Case Report: A novel PHOX2B p.Ala248_Ala266dup variant causing congenital central hypoventilation syndrome
 Irina N. Artamonova1*
Irina N. Artamonova1*  Anna M. Zlotina2
Anna M. Zlotina2  Olga R. Ismagilova3
Olga R. Ismagilova3  Tatyana A. Levko4
Tatyana A. Levko4  Natalia Yu Kolbina5
Natalia Yu Kolbina5  Aleksandr V. Bryzzhin6
Aleksandr V. Bryzzhin6  Andrey P. Smorodin7
Andrey P. Smorodin7  Alexandr V. Borodin8 Ekaterina A. Mamaeva9
Alexandr V. Borodin8 Ekaterina A. Mamaeva9  Anna A. Sukhotskaya10
Anna A. Sukhotskaya10  Ilya M. Kagantsov11
Ilya M. Kagantsov11  Daria A. Malysheva12
Daria A. Malysheva12  Elena S. Vasichkina13
Elena S. Vasichkina13  Tatiana M. Pervunina14
Tatiana M. Pervunina14  Natalia A. Petrova15*
Natalia A. Petrova15*
- 1Institute of Perinatology and Pediatrics, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 2Institute of Molecular Biology and Genetics, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 3Federal State Budgetary Scientific Institution, Research Centre for Medical Genetics (RCMG), Moscow, Russia
- 4Department of Pediatric and Medical Rehabilitation, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 5Department of Pediatric and Medical Rehabilitation, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 6Pediatric Anesthesiology and Intensive Care Unit, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 7Pediatric Surgery Anesthesiology and Intensive Care Unit Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 8World-Class Research Centre for Personalized Medicine, Research Centre of Unknown, Rare and Genetically Determined Diseases, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 9Institute of Perinatology and Pediatrics, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 10Department of Pediatric Surgery for Congenital Malformations, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 11Department of Pediatric Surgery for Congenital Malformations, Institute of Perinatology and Pediatrics, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 12Department of Pediatric Surgery for Congenital Malformations, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 13World-Class Research Centre for Personalized Medicine, Research Centre of Unknown, Rare and Genetically Determined Diseases, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 14Institute of Perinatology and Pediatrics, World-Class Research Centre for Personalized Medicine, Research Centre of Unknown, Rare and Genetically Determined Diseases, Almazov National Medical Research Centre, Saint-Petersburg, Russia
- 15World-Class Research Centre for Personalized Medicine, Research Centre of Unknown, Rare and Genetically Determined Diseases, Institute of Perinatology and Pediatrics, Almazov National Medical Research Centre, Saint-Petersburg, Russia
Introduction: Congenital central hypoventilation syndrome (CCHS) is a rare disease characterized by central alveolar hypoventilation and impaired autonomic regulation, caused by pathogenic variants of PHOX2B gene. More than 90% of patients have a polyalanine repeat mutation (PARM) in the heterozygous state, characterized by the expansion of GCN repeats and an increase in the number of alanine repeats, so that genotypes 20/24–20/33 are formed (the normal genotype is 20/20). The remaining 10% of patients harbor non-PARMs.
Case description: We present a clinical case of a girl with a novel PHOX2B heterozygous genetic variant in the exon 3: NM_003924.4: c.735_791dup, p.Ala248_Ala266dup. The duplication includes 16 GCN (alanine) repeats and 3 adjacent amino acids. Both clinically healthy parents demonstrated a normal PHOX2B sequence. In addition, the girl has a variant of unknown significance in RYR1 gene and a variant of unknown significance in NKX2-5 gene. The child's phenotype is quite special. She needs ventilation during sleep, and has Hirschsprung's disease type I, arteriovenous malformation S4 of the left lung, ventricular and atrium septal defects, coronary right ventricular fistula, hemodynamically nonsignificant, episodes of sick sinus and atrioventricular dissociation with bradycardia, divergent alternating strabismus, and oculus uterque (both eyes) (OU) retinal angiopathy. Two episodes of hypoglycemic seizures were also registered. Severe pulmonary hypertension resolved after appropriate ventilation adjustment. Diagnostic odyssey was quite dramatic.
Conclusion: Detection of a novel PHOX2B variant expands the understanding of molecular mechanisms of CCHS and genotype–phenotype correlations.
Introduction
Congenital central hypoventilation syndrome (CCHS) is a rare disease characterized by central alveolar hypoventilation and impaired autonomic regulation. In most cases, patients have normoventilation during wakefulness and hypoventilation during sleep, most pronounced in the non-rapid eye movement (non-REM) sleep phase (1–3). The estimated incidence of the disease in population is 1:200,000 (4).
CCHS is associated with the pathogenic variants of the PHOX2B gene, which encodes a highly conserved homeobox transcription factor. As a result, the expression of a number of genes is altered, which leads to a disruption in the formation of the autonomic nervous system (5–9). More than 90% of patients have a polyalanine repeat mutation (PARM) in the heterozygous state, characterized by the expansion of GCN repeats and an increase in the number of alanine repeats, so that genotypes 20/24–20/33 are formed (the normal genotype is 20/20). The remaining 10% of patients harbor non-PARMs (NPARMs) including missense, frameshift, nonsense, and splice site variants in one of three PHOX2B exons. (10). Single cases of partial or whole-gene deletions were also described (11). Pathogenic variants in the PHOX2B gene are autosomal dominant and in most cases occur de novo; however, in 5%–10% of cases, asymptomatic mosaicism is detected in one of the parents (10). The direct correlation between the number of polyalanine repeats and the severity of the disease has been proposed (10, 12, 13). Hirschsprung’s disease (detected in 20% of patients, Haddad syndrome), neural crest tumors (neuroblastoma, ganglioneuroma), glucose metabolism disorders (hypoglycemia, signs of hyperinsulinism), as well as various signs of autonomic dysfunction (thermolability, arrhythmias, fluctuations in blood pressure, sudden episodes of muscle hypotension, various ophthalmic symptoms) are more common among 20/27–20/33 and NPARM genotypes. Patients with 20/24–20/25 PARM genotypes present with mild phenotypes and late-onset variants (10, 14–17).
