 Daniel R. Principe1,2*†
Daniel R. Principe1,2*† Patrick W. Underwood3†
Patrick W. Underwood3† Murray Korc4Jose G. Trevino5
Murray Korc4Jose G. Trevino5 Hidayatullah G. Munshi6,7
Hidayatullah G. Munshi6,7 Ajay Rana2,7*
Ajay Rana2,7*- 1Medical Scientist Training Program, University of Illinois College of Medicine, Chicago, IL, United States
- 2Department of Surgery, University of Illinois at Chicago, Chicago, IL, United States
- 3Department of Surgery, University of Florida, Gainesville, FL, United States
- 4Department of Developmental and Cell Biology, University of California, Irvine, CA, United States
- 5Department of Surgery, Division of Surgical Oncology, Virginia Commonwealth University, Richmond, VA, United States
- 6Department of Medicine, Northwestern University Feinberg School of Medicine, Chicago, IL, United States
- 7Jesse Brown VA Medical Center, Chicago, IL, United States
Pancreatic ductal adenocarcinoma (PDAC) has a dismal prognosis, with a median survival time of 10-12 months. Clinically, these poor outcomes are attributed to several factors, including late stage at the time of diagnosis impeding resectability, as well as multi-drug resistance. Despite the high prevalence of drug-resistant phenotypes, nearly all patients are offered chemotherapy leading to modest improvements in postoperative survival. However, chemotherapy is all too often associated with toxicity, and many patients elect for palliative care. In cases of inoperable disease, cytotoxic therapies are less efficacious but still carry the same risk of serious adverse effects, and clinical outcomes remain particularly poor. Here we discuss the current state of pancreatic cancer therapy, both surgical and medical, and emerging factors limiting the efficacy of both. Combined, this review highlights an unmet clinical need to improve our understanding of the mechanisms underlying the poor therapeutic responses seen in patients with PDAC, in hopes of increasing drug efficacy, extending patient survival, and improving quality of life.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is currently the third leading cause of cancer death in the US. Risk factors for PDAC include tobacco smoking, germline mutations in such genes as breast cancer gene 1 (BRCA1) and BRCA2, chronic pancreatitis, obesity, long-standing type 2 diabetes (T2DM), and prolonged and excessive alcohol consumption (1–3). There has been a slow but progressive increase in PDAC incidence in the US, but the overall survival rate has also increased. Currently, the overall 5-year survival rate is approximately 10% (4). Based on recent trends in PDAC incidence and survival and the improvements in survival in cancers of the lung and breast, it has been proposed that PDAC will become the second leading cause of cancer-related death by 2030 (5). While surgical resection offers a clear survival benefit and increases 5-year survival to 25%, the majority of patients present with disseminated and/or locally advanced disease, precluding them from undergoing resection. As such, nearly all are offered conventional chemotherapy. While chemotherapy provides a survival benefit in both resectable and non-resectable forms of the disease, these benefits are modest as almost all patients harbor some degree of drug resistance (6). Further, a significant number of patients experience grade 3-4 adverse effects (6).
While several chemotherapy regimens have been approved for metastatic PDAC, the most widely used and best-studied agent is Gemcitabine, a drug was first approved by the FDA for metastatic PDAC in 1996, after showing marginal efficacy in clinical trials (7–9). It has remained in clinical use, often in combination with albumin-bound (Nab) Paclitaxel, which improved survival time compared to Gemcitabine monotherapy (10). The multi-drug regimen FOLFIRINOX (5-Fluorouracil, Leucovorin, Irinotecan, and Oxaliplatin) has also shown efficacy in metastatic PDAC. In fact, FOLFIRINOX offers improved disease-free survival compared to Gemcitabine (21.6 v 12.8 months), though FOLFINIROX is associated with a higher rate of serious adverse effects (75.9 v 52.9%) (11). Despite their spectrum of toxicities, FOLFIRINOX and Gemcitabine with Nab-Paclitaxel remain the best and most widely prescribed medications for patients seeking treatment.
Standard of Care Treatment Overview
The clinical management of patients with pancreatic cancer varies depending on several factors, ranging from overall health and wellness to the wishes of the patient and family (Figure 1). During the initial patient assessment, a physician generally orders at minimum a computed tomography (CT) scan of the chest, abdomen, and pelvis in order to assess the extent of the disease. Before administering therapy, further steps are taken to determine patient performance status (PS), symptom burden, and comorbidity profile. Based on this information, as well as a discussion with the patient and their family, healthcare providers work to determine the overall goals of care and formulate a comprehensive treatment plan (12).
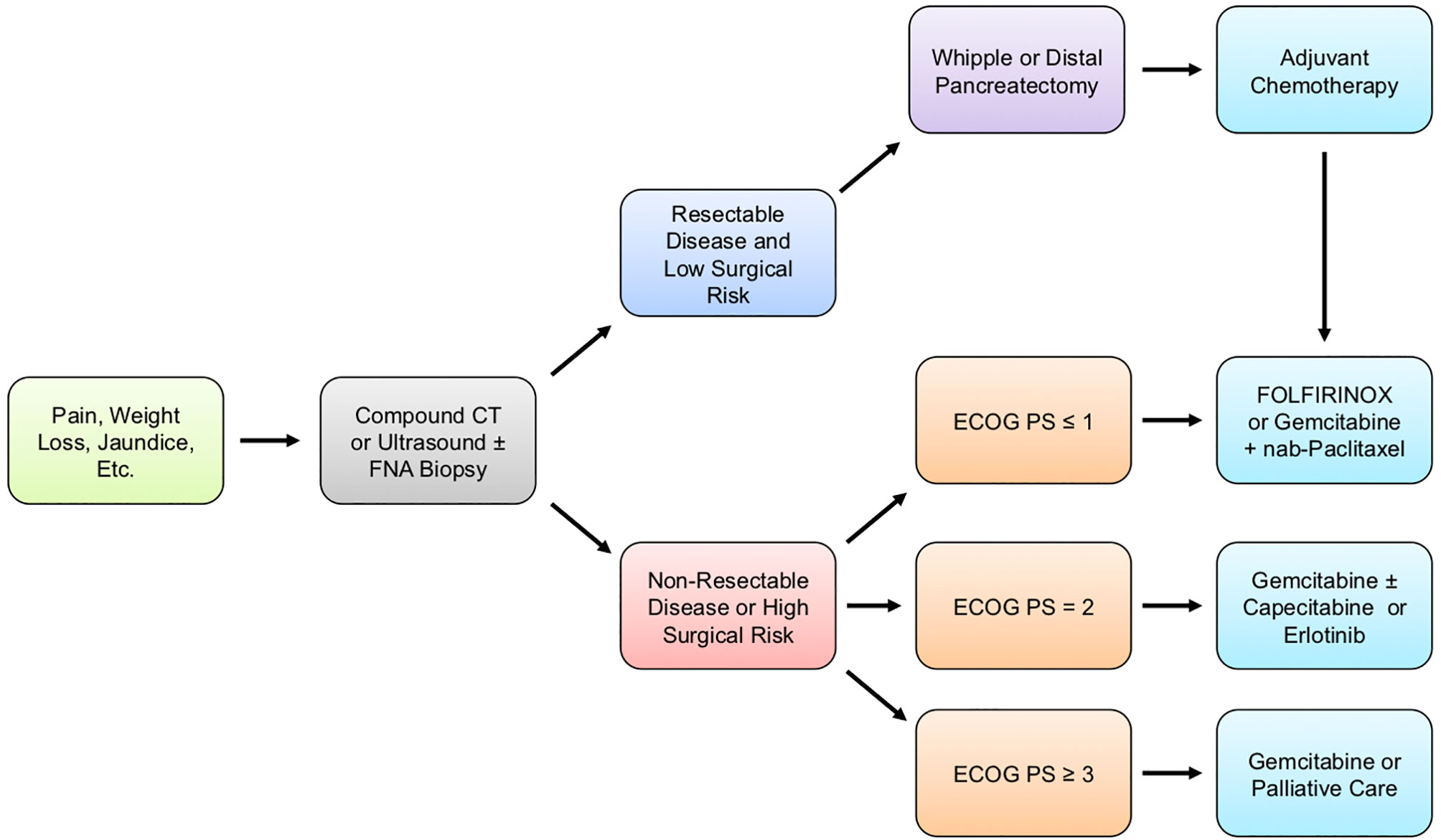
Figure 1 Generalized treatment guidelines for PDAC patients. Pancreatic ductal adenocarcinoma (PDAC) typically presents with vague clinical symptoms, including poorly localized pain, jaundice, or unintended weight loss. When PDAC is suspected, patients are typically diagnosed through computed tomography (CT) scan of the chest, abdomen, and pelvis to assess the extent of disease or ultrasound with or without a fine-needle aspiration (FNA) biopsy. Following confirmatory diagnosis, the patient’s surgical candidacy is determined based on a combination of imaging studies, Eastern Cooperative Oncology Group performance status (ECOG PS), symptom burden, surgical risk, and comorbidity profile. For operable disease, the type of surgery is determined based on the anatomical location of the tumor, as well as several additional factors described in this review article, with most patients receiving either a Whipple procedure or distal pancreatectomy. Regardless of whether a patient is treated with surgery, the current guidelines recommend chemotherapy, and the precise regimen given is based mostly on ECOG PS and comorbidity profile.
For metastatic disease, first-line treatment varies and is influenced largely by the Eastern Cooperative Oncology Group performance status (ECOG PS). For those seeking care with an ECOG PS of either 0 (Fully active, able to carry on all pre-disease performance without restriction) or 1 (Restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature, e.g., light housework, office work) (13), patients are typically offered either FOLFIRINOX or Gemcitabine plus Nab-Paclitaxel. For patients seeking treatment with an ECOG PS of 2 (Ambulatory and capable of all self-care but unable to carry out any work activities; up and about more than 50% of waking hours) or comorbidity profile that prevents the use of a more aggressive regimen, Gemcitabine is recommended in monotherapy, though agents such as Capecitabine can be offered in combination (12). For more severely disabled patients with an ECOG PS of 3 (Capable of only limited self-care; confined to a bed or chair more than 50% of waking hours) or 4 (Completely disabled; cannot carry on any self-care; totally confined to a bed or chair), or those with severe comorbidity, therapy is only offered on a case-by-case basis.
Second-line therapies are more varied and depend on additional factors, including patient preference and overall wellness. For patients who failed on first-line FOLFIRINOX and have an ECOG PS of <1 with the appropriate comorbidity profile, Gemcitabine plus Nab-Paclitaxel can be offered. Gemcitabine can also be offered alone as second-line therapy in patients with an ECOG PS of 2 or those with a substantial comorbidity profile that prevents the use of more aggressive regimens (12).
When indicated, Gemcitabine is administered via intravenous (IV) infusion at 1000 mg/m2 in four-week cycles, consisting of three once-weekly therapy followed by a break in the fourth week. While the number of cycles can vary, generally, postoperative patients undergo six cycles. Though the efficacy of Gemcitabine is significantly improved when used in combination with Nab-Paclitaxel, increasing 2-year-survival from 4% to 9%, this approach has significantly higher toxicities that must be considered. Patients treated with Gemcitabine and Nab-Paclitaxel had an increased rate of Grade 3 neutropenia (38% vs. 27%), febrile neutropenia (3% vs. 1%), fatigue (17% vs. 7%), and neuropathy (17% vs. 1%) (10). While these side effects are largely reversible, they often present a significant clinical challenge, namely a high risk of infection and reduced quality of life.
The combination of 5-FU/Leucovorin and nanoliposomal (Nal) Irinotecan has been approved by the FDA for patients who have been previously treated with Gemcitabine-based chemotherapy. This is based on recent clinical evidence showing that, in the second line, 5-FU/Leucovorin and Nal-Irinotecan offer a significant benefit to previously treated PDAC patients, extending median overall survival from 6.1 months compared to 4.2 months using 5-FU/Leucovorin (14). Importantly, this combination was well tolerated, and this study reported no new safety concerns, affirmed by a subsequent study in elderly patients (14, 15).
For patients with BRCA1/2 or PALB2-mutated PDAC, front line therapy varies significantly. While only 5-9% of PDAC patients harbor such mutations, these patients appear highly sensitive to the combination of Gemcitabine and Cisplatin. This approach led to encouraging 2 and 3-year survival rates of 31% and 18%, respectively. As PARP inhibition can cause synthetic lethality in tumors with loss of high-fidelity double-strand break homologous recombination, the authors also explored the addition of the PARP inhibitor Veliparib, though this failed to further improve clinical outcomes. Though effective, the combination of Gemcitabine and Cisplatin was associated with a relatively high rate of Grade 3 to 4 hematologic toxicities, with 48% experiencing neutropenia, 55% thrombocytopenia, and 52% anemia (16). Also for BRCA-mutated patients, the PARP inhibitor Olaparib has shown significant efficacy as maintenance therapy, specifically for patients who had not progressed during first-line platinum-based chemotherapy (17). This approach extended median progression survival to 7.4 months compared to 3.8 months on placebo, with no difference in health-related quality of life (17).
Finally, patients deficient in DNA mismatch repair (dMMR) with high microsatellite instability (MSI-H) can respond to the immune checkpoint inhibitor Pembrolizumab. The KEYNOTE-158 trial reported that in dMMR/MSI-H PDAC patients, single agent Pembrolizumab had an overall response rate of 18.2%, with a median overall survival of 4 months, median progression-free survival of 2.1 months, and a median duration of response of 13.4 months (18). However, it is important to note that only ~1% of PDAC patients are dMMR/MSI-H (19), and that immunotherapy is not widely used in PDAC treatment at this time (20).
Additional precision medicine approaches are also beginning to show promise for PDAC, particularly for patients with NTRK1–3 or ROS1 gene fusions (21). These patients have shown increased sensitivity to selective tropomyosin receptor kinase (TRK) and ROS1 inhibitors larotrectinib and entrectinib (22). For example, the STARTRK-2 trial included two PDAC patients with a TPR-NTRK gene fusion, and one with an SCL4-ROS1 gene fusion. These three patients derived substantial clinical benefit from entrectinib, showing either partial radiographic responses or stable disease (23). Similarly, the ongoing NAVIGATE trial evaluating larotrectinib in NTRK fusion-positive tumors also included one PDAC patient who showed a partial response by Response Evaluation Criteria in Solid Tumors (RECIST) criteria. Finally, select small molecule KRAS inhibitors are also emerging, particularly for tumors harboring a KRASG12C mutation (24, 25). However, it is important to note that KRASG12C mutations are exceptionally rare in PDAC, representing only 1% of all KRAS mutations (21, 26). An alternative strategy to target more common KRAS mutations is also under clinical investigation. This approach uses exosomes loaded with small interfering RNAs against KRASG12D, the most common KRAS mutation in PDAC, and has shown encouraging preclinical efficacy (27).
Surgery
Surgery offers a significant survival benefit to eligible patients, particularly when combined with adjuvant chemotherapy (28). In contrast to the overall 5-year survival rate of ~10% for all patients, patients undergoing resection for stage I PDAC have an overall 5-year survival of 38.2% compared to 2.9% for patients who did not (29, 30). Globally, rates of resection for Stage I/II disease vary, ranging from 35% to 69% (31). These variances appear to be due to several added factors, including performance status, tumor size and location (31), and socioeconomic factors such as race (32). Next, we provide a high-level overview of the surgical management of operable PDAC and emerging barriers that may hinder the utility of surgery in the clinical management of PDAC.
Surgical Candidacy
Surgery with perioperative chemotherapy remains the only treatment option for achieving long-term survival for patients with PDAC. Unfortunately, only 15-20% of patients are candidates for upfront surgical resection, while 30-40% have unresectable/locally advanced disease and 50-60% have distant metastases at presentation (33). Even for those who do undergo surgery, 5-year survival remains dismal at 24% (29). Given these observations, at the time of diagnosis, patient disease state is classified as resectable, borderline resectable, locally advanced, or metastatic.
Patients are considered resectable if there is no evidence of distance metastasis, no arterial tumor contact with the celiac axis (CA), superior mesenteric artery (SMA), or common hepatic artery (CHA), and no tumor contact greater than 180° with the superior mesenteric vein (SMV) or portal vein (PV) (Figure 2). Additionally, there should be no vein contour irregularity for patients with less than 180° contact with the SMV or PV (34). For these patients, the standard-of-care has been upfront surgery followed by adjuvant chemotherapy. Even for those who undergo surgery for resectable disease, recurrence rates are 76.7% after two years (35).
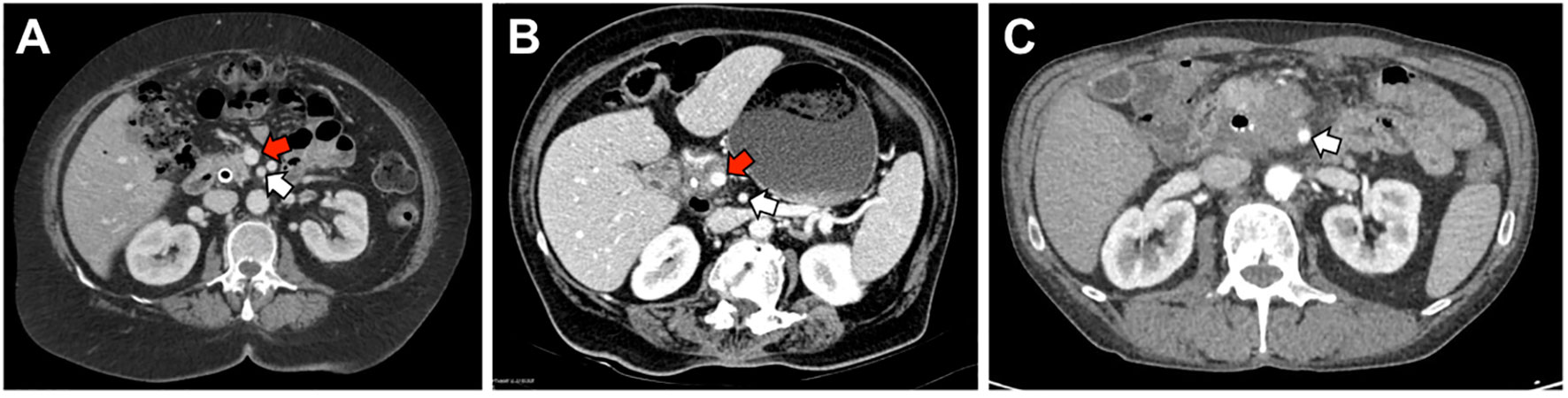
Figure 2 Representative images of resectable, borderline-resectable, and non-resectable PDAC. (A) Representative image of a patient with resectable disease as defined by NCCN guidelines. There is no tumor contact with the superior mesenteric artery (SMA, white arrow) and <180° of tumor contact with the superior mesenteric vein (SMV, red arrow). (B) Patient with borderline resectable disease due to >180° of tumor contact with the SMV but no involvement of the SMA. (C) Patient with locally advanced disease due to >180° of tumor contact with the SMA.
More recently, there is an emerging hypothesis that response to neoadjuvant chemotherapy can be used as a test of underlying tumor biology to identify patients who would benefit from surgical resection. A recent meta-analysis of eight cohort studies and 3 randomized controlled trials (RCT) of neoadjuvant therapy in resectable PDAC found an increased R0 resection rate but no survival benefit (36). A sub-analysis of patients receiving neoadjuvant gemcitabine found a survival benefit compared to upfront resection (HR 0.75, 95%CI 0.73-1.03). A more recent randomized phase III trial in the Netherlands found a lack of survival benefit in patients receiving neoadjuvant gemcitabine compared to upfront resection but improvement in secondary outcomes such as R0 resection (71% vs. 40%) and disease-free survival (37). Other clinical trials are ongoing and will help determine the appropriateness of neoadjuvant therapy for resectable PDAC (38). Additionally, neoadjuvant therapy should also be considered for patients with high-risk features such as high levels of serum CA 19-9, a large primary tumor, large regional lymph nodes, excessive weight loss, or extreme pain (34, 39, 40).
There is a category of patients who are resectable by imaging criteria but considered to be physiologically unresectable based on advanced age, frailty, comorbidities, and performance status. As might be expected, patients over the age of 80 have a significantly increased risk of mortality after surgery, although surgery continues to provide a survival benefit in these patients (41). Several studies have suggested that frail patients and those with poor performance status have worse morbidity, mortality, and survival after surgery for PDAC (42–44). However, there should be no strict age limit for surgical resection, and appropriateness for resection is best determined by evaluating patient factors, life expectancy, and properly counseling the patient.
Patients with borderline resectable disease have solid tumor contact with CHA or SMA < 180°, solid tumor contact with SMV or PV >180°, solid tumor contact with SMV or PV <180° with contour irregularity or vein thrombosis that is suitable for vessel resection, or solid tumor contact with the IVC (34). These patients typically require upfront tissue diagnosis and proceed to neoadjuvant chemotherapy after the staging is complete. Patients who respond to therapy may go on to surgical resection while those with disease progression continue with non-operative management of their disease. The optimal neoadjuvant regimen is unknown but typically involves FOLFIRINOX or Gemcitabine-based regimens. After completion of neoadjuvant chemotherapy, resection may be considered if there is no evidence of metastatic disease, no progression of disease, no more than mild increase in perivascular soft tissue, and stable or decreasing CA 19-9 (34). Neoadjuvant therapy in this population results in an approximate rate of resection of 60-70% (45–47). Neoadjuvant therapy results in an improved R0 resection rate, reduced nodal disease, and improved overall survival in this patient population (45, 48). Additionally, it may reduce the rate of futile surgery in those who progress while on chemotherapy.
