 Joana Garmendia1
,2
Joana Garmendia1
,2
 Garazi Labayru1
,2
,3
Garazi Labayru1
,2
,3
 Jone Aliri1
Jone Aliri1
 Adolfo López de Munain2
,3
,4
,5
Adolfo López de Munain2
,3
,4
,5
 Andone Sistiaga1
,2
,3
*
Andone Sistiaga1
,2
,3
*
- 1Department of Clinical and Health Psychology and Research Methodology, Psychology Faculty, University of the Basque Country (UPV/EHU), Donostia-San Sebastián, Gipuzkoa, Spain
- 2Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), Institute Carlos III, Madrid, Spain
- 3Neuroscience Area, Biogipuzkoa Health Research Institute, Donostia-San Sebastián, Gipuzkoa, Spain
- 4Neurology Department, Donostia University Hospital, Donostia-San Sebastián, Gipuzkoa, Spain
- 5Neuroscience Department, University of the Basque Country (UPV/EHU), Donostia-San Sebastián, Gipuzkoa, Spain
Introduction: Myotonic dystrophy type 1 (DM1) is a hereditary neuromuscular disorder affecting the central nervous system (CNS). Although sex differences have been explored in other neuromuscular disorders, research on this topic in DM1 remains limited. The present study aims to analyze sex differences (both the patient’s and disease-transmitting parent’s sex) with a focus on CNS outcomes.
Methods: Retrospective data from 146 non-congenital DM1 patients were analyzed, including clinical, molecular, neuropsychological, and neuroradiological data. Sex and inheritance pattern differences were analyzed using t-tests, and ANOVA analyses were conducted to address the interactions.
Results: Overall, no significant sex differences were observed except in certain cognitive domains. However, individuals with maternal inheritance showed larger CTG expansion size, lower estimated IQs, and poorer performance on visual memory, executive functions, and language domains than those with paternal inheritance. Notably, IQ performance was independently influenced by inheritance pattern and CTG expansion.
Discussion: This study is the first to delve into sex differences in DM1 with a focus on CNS outcomes. While the results revealed the absence of a sex-specific clinic-molecular profile, more substantial CNS differences were observed between patients with maternal and paternal inheritance patterns. The hypothetical existence of genomic imprinting and its potential mechanism are discussed. These findings hold potential implications for aiding clinical management by improving genetic counseling and predicting disease severity and prognosis.
1 Introduction
Myotonic dystrophy type 1 (DM1) is a multisystemic disease that affects various body systems, including muscular, ophthalmological, cardiac, endocrine, gastrointestinal, respiratory, and central nervous systems. This disorder is the most prevalent form of muscular dystrophy in adults, characterized by distal muscle weakness and myotonia, which patients typically present (1).
DM1 is a hereditary disease transmitted in an autosomal dominant manner and is diagnosed by detecting an expansion length of the trinucleotide CTG (cytosine, thymine, guanine) that exceeds 50 repetitions. This neuromuscular disorder is characterized by phenotypical variability in its clinical signs/symptoms and severity. However, it is well-established that CTG expansion size correlates positively with disease severity (1–3). Like other repeat expansion diseases, DM1 patients typically present a phenomenon known as clinical anticipation, which involves an earlier disease onset and more severe symptoms in successive generations of a family (4). Research has also investigated genomic imprinting in DM1, which consists of the differential phenotypic expression of genetic material depending on whether it has been inherited from the male or female parent (5). However, this line of inquiry is not currently being pursued. DM1 is commonly classified into different phenotypes based on the age of onset of the disease: congenital (present at birth), childhood (onset between1-10 years of age), juvenile (onset between 10–20 years of age), adult (onset between 20–40 years of age) and late-onset (onset >40 years) (6).
To date, limited research has explored sex differences within the various systems affected by DM1. Some previous studies have focused on ophthalmological aspects (7), endocrinal differences (8), as well as pain and motor function (9, 10). Additionally, more general studies have been conducted on sex differences in clinical conditions or comorbidities in DM1 (11, 12). The latter study has revealed that sex influences the clinical profile and severity of the disease in DM1 patients. While male patients tend to present higher morbidity and mortality, including more pronounced muscular, cardiac, and respiratory impairments, female patients are more prone to experiencing ophthalmological, gastrointestinal, and endocrine-related issues.
However, no studies have focused on sex differences regarding CNS involvement in DM1. In addition to considering the sex differences between patients, it is important to explore the implications of the sex of the disease-transmitting parent, known as the patient’s inheritance pattern, whether the aberrant gene has been inherited from the mother (maternal) or father (paternal).
The scientific literature highlights that maternal inheritance has been observed at a higher percentage in DM1 patients with the congenital form (6, 13, 14), considered the most severe DM1 phenotype; indeed, the literature recognizes it as a clinically different form (13, 15). Therefore, it is important to investigate whether patients with maternal inheritance present greater disease severity in different DM1 phenotypes beyond the congenital form.
To date, studies have examined the influence of the inheritance pattern on the molecular instability of the disease (CTG expansion size) (16). However, no studies have explored the direct implications of the inheritance pattern for disease severity, specifically CNS affectation.
The present study aims to (1) analyze sex differences in terms of CNS, (2) examine differences between patients with maternal and paternal inheritance in terms of CNS involvement, and (3) analyze the interactions between inheritance pattern and CTG in relation to CNS involvement.