We present a clinical case of a girl with a newly described PHOX2B variant including duplication of 16 GCN (alanine) repeats and 3 adjacent amino acids. The child's phenotype–genotype correlation is quite special.
Case description
A 3.5-year-old girl freshly diagnosed with CCHS was transferred to our hospital for further evaluation.
The girl was born at term by cesarean section from healthy non-consanguineous parents. She developed respiratory insufficiency during the first hours of life requiring constant lung ventilation. Extubation attempts were unsuccessful due to bradypnea (10–15 breaths per minute), desaturations (SpO2 up to 60%–75%), and hypercapnia (values are not available) during sleep. After one of the extubations, lung bleeding occurred, and arteriovenous malformation S4 of the left lung was diagnosed on a CT scan. The girl developed clonic seizures at the age of 3 months, which resolved after phenobarbital administration. She required prolonged ventilation, extubation attempts were still unsuccessful, with an episode of cardiorespiratory arrest, and was tracheostomized at the age of 4 months.
Subsequently, being undiagnosed, the girl was not ventilated appropriately with long periods of self-breathing during sleep, which resulted in desaturations. Decannulation to mask ventilation at 36 months failed due to intolerance, followed by re-tracheostomy. The spontaneous breathing during sleep was insufficient; however, ventilation was still sporadic. The girl suffered frequent pneumonias and tracheitis with purulent bloody sputum.
On echocardiography, ventricular septal defect, 4 mm atrial septal defects, and coronary right ventricular fistula (CAF) were diagnosed, and considered hemodynamically nonsignificant. However, chronic heart failure developed: dilation of the right chambers of the heart, right ventricle hypertrophy, pulmonary hypertension (calculated systolic pressure in the right ventriculum 70 mmHg), hepatomegaly developed by the age of 18 months, and ejection fraction was 64%–72% by Teichholz. By the age of 42 months, ejection fraction decreased to 49% and ascites developed. Episodes of sick sinus and atrioventricular dissociation with bradycardia (37–51 beats per minute, pauses up to 2,255 ms) were first diagnosed at the age of 19 months. At further evaluations, heart rhythm improved; however, episodes of bradycardia during daytime persisted.
The girl suffered constipation since birth and megacolon or dolichosigma was suspected due to ultrasound and irrigography findings.
Apart from that, thrombocytopenia persisted (80–124 × 10 × 9/L at 12–36 months).
At the age of 42 months, first episode of hypoglycemic seizures took place (blood glucose level 1.38 mmol/L, sodium 120 mmol/L, and chloride 77 mmol/L).
Genetic test was done only at the age of 3.5 years. Initially, a blood sample of the patient was sent to a commercial genetic testing laboratory for whole-exome sequencing (WES) analysis. WES was carried out using SureSelect All Exon V7 target enrichment kit (Agilent Technologies, CA, United States) and Illumina NovaSeq 6000 instrument with average target region coverage of ∼170× (98.8% of targeted nucleotides with coverage >10×). The laboratory provided us with a report with WES results including genetic variants possibly related with the clinical phenotype and secondary incidental findings in genes recommended by the ACMG (18). Based on WES data, the girl has a missense variant of uncertain significance—chr19:g.38993563 G>C, NM_000540.3:c.7879G>C (p.Val2627Leu) (rs914804033)—in RYR1 gene, in which pathogenic variants are known to be associated with malignant hyperthermia susceptibility (OMIM # 145600) but seemingly not with a CCHS condition. Moreover, WES allowed us to reveal a rare genetic variant in cardiac homeobox gene NKX2-5: chr5:g.172661909 C>G, NM_004387.4: c.178G>C, (p.Glu60Gln), (rs766199339). Notably, no PHOX2B variations were mentioned in the report.
Then, PHOX2B sequencing was performed in the Research Centre for Medical Genetics, Moscow; afterward, results were validated in our institution by bidirectional Sanger sequencing. The sequencing procedure was carried out using the BigDye Terminator Sequencing Kit (Applied Biosystems) and Genetic Analyzer AB3100 (Applied Biosystems/Hitachi, Japan). The primers were designed using the NCBI Primer Blast tool (Gene ID: 8929, NG_008243.1; exon 1: F 5′-AATTTTGTTGGCGGTTCGGG-3′, R 5′-TAGGCTCTGCTGGTAGTAAGGA-3′; exon 2: F 5′-AATCCAGTATTTCTGATCGGCCA-3′, F 5′-TGAAAGCACTATCTCAAGTCCGT-3′; exon 3a F 5′-CATACTGCTCTTCACTAAGGCG-3′, R 5′-GAGGGTGTTAAAACAAGCCGA-3′; exon 3b F 5′-GGCCCTCAATGAAAAAGCCA-3′, R 5′-TCCTCGGGCAAAAAGTCTGA-3′).
Target sequencing of PHOX2B protein-coding regions allowed us to identify a novel heterozygous genetic variant in the exon 3: NM_003924.4: c.735_791dup, (p.Ala248_Ala266dup) (Figure 1). The 57-bp duplication corresponds to 13 GCN (alanine) repeats plus 6 adjacent amino acids (Gly-Gly-Leu-Ala-Ala-Ala). It represents a non-frameshift duplication leading to the protein elongation (+19 amino acids).