Patients with locally advanced disease have >180° contact with the SMA or CA, aortic involvement, or unreconstructable involvement of the SMV/PV (34). For those with poor performance status, palliative and supportive care are best. For those with good performance status, NCCN guidelines recommend enrollment in a clinical trial of neoadjuvant chemoradiation (34). Patients should be considered for surgery after neoadjuvant therapy if there is >50% decrease in CA 19-9 and improvement in patient factors such as pain, early satiety, nutritional status, and performance status (34). The resection rate in this population is much lower than for those with the borderline resectable disease at about 28%, with a reported range of 0-40% (39, 49, 50). Patients with distant metastasis involving the liver, peritoneum, or omentum are generally considered unresectable. While there are centers exploring metastectomy for patients with oligometastatic PDAC in select patients, these data are limited to cohort studies (51, 52).
Disparities and the Underutilization of Surgery
Determining candidacy for surgical resection is best done using a multi-disciplinary approach at an experienced center, as variability in treatment and surgical utilization contributes to poor outcomes in PDAC. For instance, a 2007 study of the National Cancer Database found that a staggering 71.4% of patients with Stage I disease did not undergo surgery. Patients above the age of 65, African American patients, and patients on Medicare or Medicaid were less likely to undergo surgery (29). Other studies have confirmed that socioeconomic variables such as income, education, insurance, and treatment facility are associated with failure to receive standard treatment and worse clinical outcomes (53–55).
Patients receive expected treatment more often and have improved outcomes with treatment at medical centers with surgeons who perform a high volume of pancreatic surgeries (56–58). This raises the issue of geographic disparity, as patients who travel farther to high volume centers have improved survival than those who stay closer to home at lower volume centers, despite having more advanced disease (59). Health disparities also exist in the referral of patients to these high-volume centers (57, 60, 61). For example, patients from socioeconomically disadvantaged backgrounds and minority patients are less likely to be referred to high-volume centers, thereby contributing to their poorer outcomes (57, 60). Accordingly, centralization of PDAC care to high volume centers may address this disparity and improve outcomes, and efforts are currently ongoing to improve access to care at high volume hospitals (62).
Preoperative Biliary Drainage
Obstructive jaundice is a frequent complication for patients with pancreatic head cancers and can be relieved by preoperative biliary drainage. However, for patients going to surgery, a multicenter RCT demonstrated an increased risk of serious complications for those who underwent preoperative biliary drainage (74%) compared to those who went directly to surgery (39%) within 1 week of diagnosis (63), a finding corroborated by multiple meta-analyses (64, 65). As mentioned, the use of neoadjuvant chemotherapy is rising for patients with resectable PDAC. However, these patients often require biliary drainage for symptom palliation during neoadjuvant treatment. Therefore, the number of patients with biliary stents in place at the time of surgery is likely to increase, and this topic warrants continued exploration.
Perioperative Nutrition Management
A significant proportion of PDAC patients will develop cancer-associated cachexia and malnutrition (66), conditions associated with poor clinical outcomes (42, 67). Despite this observation, there is a lack of data evaluating preoperative nutritional interventions to improve postoperative outcomes in patients with PDAC, though the role of postoperative nutritional interventions is more established. Interestingly, there is little data to support routine enteral or parenteral nutrition in patients following pancreatic resection. Somewhat related, a multicenter European RCT compared early enteral feeding to parenteral feeding in patients after pancreaticoduodenectomy (PD) (68). Early enteral feeding was associated with increased frequency and severity of postoperative pancreatic fistula (POPF), while parenteral feeding patients had earlier recovery of oral feeding. Unfortunately, this study did not include an oral feeding group, which is supported by the Enhanced Recovery After Surgery Society recommendations (69). Several studies have reported successful resumption of oral diet early in the post-operative course without the need for enteral or parenteral nutrition (70–72). Hence, though a randomized control trial is needed, early oral feeding appears safe and does not worsen the duration or grade of POPF (72).
Staging Laparoscopy
The decision to perform staging laparoscopy is made on a case-by-case basis. Patients at the highest risk of unresectable disease are most likely to benefit from staging laparoscopy, which detects occult metastatic disease, such as small liver or peritoneal metastasis, not appreciated on preoperative imaging. Staging laparoscopy identifies occult metastatic disease in 8-26% of patients, saving a need for exploratory laparotomy (73, 74). As risk factors for the occult metastatic disease include abdominal pain, tumor size > 30 mm, indeterminate liver lesions, and CA 19-9 level > 192 U/ml (40, 73), the presence of such risk factors indicates a need for staging laparoscopy. As neoadjuvant approaches are increasingly used for patients with borderline resectable and locally advanced disease, patients at higher risk for occult metastatic disease are more likely to undergo surgery. The use of staging laparoscopy in this population will be important, however, with some suggesting staging laparoscopy before neoadjuvant therapy as a mechanism to diagnose occult metastatic disease earlier in the disease course (75).
The addition of laparoscopic ultrasound to staging laparoscopy has been proposed as a possible adjunct to staging laparoscopy. It appears to provide additional prognostic information in a minority of patients and should not be routinely performed (76). Novel imaging techniques, such as near-infrared fluorescence imaging, to detect occult metastasis or unresectable disease are now being developed (77, 78), though further evidence is required before the implementation of these techniques.
Palliative Surgery
For patients who are found to have inoperable disease on staging laparoscopy or laparotomy, the surgeon must decide whether to perform surgical palliation. Classically, a hepaticojejunostomy and gastrojejunostomy were performed for the prevention of biliary and gastric outlet obstruction. As advanced endoscopic techniques have improved, the need for these operations has come into question. For patients with obstructive jaundice, placement of self-expanding metals stents (SEMS) has become the gold standard due to its lower morbidity, but there is a higher rate of recurrent obstruction and need for repeat intervention than surgical bypass (79, 80). For the patient with obstructive jaundice and found to be inoperable at the time of surgery, a surgical biliary bypass is recommended (34). The role of a prophylactic biliary bypass is unclear and should be at the discretion of the surgeon and patient.
For patients with malignant gastric outlet obstruction (GOO) and a life expectancy >2 months, there is better long-term relief of symptoms with gastrojejunostomy (GJJ) than endoscopic stenting (81). Again, the role of prophylactic GJJ is less clear. Early literature suggested an improvement in future GOO symptoms with GJJ (82, 83). As advanced endoscopic techniques and stents have improved, a “wait-and-see” strategy has been developed for both obstructive jaundice and GOO, wherein patients receive SEMS at the time of biliary obstruction and treats only symptomatic GOO (84). The wait-and-see approach was associated with lower morbidity and hospital length of stay and a similar need for reintervention compared to prophylactic surgery of both conditions. Prophylactic GJJ did not prevent future GOO in this study. Patient factors and patient counseling play a vital role in surgical decision-making for these patients.
Minimally Invasive Surgery
Recent advances in minimally invasive surgery have prompted the use of laparoscopic and robotic techniques for pancreatic resections. Compared to many other surgical procedures, any benefit of minimally invasive techniques in pancreatic head resections is subtle. There is a significant learning curve associated with the minimally invasive techniques. For PD, the learning curve is between 40-80 cases (85, 86). Even at high volume centers, this is a time-consuming and challenging endeavor, and outcomes early in the learning curve are worsened (87). In experienced hands, minimally invasive PD appears to have lower EBL, shorter length of stay, similar morbidity/mortality, and similar oncologic outcomes (88–91).
A recent RCT compared minimally invasive techniques to open distal pancreatectomy (OPD). The LEOPARD trial found shorter length of stay, less delayed gastric emptying (DGE), better quality of life scores, and similar morbidity in patients undergoing minimally invasive distal pancreatectomy (MIPD) (92). The DIPLOMA study was a score-matched study of 1,212 patients undergoing MIPD or OPD. Similarly, they found a shorter length of stay, comparable morbidity/mortality, and similar overall survival between the two groups (93). The majority of patients undergoing MIPD are undergoing laparoscopic surgery. There is insufficient evidence to appropriately evaluate robotic vs. laparoscopic distal pancreatectomy at this time.
Vascular Resection and Reconstruction
Patients with PDAC and PV or SMV involvement were previously felt to be unresectable. Recent literature has called this into question. Although patients undergoing PD with PV/SMV resection likely have increased morbidity and mortality (94, 95), two meta-analyses found comparable survival to those that did not require resection (96, 97). At high-volume pancreatic surgery centers, venous resection and reconstruction appears safe and may prolong survival. As these patients are considered locally advanced, patients should undergo neoadjuvant therapy before surgical resection (34).
As patients requiring venous resection/reconstruction are now considered resectable, some have advocated for an “artery-first” approach to PD (98). This technique involves up-front careful dissection of involved arterial planes to determine resectability. A meta-analysis of this approach compared to conventional PD found increased R0 resection rates and survival (99). This study was limited primarily to retrospective cohort studies or case-control studies. A more recent multicenter RCT was found no difference in R0 resection rate, morbidity, or mortality. Further evidence is required to support an artery-first approach.
Arterial resection during PD for PDAC remains highly controversial but is performed for select patients with limited involvement of hepatic arteries at some centers (100–102). Patients requiring arterial resection appear to have poor short and long-term survival (101). Further evidence is needed to determine any survival benefit in patients requiring arterial resection compared to no surgery. For patients undergoing distal pancreatectomy (DP), resection of the celiac axis can be performed when there is no tumor involvement of the celiac artery origin or the confluence of the gastroduodenal artery (GDA) and CHA. A meta-analysis revealed that DP with resection of the celiac axis is associated with similar morbidity/mortality and survival compared to DP (103). This strategy should be reserved for select patients performed at high volume centers.
Lymphadenectomy
The extent of lymphadenectomy for PD has been debated. Standard lymphadenectomy includes lymph node stations 5, 6, 8a, 12b1, 12b2, 12c, 13a, 13b, 14a, 14b, 17a, and 17b (104). Multiple RCTs and meta-analyses have failed to show any survival benefit for patients undergoing extended lymphadenectomy compared to standard lymphadenectomy and may have associated morbidity (105–109). Morbidity included an increase in POPF, DGE, length of stay, and a decrease in quality of life scores (105, 107, 108). Resultantly, current evidence does not support performing an extended lymphadenectomy.
Total Pancreatectomy
Total pancreatectomy is occasionally necessary for more advanced cancers or large central tumors to obtain negative margins. There are obvious clinical implications of removing the entire pancreas, including brittle diabetes and pancreatic exocrine insufficiency. Patients who undergo total pancreatectomy compared to partial pancreatectomy appear to have similar mortality and long-term survival with an increased rate of margin-negative resection (110–112). However, the quality of life after total pancreatectomy is slightly lower than the general population or those undergoing partial pancreatectomy (113, 114). This is driven by diabetes-associated factors as well as diarrhea. Given the mortality and survival in patients undergoing total pancreatectomy, the operation is only warranted when absolutely necessary to obtain negative margins.
Adjuvant Therapy
The ability to complete adjuvant chemotherapy becomes an important consideration after surgery. Completion of adjuvant therapy after R0 resection is associated with improved survival (115, 116). Unfortunately, many patients are unable to complete adjuvant chemotherapy. A study of the SEER database determined that only 7% of Medicare patients completed adjuvant chemotherapy after upfront resection (115). A retrospective study of 309 patients from a single center found 81% initiated adjuvant therapy while 65% completed the recommend course (117). A randomized trial comparing 6 cycles of gemcitabine to observation found that 61% of patients randomized to gemcitabine received all 6 cycles (116). Many patients have delayed postoperative courses due to the morbidity of pancreatic resections. Interestingly, the time to initiation of adjuvant therapy does not affect survival (118–121). Patients with delays in receiving adjuvant treatment should still be considered candidates for therapy.
Adjuvant chemotherapy can vary significantly. Several recent studies have explored the comparative efficacy of standard of care approaches for PDAC. For instance, a recent phase III trial of 493 patients explored the comparative efficacy of a modified FOLFIRINOX regimen to Gemcitabine monotherapy in patients with resected PDAC. After a median follow-up of 33.6 months, modified-FOLFIRINOX led to a median disease-free survival of 21.6 months compared to 12.8 months in with Gemcitabine. However, modified-FOLFIRINOX was associated with a higher incidence of adverse effects, with 75.9% of patients in the modified-FOLFIRINOX group experiencing a Grade 3/4 toxicity compared to 52.9% in the Gemcitabine group (11). The phase III ESPAC-4 trial also explored the combination of adjuvant Gemcitabine plus Capecitabine with Gemcitabine monotherapy in 732 patients with recently resected PDAC. The authors reported a median overall survival of 28 months in patients treated with Gemcitabine plus Capecitabine, and 25.5 months in the Gemcitabine group. Grade 3/4 adverse events were observed in 63% of the Gemcitabine plus Capecitabine arm, and 53.5% of the Gemcitabine arm (122). Hence, additional study is warranted to better determine the optimal treatment in the adjuvant setting.
Pancreatic Cancer Chemotherapy
As mentioned, nearly all intent-to-treat patients will receive multi-agent chemotherapy. This has long been the backbone of pancreatic cancer management. While a variety of small molecule and immune checkpoint inhibitors have shown tremendous efficacy in other cancer types, such approaches generally lack supportive evidence in PDAC and most patients are managed through chemotherapy alone. For all the advances in our understanding of PDAC pathobiology, the chemotherapy agents used in PDAC patients are several decades old, with the most recent breakthroughs consisting of new combinations of existing medications. Here, we describe the discovery and mechanism of action for the most frequently used PDAC medications, as well as any emerging mechanisms of resistance in hopes that these may be used to identify more novel, targeted approaches to improve clinical response rates.
Gemcitabine
Gemcitabine is among the most widely used medications in the management of PDAC. It was first developed in the 1980s by Eli Lilly as an antiviral drug. After showing potent anti-neoplastic effects in vitro, Gemcitabine soon entered various clinical trials in the 1990s. Showing clear efficacy in pancreatic cancer patients, Gemcitabine was first approved for use in PDAC in the United Kingdom in 1995 and then by the FDA in 1996. In 1998, Gemcitabine was approved in combination with Cisplatin for first-line treatment of patients with inoperable non-small cell lung cancer. In 2004, Gemcitabine was next approved for the treatment of metastatic breast cancer following failure of prior anthracycline-containing adjuvant chemotherapy and in 2010, approved in combination with carboplatin for use in select ovarian cancer patients. While Gemcitabine is indeed approved for these and other malignancies, it is most commonly used in combination with Nab-Paclitaxel in advanced PDAC.
Gemcitabine (2’, 2’-difluoro 2’deoxycytidine or dFdC) is a nucleoside analog that is identical in structure to deoxycytidine apart from two fluoride molecules at the 2’ position (Figure 3). Gemcitabine enters cells via several nucleotide transporters, including SLC29A1, SLC28A1, and SLC28A3. However, most appear to be taken into the cell via Human Equilibrative Nucleoside Transporter 1 (hENT1). Once in the cytoplasm, Gemcitabine is phosphorylated by Deoxycytidine Kinase (dCK) to form dFdC monophosphate (dFdCMP), the substrate for two distinct reactions. This initial phosphorylation by dCK is the rate-limiting step in Gemcitabine metabolism. dFdCMP is then either deaminated by Deoxycytidylate Deaminase and converted to dFdUMP, or phosphorylated by Nucleoside Monophosphate Kinase to become dFdC diphosphate (dFdCDP). dFdUMP is a potent inhibitor of the enzyme Thymidylate Synthase. Classically, Thymidylate Synthase catalyzes the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP). dFdUMP competitively inhibits Thymidylate Synthase, thereby causing an imbalance between dUMP and dTMP, impeding DNA synthesis and promoting DNA damage, culminating in programmed cell death (123).
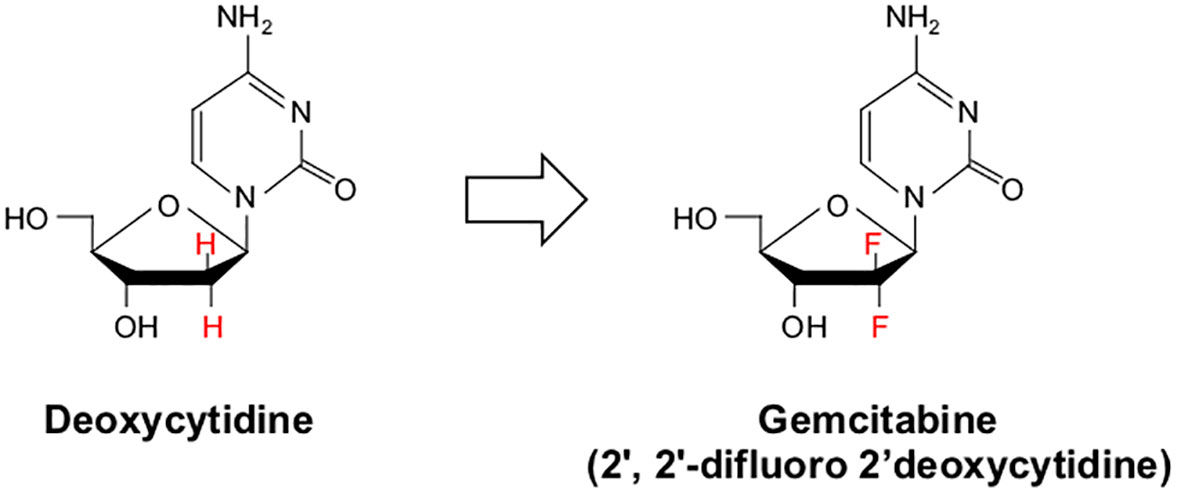
Figure 3 Chemical structure of deoxycytidine and its halogenated chemical mimic Gemcitabine. Gemcitabine (2’, 2’-difluoro 2’deoxycytidine or dFdC) is a nucleoside analog identical in structure to deoxycytidine apart from two fluoride molecules at the 2’-position.
Alternatively, dFdCDP impairs the function of Ribonucleotide Reductase (RNR). RNR is required for DNA synthesis, reducing the 2’-hydroxyl group of the ribose ring of ribonucleoside 5’-diphosphates (ADP, CDP, GDP, and UDP) for de novo synthesis of deoxyribonucleotides. This is critical for DNA synthesis and cell proliferation, and RNR is a predictor of poor outcomes in PDAC patients (124). Additionally, dFdCDP can be further phosphorylated by Nucleoside Monophosphate Kinase to become dFdC triphosphate (dFdCTP). dFdCTP is considered the most important Gemcitabine metabolite and acts by incorporation into genomic DNA. This results in the inhibition of DNA polymerases, preventing DNA synthesis and repair (125). These events are summarized in Figure 4. Gemcitabine is primarily detoxified in the liver. Upon entry into hepatocytes, dFdC is deaminated by Cytidine Deaminase (CDA) to form the inactive 2’-difluoro 2’deoxyuracil (dFdU) metabolite. Following its conversion, dFdU is then eliminated by both renal and biliary excretion (126).
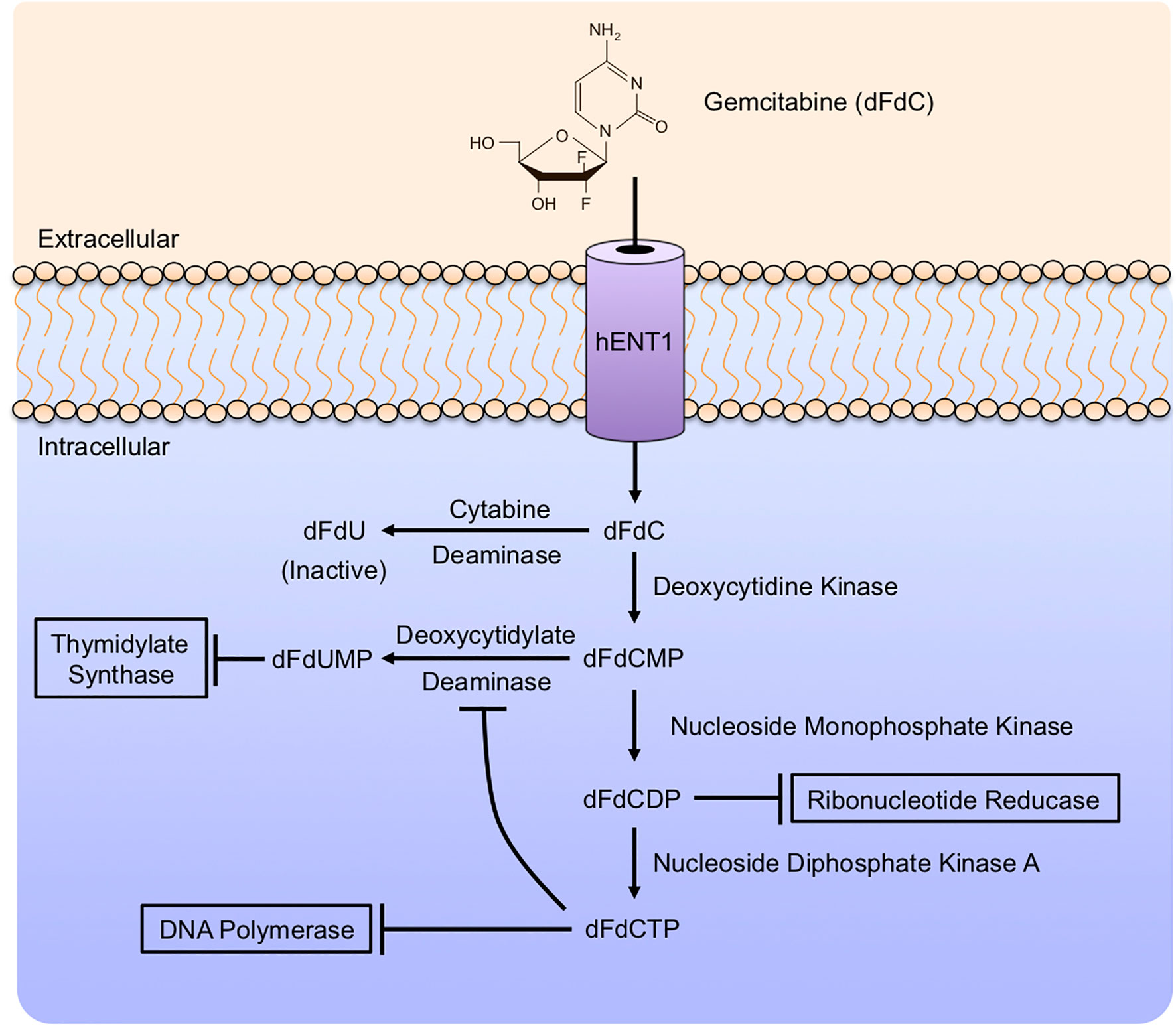
Figure 4 Gemcitabine mechanism of action. Gemcitabine enters cells via several nucleotide transporters, primarily Human Equilibrative Nucleoside Transporter 1 (hENT1). In the cytoplasm, Gemcitabine is modified extensively by a series of enzymatic reactions. Gemcitabine is phosphorylated by Deoxycytidine Kinase (dCK) to form dFdC monophosphate (dFdCMP), the rate-limiting step in Gemcitabine metabolism. Subsequently, dFdCMP can be deaminated by Deoxycytidylate Deaminase to form dFdUMP, a potent inhibitor of Thymidylate Synthase. Alternatively, dFdCMP can be phosphorylated by Nucleoside Monophosphate Kinase to become dFdC diphosphate (dFdCDP), inhibiting Ribonucleotide Reductase. dFdCDP can be further phosphorylated by Nucleoside Monophosphate Kinase A to form dFdCTP, which inhibits DNA polymerases. As an alternative to these activating modifications, Gemcitabine can be deaminated by Deoxycytidylate Deaminase to form dFdU, an inactive metabolite with no known anti-neoplastic effects.
