (DTNBP1) Variants Are Associated with Hallucinations in Schizophrenia. Eur. Psychiatry 30 (4), 486–491. doi:10.1016/j.eurpsy.2015.01.008
Christensen, S., Wagner, L., Coleman, M. M., and Appell, D. (2017). The Lived Experience of Having a Rare Medical Disorder: Hermansky-Pudlak Syndrome. Chronic Illn 13 (1), 62–72. doi:10.1177/1742395316655854
Di Pietro, S. M., Falcón-Pérez, J. M., and Dell'Angelica, E. C. (2004). Characterization of BLOC-2, a Complex Containing the Hermansky-Pudlak Syndrome Proteins HPS3, HPS5 and HPS6. Traffic 5 (4), 276–283. doi:10.1111/j.1600-0854.2004.0171.x
Enders, A., Zieger, B., Schwarz, K., Yoshimi, A., Speckmann, C., Knoepfle, E. M., et al. (2006). Lethal Hemophagocytic Lymphohistiocytosis in Hermansky-Pudlak Syndrome Type II. Blood 108 (1), 81–87. doi:10.1182/blood-2005-11-4413
Fischer, A., Latour, S., and de Saint Basile, G. (2007). Genetic Defects Affecting Lymphocyte Cytotoxicity. Curr. Opin. Immunol. 19 (3), 348–353. doi:10.1016/j.coi.2007.04.006
Fontana, S., Parolini, S., Vermi, W., Booth, S., Gallo, F., Donini, M., et al. (2006). Innate Immunity Defects in Hermansky-Pudlak Type 2 Syndrome. Blood 107 (12), 4857–4864. doi:10.1182/blood-2005-11-4398
Gahl, W. A., Brantly, M., Kaiser-Kupfer, M. I., Iwata, F., Hazelwood, S., Shotelersuk, V., et al. (1998). Genetic Defects and Clinical Characteristics of Patients with a Form of Oculocutaneous Albinism (Hermansky-Pudlak Syndrome). N. Engl. J. Med. 338 (18), 1258–1264. doi:10.1056/NEJM199804303381803
Gautam, R., Chintala, S., Li, W., Zhang, Q., Tan, J., Novak, E. K., et al. (2004). The Hermansky-Pudlak Syndrome 3 (cocoa) Protein Is a Component of the Biogenesis of Lysosome-Related Organelles Complex-2 (BLOC-2). J. Biol. Chem. 279 (13), 12935–12942. doi:10.1074/jbc.M311311200
Ghiani, C. A., Starcevic, M., Rodriguez-Fernandez, I. A., Nazarian, R., Cheli, V. T., Chan, L. N., et al. (2010). The Dysbindin-Containing Complex (BLOC-1) in Brain: Developmental Regulation, Interaction with SNARE Proteins and Role in Neurite Outgrowth. Mol. Psychiatry 15 (2115), 115–15. doi:10.1038/mp.2009.58
Hengst, M., Naehrlich, L., Mahavadi, P., Grosse-Onnebrink, J., Terheggen-Lagro, S., Skanke, L. H., et al. (2018). Hermansky-Pudlak Syndrome Type 2 Manifests with Fibrosing Lung Disease Early in Childhood. Orphanet J. Rare Dis. 13 (1), 42. doi:10.1186/s13023-018-0780-z
Hermansky, F., and Pudlak, P. (1959). Albinism Associated with Hemorrhagic Diathesis and Unusual Pigmented Reticular Cells in the Bone Marrow: Report of Two Cases with Histochemical Studies. Blood 14 (2), 162–169. doi:10.1182/blood.v14.2.162.162
Huizing, M., Anikster, Y., Fitzpatrick, D. L., Jeong, A. B., D'Souza, M., Rausche, M., et al. (2001). Hermansky-Pudlak Syndrome Type 3 in Ashkenazi Jews and Other Non-Puerto Rican Patients with Hypopigmentation and Platelet Storage-Pool Deficiency. Am. J. Hum. Genet. 69 (5), 1022–1032. doi:10.1086/324168
Huizing, M., Hess, R., Dorward, H., Claassen, D. A., Helip-Wooley, A., Kleta, R., et al. (2004). Cellular, Molecular and Clinical Characterization of Patients with Hermansky-Pudlak Syndrome Type 5. Traffic 5 (9), 711–722. doi:10.1111/j.1600-0854.2004.00208.x
Huizing, M., Malicdan, M. C. V., Wang, J. A., Pri-Chen, H., Hess, R. A., Fischer, R., et al. (2020). Hermansky-Pudlak Syndrome: Mutation Update. Hum. Mutat. 41 (3), 543–580. doi:10.1002/humu.23968