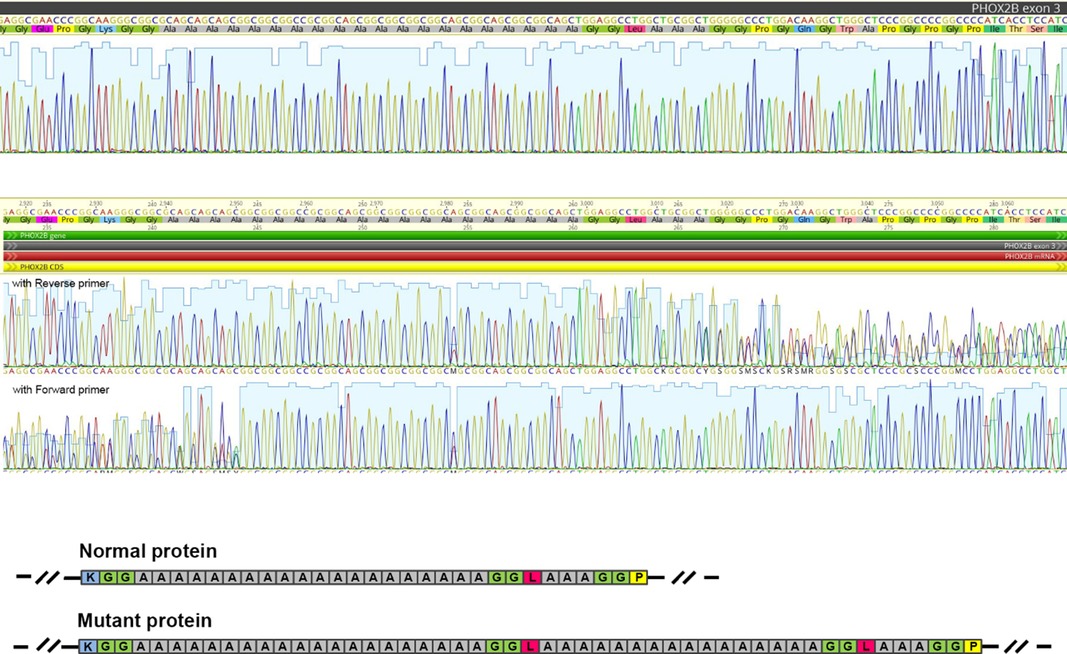
Figure 1. The PHOX2B genetic variant (p.Ala248_Ala266dup) was revealed by Sanger sequencing. The figure shows a fragment of Sanger sequencing chromatogram corresponding to the polyalanine repeat and flaking regions in a control (“reference”) sample (A) and in the reported patient (B). (C) The scheme illustrating the impact of the duplication on the amino acid sequence in the mutant protein. Colored boxes represent amino acid residues (blue—lysin, green—glycine, grey—alanine, magenta—leucine, and yellow—proline).
Both clinically healthy parents demonstrated a normal PHOX2B sequence.
After the diagnosis had been made at the age of 44 months, the girl was transported to our hospital. By admission, she was ventilated through a tracheostomy tube 3–5 h per night followed by awakening and subsequent unsuccessful attempts to resume ventilation caused by overventilation in REM sleep. The girl's height was 87 cm (−3.14 SD), weight was 11 kg (−2.55 SD), and weight-to-height ratio was −0.89 SD. Respiratory support was adjusted under tcCO2 monitoring: ST rate 25/min, Pi 15 cm Н2О, Pimax 21, EPAP 5 сm Н2О, FiO2-21%, Tin-0,7 s. The vital need for mechanical ventilation during sleep was explained to the parents.
Echocardiography showed muscular ventricular septal defect (2 mm), atrial septal defect (2–3 mm), ejection fraction of 62.5% (by Teichholz), and systolic pulmonary artery pressure of 36 mmHg. On 48-h Holter ECG, heart rate appeared normal, but there was sinus arrhythmia with pauses up to 1,248 ms, QTc prolongation up to 511 ms, and a decrease in heart rate variability with no nighttime increase in the high-frequency component of variability. Ophthalmologic examination discovered divergent alternating strabismus OU and retinal angiopathy OU.
Neurologic status was as follows. The girl maintained head upright all the time, sat and stood without support, walked independently well, kicked a ball forward, and threw a ball over hand. The girl could not run, jump, and walk upstairs. Her understanding of the addressed speech was full. She used pincer grasp, held the pen, but could not copy shapes (circle, square, etc.) or imitate vertical line. Denver Developmental Screening test at age of 4 years showed MQ = 0.62 (N ≥ 0.75) and DQ = 0.57 (N ≥ 0.7). Her cranial innervation was intact. She had muscle hypotonia, with muscle strength of 5 points in limbs according to the MRС scale (Medical Research Council scale for power of muscle), valgus flat foot. Tendon reflexes were normal. There were no meningeal and cerebral symptoms.
Hirschsprung's disease was suspected due to chronic constipation, an increase in the volume of the abdomen (Figure 2). Irrigography showed narrowing of the rectum and sigmoid colon with pronounced suprastenotic expansion (Figure 3).
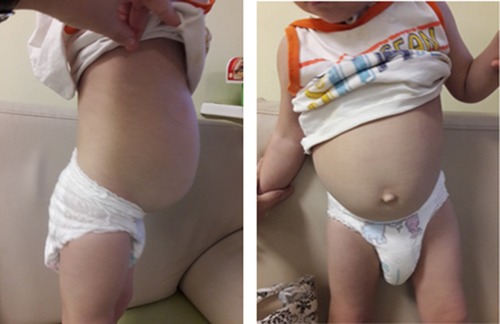
Figure 2. Hirschsprung's disease: abdomen distension.