Gemcitabine Resistance
Gemcitabine has been a standard-of-care agent in the management of advanced PDAC for decades. While Gemcitabine offers a survival benefit both as a monotherapy and in combination with other medications, these responses are seldom complete, and nearly all tumors display or will develop some degree of Gemcitabine resistance, with only 4% of patients surviving two years on Gemcitabine alone (10). Though Gemcitabine resistance is well documented clinically, the cellular and molecular mechanisms that underlie Gemcitabine resistant phenotypes remain unclear. Several studies have attempted to predict for Gemcitabine responses, though there are no such biomarkers clinically used at this time.
As discussed, the main method of Gemcitabine uptake is via the hENT1 transport protein. Accordingly, hENT1 expression has been suggested to predict for response to Gemcitabine therapy in the adjuvant setting. A retrospective meta-analysis compiled 10 studies and determined that hENT1 positively associated with improved overall survival with a hazard ratio of 0.52 in patients treated with Gemcitabine (127). The majority of trials evaluated in meta-analysis used the 10D7G2 mouse monoclonal antibody to determine hENT1 status by immunohistochemistry. Unfortunately, this antibody has no commercially available source for larger trials. To this end, a similar SP120 rabbit monoclonal anti-hENT1 antibody was generated to prospectively predict Gemcitabine responsiveness in metastatic disease. However, positive staining with SP120 showed no correlation between hENT1 expression and response to Gemcitabine. Additionally, the 11337-1-AP antibody also had a low success rate, suggesting that the predictive value of hENT1 is highly antibody-dependent (128).
While incomplete Gemcitabine uptake via loss of hENT1 may partially explain Gemcitabine resistance, emerging evidence suggests that there are several other factors that may contribute to drug responses. For instance, while the Gemcitabine inactivating enzyme CDA is mainly expressed in the liver, it is also expressed in various tissues, including the bone marrow, prostate, pancreas, and spleen (129). Genomic sequencing further suggests that pharmacogenomic variation in CDA may contribute to the therapeutic efficacy of Gemcitabine, though this requires further study and is not currently used to predict Gemcitabine responses in the clinic (129). Additionally, in some patients, Gemcitabine resistance appears to be mediated by efflux pumps, namely the ABC transporter family proteins ABCB1/MDR1, ABCC1/MRP1, and ABCG2/BCRP (130).
Other genomic alterations affecting Gemcitabine transport and metabolism have also been identified, provided further insight into Gemcitabine resistant phenotypes. For instance, Cytosolic 5’-nucleotidase 1A (NT5C1A) is a phosphatase that targets non-cyclic nucleoside monophosphates to produce nucleosides and inorganic phosphates. NT5C1A is robustly expressed in tumor cells of resected PDAC patients. Interestingly, NT5C1A promotes Gemcitabine resistance by decreasing the amount of intracellular dFdCTP and limits therapeutic responses to Gemcitabine in mice (131). Additionally, expression of the tumor suppressor Hepatocyte nuclear factor 1α (HNF1A) seems to associate with Gemcitabine sensitivity in PDAC cells, regulating the expression of ABCB1 and ABCC1. The authors further demonstrated that HNF1A regulates ABCB1 gene expression by binding its promoter region and suppressing its transcription, suggesting that HNF1A warrants exploration as a potential biomarker for Gemcitabine responses in the clinic.
Beyond alterations to nucleoside metabolism enzymes and drug transport, myriad signaling events have also been linked to Gemcitabine-resistant phenotypes. These include several cell survival pathways, namely AKT (132, 133), ERK/MAPK (134–136), HIF1α (137), GLI and SOX2 (138), NFκB (132, 139, 140), Sonic Hedgehog (141), and WNT (142, 143). Additionally, several recent preclinical studies have identified select microRNAs that may also contribute to Gemcitabine-resistance. For example, miR-21 is upregulated in Gemcitabine-refractory PDAC and is believed to contribute to drug-resistant phenotypes through a variety of mechanisms (144). Conversely, members of the miR-200 family are generally repressed in Gemcitabine-resistant cells and, when restored, promotes the reversion of post-EMT tumor cells to a more epithelial state (145, 146). Interestingly, inactivation of the Hippo tumor suppressor gene has been suggested to regulate cell-density-dependent miRNA suppression in cancer via de-repression of YAP (147). However, additional evidence suggests that YAP enhances the actions of Gemcitabine, largely by down-regulating multi-drug transporters. Similarly, cell lines with genetic ablation of Hippo signaling had increased sensitivity to Gemcitabine (148). These data suggest that the contributions of Hippo-mediated microRNA biogenesis to Gemcitabine resistance are complex and highly varied and warrant further study. Several additional mechanisms of resistance have also been suggested, including aberrations to the epigenome. For instance, one study determined that while Gemcitabine-resistant PDAC cell lines acquire an invasive phenotype and associated upregulation of CDA, these cell lines also displayed collateral hypersensitivity to histone deacetylase (HDAC) inhibitors and decreased expression of heterochromatin marks H4K20me3, H3K9me3, and H3K27me3 (149).
Recently, a group of investigators have conducted genome-wide RNA profiling of primary PDAC cell cultures and patient-derived xenografts, and related them to Gemcitabine sensitivity. Using this approach, the authors identified a unique gene expression signature associated with Gemcitabine sensitivity, which they designated GemPred. They then tested the GemPred RNA signature first in a monocentric cohort of 67 patients, then in a multicentric cohort of 368 patients. In both, GemPred+ patients who received Gemcitabine had significantly longer overall survival than those who were GemPred-. Additionally, GemPred stratification was not associated with a survival benefit in patients not receiving Gemcitabine. The authors therefore concluded that GemPred stratification predicts the benefit of adjuvant Gemcitabine in PDAC, and this approach warrants continued exploration in prospective studies (150).
Emerging evidence also suggests that the glycan biomarker sialylated tumor-related antigen (sTRA) may have utility in predicting responses to Gemcitabine, as well as several other chemotherapeutics used in PDAC. For instance, sTRA-expressing cell lines were associated with increased resistance to seven different chemotherapeutics used in PDAC, and patients with primary tumors were positive for a gene expression classifier for sTRA demonstrated no statistically significant benefit from adjuvant chemotherapy. Similarly, plasma levels of sTRA identified the PDACs that showed rapid relapse following neoadjuvant chemotherapy. Hence, pending further evaluation, sTRA may be a useful means of predicting for drug responses in PDAC patents (151).
While the above events have been well characterized, it has also been suggested that the tumor microenvironment (TME) may have many important roles in drug responses (152). PDAC is associated with a dense, reactive tumor stroma comprised of activated stellate cells, cancer-associated fibroblasts, and leukocytes (153, 154), and tumors from Gemcitabine-resistant patients appear to generally be enriched in stromal pathways (155). In addition to its role in producing a variety of growth factors and cytokines, such as hepatocyte growth factor (HGF) and transforming growth factor β (TGFβ) (156, 157), the PDAC stroma is known to impede drug delivery through several mechanisms, namely by exerting considerable mechanical force on the intratumoral vasculature and reducing patency (158). Both functions have been suggested as potential drug targets to enhance the effects of Gemcitabine.
For instance, TP53 is mutated in 75% of PDAC tumors and is implicated in a number of hallmark features of tumorigenesis (159). In mice, loss-of-function TP53 mutations were associated with hyper-activation of the JAK2/STAT3 signaling cascade, leading to extensive stromal remodeling and Gemcitabine-resistant phenotypes (160). Similarly, STAT3 activation associated with poor survival in human PDAC and inhibition of this pathway reduced the tumor stroma and sensitized tumor-bearing mice to Gemcitabine (160). Additionally, Stromal Cell-Derived Factor 1 (SDF-1 or CXCL12) and its receptor CXCR4 may also have a potential role in Gemcitabine resistance. Stromal-derived SDF-1 appears to bind CXCR4 on the surface of the tumor epithelium, thereby activating AKT, ERK, and IL6-mediated survival pathways (161). Several other studies have suggested a role for such paracrine signaling events in Gemcitabine resistance, namely as blocking stromal protein synthesis via mTOR/4E-BP1 inhibition led to a reduction in tumor IL6, as well as the loss of drug-resistant phenotypes (162).
Additional evidence suggests that the desmoplastic component of the tumor stroma may also contribute to Gemcitabine resistance. In in vitro systems, tumor cells grown in 3D collagen culture are able to override Gemcitabine-induced cell cycle arrest through MMP-14-induced expression of HMGA2 (163). However, while the biologic contribution of the stroma to Gemcitabine resistance is well established, the clinical utility of stromal depletion remains unclear. For example, catabolism of the tumor stroma induced by the Hedgehog inhibitor IPI-926 led to transient increases in intratumoral vascular density and improved the uptake of Gemcitabine (141). However, additional evidence suggests that tumors with genetic deletion of Hedgehog signals are highly aggressive, with poor differentiation and increased vascularity. These events suggest that the cancer stroma may restrain tumor growth, in part by impeding tumor angiogenesis (164). This is supported by clinical data, as the Sonic Hedgehog antagonist Vismodegib failed to improve either Gemcitabine response rates or survival in patients with metastatic PDAC (165).
While most research efforts have focused on pancreatic cancer-associated fibroblasts and stellate cells, emerging evidence appears to suggest that a variety of additional cell types within the tumor microenvironment may also contribute to Gemcitabine resistance. While PDAC is generally non-immunogenic, tumors are associated with robust macrophage infiltration. Tumor-educated macrophages appear to release several pyrimidine species, including deoxycytidine, which appear to inhibit the uptake and action of Gemcitabine in tumor cells. Similarly, genetic or pharmacologic depletion of macrophages sensitized tumor-bearing mice to Gemcitabine, further substantiating a potential role for the immune infiltrate in Gemcitabine resistance (166).
Interestingly, a recent study determined that select species of proteobacter expressing CDA internalize and metabolize Gemcitabine to its inactive form (167). These bacterial species were found in a significant fraction of tumor specimens, particularly those with intervention of the main pancreatic duct. As these are commensal duodenal flora, it is likely that the manipulation of the main pancreatic duct via the duodenum introduces CDA-expressing bacteria to the pancreas, potentially limiting the efficacy of Gemcitabine. Both in vitro and in vivo studies suggest that the addition of select antibiotics may augment the tumoricidal action of Gemcitabine in infected tumor cells, though this has yet to be explored in large scale, randomized testing (167).
Strategies to Overcome Gemcitabine Resistance
For patients receiving Gemcitabine-based chemotherapy, the eventual development of drug-resistant phenotypes is a significant issue. While several of the mechanisms described above have been proposed as potential targets for therapy, far fewer have been studied in combination with Gemcitabine to overcome that resistance in clinical trials. For example, PDAC tumors frequently overexpress receptor tyrosine kinases such as Epidermal Growth Factor Receptor (EGFR) that activate downstream PI3K/AKT signaling to suppress Gemcitabine-induced apoptosis (168). The combination of Gemcitabine and the selective EGFR inhibitor Erlotinib was evaluated in a phase III randomized controlled trial, improving median overall survival by less than two weeks, leading to its approval by the FDA (169). A recent phase II trial showed that the addition of the EGFR-neutralizing antibody panitumumab (formerly ABX-EGF) to Gemcitabine and Erlotinib nearly doubled overall survival time when compared to Gemcitabine and Erlotinib alone (8.3 v 4.2 months), though this was associated with a higher incidence of grade 3 non-hematologic toxicities (82.6 vs. 52.2%) and no patient had a complete response (170).
Several other targeted therapies with sound basic supporting data have been combined with Gemcitabine, though none have shown a significant benefit beyond the marginal improvement seen with Erlotinib. Interestingly, despite sharing a similar mechanism of action to Erlotinib, the EGFR inhibitor Cetuximab failed to significantly improve survival (171). Additionally, as discussed previously, the Smoothened inhibitor Vismodegib also failed to improve outcomes when added to Gemcitabine in a clinical trial (165).
Vascular Endothelial Growth Factor (VEGF) overexpression is commonly seen in malignancies and is associated with a poor prognosis. Pancreatic cancer is generally avascular compared to other cancer types, though studies have shown a correlation with blood vessel density, tumor VEGF-A, and disease progression (172). Several studies have evaluated targeting VEGF receptors in combination with chemotherapy, including Bevacizumab and Axitinib, though neither combination showed benefit in a phase III trial despite promising phase II data (173, 174).
Additionally, high intratumoral expression of Hyaluronan (HA) in the pancreatic tumor microenvironment significantly impairs perfusion, thereby limiting drug delivery. In murine PDAC, Pegvorhyaluronidase Alfa (PEGPH20) degrades HA in vivo, increasing drug delivery. While a phase II trial combining Gemcitabine and PEGPH20 improved progression-free survival in patients with high HA tumor expression, there was a significant increase in thromboembolic events resulting in a clinical hold in the study and re-initiation with prophylactic Enoxaparin. The phase III trial was discontinued due to failure to improve overall survival (175). Interestingly, expression of Connective Tissue Growth Factor (CTGF) also correlates with fibrosis in PDAC. In preclinical models, the anti-CTGF antibody Pamrevlumab impaired fibrous tissue adhesion. Pamrevlumab was combined with Gemcitabine and Nab-Paclitaxel in a phase I/II study of locally advanced disease, which increased the rate of surgical resection in the study arm and improved survival irrespective of whether patients were resected. Based on these findings, the drug was granted Fast Track Designation and is currently enrolling in a phase III trial in advanced PDAC (176).
Additional studies have attempted to circumvent Gemcitabine resistance via the addition of another cytotoxic agent. As discussed, Gemcitabine is now used first-line in combination with Nab-Paclitaxel, which extended 2-year-survival from 4 to 9% in a clinical trial (10). The combination of Gemcitabine and Cisplatin is currently approved for patients with advanced PDAC and underlying BRCA mutations due to the excellent responses to platinum-based alkylating agents in tumors with deficient DNA repair mechanisms (177). This concept was expanded to include unknown DNA repair deficiencies that are likely present in all PDAC patients, and Cisplatin was added to the combination Gemcitabine and Nab-Paclitaxel in a phase I/II trial. Although the sample size was small (N=25), this combination was associated with significant improvement in response rates, including two patients with complete responses and a median overall survival of nearly double that of Gemcitabine and Nab-Paclitaxel alone. This multi-drug combination is now being investigated in other stages of the disease (178). However, BRCA mutations have long been suggested as a biomarker for sensitivity to PARP inhibitors such as Olaparib. This approach is based on the long-standing hypothesis that PARP inhibition will impair the repair of single-stranded breaks, causing synthetic lethality in tumors with loss of high-fidelity double-strand break homologous recombination (179). As Olaparib is now showing clear efficacy as maintenance therapy in BRCA-mutated PDAC (17), the addition of Olaparib to Gemcitabine and Cisplatin should be considered.
In addition to the above approaches, a European study has explored the combination of adjuvant Gemcitabine and Capecitabine. This combination extended median overall survival to 28 compared to 25.5 in the Gemcitabine group (122). However, earlier reports have not identified a significant difference between Gemcitabine with Capecitabine and Gemcitabine monotherapy. Hence, Capecitabine warrants continued exploration as a Gemcitabine adjuvant (180).
Emerging preclinical evidence suggests that long-term administration of Gemcitabine leads to extensive reprogramming of the pancreatic tumor microenvironment, enhancing a number of immune cell processes, including antigen presentation, PD-L1 expression, but promoting TGFβ biosynthesis which can act to suppress cancer-directed immune mechanisms. The combination of Gemcitabine, PD-1 inhibition, and TGFβ signal inhibition was highly effective in controlling disease in transgenic models of PDAC, far surpassing the efficacy of concomitant PD-1 and TGFβ signal inhibition without Gemcitabine (181, 182). Additionally, a recent preclinical study has identified signaling by the hepatocyte growth factor receptor MET as a potential driver of Gemcitabine resistance in PDAC. In this work, the authors show that MET is highly expressed at the plasma membrane of pancreatic cancer cells, and that TR1801-ADC, a novel antibody drug conjugate composed of a MET antibody conjugated to the highly potent pyrrolobenzodiazepine toxin-linker Tesirin was highly effective in MET-overexpressing patient derived xenografts, and synergized with Gemcitabine, even in tumors previously demonstrated to be resistant to Gemcitabine (183). Hence, these and other combination strategies warrant continued exploration in the treatment of Gemcitabine-refractory PDAC.
Paclitaxel/Nab-Paclitaxel
As part of a USDA initiative to identify natural compounds with therapeutic potential, researchers determined that Pacific yew tree bark extract had significant anti-neoplastic effects in vitro. Over the next five years, this sample was fractionated, and the most active component was identified. This compound was named Taxol and soon entered large scale biological testing. Taxol was then transferred from the USDA to Bristol-Myers Squibb for commercial development under the generic name Paclitaxel (Figure 5). Paclitaxel soon entered clinical evaluation in combination with Cisplatin, which was approved for use in ovarian cancer patients in 1992. Paclitaxel was next explored as monotherapy in breast cancer and was approved in patients with axillary node involvement in 1994. Paclitaxel was approved as a second-line treatment of AIDS-related Kaposi’s sarcoma in 1997 and in combination with Cisplatin for select cases of non-small cell lung cancer in 2006. Paclitaxel is also used off label to treat several other cancers, including those of the esophagus, stomach, endometrium, cervix, prostate, head and neck, as well as sarcomas, leukemias, and lymphomas (184).
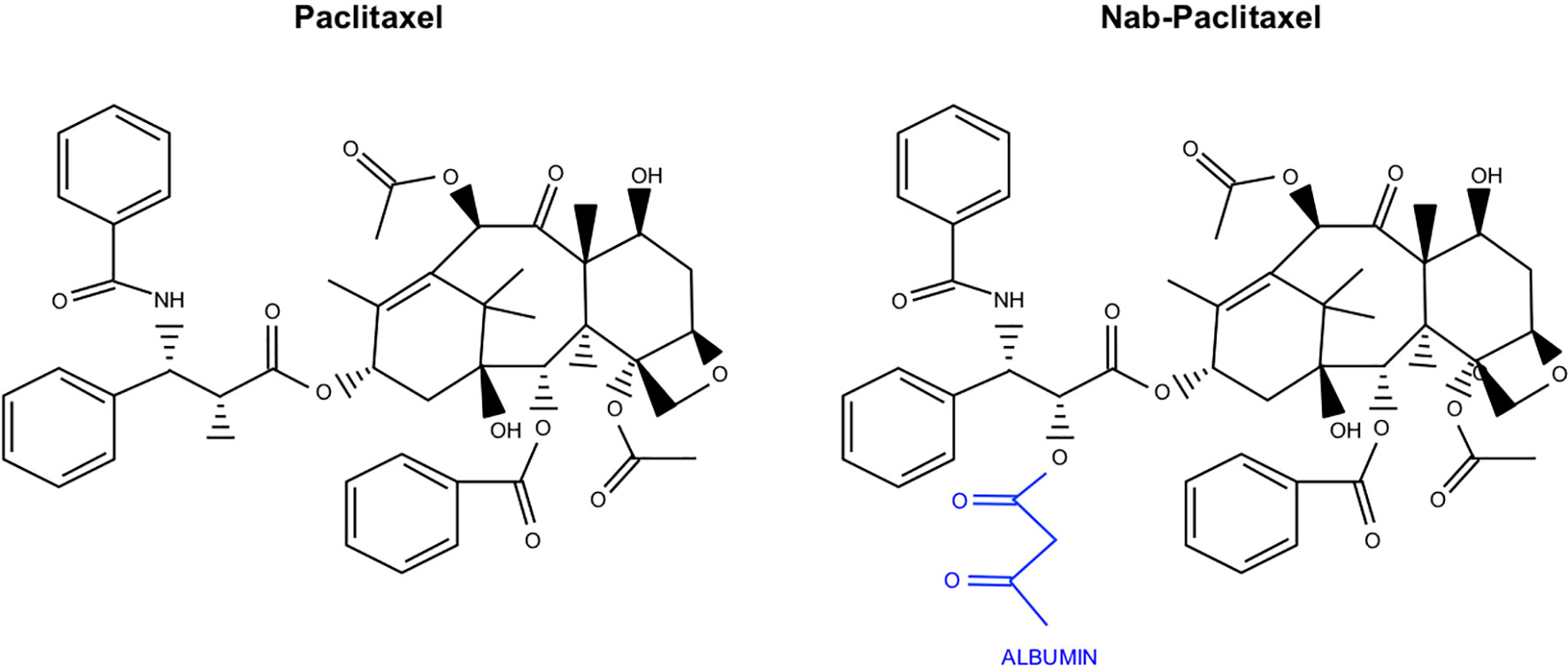
Figure 5 Chemical structure of Paclitaxel and the albumin conjugated variant Nab-Paclitaxel. Nab-Paclitaxel, used in combination with Gemcitabine in PDAC, varies from Paclitaxel in that it is conjugated to albumin as a delivery vehicle.