Hussain, N., Quezado, M., Huizing, M., Geho, D., White, J. G., Gahl, W., et al. (2006). Intestinal Disease in Hermansky-Pudlak Syndrome: Occurrence of Colitis and Relation to Genotype. Clin. Gastroenterol. Hepatol. 4 (1), 73–80. doi:10.1016/s1542-3565(05)00858-x
Jessen, B., Bode, S. F., Ammann, S., Chakravorty, S., Davies, G., Diestelhorst, J., et al. (2013). The Risk of Hemophagocytic Lymphohistiocytosis in Hermansky-Pudlak Syndrome Type 2. Blood 121 (15), 2943–2951. doi:10.1182/blood-2012-10-463166
Jessen, B., Maul-Pavicic, A., Ufheil, H., Vraetz, T., Enders, A., Lehmberg, K., et al. (2011). Subtle Differences in CTL Cytotoxicity Determine Susceptibility to Hemophagocytic Lymphohistiocytosis in Mice and Humans with Chediak-Higashi Syndrome. Blood 118 (17), 4620–4629. doi:10.1182/blood-2011-05-356113
Lahav, J., Jurk, K., Hess, O., Barnes, M. J., Farndale, R. W., Luboshitz, J., et al. (2002). Sustained Integrin Ligation Involves Extracellular Free Sulfhydryls and Enzymatically Catalyzed Disulfide Exchange. Blood 100 (7), 2472–2478. doi:10.1182/blood-2001-12-0339
Lasseaux, E., Plaisant, C., Michaud, V., Pennamen, P., Trimouille, A., Gaston, L., et al. (2018). Molecular Characterization of a Series of 990 index Patients with Albinism. Pigment Cell Melanoma Res 31 (4), 466–474. doi:10.1111/pcmr.12688
Lecchi, A., La Marca, S., Femia, E. A., Lenz, A., Boeckelmann, D., Artoni, A., et al. (2020). Novel Variant in HPS3 Gene in a Patient with Hermansky Pudlak Syndrome (HPS) Type 3. Platelets 31 (7), 960–963. doi:10.1080/09537104.2019.1704716
Li, W., Zhang, Q., Oiso, N., Novak, E. K., Gautam, R., O'Brien, E. P., et al. (2003). Hermansky-Pudlak Syndrome Type 7 (HPS-7) Results from Mutant Dysbindin, a Member of the Biogenesis of Lysosome-Related Organelles Complex 1 (BLOC-1). Nat. Genet. 35 (1), 84–89. doi:10.1038/ng1229
Liu, T., Yuan, Y., Bai, D., Qi, Z., Yang, L., Zhang, T., et al. (2021). Genetic Variants and Mutational Spectrum of Chinese Hermansky-Pudlak Syndrome Patients. Pigment Cell Melanoma Res 34 (1), 111–121. doi:10.1111/pcmr.12916
Lo, B., Li, L., Gissen, P., Christensen, H., McKiernan, P. J., Ye, C., et al. (2005). Requirement of VPS33B, a Member of the Sec1/Munc18 Protein Family, in Megakaryocyte and Platelet Alpha-Granule Biogenesis. Blood 106 (13), 4159–4166. doi:10.1182/blood-2005-04-1356
Lowe, G. C., Sánchez Guiu, I., Chapman, O., Rivera, J., Lordkipanidzé, M., Dovlatova, N., et al. (2013). Microsatellite Markers as a Rapid Approach for Autozygosity Mapping in Hermansky-Pudlak Syndrome: Identification of the Second HPS7 Mutation in a Patient Presenting Late in Life. Thromb. Haemost. 109 (4), 766–768. doi:10.1160/TH12-11-0876
Michaud, V., Lasseaux, E., Plaisant, C., Verloes, A., Perdomo-Trujillo, Y., Hamel, C., et al. (2017). Clinico-molecular Analysis of Eleven Patients with Hermansky-Pudlak Type 5 Syndrome, a Mild Form of HPS. Pigment Cell Melanoma Res 30 (6), 563–570. doi:10.1111/pcmr.12608
Mohammed, M., Al-Hashmi, N., Al-Rashdi, S., Al-Sukaiti, N., Al-Adawi, K., Al-Riyami, M., et al. (2018). Biallelic Mutations in AP3D1 Cause Hermansky-Pudlak Syndrome Type 10 Associated with Immunodeficiency and Seizure Disorder. Eur. J. Med. Genet. 62 (11), 103583.