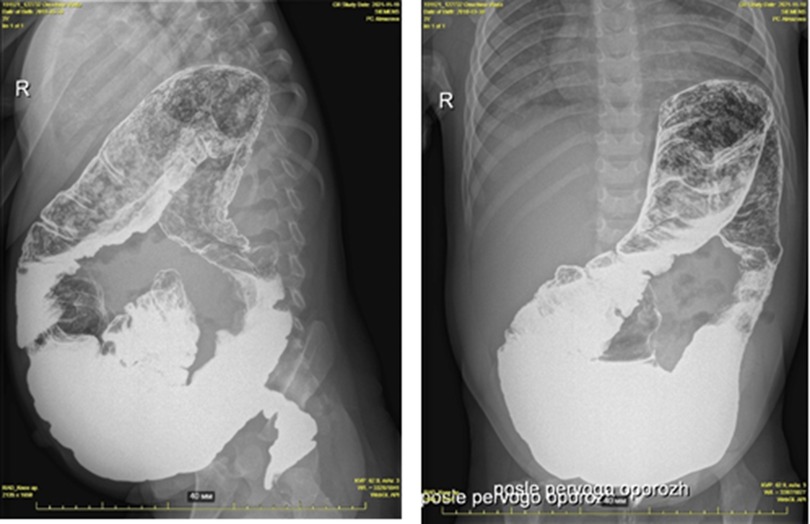
Figure 3. Hirschsprung's disease. Barium enema showing reduced caliber of the rectum, followed by a transition zone to an enlarged-caliber sigmoid.
A laparoscopy with a biopsy of the colon was performed, histology showed aganglionosis, which indicates Hirschsprung’s type I disease. A colostomy was done on the descending colon. After the surgery, bloating decreased, and ventilation improved.
Five months later, the girl was readmitted. Her height was 89 cm (−3.21 SD), weight was 14.25 kg (−0.9 SD), and weight-to-height ratio was +1.67 SD. LS Swenson pull-through was performed. After surgery, the ventilation settings were adjusted with the lower settings needed during REM sleep. Heart rate normalized with maximum QTc of 470 ms. Echocardiography data slightly improved: ejection fraction: 64.6% (by Teichholz) and systolic pulmonary artery pressure: 25 mmHg.
Another hypoglycemic episode took place at 48 months of age (glucose 2.1–2.35 mmol/L without electrolyte disorders). Afterward, glucose levels were kept under dynamic control; no more hypoglycemic episodes were registered.
The disease course is showed in Figure 4.
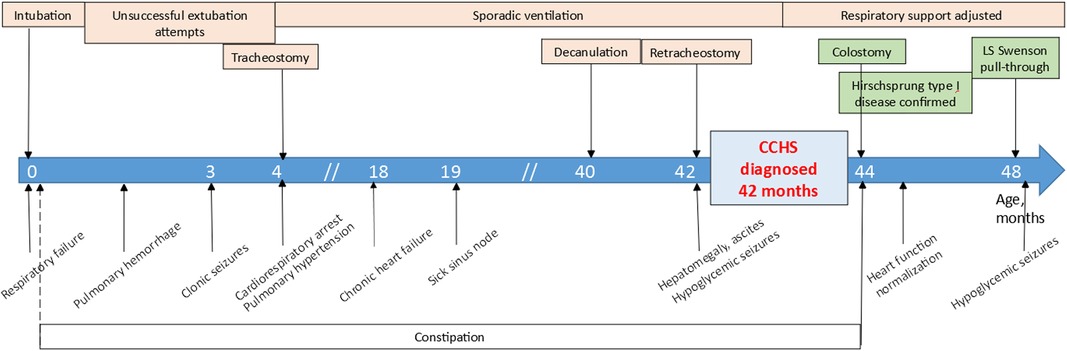
Figure 4. The disease course of the patient.
Discussion
CCHS is generally associated with PARM and NPARM pathogenic variants in PHOX2B gene (19). In the clinical case reported here, we revealed a novel heterozygous variant (p.Ala248_Ala266dup) in the exon 3 of PHOX2B. Notably, the initial whole-exome sequencing analysis did not reveal this genetic variation. Such results are in accordance with the accumulated data pointing to ineffectiveness of new generation sequencing (NGS) approaches such as targeted gene panel sequencing and exome sequencing for polyalanine repeat expansion detection (20). Here, we once again confirm that Sanger sequencing is an adequate and relevant tool for detection of genetic variants affecting PHOX2B polyalanine repeat region, which is necessary for early and correct diagnosis of CCHS conditions.
As for classification of the genetic variant, it is not so straightforward. Taking into account its size and content, the duplication should not be regarded as a traditional PARM, although it overlaps with the polyalanine repeat region. Anyway, the genetic variant results in a change in the extent and structure of the polyalanine repeat region, apparently leading to the altered protein organization. Previously, functional effects of missense, frameshift, and alanine expansion pathogenic variants in PHOX2B sequence were carefully evaluated (21). Trochet et al. showed the declined transactivation of the dopamine beta-hydroxylase promoter (a direct PHOX2B transcriptional target) by PHOX2B with polyalanine expansions. In addition, DNA binding was consistently decreased in the longest expansions (+9 alanines and above). Interestingly, the authors tested not only traditional expansions ranging from +5 to +13 alanines but also an alanine expansion interrupted by a threonine residue (Ala)11(Thr)(Ala)15. Such a variant demonstrated transactivation and DNA binding properties within the normal range (21). It implies that different kinds of alterations in the polyalanine repeat region could have varying impact on PHOX2B protein function, and, therefore, on the severity of the clinical phenotype. Further in silico and/or in vitro functional studies could specify in more detail the molecular consequences of the (p.Ala248_Ala266dup) variant.
PHOX2B is essential for normal function of autonomous nervous system (22), and hence all autonomic processes.