2 Methodology
2.1 Participants
For this study, retrospective data from a cohort of 146 DM1 patients recruited at the Neurology Department of the Donostia University Hospital (Gipuzkoa, Spain) were analyzed. Inclusion criteria included a molecular confirmation of the disease and being aged 18 years or above. Exclusion criteria were having the congenital form of DM1 and a history of major psychiatric or somatic illness, acquired brain injury, or drug abuse.
This study was approved by the Ethics Committee for Clinical Investigation of the Health Department of Gipuzkoa (DMRM-2017-01), and all participants provided signed informed consent.
The data were compiled using the method outlined below and were analyzed retrospectively for this study.
2.2 Clinical data
Clinical data, such as phenotype and inheritance pattern, were obtained through the patients’ medical records. An estimated disease duration was derived from the range of age of onset and the age at assessment. An experienced neurologist recorded data on muscular impairment using the Muscular Impairment Rating Scale (MIRS).
2.3 Data on genetic determination
The participants’ cytosine thymine guanine (CTG) expansion size was obtained from clinical data. Genetic assessment was conducted using blood sample analysis (polymerase chain reaction in DMPK alleles up to approximately 100 CTG and southern blot analysis for larger expansions).
2.4 Neuropsychological data
Data on the neuropsychological performance of the participants was accessible. The assessment included IQ estimation and performance on six cognitive domains (attention/processing speed, verbal memory, visual memory, visuoconstruction, executive functions, and language). Table 1 shows the tests employed for IQ estimation and the cognitive domains assessed. These tests were administered by an experienced neuropsychologist blind to the clinical condition of the participants (i.e., disease form, inheritance pattern, CTG repeats, or MIRS).
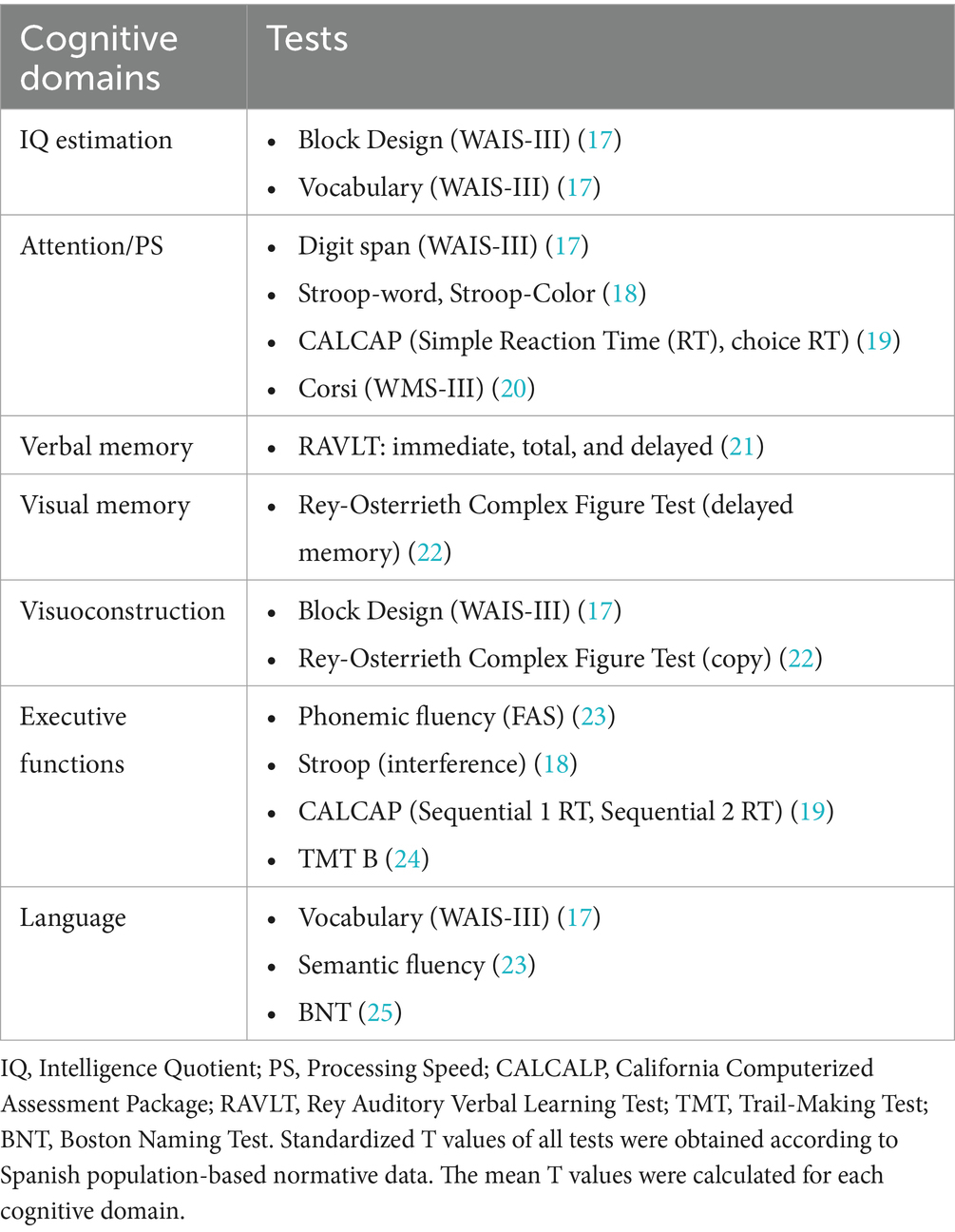
Table 1. Tests employed for assessing IQ and performance in cognitive domains.