Nurden, P., Stritt, S., Favier, R., and Nurden, A. T. (2020). Inherited Platelet Diseases with normal Platelet Count: Phenotypes, Genotypes and Diagnostic Strategy. Haematologica 106 (2), 337–350. Online ahead of print. doi:10.3324/haematol.2020.248153
Okamura, K., Hayashi, M., Abe, Y., Kono, M., Nakajima, K., Aoyama, Y., et al. (2019). NGS-based Targeted Resequencing Identified Rare Subtypes of Albinism: Providing Accurate Molecular Diagnosis for Japanese Patients with Albinism. Pigment Cell Melanoma Res 32 (6), 848–853. doi:10.1111/pcmr.12800
Pennamen, P., Le, L., Tingaud-Sequeira, A., Fiore, M., Bauters, A., Van Duong Béatrice, N., et al. (2020). BLOC1S5 Pathogenic Variants Cause a New Type of Hermansky-Pudlak Syndrome. Genet. Med. 22 (10), 1613–1622. doi:10.1038/s41436-020-0867-5
Ramadass, M., and Catz, S. D. (2016). Molecular Mechanisms Regulating Secretory Organelles and Endosomes in Neutrophils and Their Implications for Inflammation. Immunol. Rev. 273 (1), 249–265. doi:10.1111/imr.12452
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Sandrock-Lang, K., Bartsch, I., Buechele, N., Koehler, U., Simon-Gabriel, C. P., Eckenweiler, M., et al. (2017). Novel Mutation in Two brothers with Hermansky Pudlak Syndrome Type 3. Blood Cell Mol Dis 67. 75–80. doi:10.1016/j.bcmd.2017.03.001
Tang, T. T., Yang, F., Chen, B. S., Lu, Y., Ji, Y., Roche, K. W., et al. (2009). Dysbindin Regulates Hippocampal LTP by Controlling NMDA Receptor Surface Expression. Proc. Natl. Acad. Sci. U S A. 106 (50), 21395–21400. doi:10.1073/pnas.0910499106
Vincent, L. M., Adams, D., Hess, R. A., Ziegler, S. G., Tsilou, E., Golas, G., et al. (2009). Hermansky-Pudlak Syndrome Type 1 in Patients of Indian Descent. Mol. Genet. Metab. 97 (3), 227–233. doi:10.1016/j.ymgme.2009.03.011
Wang, H., Xu, J., Lazarovici, P., and Zheng, W. (2017). Dysbindin-1 Involvement in the Etiology of Schizophrenia. Int. J. Mol. Sci. 18 (10). doi:10.3390/ijms18102044
Wei, M. L. (2006). Hermansky-Pudlak Syndrome: a Disease of Protein Trafficking and Organelle Function. Pigment Cell Res 19 (1), 19–42. doi:10.1111/j.1600-0749.2005.00289.x
Keywords: Hermansky-Pudlak syndrome (HPS), HPS-3, HPS-5, HPS-7, BLOC-1, BLOC-2
Citation: Boeckelmann D, Wolter M, Neubauer K, Sobotta F, Lenz A, Glonnegger H, Käsmann-Kellner B, Mann J, Ehl S and Zieger B (2022) Hermansky-Pudlak Syndrome: Identification of Novel Variants in the Genes HPS3, HPS5, and DTNBP1 (HPS-7). Front. Pharmacol. 12:786937. doi: 10.3389/fphar.2021.786937
Received: 30 September 2021; Accepted: 24 December 2021;
Published: 19 January 2022.
Edited by:
Shenghui Zhang, First Affiliated Hospital of Wenzhou Medical University, ChinaReviewed by:
Rong He, Mayo Clinic, United StatesLluis Montoliu, National Center for Biotechnology (CSIC), Spain
Copyright © 2022 Boeckelmann, Wolter, Neubauer, Sobotta, Lenz, Glonnegger, Käsmann-Kellner, Mann, Ehl and Zieger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Zieger, barbara.zieger@uniklinik-freiburg.de
†These authors have contributed equally to this work