In PARMs, the severity of the disease almost always depends on the number of GCN repeats, with 20/24–20/26 genotypes presenting with mild hypoventilation and respiratory support during sleep only, while in patients with 20/27–20/33 genotypes, ventilation requirements may include daytime needs and vary up to 24/7 ventilation. These high-grade PARMs are also associated with higher Hirschprung's disease (HD), autonomic nervous system dysfunction (ANSD), and neural crest tumor (NCT) risk (10, 19, 23–25). Few exceptions were presented so far: Kasi et al. described a late-onset case with 20/27 genotype (26), while Lee-Kelland et al. could not find significant correlation between the length of GCN expansion and minute ventilation during either REM or non-REM sleep (27). NPARMs, on the other hand, are not so straightforward (24, 28). They are in general considered more pathogenic than PARMs and are highly associated with HD and NCT (10, 28, 29). However, there are several reports of NPARM genotypes with relatively mild phenotypes (28, 30, 31). It was speculated that the severity of NPARM depends on the PHOX2B exon with severe variants linked to exon 3, while pathogenic variants in exon 1 present with mild phenotype (23). Some cases of NPARM as well as 20/24 PARM ones present as late-onset CCHS in adolescence or even adulthood or even have no clinical signs at all (28, 32). Zhou et al. supposed that exon 1 and 2 loss-of-function NPARM variants activate nonsense-mediated mRNA decay (NMD), leading to absence of the truncated protein, like full-gene deletions do, and thus resulting in more mild phenotype (12). Exon 3 variants, being in the last exon of the PHOX2B gene, are less likely to trigger NMD control pathway, so the truncated protein is produced, having a potentially detrimental dominant-negative effect (12).
Of rare cases, compound heterozygous and homozygous variants were described (28). However, to our knowledge, there are no descriptions of such atypical PARM cases with prolonged GCN repeats interrupted by additional non-alanine amino acids.
As mentioned above, children with high-grade PARMs are supposed to have severe hypoventilation and prolonged need for ventilation which was not the case in our patient, who required respiratory support only during sleep.
From the study by Trochet et al., we can speculate that interruption of polyAla tract by different amino acids may have “protective” effect on phenotype (21). While Trochet et al. divided polyalanine expansion into two short parts (11 and 15 alanines), which led to normalization of transactivation and DNA binding properties, in our case we have a prolonged interrupted expansion of 20 + 16 alanines and a phenotype of nighttime (mostly non-rapid eye movement (NREM)) hypoventilation, which is not as severe as could be expected, in combination with HD, and autonomic dysregulation signs: sick sinus and atrioventricular dissociation with bradycardia, and divergent alternating strabismus (21). Further in silico and/or in vitro functional studies could specify in more detail the molecular consequences of the variant. The effect of expanded polyAla tract interruption on clinical presentation could be confirmed if other similar cases were described.
The multisystem presentation of CCHS requires a multidisciplinary approach. Apart from the evident need of respiratory support and regular respiratory physician's consultations with polysomnography and/or blood gas evaluations, full assessment must include testing of ANSD (neurologist, ophthalmologist, fasting, and postprandial glucose monitoring) and cardiac evaluation (including Holter monitoring, echocardiography, and arterial blood pressure measurement) (33–37). In 20/27-20/33 PARM, and NPARM genotypes oncology search is required (chest radiography, abdomen ultrasound, and brain MRI in some cases) (19). This approach was first proposed in 2010 by Weese-Mayer et al. (10) and further revised in 2020 (38) and 2022 (19).
Cardiovascular abnormalities usually described in CCHS patients are sinus node dysfunction, QTc elongation, and blood pressure dysregulation due to ANSD. Hypoventilation and hypoxia result in pulmonary hypertension and cor pulmonale (32). In our patient, apart from arrhythmia and pulmonary hypertension, we saw multiple structural defects: small septal defects, CAF, and pulmonary arteriovenous malformation (PAVM) S4 of the left lung. Association of CCHS and congenital heart defects (CHDs) was shown in two reports. Lombardo et al. reviewed single-institution population of CCHS patients and found a 30% (6/20) prevalence of CHD. CHDs were found in five children with NPARMs (multiple atrial septal defect (ASD), patent ductus arteriosus, vertebral, coronary arteries anomalies, complete vascular ring), and a secundum ASD in a patient with a 20/33 PARM (39). Similar prevalence of CHD was found in the study of Mei et al.; however, exact CHD types were not provided and patent ductus arteriosus and atrial septal defect were prevalent (40).
At the same time, WES revealed a rare genetic variant in cardiac homeobox gene NKX2-5: chr5:g.172661909 C>G, NM_004387.4: c.178G>C, (p.Glu60Gln), (rs766199339). To our knowledge, this variant had not been previously reported as a genetic cause of any congenital heart disease cases in the literature. It is present in population allele frequency databases (GnomAD 0.03%), and there are contradictory computational predictions regarding its effect on protein structure and function. Thus, currently available data are insufficient to specify the contribution of the NKX2-5 p.Glu60Gln variant to the patient's cardiac phenotype, and according to ACMG (41), it should be classified as a variant of unknown significance (VUS).
Unusual for CCHS, vascular anomalies were found in our patient: CAF, PAVM, and retinal angiopathy. It is known, that CAF and PAVM (42) are hereditary in up to 90% of cases and are often associated with telangiectasia (43), but such pathogenic variants were not discovered in our patient, as well as telangiectasia. Many cases of CAF and PAVM are asymptomatic (43, 44). The complications of PAVM may include hypoxemia, hemoptysis, pulmonary hypertension, high-output heart failure, and cerebrovascular accidents (42), but hypoventilation was not described. At that point, we believe that structural cardiovascular abnormalities in our patient do have genetic origin, but we cannot definitely attribute them to either PHOX2B or NKX2-5.