While the initial formulation of Paclitaxel is still in clinical use, researchers soon identified that many of its adverse effects are caused by its solvent (185). This led to the conjugation of Paclitaxel to albumin (Nab-Paclitaxel), decreasing hydrophobicity and improving bioavailability (186) (Figure 5). This new formulation was approved for metastatic or refractory breast cancer in 2005, for non-small cell lung cancer in 2012, and for advanced pancreatic cancer in combination with Gemcitabine in 2013 (186–188). In pancreatic cancer, Nab-Paclitaxel is used exclusively. Compared to unconjugated Paclitaxel, Nab-Paclitaxel has increased solubility without many of the solvent associated toxicities. The albumin conjugate may have additional roles in facilitating transcytosis, namely by associating with the gp60 cell surface receptor, resulting in Caveolin-1 mediated invagination (189). Pharmacokinetic studies suggest that Nab-Paclitaxel has an improved volume of distribution and more rapid clearance than unconjugated Paclitaxel (190). Additionally, tumors generally have increased concentrations of Nab-Paclitaxel when compared with unconjugated Paclitaxel, suggesting the solvents used to deliver Paclitaxel may sequester the drug in micelles and inhibit its transport (191). Interestingly, the combination of Cremophor EL and dehydrated ethanol has been shown to inhibit the binding of Paclitaxel to albumin, suggesting that these solvents may hinder albumin-associated transport of Paclitaxel to the tumor microenvironment (190).
Regardless of entry, once inside the target cells, Paclitaxel appears to exert its antineoplastic effects in a concentration-dependent manner. In vitro experiments suggest that at low concentrations, Paclitaxel inhibits the formation of mitotic spindles without affecting the function of preformed spindles or arresting cells in mitosis (192). After prolonged treatment with low nanomolar concentrations, intracellular Paclitaxel will bind β-tubulin, thereby stabilizing the mitotic spindle. This prevents the segregation of chromatids, resulting in cells with multiple micronuclei that form around clusters of chromosomes (192). These cells are not arrested in mitosis but continue to S-phase where they undergo cytokinesis due to this extra DNA content (192). At higher concentrations, Paclitaxel associates with microtubules at the taxane-binding site. This induces their polymerization into stable bundles that are resistant to de-polymerization, arresting the cells in mitosis (193). While both of these mechanisms are cytotoxic, the mechanism observed at lower concentrations is more likely the main cause of Paclitaxel-induced cell death in human tumors, as these concentrations are more akin to those used in the clinic.
An added advantage of Nab-Paclitaxel in PDAC therapy may be due to its uptake by macrophages through a process called macropinocytosis, leading to activation of Toll-like receptor 4 (TLR4) pathways and M1 polarization of the macrophages that promotes their immunostimulatory potential (194). Importantly, in an in vivo orthotopic model using KPC pancreatic cancer cells in syngeneic C57BL/6 mice, Nab-Paclitaxel alone or in combination with Gemcitabine induced an increase in M1 macrophages within the orthotopic tumors (194).
Paclitaxel Resistance
To date, there have been no mechanistic studies evaluating Nab-Paclitaxel resistance in PDAC, and clinical trial data is similarly limited. However, as Paclitaxel has been used for decades in the management of other malignancies, there are several studies in these tumor types that may offer insight into clinically useful strategies to augment the effects of Nab-Paclitaxel in PDAC. For instance, Multidrug Resistance Protein 1 (also known as MDR1, p-glycoprotein, ABCB1, or CD243) is a transmembrane ATP-dependent efflux pump. In the intestinal mucosa, MDR1 limits the oral bioavailability of Paclitaxel by promoting its export from epithelial cells (195). Several cancers, including PDAC, appear to have frequent overexpression of MDR1 or closely related genes, e.g., ABCC1/Multidrug resistance-associated protein 1 (MRP1), and their utility as predictive biomarkers warrants clinical investigation (195, 196).
Beyond its export, there are several potential mechanisms of Paclitaxel/Nab-Paclitaxel resistance that are directly related to the microtubules (197). Many Paclitaxel resistant cells possess alterations or mutations to β-Tubulin family members, which decreases the ability of the drug to stabilize microtubules (197–199). While this has yet to be thoroughly investigated in PDAC, a recent study suggests that βIII-Tubulin is strongly expressed in pancreatic cancer tissues and has important roles in tumor cell growth and survival (200). Further, silencing βIII-Tubulin enhanced the action of both Gemcitabine and Paclitaxel in PDAC cells (200). However, in ovarian cancer βII and βIII-Tubulin mutations do not appear to have a role in determining Paclitaxel sensitivity (201). Interestingly, βIVb-Tubulin inhibition appeared to sensitize PDAC Cells to the vinca alkaloids Vincristine, Vinorelbine, and Vinblastine, which also target the microtubules (202). In lung cancer cells, ERK-mediated upregulation of βIVb-Tubulin appears to promote Paclitaxel resistance (203). As there is a preclinical inhibitor (VERU-111) of βIII/βIV-tubulin that has already shown efficacy in PDAC xenografts (204), the addition of VERU-111 to Gemcitabine and Nab-Paclitaxel warrants consideration.
Several additional signaling events and cell processes have been linked to Paclitaxel resistance, particularly in breast cancer. In MCF7 cells, Paclitaxel resistance is associated with a function shift away from apoptosis and towards autophagic cell death (205). A similar study using the same cell line determined that overexpression of Bcl-XL inhibits the intrinsic pathway of programmed cell death, contributing to Paclitaxel resistant phenotypes (206). Interestingly, HER2/neu overexpressing breast cancer cells appear to have diminished responses to Paclitaxel (207). While HER2 is overexpressed in a small subset of PDAC patients (208), the HER2-inhibiting antibody Trastuzumab failed to improve outcomes in HER2 overexpressing PDAC patients (209). Hence, while HER2 may be a poor target for therapy, HER2 may be a useful biomarker in determining Nab-Paclitaxel sensitivity.
Beyond the well-characterized role of aberrant apoptotic and autophagy-associated signals (210–214), several other cellular processes also appear to contribute to Paclitaxel responses. These include hypoxia, which induces mTOR-mediated autophagy and concurrent JNK-mediated pro-survival signaling (215). Additionally, overexpression of the centrosomal ninein-like protein (NLP) also appears to impede Paclitaxel-induced cell death (215). Finally, there is a growing body of evidence suggesting that miRNA dysregulation may also have important roles in Paclitaxel responses. Overexpression of miR100 sensitized MCF7 cells to Paclitaxel-induced cell death in part by targeting mTOR (216). Similarly, overexpression of miR-16 and miR-17 synergized to reduce Bcl2 and autophagy, respectively, sensitizing tumor cells to Paclitaxel in vitro (217). These mechanisms are largely uncharacterized in PDAC and should be carefully explored to identify those most pertinent to therapy.
5-Fluorouracil
Initially patented in 1956, 5-FU has been a mainstay in cancer treatment since its FDA approval in 1962. 5-FU is currently used in several cancers, including breast, colon, esophagus, stomach, and pancreatic cancer, as part of the multidrug regimen FOLFIRINOX. In 1957, the Heidelberger group at the University of Wisconsin reported that rat hepatoma cells displayed increased uptake of Uracil when compared to normal cells (218). Based on these observations, they synthesized a modified Uracil with a fluoride substitution at the 5-position, fluoropyrimidine 5-fluorouracil or 5-FU (218) (Figure 6). This compound was wildly successful, showing anti-neoplastic activity against various cancer cells, and was immediately introduced to the clinic before obtaining a patent.
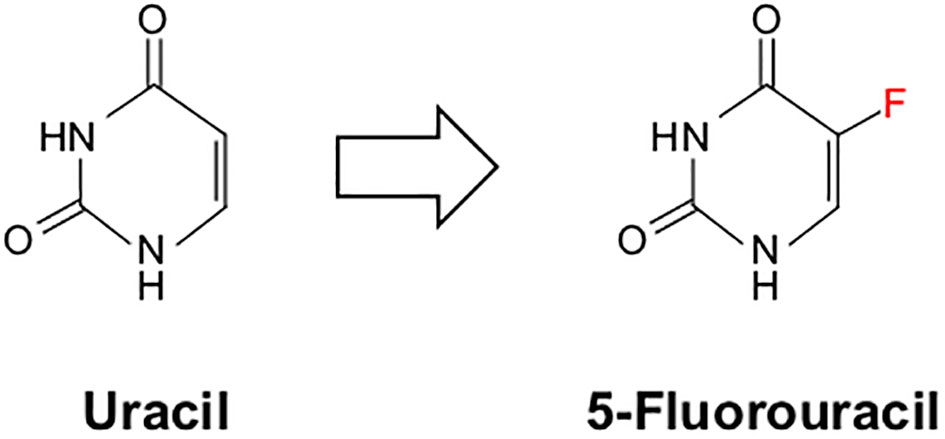
Figure 6 Chemical structure of uracil and its chemical mimic 5-Fluorouracil. 5-Fluorouracil (5-FU) is a nucleoside analog identical in structure to uracil apart from a single fluoride molecule at the 5’-position.
In their 1958 trial involving 103 cases of solid and hematologic malignancies, this group observed an approximate 25% response rate, though regression was only seen in those with severe toxicity (219). These observations, combined with the lack of 5-FU resistance seen in this cohort, eventually led to the addition of 5-FU to cyclophosphamide, methotrexate, and prednisone in 1972 to form the CMFP combination regiment for metastatic breast cancer. This combined approach had a complete remission rate of 20% and a partial remission rate of 40%. By omitting prednisone to create the CMF regimen still used today, physicians observed response of 57%. These data led to several additional studies centered on 5-FU, which would eventually be approved for the many other cancer types described, including pancreatic cancer, as part of the FOLFIRINOX regimen. As discussed, this approach showed improved efficacy when compared to single agent Gemcitabine in metastatic PDAC (220), and as a result, FOLFIRINOX was soon approved and is now a first-line treatment.
5-FU appears to have a variety of effects in the target cells, many of which are not entirely clear. 5-FU is taken into cells in the same manner as Uracil, both sharing a saturable, facilitated transport mechanism (221). Similar to Gemcitabine, intracellular 5-FU interacts with a variety of modifying enzymes, converting it to several active metabolites. These include Fluorodeoxyuridine monophosphate (FdUMP), Fluorodeoxyuridine triphosphate (FdUTP), and Fluorouridine triphosphate (FUTP), many of which are responsible for the anti-neoplastic effects of 5-FU, mainly via the inhibition of Thymidylate Synthase (Figure 7).
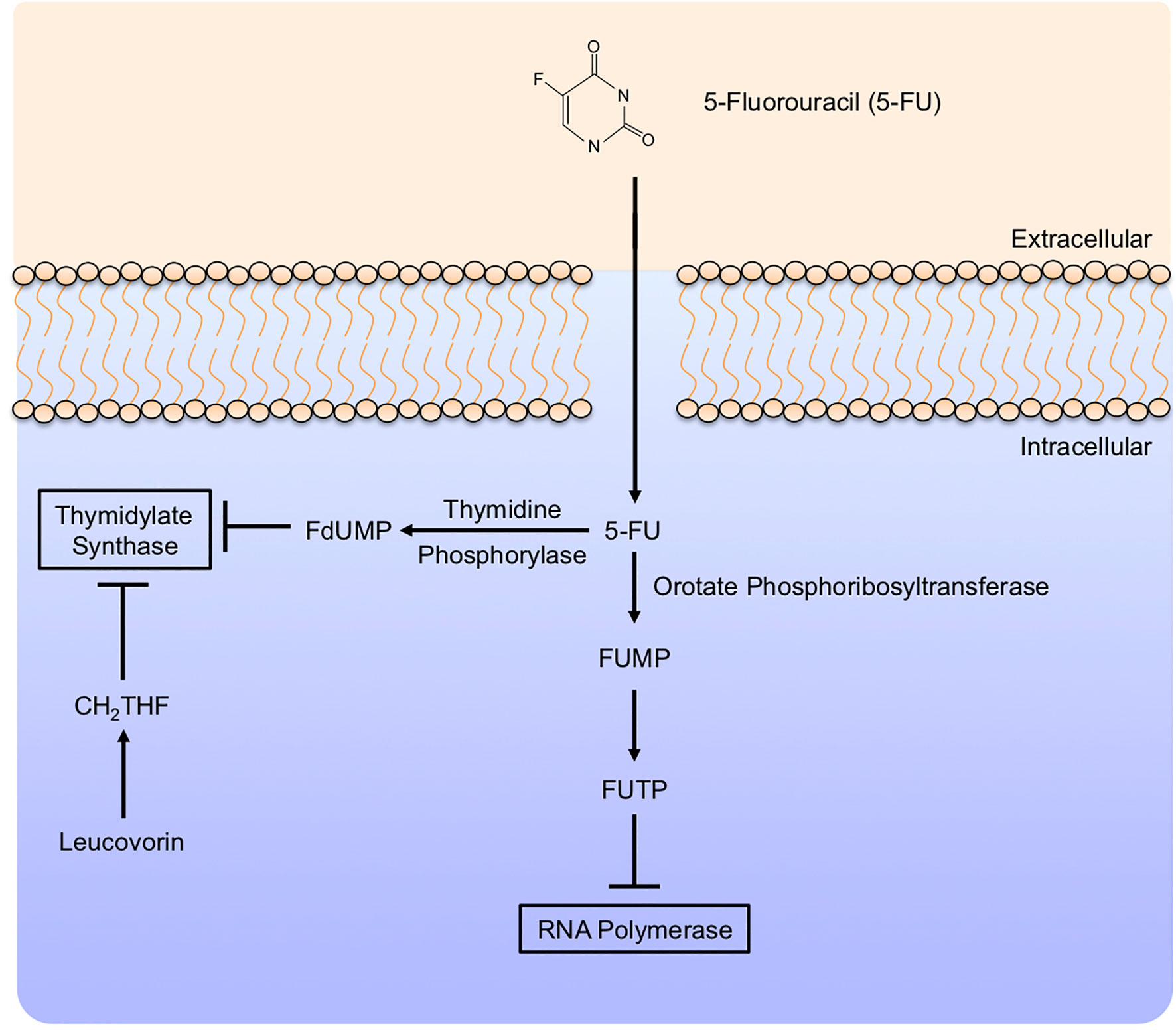
Figure 7 5-Fluorouracil mechanism of action. 5-Fluorouracil (5-FU) enters target cells in a similar manner to uracil, mainly by facilitated transport. Like Gemcitabine, 5-Fluorouracil (5-FU) is modified extensively in the cytoplasm. This includes conversion to Fluorodeoxyuridine monophosphate (FdUMP) by Thymidine Phosphorylase, inhibiting Thymidylate Synthase and potentiated by the addition of Leucovorin. In a parallel mechanism, 5-FU can be further modified to Fluorodeoxyuridine triphosphate, which subsequently inhibits RNA Polymerase.
5-FU may have tumoricidal effects extending beyond Thymidylate Synthase inhibition, though these are less clear. 5-FU appears to have additional effects on transcription, namely via incorporating its metabolite FUTP into RNA (222). The incorporation of 5-FU metabolites into RNA is itself cytotoxic, suppressing the growth of both breast (223) and colon (224) cancer cells. 5-FU also appears to interfere with the maturation processing of both mRNA and tRNAs through various alternate mechanisms, all of which appear to further dysregulate critical cell processes and reduce viability (222).
5-FU Resistance
Despite being used in cancer treatment for nearly 60 years, there is little known regarding 5-FU resistance, particularly in PDAC. While some studies suggest that PDAC cells acquire a means to resist 5-FU through altered expression of select apoptotic regulators, or aberrant cell processes, including autophagy, none has translated into a successful adjuvant therapy for pancreatic cancer patients (225, 226). The majority of clinical studies exploring 5-FU have been conducted in colon cancer. As single-agent response rates are low (227), investigators have long been exploring combination strategies to enhance the efficacy of 5-FU based chemotherapy. The addition of other anti-cancer agents to 5-FU has helped to achieve response rates as high as 80% (228), and this approach is now the standard-of-care in several tumor types. However, these approaches often predispose patients to additive toxicities. Therefore, like the other drugs described in this review, there is a need to better understand the mechanisms through which tumor cells resist 5-FU in hopes that these events can be reversed, and drug responses can be achieved without the use of higher doses or additional chemotherapies.
As mentioned, no studies have identified a precise, clinically actionable mechanism of 5-FU resistance in PDAC. However, several studies in other cancers have suggested that several aberrant cell processes can limit the anti-tumor efficacy of 5-FU in other cancers (229). For instance, in a study of 13 colon cancer cell lines, though there was no predictive mechanism with respect to drug transport, accumulation, or Thymidylate Synthase kinetics, increased expression of Thymidylate Synthase was strongly associated with reduced 5-FU sensitivity (230). Accordingly, the induction of Thymidylate Synthase has similarly been shown to impede 5-FU responses in vitro, and in patients, Thymidylate Synthase expression was directly related to the efficacy of 5-FU (230). Hence, Thymidylate Synthase inhibition has long been suggested as a potential means of overcoming 5-FU resistance (230–232).
Several additional mechanisms of 5-FU resistance have also been suggested and have been reviewed extensively (229). Notably, cells with diminished P53 expression had increased 5-FU sensitivity, suggestive of a role for defective DNA repair processes in mediating drug responses (230). Should this be true of PDAC tumors, this may be of some clinical utility given the high frequency of TP53 mutations. Other recent evidence appears to implicate WNT signaling in 5-FU resistance, namely via suppressing the checkpoint kinase 1 (CHK1) pathway, particularly in P53 wild-type cells (233). Hence, these and other mechanisms must be exhaustively characterized in PDAC to further enhance the therapeutic efficacy of the FOLFIRINOX regimen.
Irinotecan
Like Paclitaxel, the commercial development of Irinotecan can be traced back to a screen of natural products (234). In 1966, investigators isolated Camptothecin from the bark/stem of the Camptotheca acuminate or Happy tree. This tree is indigenous to China, and its extract had long been used in traditional Chinese medicine as an anti-cancer agent (234). This compound was found to have significant anti-tumor activity but was associated with severe toxicity in preclinical testing (235). In an effort to develop a similar compound with a more favorable adverse effect profile, the water-soluble Camptothecin analog Irinotecan was synthesized. While Irinotecan demonstrated little anti-tumor activity in vitro, its metabolite 7-Ethyl-10-Hydroxycamptothecin (SN-38) displayed up to a 2000-fold increase in potency (236, 237). Irinotecan soon entered clinical trial and was approved by the FDA as a second-line monotherapy for metastatic colorectal cancer in 1996 (238). In a subsequent trial, the addition of Irinotecan to 5-FU/Leucovorin resulted in twice the response rate compared to 5-FU/Leucovorin monotherapy, resulting in the approval of Irinotecan as first-line therapy in 1998. Irinotecan was approved for metastatic PDAC in 2013, where it is currently used as part of the multidrug regimen FOLFIRINOX (239).
Like Camptothecin, Irinotecan functions as a topoisomerase inhibitor that functions by primarily impeding DNA synthesis during S-phase. Structurally, the two are quite similar, containing a pentacyclic ring structure with a pyrrole (3, 4 β) quinoline moiety, an S-configured lactone form, and a carboxylate form. However, Irinotecan has an additional ethyl group, and a bipiperidinocarbonyloxy group in place of two hydrogen molecules, improving water solubility and reducing many of the severe toxicities observed in response to Camptothecin. In the intestinal mucosa, liver, and plasma, Irinotecan undergoes extensive modification, namely carboxylesterase-mediated cleavage of the carbamate bond between the camptothecin moiety and the dipiperidino side chain. This converts water-soluble Irinotecan to lipid-soluble SN-38 (Figure 8). SN-38 is roughly 1000 times more potent than Irinotecan, and is believed to be responsible for most anti-tumor effects (240).