2.5 Neuroradiological data
All magnetic resonance scans were conducted using a 1.5 T scanner (Achieva Nova, Philips). The current results were obtained from a high-resolution volumetric turbo field echo series (sagittal 3D T1 weighted acquisition, repetition time 7.2, echo time 3.3, flip angle 8, matrix 256 × 232, slice thickness 1 mm, voxel dimensions 1 × 1 × 1 mm, number of signal averages 1, no slices 160, gap 0, and a total scan duration 5 min 34 s).
Grey matter (GM) and White matter (WM) volumes were determined using voxel-based morphometry via the FMRIB Software Library (FSL version 6.01) (26). White matter lesions (WML) were assessed according to the Wahlund scale (27). When lesions >5 mm were identified, severity was rated from 0 (no lesions) to 3 (diffuse involvement).
2.6 Statistical analyses
Data were analyzed using the SPSS statistical package (version 27).
To explore sex differences for both patients and the transmitting parent (inheritance pattern), contingency analysis (chi-square) was used for categorical data, while parametric tests (t-test) were employed for interval data. Effect sizes were reported using Cohen’s d, categorized as small (d ≤ 0.49), moderate (d = 0.50–0.79), or high (d ≥ 0.80).
In addition, given the well-established association between inheritance pattern and CTG expansion size and the correlation between CTG and CNS involvement, an ANOVA was conducted. This analysis aimed to examine the independent effects of these two variables (CTG and inheritance pattern) and to evaluate the impact of their interaction on the CNS variables (cognitive and structural brain outcomes) under study.
3 Results
3.1 Participants
The retrospective data analysis included 146 DM1 patients. Table 2 illustrates the distribution of sex, inheritance pattern, and phenotype among these patients. The sample is equally distributed in terms of sex (50/50), with a notable prevalence of paternal transmission. Regarding phenotype, most patients were adult-onset DM1 patients, followed by juvenile-onset, late/partial-onset, and finally, childhood onset.
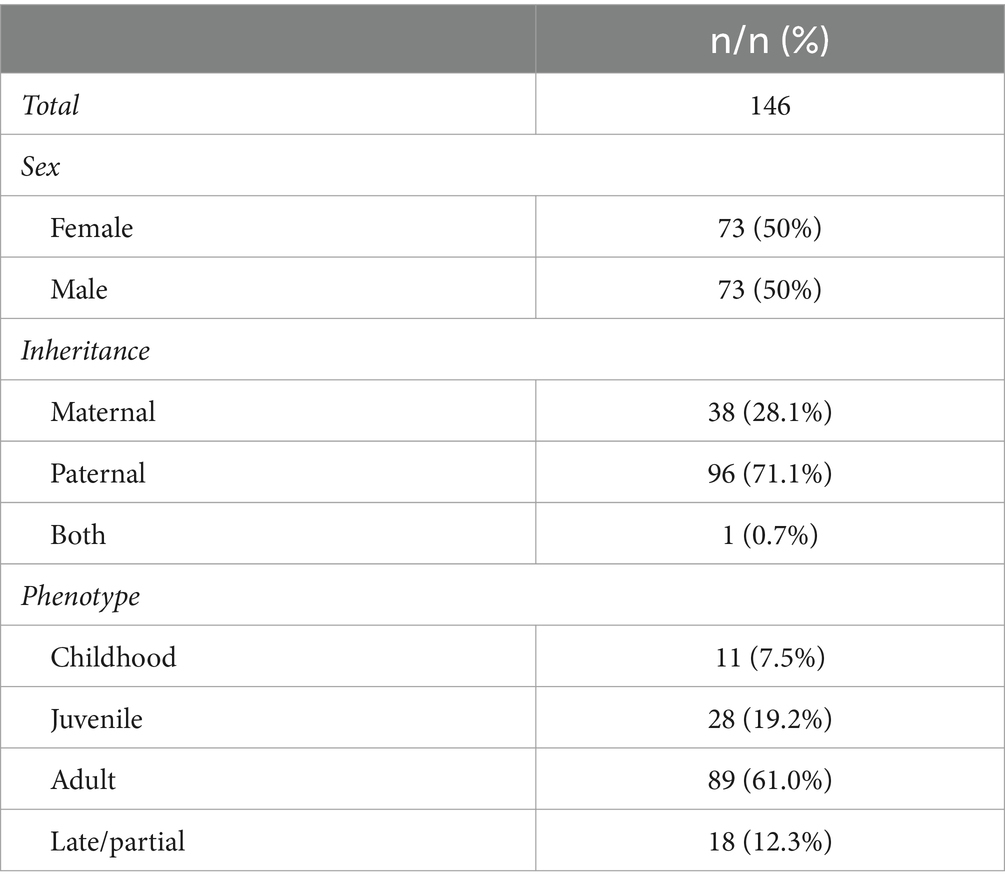
Table 2. Sex, inheritance pattern, and phenotype of the DM1 participants.
3.2 Sex differences in DM1
Clinical, cognitive, and structural brain data were used to explore differences between female and male patients, and the findings are summarized in Table 3.

Table 3. Sex differences in clinical, cognitive, and brain structure outcomes.
Regarding clinical data, both female and male patients presented a similar phenotype distribution (χ2 (1) = 1.28; p = 0.733), and no sex differences were found in CTG, MIRS, and disease duration among DM1 patients.
