 Leon F. McSwain1Kiran K. Parwani2,3Shubin W. Shahab2Dolores Hambardzumyan4Tobey J. MacDonald1,2
Leon F. McSwain1Kiran K. Parwani2,3Shubin W. Shahab2Dolores Hambardzumyan4Tobey J. MacDonald1,2 Jennifer M. Spangle2,3
Jennifer M. Spangle2,3 Anna Marie Kenney1*
Anna Marie Kenney1*- 1Department of Pediatrics, Emory University, Atlanta, GA, United States
- 2Winship Cancer Institute, Emory University, Atlanta, GA, United States
- 3Department of Radiation Oncology, Emory University, Atlanta, GA, United States
- 4Departments of Neurosurgery and Oncological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Medulloblastoma (MB) is the most common malignant brain tumor in children with standard of care consisting of surgery, radiation, and chemotherapy. Recent molecular profiling led to the identification of four molecularly distinct MB subgroups – Wingless (WNT), Sonic Hedgehog (SHH), Group 3, and Group 4. Despite genomic MB characterization and subsequent tumor stratification, clinical treatment paradigms are still largely driven by histology, degree of surgical resection, and presence or absence of metastasis rather than molecular profile. Patients usually undergo resection of their tumor followed by craniospinal radiation (CSI) and a 6 month to one-year multi-agent chemotherapeutic regimen. While there is clearly a need for development of targeted agents specific to the molecular alterations of each patient, targeting proteins responsible for DNA damage repair could have a broader impact regardless of molecular subgrouping. DNA damage response (DDR) protein inhibitors have recently emerged as targeted agents with potent activity as monotherapy or in combination in different cancers. Here we discuss the molecular underpinnings of genomic instability in MB and potential avenues for exploitation through DNA damage response inhibition.
Current Clinical Approaches to Medulloblastoma
Medulloblastoma (MB) is the most common malignant brain tumor affecting children. Standard treatment of non-infant medulloblastoma requires maximal safe surgical resection followed by radiation and chemotherapy (1). Within the last ten years, microarray and methylome profiling from multiple groups led to the identification of four major molecularly distinct MB subgroups – Wingless (WNT), Sonic Hedgehog (SHH), Group 3, and Group 4 (Figure 1) (3–5). While WNT and SHH MB are driven by alterations in WNT and SHH pathways respectively, oncogenic drivers of Group 3 and Group 4 tumors are less clear (6, 7). The amplification of cMYC, MYCN, and mutations in TP53, are common molecular alterations driving DNA damage induced apoptotic resistance and frequently enriched upon relapse (6, 8). While cMYC amplification is predominant in Group 3 MB and is associated with poor patient prognosis, this occurs in other subgroups as well (9). Furthermore, following SHH stratification into 4 molecularly distinct categories (SHHα SHHβ SHHδ SHHγ) it was found that p53 mutations are enriched in SHHα and patients have a worse prognosis (10). Thus, prognosis varies based on subgrouping and in fact WNT patients have the best outcome compared to those of other subgroups with MYC amplification or TP53 mutation (11).
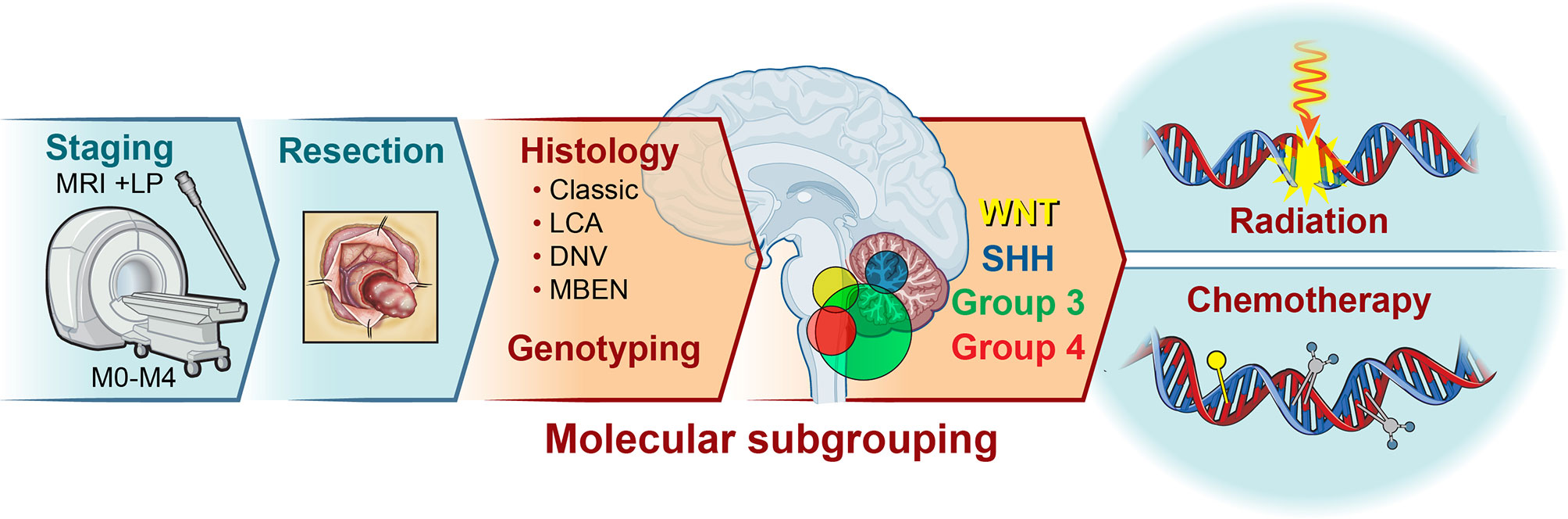
Figure 1 Medulloblastoma is a tumor of the cerebellum. Medulloblastoma constitutes the most common childhood malignant brain tumor, accounting for approximately 20% of all central nervous system tumors. Patient stratification consists of categorization based on age and presence of metastasis, histology, and genotyping. Following initial staging*, maximal surgical resection is typically attempted for all patients. Subsequent histology and molecular profiling introduces subgroup classification and potential alterations to the treatment protocol. While the vast majority of patients receive some combination of radiation and chemotherapy, the age, location of tumor (2), extent of spread, histology, and subgroup inform the intensity and duration of treatment. MRI, Magnetic Resonance Imaging; STR, Subtotal Resection; GTR, Gross Total Resection; LCA, Large Cell Anaplastic; DNV, Desmoplastic/Nodular; MBEN, medulloblastoma with extensive nodularity; LP, Lumbar Puncture *may be delayed to post-surgery.
Despite recent genomic MB characterization and subsequent tumor stratification, clinical treatment paradigms are still largely driven by histology, degree of surgical resection, and presence or absence of metastasis rather than molecular profile (12). Patients usually undergo resection of their tumor followed by craniospinal radiation (CSI) and a 6 month to one-year multi-agent chemotherapeutic regimen. Traditionally, radiation has been recognized as the mainstay of treatment for non-infant MB patients (>3 yo). Prior to the 1950s, MB was considered a universally fatal diagnosis. In 1953, Patterson and Farr published a case series of 27 patients treated with CSI and observed a 3-year survival of 65% (13). This landmark paper led to the widespread acceptance of CSI as the therapy for MB. However, the long-term neurocognitive sequelae of radiation therapy, along with the advent of cytotoxic agents, propelled the addition of chemotherapy in the 1970s (14). Since then improvement in survival has been incremental, comprising better surgical and radiation techniques, better supportive care, and combination/intensification of agents.
Over the last decade, modifications to the above therapeutic regimen have been primarily driven by molecular subgrouping. Given the good outcomes of patients with WNT tumors, there is an effort to reduce the radiation dose for these patients (12). Additionally, administration of carboplatin concomitant with radiation was recently found to be beneficial specifically for Group 3 patients (15). Multiple clinical trials are underway to evaluate the efficacy of targeted therapies, particularly in SHH MB, although none have made it to clinical practice yet. While there is clearly a need for development of targeted agents specific to the molecular alterations of each patient, targeting proteins responsible for DNA damage repair could have a broader impact regardless of molecular subgrouping. DDR protein inhibitors have recently emerged as targeted agents with potent activity as monotherapy or in combination in different cancers. Historically, the synthetic lethality achieved through PARP inhibition in BRCA1/2 mutated breast cancer represents the ideal scenario for DDR inhibitor monotherapy; however, PARP inhibition is applicable outside of BRCA1/2 mutated cancers (16). In the setting of MB, the use of DDR inhibitors in conjunction with standard of care therapies represents an avenue for sensitization to lower doses of standard of care therapies even without synthetically lethal combinations. In this review we discuss the components of the DDR as they relate to MB and the potential for therapeutic benefit using DDR inhibitors, either alone or in rational combinations.
DNA Damage Signaling
Since the standardization of medulloblastoma treatment from the mid-1980s to the mid-2000s, surgical resection, radiation and chemotherapy continue to serve as the primary treatment modalities for MB patients (17, 18). While radiation delivery optimization and dose reduction preserves vital tissue proximal to the primary tumor site, posterior fossa irradiation and on-target off-tumor effects of chemotherapy remain important considerations for therapeutic development (19, 20). Targeting proteins involved in resolving DNA damage from either ionizing radiation (IR) induced single and double strand breaks (SSBs and DSBs) or adducts formed by chemotherapies could permit lower therapeutic dosing and more effective targeting of tumor cells (21).
Targeting Proteins Involved in the DNA Damage Response to Radiation and Chemotherapy
The DNA damage responses to IR-induced SSBs and DSBs, and adducts formed by chemotherapies, are extensively reviewed elsewhere (22, 23). Here we will discuss the central tenets of the DDR, including detection, downstream signaling, and repair, as they pertain to MB therapy resistance and potentially targetable DDR proteins. Firstly, exposure to ionizing radiation results in the accumulation of SSBs and DSBs, which the cell will repair through one of the two methods: homologous recombination (HR) or the more error prone non-homologous end joining (NHEJ) (Figure 2) (25). Comparatively, DNA adduct forming agents, such as those commonly used to treat MB including cisplatin, lomustine, cyclophosphamide, and temozolomide, require alternate repair proteins to perform base excision repair (BER) or nucleotide excision repair (NER) (Figure 2). Proteins important for detection, signaling, and repair, including ATM/ATR, CHK1/2, and PARP function in both types of damage repair while APE1 and CK2 facilitate DNA adduct repair, specifically. Other proteins discussed include AKT, a mediator of radiation resistance in the MB stem cell niche, p53 the “guardian of the genome,” which when mutated, results in substantially worse standard of care response primarily due to defective cell death signaling downstream of damage recognition, and proteins that regulate p53.
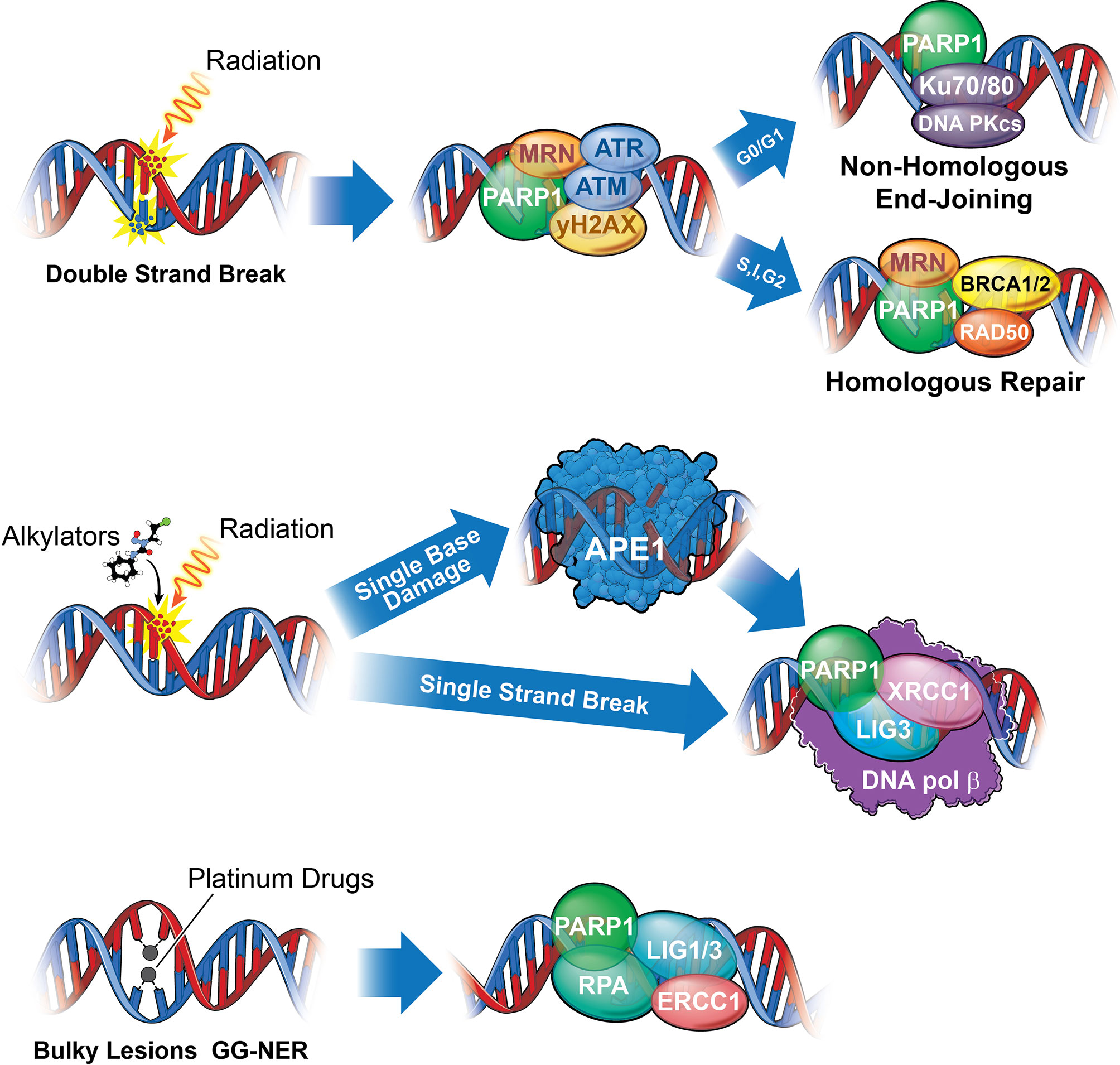
Figure 2 A summary of the DNA damage response in medulloblastoma. Numerous DNA damage response signaling axes mediate DNA repair following medulloblastoma standard of care. Chemotherapies utilized, including temozolomide, cisplatin, lomustine, and cyclophosphamide (with cisplatin and cyclophosphamide constituting standard of care), bind directly to DNA to create bulky lesions repaired through a combination of either Base Excision Repair (BER), Nucleotide Excision Repair (NER), MGMT (Not shown), and HR or NHEJ, while IR mediated DSBs are repaired through NHEJ and HR. (Top) The deleterious effects of Ionizing radiation results primarily from DNA double strand breaks (DSBs) repaired either through Homologous Repair (HR) or Non-Homologous End Joining (NHEJ). While PARP, M/R/N (a complex of 3 proteins: MRE-11, RAD50, NBS), and ATM function universally to recognize strand breaks, cells in G0/G1 lack sister chromatids and will repair through a more error prone NHEJ, while those in S, Interphase, or G2 will repair through HR, utilizing the sister chromatid to replace the missing bases. Proteins directly involved in repair of double strand breaks include Ku70/80 and DNA-PKcs for NHEJ and M/R/N, BRCA1/2, and RAD50 for HR. Upstream effectors, ATM and ATR, will signal through Chk1 and 2 to arrest the cell and either repair or commit apoptosis (Figure 4). (Middle) Single strand breaks resulting from radiation or excision of damaged base pairs by APE1 (such as those resulting from alkylation) are identified by PARP1 and repaired by XRCC1, DNA Ligase III (LIG3), and DNA Polymerase β. (Bottom) Bulky lesions resulting from platinum-based drugs such as cisplatin or carboplatin can be repaired either through transcription coupled NER (TC-NER) or global genomic NER (GG-NER). While it remains unclear what role PARP may play in TC-NER, it serves as a recognition and recruitment protein for GG-NER, functioning alongside DNA Ligase I or III (LIG1/3), RPA, and ERCC1 proteins. Bifunctional alkylating agents and chloro-ethylating agents, such as cyclophosphamide or lomustine, respectively, can methylate guanine. Methylated guanine can be repaired through MGMT (not shown), through direct removal of guanine methyl group, potentially resulting intrastrand cross-linking (ICL) requiring a combination of NER and HR or NHEJ to repair) or interstrand cross-linking, repaired through NER. Platinum drugs mediate interstrand cross-linking, repaired predominantly through NER. Finally, temozolomide can be repaired either through BER (requiring APE1) or MGMT (24).
ATR-Chk1
Dozens of proteins function together to recognize and initiate repair of damaged DNA; however, ATR and ATM can initiate cell cycle arrest, damage repair, and apoptosis by signaling through Checkpoint Kinase 1 and 2 (Figures 1, 4) (26). More specifically, the ATR-Chk1 signaling axis has emerged as a key player in postnatal cerebellar development and MB therapeutic resistance, and Chk1 is upregulated across MB subgroups (27). Saran and colleagues investigated SHH signaling in a patched 1 receptor heterozygous IR inducible SHH MB model (hereafter referred to as the PTCH+/- model), where radiation induces a “second hit” to the smoothened receptor inhibitor, PTCH, resulting in constitutive SHH signaling and tumorigenic transformation. The group uncovered a link between PTCH heterozygosity and tumor formation following IR exposure in postnatal day 4 (P4) but not P10 mice as a result of differences in p53 activation (28, 29). Subsequently, SHH pathway dysregulation was linked to ATR-Chk1 pathway inactivation in the PTCH+/- model (30). Through overactivation of the SHH signaling cascade via PTCH inactivation and overexpression of Gli1 protein, the phosphorylation and activation of Chk1 is attenuated, resulting in abrogation of the S-phase cell cycle checkpoint and chromosomal aberrations. In an ATR-deleted mouse model (ATRhGFAP-Cre) of cerebellar granular neural precursor cells (CGNPs), the putative cell of origin for SHH MB, cells develop extensive chromosomal abnormalities leading to cerebellar hypoplasia (31). From the same study, ATR deletion results in abrogation of cell cycle checkpoint activation, evidenced by PCNA and pHH3 positive staining, and accumulation of γH2AX, a marker of DNA damage, ultimately resulting in p53 accumulation, caspas-3 cleavage, and apoptosis. Additionally, ATR deletion from a smoothened overexpressing SHH model through cre-recombination (SmoM2;AtrG-cre), inhibits tumor formation; all of which suggest a requirement for ATR in maintaining genomic stability during cerebellar development and tumor formation, nominating the ATR-Chk1 axis as a potential therapeutic target.
What we ultimately seek to therapeutically exploit by targeting the ATR-Chk1 pathway are cells with unstable genomes; in fact, many cells amenable to ATR-Chk1 signaling inhibition are MYCN or cMYC amplified or overexpressing. SHH and WNT subgroups overexpress MYCN while Group3 and WNT subgroups overexpress cMYC (9). MYCN and cMYC increase the number of firing replication origins, causing collision between replication and transcriptional complexes, resulting in fork stalling and collapse and the accumulation of DDR marks at the sites of active DNA replication (32–34). However, the simultaneous regulation of cell cycle check-points and DDR allows cells to maintain a complex balancing act between genomic instability and tumor maintenance, which depends upon the ATR-Chk1 signaling axis (34). An example of this can be found in MYCN expressing SHH MB. Sonic Hedgehog signaling drives MYCN expression in both CGNPs and MBs of the PTCH+/- model, and MYCN overexpression can drive cell proliferation independent of SHH signaling (35). Coincidently, the genomic instability resulting from PTCH1 deletion can lead to the amplification of regions of chromosome 12, including the MYCN gene (36). Through transcriptional upregulation of the MRE11/RAD50/NBS1 (M/R/N) complex downstream of ATR-Chk1 in CGNPs, MYCN drives not only a genomically unstable, highly replicative state, but also an increase in proteins required for recognition and repair to counter the instability (32).
The lack of enzymatic activity and hydrophobic pockets paired with an intrinsically-disordered N-terminal make MYCN a challenging drug target (37). Given these challenges, alongside the potentially broader impact of DNA repair protein inhibition, much of the research has focused on sensitization of cells to genomic stress through inhibition of either Chk1 or Wee1, a target of Chk1 (38). The correlative relationship between protein expression and cell sensitivity to protein inhibition is not applicable in the case of Chk1. While all MB subgroups demonstrate elevated Chk1 expression and worse prognosis with high Chk1 expression, as mentioned previously, the relative expression of cMYC is a greater predictor of responsiveness to Chk1 inhibition (27). In earlier studies utilizing AZD-7762, a Chk1 inhibitor, numerous cell lines demonstrated response as measured through reductions in cell viability, accumulation of γH2AX, and increase in apoptotic markers. Daoy, D283, D425, UW-228, and HD-MB03 all show varying degrees of Chk1 inhibitor response; however, high cMYC-expressing cells such as D283 and HD-MB03, both Group 3 cell lines, are more sensitive to damage as seen through a higher ratio of reduced viability to γH2AX foci formation (27, 39). Lower levels of damage accumulation are required to push the cell fate towards apoptosis. In vivo and synergism studies subsequently emerged using AZD-7762, MK-8776, Rabusertib, or Prexasertib (all Chk1 inhibitors) in combination with cisplatin or gemcitabine. Inhibition of Chk1 in Group 3 cells, including D425, D283, and SU-MB002, but not SHH PDX models, results in cell cycle checkpoint abrogation and a greater accumulation of DNA damage and subsequent cell death following simultaneous exposure to cisplatin or gemcitabine (40).
Genomic surveillance is crucial for cerebellar development, tumorigenesis, and tumor maintenance. While ablation of the ATR-Chk1 axis results in cerebellar hypoplasia in non-tumor models, downregulation during SHH tumor development leads to extensive chromosomal abnormalities and abrogation of tumor development. This complex balancing act is a requirement for maintaining a highly proliferative, tumorigenic state. The cMYC amplification and overexpression of Group 3 models creates a reliance on the ATR-Chk1 axis for viability where inhibition could result in cell death, a phenotype potentially exploitable in WNT tumors given their MYC status. And even though SHH PDX models do not appear responsive to CHK1 inhibition in recent studies, inhibition of MYCN → M/R/N signaling is unexplored. MYC amplification alongside Chk1 upregulation may serve as reliable biomarkers for Chk1 pathway inhibition, particularly in Group 3 patients who have the worst prognosis. Currently, Prexasertib in combination with Cyclophosphamide or gemcitabine is in a phase 1 clinical trial for refractory Group 3 and Group 4 patients.1ct
PARP
Poly-ADP-ribose Polymerase, or PARP, plays numerous roles in the DNA damage response, but is most notable for damage recognition and repair protein recruitment through ribosylation of damaged DNA and auto-ribosylation. While PARP1 is not typically required for cell survival, because it facilitates repair of IR induced strand breaks through both NHEJ and HR, and repair of bulky adducts through NER (Figure 2), inhibition could sensitize MB cells to existing therapies and even synergize with the aforementioned Chk1 inhibitors (41–43). In fact, while BRCA1 and BRCA2 mutation status are positive predictors of PARP1 inhibitor response due to synthetic lethality in the setting of other tumor types, this is not a requirement and other gene signatures can serve as predictors of therapeutic response where the use of single molecular markers prove to be insufficient (44). Additionally, the BRCA1/2 mutation status across MB patients remains unclear, though RNAseq data have shown upregulation of gene signatures associated with BRCA1/2 mutation in Group3 and 4 and a subset of SHH patients (45).
A role for PARP1 in MB tumor formation emerged through studies of p53 null mice. While TP53 ablation alone is not sufficient for brain tumor formation, in TP53-/-; PARP1-/- mice, neuronal cells are predisposed to malignant transformation (46, 47). Saran and colleagues, using a p53 wild type PTCH+/-;PARP1-/- model of SHH MB, demonstrated that PARP1 ablation leads to increased frequency of preneoplastic lesions, accumulation of γH2AX foci, and CGNP genomic instability (48). Chromosomal rearrangements resulting from PARP1 abrogation lead to a second hit in the PTCH1 allele, increasing the incidence of tumor formation. The absence of PARP also increases phosphorylation of Ser18-p53, suggesting that the majority of genetically unstable cells undergo apoptosis while few cells escape to accelerate tumor formation. Pre-treatment of in vitro MB models, D283, D556, and UW228-2, with olaparib prior to irradiation results in greater accumulation of γH2AX foci that are sustained longer than control (49). In fact, olaparib is blood brain barrier (BBB) penetrant and could serve as an effective brain tumor therapy (50) Considering the genomic instability of CGNPs following PARP1 and ATR ablation, there is also a potential for synergism between PARP and ATR inhibition. While not yet studied in the setting of MB, in glioma-bearing mice the combination of VE822, an ATR inhibitor, and olaparib leads to a 60% increase in survival compared to control-treated (51).
In addition to PARP’s role in radiation induced strand break repair, it also mediates base excision repair. As mentioned above, PARP1 is critical for resolution of adducts by functioning as a component of the BER complex, consisting of DNA ligase III (LIG3), DNA Polymerase β (POLB), and XRCC1 (41, 52). Adducts formed by lomustine, cisplatin, and cyclophosphamide, which serve as cytotoxic chemotherapies to treat MB, require functional BER or NER for resolution (53, 54). Unfortunately, the combination of PARP inhibitors with cyclophosphamide does not improve response rate over cyclophosphamide alone, at least in the setting of breast cancer, and combinations with lomustine are understudied (55). However, the administration of PARP inhibitors alongside platinum-based drugs like cisplatin has the potential to enhance targeting of tumor cells while reducing secondary cytotoxicities (56). In fact, compared to veliparib-mediated catalytic inhibition of PARP1, which abrogates PARylation, olaparib demonstrates superior DNA-PARP trapping, resulting in not only a lack of repair, but also replication fork stalling and double strand breaks. Olaparib synergizes with cisplatin and temozolomide, an emerging MB therapeutic for recurrent patients (57–60). In Group 3 and 4 xenograft models generated using D384, D425, and D283 cell lines, D384 and D425 show a robust response to combination therapy with temozolomide and another PARP inhibitor rucaparib, whose DNA trapping kinetics are comparable to those of olaparib (61, 62).
Historically, PARP inhibition induced apoptosis through synthetic lethality when combined with BRCA1/2 mutations in breast cancer patients resulting from unrepaired strand breaks. However, BRCA1/2 mutation is not a requirement for the use of PARP inhibitors. PARP1 is crucial to DDR activation through its PAR catalytic activity, which is exemplified in developmental MB models where ablation leads to genomic instability and cell death following radiation. While there is a reliance of MYC amplified cells on the ATR-Chk1 signaling axis, PARP inhibition appears to be relevant across all subgroups not only in the context of radiation but also when combined with platinum-based drugs and temozolomide. Inhibiting PARP could have a broader impact compared to ATR-CHK1 in cells without MYC amplification or overexpression in combination with standard of care therapies and could afford dose decreasing to ameliorate toxicities from chemotherapy, namely cisplatin. Currently, olaparib is in a phase 2 clinical trial for patients with advanced or refractory solid tumors, including MB.2ct
APE1
Apurinic/apyrimidinic endonuclease 1 (APE1) not only regulates DNA binding of transcription factors through cysteine residue redox regulation, it is also responsible for DNA incision proximal to adducts formed by platinum-based drugs and alkylating agents such as cisplatin and temozolomide (Figure 2) (63, 64). The production of oxygen free radicals from ionizing radiation also produces abasic sites in the DNA reparable through APE1, the abrogation of which leads to unrepaired DNA and cell death (65–67). Additionally, due to the potential for PARP inhibitor resistance, targeting APE1 could sensitize PARP inhibitor resistant cells to chemotherapy and radiation (68).