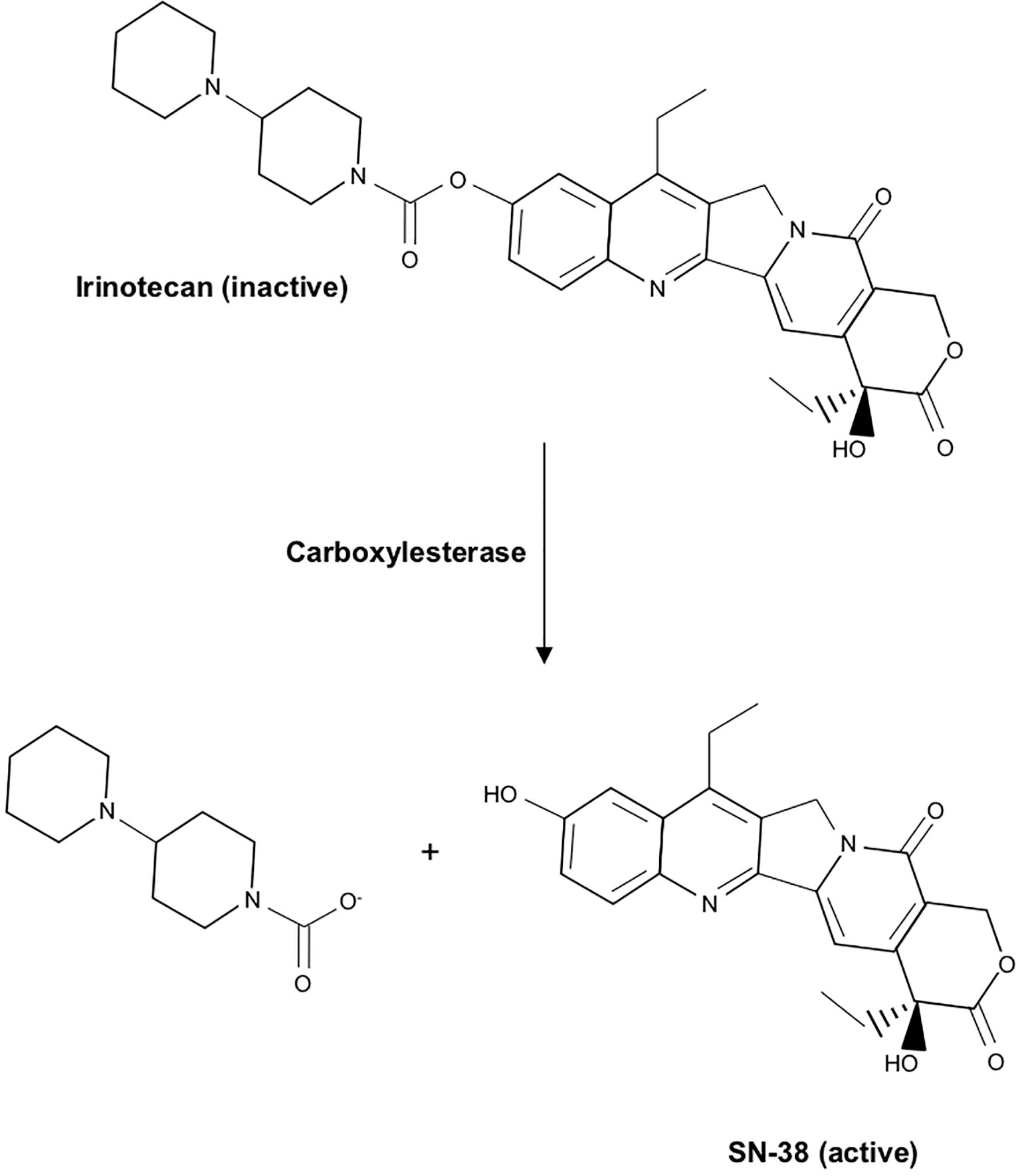
Figure 8 Chemical structure of Irinotecan and conversion to its active metabolite SN-38. In the intestinal mucosa, liver, and plasma, Irinotecan undergoes carboxylesterase-mediated cleavage of the carbamate bond between the camptothecin moiety and the dipiperidino side chain, converting water-soluble Irinotecan to lipid-soluble SN-38, its active metabolite.
The mechanisms through which SN-38 induces cell death have been well described. During DNA replication, Topoisomerase I introduces a single strand knick into the DNA backbone in order to relieve tension (235). While these strands would normally be rejoined later in the DNA replication process, SN-38 stabilizes the association of Topoisomerase I to the 3’-phosphate at the DNA break site. This inhibits the ligating function of Topoisomerase I, resulting in persistent single-strand DNA breaks (235). These breaks interfere with further DNA synthesis, causing double-strand DNA breaks, the arrest of the S-phase, and apoptosis (235).
Irinotecan Resistance
Like many other medications described in this review, there is little data available regarding Irinotecan resistance in PDAC. Like 5-FU, Irinotecan has been used longer in the clinical management of colon cancer. Hence, there are more insights into Irinotecan resistance in colon cancer cells, though none has translated into a successful adjuvant therapy at this time. In colon cancers, Irinotecan resistance appears to be mediated through several mechanisms, including reduced intracellular concentrations of SN-38. This is often mediated by active transport of either Irinotecan of SN-38 by the ATP-binding cassette gene multidrug resistance protein or MRP (241). However, several other factors appear to have driving roles in Irinotecan resistance, including variable levels of converting, reduced Topoisomerase I expression, Topoisomerase I mutation, alterations in the cellular response to Camptothecin-Topoisoomerase I-DNA complex formation, and the activation of multiple signaling cascades including NFκB, EGFR, and SRC (242, 243). However, these are almost entirely unexplored in pancreatic cancer and warrants further evaluation given the advent of the FOLFIRINOX regimen.
Oxaliplatin
Platinum-based agents have been used successfully utilized in cancer therapy for decades. The first platinum-based anti-neoplastic Cisplatin was synthesized in 1845, though it was not approved for cancer patients until 1978 (244). In the decades since, Cisplatin has been used in several cancer types and remains one of the most widely used anti-cancer medications in the developed world. Its success led to the discovery of several similar agents, including Oxaliplatin. Stemming from efforts to improve the toxicity, solubility, and resistance profiles of Cisplatin, Oxaliplatin was first approved for clinical use in Europe in 1996, and the first New Drug Application (NDA) for Oxaliplatin was submitted to the FDA in 1999.
In early trials, while Oxaliplatin demonstrated clear anti-tumor activity in colorectal cancer, it failed to show an increased survival benefit compared to the then standard-of-care treatment, and it was not approved for use. However, subsequent trials examining the combination of Oxaliplatin, 5-FU/Leucovorin, and Irinotecan in metastatic colon cancer showed that the combination had an increased response rate compared to either Oxaliplatin or 5-FU monotherapy and offered a 2-month increase in progression-free survival (245). This led to 2002 FDA approval of the combined regimen in metastatic colorectal cancers that had either recurred or progressed during or within six months of completion of first-line therapy (246). Oxaliplatin has since been approved for use in metastatic PDAC, where it is currently used as part of the multidrug regimen FOLFIRINOX.
Oxaliplatin is structurally similar to Cisplatin, differing in the replacement of amine groups with diaminocyclohexane (DACH) and the chlorine ligands with oxalate (247) (Figure 9). These structural modifications have been suggested to both increase antitumor activity and water solubility (248). Oxaliplatin is thought to enter tumor cells through a variety of transporters, namely cation transporters hOCT2 and hOCT3 (249, 250). Once inside the target cells, Oxaliplatin is converted to its active derivatives via several successive reactions, including non-enzymatic displacement of the oxalate ligand, its replacement with chloride ions, and subsequent hydrolysis. These transient reactive species bind macromolecules, specifically neighboring guanine moieties of DNA, resulting in intrastrand crosslinks (248). The current consensus is that Oxaliplatin-mediated cytotoxicity directly results from the formation of these DNA crosslinks, and the subsequent effects on DNA replication and transcription (251). Interestingly, Oxaliplatin has also been shown to initiate interstrand cross-links at a lower frequency, which may also have a role in its tumoricidal activity (251).
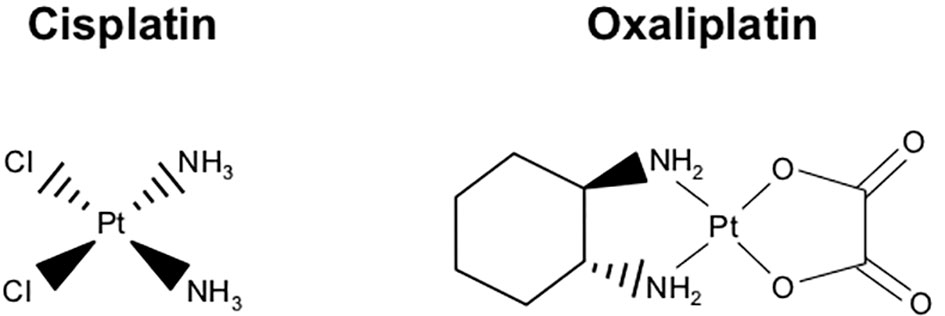
Figure 9 Chemical structure of Oxaliplatin and structural similarity to Cisplatin. Oxaliplatin is structurally similar to Cisplatin, though it differs in the replacement of amine groups with diaminocyclohexane and the chlorine ligands with oxalate, thereby increasing antitumor activity and water solubility.
Oxaliplatin Resistance
Resistance to Oxaliplatin has been extensively evaluated in several cancers and appears to be largely mediated by a variety of transport proteins. For instance, overexpression of multidrug resistance-associated protein 1 (MRP1), a unidirectional efflux transporter also known as ABCC1, is associated with decreased drug accumulation and increased resistance in ovarian cancer cells (252). Additional studies suggest that glutathione-mediated export may also contribute to Oxaliplatin resistance, though this is less established and likely context-specific. For instance, select studies have determined that increased levels of glutathione are associated with poor responses Oxaliplatin (253); however, others have found no association between glutathione and Oxaliplatin or even a positive association between glutathione levels and Oxaliplatin sensitivity (254, 255). Hence, these and other export mechanism warrants continued exploration, particularly in PDAC.
Several other additional resistance mechanisms have also been identified. For instance, in addition to diminished drug uptake, several Oxaliplatin-resistant cell lines demonstrate decreased platinum accumulation in their DNA and increased tolerance of DNA adducts (256). This has been suggested to stem from an increased propensity for base excision repair (BER), as colorectal cancers with low expression of the BER mediator excision repair cross-complementing (ERCC1) have been shown to have increased sensitivity to Oxaliplatin (257). Other potential resistance mechanisms appear to stem from alterations to the apoptosis program. Potential mediators include pyruvate kinase isoform M2, or loss of Bax-mediated apoptosis, though these have yet to be fully characterized in PDAC (258–261).
Summary
There is currently no effective treatment for PDAC. Given the lack of early detection or chemoprevention strategies, most patients will present with advanced disease. For those with operable cancer, surgery can significantly extend survival, though overall outcomes remain poor. As discussed, there are many factors limiting the efficacy of both surgical and medical treatments of PDAC. While alternate approaches such as immunotherapy are beginning to show promise in clinical trials, there is an urgent need to better understand the limitations of the current standard of care approach for PDAC. With respect to chemoresistance, this is largely unexplored in PDAC, particularly for non-Gemcitabine agents. Thus, it is essential to improve our collective understanding of the mechanisms that underlie the near-universally poor therapeutic responses seen in PDAC patients to enhance drug efficacy, extending patient survival, and improve quality of life.
Author Contributions
DP and PU drafted the manuscript. DP assembled figures. MK, JT, HM, and AR edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Veterans Affairs Merit Award I01BX004903 and Career Scientist Award IK6 BX004855 to AR, by NIH R01CA07059 to M. Korc, by NIH F30CA236031 and UIC Award for Graduate Research to DP, by NIH R01CA242003 and the Joseph and Ann Matella Fund for Pancreatic Cancer Research to JT, and by NIH R01CA217907, NIH R21CA255291 and Veterans Affairs Merit Award I01BX002922 to HM.
Disclaimer
Per the funding policy of the U.S. Department of Veterans Affairs, we are required to state that these contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic Cancer. Nat Rev Dis Primers (2016) 2:16022. doi: 10.1038/nrdp.2016.22
2. Korc M, Jeon CY, Edderkaoui M, Pandol SJ, Petrov MS, , Consortium for the Study of Chronic Pancreatitis D, et al. Tobacco and Alcohol as Risk Factors for Pancreatic Cancer. Best Pract Res Clin Gastroenterol (2017) 31:529–36. doi: 10.1016/j.bpg.2017.09.001
3. Principe DR, Rana A. Updated Risk Factors to Inform Early Pancreatic Cancer Screening and Identify High Risk Patients. Cancer Lett (2020) 485:56–65. doi: 10.1016/j.canlet.2020.04.022
4. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
5. Quante AS, Ming C, Rottmann M, Engel J, Boeck S, Heinemann V, et al. Projections of Cancer Incidence and Cancer-Related Deaths in Germany by 2020 and 2030. Cancer Med (2016) 5:2649–56. doi: 10.1002/cam4.767
6. Sultana A, Smith CT, Cunningham D, Starling N, Neoptolemos JP, Ghaneh P. Meta-Analyses of Chemotherapy for Locally Advanced and Metastatic Pancreatic Cancer. J Clin Oncol (2007) 25:2607–15. doi: 10.1200/JCO.2006.09.2551
7. Moore M. Activity of Gemcitabine in Patients With Advanced Pancreatic Carcinoma. A Rev Cancer (1996) 78:633–8. doi: 10.1002/(SICI)1097-0142(19960801)78:3+<633::AID-CNCR8>3.0.CO;2-B
8. Carmichael J, Fink U, Russell RC, Spittle MF, Harris AL, Spiessi G, et al. Phase II Study of Gemcitabine in Patients With Advanced Pancreatic Cancer. Br J Cancer (1996) 73:101–5. doi: 10.1038/bjc.1996.18
9. Casper ES, Green MR, Kelsen DP, Heelan RT, Brown TD, Flombaum CD, et al. Phase II Trial of Gemcitabine (2,2’-Difluorodeoxycytidine) in Patients With Adenocarcinoma of the Pancreas. Invest New Drugs (1994) 12:29–34. doi: 10.1007/BF00873232
10. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased Survival in Pancreatic Cancer With Nab-Paclitaxel Plus Gemcitabine. N Engl J Med (2013) 369:1691–703. doi: 10.1056/NEJMoa1304369
11. Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med (2018) 379:2395–406. doi: 10.1056/NEJMoa1809775
12. Sohal DP, Mangu PB, Laheru D. Metastatic Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline Summary. J Oncol Pract (2017) 13:261–4. doi: 10.1200/JOP.2016.017368
13. Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, et al. Toxicity and Response Criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol (1982) 5:649–55. doi: 10.1097/00000421-198212000-00014
14. Wang-Gillam A, Hubner RA, Siveke JT, Von Hoff DD, Belanger B, de Jong FA, et al. NAPOLI-1 Phase 3 Study of Liposomal Irinotecan in Metastatic Pancreatic Cancer: Final Overall Survival Analysis and Characteristics of Long-Term Survivors. Eur J Cancer (2019) 108:78–87. doi: 10.1016/j.ejca.2018.12.007
15. Macarulla T, Blanc JF, Wang-Gillam A, Chen LT, Siveke JT, Mirakhur B, et al. Liposomal Irinotecan and 5-Fluorouracil/Leucovorin in Older Patients With Metastatic Pancreatic Cancer - A Subgroup Analysis of the Pivotal NAPOLI-1 Trial. J Geriatr Oncol (2019) 10:427–35. doi: 10.1016/j.jgo.2019.02.011
16. O’Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J Clin Oncol (2020) 38:1378–88. doi: 10.1200/JCO.19.02931
17. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med (2019) 381:317–27. doi: 10.1056/NEJMoa1903387
18. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol (2020) 38:1–10. doi: 10.1200/JCO.19.02105
19. Hu ZI, Shia J, Stadler ZK, Varghese AM, Capanu M, Salo-Mullen E, et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin Cancer Res (2018) 24:1326–36. doi: 10.1158/1078-0432.CCR-17-3099
20. Principe DR, Korc M, Kamath SD, Munshi HG, Rana A. Trials and Tribulations of Pancreatic Cancer Immunotherapy. Cancer Lett (2021) 504:1–14. doi: 10.1016/j.canlet.2021.01.031
21. Nevala-Plagemann C, Hidalgo M, Garrido-Laguna I. From State-of-the-Art Treatments to Novel Therapies for Advanced-Stage Pancreatic Cancer. Nat Rev Clin Oncol (2020) 17:108–23. doi: 10.1038/s41571-019-0281-6
22. Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results From Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov (2017) 7:400–9. doi: 10.1158/2159-8290.CD-16-1237
23. Pishvaian MJ, Rolfo CD, Liu SV, Multani PS, Maneval EC, Garrido-Laguna I. Clinical Benefit of Entrectinib for Patients With Metastatic Pancreatic Cancer Who Harbor NTRK and ROS1 Fusions. J Clin Oncol (2018) 36:521. doi: 10.1200/JCO.2018.36.4_suppl.521
24. Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers With a Covalent G12C-Specific Inhibitor. Cell (2018) 172:578–89.e17. doi: 10.1016/j.cell.2018.01.006
25. Gillson J, Ramaswamy Y, Singh G, Gorfe AA, Pavlakis N, Samra J, et al. Small Molecule KRAS Inhibitors: The Future for Targeted Pancreatic Cancer Therapy? Cancers (Basel) (2020) 12(5):1341. doi: 10.3390/cancers12051341
26. Waters AM, Der CJ. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med (2018) 8(9):a031435. doi: 10.1101/cshperspect.a031435
27. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes Facilitate Therapeutic Targeting of Oncogenic KRAS in Pancreatic Cancer. Nature (2017) 546:498–503. doi: 10.1038/nature22341
28. Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, et al. A Randomized Trial of Chemoradiotherapy and Chemotherapy After Resection of Pancreatic Cancer. N Engl J Med (2004) 350:1200–10. doi: 10.1056/NEJMoa032295
29. Bilimoria KY, Bentrem DJ, Ko CY, Stewart AK, Winchester DP, Talamonti MS. National Failure to Operate on Early Stage Pancreatic Cancer. Ann Surg (2007) 246:173–80. doi: 10.1097/SLA.0b013e3180691579
30. Strobel O, Neoptolemos J, Jager D, Buchler MW. Optimizing the Outcomes of Pancreatic Cancer Surgery. Nat Rev Clin Oncol (2019) 16:11–26. doi: 10.1038/s41571-018-0112-1
31. Huang L, Jansen L, Balavarca Y, Molina-Montes E, Babaei M, van der Geest L, et al. Resection of Pancreatic Cancer in Europe and USA: An International Large-Scale Study Highlighting Large Variations. Gut (2019) 68:130–9. doi: 10.1136/gutjnl-2017-314828
32. Shah A, Chao KS, Ostbye T, Castleberry AW, Pietrobon R, Gloor B, et al. Trends in Racial Disparities in Pancreatic Cancer Surgery. J Gastrointest Surg (2013) 17:1897–906. doi: 10.1007/s11605-013-2304-4
33. Shinde RS, Bhandare M, Chaudhari V, Shrikhande SV. Cutting-Edge Strategies for Borderline Resectable Pancreatic Cancer. Ann Gastroenterol Surg (2019) 3:368–72. doi: 10.1002/ags3.12254
35. Groot VP, Rezaee N, Wu W, Cameron JL, Fishman EK, Hruban RH, et al. Patterns, Timing, and Predictors of Recurrence Following Pancreatectomy for Pancreatic Ductal Adenocarcinoma. Ann Surg (2018) 267:936–45. doi: 10.1097/SLA.0000000000002234
36. Ye M, Zhang Q, Chen Y, Fu Q, Li X, Bai X, et al. Neoadjuvant Chemotherapy for Primary Resectable Pancreatic Cancer: A Systematic Review and Meta-Analysis. HPB (Oxf) (2020) 22(6):821–32. doi: 10.1016/j.hpb.2020.01.001
37. Versteijne E, Suker M, Groothuis K, Akkermans-Vogelaar JM, Besselink MG, Bonsing BA, et al. Preoperative Chemoradiotherapy Versus Immediate Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Results of the Dutch Randomized Phase III PREOPANC Trial. J Clin Oncol (2020) 38(16):1763–73. doi: 10.1200/JCO.19.02274
38. Motoi F, Kosuge T, Ueno H, Yamaue H, Satoi S, Sho M, et al. Randomized Phase II/III Trial of Neoadjuvant Chemotherapy With Gemcitabine and S-1 Versus Upfront Surgery for Resectable Pancreatic Cancer (Prep-02/Jsap05). Jpn J Clin Oncol (2019) 49:190–4. doi: 10.1093/jjco/hyy190
39. Gemenetzis G, Groot VP, Blair AB, Laheru DA, Zheng L, Narang AK, et al. Survival in Locally Advanced Pancreatic Cancer After Neoadjuvant Therapy and Surgical Resection. Ann Surg (2019) 270:340–7. doi: 10.1097/SLA.0000000000002753
40. Karachristos A, Scarmeas N, Hoffman JP. CA 19-9 Levels Predict Results of Staging Laparoscopy in Pancreatic Cancer. J Gastrointest Surg (2005) 9:1286–92. doi: 10.1016/j.gassur.2005.06.008
41. Li X, Liu Z, Ye Z, Gou S, Wang C. Impact of Age on Survival of Patients With Pancreatic Cancer After Surgery: Analysis of SEER Data. Pancreatology (2018) 18:133–8. doi: 10.1016/j.pan.2017.11.008
42. Delitto D, Judge SM, George TJ Jr., Sarosi GA, Thomas RM, Behrns KE, et al. A Clinically Applicable Muscular Index Predicts Long-Term Survival in Resectable Pancreatic Cancer. Surgery (2017) 161:930–8. doi: 10.1016/j.surg.2016.09.038
43. Mogal H, Vermilion SA, Dodson R, Hsu FC, Howerton R, Shen P, et al. Modified Frailty Index Predicts Morbidity and Mortality After Pancreaticoduodenectomy. Ann Surg Oncol (2017) 24:1714–21. doi: 10.1245/s10434-016-5715-0
44. Namm JP, Thakrar KH, Wang CH, Stocker SJ, Sur MD, Berlin J, et al. A Semi-Automated Assessment of Sarcopenia Using Psoas Area and Density Predicts Outcomes After Pancreaticoduodenectomy for Pancreatic Malignancy. J Gastrointest Oncol (2017) 8:936–44. doi: 10.21037/jgo.2017.08.09
45. Jang JY, Han Y, Lee H, Kim SW, Kwon W, Lee KH, et al. Oncological Benefits of Neoadjuvant Chemoradiation With Gemcitabine Versus Upfront Surgery in Patients With Borderline Resectable Pancreatic Cancer: A Prospective, Randomized, Open-Label, Multicenter Phase 2/3 Trial. Ann Surg (2018) 268:215–22. doi: 10.1097/SLA.0000000000002705
46. Janssen QP, Buettner S, Suker M, Beumer BR, Addeo P, Bachellier P, et al. Neoadjuvant FOLFIRINOX in Patients With Borderline Resectable Pancreatic Cancer: A Systematic Review and Patient-Level Meta-Analysis. J Natl Cancer Inst (2019) 111:782–94. doi: 10.1093/jnci/djz073