In terms of cognitive performance, female patients showed significantly poorer performance in the attention/processing speed and visual memory domain. In contrast, male patients performed significantly worse in the verbal memory domain. However, the effect size was small to moderate in all cases.
Concerning structural brain outcomes, although not statistically significant, female patients tended to have lower WM volume, while male patients more WML, with moderate and high effect sizes, respectively.
3.3 Differences regarding inheritance pattern in DM1
Differences between patients with maternal and paternal inheritance patterns are summarized in Table 4. Notably, the patient with inheritance transmission from both parents was excluded from further analysis.
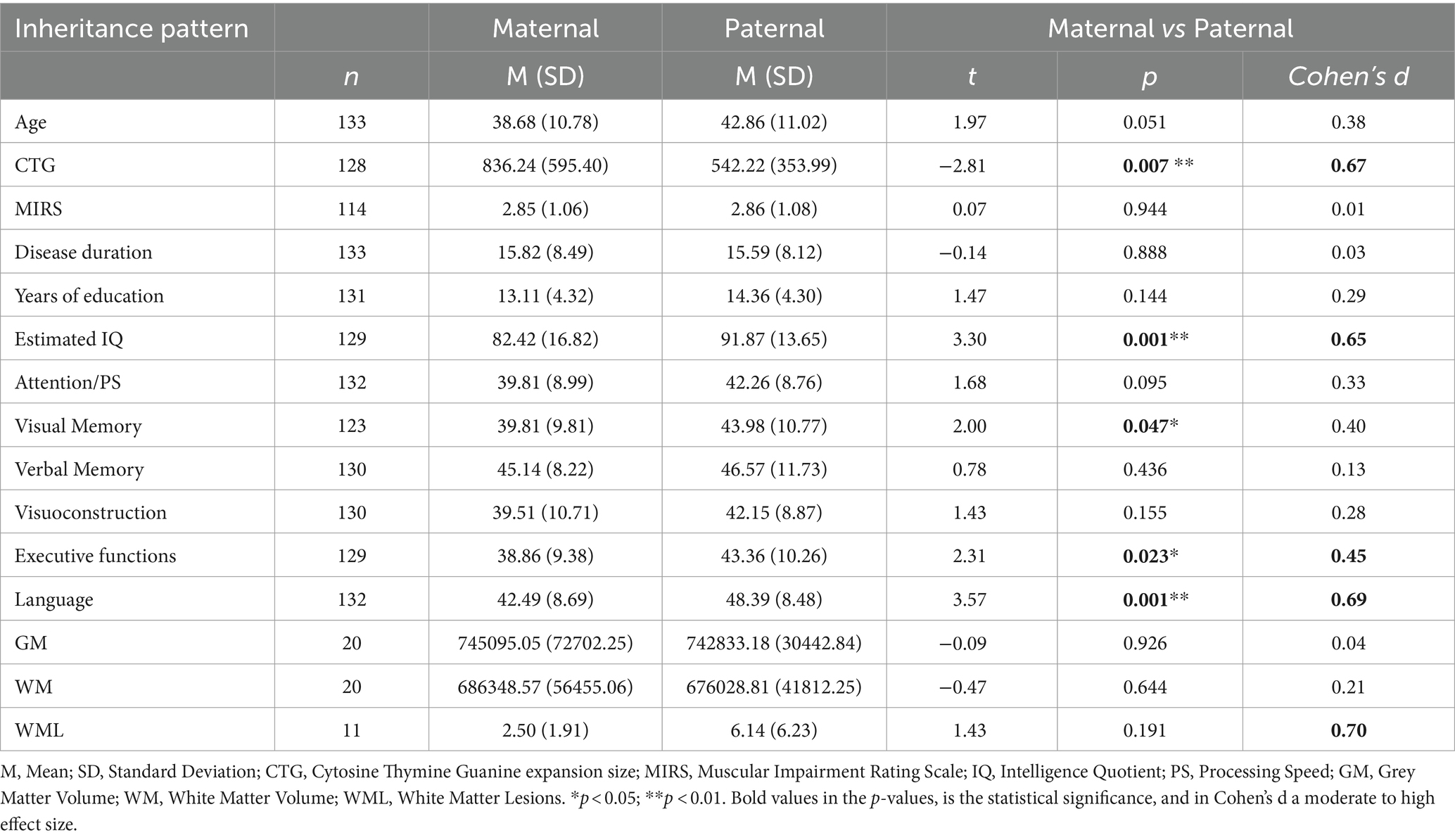
Table 4. Differences in clinical, cognitive, and brain structure outcomes according to inheritance pattern.
Regarding clinical data, there was a significant difference in phenotype distribution between maternal and paternal inheritance patients (χ2 (1) = 39.07; p < 0.001). Specifically, childhood and juvenile-onset phenotypes were more prevalent among patients with maternal inheritance, while those with paternal inheritance presented more adult and late-onset phenotypes.
Furthermore, patients with maternal inheritance showed significantly larger CTG expansion (moderate-high effect size). However, no significant muscular and disease duration differences were observed between maternal and paternal inheritance.
In relation to cognition, as shown in Table 4, DM1 patients with maternal inheritance demonstrated poorer cognitive performance than those with paternal inheritance. Specifically, when examining statistically significant differences, DM1 patients with maternal inheritance presented a lower estimated IQ and exhibited inferior performance in the cognitive domains of visual memory, executive functions, and language (moderate-high effect sizes) compared to patients with paternal inheritance.
In terms of brain structure, volume differences were not statistically significant. However, there was a moderate to high effect size in the difference between patients with paternal and maternal inheritance in WML, with paternal inheritance patients showing more lesions.
3.4 Analysis of interaction between inheritance pattern and CTG for CNS variables
Upon further analysis of the differences in CNS outcomes between maternal and paternal inheritance, the following results emerged.
Notably, the reported differences in IQ were independently influenced by both the inheritance pattern and CTG expansion size (independent effect), with no interaction between these variables. Specifically, maternal inheritance and larger CTG expansion size were associated with a lower IQ estimation. To better illustrate the independent effects of inheritance pattern and CTG expansion size on estimated IQ, a graph is displayed in Figure 1. This graph shows that patients with maternal inheritance typically present lower IQ and that the effect of CTG is greater than in paternal inheritance. This finding implies that for the same CTG expansion size, for example, a CTG expansion size of 300, the expected IQ in a patient with maternal inheritance would be 90.04, whereas in a paternal inheritance patient, it would be 94.96 (Maternal inheritance: 96.04–0.02*300 = 90.04; Paternal inheritance: 97.96–0.01 * 300 = 94.96).
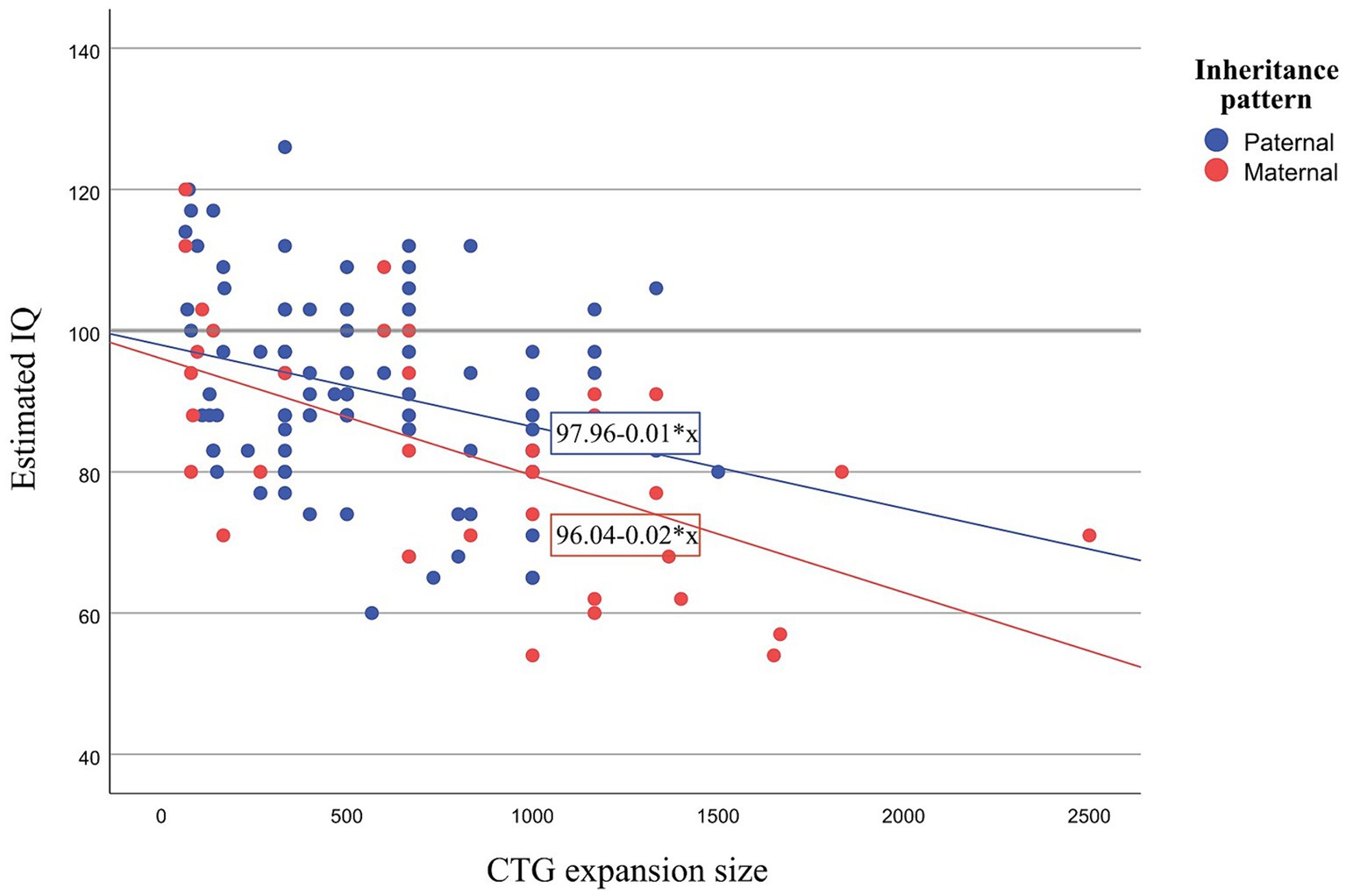
Figure 1. Scatter plot showing estimated IQ and CTG for paternal and maternal inheritance. IQ, Intelligence Quotient; CTG, Cytosine Thymine Guanine.