Based on WES results, the patient has a genetic variant c.7879G>C (p.Val2627Leu), rs914804033, in RYR1 gene. The RYR1 gene encodes the skeletal muscle ryanodine receptor playing a role of a calcium release channel of the sarcoplasmic reticulum and ensures connection between the sarcoplasmic reticulum and transverse tubule. To date, a spectrum of different autosomal dominant and recessive clinical conditions has been described in association with RYR1 genetic variants. In particular, heterozygous, homozygous, or compound heterozygous RYR1 variants can cause central core disease (OMIM #117000). Biallelic variants were also revealed in cases of minicore myopathy with external ophthalmoplegia (OMIM #255320). Susceptibility to malignant hyperthermia is another autosomal dominant skeletal muscle disorder caused by heterozygous pathogenic variants in RYR1 (OMIM #145600). In addition, heterozygous RYR1 variants can be a genetic basis of the King–Denborough syndrome (KDS), a clinical condition characterized by the triad of congenital myopathy, dysmorphic features, and susceptibility to malignant hyperthermia (45). The revealed RYR1 genetic variant is present in population allele frequency databases (GnomAD Genomes f = 0.0000319), and it has been reported in individuals with a history of a malignant hyperthermia reaction. Our patient did not show clinical signs of neuromuscular disease. In her neurological state, we noted muscle hypotonia, most likely central, but there were no signs of muscle weakness.
Hypoglycemia in our case is consistent with well-described spectrum of glucose metabolism disturbances in CCHS (34) with no phenotype–genotype correlation.
The diagnosis and treatment of patients with CCHS are quite complicated and may be accompanied by late diagnosis of Hirschsprung's disease (10). The inclusion of a pediatric surgeon in a multidisciplinary team is important for appropriate diagnosis and treatment of HD. In the case we describe, HD was not diagnosed in time. We believe that all children with CCHS and constipation should be screened for HD, as well as all patients with HD and respiratory issues should be screened for CCHS.
Neurodevelopmental delay is an issue in CCHS patients. Its causes are disputable with input of recurrent or acute hypoxemia and hypoventilation, on the one hand, and PHOX2B dysfunction itself, on the other hand (19). Neurologic consequences may vary from severe headache and daytime drowsiness to status epilepticus (32).
Presence of mild cases along with mosaicism (both somatic and germinogene) and incomplete penetrance makes it necessary to examine all the relatives of CCHS patients.
In our case, in the course of diagnostic odyssey while hypoventilation was not appropriately adjusted, it resulted in heart failure, pulmonary hypertension, and arrhythmia, which at that time were addressed not to respiratory dysfunction but to heart and vascular structural anomalies (septal defects and coronary right ventricular fistula). A significant improvement was achieved after respiratory support correction, which makes us speculate that it was hypoventilation that caused these cardiovascular consequences. However, with late diagnosis, the changes may be irreversible (32).
Conclusion
The detection of a novel PHOX2B variant expands the understanding of molecular mechanisms of CCHS and genotype–phenotype correlations.
Our case seems to combine clinical features of both nonsevere PARMs and NPARMs: the girl has HD, ANSD, and structural cardiovascular abnormalities, like severe NPARM cases do (24, 40, 46), but her hypoventilation is relatively mild, requiring non-invasive lung ventilation (NLV) only during sleep. It was proposed that PHOX2B pathogenic variants have variable penetrance depending on unknown epigenetic factors (28, 46), and further evaluation is needed. The understanding of molecular effects of different kinds of alterations in the polyalanine repeat region could provide more information on estimated phenotype changes. Association of PHOX2B pathogenic variants with cardiovascular anomalies needs extra research. The diagnostic odyssey also illustrates an insufficient awareness of CCHS and pathophysiology of autonomic regulation disorders among medical society.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
INA is the first author and attending physician, and wrote the draft. AMZ performed gene sequencing and described genetic issues. ORI performed gene sequencing and revised the manuscript. TAL was the attending physician and contributed to case description and manuscript revision. NYK contributed to discussion and manuscript revision. AVBr and AVBo adjusted respiratory support and revised the manuscript. APS was the attending physician during postoperation period. EAM contributed to neurologic issues description and discussion. AAS and DAM were the operating surgeons and contributed to case description and surgical issues. IMK contributed to discussion and revised the manuscript. ESV revised the manuscript. NAP is the senior author and supervised and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075-15-2022-301).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Paton JY, Swaminathan S, Sargent CW, Keens TG. Hypoxic and hypercapnic ventilatory responses in awake children with congenital central hypoventilation syndrome. Am Rev Respir Dis. (1989) 140(2):368–72. doi: 10.1164/ajrccm/140.2.368