Similar to a requirement for PARP and ATR in maintaining genome integrity during cerebellar development, due to the high CNS oxidative stress in postnatal mice, APE1 can protect cells against postnatal oxidative DNA damage (69). 100% of Mice lacking APE1 die within 30 days of birth concomitant with accumulation of extensive γH2AX accumulation. And while p53 ablation in these mice rescues their viability, the absence of p53 alone does not result in tumor burden by post-natal day 20 whereas 100% of (Ape1L/L;p53L/L)Nes-cre mice develop tumors by day 15. From the same study, APE1 deficient astrocytes maintain radiation induced DNA damage compared to controls and are sensitized to cisplatin. Similarly, after uncovering a role for APE1 in mediating resistance of glioma to adjuvant radiation and alkylating agent based chemotherapy, Silber and colleagues extended these findings to MB and primitive neuroectodermal tumors (70–72). In patients deemed high risk as indicated by tumor invasion into the surrounding brain, APE1 endonuclease activity is elevated (73). In UW228-2 cells treated with siRNA against APE1 in combination with temozolomide, survival is decreased compared to control. These data point to a requirement for APE1 to mediate cisplatin adduct resolution in MB. Given the barriers to small molecule inhibitor development for some targets and potential lack of BBB penetrance, the utilization of siRNA conjugated nanoparticles has emerged as a viable alternative, potentially alleviating inhibitor resistance and issues with BBB penetrance (74). UW228 cells exposed to nanoparticle conjugated siRNA against APE1 in vitro sustain radiation induced DNA damage (69, 75). Only recently did a selective APE1 inhibitor emerge in high throughput drug screening of non-small cell lung cancer (NSCLC) (76). The application of NO.0449-0145 induces DNA damage in vitro and in an in vivo xenograft model and overcomes cisplatin and erlotinib resistance; however, the BBB penetrance and applicability in MB would require further investigations.
CK2 and MGMT
Casein Kinase II (CK2), a pleiotropic protein with diverse functions from Epithelial to Mesenchymal Transition (EMT) regulation to DNA damage repair, has become a potential therapeutic target in MB (77). While CK2 regulates redox activity of APE1 through post-translational modification, its role in DNA repair comes from signaling through O6-MethylGuanine-DNA Methyltransferase (MGMT), a DDR protein capable of mediating temozolomide resistance (78–80). Unlike protein complexes that mediate BER and NER, MGMT can function alone to repair O6-AG and O4-alkylthymine adducts formed by alkylating agents (81). Even though MGMT promoter methylation in CpG rich sequences and subsequent overexpression is well known for mediating temozolomide resistance in glioblastoma, the clinical relevance of targeting MGMT in MB has only recently emerged (82).
In 2018 Li and colleagues uncovered a CK2 → β-Catenin → MGMT signaling axis in SHH MB (80). Knockout of either isoform, CSNK2A1 or CSNK2B, results in decreased tumorigenic potential. In a high throughput screen using CX-4945, an orally bioavailable and BBB penetrant selective inhibitor of CK2, temozolomide was found to synergize with CK2 inhibition (83, 84). Indeed, CK2 KO leads to a decrease in MGMT and β-Catenin and an increase in apoptosis following temozolomide exposure, which was rescued by re-expression of β-Catenin. These data point to CK2 as a modulator of MGMT activity and temozolomide resistance in MB. There are other DDR proteins involved in recognition, repair, and signaling found to be CK2 substrates, including p53, BRCA1, XPB, XRCC1, XRCC4, Histone H1, and Rad51 (85). CK2 is also implicated in driving proliferative and migratory phenotypes and inhibition of apoptosis in multiple cancers, including MB (85). Initial studies showed binding of CK2 to the smoothened receptor and promotion of SHH signaling (86). Proteomic analysis of CGNPs showed increased phosphorylation of CK2 motifs in postnatal day 7 mice, implicating CK2 as a driver of cerebellar developmental programming (87). Subsequent CK2 knockdown or treatment with the CK2 inhibitor 4,5,6,7-Tetrabromobenzotriazole (TBB) destabilizes Gli2 and decreases Gli1 expression, downstream mediators of SHH signaling. More importantly, D175N mutation in CK2 confers resistance to TBB but not CX-4945 likely due to ATP-binding cavity enlargement, further demonstrating CX-4945 robustness. These findings point to a broader impact of CK2 targeting and the potential for radiation and chemotherapy sensitization and inhibition of oncogenic phenotypes. CX4945 is currently in a multi-phase clinical trial for recurrent, SHH subgroup MB.3ct
AKT and PI3K
AKT, or protein kinase B (PKB), is a member of the AGC serine/threonine kinase family that plays major roles in cellular growth, survival, and DNA repair (88, 89). Receptor tyrosine kinases (RTKs) such as PDGFR, VEGFR, and IGF-1R are some of the primary drivers of AKT phosphorylation in tumors, including MB, typically due to the amplification of the receptors or ligands, or the presence of an autocrine feedback loops (90–94). Overexpression or following ligand binding to these receptors leads to dimerization, autophosphorylation, and activation of phosphoinositide 3-kinase (PI3K) which is recruited to the receptor and activated (95). PI3K then catalyzes the conversion of PIP2 to PIP3, recruiting AKT via its pleckstrin homology domain to be phosphorylated by PDK1, on Thr308, and mTORC2, on Ser473 (Figure 3) (96, 97). Dual phosphorylation at these marks is a requirement for full activation of AKT, leading to downstream activation of mTORC1 and modulation of cell growth and proliferation (98).
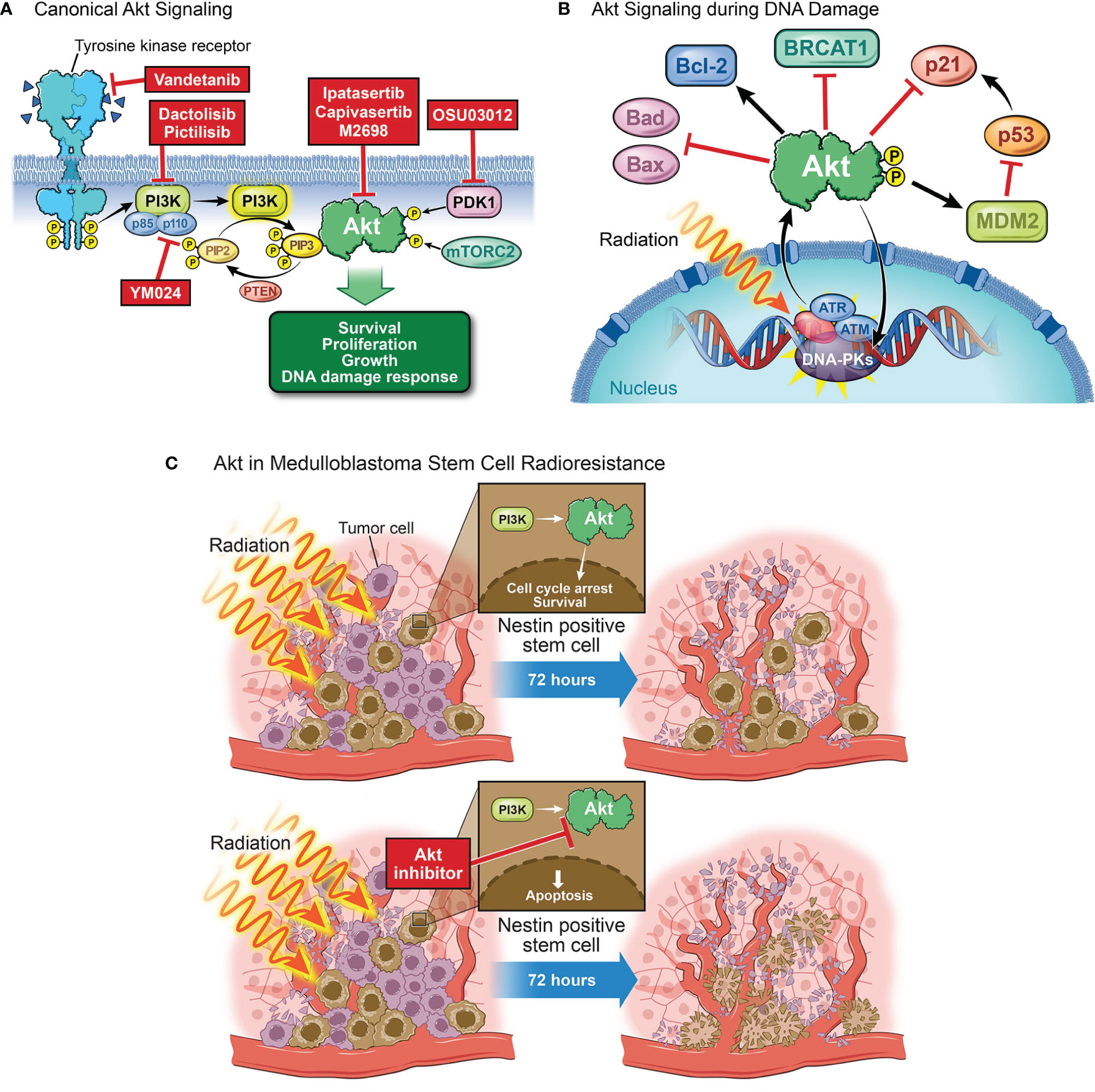
Figure 3 The role of AKT in DNA damage. (A) Canonical AKT signaling begins with the activation of a receptor tyrosine kinase (e.g., VEGFR, EGFR, or IGFR). Receptor activation, by binding of a ligand, dimerization, and autophosphorylation, causes binding and activation of p85 and p110, the respective regulatory and catalytic subunits of PI3K. Activated PI3K will go on to catalyze the conversion of PIP2 to PIP3, a process which can be reversed by the tumor suppressor PTEN. If PIP3 is formed, it will recruit AKT to the membrane at which point AKT will be phosphorylated by PDK1 and mTORC2. This dual phosphorylation fully activates AKT, allowing it to affect several downstream processes such as cell survival, proliferation, growth, and the DNA damage response. (B) Following exposure to radiation, repair proteins (e.g. ATM, ATR, DNA-PKs) are recruited to the damaged site to facilitate repair. The activation and recruitment of these proteins correlates with an increase in phosphorylated AKT. p-AKT then inhibits apoptosis through Bad and Bax inhibition and Bcl-2 activation. p-AKT will also inhibit the downstream target of p53, p21, to inhibit cell cycle arrest. A feedback loop exists in that AKT can also phosphorylate and activate these proteins to aid in DNA repair. AKT facilitates NHEJ by phosphorylating DNA-PKs and inhibits HR by promoting cytoplasmic localization of BRCA1 and Rad51. (C) Irradiation decreases the population of tumor cells at the perivascular niche with the exception of stem cells which are largely radioresistant. Nestin-positive stem cells in the perivascular niche treated with radiation activate the PI3K/AKT pathway to undergo cell cycle arrest and re-enter the cycle 72 hours later, driving MB survival and recurrence. AKT inhibition prior to irradiation abrogates perivascular stem cell radiation resistance, leading to apoptosis of these cells.
In the context of DNA damage, ATM, ATR, and DNA-PKs can all phosphorylate and activate AKT even in the absence of upstream RTK activation; and while the mechanism through which ATM and ATR activate AKT is unclear, DNA-PKs can directly phosphorylate AKT at serine 473 (99). Additionally, PARP indirectly releases AKT from SIRT1 inhibition and accumulates adenosine monophosphate (AMP) driving AMP-activated protein kinase (AMPK) mediated activation of AKT. AKT can also modulate DDR through feedback mechanisms involving DNA damage signaling sensors and effectors (99). Activated AKT increases cMYC transcription and inhibits the cyclin-dependent kinase inhibitor p21Cip1, a target of p53, both of which promote cell cycle progression (100, 101). Additionally, AKT suppresses ATR/Chk1 signaling and subsequent HR, yet AKT can be activated in an ATM/ATR-dependent manner and repair DSBs through DNA-PKcs mediated NHEJ. As many DNA damaging agents target dividing cells (102), it is important to highlight that potential resistance to these agents can occur often due to AKT driven NHEJ (103). p-AKT accumulates in irradiated lung carcinoma and prostate cancer cells and activates DNA-PKs to induce NHEJ (104). Conversely, AKT inhibits HR by mediating BRCA1 and RAD51 cytoplasmic retention (105). Active AKT phosphorylates Bad, Bax, and Bcl-2, inhibiting apoptosis and promoting cell survival (106). Together, these signaling roles drive a pro-repair, pro-survival phenotype that could be exploited therapeutically.
Many of these AKT signaling phenotypes are conserved in the setting of medulloblastoma, including both pro-survival and growth and the DNA damage response. In the CGNP developmental model of SHH MB, SHH signaling drives expression of insulin growth factor (IGF), resulting in an autocrine feedback loop between IGF and IGF1R to promote CGNP proliferation, supporting a role for AKT as a developmental protein (107). Additionally, irradiated P1 and P10 mice harbor increased p-AKT in non-irradiated and irradiated cerebella at P1 compared to P10 (108). There is also interplay between p53 levels and p-AKT activity in that P10 cerebella are more resistant to MB formation and sensitive to radiation-induced cell death due to an increase in p53 expression and lower p-AKT activity. Furthermore, AKT constitutes a source of resistance to smoothened inhibitors and is strongly associated with poor outcomes (109). Indeed, smoothened receptor inhibition alongside PI3K through Vismodegib and Dactolisib, respectively, synergizes to decrease cell viability in both SHH and Group 3 cell models (110). From the same study, combination of cisplatin and Dactolisib, but not Vismodegib, results in significant decrease in viability compared to single agent supporting a role for the PI3K/AKT signaling axis in post-DNA damage survival. Inhibition of the upstream AKT kinase, PKD1, with OSU03012 results in a decrease in p-AKT S473 and a sensitization to both doxorubicin and cyclophosphamide but not temozolomide in Group 3 models (111). Subsequently, it was found that many MB cells and patient samples overexpress the oncogenic PI3K catalytic subunit p110α isoform responsible for phosphatidylinositol phosphorylation. YM024-mediated inhibition of the p110α subunit abrogates doxorubicin-induced AKT phosphorylation at S473, sensitizing cells to doxorubicin (112). Inhibition of AKT phosphorylation by targeting upstream RTKs can also sensitize MB to DNA damage. Treatment of SHH-TP53-mutated MB and MYC-amplified MB cells with Vandetanib, a multi-kinase inhibitor targeting RET, VEGFR2, and EGFR, reduces cell migration and cell viability (113). When combined with Pictilisib, a PI3K-inhibitor, AKT phosphorylation and protein levels are decreased compared to monotherapy. Following combination, cells are sensitized to etoposide, a chemotherapy which binds and forms a ternary complex between topoisomerase II and DNA. Thus, targeting AKT directly or indirectly would not only decrease tumor growth but will also sensitize cells to DNA damage.
There is also a role for AKT in mediating post-radiation survival of cancer stem cells residing in the perivascular niche (PVN), the putative cells of recurrence for many brain tumors (114). Following radiation, AKT is phosphorylated and activated in nestin-positive mouse MB stem cells (115). The PI3K/AKT/mTOR axis also regulates hypoxia-inducible factor 1-alpha (HIF-1α) mediated expression of VEGF, leading to the vascularization of the tumor, supporting a role for AKT in the development and maintenance of the PVN stem cell niche (116). These findings are further supported by Hambardzumyan et al. who demonstrated that radiation-treated PVN resident nestin-positive stem cells activate the PI3K/AKT pathway, undergo cell cycle arrest, and re-enter cell cycle 72 hours later (117). Furthermore, inhibiting AKT prior to irradiation sensitizes cells in this niche to treatment, demonstrating that these cells’ inherent radiation resistance can be partially overcome through AKT inhibition.
AKT signaling is vital in cell cycling and proliferation which drive tumorigenesis. These processes render cells resistant to DNA damaging agents by the additional upregulation of DNA damage repair proteins which can result in a feedback loop that activates AKT. Previous studies have clearly demonstrated that inhibiting AKT sensitizes MB to DNA damage in vitro, making AKT a compelling target in vivo that has the potential to be combined with irradiation and chemotherapy. The challenge in AKT targeting lies in ensuring that inhibitors are potent enough to overcome the activation of upstream proteins, such as the receptor tyrosine kinases that activate AKT. Currently, there are several AKT inhibitors in clinical trials as both monotherapy and combination therapy for other solid tumors. Ipatasertib and Capivasertib treatments result in an increase in progression free survival in patients harboring solid tumors such as breast and ovarian cancers (118). However, despite these treatment options, minor toxicities such as hyperglycemia and skin rash still occurred with nausea and fatigue presented when AKT inhibition was combined with chemotherapy (119). M2698 is a BBB penetrant, ATP-competitive inhibitor of AKT1/3 and p70S6K, a downstream target of AKT, that exhibits significant growth arrest in many solid tumor cell lines. M2698 treated mice orthotopically-implanted with GBM cells exhibit increased survival and reduced brain tumor burden (120). Samotolisib, a PI3K isoform inhibitor, is currently in phase 2 clinical trials for patients with relapsed or refractory advanced solid tumors, including medulloblastoma.4ct
p53
p53 expression and mutation status emerged in the early 1990s as a biomarker for poor therapeutic response and shorter overall survival in MB due to the role of p53 in DNA repair and cell fate following genomic insult (Table 1) (123–125). In the absence of external cell stressors, p53 is degraded to prevent cell cycle inhibition or inappropriate apoptotic induction; thus, any dysregulation of the p53 pathway resulting from mutations in TP53 or alterations to proteins responsible for p53 degradation, namely WIP1 and MDM2, is made apparent by atypical alterations to p53 levels (126–128). While TP53 mutations are infrequent in MB, occurring in approximately 10%-15% of patients, p53 status and the potential for p53 therapeutics even outside of TP53 mutant patients remain important considerations (8, 129–131). When activated downstream of ATM-Chk2 and ATR-Chk1 following DNA damage, p53 signals through BAX, NOVA, and PUMA to initiate cell death and p21 to inhibit cell cycling, bringing about a potential therapeutic window and an explanation as to why TP53 mutant patients have a substantially worse prognosis (Figure 4) (132).
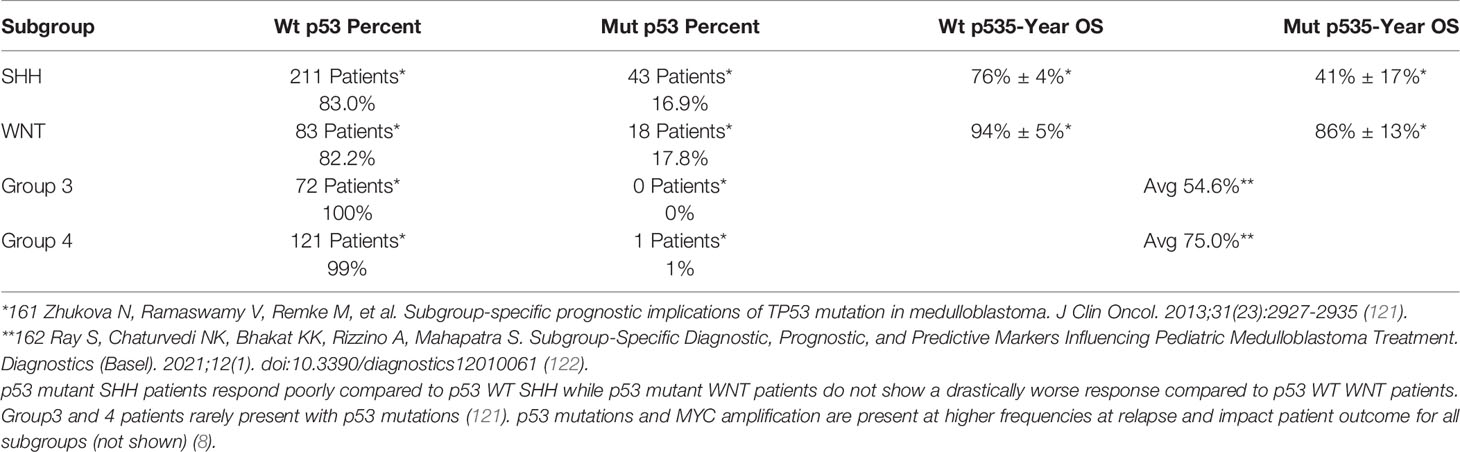
Table 1 p53 mutation status and effect on medulloblastoma patient survival.
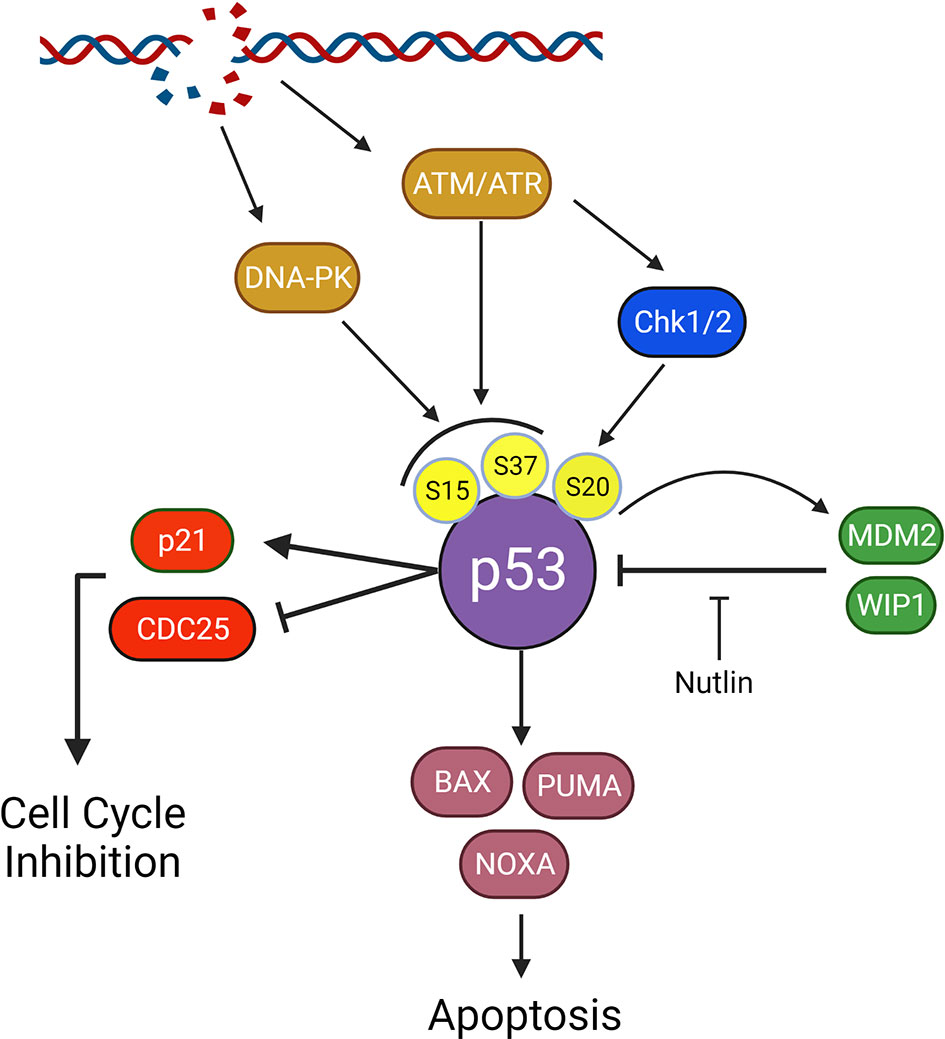
Figure 4 Impact of p53 on survival and p53 signaling in the DNA damage response. P53 is a downstream effector following double strand DNA breaks and plays a crucial role in tumorigenesis. Following Damage detection by ATM/ATR or activation of DNA-PKcs, p53 is phosphorylated at Ser15 or Ser37. ATM and ATR will also activate Chk1 and 2 resulting in phosphorylation of p53 at Ser20. Together these phospho-sites inhibit p53 degradation by preventing MDM2 binding and facilitate p53 tetramerization, a crucial step in p53 activation. p53 will activate p21 and inhibit CDC25 A,B, and C to activate the cell cycle checkpoint and inhibit cycling. p53 will also transcriptionally upregulate pro-death proteins, BAX, PUMA, and NOXA. Upon overcoming inhibition by BCL-2 family apoptotic inhibitors (not shown), BAX, PUMA, and NOXA will activate apoptosis. Inhibition of the MDM2-p53 interaction with nutlin allows for p53 accumulation and apoptosis, ideally in cells heavily reliant on p53 suppression for survival.
TP53 Status as a Predictor of Chk1 and PARP Inhibition
Given the emergence of DDR specific therapeutics and the role of p53 in mediating cell death and cell cycle checkpoint activation, TP53 mutation status should be considered as a potential predictor for patient response to inhibition of the previously discussed DDR proteins. While TP53 wild type CGNPs and mutant Daoy cells demonstrate Chk1 signaling axis requirements, Daoy cells may no longer represent MB patients and targeting Chk1 in novel SHH PDX models shows no survival advantage, regardless of p53 status (27, 32, 39, 40, 133). However, Group 3 PDX models, both TP53 wildtype, D283, TKMB870, and SU-MB002, and TP53 mutant cells, D425, in addition to a MYC amplified p53 dominant negative syngeneic mouse model, respond to Chk1 inhibition (40). These data suggest that p53 status is not a predictor of Chk1 response in the setting of Group 3 MB, which is supported by findings in other cancers (134). However, a requirement for p53 functionality in PARP inhibitor mediated cell death in MB is less clear. p53 mediated apoptosis in PTCH+/- model following PARP ablation suggests that p53 is required for apoptotic induction in the absence of PARP (48). Additionally, the Group 3 models tested for combination of olaparib and radiation, D283 and D556, are TP53 wild type; and while UW228 is TP53 mutant and responsive to olaparib and radiation, it may no longer be representative of SHH MB (49, 133). However, when testing combinations of PARP inhibitor and temozolomide in D425 and D384, both Group 3 models, p53 status does not predict PARP inhibitor response, and in fact D425, a TP53 mutant model, tumor growth inhibition is greater compared to D384 and D283 from the same study (61). Thus, p53 may be required for therapeutic response following PARPi combined with radiation whereas for PARPi plus temozolomide, p53 may not be required.
p53 and Medulloblastoma Development
p53 plays a significant role in mediating cellular response to radiation and chemotherapy, including induction of apoptosis through BCL-2 and BCL-XL inhibition and transcriptional activation of BAX, NOXA, and PUMA and regulation of G1 and G2 cell cycle checkpoints through numerous targets including p21 and 14-3-3-σ (135, 136). The dysregulation of p53 signaling pathway components introduces mechanisms of therapeutic resistance to standard of care therapy, and, potentially, any of the aforementioned DDR therapeutics. The importance of p53 for apoptosis of pre-neoplastic cerebellum is made apparent through developmental SHH MB modeling where inactivation of p53 alongside other tumor suppressors predisposes to MB development. The absence of p53 and the G1 restriction point protein Rb in CGNPs of the external granular layer leads to induction of MB via unabated cell cycling (47). The genomic instability resulting from ablation of these tumor suppressors drives MB development through the amplification of MYCN and PTCH2, an isoform of the frequently mutated PTCH1 of SHH MB possessing smoothened inhibitory features (137, 138). Furthermore, in the absence of PTCH1, Rb inhibition mediated through ablation of CDKN2C (Ink4c or p18), an inhibitor of proteins responsible for Rb inhibition (CDK4, CDK6, and CyclinD1), leads to the formation of MB even in the presence of wild type TP53 (139). These data provide a mechanism by which loss of p53 or restriction point inactivation in combination with SHH mitogenic signaling would lead to unabated cell cycling and tumor development. Overall, in the absence of restriction point and cell cycle checkpoint safeguards, the effects of DNA damage are exacerbated as the cell cannot appropriately respond to damage.
More direct evidence for the importance of intact DDR for MB suppression follows from p53 and DDR protein ablation studies. Mice deficient for KU80 or DNA Ligase IV, proteins required for NHEJ mediated DSB repair, in a TP53 null background, accelerates MB formation leading to shorter overall survival (140, 141). Tumorigenesis is also achieved through combined loss of p53 and other DDR pathways, including HR and BER. The absence of XRCC2, which forms a complex with Rad51 paralogs to prepare DNA filaments for HR repair, results in embryonic lethality that is rescued by TP53 ablation, driving MB formation (142, 143). Furthermore, requirements for BER and SSB repair, pathways crucial for cisplatin adduct resolution, is made evident again by the simultaneous ablation of TP53 and DNA Polymerase β or XRCC1 (144). POLB-/-;TP53-/- and XRCC1-/-;TP53-/- mice overexpress MYCN and CDK6 and develop aggressive classical MBs resembling the SHHα subtype. The absence of genomic guardians in CGNPs amplifies their tumorigenic potential and further delineates a requirement for intact DNA damage signaling in the developing cerebellum. p53 loss is required for MB development when DNA repair mechanisms are no longer present. Where DNA damaging reagents are utilized for pediatric MB patients with defective p53, DDR signaling pathways cannot activate p53 mediated apoptosis or senescence, creating potential opportunities for therapeutic resistance.
Targeting the p53 Response
While the p53 inactivating mutations are the most apparent method for p53 pathway dysregulation, alterations to p53 regulators or signaling effectors are also described in MB and can be therapeutically harnessed for p53 signaling modulation when combined with DNA damaging reagents. MDM2, or Double Minute 2, is an E3 ubiquitin ligase responsible for the degradation of p53 in the absence of cell stress; thus, MDM2 dysregulation may lead to inappropriate p53 degradation, resulting in cell cycle checkpoint abrogation or apoptotic resistance. And while MDM2 is only amplified in a small percentage of adult MB, manipulation of p53 levels through MDM2 inhibition could be viable in patients without MDM2 overexpression and enhance the effects of DNA damage therapies (131, 145). MDM2 ablation from the PTCH+/- model results in p53 accumulation, decreased CGNP expansion, and aberrant cerebellar foliation (146). It follows from this reasoning that disrupting the interaction between MDM2 and p53 could enhance p53 mediated apoptosis and cell cycle inhibition, made possible by MDM2 inhibitors such as nutlin (147). MDM2 inhibition by nutlin in TP53 wild type MB cells, HDMB03, ON2-76, D341, and D283, results in the accumulation of p53, p21, and sub G1 population size; however, TP53 mutant cells, Daoy and UW228, are unaffected (148). Additionally, when combined with a DNA intercalating agent, doxorubicin, nutlin further enhances the apoptotic and cell cycle inhibitory activity of p53 (149).
The interaction between MDM2 and p53 can also be modulated by other proteins, including BAI1, I2PP2A, and WIP1. BAI1, or Brain-specific angiogenesis inhibitor 1, inhibits angiogenesis and upregulates p53 activity through MDM2 inhibition (150, 151). Therein, ADGRB1 (BAI1) ablation in the PTCH+/- model results in substantial decreases in p53 and p21, and an approximately 50% increase in MB tumor formation. Furthermore, epigenetic reactivation of BAI1 through KCC-07, a MBD2 (Methyl-CpG-binding domain protein 2) inhibitor, potentiates the activity of p53 and cell cycle inhibition, bestowing a survival advantage in mice harboring D556 xenograft tumors (151). BAI1 epigenetic reactivation can also be achieved by inhibiting EZH2, an epigenetic transcriptional repressor responsible for H3K27 methylation (152). Application of the EZH2 inhibitor, tazemetostat, yields a mild survival advantage in TP53 wild type MB xenograft models. Another MDM2 antagonist, I2PP2A or Phosphatase 2A Inhibitor 2 also known as SET, inhibits PP2A which dephosphorylates MDM2, rendering it incapable of degrading p53. The application of COG112 in primary SmoA1 SHH MB mouse cells inhibits I2PP2A, allowing PP2A to dephosphorylate MDM2, resulting in accumulation of p53, p21, and a decrease in cell viability of TP53 wild type ONS-76 cells (153). Synergism between radiation or chemotherapy and I2PP2A or EZH2 inhibition remains to be determined.
A final protein of interest, WIP1/PPMD1 or wild-type p53-induced phosphatase 2, is a serine/threonine phosphatase belonging to the PP2C family, which can positively regulate p53 degradation (154, 155). WIP1 is capable of dephosphorylating p53 at Ser15 (facilitating MDM2 binding), direct upregulation of MDM2 activity, and contributing to DDR termination through pATM and γH2AX dephosphorylation. Thus, targeting WIP could amplify the ATM-Chk2 signaling axis, augmenting DNA damage recognition and p53 activation, and potentially sensitizing to DNA damaging reagents. WIP1 is overexpressed in a considerable population of MB cell lines and patient tumors as a result of chromosome 17q copy number gains, present in ~46% of patients sampled (156). And while the overexpression of WIP1 mildly abrogates accumulation of p53Ser15 phosphorylation following UV exposure and increases the expression of p21 in MB, and knockdown sensitizes to ionizing radiation in Diffuse Intrinsic Pontine Glioma (DIPG), the level of sensitization achievable in MB is not clear (157). Secondarily, similar to the pleiotropic nature of CK2, WIP1 is known to influence not only DDR, but also proliferation and invasiveness. Utilizing the RCAS nestin-tva mouse model, when co-infected with transgenes overexpressing SHH ligand and WIP1, 34% of mice develop spontaneous tumors compared to 8% of mice with SHH overexpression (158). Importantly, WIP1 overexpression alone was not sufficient to induce spontaneous tumor formation. WIP1 overexpression also drives expression of SHH signaling targets, Gli1 and Ptch1, and enhances proliferation in CGNPs independent of p53 inhibition (159). Finally, in both smoothened-overexpressing and PTCH+/- MB models, PPM1D knockout dramatically suppressed de novo tumor formation. Thus, targeting WIP1 could not only reduce proliferative and tumorigenic phenotypes, but also sensitize to MB DNA damaging therapies.
Historically, MB patients harboring TP53 mutations have had a substantially worse prognosis than their WT counterparts likely due to ineffective apoptotic signaling following standard of care therapies. And while the vast majority of cancer therapies focus on inhibiting genes mediating resistance, the activation of tumor suppressors is beginning to emerge as a potential alternative. Inhibition of MDM2 oncogenes as a way to reactivate p53 is being tested in clinical trials for hematologic and solid tumors.5ct Results for preclinical research for p53 activation through MDM2 and WIP1 inhibition is mixed, however, and the efficacy of these inhibitors in patients is to be determined. Secondarily, for patients with TP53 mutations, MDM2 and WIP1 inhibition will likely have no effect because mutant p53 is dominant negative over any endogenous WT p53. Over the last two decades researchers have turned to gene therapy as a way to reintroduce p53 through recombinant viruses, a more relevant approach for TP53 mutant tumors.6,7ct This localized method to protein expression may not carry the same systemic issues as administering an MDM2 or WIP1 inhibitor, however, gene therapy is challenging and carries potentially dangerous side effects for constructs capable of mutating into self-replicating viruses. Presumably, given the role of a p53 virus would be to transiently express p53, this rids the potential for inappropriate genomic integration and cellular transformation. Thus, given the rapidly developing landscape of gene therapy, WT p53 expression could develop into a viable therapeutic option.
Conclusions
Given the lack of success with molecular therapy in medulloblastoma and the only recently emerging impact of molecular subgrouping on patient treatment, DNA damaging therapeutics will remain the backbone therapy in MB patients for the foreseeable future. DNA damage proteins are highly conserved in their response to genotoxic therapies across patients; however, the responsiveness may be contingent upon numerous factors, including underlying genomic instability driven by MYC amplification, or the presence of secondary mutations or tumor suppressor inhibition such as TP53 loss or mutation. These factors may drive apoptotic resistance and potentially advantageous mutagenesis. Alternatively, variations to proteins playing either a direct or indirect role in the DDR, such as increased Chk1 expression, elevated APE1 activity, or phosphorylation of AKT, can prime tumors for responsiveness to DNA damage. Together, these alterations may lead to the clonal evolution of therapeutically resistant cells from a once minority cell population such as those of the perivascular niche, ultimately resulting in patient relapse. The targeting of DDR proteins is broadly applicable even outside of a synthetically lethal context, such as with PARP dependence in BRCA1/2 mutation positive cancers, and numerous inhibitors against PARP1, ATR, Chk1, and MDM2 are being tested in combination with standard of care radiation and chemotherapy in the setting of medulloblastoma and beyond (160–162).
Clinical Trials Bibliography
1ct. National Library of Medicine (US). (2019, July -). Evaluation of LY2606368 Therapy in Combination With Cyclophosphamide or Gemcitabine for Children and Adolescents With Refractory or Recurrent Group 3/Group 4 or SHH Medulloblastoma Brain Tumors. ClinicalTrials.gov Identifier: NCT04023669. https://clinicaltrials.gov/ct2/show/NCT04023669
2ct. National Library of Medicine (US). (2017, July -). Olaparib in Treating Patients With Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphoma, or Histiocytic Disorders With Defects in DNA Damage Repair Genes (A Pediatric MATCH Treatment Trial). ClinicalTrials.gov Identifier: NCT03233204. https://clinicaltrials.gov/ct2/show/NCT03233204
3ct. National Library of Medicine (US). (2019, April -). Testing the Safety and Tolerability of CX-4945 in Patients With Recurrent Medulloblastoma Who May or May Not Have Surgery. ClinicalTrials.gov Identifier: NCT03904862. https://www.clinicaltrials.gov/ct2/show/NCT03904862
4ct. National Library of Medicine (US). (2017, July -). Samotolisib in Treating Patients With Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphoma, or Histiocytic Disorders With TSC or PI3K/MTOR Mutations (A Pediatric MATCH Treatment Trial). ClinicalTrials.gov Identifier: NCT03213678. https://clinicaltrials.gov/ct2/show/NCT03213678
5ct. National Library of Medicine (US). (2018, August -). Phase 1 Study of the Dual MDM2/MDMX Inhibitor ALRN-6924 in Pediatric Cancer. ClinicalTrials.gov Identifier: NCT03654716. https://clinicaltrials.gov/ct2/show/NCT03654716
6ct. National Library of Medicine (US). (2004, August - 2009, February). Gene Therapy in Treating Patients With Recurrent or Progressive Brain Tumors. ClinicalTrials.gov Identifier: NCT00004080. https://clinicaltrials.gov/ct2/show/NCT00004080
7ct National Library of Medicine (US). (2004, July - 2018, June). Gene Therapy in Treating Patients With Recurrent Malignant Gliomas. ClinicalTrials.gov Identifier: NCT00004041. https://clinicaltrials.gov/ct2/show/NCT00004041
Author Contributions
LM and AK developed the concept of the review. SS provided clinical contextual information. DH provided radiation response in medulloblastoma expertise and advised on figure preparation. KP and JS provided Akt and DNA damage response expertise. All authors contributed to writing and approval of the final version.
Funding
JS is supported by NCI 4R00CA204601. AK is supported by NINDS R01NS110386 and Alex’s Lemonade Stand Foundation for Childhood Cancer Research. Dr.s JS and AK are members of the Winship Cancer Institute (P30 Center Grant CA138292).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
WNT, Wingless; SHH, Sonic Hedgehog; DDR, DNA Damage Response; MB, Medulloblastoma; DSB, Double Strand Break; SSB, Single Strand Break; NHEJ, Non-Homologous End Joining; HR, Homologous Repair; BER, Base Excision Repair; NER, Nucleotide Excision Repair; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related protein; Chk1, Checkpoint Kinase 1; Chk2, Checkpoint Kinase 2; PTCH, Patched; SMO, Smoothened; PARP, Poly ADP Ribose Polymerase 1; BBB, Blood Brain Barrier; CGNP, Cerebellar Granular Neural Precursor; PVN, Perivascular Niche; APE1, Apurinic/apyrimidinic endonuclease 1; CK2, Casein Kinase 2; MGMT, O6-MethylGuanine-DNA Methyltransferase; KD, Knockdown; KO, Knockout; OE, Overexpression; MRI, Magnetic Resonance Imaging; STR, Subtotal Resection; GTR, Gross Total Resection; LCA, Large Cell Anaplastic; DNV, Desmoplastic/Nodular; MBEN, medulloblastoma with extensive nodularity; LP, Lumbar Puncture; CSI, Craniospinal Irradiation; IR, Ionizing Radiation; P(n), Post-natal day (n); TBB, 4,5,6,7-Tetrabromobenzotriazole; NSCLC, non-small cell lung cancer; RTK, Receptor Tyrosine Kinase; PI3K, Phosphoinositide 3-kinase; EMT, Epithelial to Mesenchymal Transition; (M/R/N), MRE11/RAD50/NBS1.
References
1. Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2010–2014. Neuro Oncol (2017) pp. v1–v88. doi: 10.1093/neuonc/nox158
2. Wefers AK, Warmuth-Metz M, Pöschl J, von Bueren AO, Monoranu C-M, Seelos K, et al. Subgroup-Specific Localization of Human Medulloblastoma Based on Pre-Operative MRI. Acta Neuropathol (2014) 127(6):931–3. doi: 10.1007/s00401-014-1271-5
3. Kool M, Korshunov A, Remke M, Jones DTW, Schlanstein M, Northcott PA, et al. Molecular Subgroups of Medulloblastoma: An International Meta-Analysis of Transcriptome, Genetic Aberrations, and Clinical Data of WNT, SHH, Group 3, and Group 4 Medulloblastomas. Acta Neuropathol (2012) 123(4):473–84. doi: 10.1007/s00401-012-0958-8
4. Northcott PA, Dubuc AM, Pfister S, Taylor MD. Molecular Subgroups of Medulloblastoma. Expert Rev Neurother (2012) 12(7):871–84. doi: 10.1586/ern.12.66
5. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho Y-J, Clifford SC, et al. Molecular Subgroups of Medulloblastoma: The Current Consensus. Acta Neuropathol (2012) 123(4):465–72. doi: 10.1007/s00401-011-0922-z
6. Juraschka K, Taylor MD. Medulloblastoma in the Age of Molecular Subgroups: A Review: JNSPG 75th Anniversary Invited Review Article. J Neurosurg Pediatr (2019) 24(4):353–63. doi: 10.3171/2019.5.PEDS18381
7. Dhanyamraju PK, Patel TN, Dovat S. Medulloblastoma: “Onset of the Molecular Era.”Mol Biol Rep (2020) 47(12):9931–7. doi: 10.1007/s11033-020-05971-w
8. Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, et al. Combined MYC and P53 Defects Emerge at Medulloblastoma Relapse and Define Rapidly Progressive, Therapeutically Targetable Disease. Cancer Cell (2015) 27(1):72–84. doi: 10.1016/j.ccell.2014.11.002
9. Roussel MF, Robinson GW. Role of MYC in Medulloblastoma. Cold Spring Harb Perspect Med (2013) 3(11). doi: 10.1101/cshperspect.a014308
10. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral Heterogeneity Within Medulloblastoma Subgroups. Cancer Cell (2017) 31(6):737–754.e6.
11. Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F, et al. Risk Stratification of Childhood Medulloblastoma in the Molecular Era: The Current Consensus. Acta Neuropathol (2016) 131(6):821–31. doi: 10.1007/s00401-016-1569-6
12. Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, et al. Medulloblastoma. Nat Rev Dis Primers (2019) 5(1):11. doi: 10.1038/s41572-019-0063-6
13. Paterson E, Farr RF. Cerebellar Medulloblastoma: Treatment by Irradiation of the Whole Central Nervous System. Acta Radiologica (1953) 39(4):323–36. doi: 10.3109/00016925309136718
14. Rutka JT, Hoffman HJ. Medulloblastoma: A Historical Perspective and Overview. J Neurooncol (1996) 29(1):1–7. doi: 10.1007/BF00165513
15. Leary S, Packer R, Jaju A, Heier L, Burger P, Smith K, et al. Mbcl-16. Efficacy of Carboplatin Given Concomitantly With Radiation and Isotretinoin as a Pro-Apoptotic Agent in Maintenance Therapy in High-Risk Medulloblastoma: A Report From the Children’s Oncology Group. Neuro-Oncology (2020) 22(Supplement_3):iii391–1. doi: 10.1093/neuonc/noaa222.492
16. Yap TA, Plummer R, Azad NS, Helleday T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am Soc Clin Oncol Educ Book (2019) 39:185–95. doi: 10.1200/EDBK_238473
17. Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, et al. Phase III Study of Craniospinal Radiation Therapy Followed by Adjuvant Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J Clin Oncol (2006) 24(25):4202–8. doi: 10.1200/JCO.2006.06.4980
18. Friedman HS, Schold SC Jr. Rational Approaches to the Chemotherapy of Medulloblastoma. Neurol Clin (1985) 3(4):843–53. doi: 10.1016/S0733-8619(18)31013-2
19. Ruggiero A, Trombatore G, Triarico S, Arena R, Ferrara P, Scalzone M, et al. Multi-Institution Prospective Trial of Reduced-Dose Craniospinal Irradiation (23.4 Gy) Followed by Conformal Posterior Fossa (36 Gy) and Primary Site Irradiation (55.8 Gy) and Dose-Intensive Chemotherapy for Average-Risk Medulloblastoma. Int J Radiat Oncol Biol Phys (2008) 70(3):782–7.
20. Merchant TE, Kun LE, Krasin MJ, Jones-Wallace D, Chintagumpala MM, Woo SY, et al. Platinum Compounds in Children With Cancer: Toxicity and Clinical Management. Anticancer Drugs (2013) 24(10):1007–19. doi: 10.1097/CAD.0b013e3283650bda
21. Sadoughi F, Mirsafaei L, Dana PM, Hallajzadeh J, Asemi Z, Mansournia MA, et al. The Role of DNA Damage Response in Chemo- and Radio-Resistance of Cancer Cells: Can DDR Inhibitors Sole the Problem? DNA Repair (2021) 101:103074. doi: 10.1016/j.dnarep.2021.103074
22. Santivasi WL, Xia F. Ionizing Radiation-Induced DNA Damage, Response, and Repair. Antioxid Redox Signal (2014) 21(2):251–9. doi: 10.1089/ars.2013.5668
23. Goldstein M, Kastan MB. The DNA Damage Response: Implications for Tumor Responses to Radiation and Chemotherapy. Annu Rev Med (2015) 66:129–43. doi: 10.1146/annurev-med-081313-121208
24. Basu A, Broyde S, Iwai S, Kisker C. DNA Damage, Mutagenesis, and DNA Repair. J Nucleic Acids (2010) 2010:182894. doi: 10.4061/2010/182894
25. Hong B-J, Kim J, Jeong H, Bok S, Kim Y-E, Ahn G-O. Tumor Hypoxia and Reoxygenation: The Yin and Yang for Radiotherapy. Radiat Oncol J (2016) 34(4):239–49. doi: 10.3857/roj.2016.02012
26. Bartek J, Lukas J. Chk1 and Chk2 Kinases in Checkpoint Control and Cancer. Cancer Cell (2003) 3(5):421–9. doi: 10.1016/S1535-6108(03)00110-7
27. Prince EW, Balakrishnan I, Shah M, Mulcahy Levy JM, Griesinger AM, Alimova I, et al. Checkpoint Kinase 1 Expression is an Adverse Prognostic Marker and Therapeutic Target in MYC-Driven Medulloblastoma. Oncotarget (2016) 7(33):53881–94. doi: 10.18632/oncotarget.10692
28. Pazzaglia S, Mancuso M, Atkinson MJ, Tanori M, Rebessi S, Majo VD, et al. High Incidence of Medulloblastoma Following X-Ray-Irradiation of Newborn Ptc1 Heterozygous Mice. Oncogene (2002) 21(49):7580–4. doi: 10.1038/sj.onc.1205973
29. Pazzaglia S, Tanori M, Mancuso M, Gessi M, Pasquali E, Leonardi S, et al. Two-Hit Model for Progression of Medulloblastoma Preneoplasia in Patched Heterozygous Mice. Oncogene (2006) 25(40):5575–80. doi: 10.1038/sj.onc.1209544
30. Leonard JM, Ye H, Wetmore C, Karnitz LM. Sonic Hedgehog Signaling Impairs Ionizing Radiation–Induced Checkpoint Activation and Induces Genomic Instability. J Cell Biol (2008) 183(3):385–91. doi: 10.1083/jcb.200804042
31. Lang PY, Nanjangud GJ, Sokolsky-Papkov M, Shaw C, Hwang D, Parker JS, et al. ATR Maintains Chromosomal Integrity During Postnatal Cerebellar Neurogenesis and is Required for Medulloblastoma Formation. Development (2016) 143(21):4038–52. doi: 10.1242/dev.139022
32. Petroni M, Giannini G. A MYCN-MRN Complex Axis Controls Replication Stress for the Safe Expansion of Neuroprogenitor Cells. Mol Cell Oncol (2016) 3(2):e1079673. doi: 10.1080/23723556.2015.1079673
33. Kuzyk A, Mai S. C-MYC-Induced Genomic Instability. Cold Spring Harb Perspect Med (2014) 4(4):a014373. doi: 10.1101/cshperspect.a014373
34. Campaner S, Amati B. Two Sides of the Myc-Induced DNA Damage Response: From Tumor Suppression to Tumor Maintenance. Cell Div (2012) 7(1):6. doi: 10.1186/1747-1028-7-6
35. Kenney AM, Cole MD, Rowitch DH. Nmyc Upregulation by Sonic Hedgehog Signaling Promotes Proliferation in Developing Cerebellar Granule Neuron Precursors. Development (2003) 130(1):15–28. doi: 10.1242/dev.00182
36. Frappart P-O, Lee Y, Russell HR, Chalhoub N, Wang Y-D, Orii KE, et al. Recurrent Genomic Alterations Characterize Medulloblastoma Arising From DNA Double-Strand Break Repair Deficiency. Proc Natl Acad Sci U S A (2009) 106(6):1880–5. doi: 10.1073/pnas.0806882106
37. Wolpaw AJ, Bayliss R, Büchel G, Dang CV, Eilers M, Gustafson WC, et al. Drugging the “Undruggable” MYCN Oncogenic Transcription Factor: Overcoming Previous Obstacles to Impact Childhood Cancers. Cancer Res (2021) 81(7):1627–32. doi: 10.1158/0008-5472.CAN-20-3108
38. Moreira DC, Venkataraman S, Subramanian A, Desisto J, Balakrishnan I, Prince E, et al. Targeting MYC-Driven Replication Stress in Medulloblastoma With AZD1775 and Gemcitabine. J Neurooncol (2020) 147(3):531–45. doi: 10.1007/s11060-020-03457-0
39. Krüger K, Geist K, Stuhldreier F, Schumacher L, Blümel L, Remke M, et al. Multiple DNA Damage-Dependent and DNA Damage-Independent Stress Responses Define the Outcome of ATR/Chk1 Targeting in Medulloblastoma Cells. Cancer Lett (2018) 430:34–46. doi: 10.1016/j.canlet.2018.05.011
40. Endersby R, Whitehouse J, Pribnow A, Kuchibhotla M, Hii H, Carline B, et al. Small-Molecule Screen Reveals Synergy of Cell Cycle Checkpoint Kinase Inhibitors With DNA-Damaging Chemotherapies in Medulloblastoma. Sci Transl Med (2021) 13(577). doi: 10.1126/scitranslmed.aba7401
41. Ray Chaudhuri A, Nussenzweig A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat Rev Mol Cell Biol (2017) 18(10):610–21. doi: 10.1038/nrm.2017.53
42. Chan CY, Tan KV, Cornelissen B. PARP Inhibitors in Cancer Diagnosis and Therapy. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-20-2766
43. Parmar K, Kochupurakkal BS, Lazaro J-B, Wang ZC, Palakurthi S, Kirschmeier PT, et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin Cancer Res (2019) 25(20):6127–40. doi: 10.1158/1078-0432.CCR-19-0448
44. McGrail DJ, Lin CC-J, Garnett J, Liu Q, Mo W, Dai H, et al. Improved Prediction of PARP Inhibitor Response and Identification of Synergizing Agents Through Use of a Novel Gene Expression Signature Generation Algorithm. NPJ Syst Biol Appl (2017) 3:8. doi: 10.1038/s41540-017-0011-6
45. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The Whole-Genome Landscape of Medulloblastoma Subtypes. Nature (2017) 547(7663):311–7. doi: 10.1038/nature22973
46. Tong W-M, Ohgaki H, Huang H, Granier C, Kleihues P, Wang Z-Q. Null Mutation of DNA Strand Break-Binding Molecule Poly(ADP-Ribose) Polymerase Causes Medulloblastomas in P53–/– Mice. Am J Pathol (2003) 162(1):343–52. doi: 10.1016/s0002-9440(10)63825-4
47. Marino S, Vooijs M, van der Gulden H, Jonkers J, Berns A. Induction of Medulloblastomas in P53-Null Mutant Mice by Somatic Inactivation of Rb in the External Granular Layer Cells of the Cerebellum. Genes Dev (2000) 14(8):994–1004. doi: 10.1101/gad.14.8.994
48. Tanori M, Mancuso M, Pasquali E, Leonardi S, Rebessi S, Di Majo V, et al. PARP-1 Cooperates With Ptc1 to Suppress Medulloblastoma and Basal Cell Carcinoma. Carcinogenesis (2008) 29(10):1911–9. doi: 10.1093/carcin/bgn174
49. van Vuurden DG, Hulleman E, Meijer OLM, Wedekind LE, Kool M, Witt H, et al. PARP Inhibition Sensitizes Childhood High Grade Glioma, Medulloblastoma and Ependymoma to Radiation. Oncotarget (2011) 2(12):984–96. doi: 10.18632/oncotarget.362
50. Carruthers R, Chalmers AJ. The Potential of PARP Inhibitors in Neuro-Oncology. CNS Oncol (2012) 1(1):85–97. doi: 10.2217/cns.12.13
51. Ning J-F, Stanciu M, Humphrey MR, Gorham J, Wakimoto H, Nishihara R, et al. Myc Targeted CDK18 Promotes ATR and Homologous Recombination to Mediate PARP Inhibitor Resistance in Glioblastoma. Nat Commun (2019) 10(1):2910. doi: 10.1038/s41467-019-10993-5
52. Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, et al. Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit Rev Eukaryot Gene Expr (2014) 24(1):15–28. doi: 10.1615/CritRevEukaryotGeneExpr.2013006875
53. Kondo N, Takahashi A, Ono K, Ohnishi T. DNA Damage Induced by Alkylating Agents and Repair Pathways. J Nucleic Acids (2010) 2010:1–7. doi: 10.4061/2010/543531
54. Reardon JT, Vaisman A, Chaney SG, Sancar A. Efficient Nucleotide Excision Repair of Cisplatin, Oxaliplatin, and Bis-Aceto-Ammine-Dichloro-Cyclohexylamine-Platinum(IV) (JM216) Platinum Intrastrand DNA Diadducts. Cancer Res (1999) 59(16):3968–71.
55. Kummar S, Wade JL, Oza AM, Sullivan D, Chen AP, Gandara DR, et al. Randomized Phase II Trial of Cyclophosphamide and the Oral Poly (ADP-Ribose) Polymerase Inhibitor Veliparib in Patients With Recurrent, Advanced Triple-Negative Breast Cancer. Invest New Drugs (2016) 34(3):355–63. doi: 10.1007/s10637-016-0335-x
56. McQuade RM, Stojanovska V, Bornstein JC, Nurgali K. PARP Inhibition in Platinum-Based Chemotherapy: Chemopotentiation and Neuroprotection. Pharmacol Res (2018) 137:104–13. doi: 10.1016/j.phrs.2018.09.031
57. Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for Poly(ADP-Ribose) Polymerase (PARP) Inhibitors in Combination Therapy With Camptothecins or Temozolomide Based on PARP Trapping Versus Catalytic Inhibition. J Pharmacol Exp Ther (2014) 349(3):408–16. doi: 10.1124/jpet.113.210146
58. Cefalo G, Massimino M, Ruggiero A, Barone G, Ridola V, Spreafico F, et al. Temozolomide is an Active Agent in Children With Recurrent Medulloblastoma/Primitive Neuroectodermal Tumor: An Italian Multi-Institutional Phase II Trial. Neuro Oncol (2014) 16(5):748–53. doi: 10.1093/neuonc/not320
59. Bautista F, Fioravantti V, de Rojas T, Carceller F, Madero L, Lassaletta A, et al. Medulloblastoma in Children and Adolescents: A Systematic Review of Contemporary Phase I and II Clinical Trials and Biology Update. Cancer Med (2017) 6(11):2606–24. doi: 10.1002/cam4.1171
60. Le Teuff G, Castaneda-Heredia A, Dufour C, Jaspan T, Calmon R, Devos A, et al. Phase II Study of Temozolomide and Topotecan (TOTEM) in Children With Relapsed or Refractory Extracranial and Central Nervous System Tumors Including Medulloblastoma With Post Hoc Bayesian Analysis: A European ITCC Study. Pediatr Blood Cancer (2020) 67(1):e28032. doi: 10.1002/pbc.28032
61. Daniel RA, Rozanska AL, Mulligan EA, Drew Y, Thomas HD, Castelbuono DJ, et al. Central Nervous System Penetration and Enhancement of Temozolomide Activity in Childhood Medulloblastoma Models by Poly(ADP-Ribose) Polymerase Inhibitor AG-014699. Br J Cancer (2010) 103(10):1588–96. doi: 10.1038/sj.bjc.6605946
62. Murai J, Huang S-YN, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP Trapping by BMN 673 and Comparison With Olaparib and Rucaparib. Mol Cancer Ther (2014) 13(2):433–43. doi: 10.1158/1535-7163.MCT-13-0803
63. Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The Many Functions of APE1/Ref-1: Not Only a DNA Repair Enzyme. Antioxid Redox Signal (2009) 11(3):601–20. doi: 10.1089/ars.2008.2194
64. Thakur S, Sarkar B, Cholia RP, Gautam N, Dhiman M, Mantha AK. APE1/Ref-1 as an Emerging Therapeutic Target for Various Human Diseases: Phytochemical Modulation of its Functions. Exp Mol Med (2014) 46:e106. doi: 10.1038/emm.2014.42
65. Demple B, Harrison L. Repair of Oxidative Damage to DNA: Enzymology and Biology. Annu Rev Biochem (1994) 63:915–48. doi: 10.1146/annurev.bi.63.070194.004411
66. Loeb LA, Preston BD. Mutagenesis by Apurinic/Apyrimidinic Sites. Annu Rev Genet (1986) 20:201–30. doi: 10.1146/annurev.ge.20.120186.001221
67. Boiteux S, Guillet M. Abasic Sites in DNA: Repair and Biological Consequences in Saccharomyces Cerevisiae. DNA Repair (2004) 3(1):1–12. doi: 10.1016/j.dnarep.2003.10.002
68. Li H, Liu Z-Y, Wu N, Chen Y-C, Cheng Q, Wang J. PARP Inhibitor Resistance: The Underlying Mechanisms and Clinical Implications. Mol Cancer (2020) 19(1):107. doi: 10.1186/s12943-020-01227-0
69. Dumitrache LC, Shimada M, Downing SM, Kwak YD, Li Y, Illuzzi JL, et al. Apurinic Endonuclease-1 Preserves Neural Genome Integrity to Maintain Homeostasis and Thermoregulation and Prevent Brain Tumors. Proc Natl Acad Sci U S A (2018) 115(52):E12285–94. doi: 10.1073/pnas.1809682115
70. Bobola MS, Emond MJ, Blank A, Meade EH, Kolstoe DD, Berger MS, et al. Apurinic Endonuclease Activity in Adult Gliomas and Time to Tumor Progression After Alkylating Agent-Based Chemotherapy and After Radiotherapy. Clin Cancer Res (2004) 10(23):7875–83. doi: 10.1158/1078-0432.CCR-04-1161
71. Silber JR, Bobola MS, Blank A, Schoeler KD, Haroldson PD, Huynh MB, et al. The Apurinic/Apyrimidinic Endonuclease Activity of Ape1/Ref-1 Contributes to Human Glioma Cell Resistance to Alkylating Agents and is Elevated by Oxidative Stress. Clin Cancer Res (2002) 8(9):3008–18.
72. Bobola MS, Blank A, Berger MS, Stevens BA, Silber JR. Apurinic/apyrimidinic Endonuclease Activity is Elevated in Human Adult Gliomas. Clin Cancer Res (2001) 7(11):3510–8.
73. Bobola MS, Finn LS, Ellenbogen RG, Geyer JR, Berger MS, Braga JM, et al. Apurinic/apyrimidinic Endonuclease Activity is Associated With Response to Radiation and Chemotherapy in Medulloblastoma and Primitive Neuroectodermal Tumors. Clin Cancer Res (2005) 11(20):7405–14. doi: 10.1158/1078-0432.CCR-05-1068
74. Saeedi M, Eslamifar M, Khezri K, Dizaj SM. Applications of Nanotechnology in Drug Delivery to the Central Nervous System. Biomedicine Pharmacother (2019) 111:666–75. doi: 10.1016/j.biopha.2018.12.133
75. Kievit FM, Stephen ZR, Wang K, Dayringer CJ, Sham JG, Ellenbogen RG, et al. Nanoparticle Mediated Silencing of DNA Repair Sensitizes Pediatric Brain Tumor Cells to γ-Irradiation. Mol Oncol (2015) 9(6):1071–80. doi: 10.1016/j.molonc.2015.01.006
76. Long K, Gu L, Li L, Zhang Z, Li E, Zhang Y, et al. Small-Molecule Inhibition of APE1 Induces Apoptosis, Pyroptosis, and Necroptosis in non-Small Cell Lung Cancer. Cell Death Dis (2021) 12(6):503. doi: 10.1038/s41419-021-03804-7
77. Borgo C, Ruzzene M. Role of Protein Kinase CK2 in Antitumor Drug Resistance. J Exp Clin Cancer Res (2019) 38(1):287. doi: 10.1186/s13046-019-1292-y
78. Fritz G, Kaina B. Phosphorylation of the DNA Repair Protein APE/REF-1 by CKII Affects Redox Regulation of AP-1. Oncogene (1999) 18(4):1033–40. doi: 10.1038/sj.onc.1202394
79. Carter RJ, Parsons JL. Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol Cell Biol (2016) 36(10):1426–37. doi: 10.1128/MCB.00030-16
80. Nitta RT, Bolin S, Luo E, Solow-Codero DE, Samghabadi P, Purzner T, et al. Casein Kinase 2 Inhibition Sensitizes Medulloblastoma to Temozolomide. Oncogene (2019) 38(42):6867–79. doi: 10.1038/s41388-019-0927-y
81. Gerson SL. MGMT: Its Role in Cancer Aetiology and Cancer Therapeutics. Nat Rev Cancer (2004) 4(4):296–307. doi: 10.1038/nrc1319
82. Rao AM, Quddusi A, Shamim MS. The Significance of MGMT Methylation in Glioblastoma Multiforme Prognosis. J Pak Med Assoc (2018) 68(7):1137–9.
83. Zheng Y, McFarland BC, Drygin D, Yu H, Bellis SL, Kim H, et al. Targeting Protein Kinase CK2 Suppresses Prosurvival Signaling Pathways and Growth of Glioblastoma. Clin Cancer Res (2013) 19(23):6484–94. doi: 10.1158/1078-0432.CCR-13-0265
84. Pierre F, Chua PC, O'Brien SE, Siddiqui-Jain A, Bourbon P, Haddach M, et al. Discovery and SAR of 5-(3-Chlorophenylamino)benzo[c][2,6]naphthyridine-8-Carboxylic Acid (CX-4945), the First Clinical Stage Inhibitor of Protein Kinase CK2 for the Treatment of Cancer. J Medicinal Chem (2011) 54(2):635–54. doi: 10.1021/jm101251q
85. Rabalski AJ, Gyenis L, Litchfield DW. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a Potential Target to Inhibit Survival and DNA Damage Response and Repair Pathways in Cancer Cells. Clin Cancer Res (2016) 22(12):2840–7. doi: 10.1158/1078-0432.CCR-15-1314
86. Jia H, Liu Y, Xia R, Tong C, Yue T, Jiang J, et al. Casein Kinase 2 Promotes Hedgehog Signaling by Regulating Both Smoothened and Cubitus Interruptus. J Biol Chem (2010) 285(48):37218–26. doi: 10.1074/jbc.M110.174565
87. Purzner T, Purzner J, Buckstaff T, Cozza G, Gholamin S, Rusert JM, et al. Developmental Phosphoproteomics Identifies the Kinase CK2 as a Driver of Hedgehog Signaling and a Therapeutic Target in Medulloblastoma. Sci Signal (2018) 11(547). doi: 10.1126/scisignal.aau5147
88. Leroux AE, Schulze JO, Biondi RM. AGC Kinases, Mechanisms of Regulation and Innovative Drug Development. Semin Cancer Biol (2018) 48:1–17. doi: 10.1016/j.semcancer.2017.05.011
89. Manning BD, Cantley LC. AKT/PKB Signaling: Navigating Downstream. Cell (2007) 129(7):1261–74. doi: 10.1016/j.cell.2007.06.009
90. Liu W, Zhang S, Zhang L, Cui Q, Wang J, Gui T, et al. A Prognostic Analysis of Pediatrics Central Nervous System Small Cell Tumors: Evaluation of EGFR Family Gene Amplification and Overexpression. Diagn Pathol (2014) 9:132. doi: 10.1186/1746-1596-9-132
91. Slongo ML, Molena B, Brunati AM, Frasson M, Gardiman M, Carli M, et al. Functional VEGF and VEGF Receptors are Expressed in Human Medulloblastomas. Neuro Oncol (2007) 9(4):384–92. doi: 10.1215/15228517-2007-032
92. Svalina MN, Kikuchi K, Abraham J, Lal S, Davare MA, Settelmeyer TP, et al. IGF1R as a Key Target in High Risk, Metastatic Medulloblastoma. Sci Rep (2016) 6:27012. doi: 10.1038/srep27012
93. Blom T, Roselli A, Häyry V, Tynninen O, Wartiovaara K, Korja M, et al. Amplification and Overexpression of KIT, PDGFRA, and VEGFR2 in Medulloblastomas and Primitive Neuroectodermal Tumors. J Neurooncol (2010) 97(2):217–24. doi: 10.1007/s11060-009-0014-2
94. Black P, Carroll R, Glowacka D. Expression of Platelet-Derived Growth Factor Transcripts in Medulloblastomas and Ependymomas. Pediatr Neurosurg (1996) 24(2):74–8. doi: 10.1159/000121020
95. Hoxhaj G, Manning BD. The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat Rev Cancer (2020) 20(2):74–88. doi: 10.1038/s41568-019-0216-7
96. Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell (2017) 169(3):381–405. doi: 10.1016/j.cell.2017.04.001
97. Dangelmaier C, Manne BK, Liverani E, Jin J, Bray P, Kunapuli SP. PDK1 Selectively Phosphorylates Thr(308) on Akt and Contributes to Human Platelet Functional Responses. Thromb Haemos (2014) 111(3):508–17. doi: 10.1160/TH13-06-0484
98. Xu F, Na L, Li Y, Chen L. Roles of the PI3K/AKT/mTOR Signalling Pathways in Neurodegenerative Diseases and Tumours. Cell Biosci (2020) 10:54. doi: 10.1186/s13578-020-00416-0
99. Liu Q, Turner KM, Alfred Yung WK, Chen K, Zhang W. Role of AKT Signaling in DNA Repair and Clinical Response to Cancer Therapy. Neuro Oncol (2014) 16(10):1313–23. doi: 10.1093/neuonc/nou058
100. Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: A Double-Edged Sword in Cell Proliferation and Genome Stability. J Oncol (2012) 2012:951724. doi: 10.1155/2012/951724
101. Benson EK, Mungamuri SK, Attie O, Kracikova M, Sachidanandam R, Manfredi JJ, et al. P53-Dependent Gene Repression Through P21 is Mediated by Recruitment of E2F4 Repression Complexes. Oncogene (2014) 33(30):3959–69. doi: 10.1038/onc.2013.378
102. Woods D, Turchi JJ. Chemotherapy Induced DNA Damage Response: Convergence of Drugs and Pathways. Cancer Biol Ther (2013) 14(5):379–89. doi: 10.4161/cbt.23761
103. Iyama T, Wilson DM 3rd. DNA Repair Mechanisms in Dividing and non-Dividing Cells. DNA Repair (2013) 12(8):620–36. doi: 10.1016/j.dnarep.2013.04.015
104. Fraser M, Harding SM, Zhao H, Coackley C, Durocher D, Bristow RG. MRE11 Promotes AKT Phosphorylation in Direct Response to DNA Double-Strand Breaks. Cell Cycle (2011) 10(13):2218–32. doi: 10.4161/cc.10.13.16305
105. Plo I, Laulier C, Gauthier L, Lebrun F, Calvo F, Lopez BS. AKT1 Inhibits Homologous Recombination by Inducing Cytoplasmic Retention of BRCA1 and RAD51. Cancer Res (2008) 68(22):9404–12. doi: 10.1158/0008-5472.CAN-08-0861
106. Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z, et al. PI3K/AKT Pathway as a Key Link Modulates the Multidrug Resistance of Cancers. Cell Death Dis (2020) 11(9):797. doi: 10.1038/s41419-020-02998-6
107. Roussel MF, Hatten ME. Cerebellum Development and Medulloblastoma. Curr Top Dev Biol (2011) 94:235–82. doi: 10.1016/B978-0-12-380916-2.00008-5
108. Pazzaglia S, Tanori M, Mancuso M, Rebessi S, Leonardi S, Di Majo V, et al. Linking DNA Damage to Medulloblastoma Tumorigenesis in Patched Heterozygous Knockout Mice. Oncogene (2006) 25(8):1165–73. doi: 10.1038/sj.onc.1209032
109. Kool M, Jones DTW, Jäger N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell (2014) 25(3):393–405. doi: 10.1016/j.ccr.2014.02.004
110. Chaturvedi NK, Kling MJ, Coulter DW, McGuire TR, Ray S, Kesherwani V, et al. Improved Therapy for Medulloblastoma: Targeting Hedgehog and PI3K-mTOR Signaling Pathways in Combination With Chemotherapy. Oncotarget (2018) 9(24):16619–33. doi: 10.18632/oncotarget.24618
111. Baryawno N, Sveinbjörnsson B, Eksborg S, Chen C-S, Kogner P, Johnsen JI. Small-Molecule Inhibitors of Phosphatidylinositol 3-Kinase/Akt Signaling Inhibit Wnt/beta-Catenin Pathway Cross-Talk and Suppress Medulloblastoma Growth. Cancer Res (2010) 70(1):266–76. doi: 10.1158/0008-5472.CAN-09-0578
112. Guerreiro AS, Fattet S, Fischer B, Shalaby T, Jackson SP, Schoenwaelder SM, et al. Targeting the PI3K P110α Isoform Inhibits Medulloblastoma Proliferation, Chemoresistance, and Migration. Clin Cancer Res (2008) 14(21):6761–9. doi: 10.1158/1078-0432.CCR-08-0385
113. Craveiro RB, Ehrhardt M, Velz J, Olschewski M, Goetz B, Pietsch T, et al. The Anti-Neoplastic Activity of Vandetanib Against High-Risk Medulloblastoma Variants is Profoundly Enhanced by Additional PI3K Inhibition. Oncotarget (2017) 8(29):46915–27. doi: 10.18632/oncotarget.14911
114. Charles N, Holland EC. The Perivascular Niche Microenvironment in Brain Tumor Progression. Cell Cycle (2010) 9(15):3012–21. doi: 10.4161/cc.9.15.12710
115. Eckerdt F, Clymer J, Bell JB, Beauchamp EM, Blyth GT, Goldman S, et al. Pharmacological mTOR Targeting Enhances the Antineoplastic Effects of Selective PI3Kα Inhibition in Medulloblastoma. Sci Rep (2019) 9(1):12822.