Analysis of the remaining cognitive domains revealed an interaction between inheritance pattern and CTG in the visual memory domain. Additionally, an independent effect of inheritance pattern was observed in the language domain, with CTG not emerging as a significant predictor. Finally, no significant differences were reported for executive functioning, which could potentially be attributed to CTG being controlled or the lower statistical power in this analysis due to the inclusion of more variables.
The following table (Table 5) displays the results of ANOVA analyses for CNS variables where differences were identified between maternal and paternal inheritance.
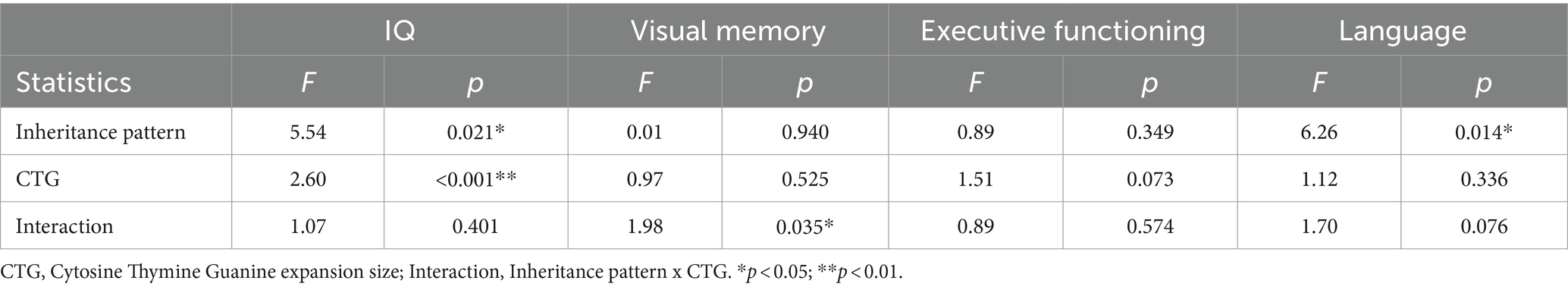
Table 5. ANOVA showing the effect of inheritance pattern, CTG and their interaction over cognitive performance.
4 Discussion
Sex differences have been studied in neuromuscular disorders, shedding light on potential differences in prevalence, onset, severity, and clinical progression of the disease between male and female patients (28). However, there is currently limited literature addressing sex differences in DM1. This study represents the first investigation focusing on sex differences in CNS involvement within this population.
Overall, the main finding of this study indicates that male and female DM1 patients show a similar clinical profile, as assessed by the variables examined in this study. Only subtle differences were found in certain cognitive domains and brain outcomes. A previous study exploring sex differences across a broad spectrum of clinical signs/symptoms in DM1 (11) indirectly assessed cognitive performance based on educational level and suggested that male patients presented more cognitive impairment than females. However, this result was not obtained in this study, in which a comprehensive cognitive assessment was conducted.
Regarding the sex of the transmitting parent (inheritance pattern), this study confirmed that the phenotype distribution differed between both groups. Specifically, patients who inherited the disease from their mother presented an earlier onset of signs and symptoms (childhood and juvenile-onset DM1), while those who inherited it from their father presented a later onset of signs and symptoms (adult and late-onset DM1). However, some authors have suggested that inheritance pattern does not impact the development of childhood-onset DM1 (6, 14). Despite earlier disease onset observed in maternally inherited patient in this study, there was no significant difference in disease duration between individuals with maternal and paternal inheritance. This might be attributed to the slight age difference, although not statistically significant, with maternally inherited patients being slightly younger.
At a molecular level, patients with maternal inheritance showed greater molecular instability (CTG expansion size) than paternally inherited patients, which could also explain the earlier onset of symptoms mentioned previously, considering that CTG typically correlates with disease severity and age of onset. Similarly, another study reported that the sex of the transmitting parent might determine the molecular defect of their offspring who inherited DM1 (16).
While maternal inheritance DM1 patients showed a greater molecular defect, they were no more muscularly affected than patients with paternal inheritance. This result could be due to the measure employed to address muscular impairment. Although the MIRS is commonly employed for this purpose, the limited range of the scale could hinder the discrimination of patients’ muscular variability.
In terms of CNS outcomes, DM1 patients with maternal inheritance presented lower IQ scores and poorer performance across various cognitive domains. It is worth noting the effect of CTG expansion size on these differences in cognitive performance between maternal and paternal inheritance patients. However, this study confirms that inheritance pattern affects certain cognitive domains regardless of CTG expansion size. For instance, when comparing two patients with identical CTG expansion sizes, the estimated IQ of the patient with maternal inheritance is expected to be lower than that of the patient with paternal inheritance.
Although recent literature has questioned the phenomenon of genomic imprinting in DM1 (13), this study suggests that the sex of the transmitting parent has phenomic significance, resulting in a differential clinical profile.
These findings open the possibility of the existence of a factor linked to the gestation period within the uterine environment of a DM1 mother, which could affect the future cognitive development of the DM1 siblings independently of the CTG size. During the premolecular stage of DM1 research, several authors speculated about the presence of a humoral factor to explain the fact that nearly all patients with congenital forms were born from affected mothers (29, 30). With the discovery of the molecular substrate of the disease and the intergenerational increase in expansion size differing by the sex of the transmitter, this hypothesis was disregarded.
However, the data from this study would allow a reconsideration of the hypothesis regarding the existence of this specific gestational factor. This factor could exhibit an epigenetic character, associated with specific changes in the function of certain genes due to processes of differential methylation or even a distinct proportion and biological effect of circulating miRNAs (31). Another non-exclusive possibility to the aforementioned is that alternative splicing of unknown maternal genes could induce aberrant proteins that traverse the placenta, generating changes in fetal brain development. Future studies with large DM1 cohorts and/or experimental studies in animal models, would be necessary to explore the existence and impact of this hypothetical maternal factor.
This study is not free of limitations. One of the main constraints when studying a rare condition is the sample size, although the sample recruited for this study was considerable. Nevertheless, not all participants underwent MRI scanning, resulting in a smaller sample size for assessing brain structural outcomes. Another issue worth considering is the high percentage of paternal inheritance (71.1%) observed in the DM1 sample, which should be taken into account when generalizing the results to the entire DM1 population. However, this higher percentage of paternal inheritance is a trend commonly observed in the literature, with reported rates of paternal transmission ranging from 42–50% for infantile DM1, 68–72% for juvenile DM1, and approximately 70% for adult and late-onset DM1 (11, 32). Furthermore, it is worth noting that congenital DM1 patients — who typically present maternal inheritance — were excluded from this study, which could partially explain the lower percentage of maternal transmission observed. Finally, the long-established genetic counseling provided by the Neurology Service in our region might have influenced the decrease in cases of maternally transmitted DM1.
Overall, does sex play a role in DM1? In general terms, while the clinic-molecular and CNS profiles of males and females with DM1 appear to be similar, the results of this study suggest that the inheritance pattern influences CNS outcomes, with a poorer prognosis observed for those with maternal inheritance. Therefore, regarding CNS involvement, sex appears to play a role, but only in relation to the sex of the transmitting parent.
The implications of these findings should be considered in clinical practice, particularly in genetic counseling and when informing patients about disease severity and prognosis. Ultimately, this knowledge can help to tailor healthcare management to meet the specific needs of each patient.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://osf.io/pzgxh.
Ethics statement
The studies involving humans were approved by Ethics Committee for Clinical Investigation of the Health Department of Gipuzkoa (DMRM-2017-01). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
JG: Conceptualization, Data curation, Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. GL: Conceptualization, Investigation, Methodology, Supervision, Validation, Writing – review & editing. JA: Data curation, Formal analysis, Supervision, Writing – review & editing. AM: Investigation, Resources, Supervision, Validation, Writing – review & editing, Conceptualization, Project administration. AS: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (Ref: 609), from the Institute of Health Carlos III co-founded by the European Union [PI17/01231 to AS; PI22/01118 to AS]; Basque Government [2022111031 to AS]; and University of the Basque Country [PIF 20/238 to JG; GU 20/057 to JG, GL, and AS].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Bigot, A, Klein, AF, Gasnier, E, Jacquemin, V, Ravassard, P, Butler-Browne, G, et al. Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am J Pathol. (2009) 174:1435–42. doi: 10.2353/ajpath.2009.080560
3. Hunter, A, Tsilfidis, C, Mettler, G, Jacob, P, Mahadevan, M, Surh, L, et al. The correlation of age of onset with CTG trinucleotide repeat amplification in myotonic dystrophy. J Med Genet. (1992) 29:774–9. doi: 10.1136/jmg.29.11.774
4. Paulson, H . Repeat expansion diseases. Handbook Clin Neurol. (2018) 147:105–23. doi: 10.1016/B978-0-444-63233-3.00009-9
5. Chatkupt, S, Antonowicz, M, and Johnson, WG. Parents do matter: genomic imprinting and parental sex effects in neurological disorders. J Neurol Sci. (1995) 130:1–10. doi: 10.1016/0022-510X(94)00284-U
6. Turner, C, and Hilton-Jones, D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry. (2010) 81:358–67. doi: 10.1136/jnnp.2008.158261
7. Scutifero, M, Lanza, M, Petillo, R, De Bernardo, M, Passamano, L, Rosa, N, et al. Gender effect on onset, prevalence and surgical treatment of cataract in patients with myotonic dystrophy type 1. Acta Myol. (2022) 41:105–10. doi: 10.36185/2532-1900-N75
8. Spaziani, M, Semeraro, A, Bucci, E, Rossi, F, Garibaldi, M, Papassifachis, MA, et al. Hormonal and metabolic gender differences in a cohort of myotonic dystrophy type 1 subjects: a retrospective, case–control study. J Endocrinol Investig. (2020) 43:663–75. doi: 10.1007/s40618-019-01156-w
9. Liguori, S, Moretti, A, Toro, G, Paoletta, M, Palomba, A, Barra, G, et al. Pain and motor function in myotonic dystrophy type 1: a cross-sectional study. IJERPH. (2023) 20:5244. doi: 10.3390/ijerph20075244
10. Solbakken, G, Løseth, S, Froholdt, A, Eikeland, TD, Nærland, T, Frich, JC, et al. Pain in adult myotonic dystrophy type 1: relation to function and gender. BMC Neurol. (2021) 21:101. doi: 10.1186/s12883-021-02124-9
11. Dogan, C, De Antonio, M, Hamroun, D, Varet, H, Fabbro, M, Rougier, F, et al. Gender as a modifying factor influencing myotonic dystrophy type 1 phenotype severity and mortality: a Nationwide multiple databases cross-sectional observational study. PLoS One. (2016) 11:e0148264. doi: 10.1371/journal.pone.0148264
12. Ouyang, L, Wang, Y, Valdez, R, Johnson, N, Gutmann, L, Street, N, et al. Gender difference in clinical conditions among hospitalized adults with myotonic dystrophy. Muscle Nerve. (2019) 59:348–53. doi: 10.1002/mus.26402
13. Lanni, S, and Pearson, CE. Molecular genetics of congenital myotonic dystrophy. Neurobiol Dis. (2019) 132:104533. doi: 10.1016/j.nbd.2019.104533
14. Stokes, M, Varughese, N, Iannaccone, S, and Castro, D. Clinical and genetic characteristics of childhood-onset myotonic dystrophy. Muscle Nerve. (2019) 60:732–8. doi: 10.1002/mus.26716
15. Meola, G, and Cardani, R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta (BBA) - Mol Basis Dis. (2015) 1852:594–606. doi: 10.1016/j.bbadis.2014.05.019
16. Brunner, HG, Brüggenwirth, HT, Nillesen, W, Jansen, G, Hamel, BC, Hoppe, RL, et al. Influence of sex of the transmitting parent as well as of parental allele size on the CTG expansion in myotonic dystrophy (DM). Am J Hum Genet. (1993) 53:1016–23.