47. Murphy JE, Wo JY, Ryan DP, Jiang W, Yeap BY, Drapek LC, et al. Total Neoadjuvant Therapy With FOLFIRINOX Followed by Individualized Chemoradiotherapy for Borderline Resectable Pancreatic Adenocarcinoma: A Phase 2 Clinical Trial. JAMA Oncol (2018) 4:963–9. doi: 10.1001/jamaoncol.2018.0329
48. Miyasaka Y, Ohtsuka T, Kimura R, Matsuda R, Mori Y, Nakata K, et al. Neoadjuvant Chemotherapy With Gemcitabine Plus Nab-Paclitaxel for Borderline Resectable Pancreatic Cancer Potentially Improves Survival and Facilitates Surgery. Ann Surg Oncol (2019) 26:1528–34. doi: 10.1245/s10434-019-07309-8
49. Herman JM, Chang DT, Goodman KA, Dholakia AS, Raman SP, Hacker-Prietz A, et al. Phase 2 Multi-Institutional Trial Evaluating Gemcitabine and Stereotactic Body Radiotherapy for Patients With Locally Advanced Unresectable Pancreatic Adenocarcinoma. Cancer (2015) 121:1128–37. doi: 10.1002/cncr.29161
50. Suker M, Beumer BR, Sadot E, Marthey L, Faris JE, Mellon EA, et al. FOLFIRINOX for Locally Advanced Pancreatic Cancer: A Systematic Review and Patient-Level Meta-Analysis. Lancet Oncol (2016) 17:801–10. doi: 10.1016/S1470-2045(16)00172-8
51. Hackert T, Niesen W, Hinz U, Tjaden C, Strobel O, Ulrich A, et al. Radical Surgery of Oligometastatic Pancreatic Cancer. Eur J Surg Oncol (2017) 43:358–63. doi: 10.1016/j.ejso.2016.10.023
52. Robinson LA, Tanvetyanon T, Springett G, Fontaine J, Toloza E, Hodul P, et al. Pulmonary Metastasectomy for Suspected Pancreaticobiliary Cancer. J Thorac Cardiovasc Surg (2016) 152:75–82. doi: 10.1016/j.jtcvs.2016.02.066
53. Lutfi W, Zenati MS, Zureikat AH, Zeh HJ, Hogg ME. Health Disparities Impact Expected Treatment of Pancreatic Ductal Adenocarcinoma Nationally. Ann Surg Oncol (2018) 25:1860–7. doi: 10.1245/s10434-018-6487-5
54. Nipp R, Tramontano AC, Kong CY, Pandharipande P, Dowling EC, Schrag D, et al. Disparities in Cancer Outcomes Across Age, Sex, and Race/Ethnicity Among Patients With Pancreatic Cancer. Cancer Med (2018) 7:525–35. doi: 10.1002/cam4.1277
55. Riner AN, Underwood PW, Yang K, Herremans KM, Cameron ME, Chamala S, et al. Disparities in Pancreatic Ductal Adenocarcinoma-The Significance of Hispanic Ethnicity, Subgroup Analysis, and Treatment Facility on Clinical Outcomes. Cancer Med (2020) 9(12):4069–82. doi: 10.1158/1538-7445.AM2020-1172
56. Fong Y, Gonen M, Rubin D, Radzyner M, Brennan MF. Long-Term Survival is Superior After Resection for Cancer in High-Volume Centers. Ann Surg (2005) 242:540–4; discussion 4-7. doi: 10.1097/01.sla.0000184190.20289.4b
57. Powers BD, Fulp W, Dhahri A, DePeralta DK, Ogami T, Rothermel L, et al. The Impact of Socioeconomic Deprivation on Clinical Outcomes for Pancreatic Adenocarcinoma at a High-Volume Cancer Center: A Retrospective Cohort Analysis. Ann Surg (2019). doi: 10.1097/SLA.0000000000003706
58. Schmidt CM, Turrini O, Parikh P, House MG, Zyromski NJ, Nakeeb A, et al. Effect of Hospital Volume, Surgeon Experience, and Surgeon Volume on Patient Outcomes After Pancreaticoduodenectomy: A Single-Institution Experience. Arch Surg (2010) 145:634–40. doi: 10.1001/archsurg.2010.118
59. Lidsky ME, Sun Z, Nussbaum DP, Adam MA, Speicher PJ, Blazer DG 3rd. Going the Extra Mile: Improved Survival for Pancreatic Cancer Patients Traveling to High-Volume Centers. Ann Surg (2017) 266:333–8. doi: 10.1097/SLA.0000000000001924
60. Chang DC, Zhang Y, Mukherjee D, Wolfgang CL, Schulick RD, Cameron JL, et al. Variations in Referral Patterns to High-Volume Centers for Pancreatic Cancer. J Am Coll Surg (2009) 209:720–6. doi: 10.1016/j.jamcollsurg.2009.09.011
61. Liu JH, Zingmond DS, McGory ML, SooHoo NF, Ettner SL, Brook RH, et al. Disparities in the Utilization of High-Volume Hospitals for Complex Surgery. JAMA (2006) 296:1973–80. doi: 10.1001/jama.296.16.1973
62. Blanco BA, Kothari AN, Blackwell RH, Brownlee SA, Yau RM, Attisha JP, et al. “Take the Volume Pledge” May Result in Disparity in Access to Care. Surgery (2017) 161:837–45. doi: 10.1016/j.surg.2016.07.017
63. van der Gaag NA, Rauws EA, van Eijck CH, Bruno MJ, van der Harst E, Kubben FJ, et al. Preoperative Biliary Drainage for Cancer of the Head of the Pancreas. N Engl J Med (2010) 362:129–37. doi: 10.1056/NEJMoa0903230
64. Lee PJ, Podugu A, Wu D, Lee AC, Stevens T, Windsor JA. Preoperative Biliary Drainage in Resectable Pancreatic Cancer: A Systematic Review and Network Meta-Analysis. HPB (Oxf) (2018) 20:477–86. doi: 10.1016/j.hpb.2017.12.007
65. Scheufele F, Schorn S, Demir IE, Sargut M, Tieftrunk E, Calavrezos L, et al. Preoperative Biliary Stenting Versus Operation First in Jaundiced Patients Due to Malignant Lesions in the Pancreatic Head: A Meta-Analysis of Current Literature. Surgery (2017) 161:939–50. doi: 10.1016/j.surg.2016.11.001
66. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-Associated Cachexia. Nat Rev Dis Primers (2018) 4:17105. doi: 10.1038/nrdp.2017.105
67. La Torre M, Ziparo V, Nigri G, Cavallini M, Balducci G, Ramacciato G. Malnutrition and Pancreatic Surgery: Prevalence and Outcomes. J Surg Oncol (2013) 107:702–8. doi: 10.1002/jso.23304
68. Perinel J, Mariette C, Dousset B, Sielezneff I, Gainant A, Mabrut JY, et al. Early Enteral Versus Total Parenteral Nutrition in Patients Undergoing Pancreaticoduodenectomy: A Randomized Multicenter Controlled Trial (Nutri-DPC). Ann Surg (2016) 264:731–7. doi: 10.1097/SLA.0000000000001896
69. Lassen K, Coolsen MM, Slim K, Carli F, de Aguilar-Nascimento JE, Schafer M, et al. Guidelines for Perioperative Care for Pancreaticoduodenectomy: Enhanced Recovery After Surgery (ERAS(R)) Society Recommendations. Clin Nutr (2012) 31:817–30. doi: 10.1016/j.clnu.2012.08.011
70. Gerritsen A, Wennink RA, Besselink MG, van Santvoort HC, Tseng DS, Steenhagen E, et al. Early Oral Feeding After Pancreatoduodenectomy Enhances Recovery Without Increasing Morbidity. HPB (Oxf) (2014) 16:656–64. doi: 10.1111/hpb.12197
71. Robertson N, Gallacher PJ, Peel N, Garden OJ, Duxbury M, Lassen K, et al. Implementation of an Enhanced Recovery Programme Following Pancreaticoduodenectomy. HPB (Oxf) (2012) 14:700–8. doi: 10.1111/j.1477-2574.2012.00521.x
72. Wu JM, Kuo TC, Chen HA, Wu CH, Lai SR, Yang CY, et al. Randomized Trial of Oral Versus Enteral Feeding for Patients With Postoperative Pancreatic Fistula After Pancreatoduodenectomy. Br J Surg (2019) 106:190–8. doi: 10.1002/bjs.11087
73. Gemenetzis G, Groot VP, Blair AB, Ding D, Thakker SS, Fishman EK, et al. Incidence and Risk Factors for Abdominal Occult Metastatic Disease in Patients With Pancreatic Adenocarcinoma. J Surg Oncol (2018) 118:1277–84. doi: 10.1002/jso.25288
74. Doucas H, Sutton CD, Zimmerman A, Dennison AR, Berry DP. Assessment of Pancreatic Malignancy With Laparoscopy and Intraoperative Ultrasound. Surg Endosc (2007) 21:1147–52. doi: 10.1007/s00464-006-9093-8
75. Peng JS, Mino J, Monteiro R, Morris-Stiff G, Ali NS, Wey J, et al. Diagnostic Laparoscopy Prior to Neoadjuvant Therapy in Pancreatic Cancer Is High Yield: An Analysis of Outcomes and Costs. J Gastrointest Surg (2017) 21:1420–7. doi: 10.1007/s11605-017-3470-6
76. Looijen GA, Pranger BK, de Jong KP, Pennings JP, de Meijer VE, Erdmann JI. The Additional Value of Laparoscopic Ultrasound to Staging Laparoscopy in Patients With Suspected Pancreatic Head Cancer. J Gastrointest Surg (2018) 22:1186–92. doi: 10.1007/s11605-018-3726-9
77. Handgraaf HJM, Sibinga Mulder BG, Shahbazi Feshtali S, Boogerd LSF, van der Valk MJM, Farina Sarasqueta A, et al. Staging Laparoscopy With Ultrasound and Near-Infrared Fluorescence Imaging to Detect Occult Metastases of Pancreatic and Periampullary Cancer. PLoS One (2018) 13:e0205960. doi: 10.1371/journal.pone.0205960
78. Shirakawa S, Toyama H, Kido M, Fukumoto T. A Prospective Single-Center Protocol for Using Near-Infrared Fluorescence Imaging With Indocyanine Green During Staging Laparoscopy to Detect Small Metastasis From Pancreatic Cancer. BMC Surg (2019) 19:165. doi: 10.1186/s12893-019-0635-0
79. Glazer ES, Hornbrook MC, Krouse RS. A Meta-Analysis of Randomized Trials: Immediate Stent Placement vs. Surgical Bypass in the Palliative Management of Malignant Biliary Obstruction. J Pain Symptom Manage (2014) 47:307–14. doi: 10.1016/j.jpainsymman.2013.03.013
80. Taylor MC, McLeod RS, Langer B. Biliary Stenting Versus Bypass Surgery for the Palliation of Malignant Distal Bile Duct Obstruction: A Meta-Analysis. Liver Transpl (2000) 6:302–8. doi: 10.1053/lv.2000.5196
81. Jeurnink SM, Steyerberg EW, van Hooft JE, van Eijck CH, Schwartz MP, Vleggaar FP, et al. Surgical Gastrojejunostomy or Endoscopic Stent Placement for the Palliation of Malignant Gastric Outlet Obstruction (SUSTENT Study): A Multicenter Randomized Trial. Gastrointest Endosc (2010) 71:490–9. doi: 10.1016/j.gie.2009.09.042
82. Lillemoe KD, Cameron JL, Hardacre JM, Sohn TA, Sauter PK, Coleman J, et al. Is Prophylactic Gastrojejunostomy Indicated for Unresectable Periampullary Cancer? A Prospective Randomized Trial Ann Surg (1999) 230:322–8; discussion 8-30. doi: 10.1097/00000658-199909000-00005
83. Van Heek NT, De Castro SM, van Eijck CH, van Geenen RC, Hesselink EJ, Breslau PJ, et al. The Need for a Prophylactic Gastrojejunostomy for Unresectable Periampullary Cancer: A Prospective Randomized Multicenter Trial With Special Focus on Assessment of Quality of Life. Ann Surg (2003) 238:894–902. doi: 10.1097/01.sla.0000098617.21801.95
84. Williamsson C, Wennerblom J, Tingstedt B, onsson C. A Wait-and-See Strategy With Subsequent Self-Expanding Metal Stent on Demand is Superior to Prophylactic Bypass Surgery for Unresectable Periampullary Cancer. HPB (Oxf) (2016) 18:107–12. doi: 10.1016/j.hpb.2015.08.009
85. Boone BA, Zenati M, Hogg ME, Steve J, Moser AJ, Bartlett DL, et al. Assessment of Quality Outcomes for Robotic Pancreaticoduodenectomy: Identification of the Learning Curve. JAMA Surg (2015) 150:416–22. doi: 10.1001/jamasurg.2015.17
86. Wang M, Meng L, Cai Y, Li Y, Wang X, Zhang Z, et al. Learning Curve for Laparoscopic Pancreaticoduodenectomy: A CUSUM Analysis. J Gastrointest Surg (2016) 20:924–35. doi: 10.1007/s11605-016-3105-3
87. Sharpe SM, Talamonti MS, Wang CE, Prinz RA, Roggin KK, Bentrem DJ, et al. Early National Experience With Laparoscopic Pancreaticoduodenectomy for Ductal Adenocarcinoma: A Comparison of Laparoscopic Pancreaticoduodenectomy and Open Pancreaticoduodenectomy From the National Cancer Data Base. J Am Coll Surg (2015) 221:175–84. doi: 10.1016/j.jamcollsurg.2015.04.021
88. Ausania F, Landi F, Martinez-Perez A, Fondevila C. A Meta-Analysis of Randomized Controlled Trials Comparing Laparoscopic vs Open Pancreaticoduodenectomy. HPB (Oxf) (2019) 21:1613–20. doi: 10.1016/j.hpb.2019.05.017
89. Nickel F, Haney CM, Kowalewski KF, Probst P, Limen EF, Kalkum E, et al. Laparoscopic Versus Open Pancreaticoduodenectomy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Ann Surg (2020) 271:54–66. doi: 10.1097/SLA.0000000000003309
90. Palanivelu C, Senthilnathan P, Sabnis SC, Babu NS, Srivatsan Gurumurthy S, Anand Vijai N, et al. Randomized Clinical Trial of Laparoscopic Versus Open Pancreatoduodenectomy for Periampullary Tumours. Br J Surg (2017) 104:1443–50. doi: 10.1002/bjs.10662
91. Poves I, Burdio F, Morato O, Iglesias M, Radosevic A, Ilzarbe L, et al. Comparison of Perioperative Outcomes Between Laparoscopic and Open Approach for Pancreatoduodenectomy: The PADULAP Randomized Controlled Trial. Ann Surg (2018) 268:731–9. doi: 10.1097/SLA.0000000000002893
92. de Rooij T, van Hilst J, van Santvoort H, Boerma D, van den Boezem P, Daams F, et al. Minimally Invasive Versus Open Distal Pancreatectomy (LEOPARD): A Multicenter Patient-Blinded Randomized Controlled Trial. Ann Surg (2019) 269:2–9. doi: 10.1097/SLA.0000000000002979
93. van Hilst J, de Rooij T, Klompmaker S, Rawashdeh M, Aleotti F, Al-Sarireh B, et al. Minimally Invasive Versus Open Distal Pancreatectomy for Ductal Adenocarcinoma (DIPLOMA): A Pan-European Propensity Score Matched Study. Ann Surg (2019) 269:10–7. doi: 10.1016/j.pan.2017.07.069
94. Castleberry AW, White RR, de la Fuente SG, Clary BM, Blazer DG 3rd, McCann RL, et al. The Impact of Vascular Resection on Early Postoperative Outcomes After Pancreaticoduodenectomy: An Analysis of the American College of Surgeons National Surgical Quality Improvement Program Database. Ann Surg Oncol (2012) 19:4068–77. doi: 10.1245/s10434-012-2585-y
95. Giovinazzo F, Turri G, Katz MH, Heaton N, Ahmed I. Meta-Analysis of Benefits of Portal-Superior Mesenteric Vein Resection in Pancreatic Resection for Ductal Adenocarcinoma. Br J Surg (2016) 103:179–91. doi: 10.1002/bjs.9969
96. Yu XZ, Li J, Fu DL, Di Y, Yang F, Hao SJ, et al. Benefit From Synchronous Portal-Superior Mesenteric Vein Resection During Pancreaticoduodenectomy for Cancer: A Meta-Analysis. Eur J Surg Oncol (2014) 40:371–8. doi: 10.1016/j.ejso.2014.01.010
97. Zhou Y, Zhang Z, Liu Y, Li B, Xu D. Pancreatectomy Combined With Superior Mesenteric Vein-Portal Vein Resection for Pancreatic Cancer: A Meta-Analysis. World J Surg (2012) 36:884–91. doi: 10.1007/s00268-012-1461-z
98. Sanjay P, Takaori K, Govil S, Shrikhande SV, Windsor JA. ‘Artery-First’ Approaches to Pancreatoduodenectomy. Br J Surg (2012) 99:1027–35. doi: 10.1002/bjs.8763
99. Ironside N, Barreto SG, Loveday B, Shrikhande SV, Windsor JA, Pandanaboyana S. Meta-Analysis of an Artery-First Approach Versus Standard Pancreatoduodenectomy on Perioperative Outcomes and Survival. Br J Surg (2018) 105:628–36. doi: 10.1002/bjs.10832
100. Katz MH, Wang H, Fleming JB, Sun CC, Hwang RF, Wolff RA, et al. Long-Term Survival After Multidisciplinary Management of Resected Pancreatic Adenocarcinoma. Ann Surg Oncol (2009) 16:836–47. doi: 10.1245/s10434-008-0295-2
101. Mollberg N, Rahbari NN, Koch M, Hartwig W, Hoeger Y, Buchler MW, et al. Arterial Resection During Pancreatectomy for Pancreatic Cancer: A Systematic Review and Meta-Analysis. Ann Surg (2011) 254:882–93. doi: 10.1097/SLA.0b013e31823ac299
102. Tseng JF, Raut CP, Lee JE, Pisters PW, Vauthey JN, Abdalla EK, et al. Pancreaticoduodenectomy With Vascular Resection: Margin Status and Survival Duration. J Gastrointest Surg (2004) 8:935–49; discussion 49-50. doi: 10.1016/j.gassur.2004.09.046
103. Gong H, Ma R, Gong J, Cai C, Song Z, Xu B. Distal Pancreatectomy With En Bloc Celiac Axis Resection for Locally Advanced Pancreatic Cancer: A Systematic Review and Meta-Analysis. Med (Baltimore) (2016) 95:e3061. doi: 10.1097/MD.0000000000003061
104. Tol JA, Gouma DJ, Bassi C, Dervenis C, Montorsi M, Adham M, et al. Definition of a Standard Lymphadenectomy in Surgery for Pancreatic Ductal Adenocarcinoma: A Consensus Statement by the International Study Group on Pancreatic Surgery (ISGPS). Surgery (2014) 156:591–600. doi: 10.1016/j.surg.2014.06.016
105. Farnell MB, Pearson RK, Sarr MG, DiMagno EP, Burgart LJ, Dahl TR, et al. A Prospective Randomized Trial Comparing Standard Pancreatoduodenectomy With Pancreatoduodenectomy With Extended Lymphadenectomy in Resectable Pancreatic Head Adenocarcinoma. Surgery (2005) 138:618–28. doi: 10.1016/j.surg.2005.06.044
106. Pedrazzoli S, DiCarlo V, Dionigi R, Mosca F, Pederzoli P, Pasquali C, et al. Standard Versus Extended Lymphadenectomy Associated With Pancreatoduodenectomy in the Surgical Treatment of Adenocarcinoma of the Head of the Pancreas: A Multicenter, Prospective, Randomized Study. Lymphadenectomy Study Group Ann Surg (1998) 228:508–17. doi: 10.1097/00000658-199810000-00007
107. Sun J, Yang Y, Wang X, Yu Z, Zhang T, Song J, et al. Meta-Analysis of the Efficacies of Extended and Standard Pancreatoduodenectomy for Ductal Adenocarcinoma of the Head of the Pancreas. World J Surg (2014) 38:2708–15. doi: 10.1007/s00268-014-2633-9
108. Yeo CJ, Cameron JL, Lillemoe KD, Sohn TA, Campbell KA, Sauter PK, et al. Pancreaticoduodenectomy With or Without Distal Gastrectomy and Extended Retroperitoneal Lymphadenectomy for Periampullary Adenocarcinoma, Part 2: Randomized Controlled Trial Evaluating Survival, Morbidity, and Mortality. Ann Surg (2002) 236:355–66; discussion 66-8. doi: 10.1097/00000658-200209000-00012
109. Dasari BV, Pasquali S, Vohra RS, Smith AM, Taylor MA, Sutcliffe RP, et al. Extended Versus Standard Lymphadenectomy for Pancreatic Head Cancer: Meta-Analysis of Randomized Controlled Trials. J Gastrointest Surg (2015) 19:1725–32. doi: 10.1007/s11605-015-2859-3
110. Nathan H, Wolfgang CL, Edil BH, Choti MA, Herman JM, Schulick RD, et al. Peri-Operative Mortality and Long-Term Survival After Total Pancreatectomy for Pancreatic Adenocarcinoma: A Population-Based Perspective. J Surg Oncol (2009) 99:87–92. doi: 10.1002/jso.21189
111. Passeri MJ, Baker EH, Siddiqui IA, Templin MA, Martinie JB, Vrochides D, et al. Total Compared With Partial Pancreatectomy for Pancreatic Adenocarcinoma: Assessment of Resection Margin, Readmission Rate, and Survival From the U S. Natl Cancer Database Curr Oncol (2019) 26:e346–e56. doi: 10.3747/co.26.4066
112. Reddy S, Wolfgang CL, Cameron JL, Eckhauser F, Choti MA, Schulick RD, et al. Total Pancreatectomy for Pancreatic Adenocarcinoma: Evaluation of Morbidity and Long-Term Survival. Ann Surg (2009) 250:282–7. doi: 10.1097/SLA.0b013e3181ae9f93
113. Scholten L, Latenstein AEJ, van Eijck C, Erdmann J, van der Harst E, Mieog JSD, et al. Outcome and Long-Term Quality of Life After Total Pancreatectomy (PANORAMA): A Nationwide Cohort Study. Surgery (2019) 166:1017–26. doi: 10.1016/j.surg.2019.07.025
114. Scholten L, Stoop TF, Del Chiaro M, Busch OR, van Eijck C, Molenaar IQ, et al. Systematic Review of Functional Outcome and Quality of Life After Total Pancreatectomy. Br J Surg (2019) 106:1735–46. doi: 10.1002/bjs.11296
115. Altman AM, Wirth K, Marmor S, Lou E, Chang K, Hui JYC, et al. Completion of Adjuvant Chemotherapy After Upfront Surgical Resection for Pancreatic Cancer Is Uncommon Yet Associated With Improved Survival. Ann Surg Oncol (2019) 26:4108–16. doi: 10.1245/s10434-019-07602-6