116. Crespo S, Kind M, Arcaro A. The Role of the PI3K/AKT/mTOR Pathway in Brain Tumor Metastasis. J Cancer Metastasis Treat (2016) 2(3):80. doi: 10.20517/2394-4722.2015.72
117. Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K Pathway Regulates Survival of Cancer Stem Cells Residing in the Perivascular Niche Following Radiation in Medulloblastoma In Vivo. Genes Dev (2008) 22(4):436–48. doi: 10.1101/gad.1627008
118. Shariati M, Meric-Bernstam F. Targeting AKT for Cancer Therapy. Expert Opin Investig Drugs (2019) 28(11):977–88. doi: 10.1080/13543784.2019.1676726
119. Martorana F, Motta G, Pavone G, Motta L, Stella S, Vitale SR, et al. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front Pharmacol (2021) 12:662232. doi: 10.3389/fphar.2021.662232
120. Machl A, Wilker EW, Tian H, Liu X, Schroeder P, Clark A, et al. M2698 is a Potent Dual-Inhibitor of P70s6k and Akt That Affects Tumor Growth in Mouse Models of Cancer and Crosses the Blood-Brain Barrier. Am J Cancer Res (2016) 6(4):806–18.
121. Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJH, Martin DC, et al. Subgroup-Specific Prognostic Implications of TP53 Mutation in Medulloblastoma. J Clin Oncol (2013) 31(23):2927–35. doi: 10.1200/JCO.2012.48.5052
122. Ray S, Chaturvedi NK, Bhakat KK, Rizzino A, Mahapatra S. Subgroup-Specific Diagnostic, Prognostic, and Predictive Markers Influencing Pediatric Medulloblastoma Treatment. Diagnost (Basel) (2021) 12(1). doi: 10.3390/diagnostics12010061
123. Badiali M, Iolascon A, Loda M, Scheithauer BW, Basso G, Trentini GP, et al. P53 Gene Mutations in Medulloblastoma. Immunohistochemistry, Gel Shift Analysis, and Sequencing. Diagn Mol Pathol (1993) 2(1):23–8. doi: 10.1097/00019606-199303000-00004
124. Pomeroy SL. The P53 Tumor Suppressor Gene and Pediatric Brain Tumors. Curr Opin Pediatr (1994) 6(6):632–5. doi: 10.1097/00008480-199412000-00003
125. Dee S, Haas-Kogan DA, Israel MA. Inactivation of P53 is Associated With Decreased Levels of Radiation-Induced Apoptosis in Medulloblastoma Cell Lines. Cell Death Differ (1995) 2(4):267–75.
126. Lu X, Ma O, Nguyen T-A, Jones SN, Oren M, Donehower LA. The Wip1 Phosphatase Acts as a Gatekeeper in the P53-Mdm2 Autoregulatory Loop. Cancer Cell (2007) 12(4):342–54. doi: 10.1016/j.ccr.2007.08.033
127. Chao CC-K. Mechanisms of P53 Degradation. Clin Chim Acta (2015) 438:139–47. doi: 10.1016/j.cca.2014.08.015
128. Woodburn RT, Azzarelli B, Montebello JF, Goss IE. Intense P53 Staining is a Valuable Prognostic Indicator for Poor Prognosis in Medulloblastoma/Central Nervous System Primitive Neuroectodermal Tumors. J Neurooncol (2001) 52(1):57–62. doi: 10.1023/A:1010691330670
129. Adesina AM, Nalbantoglu J, Cavenee WK. P53 Gene Mutation and Mdm2 Gene Amplification are Uncommon in Medulloblastoma. Cancer Res (1994) 54(21):5649–51.
130. Saylors RL 3rd, Sidransky D, Friedman HS, Bigner SH, Bigner DD, Vogelstein B, et al. Infrequent P53 Gene Mutations in Medulloblastomas. Cancer Res (1991) 51(17):4721–3.
131. Giordana MT, Duó D, Gasverde S, Trevisan E, Boghi A, Morra I, et al. MDM2 Overexpression is Associated With Short Survival in Adults With Medulloblastoma. Neuro Oncol (2002) 4(2):115–22. doi: 10.1093/neuonc/4.2.115
132. Harris SL, Levine AJ. The P53 Pathway: Positive and Negative Feedback Loops. Oncogene (2005) 24(17):2899–908. doi: 10.1038/sj.onc.1208615
133. Dharia NV, Kugener G, Guenther LM, Malone CF, Durbin AD, Hong AL, et al. A First-Generation Pediatric Cancer Dependency Map. Nat Genet (2021) 53(4):529–38. doi: 10.1038/s41588-021-00819-w
134. Zenvirt S, Kravchenko-Balasha N, Levitzki A. Status of P53 in Human Cancer Cells Does Not Predict Efficacy of CHK1 Kinase Inhibitors Combined With Chemotherapeutic Agents. Oncogene (2010) 29(46):6149–59. doi: 10.1038/onc.2010.343
135. Williams AB, Schumacher B. P53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med (2016) 6(5). doi: 10.1101/cshperspect.a026070
136. Joerger AC, Fersht AR. The P53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu Rev Biochem (2016) 85:375–404. doi: 10.1146/annurev-biochem-060815-014710
137. Zhulyn O, Nieuwenhuis E, Liu YC, Angers S, Hui C-C. Ptch2 Shares Overlapping Functions With Ptch1 in Smo Regulation and Limb Development. Dev Biol (2015) 397(2):191–202. doi: 10.1016/j.ydbio.2014.10.023
138. Shakhova O, Leung C, van Montfort E, Berns A, Marino S. Lack of Rb and P53 Delays Cerebellar Development and Predisposes to Large Cell Anaplastic Medulloblastoma Through Amplification of N-Myc and Ptch2. Cancer Res (2006) 66(10):5190–200. doi: 10.1158/0008-5472.CAN-05-3545
139. Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, et al. The Tumor Suppressors Ink4c and P53 Collaborate Independently With Patched to Suppress Medulloblastoma Formation. Genes Dev (2005) 19(22):2656–67. doi: 10.1101/gad.1368605
140. Holcomb VB, Vogel H, Marple T, Kornegay RW, Hasty P. Ku80 and P53 Suppress Medulloblastoma That Arise Independent of Rag-1-Induced DSBs. Oncogene (2006) 25(54):7159–65. doi: 10.1038/sj.onc.1209704
141. Lee Y, McKinnon PJ. DNA Ligase IV Suppresses Medulloblastoma Formation. Cancer Res (2002) 62(22):6395–9.
142. Orii KE, Lee Y, Kondo N, McKinnon PJ. Selective Utilization of Nonhomologous End-Joining and Homologous Recombination DNA Repair Pathways During Nervous System Development. Proc Natl Acad Sci U S A (2006) 103(26):10017–22. doi: 10.1073/pnas.0602436103
143. Chun J, Buechelmaier ES, Powell SN. Rad51 Paralog Complexes BCDX2 and CX3 Act at Different Stages in the BRCA1-BRCA2-Dependent Homologous Recombination Pathway. Mol Cell Biol (2013) 33(2):387–95. doi: 10.1128/MCB.00465-12
144. Kim J, Kim J, Lee Y. DNA Polymerase β Deficiency in the P53 Null Cerebellum Leads to Medulloblastoma Formation. Biochem Biophys Res Commun (2018) 505(2):548–53. doi: 10.1016/j.bbrc.2018.09.166
145. Konopleva M, Martinelli G, Daver N, Papayannidis C, Wei A, Higgins B, et al. MDM2 Inhibition: An Important Step Forward in Cancer Therapy. Leukemia (2020) 34(11):2858–74. doi: 10.1038/s41375-020-0949-z
146. Malek R, Matta J, Taylor N, Perry ME, Mendrysa SM. The P53 Inhibitor MDM2 Facilitates Sonic Hedgehog-Mediated Tumorigenesis and Influences Cerebellar Foliation. PloS One (2011) 6(3):e17884. doi: 10.1371/journal.pone.0017884
147. Van Maerken T, Rihani A, Van Goethem A, De Paepe A, Speleman F, Vandesompele J. Pharmacologic Activation of Wild-Type P53 by Nutlin Therapy in Childhood Cancer. Cancer Lett (2014) 344(2):157–65. doi: 10.1016/j.canlet.2013.11.002
148. Künkele A, De Preter K, Heukamp L, Thor T, Pajtler KW, Hartmann W, et al. Pharmacological Activation of the P53 Pathway by Nutlin-3 Exerts Anti-Tumoral Effects in Medulloblastomas. Neuro Oncol (2012) 14(7):859–69. doi: 10.1093/neuonc/nos115
149. Ghassemifar S, Mendrysa SM. MDM2 Antagonism by Nutlin-3 Induces Death in Human Medulloblastoma Cells. Neurosci Lett (2012) 513(1):106–10. doi: 10.1016/j.neulet.2012.02.022
150. Nishimori H, Shiratsuchi T, Urano T, Kimura Y, Kiyono K, Tatsumi K, et al. A Novel Brain-Specific P53-Target Gene, BAI1, Containing Thrombospondin Type 1 Repeats Inhibits Experimental Angiogenesis. Oncogene (1997) 15(18):2145–50. doi: 10.1038/sj.onc.1201542
151. Zhu D, Osuka S, Zhang Z, Reichert ZR, Yang L, Kanemura Y, et al. BAI1 Suppresses Medulloblastoma Formation by Protecting P53 From Mdm2-Mediated Degradation. Cancer Cell (2018) 33(6):1004–1016.e5. doi: 10.1016/j.ccell.2018.05.006
152. Zhang H, Zhu D, Zhang Z, Kaluz S, Yu B, Devi NS, et al. EZH2 Targeting Reduces Medulloblastoma Growth Through Epigenetic Reactivation of the BAI1/p53 Tumor Suppressor Pathway. Oncogene (2020) 39(5):1041–8. doi: 10.1038/s41388-019-1036-7
153. Wei Y, Maximov V, Morrissy SA, Taylor MD, Pallas DC, Kenney AM. P53 Function Is Compromised by Inhibitor 2 of Phosphatase 2A in Sonic Hedgehog Medulloblastoma. Mol Cancer Res (2019) 17(1):186–98. doi: 10.1158/1541-7786.MCR-18-0485
154. Pecháčková S, Burdová K, Macurek L. WIP1 Phosphatase as Pharmacological Target in Cancer Therapy. J Mol Med (2017) 95(6):589–99. doi: 10.1007/s00109-017-1536-2
155. Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, et al. Wip1, a Novel Human Protein Phosphatase That is Induced in Response to Ionizing Radiation in a P53-Dependent Manner. Proc Natl Acad Sci U S A (1997) 94(12):6048–53. doi: 10.1073/pnas.94.12.6048
156. Castellino RC, De Bortoli M, Lu X, Moon S-H, Nguyen T-A, Shepard MA, et al. Medulloblastomas Overexpress the P53-Inactivating Oncogene WIP1/PPM1D. J Neurooncol (2008) 86(3):245–56. doi: 10.1007/s11060-007-9470-8
157. Akamandisa MP, Nie K, Nahta R, Hambardzumyan D, Castellino RC. Inhibition of Mutant PPM1D Enhances DNA Damage Response and Growth Suppressive Effects of Ionizing Radiation in Diffuse Intrinsic Pontine Glioma. Neuro Oncol (2019) 21(6):786–99. doi: 10.1093/neuonc/noz053
158. Doucette TA, Yang Y, Pedone C, Kim JYH, Dubuc A, Northcott PD, et al. WIP1 Enhances Tumor Formation in a Sonic Hedgehog–Dependent Model of Medulloblastoma. Neurosurgery (2012) 70(4):1003–10. doi: 10.1227/neu.0b013e31823e5332
159. Wen J, Lee J, Malhotra A, Nahta R, Arnold AR, Buss MC, et al. WIP1 Modulates Responsiveness to Sonic Hedgehog Signaling in Neuronal Precursor Cells and Medulloblastoma. Oncogene (2016) 35(42):5552–64. doi: 10.1038/onc.2016.96
160. Yi M, Dong B, Qin S, Chu Q, Wu K, Luo S. Advances and Perspectives of PARP Inhibitors. Exp Hematol Oncol (2019) 8:29. doi: 10.1186/s40164-019-0154-9
161. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol (2016) 6:7. doi: 10.3389/fonc.2016.00007
Keywords: medulloblastoma, radiation oncology, pediatrics, therapeutic targeting, p53 status
Citation: McSwain LF, Parwani KK, Shahab SW, Hambardzumyan D, MacDonald TJ, Spangle JM and Kenney AM (2022) Medulloblastoma and the DNA Damage Response. Front. Oncol. 12:903830. doi: 10.3389/fonc.2022.903830
Received: 24 March 2022; Accepted: 10 May 2022;
Published: 07 June 2022.
Edited by:
Anat Erdreich-Epstein, Children’s Hospital of Los Angeles, United StatesReviewed by:
Lucia Di Marcotullio, Sapienza University of Rome, ItalyAtsushi Makimoto, Tokyo Metropolitan Children’s Medical Center, Japan
Copyright © 2022 McSwain, Parwani, Shahab, Hambardzumyan, MacDonald, Spangle and Kenney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Marie Kenney, anna.kenney@emory.edu