17. Wechsler, D . WAIS-III: Escala de Inteligencia de Wechsler para Adultos III. Madrid: TEA Ediciones (1999).
19. Miller, EN . CalCAP: California computerized assessment package. Los Angeles: Norland Software (1990).
20. Tulsky, DS, Chiaravalloti, ND, Palmer, BW, and Chelune, GJ. The Wechsler memory scale In: Clinical interpretation of the WAIS-III and WMS-III. 3rd ed. San Diego: Academic Press (2003). 93–139.
21. Lezak, M, Howieson, H, and Loring, D. Neuropsychological assessment. 4th ed. New York: Oxford University Press (2004).
22. Osterrieth, PA . Le test de copie d’une figure complexe; contribution à l’étude de la perception et de la mémoire [Test of copying a complex figure; contribution to the study of perception and memory]. Arch Psychol. (1944) 30:206–356.
23. Casals-Coll, M, Sánchez-Benavides, G, Quintana, M, Manero, RM, Rognoni, T, Calvo, L, et al. Estudios normativos españoles en población adulta joven (proyecto NEURONORMA jóvenes): normas para los test de fluencia verbal. Neurologia. (2013) 28:33–40. doi: 10.1016/j.nrl.2012.02.010
24. Pena-Casanova, J, Quinones-Ubeda, S, Quintana-Aparicio, M, Aguilar, M, Badenes, D, Molinuevo, JL, et al. Spanish multicenter normative studies (NEURONORMA project): norms for verbal span, visuospatial span, letter and number sequencing, trail making test, and symbol digit modalities test. Arch Clin Neuropsychol. (2009) 24:321–41. doi: 10.1093/arclin/acp038
25. Pena-Casanova, J, Quinones-Ubeda, S, Gramunt-Fombuena, N, Aguilar, M, Casas, L, Molinuevo, JL, et al. Spanish multicenter normative studies (NEURONORMA project): norms for Boston naming test and token test. Arch Clin Neuropsychol. (2009) 24:343–54. doi: 10.1093/arclin/acp039
26. Andersson, JL, Jenkinson, M, and Smith, S. Non-linear registration aka spatial normalisation FMRIB Technial report TR07JA2. Oxford: FMRIB Analysis Group of the University of Oxford (2007).
27. Wahlund, LO, Barkhof, F, Fazekas, F, Bronge, L, Augustin, M, Sjögren, M, et al. A new rating scale for age-related white matter changes applicable to MRI and CT. Stroke. (2001) 32:1318–22. doi: 10.1161/01.STR.32.6.1318
28. Vinciguerra, C, Iacono, S, Bevilacqua, L, Landolfi, A, Piscosquito, G, Ginanneschi, F, et al. Sex differences in neuromuscular disorders. Mech Ageing Dev. (2023) 211:111793. doi: 10.1016/j.mad.2023.111793
29. Farkas-Bargeton, E, Barbet, JP, Dancea, S, Wehrle, R, Checouri, A, and Dulac, O. Immaturity of muscle fibers in the congenital form of myotonic dystrophy: its consequences and its origin. J Neurol Sci. (1988) 83:145–59. doi: 10.1016/0022-510X(88)90064-0
30. Harper, P, and Dyken, P. Early-onset dystrophia myotonica evidence supporting a maternal environmental factor. Lancet. (1972) 300:53–5. doi: 10.1016/S0140-6736(72)91548-6
31. Perfetti, A, Greco, S, Cardani, R, Fossati, B, Cuomo, G, Valaperta, R, et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci Rep. (2016) 6:38174. doi: 10.1038/srep38174
Keywords: myotonic dystrophy type 1, DM1, sex, inheritance pattern, CTG, genomic imprinting, CNS, cognition
Citation: Garmendia J, Labayru G, Aliri J, López de Munain A and Sistiaga A (2024) CNS involvement in myotonic dystrophy type 1: does sex play a role? Front. Neurol. 15:1399898. doi: 10.3389/fneur.2024.1399898
Edited by:
Corrado Italo Angelini, University of Padua, ItalyReviewed by:
Roberto Massa, University of Rome Tor Vergata, ItalyGiuseppe Vita, University of Messina, Italy
Copyright © 2024 Garmendia, Labayru, Aliri, López de Munain and Sistiaga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andone Sistiaga, andone.sistiaga@ehu.eus