116. Oettle H, Neuhaus P, Hochhaus A, Hartmann JT, Gellert K, Ridwelski K, et al. Adjuvant Chemotherapy With Gemcitabine and Long-Term Outcomes Among Patients With Resected Pancreatic Cancer: The CONKO-001 Randomized Trial. JAMA (2013) 310:1473–81. doi: 10.1001/jama.2013.279201
117. DePeralta DK, Ogami T, Zhou JM, Schell MJ, Powers BD, Hodul PJ, et al. Completion of Adjuvant Therapy in Patients With Resected Pancreatic Cancer. HPB (Oxf) (2020) 22:241–8. doi: 10.1016/j.hpb.2019.07.008
118. Mirkin KA, Greenleaf EK, Hollenbeak CS, Wong J. Time to the Initiation of Adjuvant Chemotherapy Does Not Impact Survival in Patients With Resected Pancreatic Cancer. Cancer (2016) 122:2979–87. doi: 10.1002/cncr.30163
119. Mirkin KA, Greenleaf EK, Hollenbeak CS, Wong J. Minimally Invasive Surgical Approaches Offer Earlier Time to Adjuvant Chemotherapy But Not Improved Survival in Resected Pancreatic Cancer. Surg Endosc (2018) 32:2387–96. doi: 10.1007/s00464-017-5937-7
120. Nussbaum DP, Adam MA, Youngwirth LM, Ganapathi AM, Roman SA, Tyler DS, et al. Minimally Invasive Pancreaticoduodenectomy Does Not Improve Use or Time to Initiation of Adjuvant Chemotherapy for Patients With Pancreatic Adenocarcinoma. Ann Surg Oncol (2016) 23:1026–33. doi: 10.1245/s10434-015-4937-x
121. Xia BT, Ahmad SA, Al Humaidi AH, Hanseman DJ, Ethun CG, Maithel SK, et al. Time to Initiation of Adjuvant Chemotherapy in Pancreas Cancer: A Multi-Institutional Experience. Ann Surg Oncol (2017) 24:2770–6. doi: 10.1245/s10434-017-5918-z
122. Neoptolemos JP, Palmer DH, Ghaneh P, Psarelli EE, Valle JW, Halloran CM, et al. Comparison of Adjuvant Gemcitabine and Capecitabine With Gemcitabine Monotherapy in Patients With Resected Pancreatic Cancer (ESPAC-4): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet (2017) 389:1011–24. doi: 10.1016/S0140-6736(16)32409-6
123. Honeywell RJ, Ruiz van Haperen VW, Veerman G, Smid K, Peters GJ. Inhibition of Thymidylate Synthase by 2’,2’-Difluoro-2’-Deoxycytidine (Gemcitabine) and its Metabolite 2’,2’-Difluoro-2’-Deoxyuridine. Int J Biochem Cell Biol (2015) 60:73–81. doi: 10.1016/j.biocel.2014.12.010
124. Han QL, Zhou YH, Lyu Y, Yan H, Dai GH. Effect of Ribonucleotide Reductase M1 Expression on Overall Survival in Patients With Pancreatic Cancer Receiving Gemcitabine Chemotherapy: A Literature-Based Meta-Analysis. J Clin Pharm Ther (2018) 43:163–9. doi: 10.1111/jcpt.12655
125. Smid K, Van Moorsel CJ, Noordhuis P, Voorn DA, Peters GJ. Interference of Gemcitabine Triphosphate With the Measurements of Deoxynucleotides Using an Optimized DNA Polymerase Elongation Assay. Int J Oncol (2001) 19:157–62. doi: 10.3892/ijo.19.1.157
126. Wickremsinhe E, Bao J, Smith R, Burton R, Dow S, Perkins E. Preclinical Absorption, Distribution, Metabolism, and Excretion of an Oral Amide Prodrug of Gemcitabine Designed to Deliver Prolonged Systemic Exposure. Pharmaceutics (2013) 5:261–76. doi: 10.3390/pharmaceutics5020261
127. Nordh S, Ansari D, Andersson R. Hent1 Expression is Predictive of Gemcitabine Outcome in Pancreatic Cancer: A Systematic Review. World J Gastroenterol (2014) 20:8482–90. doi: 10.3748/wjg.v20.i26.8482
128. Yusuf D, Lui A, Abdelaziz Z, Spratlin JL, Mackey JR. Predictive Value of Hent1 Immunohistochemistry for Gemcitabine-Treated Pancreatic Cancer is Antibody Dependent. J Clin Oncol (2017) 35:e15746–e. doi: 10.1200/JCO.2017.35.15_suppl.e15746
129. Gilbert JA, Salavaggione OE, Ji Y, Pelleymounter LL, Eckloff BW, Wieben ED, et al. Gemcitabine Pharmacogenomics: Cytidine Deaminase and Deoxycytidylate Deaminase Gene Resequencing and Functional Genomics. Clin Cancer Res (2006) 12:1794–803. doi: 10.1158/1078-0432.CCR-05-1969
130. Chen M, Xue X, Wang F, An Y, Tang D, Xu Y, et al. Expression and Promoter Methylation Analysis of ATP-Binding Cassette Genes in Pancreatic Cancer. Oncol Rep (2012) 27:265–9. doi: 10.3892/or.2011.1475
131. Patzak MS, Kari V, Patil S, Hamdan FH, Goetze RG, Brunner M, et al. Cytosolic 5’-Nucleotidase 1A Is Overexpressed in Pancreatic Cancer and Mediates Gemcitabine Resistance by Reducing Intracellular Gemcitabine Metabolites. EBioMedicine (2019) 40:394–405. doi: 10.1016/j.ebiom.2019.01.037
132. Arlt A, Gehrz A, Muerkoster S, Vorndamm J, Kruse ML, Folsch UR, et al. Role of NF-kappaB and Akt/PI3K in the Resistance of Pancreatic Carcinoma Cell Lines Against Gemcitabine-Induced Cell Death. Oncogene (2003) 22:3243–51. doi: 10.1038/sj.onc.1206390
133. Simon PO Jr., McDunn JE, Kashiwagi H, Chang K, Goedegebuure PS, Hotchkiss RS, et al. Targeting AKT With the Proapoptotic Peptide, TAT-CTMP: A Novel Strategy for the Treatment of Human Pancreatic Adenocarcinoma. Int J Cancer (2009) 125:942–51. doi: 10.1002/ijc.24424
134. Jin X, Pan Y, Wang L, Ma T, Zhang L, Tang AH, et al. Fructose-1,6-Bisphosphatase Inhibits ERK Activation and Bypasses Gemcitabine Resistance in Pancreatic Cancer by Blocking IQGAP1-MAPK Interaction. Cancer Res (2017) 77:4328–41. doi: 10.1158/0008-5472.CAN-16-3143
135. Li Y, Xi Z, Chen X, Cai S, Liang C, Wang Z, et al. Natural Compound Oblongifolin C Confers Gemcitabine Resistance in Pancreatic Cancer by Downregulating Src/MAPK/ERK Pathways. Cell Death Dis (2018) 9:538. doi: 10.1038/s41419-018-0574-1
136. Zheng C, Jiao X, Jiang Y, Sun S. ERK1/2 Activity Contributes to Gemcitabine Resistance in Pancreatic Cancer Cells. J Int Med Res (2013) 41:300–6. doi: 10.1177/0300060512474128
137. Wang R, Cheng L, Xia J, Wang Z, Wu Q, Wang Z. Gemcitabine Resistance is Associated With Epithelial-Mesenchymal Transition and Induction of HIF-1alpha in Pancreatic Cancer Cells. Curr Cancer Drug Targets (2014) 14:407–17. doi: 10.2174/1568009614666140226114015
138. Jia Y, Gu D, Wan J, Yu B, Zhang X, Chiorean EG, et al. The Role of GLI-SOX2 Signaling Axis for Gemcitabine Resistance in Pancreatic Cancer. Oncogene (2019) 38:1764–77. doi: 10.1038/s41388-018-0553-0
139. Pan X, Arumugam T, Yamamoto T, Levin PA, Ramachandran V, Ji B, et al. Nuclear Factor-kappaB P65/relA Silencing Induces Apoptosis and Increases Gemcitabine Effectiveness in a Subset of Pancreatic Cancer Cells. Clin Cancer Res (2008) 14:8143–51. doi: 10.1158/1078-0432.CCR-08-1539
140. Jung H, Kim JS, Kim WK, Oh KJ, Kim JM, Lee HJ, et al. Intracellular Annexin A2 Regulates NF-KappaB Signaling by Binding to the P50 Subunit: Implications for Gemcitabine Resistance in Pancreatic Cancer. Cell Death Dis (2015) 6:e1606. doi: 10.1038/cddis.2014.558
141. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science (2009) 324:1457–61. doi: 10.1126/science.1171362
142. Zhang HH, Zhang ZY, Che CL, Mei YF, Shi YZ. Array Analysis for Potential Biomarker of Gemcitabine Identification in Non-Small Cell Lung Cancer Cell Lines. Int J Clin Exp Pathol (2013) 6:1734–46.
143. Humbert M, Casteran N, Letard S, Hanssens K, Iovanna J, Finetti P, et al. Masitinib Combined With Standard Gemcitabine Chemotherapy: In Vitro and In Vivo Studies in Human Pancreatic Tumour Cell Lines and Ectopic Mouse Model. PLoS One (2010) 5:e9430. doi: 10.1371/journal.pone.0009430
144. Moriyama T, Ohuchida K, Mizumoto K, Yu J, Sato N, Nabae T, et al. MicroRNA-21 Modulates Biological Functions of Pancreatic Cancer Cells Including Their Proliferation, Invasion, and Chemoresistance. Mol Cancer Ther (2009) 8:1067–74. doi: 10.1158/1535-7163.MCT-08-0592
145. Howe EN, Cochrane DR, Richer JK. The miR-200 and miR-221/222 microRNA Families: Opposing Effects on Epithelial Identity. J Mammary Gland Biol Neoplasia (2012) 17:65–77. doi: 10.1007/s10911-012-9244-6
146. Ma J, Fang B, Zeng F, Ma C, Pang H, Cheng L, et al. Down-Regulation of miR-223 Reverses Epithelial-Mesenchymal Transition in Gemcitabine-Resistant Pancreatic Cancer Cells. Oncotarget (2015) 6:1740–9. doi: 10.18632/oncotarget.2714
147. Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, et al. Hippo Signaling Regulates Microprocessor and Links Cell-Density-Dependent miRNA Biogenesis to Cancer. Cell (2014) 156:893–906. doi: 10.1016/j.cell.2013.12.043
148. Gujral TS, Kirschner MW. Hippo Pathway Mediates Resistance to Cytotoxic Drugs. Proc Natl Acad Sci USA (2017) 114:E3729–38. doi: 10.1073/pnas.1703096114
149. Samulitis BK, Pond KW, Pond E, Cress AE, Patel H, Wisner L, et al. Gemcitabine Resistant Pancreatic Cancer Cell Lines Acquire an Invasive Phenotype With Collateral Hypersensitivity to Histone Deacetylase Inhibitors. Cancer Biol Ther (2015) 16:43–51. doi: 10.4161/15384047.2014.986967
150. Nicolle R, Gayet O, Duconseil P, Vanbrugghe C, Roques J, Bigonnet M, et al. A Transcriptomic Signature to Predict Adjuvant Gemcitabine Sensitivity in Pancreatic Adenocarcinoma. Ann Oncol (2021) 32:250–60. doi: 10.1016/j.annonc.2020.10.601
151. Gao C, Wisniewski L, Liu Y, Staal B, Beddows I, Plenker D, et al. Detection of Chemotherapy-Resistant Pancreatic Cancer Using a Glycan Biomarker, sTRA. Clin Cancer Res (2021) 27:226–36. doi: 10.1158/1078-0432.CCR-20-2475
152. Kota J, Hancock J, Kwon J, Korc M. Pancreatic Cancer: Stroma and its Current and Emerging Targeted Therapies. Cancer Lett (2017) 391:38–49. doi: 10.1016/j.canlet.2016.12.035
153. Rasheed ZA, Matsui W, Maitra A. Pathology of Pancreatic Stroma in PDAC. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment, Chapter 1. Trivandrum (India): Transworld Research Network (2012).
154. Rucki AA, Zheng L. Pancreatic Cancer Stroma: Understanding Biology Leads to New Therapeutic Strategies. World J Gastroenterol (2014) 20:2237–46. doi: 10.3748/wjg.v20.i9.2237
155. Garrido-Laguna I, Uson M, Rajeshkumar NV, Tan AC, de Oliveira E, Karikari C, et al. Tumor Engraftment in Nude Mice and Enrichment in Stroma- Related Gene Pathways Predict Poor Survival and Resistance to Gemcitabine in Patients With Pancreatic Cancer. Clin Cancer Res (2011) 17:5793–800. doi: 10.1158/1078-0432.CCR-11-0341
156. Pothula SP, Xu Z, Goldstein D, Biankin AV, Pirola RC, Wilson JS, et al. Hepatocyte Growth Factor Inhibition: A Novel Therapeutic Approach in Pancreatic Cancer. Br J Cancer (2016) 114:269–80. doi: 10.1038/bjc.2015.478
157. Principe DR, DeCant B, Mascarinas E, Wayne EA, Diaz AM, Akagi N, et al. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res (2016) 76:2525–39. doi: 10.1158/0008-5472.CAN-15-1293
158. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan Impairs Vascular Function and Drug Delivery in a Mouse Model of Pancreatic Cancer. Gut (2013) 62:112–20. doi: 10.1136/gutjnl-2012-302529
159. Weissmueller S, Manchado E, Saborowski M, Morris J, Wagenblast E, Davis CA, et al. Mutant P53 Drives Pancreatic Cancer Metastasis Through Cell-Autonomous PDGF Receptor Beta Signaling. Cell (2014) 157:382–94. doi: 10.1016/j.cell.2014.01.066
160. Wormann SM, Song L, Ai J, Diakopoulos KN, Kurkowski MU, Gorgulu K, et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology (2016) 151:180–93.e12. doi: 10.1053/j.gastro.2016.03.010
161. Zhang H, Wu H, Guan J, Wang L, Ren X, Shi X, et al. Paracrine SDF-1alpha Signaling Mediates the Effects of PSCs on GEM Chemoresistance Through an IL-6 Autocrine Loop in Pancreatic Cancer Cells. Oncotarget (2015) 6:3085–97. doi: 10.18632/oncotarget.3099
162. Duluc C, Moatassim-Billah S, Chalabi-Dchar M, Perraud A, Samain R, Breibach F, et al. Pharmacological Targeting of the Protein Synthesis mTOR/4e-BP1 Pathway in Cancer-Associated Fibroblasts Abrogates Pancreatic Tumour Chemoresistance. EMBO Mol Med (2015) 7:735–53. doi: 10.15252/emmm.201404346
163. Dangi-Garimella S, Krantz SB, Barron MR, Shields MA, Heiferman MJ, Grippo PJ, et al. Three-Dimensional Collagen I Promotes Gemcitabine Resistance in Pancreatic Cancer Through MT1-MMP-Mediated Expression of HMGA2. Cancer Res (2011) 71:1019–28. doi: 10.1158/0008-5472.CAN-10-1855
164. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
165. Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol (2015) 33:4284–92. doi: 10.1200/JCO.2015.62.8719
166. Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab (2019) 29(6):1390–9.e6. doi: 10.1101/463125
167. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential Role of Intratumor Bacteria in Mediating Tumor Resistance to the Chemotherapeutic Drug Gemcitabine. Science (2017) 357:1156–60. doi: 10.1126/science.aah5043
168. Ng SS, Tsao MS, Nicklee T, Hedley DW. Effects of the Epidermal Growth Factor Receptor Inhibitor OSI-774, Tarceva, on Downstream Signaling Pathways and Apoptosis in Human Pancreatic Adenocarcinoma. Mol Cancer Ther (2002) 1:777–83.
169. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol (2007) 25:1960–6. doi: 10.1200/JCO.2006.07.9525
170. Halfdanarson TR, Foster NR, Kim GP, Meyers JP, Smyrk TC, McCullough AE, et al. A Phase II Randomized Trial of Panitumumab, Erlotinib, and Gemcitabine Versus Erlotinib and Gemcitabine in Patients With Untreated, Metastatic Pancreatic Adenocarcinoma: North Central Cancer Treatment Group Trial N064B (Alliance). Oncologist (2019) 24:589–e160. doi: 10.1634/theoncologist.2018-0878
171. Philip PA, Benedetti J, Corless CL, Wong R, O’Reilly EM, Flynn PJ, et al. Phase III Study Comparing Gemcitabine Plus Cetuximab Versus Gemcitabine in Patients With Advanced Pancreatic Adenocarcinoma: Southwest Oncology Group-Directed Intergroup Trial S0205. J Clin Oncol (2010) 28:3605–10. doi: 10.1200/JCO.2009.25.7550
172. Korc M. Pathways for Aberrant Angiogenesis in Pancreatic Cancer. Mol Cancer (2003) 2:8. doi: 10.1186/1476-4598-2-8
173. Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, et al. Gemcitabine Plus Bevacizumab Compared With Gemcitabine Plus Placebo in Patients With Advanced Pancreatic Cancer: Phase III Trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol (2010) 28:3617–22. doi: 10.1200/JCO.2010.28.1386
174. Kindler HL, Ioka T, Richel DJ, Bennouna J, Letourneau R, Okusaka T, et al. Axitinib Plus Gemcitabine Versus Placebo Plus Gemcitabine in Patients With Advanced Pancreatic Adenocarcinoma: A Double-Blind Randomised Phase 3 Study. Lancet Oncol (2011) 12:256–62. doi: 10.1016/S1470-2045(11)70004-3
175. Hakim N, Patel R, Devoe C, Saif MW. Why HALO 301 Failed and Implications for Treatment of Pancreatic Cancer. Pancreas (Fairfax) (2019) 3:e1–4. doi: 10.17140/POJ-3-e010
176. Carrier E, Kouchakji E, Lipson K, Porter S, Picozzi V, Pishvaian MJ, et al. 1734pdanti-CTGF Human Recombinant Monoclonal Antibody Pamrevlumab Increases Resectability and Resection Rate When Combined With Gemcitabine/Nab-Paclitaxel in the Treatment of Locally Advanced Pancreatic Cancer Patients. Ann Oncol (2017) 28(S5):V268. doi: 10.1093/annonc/mdx369.166
177. Fogelman D, Sugar EA, Oliver G, Shah N, Klein A, Alewine C, et al. Family History as a Marker of Platinum Sensitivity in Pancreatic Adenocarcinoma. Cancer Chemother Pharmacol (2015) 76:489–98. doi: 10.1007/s00280-015-2788-6
178. Jameson GS, Borazanci EH, Babiker HM, Poplin E, Niewiarowska AA, Gordon MS, et al. A Phase Ib/II Pilot Trial With Nab-Paclitaxel Plus Gemcitabine Plus Cisplatin in Patients (Pts) With Stage IV Pancreatic Cancer. J Clin Oncol (2017) 35:341. doi: 10.1200/JCO.2017.35.4_suppl.341
179. Principe DR, Narbutis M, Koch R, Rana A. Frequency and Prognostic Value of Mutations Associated With the Homologous Recombination DNA Repair Pathway in a Large Pan Cancer Cohort. Sci Rep (2020) 10:20223. doi: 10.1038/s41598-020-76975-6
180. Herrmann R, Bodoky G, Ruhstaller T, Glimelius B, Bajetta E, Schuller J, et al. Gemcitabine Plus Capecitabine Compared With Gemcitabine Alone in Advanced Pancreatic Cancer: A Randomized, Multicenter, Phase III Trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. J Clin Oncol (2007) 25:2212–7. doi: 10.1200/JCO.2006.09.0886
181. Principe DR, Park A, Dorman MJ, Kumar S, Viswakarma N, Rubin J, et al. TGFbeta Blockade Augments PD-1 Inhibition to Promote T-Cell-Mediated Regression of Pancreatic Cancer. Mol Cancer Ther (2019) 18:613–20. doi: 10.1158/1535-7163.MCT-18-0850
182. Principe DR, Narbutis M, Kumar S, Park A, Viswakarma N, Dorman MJ, et al. Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination Immunotherapy. Cancer Res (2020) 80(15):3101–15. doi: 10.1158/1538-7445.AM2020-955
183. Cazes A, Betancourt O, Esparza E, Mose ES, Jaquish D, Wong E, et al. A MET Targeting Antibody-Drug Conjugate Overcomes Gemcitabine Resistance in Pancreatic Cancer. Clin Cancer Res (2021) 27(7):2100–10. doi: 10.1158/1078-0432.CCR-20-3210
184. Weaver BA. How Taxol/Paclitaxel Kills Cancer Cells. Mol Biol Cell (2014) 25:2677–81. doi: 10.1091/mbc.e14-04-0916
185. Yang CH, Horwitz SB. Taxol((R)): The First Microtubule Stabilizing Agent. Int J Mol Sci (2017) 18(8):1733. doi: 10.3390/ijms18081733
186. Viudez A, Ramirez N, Hernandez-Garcia I, Carvalho FL, Vera R, Hidalgo M. Nab-Paclitaxel: A Flattering Facelift. Crit Rev Oncol Hematol (2014) 92:166–80. doi: 10.1016/j.critrevonc.2014.06.001
187. Blair HA, Deeks ED. Albumin-Bound Paclitaxel: A Review in Non-Small Cell Lung Cancer. Drugs (2015) 75:2017–24. doi: 10.1007/s40265-015-0484-9
188. Zong Y, Wu J, Shen K. Nanoparticle Albumin-Bound Paclitaxel as Neoadjuvant Chemotherapy of Breast Cancer: A Systematic Review and Meta-Analysis. Oncotarget (2017) 8:17360–72. doi: 10.18632/oncotarget.14477
189. Gradishar WJ. Albumin-Bound Paclitaxel: A Next-Generation Taxane. Expert Opin Pharmacother (2006) 7:1041–53. doi: 10.1517/14656566.7.8.1041
190. Gardner ER, Dahut WL, Scripture CD, Jones J, Aragon-Ching JB, Desai N, et al. Randomized Crossover Pharmacokinetic Study of Solvent-Based Paclitaxel and Nab-Paclitaxel. Clin Cancer Res (2008) 14:4200–5. doi: 10.1158/1078-0432.CCR-07-4592
191. Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, et al. Increased Antitumor Activity, Intratumor Paclitaxel Concentrations, and Endothelial Cell Transport of Cremophor-Free, Albumin-Bound Paclitaxel, ABI-007, Compared With Cremophor-Based Paclitaxel. Clin Cancer Res (2006) 12:1317–24. doi: 10.1158/1078-0432.CCR-05-1634
192. Long BH, Fairchild CR. Paclitaxel Inhibits Progression of Mitotic Cells to G1 Phase by Interference With Spindle Formation Without Affecting Other Microtubule Functions During Anaphase and Telephase. Cancer Res (1994) 54:4355–61.
193. Schiff PB, Fant J, Horwitz SB. Promotion of Microtubule Assembly In Vitro by Taxol. Nature (1979) 277:665–7. doi: 10.1038/277665a0
194. Cullis J, Siolas D, Avanzi A, Barui S, Maitra A, Bar-Sagi D. Macropinocytosis of Nab-Paclitaxel Drives Macrophage Activation in Pancreatic Cancer. Cancer Immunol Res (2017) 5:182–90. doi: 10.1158/2326-6066.CIR-16-0125
195. Sparreboom A, van Asperen J, Mayer U, Schinkel AH, Smit JW, Meijer DK, et al. Limited Oral Bioavailability and Active Epithelial Excretion of Paclitaxel (Taxol) Caused by P-Glycoprotein in the Intestine. Proc Natl Acad Sci USA (1997) 94:2031–5. doi: 10.1073/pnas.94.5.2031
196. O’Driscoll L, Walsh N, Larkin A, Ballot J, Ooi WS, Gullo G, et al. Mdr1/P-Glycoprotein and MRP-1 Drug Efflux Pumps in Pancreatic Carcinoma. Anticancer Res (2007) 27:2115–20.
197. Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of Taxol Resistance Related to Microtubules. Oncogene (2003) 22:7280–95. doi: 10.1038/sj.onc.1206934
198. Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, et al. Paclitaxel-Resistant Human Ovarian Cancer Cells Have Mutant Beta-Tubulins That Exhibit Impaired Paclitaxel-Driven Polymerization. J Biol Chem (1997) 272:17118–25. doi: 10.1074/jbc.272.27.17118
199. Monzo M, Rosell R, Sanchez JJ, Lee JS, O’Brate A, Gonzalez-Larriba JL, et al. Paclitaxel Resistance in Non-Small-Cell Lung Cancer Associated With Beta-Tubulin Gene Mutations. J Clin Oncol (1999) 17:1786–93. doi: 10.1200/JCO.1999.17.6.1786
200. McCarroll JA, Sharbeen G, Liu J, Youkhana J, Goldstein D, McCarthy N, et al. betaIII-Tubulin: A Novel Mediator of Chemoresistance and Metastases in Pancreatic Cancer. Oncotarget (2015) 6:2235–49. doi: 10.18632/oncotarget.2946
201. Kanakkanthara A, Northcote PT, Miller JH. betaII-Tubulin and betaIII-Tubulin Mediate Sensitivity to Peloruside A and Laulimalide, But Not Paclitaxel or Vinblastine, in Human Ovarian Carcinoma Cells. Mol Cancer Ther (2012) 11:393–404. doi: 10.1158/1535-7163.MCT-11-0614
202. Sharbeen G, McCarroll J, Liu J, Youkhana J, Limbri LF, Biankin AV, et al. Delineating the Role of betaIV-Tubulins in Pancreatic Cancer: betaIVb-Tubulin Inhibition Sensitizes Pancreatic Cancer Cells to Vinca Alkaloids. Neoplasia (2016) 18:753–64. doi: 10.1016/j.neo.2016.10.011
203. Atjanasuppat K, Lirdprapamongkol K, Jantaree P, Svasti J. Non-Adherent Culture Induces Paclitaxel Resistance in H460 Lung Cancer Cells via ERK-Mediated Up-Regulation of betaIVa-Tubulin. Biochem Biophys Res Commun (2015) 466:493–8. doi: 10.1016/j.bbrc.2015.09.057
204. Kashyap VK, Wang Q, Setua S, Nagesh PKB, Chauhan N, Kumari S, et al. Therapeutic Efficacy of a Novel betaIII/betaIV-Tubulin Inhibitor (VERU-111) in Pancreatic Cancer. J Exp Clin Cancer Res (2019) 38:29. doi: 10.1186/s13046-018-1009-7
205. Ajabnoor GM, Crook T, Coley HM. Paclitaxel Resistance is Associated With Switch From Apoptotic to Autophagic Cell Death in MCF-7 Breast Cancer Cells. Cell Death Dis (2012) 3:e260. doi: 10.1038/cddis.2011.139
206. Wang Z, Goulet R 3rd, Stanton KJ, Sadaria M, Nakshatri H. Differential Effect of Anti-Apoptotic Genes Bcl-xL and C-FLIP on Sensitivity of MCF-7 Breast Cancer Cells to Paclitaxel and Docetaxel. Anticancer Res (2005) 25:2367–79.
207. Witters LM, Santala SM, Engle L, Chinchilli V, Lipton A. Decreased Response to Paclitaxel Versus Docetaxel in HER-2/Neu Transfected Human Breast Cancer Cells. Am J Clin Oncol (2003) 26:50–4. doi: 10.1097/00000421-200302000-00011
208. Chou A, Waddell N, Cowley MJ, Gill AJ, Chang DK, Patch AM, et al. Clinical and Molecular Characterization of HER2 Amplified-Pancreatic Cancer. Genome Med (2013) 5:78. doi: 10.1186/gm482
209. Harder J, Ihorst G, Heinemann V, Hofheinz R, Moehler M, Buechler P, et al. Multicentre Phase II Trial of Trastuzumab and Capecitabine in Patients With HER2 Overexpressing Metastatic Pancreatic Cancer. Br J Cancer (2012) 106:1033–8. doi: 10.1038/bjc.2012.18
210. Kutuk O, Letai A. Displacement of Bim by Bmf and Puma Rather Than Increase in Bim Level Mediates Paclitaxel-Induced Apoptosis in Breast Cancer Cells. Cell Death Differ (2010) 17:1624–35. doi: 10.1038/cdd.2010.41
211. Miller AV, Hicks MA, Nakajima W, Richardson AC, Windle JJ, Harada H. Paclitaxel-Induced Apoptosis Is BAK-Dependent, But BAX and BIM-Independent in Breast Tumor. PLoS One (2013) 8:e60685. doi: 10.1371/journal.pone.0060685
212. Sharifi S, Barar J, Hejazi MS, Samadi N. Roles of the Bcl-2/Bax Ratio, Caspase-8 and 9 in Resistance of Breast Cancer Cells to Paclitaxel. Asian Pac J Cancer Prev (2014) 15:8617–22. doi: 10.7314/APJCP.2014.15.20.8617
213. Sunters A, Fernandez de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, et al. FoxO3a Transcriptional Regulation of Bim Controls Apoptosis in Paclitaxel-Treated Breast Cancer Cell Lines. J Biol Chem (2003) 278:49795–805. doi: 10.1074/jbc.M309523200
214. Zhang SF, Wang XY, Fu ZQ, Peng QH, Zhang JY, Ye F, et al. TXNDC17 Promotes Paclitaxel Resistance via Inducing Autophagy in Ovarian Cancer. Autophagy (2015) 11:225–38. doi: 10.1080/15548627.2014.998931
215. Notte A, Ninane N, Arnould T, Michiels C. Hypoxia Counteracts Taxol-Induced Apoptosis in MDA-MB-231 Breast Cancer Cells: Role of Autophagy and JNK Activation. Cell Death Dis (2013) 4:e638. doi: 10.1038/cddis.2013.167
216. Zhang B, Zhao R, He Y, Fu X, Fu L, Zhu Z, et al. MicroRNA 100 Sensitizes Luminal A Breast Cancer Cells to Paclitaxel Treatment in Part by Targeting mTOR. Oncotarget (2016) 7:5702–14. doi: 10.18632/oncotarget.6790
217. Chatterjee A, Chattopadhyay D, Chakrabarti G. MiR-16 Targets Bcl-2 in Paclitaxel-Resistant Lung Cancer Cells and Overexpression of miR-16 Along With miR-17 Causes Unprecedented Sensitivity by Simultaneously Modulating Autophagy and Apoptosis. Cell Signal (2015) 27:189–203. doi: 10.1016/j.cellsig.2014.11.023
218. Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, et al. Fluorinated Pyrimidines, a New Class of Tumour-Inhibitory Compounds. Nature (1957) 179:663–6. doi: 10.1038/179663a0
219. Curreri AR, Ansfield FJ, Mc IF, Waisman HA, Heidelberger C. Clinical Studies With 5-Fluorouracil. Cancer Res (1958) 18:478–84.
220. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX Versus Gemcitabine for Metastatic Pancreatic Cancer. N Engl J Med (2011) 364:1817–25. doi: 10.1056/NEJMoa1011923
221. Wohlhueter RM, McIvor RS, Plagemann PG. Facilitated Transport of Uracil and 5-Fluorouracil, and Permeation of Orotic Acid Into Cultured Mammalian Cells. J Cell Physiol (1980) 104:309–19. doi: 10.1002/jcp.1041040305
222. Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat Rev Cancer (2003) 3:330–8. doi: 10.1038/nrc1074
223. Kufe DW, Major PP. 5-Fluorouracil Incorporation Into Human Breast Carcinoma RNA Correlates With Cytotoxicity. J Biol Chem (1981) 256:9802–5. doi: 10.1016/S0021-9258(19)68695-3
224. Glazer RI, Lloyd LS. Association of Cell Lethality With Incorporation of 5-Fluorouracil and 5-Fluorouridine Into Nuclear RNA in Human Colon Carcinoma Cells in Culture. Mol Pharmacol (1982) 21:468–73.
225. Lund K, Olsen CE, Wong JJW, Olsen PA, Solberg NT, Hogset A, et al. 5-FU Resistant EMT-Like Pancreatic Cancer Cells are Hypersensitive to Photochemical Internalization of the Novel Endoglin-Targeting Immunotoxin CD105-Saporin. J Exp Clin Cancer Res (2017) 36:187. doi: 10.1186/s13046-017-0662-6
226. Shi X, Liu S, Kleeff J, Friess H, Buchler MW. Acquired Resistance of Pancreatic Cancer Cells Towards 5-Fluorouracil and Gemcitabine is Associated With Altered Expression of Apoptosis-Regulating Genes. Oncology (2002) 62:354–62. doi: 10.1159/000065068
227. Giacchetti S, Perpoint B, Zidani R, Le Bail N, Faggiuolo R, Focan C, et al. Phase III Multicenter Randomized Trial of Oxaliplatin Added to Chronomodulated Fluorouracil-Leucovorin as First-Line Treatment of Metastatic Colorectal Cancer. J Clin Oncol (2000) 18:136–47. doi: 10.1200/JCO.2000.18.1.136
228. Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, et al. Irinotecan Combined With Fluorouracil Compared With Fluorouracil Alone as First-Line Treatment for Metastatic Colorectal Cancer: A Multicentre Randomised Trial. Lancet (2000) 355:1041–7. doi: 10.1016/S0140-6736(00)02034-1
229. Zhang N, Yin Y, Xu SJ, Chen WS. 5-Fluorouracil: Mechanisms of Resistance and Reversal Strategies. Molecules (2008) 13:1551–69. doi: 10.3390/molecules13081551
230. Peters GJ, Backus HH, Freemantle S, van Triest B, Codacci-Pisanelli G, van der Wilt CL, et al. Induction of Thymidylate Synthase as a 5-Fluorouracil Resistance Mechanism. Biochim Biophys Acta (2002) 1587:194–205. doi: 10.1016/S0925-4439(02)00082-0
231. Papamichael D. The Use of Thymidylate Synthase Inhibitors in the Treatment of Advanced Colorectal Cancer: Current Status. Oncologist (1999) 4:478–87. doi: 10.1634/theoncologist.4-6-478
232. van Triest B, Pinedo HM, van Hensbergen Y, Smid K, Telleman F, Schoenmakers PS, et al. Thymidylate Synthase Level as the Main Predictive Parameter for Sensitivity to 5-Fluorouracil, But Not for Folate-Based Thymidylate Synthase Inhibitors, in 13 Nonselected Colon Cancer Cell Lines. Clin Cancer Res (1999) 5:643–54.
233. He L, Zhu H, Zhou S, Wu T, Wu H, Yang H, et al. Wnt Pathway is Involved in 5-FU Drug Resistance of Colorectal Cancer Cells. Exp Mol Med (2018) 50:101. doi: 10.1038/s12276-018-0128-8
234. Efferth T, Fu YJ, Zu YG, Schwarz G, Konkimalla VS, Wink M. Molecular Target-Guided Tumor Therapy With Natural Products Derived From Traditional Chinese Medicine. Curr Med Chem (2007) 14:2024–32. doi: 10.2174/092986707781368441
235. Gerrits CJ, de Jonge MJ, Schellens JH, Stoter G, Verweij J. Topoisomerase I Inhibitors: The Relevance of Prolonged Exposure for Present Clinical Development. Br J Cancer (1997) 76:952–62. doi: 10.1038/bjc.1997.491
236. Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular Roles of SN-38, a Metabolite of the Camptothecin Derivative CPT-11, in the Antitumor Effect of CPT-11. Cancer Res (1991) 51:4187–91.
237. Kunimoto T, Nitta K, Tanaka T, Uehara N, Baba H, Takeuchi M, et al. Antitumor Activity of 7-Ethyl-10-[4-(1-Piperidino)-1-Piperidino]Carbonyloxy-Camptothec in, a Novel Water-Soluble Derivative of Camptothecin, Against Murine Tumors. Cancer Res (1987) 47:5944–7.
238. Fujita K, Kubota Y, Ishida H, Sasaki Y. Irinotecan, a Key Chemotherapeutic Drug for Metastatic Colorectal Cancer. World J Gastroenterol (2015) 21:12234–48. doi: 10.3748/wjg.v21.i43.12234
239. Thota R, Pauff JM, Berlin JD. Treatment of Metastatic Pancreatic Adenocarcinoma: A Review. Oncol (Williston Park) (2014) 28:70–4.
240. de Bruijn P, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A. Determination of Irinotecan (CPT-11) and Its Active Metabolite SN-38 in Human Plasma by Reversed-Phase High-Performance Liquid Chromatography With Fluorescence Detection. J Chromatogr B BioMed Sci Appl (1997) 698:277–85. doi: 10.1016/S0378-4347(97)00290-9
241. Longley DB, Johnston PG. Molecular Mechanisms of Drug Resistance. J Pathol (2005) 205:275–92. doi: 10.1002/path.1706
242. Xu Y, Villalona-Calero MA. Irinotecan: Mechanisms of Tumor Resistance and Novel Strategies for Modulating its Activity. Ann Oncol (2002) 13:1841–51. doi: 10.1093/annonc/mdf337
243. Petitprez A, Larsen AK. Irinotecan Resistance is Accompanied by Upregulation of EGFR and Src Signaling in Human Cancer Models. Curr Pharm Des (2013) 19:958–64. doi: 10.2174/138161213804547204
244. Florea AM, Busselberg D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers (Basel) (2011) 3:1351–71. doi: 10.3390/cancers3011351
245. Graham J, Mushin M, Kirkpatrick P. Oxaliplatin. Nat Rev Drug Discov (2004) 3:11–2. doi: 10.1038/nrd1287
246. Ibrahim A, Hirschfeld S, Cohen MH, Griebel DJ, Williams GA, Pazdur R. FDA Drug Approval Summaries: Oxaliplatin. Oncologist (2004) 9:8–12. doi: 10.1634/theoncologist.9-1-8
247. Alcindor T, Beauger N. Oxaliplatin: A Review in the Era of Molecularly Targeted Therapy. Curr Oncol (2011) 18:18–25. doi: 10.3747/co.v18i1.708
248. Di Francesco AM, Ruggiero A, Riccardi R. Cellular and Molecular Aspects of Drugs of the Future: Oxaliplatin. Cell Mol Life Sci (2002) 59:1914–27. doi: 10.1007/PL00012514
249. Burger H, Zoumaro-Djayoon A, Boersma AW, Helleman J, Berns EM, Mathijssen RH, et al. Differential Transport of Platinum Compounds by the Human Organic Cation Transporter Hoct2 (Hslc22a2). Br J Pharmacol (2010) 159:898–908. doi: 10.1111/j.1476-5381.2009.00569.x
250. Yokoo S, Masuda S, Yonezawa A, Terada T, Katsura T, Inui K. Significance of Organic Cation Transporter 3 (SLC22A3) Expression for the Cytotoxic Effect of Oxaliplatin in Colorectal Cancer. Drug Metab Dispos (2008) 36:2299–306. doi: 10.1124/dmd.108.023168
251. Woynarowski JM, Chapman WG, Napier C, Herzig MC, Juniewicz P. Sequence- and Region-Specificity of Oxaliplatin Adducts in Naked and Cellular DNA. Mol Pharmacol (1998) 54:770–7. doi: 10.1124/mol.54.5.770
252. Beretta GL, Benedetti V, Cossa G, Assaraf YG, Bram E, Gatti L, et al. Increased Levels and Defective Glycosylation of MRPs in Ovarian Carcinoma Cells Resistant to Oxaliplatin. Biochem Pharmacol (2010) 79:1108–17. doi: 10.1016/j.bcp.2009.12.002
253. Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal Control of Cystine Metabolism Promotes Cancer Cell Survival in Chronic Lymphocytic Leukaemia. Nat Cell Biol (2012) 14:276–86. doi: 10.1038/ncb2432
254. Arnould S, Hennebelle I, Canal P, Bugat R, Guichard S. Cellular Determinants of Oxaliplatin Sensitivity in Colon Cancer Cell Lines. Eur J Cancer (2003) 39:112–9. doi: 10.1016/S0959-8049(02)00411-2
255. Tozawa K, Oshima T, Kobayashi T, Yamamoto N, Hayashi C, Matsumoto T, et al. Oxaliplatin in Treatment of the Cisplatin-Resistant MKN45 Cell Line of Gastric Cancer. Anticancer Res (2008) 28:2087–92.
256. Mishima M, Samimi G, Kondo A, Lin X, Howell SB. The Cellular Pharmacology of Oxaliplatin Resistance. Eur J Cancer (2002) 38:1405–12. doi: 10.1016/S0959-8049(02)00096-5
257. Shirota Y, Stoehlmacher J, Brabender J, Xiong YP, Uetake H, Danenberg KD, et al. ERCC1 and Thymidylate Synthase mRNA Levels Predict Survival for Colorectal Cancer Patients Receiving Combination Oxaliplatin and Fluorouracil Chemotherapy. J Clin Oncol (2001) 19:4298–304. doi: 10.1200/JCO.2001.19.23.4298
258. Stetak A, Veress R, Ovadi J, Csermely P, Keri G, Ullrich A. Nuclear Translocation of the Tumor Marker Pyruvate Kinase M2 Induces Programmed Cell Death. Cancer Res (2007) 67:1602–8. doi: 10.1158/0008-5472.CAN-06-2870
259. Martinez-Balibrea E, Plasencia C, Gines A, Martinez-Cardus A, Musulen E, Aguilera R, et al. A Proteomic Approach Links Decreased Pyruvate Kinase M2 Expression to Oxaliplatin Resistance in Patients With Colorectal Cancer and in Human Cell Lines. Mol Cancer Ther (2009) 8:771–8. doi: 10.1158/1535-7163.MCT-08-0882
260. Gourdier I, Del Rio M, Crabbe L, Candeil L, Copois V, Ychou M, et al. Drug Specific Resistance to Oxaliplatin Is Associated With Apoptosis Defect in a Cellular Model of Colon Carcinoma. FEBS Lett (2002) 529:232–6. doi: 10.1016/S0014-5793(02)03347-1
261. National Center for Biotechnology Information. PubChem Database. Irinotecan, CID=60838. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/60838 (Accessed on Apr. 19, 2020).
Keywords: pancreatic cancer, chemotherapy, radiation, surgery, immunotherapy, targeted therapy, treatment
Citation: Principe DR, Underwood PW, Korc M, Trevino JG, Munshi HG and Rana A (2021) The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol. 11:688377. doi: 10.3389/fonc.2021.688377
Received: 30 March 2021; Accepted: 29 June 2021;
Published: 15 July 2021.
Edited by:
Alessio G. Morganti, University of Bologna, ItalyReviewed by:
Savio George Barreto, Medanta The Medicity Hospital, IndiaMariacristina Di Marco, University of Bologna, Italy
Milly Buwenge, University of Bologna, Italy
Copyright © 2021 Principe, Underwood, Korc, Trevino, Munshi and Rana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel R. Principe, principe@illinois.edu; Ajay Rana, arana@uic.edu
†These authors share first authorship