2. Shea SA, Andres LP, Shannon DC, Guz A, Banzett RB. Respiratory sensations in subjects who lack a ventilatory response to CO2. Respir Physiol. (1993) 93(2):203–19. doi: 10.1016/0034-5687(93)90006-V
3. Marcus CL, Bautista DB, Amihyia A, Ward SL, Keens TG. Hypercapneic arousal responses in children with congenital central hypoventilation syndrome. Pediatrics. (1991) 88(5):993–8.1945641
4. Trang H, Dehan M, Beaufils F, Zaccaria I, Amiel J, Gaultier C, et al. The French Congenital Central Hypoventilation Syndrome Registry: general data, phenotype, and genotype. Chest. (2005) 127(1):72–9. doi: 10.1378/chest.127.1.72
5. Bishara J, Keens TG, Perez IA. The genetics of congenital central hypoventilation syndrome: clinical implications. Appl Clin Genet. (2018) 11:135–44. doi: 10.2147/TACG.S140629
6. Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. (1999) 399(6734):366–70. doi: 10.1038/20700
7. Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development. (1997) 124(20):4065–75. doi: 10.1242/dev.124.20.4065
8. Sasaki A, Kanai M, Kijima K, Akaba K, Hashimoto M, Hasegawa H, et al. Molecular analysis of congenital central hypoventilation syndrome. Hum Genet. (2003) 114(1):22–6. doi: 10.1007/s00439-003-1036-z
9. Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Silvestri JM, Curran ME, et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am J Med Genet A. (2003) 123A(3):267–78. doi: 10.1002/ajmg.a.20527
10. Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H, et al. An official ATS clinical policy statement: congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med. (2010) 181(6):626–44. doi: 10.1164/rccm.200807-1069ST
11. Jennings LJ, Yu M, Rand CM, Kravis N, Berry-Kravis EM, Patwari PP, et al. Variable human phenotype associated with novel deletions of the PHOX2B gene. Pediatr Pulmonol. (2012) 47(2):153–61. doi: 10.1002/ppul.21527
12. Zhou A, Rand CM, Hockney SM, Niewijk G, Reineke P, Speare V, et al. Paired-like homeobox gene (PHOX2B) nonpolyalanine repeat expansion mutations (NPARMs): genotype–phenotype correlation in congenital central hypoventilation syndrome (CCHS). Genet Med. (2021) 23(9):1656–63. doi: 10.1038/s41436-021-01178-x
13. Matera I, Bachetti T, Puppo F, Di Duca M, Morandi F, Casiraghi GM, et al. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset central hypoventilation syndrome. J Med Genet. (2004) 41(5):373–80. doi: 10.1136/jmg.2003.015412
14. Congenital central hypoventilation syndrome clinical presentation: history, physical examination. Available at: https://emedicine.medscape.com/article/1002927-clinical?src=refgatesrc1 (Accessed September 29, 2022).
15. Marics G, Amiel J, Vatai B, Lódi C, Mikos B, Tóth-Heyn P. Autonomic dysfunction of glucose homoeostasis in congenital central hypoventilation syndrome. Acta Paediatr. (2013) 102(4):e178–80. doi: 10.1111/apa.12125
16. Haddad GG, Mazza NM, Defendini R, Blanc WA, Driscoll JM, Epstein MA, et al. Congenital failure of automatic control of ventilation, gastrointestinal motility and heart rate. Medicine (Baltimore). (1978) 57(6):517–26. doi: 10.1097/00005792-197811000-00003
17. Sandoval RL, Zaconeta CM, Margotto PR, de Cardoso MTO, França EMS, Medina CTN, et al. Congenital central hypoventilation syndrome associated with Hirschsprung's disease: case report and literature review. Rev Paul Pediatr Orgao Soc Pediatr Sao Paulo. (2016) 34(3):374–8. doi: 10.1016/j.rpped.2015.10.009
18. Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. (2017) 19(2):249–55. doi: 10.1038/gim.2016.190
19. Kasi AS, Li H, Harford KL, Lam HV, Mao C, Landry AM, et al. Congenital central hypoventilation syndrome: optimizing care with a multidisciplinary approach. J Multidiscip Healthc. (2022) 15:455–69. doi: 10.2147/JMDH.S284782
20. Weese-Mayer DE, Rand CM, Khaytin I, Slattery SM, Yap KL, Marazita ML, et al. Congenital central hypoventilation syndrome. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al., editors. Genereviews®. Seattle, WA: University of Washington, Seattle (1993). Available at: http://www.ncbi.nlm.nih.gov/books/NBK1427/ (Accessed November 12, 2022).
21. Trochet D, Hong SJ, Lim JK, Brunet JF, Munnich A, Kim KS, et al. Molecular consequences of PHOX2B missense, frameshift and alanine expansion mutations leading to autonomic dysfunction. Hum Mol Genet. (2005) 14(23):3697–708. doi: 10.1093/hmg/ddi401
22. Amiel J, Laudier B, Attié-Bitach T, Trang H, de Pontual L, Gener B, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. (2003) 33(4):459–61. doi: 10.1038/ng1130
23. Bachetti T, Ceccherini I. Causative and common PHOX2B variants define a broad phenotypic spectrum. Clin Genet. (2020) 97(1):103–13. doi: 10.1111/cge.13633
24. Katwa U, D’Gama AM, Qualls AE, Donovan LM, Heffernan J, Shi J, et al. Atypical presentations associated with non-polyalanine repeat PHOX2B mutations. Am J Med Genet A. (2018) 176(7):1627–31. doi: 10.1002/ajmg.a.38720
25. Weese-Mayer DE, Rand CM, Zhou A, Carroll MS, Hunt CE. Congenital central hypoventilation syndrome: a bedside-to-bench success story for advancing early diagnosis and treatment and improved survival and quality of life. Pediatr Res. (2017) 81(1–2):192–201. doi: 10.1038/pr.2016.196
26. Kasi AS, Kun SS, Keens TG, Perez IA. Adult with PHOX2B mutation and late-onset congenital central hypoventilation syndrome. J Clin Sleep Med. (2018) 14(12):2079–81. doi: 10.5664/jcsm.7542
27. Lee-Kelland R, Heraghty J, Flemming P, Williams M, Smith H, Henderson J. Respiratory. Arch Dis Child. (2009) 94(Suppl 1):A38–40. https://adc.bmj.com/content/94/Suppl_1/A38
28. Sivan Y, Zhou A, Jennings LJ, Berry-Kravis EM, Yu M, Zhou L, et al. Congenital central hypoventilation syndrome: severe disease caused by co-occurrence of two PHOX2B variants inherited separately from asymptomatic family members. Am J Med Genet A. (2019) 179(3):503–6. doi: 10.1002/ajmg.a.61047
29. Berry-Kravis EM, Zhou L, Rand CM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med. (2006) 174(10):1139–44. doi: 10.1164/rccm.200602-305OC
30. Amimoto Y, Okada K, Nakano H, Sasaki A, Hayasaka K, Odajima H. A case of congenital central hypoventilation syndrome with a novel mutation of the PHOX2B gene presenting as central sleep apnea. J Clin Sleep Med. (2014) 10(3):327–9. doi: 10.5664/jcsm.3542
31. Byers HM, Chen M, Gelfand AS, Ong B, Jendras M, Glass IA. Expanding the phenotype of congenital central hypoventilation syndrome impacts management decisions. Am J Med Genet A. (2018) 176(6):1398–404. doi: 10.1002/ajmg.a.38726
32. Fine-Goulden MR, Manna S, Durward A. Cor pulmonale due to congenital central hypoventilation syndrome presenting in adolescence. Pediatr Crit Care Med. (2009) 10(4):e41–2. doi: 10.1097/PCC.0b013e318198b219
33. Cain JT, Kim DI, Quast M, Shivega WG, Patrick RJ, Moser C, et al. Nonsense pathogenic variants in exon one of PHOX2B lead to translational reinitiation in congenital central hypoventilation syndrome. Am J Med Genet A. (2017) 173(5):1200–7. doi: 10.1002/ajmg.a.38162
34. Musthaffa YM, Goyal V, Harris MA, Kapur N, Leger J, Harris M. Dysregulated glucose homeostasis in congenital central hypoventilation syndrome. J Pediatr Endocrinol Metab. (2018) 31(12):1325–33. doi: 10.1515/jpem-2018-0086
35. Gronli JO, Santucci BA, Leurgans SE, Berry-Kravis EM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B genotype determines risk for sudden death. Pediatr Pulmonol. (2008) 43(1):77–86. doi: 10.1002/ppul.20744
36. Laifman E, Keens TG, Bar-Cohen Y, Perez IA. Life-threatening cardiac arrhythmias in congenital central hypoventilation syndrome. Eur J Pediatr. (2020) 179(5):821–5. doi: 10.1007/s00431-019-03568-5
37. Amin R. Beyond the retrotrapezoid nucleus in congenital central hypoventilation syndrome. Am J Respir Crit Care Med. (2022) 205(3):271–2. doi: 10.1164/rccm.202111-2602ED
38. Trang H, Samuels M, Ceccherini I, Frerick M, Garcia-Teresa MA, Peters J, et al. Guidelines for diagnosis and management of congenital central hypoventilation syndrome. Orphanet J Rare Dis. (2020) 15(1):252. doi: 10.1186/s13023-020-01460-2
39. Lombardo RC, Porollo A, Cnota JF, Hopkin RJ. Congenital heart disease and aortic arch variants associated with mutation in PHOX2B. Genet Med. (2018) 20(12):1538–43. doi: 10.1038/gim.2018.34
40. Mei M, Yang L, Lu Y, Wang L, Cheng G, Cao Y, et al. Congenital central hypoventilation syndrome in neonates: report of fourteen new cases and a review of the literature. Transl Pediatr. (2021) 10(4):733–45. doi: 10.21037/tp-20-303
41. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
42. http://fyra.io. Pulmonary arteriovenous malformations and coronary artery fistulas. cardiac interventions today. Bryn Mawr Communications. Available at: https://citoday.com/articles/2009-nov/1109_04-php (Accessed October 7, 2022).
43. Virmani R, Carter-Monroe N, Taylor AJ. Chapter 63—congenital anomalies and malformations of the vasculature. In: Creager MA, Beckman JA, Loscalzo J, editors. Vascular medicine: a companion to Braunwald's heart disease (second edition). Philadelphia, PA: W.B. Saunders (2013). p. 771–89. Available at: https://www.sciencedirect.com/science/article/pii/B978143772930600063X (Accessed October 7, 2022).
44. Osten MD, Horlick EM. Pulmonary arteriovenous malformations and coronary artery fistulas.' (2009). p. 29–33. https://citoday.com/articles/2009-nov/1109_04-php (Accessed October 7, 2022).
45. Dowling JJ, Lillis S, Amburgey K, Zhou H, Al-Sarraj S, Buk SJA, et al. King–Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord NMD. (2011) 21(6):420–7. doi: 10.1016/j.nmd.2011.03.006
Keywords: genotype–phenotype correlation, congenital central hypoventilation syndrome (CCHS), PHOX2B, novel mutation, Hirschsprung disease, polyalanine sequence
Citation: Artamonova IN, Zlotina AM, Ismagilova OR, Levko TA, Kolbina NY, Bryzzhin AV, Smorodin AP, Borodin AV, Mamaeva EA, Sukhotskaya AA, Kagantsov IM, Malysheva DA, Vasichkina ES, Pervunina TM and Petrova NA (2023) Case Report: A novel PHOX2B p.Ala248_Ala266dup variant causing congenital central hypoventilation syndrome. Front. Pediatr. 10:1070303. doi: 10.3389/fped.2022.1070303
Received: 14 October 2022; Accepted: 30 December 2022;
Published: 15 February 2023.
Edited by:
Lawrence Todd Reiter, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Kai Lee Yap, Ann & Robert H. Lurie Children's Hospital of Chicago, United StatesIsabella Ceccherini, Giannina Gaslini Institute (IRCCS), Italy
Sarah Barclay, University of Calgary, Canada
© 2023 Artamonova, Zlotina, Ismagilova, Levko, Kolbina, Bryzzhin, Smorodin, Borodin, Mamaeva, Sukhotskaya, Kagantsov, Malysheva, Vasichkina, Pervunina and Petrova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irina N. Artamonova irina.artamonova167@gmail.com Natalia A. Petrova natalja5@yandex.ru
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics