 Udaya Geetha Vijayakumar†
Udaya Geetha Vijayakumar† Vanessa Milla†
Vanessa Milla† Mei Yu Cynthia Stafford
Mei Yu Cynthia Stafford Anthony J. BjoursonWilliam Duddy
Anthony J. BjoursonWilliam Duddy Stephanie Marie-Rose Duguez*
Stephanie Marie-Rose Duguez*- Northern Ireland Center for Stratified Medicine, Biomedical Sciences Research Institute, Londonderry, United Kingdom
Amyotrophic lateral sclerosis (ALS), also known as motor neuron disease, is an incurable neurodegenerative condition, characterized by the loss of upper and lower motor neurons. It affects 1–1.8/100,000 individuals worldwide, and the number of cases is projected to increase as the population ages. Thus, there is an urgent need to identify both therapeutic targets and disease-specific biomarkers–biomarkers that would be useful to diagnose and stratify patients into different sub-groups for therapeutic strategies, as well as biomarkers to follow the efficacy of any treatment tested during clinical trials. There is a lack of knowledge about pathogenesis and many hypotheses. Numerous “omics” studies have been conducted on ALS in the past decade to identify a disease-signature in tissues and circulating biomarkers. The first goal of the present review was to group the molecular pathways that have been implicated in monogenic forms of ALS, to enable the description of patient strata corresponding to each pathway grouping. This strategy allowed us to suggest 14 strata, each potentially targetable by different pharmacological strategies. The second goal of this review was to identify diagnostic/prognostic biomarker candidates consistently observed across the literature. For this purpose, we explore previous biomarker-relevant “omics” studies of ALS and summarize their findings, focusing on potential circulating biomarker candidates. We systematically review 118 papers on biomarkers published during the last decade. Several candidate markers were consistently shared across the results of different studies in either cerebrospinal fluid (CSF) or blood (leukocyte or serum/plasma). Although these candidates still need to be validated in a systematic manner, we suggest the use of combinations of biomarkers that would likely reflect the “health status” of different tissues, including motor neuron health (e.g., pNFH and NF-L, cystatin C, Transthyretin), inflammation status (e.g., MCP-1, miR451), muscle health (miR-338-3p, miR-206) and metabolism (homocysteine, glutamate, cholesterol). In light of these studies and because ALS is increasingly perceived as a multi-system disease, the identification of a panel of biomarkers that accurately reflect features of pathology is a priority, not only for diagnostic purposes but also for prognostic or predictive applications.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurological disorder with an adult onset around 54–67 years old (1). Its clinical hallmark is the degeneration of both upper and lower motor neurons (2, 3), leading to progressive muscle atrophy and weakness, and ultimately to paralysis. Death, often resulting from swallowing problems and respiratory failure (4, 5), generally occurs within 2–4 years from disease onset (6–8), although 5–10% of ALS patients survive over 10 years (7). ALS has a median incidence of about 2.8 cases per 100,000 persons per year and a median prevalence about 5.4 cases per 100,000 persons for a median age at 61.8 ± 3.8 years (1). The incidence and prevalence thus increases with age and reaches a cumulative lifetime risk of 1 in 400 after 80 years old (9, 10). Due to the projected aging of the global population, ALS cases are expected to increase by 69% in the next 25 years (11), underlining the urgent need to identify causes, biomarkers and therapeutic targets for ALS.
The causes of ALS are largely unknown, with ~90% of cases being sporadic (sALS) while only ~10% are familial ALS (fALS) (12). Intensive research since the 1990's has aimed to unravel the mechanisms involved in motor neuron degeneration. These studies suggest that ALS is a complex disease driven by a combination of several systemic parameters. To date, up to 30 genes (Figure 1) are described as monogenic causes of ALS, with the most frequent being C9orf72, SOD1, FUS, and TARDBP/TDP43 (13–15). In motor neurons, these identified mutations are functionally associated with an alteration of electrophysiological properties (16), accumulation of stress marks (17) and sensitivity to stress (18) (Figure 2). However, these monogenic forms explain only 15% of sporadic cases and 66% of familial cases (12) (Figure 1).
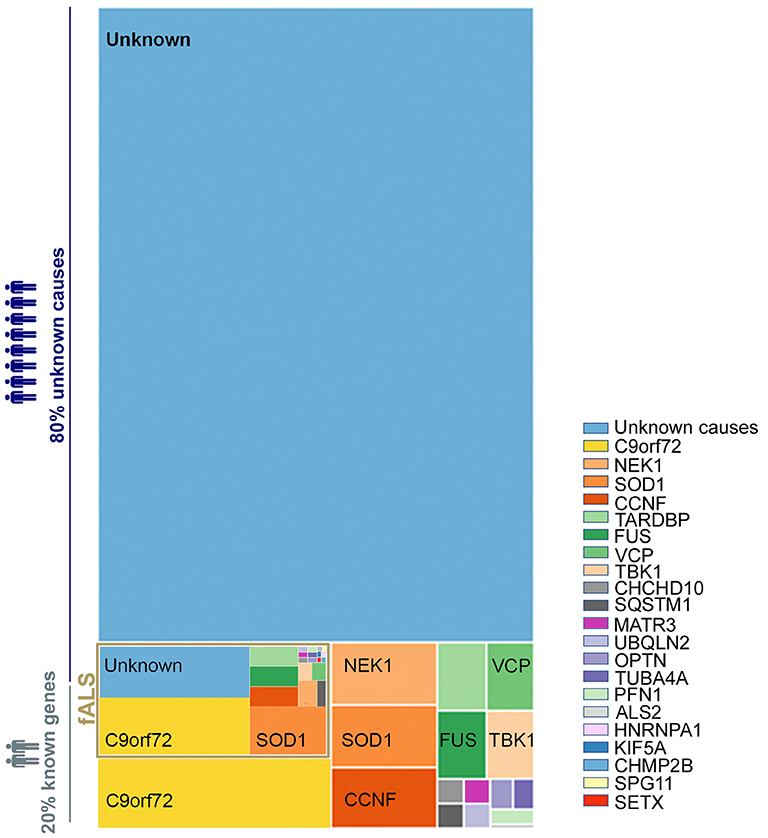
Figure 1. Distribution of genetic basis among the ALS population. A treemap representation of the proportion of ALS patients carrying known causative mutation. The full rectangle represents 100% of all ALS cases. The fALS are highlighted in gold with a frequency adjusted to represent 7.5% of the total (as fALS is estimated at 5–10% of all ALS cases). The two light blue blocks represent those with no known ALS-associated gene mutation among sporadic and familial cases. Cases with known mutations are represented in the other blocks, broken down by affected gene. The color code for each gene is preserved between familial and sporadic cases. The size of each block is proportional to the percentage of ALS associated to the considered genes–proportions given in Volk et al. (13). Overall, some 80% of ALS cases (sALS and fALS combined) are not explained by a known mutation.
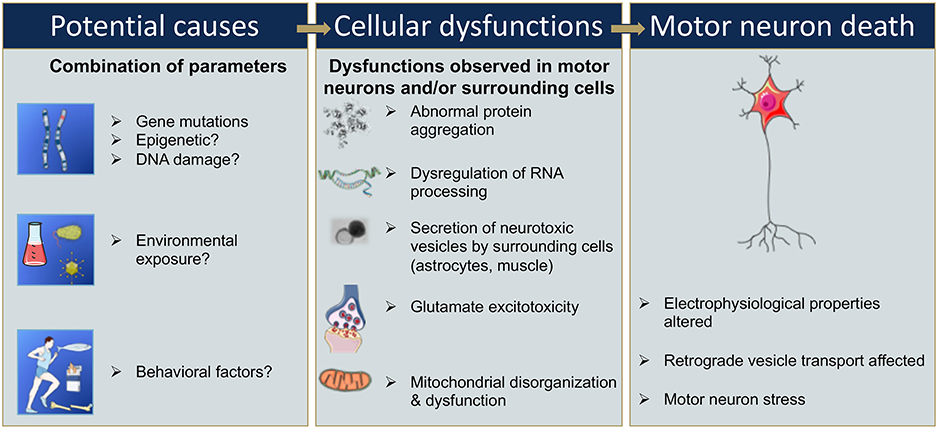
Figure 2. Sequential events that could be involved in motor neuron death in ALS. Gene mutations, epigenetic changes, or DNA damage that occur either spontaneously or due to environmental risk factors such as exposure to toxins or infectious agents, or behavioral factors, have all been proposed as potentially leading to cellular dysfunction (9, 13, 14, 20–23). Cellular dysfunction could include abnormal protein aggregations, alteration of RNA processing, secretion of neurotoxic vesicles by surrounding cells such as astrocyte, muscle cells, glutamate excitotoxicity, and mitochondrial disorganization and dysfunction leading to oxidative stress (24–30). These cellular dysfunctions may take place in motor neurons and/or surrounding cells and, combined or alone, could lead to an alteration of the electrophysiological properties of the motor neuron, and/or to an induction of secretion of neurotoxic elements by surrounding cells, in either case ultimately leading to motor neuron death (16–18).
Furthermore, the penetrance of these disease-associated mutations is quite variable and can increase with age (12, 19). The variability in penetrance as well as the lack of identification of a single associated gene mutations in 85% of sALS suggests that some ALS cases have a multigenic component, and/or involve epigenetic modification, and/or result from DNA damage, environmental risk factors, or viral infections (9, 14, 20–23) (Figure 2). In these cases, it is likely a combination of these factors that leads to cellular dysfunction such as glutamate-mediated excitotoxicity (24), abnormal protein aggregation (25), mitochondrial disorganization and dysfunction (26, 27) contributing to the oxidative stress (28–30) (Figure 2). Adding to the complexity of ALS, several studies suggest that not only the motor neurons are affected but also the surrounding cells, and that these cells participate in the propagation and burden of the disease. For instance, activated microglia cells release superoxide and nitric oxide metabolites, elements that are toxic to neuronal cells (31). Astrocytes can also participate in the propagation of neurotoxic elements (32, 33) such as SOD1 aggregates (34–36), and a failure of astrocytes to remove extracellular glutamate may mediate excitotoxicity (37–39). Ultimately, the intracellular dysfunction of the motor neuron combined with aberrant secretion of neurotoxic elements of surrounding cells leads to motor neuron stress, aberrant electrophysiological properties, and consequently to motor neuron death (Figure 2).
In the absence of a reliable diagnostic test for ALS, diagnosis is based on clinical and electrophysiological criteria such as evidence for progressive involvement of both upper and lower motor neurons and exclusion of diseases mimicking ALS as set out in the Revised El Escorial Criteria (REEC), Airlie House criteria (AHC) and Awaji criteria (2, 40). The process of diagnosis can be lengthy and there is a typical diagnostic delay of 9-15 months from onset to diagnostic confirmation (41). Considering that the average survival from onset is 2–4 years (6–8) and that efficacy of Riluzole is improved by early treatment (42), there is an urgent need to improve diagnostic speed and accuracy for ALS. One way of achieving this is the identification of biomarkers specific to ALS pathology, to enable the development a reliable fast diagnostic test. As well as diagnostics, it is also important to identify prognostic biomarkers that can be used to monitor the status of the pathology–various candidates may serve both these purposes. The identification of ALS biomarkers will contribute to a better understanding of the disease pathogenesis, and permit targeted drug development and patient stratification for more efficient clinical trials, assuming that different sub-cohorts of ALS patients respond differently to treatments. Biomarker discovery can be achieved by examining the “omics” contents of ALS patient tissues.
The present review has two aims: (1) to identify pathways commonly affected in genetic forms of ALS, and stratify the patients accordingly, and (2) to explore previous genomic, transcriptomic, proteomic, metabolomic and miRNomic studies of ALS published during the last decade, and summarize the findings, highlighting potential biomarker candidates for ALS disease management and treatment.
Genetic Markers for ALS Patient Stratification
The first gene identified to be associated with ALS was SOD1 in 1993 (43). Since then 29 new genes have been identified (13–15), representing the most frequent genetic mutations included in current diagnostic processes (13, 44) (Figure 1). These 30 genes offer crucial clues in understanding the pathogenesis of ALS—some of the gene products interact with each other (14)—and enable the identification of diverse cellular pathways that are disrupted in ALS patients (Table 1). Even if most ALS cases are sporadic, the pathways disrupted in familial cases may also be affected in sporadic cases, as both sALS and fALS can share common molecular signatures or functional biological effects such as FUS or TDP43 protein aggregations or accumulation of stress granules formation (45), disruption in RNA processing (46), or disruption of autophagy and mitochondrial functions (47). When sorting the genes associated to ALS according to their primary cellular functions, several categories of dominantly affected pathway can be highlighted, such as (1) mitochondrial metabolism and turnover, (2) axonal transport and the cytoskeleton, (3) autophagy and proteostasis, (4) endosomal and vesicular trafficking, (5) DNA repair, and (6) ribostasis/RNA alteration/Nucleocytoplasmic transport—with most of the genes being involved in multiple pathways. It may be possible to group patients into strata depending on which combination of pathways is dysregulated, and to recruit patients accordingly for translational research and clinical trials. We have cautiously assigned each causal gene to one of 14 strata, depending on the profile of its affected pathways (Table 1). These groupings represent our effort to summarize current understanding and are not intended to be definitive—indeed, it will be important to modify and update them on an ongoing basis with improvements in the knowledge of protein function and the impact of mutations. Although these 14 strata are directly applicable to only 20% of total ALS cases (Figure 1), future work may determine whether (and which of) these molecular signatures are implicated in the remaining cases.
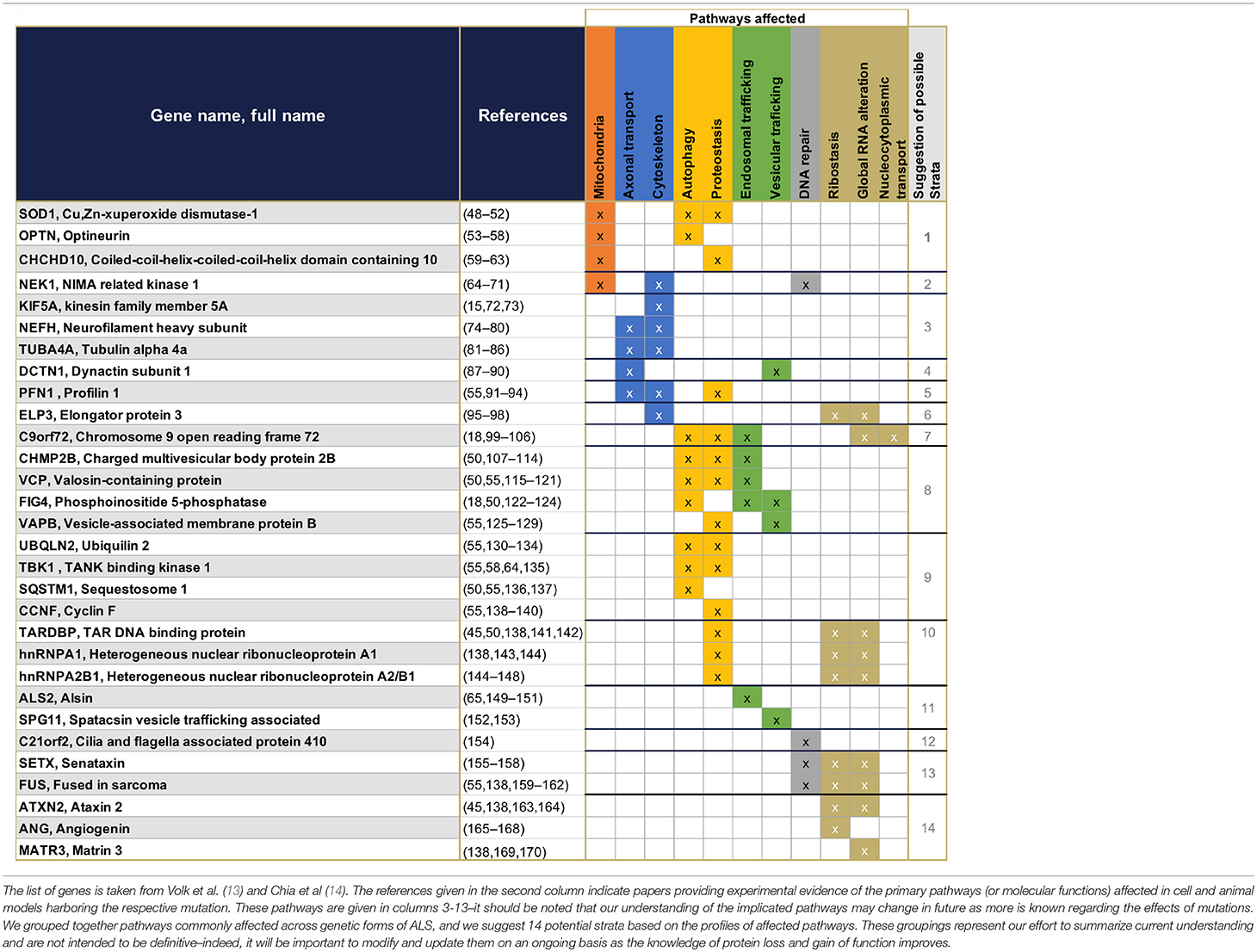
Table 1. Summary of the 30 genes presently known to have monogenic association with ALS, and their primary functions.
The Search for Circulating Biomarkers
The identification of circulating markers associated with ALS pathology would be important tools to provide early disease diagnosis and to track progression or treatment. There has been a concerted focus aimed at identifying such biomarkers in different body fluids over the past 20 years. In Table S1, we summarized 76 studies that investigated proteins, miRs, mRNAs, and metabolites as potential biomarkers in cerebrospinal fluid (CSF) or blood (blood cells, serum or plasma). To date, little has been done investigating urine-based biomarkers, and thus urine biomarker analyses are not reported in the current review. CSF is the most frequently used sample source, and several studies (Table S1) report a consistent decrease in protein levels of transthyretin—involved in neurogenesis, nerve repair and axonal growth (171)—and cystatin c—an endogenous cysteine protease inhibitor that can protect motor neurons against neurotoxicity by stimulating autophagy and inhibition of cathepsin B (172). In addition, CSF cystatin C protein levels positively correlated with the survival of ALS patients and could be thus potentially used as a prognostic biomarker (173). However, both transthyretin and cystatin C decreases are not specific to ALS patients and a similar pattern is observed in other neurodegenerative diseases (173) such as Alzheimer's (171), suggesting that the protein levels of both transthyretin and cystatin C level are a common signature for neuron vulnerabilities and neurodegeneration. The protein levels of neurofilament light chain (NF-L) and the phosphorylated form of neurofilament heavy chain (pNFH) were also consistently found to be increased in the CSF of ALS patients across multiple studies (Table S1), with a high level of either NF-L or pNFH predicting a shorter life expectancy (174–178). NF-L and pNFH are markers for axonal damage (179). In this context, similarly to M-creatine kinase for myofiber fragility in muscular dystrophy (180), NF-L and pNFH thus directly reflect the health of the neurons –the cells specifically impacted by ALS.
Combining NF-L and pNFH with other markers that reflect the “health status” of other tissues such as glial cells, skeletal muscle, or inflammatory response, may represent a useful addition, as ALS is now perceived as a multisystemic disease. Such a multi-marker approach may represent a useful complement to a panel of biomarkers to test the efficacy of drugs in clinical trials. In this respect, miR-451—an inhibitor of microglial cell activation (181)—was consistently decreased in leukocytes of ALS patients (Table S1), while the pro-inflammatory MCP-1, secreted by the glial cells and neurons (182), was found to be increased in both serum and plasma (Table S1). Both miR-451 and MCP-1 could thus potentially inform the status of inflammatory cell recruitment and activation (181, 182). In addition, miR-206, which is essential for skeletal muscle growth and regeneration (183), as well as miR-338-3p, a regulator of neuromuscular junctions (184), are consistently upregulated in leukocytes—with miR-206 also consistently reported to be upregulated in serum and plasma samples across multiple studies (Table S1). In this context, miR-206 and miR-338-3p could be clinically useful candidate biomarkers of the health status of skeletal muscle (185).
Regarding circulating mRNAs, no obvious consistent candidates have been identified yet across previous studies (Table S1). With regard to analyses of circulating metabolite candidates, huge variation is observed between studies, though there was a general tendency for upregulation of specific metabolites in serum and plasma (Table S1), which is consistent with the hypermetabolism observed in some ALS patients (186). For instance, creatine, which is linked to cell energy metabolism, was consistently increased in CSF and plasma across studies (Table S1). Pyruvate and glucose were also found to be increased in CSF and plasma of ALS patients (Table S1), potentially reflecting a dysregulation of glycolytic metabolism as observed in SOD1-G93A motor neurons (187), and in some ALS cases (188, 189). This upregulation of glycolysis correlates with a shorter survival time and thus could be used as a prognostic biomarker (188, 189). Similarly, the upregulation of cholesterol and LDL observed in CSF and plasma across studies (Table S1) could also reflect a global dysregulation of lipid metabolism in ALS patients (190, 191). Other neurotoxic metabolites, such as homocysteine, were consistently increased in all body fluids (Table S1). Altogether, these data suggest a global dysregulation of the energy metabolism in ALS patients.
Other types of molecules could be investigated as biomarkers in ALS, such as long non-coding RNA (lncRNA), which can act in cis to either silence or enhance the expression of proximal genes (192) and which are known to have a key role in normal neuronal development, as well as in development and progression of neurodegenerative diseases [see (193) for review]. The lncRNA have also been detected in body fluids and have been suggested as potential diagnostic and/or prognostic biomarkers in, but not only, lung cancer (194), triple negative breast cancer (195) and cardiovascular diseases (196). In this context, lncRNA could be investigated as new biomarker candidates for neurodegenerative diseases (193), including ALS.
Exploring potential ALS Signatures in Tissue
Studying changes at the molecular level of specific tissues affected in ALS should improve our understanding of the disease mechanisms and multi-systemic impact.
Postmortem brain or spinal cord have been widely investigated. Accumulation of pNF-H and NF-L in brain tissue (Table S2) positively correlate with the accumulation of these markers in CSF (Table S1), and may be reflective of motor neuron breakdown (179). Similarly, miR-146a and miR-338-3p, both increased in spinal cord (Table S2), are also detected at a greater level in circulating blood cells of ALS patients (Table S1). These two miRNAs are involved in the regulation of the inflammatory response (197) and the neuromuscular junction (184, 198). In addition, miR-206, a skeletal muscle growth regulator (183), is increased in ALS muscles across studies [Table S1, 2 studies show significant increases (199, 200), the third study only shows a tendency toward an increase in levels (201)]. Together these data reinforce the suggestion that these candidate biomarkers may have utility in determining the status of motor neurons, inflammatory cells and muscle in ALS at different stages of the disease.
When looking at the proteomic and transcriptomic signature of ALS tissues, most observations have not been reproduced across studies. This lack of repeatability could be attributed to numerous factors, such as: different study populations; different types of control subject; different sample sources; different stages of the disease; and the use of different methodological strategies (Table S2).
However, when looking at the different pathways affected in nervous or muscle tissues, we can identify dominant signatures. For instance, skeletal muscle exhibits a dysregulation of pathways involved in muscle atrophy/growth, cytoskeletal maintenance and metabolism, while the central nervous system exhibits inflammatory and excitotoxicity features accompanied by disruptions in axonal transport, cell death, autophagy, metabolism, and RNA processing (Table S2). Concordantly, the systematic decrease of N-acetyl-aspartate observed in vivo by magnetic resonance spectrometry in the central nervous system across studies reflects (Table S2) neuron degeneration. These markers likely capture most strongly the endpoints of ALS disease, including degeneration processes in motor neuron death, and muscle denervation and atrophy, and it will be important for future studies to identify biomarkers that track early features of the disease.
Conclusion
The number of monogenic forms, combined with potential multisystemic contributions to ALS pathology, render it difficult first to unravel physiopathological events, and then to understand which of these events could be pharmacologically targeted. However, by taking a wide-angle view of the pathways affected in different monogenic forms of the disease, it is possible to discern patient strata, with each stratum potentially representing a separate target for therapeutic intervention. Such a strategy is directly applicable to monogenic forms of ALS—known in ~20% of current ALS cases—and future work may discover the extent to which each of these potential targets are transferrable to the 80% of cases in which causal links (genetic or otherwise) have not been identified. Identifying biomarkers to diagnose ALS patients and predict their progression (prognostic biomarkers) may also lead to the identification of patient strata in these non-causally linked forms of ALS.
Identifying such biomarkers in ALS is a significant challenge as it involves the assessment, not only of motor neuron health status, but also that of other cell types affected in ALS such as astrocytes, microglia, skeletal muscle and inflammatory cells. In this review, we collated across a large number of recently published studies on ALS biomarkers covering several different cell and tissue types (76 studies on body fluids and 42 studies on tissues), and identified only a relatively few candidates that are consistently identified as potential biomarkers across multiple independent studies. These candidate biomarkers are predominantly reflective of motor neuron health, the inflammatory status, and skeletal muscle health (Figure 3). As ALS is increasingly recognized as a multi-systemic disease, it is thus important to track the progression or the recovery of these multiple tissues during clinical trials. In addition, some of these candidates have been confirmed in murine models, e.g., miR-206 in SOD1-G93A mice reflects disease progression in the murine model (202), making them interesting candidates for assessment in pre-clinical studies. As a multi-systemic disease, it is likely that a panel of biomarkers will be needed to fully capture features of ALS pathology.
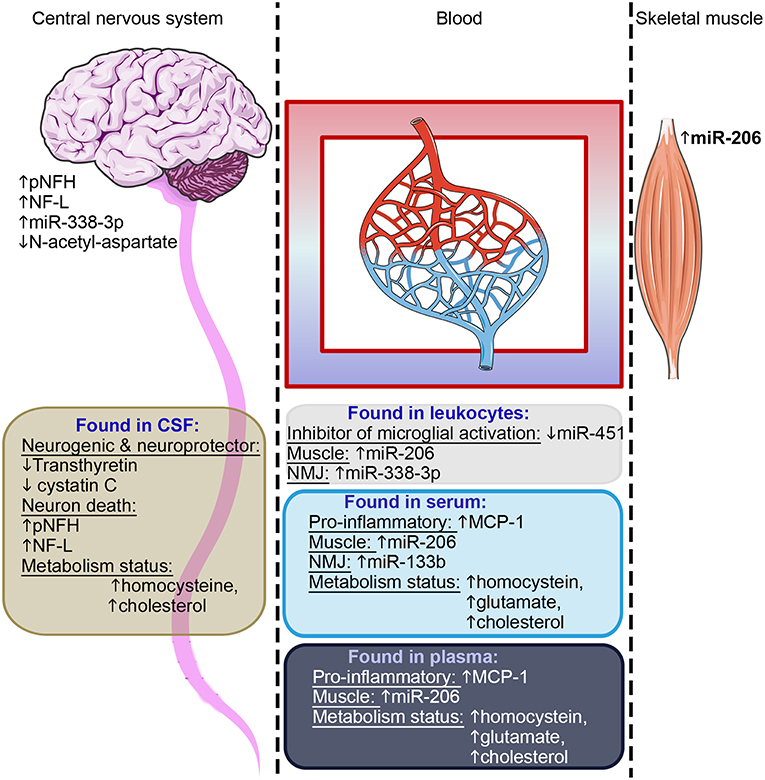
Figure 3. Summary of candidate biomarkers consistently found across studies. Candidates observed in CSF are highlighted in brown, in leukocytes in gray, in serum light blue and in plasma dark blue. These candidate biomarkers reflect the motor neuron health, the inflammatory status, skeletal muscle health, and metabolism status–as indicated in each text block. Some of these candidates were found in postmortem central nervous tissue or on muscle biopsies. NMJ, neuromuscular junction.
Considering the different source tissues and the potential implication of each of these in the pathology, our capacity to detect them in accessible fluids, and also the desire to have biomarkers that are confirmed in multiple studies, we would suggest that a useful approach to obtain an overall picture of disease progress in any given patient, may be to combine biomarker candidate molecules from across those listed in Table 2. For example, of biomarkers confirmed in multiple studies, we could suggest a panel of Cystatin C, pNFH and NF-L, all reflecting neuronal survival, MCP1 as a pro-inflammatory marker, the MiRs 206 and 133b reflecting muscle origin and neuromuscular junction, respectively, and some indicators of dysregulated metabolism such as homocysteine, glutamate, or cholesterol. Such a panel (or a variation of it with similarly diverse properties in terms of tissue origin), would be useful to assess the overall “health status” of different tissues. However, all of the biomarkers so far proposed require further validation, as would any specific combination of them.
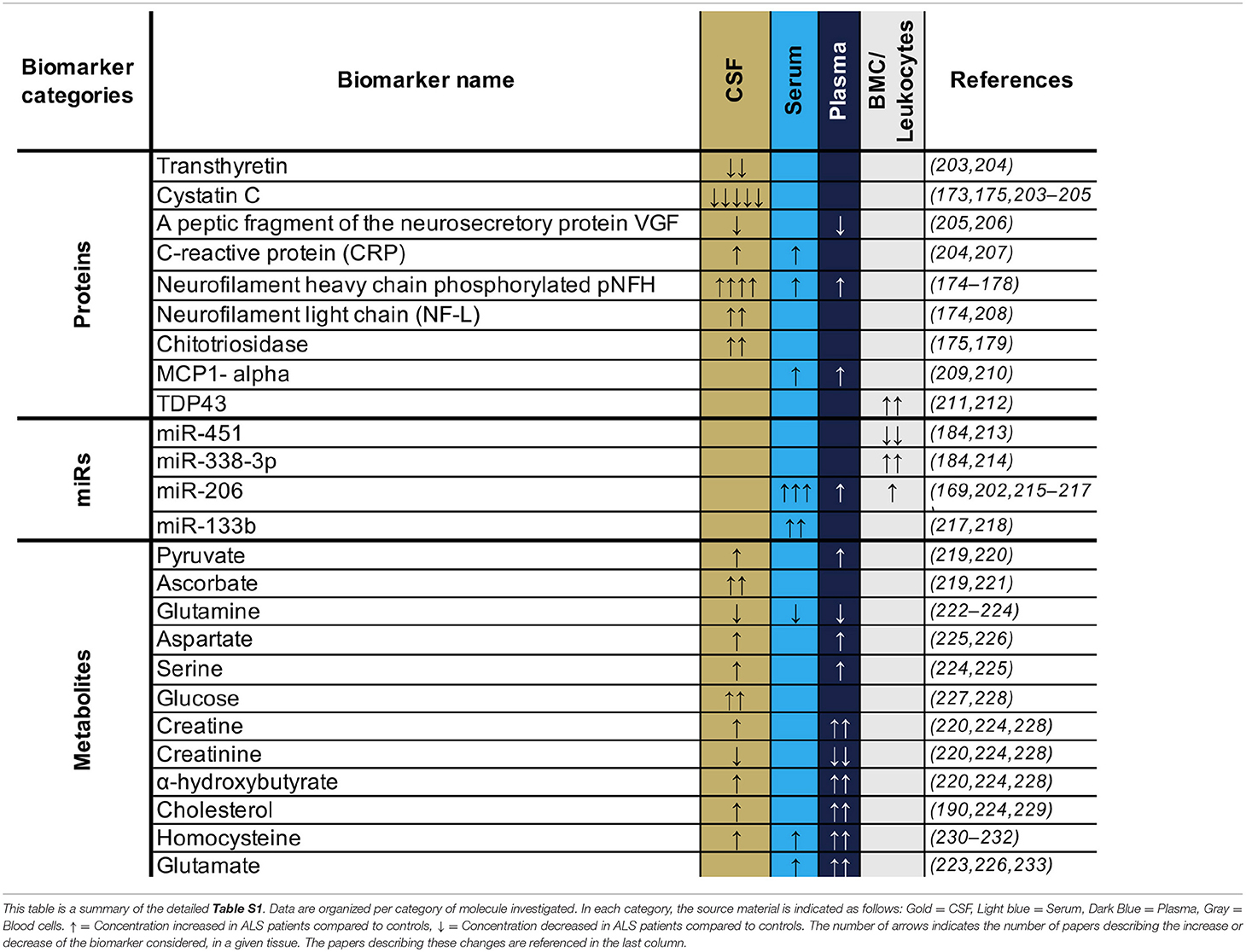
Table 2. Circulating biomarker candidates consistently observed and confirmed across studies.
The development of a heterogeneous multi-biomarker panel—likely including robust new biomarkers and the biomarkers cited in this report—could be seen as a priority, not only for diagnostic purposes but also for prognostic or predictive applications.
Author Contributions
UV, VM, and MS collated the data from the literature, and wrote the paper. WD and SD organized the data, wrote the paper. AB, WD, and SD edited the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financed by TARGET-ALS (ViTAL consortium, PI: SD), ARsLA (TEAM consortium, PI: SD), European Union Regional Development Fund (ERDF) EU Sustainable Competitiveness Programme for N. Ireland, Northern Ireland Public Health Agency (HSC R&D) & Ulster University (PI: AB). UV's post-doctoral position is financed by Target-ALS.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00400/full#supplementary-material
References
1. Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. (2013) 41:118–30. doi: 10.1159/000351153
2. Al-Chalabi A, Hardiman O, Kiernan MC, Chiò A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. (2016) 15:1182–94. doi: 10.1016/S1474-4422(16)30199-5
3. Talbot K, Feneberg E, Scaber J, Thompson AG, Turner MR. Amyotrophic lateral sclerosis: the complex path to precision medicine. J Neurol. (2018) 265:2454–62. doi: 10.1007/s00415-018-8983-8
4. Niedermeyer S, Murn M, Choi PJ. Respiratory failure in amyotrophic lateral sclerosis. Chest. (2018) 155:401–8. doi: 10.1016/j.chest.2018.06.035
5. Luchesi KF, Kitamua S, Mourão LF. Amyotrophic Lateral Sclerosis survival analysis: swallowing and non-oral feeding. NeuroRehabilitation. (2014) 35:535–42. doi: 10.3233/NRE-141149
6. Talbott EO, Malek AM, Lacomis D. The epidemiology of amyotrophic lateral sclerosis. Handb Clin Neurol. 138:225–38. doi: 10.1016/B978-0-12-802973-2.00013-6
7. Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. (2009) 10:310–23. doi: 10.3109/17482960802566824
8. del Aguila MA, Longstreth WT, McGuire V, Koepsell TD, van Belle G. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. (2003) 60:813–9. doi: 10.1212/01.WNL.0000049472.47709.3B
9. Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. (2017) 377:1602. doi: 10.1056/NEJMc1710379
10. Broussalis E, Grinzinger S, Kunz AB, Killer-Oberpfalzer M, Haschke-Becher E, Hartung H-P, et al. Late age onset of amyotrophic lateral sclerosis is often not considered in elderly people. Acta Neurol Scand. (2018) 137:329–34. doi: 10.1111/ane.12869
11. Arthur KC, Calvo A, Price TR, Geiger JT, Chiò A, Traynor BJ. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun. (2016) 7:12408. doi: 10.1038/ncomms12408
12. Turner MR, Al-Chalabi A, Chio A, Hardiman O, Kiernan MC, Rohrer JD, et al. Genetic screening in sporadic ALS and FTD. J Neurol Neurosurg Psychiatry. (2017) 88:1042–4. doi: 10.1136/jnnp-2017-315995
13. Volk AE, Weishaupt JH, Andersen PM, Ludolph AC, Kubisch C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med Genet. (2018) 30:252–8. doi: 10.1007/s11825-018-0185-3
14. Chia R, Chiò A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. (2018) 17:94–102. doi: 10.1016/S1474-4422(17)30401-5
15. Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron. (2018) 97:1268–83.e6. doi: 10.1016/j.neuron.2018.02.027
16. Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SSW, Sandoe J, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. (2014) 7:1–11. doi: 10.1016/j.celrep.2014.03.019
17. Seminary ER, Sison SL, Ebert AD. Modeling protein aggregation and the heat shock response in ALS iPSC-derived motor neurons. Front Neurosci. (2018) 12:86. doi: 10.3389/fnins.2018.00086
18. Shi Y, Lin S, Staats KA, Li Y, Chang W-H, Hung S-T, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. (2018) 24:313–25. doi: 10.1038/nm.4490
19. Majounie E, Renton AE, Mok K, Dopper EGP, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. (2012) 11:323–30. doi: 10.1016/S1474-4422(12)70043-1
20. Armon C. From snow to hill to ALS: an epidemiological odyssey in search of ALS causation. J Neurol Sci. (2018) 391:134–40. doi: 10.1016/j.jns.2018.05.016
21. Xue YC, Feuer R, Cashman N, Luo H. Enteroviral infection: the forgotten link to amyotrophic lateral sclerosis? Front Mol Neurosci. (2018) 11:63. doi: 10.3389/fnmol.2018.00063
22. Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron. (2017) 95:808–16.e9. doi: 10.1016/j.neuron.2017.07.025
23. Al-Chalabi A, Van Den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. (2017) 13:96–104. doi: 10.1038/nrneurol.2016.182
24. Blasco H, Mavel S, Corcia P, Gordon PH. The glutamate hypothesis in ALS: pathophysiology and drug development. Curr Med Chem. (2014) 21:3551–75. doi: 10.2174/0929867321666140916120118
25. Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. (2004) 10:S10–S17. doi: 10.1038/nm1066
26. Cappello V, Francolini M. Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int J Mol Sci. (2017) 18:2092. doi: 10.3390/ijms18102092
27. Delic V, Kurien C, Cruz J, Zivkovic S, Barretta J, Thomson A, Hennessey D, Joseph J, Ehrhart J, Willing AE, et al. Discrete mitochondrial aberrations in the spinal cord of sporadic ALS patients. J Neurosci Res. (2018) 96:1353–66. doi: 10.1002/jnr.24249
28. Anand A, Thakur K, Gupta PK. ALS and oxidative stress: the neurovascular scenario. Oxid Med Cell Longev. (2013) 2013:1–14. doi: 10.1155/2013/635831
29. Sharma A, Varghese AM, Vijaylakshmi K, Sumitha R, Prasanna VK, Shruthi S, et al. Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces mitochondrial and lysosomal dysfunction. Neurochem Res. (2016) 41:965–84. doi: 10.1007/s11064-015-1779-7
30. Onesto E, Colombrita C, Gumina V, Borghi MO, Dusi S, Doretti A, et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol Commun. (2016) 4:47. doi: 10.1186/s40478-016-0316-5
31. Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. (2006) 103:16021–6. doi: 10.1073/pnas.0607423103
32. Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. (2007) 10:615–22. doi: 10.1038/nn1876
33. Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. (2011) 29:824–8. doi: 10.1038/nbt.1957
34. Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L, et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J Biol Chem. (2013) 288:15699–711. doi: 10.1074/jbc.M112.425066
35. Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. (2008) 3:637–48. doi: 10.1016/j.stem.2008.09.017
36. Marchetto MCN, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non-cell-autonomous effect of human SOD1G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. (2008) 3:649–57. doi: 10.1016/j.stem.2008.10.001
37. Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. (1992) 326:1464–8. doi: 10.1056/NEJM199205283262204
38. Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. (1996) 16:675–86.
39. Medina L, Figueredo-Cardenas G, Rothstein JD, Reiner A. Differential abundance of glutamate transporter subtypes in amyotrophic lateral sclerosis (ALS)-vulnerable versus ALS-resistant brain stem motor cell groups. Exp Neurol. (1996) 142:287–95. doi: 10.1006/exnr.1996.0198
40. Okita T, Nodera H, Shibuta Y, Nodera A, Asanuma K, Shimatani Y, et al. Can Awaji ALS criteria provide earlier diagnosis than the revised El Escorial criteria? J Neurol Sci. (2011) 302:29–32. doi: 10.1016/j.jns.2010.12.007
41. Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. (2011) 7:639–49. doi: 10.1038/nrneurol.2011.153
42. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane database Syst Rev. (2012) 2012:CD001447. doi: 10.1002/14651858.CD001447.pub3
43. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. (1993) 362:59–62. doi: 10.1038/362059a0
44. Bocker MT, Hellwig I, Breiling A, Eckstein V, Ho AD, Lyko F. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood. (2011) 117:e182-9. doi: 10.1182/blood-2011-01-331926
45. Monahan Z, Shewmaker F, Pandey UB. Stress granules at the intersection of autophagy and ALS. Brain Res. (2016) 1649:189–200. doi: 10.1016/j.brainres.2016.05.022
46. Ajroud-Driss S, Siddique T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta. (2015) 1852:679–84. doi: 10.1016/j.bbadis.2014.08.010
47. Edens BM, Miller N, Ma Y-C. Impaired autophagy and defective mitochondrial function: converging paths on the road to motor neuron degeneration. Front Cell Neurosci. (2016) 10:44. doi: 10.3389/fncel.2016.00044
48. Kitamura A, Inada N, Kubota H, Matsumoto G, Kinjo M, Morimoto RI, et al. Dysregulation of the proteasome increases the toxicity of ALS-linked mutant SOD1. Genes to Cells. (2014) 19:209–24. doi: 10.1111/gtc.12125
49. An T, Shi P, Duan W, Zhang S, Yuan P, Li Z, et al. Oxidative stress and autophagic alteration in brainstem of SOD1-G93A mouse model of ALS. Mol Neurobiol. (2014) 49:1435–48. doi: 10.1007/s12035-013-8623-3
50. Otomo A, Pan L, Hadano S. Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol Res Int. (2012) 2012:498428. doi: 10.1155/2012/498428
51. Kaur SJ, McKeown SR, Rashid S. Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene. (2016) 577:109–18. doi: 10.1016/j.gene.2015.11.049
52. Lautenschläger J, Lautenschläger C, Tadic V, Süße H, Ortmann W, Denzler J, et al. Novel computer vision algorithm for the reliable analysis of organelle morphology in whole cell 3D images—A pilot study for the quantitative evaluation of mitochondrial fragmentation in amyotrophic lateral sclerosis. Mitochondrion. (2015) 25:49–59. doi: 10.1016/J.MITO.2015.10.003
53. Moore AS, Holzbaur ELF. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci USA. (2016) 113:E3349-58. doi: 10.1073/pnas.1523810113
54. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA. (2016) 113:4039–44. doi: 10.1073/pnas.1523926113
55. Shahheydari H, Ragagnin A, Walker AK, Toth RP, Vidal M, Jagaraj CJ, et al. Protein quality control and the amyotrophic lateral sclerosis/frontotemporal dementia continuum. Front Mol Neurosci. (2017) 10:119: doi: 10.3389/FNMOL.2017.00119
56. Heo J-M, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. (2015) 60:7–20. doi: 10.1016/j.molcel.2015.08.016
57. Ying H, Yue BYJT. Optineurin: the autophagy connection. Exp Eye Res. (2016) 144:73–80. doi: 10.1016/j.exer.2015.06.029
58. Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. (2017) 10:5. doi: 10.1186/s13041-017-0287-x
59. Lehmer C, Schludi MH, Ransom L, Greiling J, Junghänel M, Exner N, et al. A novel CHCHD10 mutation implicates a Mia40-dependent mitochondrial import deficit in ALS. EMBO Mol Med. (2018) 10:8558. doi: 10.15252/emmm.201708558
60. Genin EC, Plutino M, Bannwarth S, Villa E, Cisneros-Barroso E, Roy M, et al. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol Med. (2016) 8:58–72. doi: 10.15252/emmm.201505496
61. Woo J-AA, Liu T, Trotter C, Fang CC, De Narvaez E, LePochat P, et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat Commun. (2017) 8:15558. doi: 10.1038/ncomms15558
62. Anderson CJ, Bredvik K, Burstein SR, Davis C, Meadows SM, Dash J, et al. ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. (2019) doi: 10.1007/s00401-019-01989-y. [Epub ahead of print].
63. Burstein SR, Valsecchi F, Kawamata H, Bourens M, Zeng R, Zuberi A, et al. In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum Mol Genet. (2018) 27:160–77. doi: 10.1093/hmg/ddx397
64. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. (2015) 347:1436–41. doi: 10.1126/science.aaa3650
65. Walker C, El-Khamisy SF. Perturbed autophagy and DNA repair converge to promote neurodegeneration in amyotrophic lateral sclerosis and dementia. Brain. (2018) 141:1247–62. doi: 10.1093/brain/awy076
66. Melo-Hanchuk TD, Slepicka PF, Meirelles GV, Basei FL, Lovato DV, Granato DC, et al. NEK1 kinase domain structure and its dynamic protein interactome after exposure to Cisplatin. Sci Rep. (2017) 7:5445. doi: 10.1038/s41598-017-05325-w
67. Chen Y, Chen C-F, Riley DJ, Chen P-L. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle. (2011) 10:655–63. doi: 10.4161/cc.10.4.14814
68. Singh V, Connelly ZM, Shen X, De Benedetti A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle. (2017) 16:915–26. doi: 10.1080/15384101.2017.1314421
69. Liu S, Ho CK, Ouyang J, Zou L. Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc Natl Acad Sci USA. (2013) 110:2175–80. doi: 10.1073/pnas.1217781110
70. Al-Jassar C, Andreeva A, Barnabas DD, McLaughlin SH, Johnson CM, Yu M, et al. The Ciliopathy-Associated Cep104 Protein Interacts with Tubulin and Nek1 Kinase. Structure. (2017) 25:146–56. doi: 10.1016/j.str.2016.11.014
71. Chen Y, Craigen WJ, Riley DJ. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle. (2009) 8:257–67. doi: 10.4161/cc.8.2.7551
72. Füger P, Sreekumar V, Schüle R, Kern J V., Stanchev DT, Schneider CD, et al. Spastic Paraplegia Mutation N256S in the Neuronal Microtubule Motor KIF5A Disrupts Axonal Transport in a Drosophila HSP Model. PLoS Genet. (2012) 8:e1003066. doi: 10.1371/journal.pgen.1003066
73. Liu M, Nadar VC, Kozielski F, Kozlowska M, Yu W, Baas PW. Kinesin-12, a mitotic microtubule-associated motor protein, impacts axonal growth, navigation, and branching. J Neurosci. (2010) 30:14896–906. doi: 10.1523/JNEUROSCI.3739-10.2010
74. Jacquier A, Delorme C, Belotti E, Juntas-Morales R, Solé G, Dubourg O, et al. Cryptic amyloidogenic elements in mutant NEFH causing Charcot-Marie-Tooth 2 trigger aggresome formation and neuronal death. Acta Neuropathol Commun. (2017) 5:55. doi: 10.1186/s40478-017-0457-1
75. Xu Z, Henderson RD, David M, McCombe PA. Neurofilaments as biomarkers for amyotrophic lateral sclerosis: a systematic review and meta-analysis. PLoS ONE. (2016) 11:e0164625. doi: 10.1371/journal.pone.0164625
76. Figlewicz DA, Krizus A, Martinoli MG, Meininger V, Dib M, Rouleau GA, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet. (1994) 3:1757–61.
77. Rebelo AP, Abrams AJ, Cottenie E, Horga A, Gonzalez M, Bis DM, et al. Cryptic amyloidogenic elements in the 3′ UTRs of neurofilament genes trigger axonal neuropathy. Am J Hum Genet. (2016) 98:597–614. doi: 10.1016/j.ajhg.2016.02.022
78. Julien J-P. Neurofilament functions in health and disease. Curr Opin Neurobiol. (1999) 9:554–60. doi: 10.1016/S0959-4388(99)00004-5
79. Thyagarajan A, Strong MJ, Szaro BG. Post-transcriptional control of neurofilaments in development and disease. Exp Cell Res. (2007) 313:2088–97. doi: 10.1016/j.yexcr.2007.02.014
80. Lobsiger CS, Garcia ML, Ward CM, Cleveland DW. Altered axonal architecture by removal of the heavily phosphorylated neurofilament tail domains strongly slows superoxide dismutase 1 mutant-mediated ALS. Proc Natl Acad Sci USA. (2005) 102:10351–6. doi: 10.1073/pnas.0503862102
81. Howes SC, Alushin GM, Shida T, Nachury M V, Nogales E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol Biol Cell. (2014) 25:257–66. doi: 10.1091/mbc.E13-07-0387
82. Laird FM, Farah MH, Ackerley S, Hoke A, Maragakis N, Rothstein JD, et al. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci. (2008) 28:1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008
83. Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. (2014) 84:324–31. doi: 10.1016/j.neuron.2014.09.027
84. Helferich AM, Brockmann SJ, Reinders J, Deshpande D, Holzmann K, Brenner D, et al. Dysregulation of a novel miR-1825/TBCB/TUBA4A pathway in sporadic and familial ALS. Cell Mol Life Sci. (2018) 75:4301–19. doi: 10.1007/s00018-018-2873-1
85. Schäfer MK, Bellouze S, Jacquier A, Schaller S, Richard L, Mathis S, et al. Sensory neuropathy in progressive motor neuronopathy (pmn) mice is associated with defects in microtubule polymerization and axonal transport. Brain Pathol. (2017) 27:459–71. doi: 10.1111/bpa.12422
86. Clark JA, Yeaman EJ, Blizzard CA, Chuckowree JA, Dickson TC. A case for microtubule vulnerability in amyotrophic lateral sclerosis: altered dynamics during disease. Front Cell Neurosci. (2016) 10:204. doi: 10.3389/fncel.2016.00204
87. Liu X, Yang L, Tang L, Chen L, Liu X, Fan D. DCTN1 gene analysis in Chinese patients with sporadic amyotrophic lateral sclerosis. PLoS ONE. (2017) 12:e0182572. doi: 10.1371/journal.pone.0182572
88. Hafezparast M, Ahmad-Annuar A, Hummerich H, Shah P, Ford M, Baker C, et al. Paradigms for the identification of new genes in motor neuron degeneration. Amyotroph Lateral Scler Other Motor Neuron Disord. (2003) 4:249–57. doi: 10.1080/14660820310016084
89. Vohra BPS, Sasaki Y, Miller BR, Chang J, DiAntonio A, Milbrandt J. Amyloid precursor protein cleavage-dependent and -independent axonal degeneration programs share a common nicotinamide mononucleotide adenylyltransferase 1-sensitive pathway. J Neurosci. (2010) 30:13729–38. doi: 10.1523/JNEUROSCI.2939-10.2010
90. Ikenaka K, Katsuno M, Kawai K, Ishigaki S, Tanaka F, Sobue G. Disruption of axonal transport in motor neuron diseases. Int J Mol Sci. (2012) 13:1225–38. doi: 10.3390/ijms13011225
91. Henty-Ridilla JL, Juanes MA, Goode BL. Profilin directly promotes microtubule growth through residues mutated in amyotrophic lateral sclerosis. Curr Biol. (2017) 27:3535–43.e4. doi: 10.1016/j.cub.2017.10.002
92. Wu C-H, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. (2012) 488:499–503. doi: 10.1038/nature11280
93. Nekouei M, Ghezellou P, Aliahmadi A, Arjmand S, Kiaei M, Ghassempour A. Changes in biophysical characteristics of PFN1 due to mutation causing amyotrophic lateral sclerosis. Metab Brain Dis. (2018) 33:1975–84. doi: 10.1007/s11011-018-0305-4
94. Kiaei M, Balasubramaniam M, Govind Kumar V, Shmookler Reis RJ, Moradi M, Varughese KI. ALS-causing mutations in profilin-1 alter its conformational dynamics: a computational approach to explain propensity for aggregation. Sci Rep. (2018) 8:13102. doi: 10.1038/s41598-018-31199-7
95. Simpson CL, Lemmens R, Miskiewicz K, Broom WJ, Hansen VK, van Vught PWJ, et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet. (2009) 18:472–481. doi: 10.1093/hmg/ddn375
96. Bento-Abreu A, Jager G, Swinnen B, Rué L, Hendrickx S, Jones A, et al. Elongator subunit 3 (ELP3) modifies ALS through tRNA modification. Hum Mol Genet. (2018) 27:1276–89. doi: 10.1093/hmg/ddy043
97. Nguyen L, Humbert S, Saudou F, Chariot A. Elongator–an emerging role in neurological disorders. Trends Mol Med. (2010) 16:1–6. doi: 10.1016/j.molmed.2009.11.002
98. Tielens S, Huysseune S, Godin JD, Chariot A, Malgrange B, Nguyen L. Elongator controls cortical interneuron migration by regulating actomyosin dynamics. Cell Res. (2016) 26:1131–48. doi: 10.1038/cr.2016.112
99. Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. (2015) 525:56–61. doi: 10.1038/nature14973
100. Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RAK, Levina V, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. (2014) 23:3579–95. doi: 10.1093/hmg/ddu068
101. Bäumer D, Talbot K, Turner MR. Advances in motor neurone disease. J R Soc Med. (2014) 107:14–21. doi: 10.1177/0141076813511451
102. DeJesus-Hernandez M, Mackenzie IRR, Boeve BFF, Boxer ALL, Baker M, Rutherford NJJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
103. Babić Leko M, Župunski V, Kirincich J, Smilović D, Hortobágyi T, Hof PR, et al. Molecular mechanisms of neurodegeneration related to C9orf72 hexanucleotide repeat expansion. Behav Neurol. (2019) 2019:1–18. doi: 10.1155/2019/2909168
104. Burk K, Pasterkamp RJ. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. (2019) doi: 10.1007/s00401-019-01964-7. [Epub ahead of print].
105. Guo Q, Lehmer C, Martínez-Sánchez A, Rudack T, Beck F, Hartmann H, et al. In situ structure of neuronal C9orf72 Poly-GA aggregates reveals proteasome recruitment. Cell. (2018) 172:696–705.e12. doi: 10.1016/j.cell.2017.12.030
106. Ho WY, Tai YK, Chang J-C, Liang J, Tyan S-H, Chen S, et al. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy. (2019) 15:827–42. doi: 10.1080/15548627.2019.1569441
107. Han J-H, Ryu H-H, Jun M-H, Jang D-J, Lee J-A. The functional analysis of the CHMP2B missense mutation associated with neurodegenerative diseases in the endo-lysosomal pathway. Biochem Biophys Res Commun. (2012) 421:544–9. doi: 10.1016/j.bbrc.2012.04.041
108. Zaglia T, Milan G, Ruhs A, Franzoso M, Bertaggia E, Pianca N, et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J Clin Invest. (2014) 124:2410–24. doi: 10.1172/JCI66339
109. Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. (2006) 67:1074–7. doi: 10.1212/01.wnl.0000231510.89311.8b
110. Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EMC, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. (2007) 179:485–500. doi: 10.1083/jcb.200702115
111. Vandal S, Zheng X, Ahmad S. Molecular genetics of frontotemporal dementia elucidated by drosophila models—defects in endosomal–lysosomal pathway. Int J Mol Sci. (2018) 19:1714. doi: 10.3390/ijms19061714
112. Krasniak CS, Ahmad ST. The role of CHMP2BIntron5 in autophagy and frontotemporal dementia. Brain Res. (2016) 1649:151–7. doi: 10.1016/j.brainres.2016.02.051
113. Clayton EL, Mizielinska S, Edgar JR, Nielsen TT, Marshall S, Norona FE, et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol. (2015) 130:511–23. doi: 10.1007/s00401-015-1475-3
114. Tanikawa S, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K. Endosomal sorting related protein CHMP2B is localized in Lewy bodies and glial cytoplasmic inclusions in α-synucleinopathy. Neurosci Lett. (2012) 527:16–21. doi: 10.1016/j.neulet.2012.08.035
115. Franz A, Ackermann L, Hoppe T. Create and preserve: proteostasis in development and aging is governed by Cdc48/p97/VCP. Biochim Biophys Acta. (2014) 1843:205–15. doi: 10.1016/j.bbamcr.2013.03.031
116. Shaw CE. Capturing VCP: another molecular piece in the ALS jigsaw puzzle. Neuron. (2010) 68:812–14. doi: 10.1016/j.neuron.2010.11.040
117. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. (2010) 68:857–64. doi: 10.1016/j.neuron.2010.11.036
118. Yin HZ, Nalbandian A, Hsu C-I, Li S, Llewellyn KJ, Mozaffar T, et al. Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. (2012) 3:e374. doi: 10.1038/cddis.2012.115
119. Wang T, Xu W, Qin M, Yang Y, Bao P, Shen F, et al. Pathogenic mutations in the valosin-containing Protein/p97(VCP) N-domain Inhibit the SUMOylation of VCP and lead to impaired stress response. J Biol Chem. (2016) 291:14373–84. doi: 10.1074/jbc.M116.729343
120. Llewellyn KJ, Walker N, Nguyen C, Tan B, BenMohamed L, Kimonis VE, et al. A fine balance of dietary lipids improves pathology of a murine model of VCP-associated multisystem proteinopathy. PLoS ONE. (2015) 10:e0131995. doi: 10.1371/journal.pone.0131995
121. Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. (2017) 36:135–50. doi: 10.15252/embj.201695148
122. Kon T, Mori F, Tanji K, Miki Y, Toyoshima Y, Yoshida M, et al. ALS-associated protein FIG4 is localized in Pick and Lewy bodies, and also neuronal nuclear inclusions, in polyglutamine and intranuclear inclusion body diseases. Neuropathology. (2014) 34:19–26. doi: 10.1111/neup.12056
123. Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. (2009) 84:85–8. doi: 10.1016/j.ajhg.2008.12.010
124. Lenk GM, Meisler MH. Mouse models of PI(3,5)P2 deficiency with impaired lysosome function. Methods Enzymol. (2014) 534:245–60. doi: 10.1016/B978-0-12-397926-1.00014-7
125. Nishimura AL, Mitne-Neto M, Silva HCAA, Richieri-Costa A, Middleton S, Cascio D, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. (2004) 75:822–31. doi: 10.1086/425287
126. Genevini P, Colombo MN, Venditti R, Marcuzzo S, Colombo SF, Bernasconi P, et al. VAPB depletion alters neuritogenesis and phosphoinositide balance in motoneuron-like cells: relevance to VAPB-linked ALS. J Cell Sci. (2019) 2019:jcs.220061. doi: 10.1242/jcs.220061
127. Vinay Kumar C, Kumar KM, Swetha R, Ramaiah S, Anbarasu A. Protein aggregation due to nsSNP resulting in P56S VABP protein is associated with amyotrophic lateral sclerosis. J Theor Biol. (2014) 354:72–80. doi: 10.1016/j.jtbi.2014.03.027
128. Kabashi E, El Oussini H, Bercier V, Gros-Louis F, Valdmanis PN, McDearmid J, et al. Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum Mol Genet. (2013) 22:2350–60. doi: 10.1093/hmg/ddt080
129. Aliaga L, Lai C, Yu J, Chub N, Shim H, Sun L, et al. Amyotrophic lateral sclerosis-related VAPB P56S mutation differentially affects the function and survival of corticospinal and spinal motor neurons. Hum Mol Genet. (2013) 22:4293–305. doi: 10.1093/hmg/ddt279
130. Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, Johnson C, et al. UBQLN2 mediates autophagy-independent protein aggregate clearance by the proteasome. Cell. (2016) 166:935–49. doi: 10.1016/j.cell.2016.07.001
131. Blokhuis AM, Groen EJN, Koppers M, van den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. (2013) 125:777–94. doi: 10.1007/s00401-013-1125-6
132. Deng H-X, Chen W, Hong S-T, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. (2011) 477:211–15. doi: 10.1038/nature10353
133. Chen T, Huang B, Shi X, Gao L, Huang C. Mutant UBQLN2P497H in motor neurons leads to ALS-like phenotypes and defective autophagy in rats. Acta Neuropathol Commun. (2018) 6:122. doi: 10.1186/s40478-018-0627-9
134. Jantrapirom S, Lo Piccolo L, Yoshida H, Yamaguchi M. Depletion of ubiquilin induces an augmentation in soluble ubiquitinated drosophila TDP-43 to drive neurotoxicity in the fly. Biochim Biophys Acta. (2018) 1864:3038–49. doi: 10.1016/j.bbadis.2018.06.017
135. Brenner D, Sieverding K, Bruno C, Lüningschrör P, Buck E, Mungwa S, et al. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J Exp Med. (2019) 216:jem.20180729. doi: 10.1084/jem.20180729
136. Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun. (2016) 7:11253. doi: 10.1038/ncomms11253
137. Goode A, Rea S, Sultana M, Shaw B, Searle MS, Layfield R. ALS-FTLD associated mutations of SQSTM1 impact on Keap1-Nrf2 signalling. Mol Cell Neurosci. (2016) 76:52–8. doi: 10.1016/j.mcn.2016.08.004
138. Zhao M, Kim JR, van Bruggen R, Park J. RNA-binding proteins in amyotrophic lateral sclerosis. Mol Cells. (2018) 41:818–29. doi: 10.14348/molcells.2018.0243
139. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Velde CV, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. (2008) 40:572–4. doi: 10.1038/ng.132
140. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. (2008) 319:1668–72. doi: 10.1126/science.1154584
141. Kim HJ, Kim NC, Wang Y-D, Scarborough EA, Moore J, Diaz Z, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. (2013) 495:467–73. doi: 10.1038/nature11922
142. Higashi S, Kabuta T, Nagai Y, Tsuchiya Y, Akiyama H, Wada K. TDP-43 associates with stalled ribosomes and contributes to cell survival during cellular stress. J Neurochem. (2013) 126:288–300. doi: 10.1111/jnc.12194
143. Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. (2013) 4:2980. doi: 10.1038/ncomms3980
144. Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell. (2018) 173:677–92.e20. doi: 10.1016/j.cell.2018.03.002
145. Lai C, Xie C, Shim H, Chandran J, Howell BW, Cai H. Regulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsin. Mol Brain. (2009) 2:23. doi: 10.1186/1756-6606-2-23
146. Hadano S, Yanagisawa Y, Skaug J, Fichter K, Nasir J, Martindale D, et al. Cloning and characterization of three novel genes, ALS2CR1, ALS2CR2, and ALS2CR3, in the juvenile amyotrophic lateral sclerosis (ALS2) critical region at chromosome 2q33–q34: candidate genes for ALS2. Genomics. (2001) 71:200–13. doi: 10.1006/geno.2000.6392
147. Uversky VN. The roles of intrinsic disorder-based liquid-liquid phase transitions in the "Dr. Jekyll-Mr. Hyde" behavior of proteins involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Autophagy. (2017) 13:2115–2162. doi: 10.1080/15548627.2017.1384889
148. Martinez FJ, Pratt GA, Van Nostrand EL, Batra R, Huelga SC, Kapeli K, et al. Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system. Neuron. (2016) 92:780–95. doi: 10.1016/j.neuron.2016.09.050
149. van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. (2016) 48:1043–8. doi: 10.1038/ng.3622
150. Sato K, Otomo A, Ueda MT, Hiratsuka Y, Suzuki-Utsunomiya K, Sugiyama J, et al. Altered oligomeric states in pathogenic ALS2 variants associated with juvenile motor neuron diseases cause loss of ALS2-mediated endosomal function. J Biol Chem. (2018) 293:17135–53. doi: 10.1074/jbc.RA118.003849
151. Hadano S, Otomo A, Kunita R, Suzuki-Utsunomiya K, Akatsuka A, Koike M, et al. Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1H46R-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS ONE. (2010) 5:e9805. doi: 10.1371/journal.pone.0009805
152. Branchu J, Boutry M, Sourd L, Depp M, Leone C, Corriger A, et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol Dis. (2017) 102:21–37. doi: 10.1016/j.nbd.2017.02.007
153. Orlacchio A, Babalini C, Borreca A, Patrono C, Massa R, Basaran S, et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain. (2010) 133:591–598. doi: 10.1093/brain/awp325
154. Fang X, Lin H, Wang X, Zuo Q, Qin J, Zhang P. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim Biophys Sin. (2015) 47:834–41. doi: 10.1093/abbs/gmv076
155. Hirano M, Quinzii CM, Mitsumoto H, Hays AP, Roberts JK, et al. Senataxin mutations and amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2011) 12:223–7. doi: 10.3109/17482968.2010.545952
156. Becherel OJ, Yeo AJ, Stellati A, Heng EYH, Luff J, Suraweera AM, et al. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet. (2013) 9:e1003435. doi: 10.1371/journal.pgen.1003435
157. Bennett CL, La Spada AR. Senataxin, a novel helicase at the interface of RNA transcriptome regulation and neurobiology: from normal function to pathological roles in motor neuron disease and cerebellar degeneration. Adv Neurobiol. 20:265–81. doi: 10.1007/978-3-319-89689-2_10
158. Grunseich C, Wang IX, Watts JA, Burdick JT, Guber RD, Zhu Z, et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Mol Cell. (2018) 69:426–37.e7. doi: 10.1016/j.molcel.2017.12.030
159. Kwiatkowski TJ, Bosco DA, LeClerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. (2009) 323:1205–8. doi: 10.1126/science.1166066
160. Wang H, Guo W, Mitra J, Hegde PM, Vandoorne T, Eckelmann BJ, Mitra S, Tomkinson AE, Van Den Bosch L, Hegde ML. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat Commun. (2018) 9:3683. doi: 10.1038/s41467-018-06111-6
161. Devoy A, Kalmar B, Stewart M, Park H, Burke B, Noy SJ, et al. Humanized mutant FUS drives progressive motor neuron degeneration without aggregation in “FUSDelta14” knockin mice. Brain. (2017) 140:2797–805. doi: 10.1093/brain/awx248
162. Butti Z, Patten SA. RNA Dysregulation in amyotrophic lateral sclerosis. Front Genet. (2019) 9:712. doi: 10.3389/fgene.2018.00712
163. Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. (2010) 466:1069–75. doi: 10.1038/nature09320
164. Ostrowski L, Hall A, Mekhail K. Ataxin-2: from RNA control to human health and disease. Genes (Basel). (2017) 8:157. doi: 10.3390/genes8060157
165. Thiyagarajan N, Ferguson R, Subramanian V, Acharya KR. Structural and molecular insights into the mechanism of action of human angiogenin-ALS variants in neurons. Nat Commun. (2012) 3:1121. doi: 10.1038/ncomms2126
166. Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, et al. ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat Genet. (2006) 38:411–3. doi: 10.1038/ng1742
167. Cronin S, Greenway MJ, Ennis S, Kieran D, Green A, Prehn JHM, et al. Elevated serum angiogenin levels in ALS. Neurology. (2006) 67:1833–6. doi: 10.1212/01.wnl.0000244466.46020.47
168. Li S, Hu G-F. Angiogenin-mediated rRNA transcription in cancer and neurodegeneration. Int J Biochem Mol Biol. (2010) 1:26–35.
169. Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. (2014) 17:664–6. doi: 10.1038/nn.3688
170. Malik AM, Miguez RA, Li X, Ho Y-S, Feldman EL, Barmada SJ. Matrin 3-dependent neurotoxicity is modified by nucleic acid binding and nucleocytoplasmic localization. Elife. (2018) 7:eLife.35977. doi: 10.7554/eLife.35977
171. Vieira M, Saraiva MJ. Transthyretin: a multifaceted protein. Biomol Concepts. (2014) 5:45–54. doi: 10.1515/bmc-2013-0038
172. Watanabe S, Hayakawa T, Wakasugi K, Yamanaka K. Cystatin C protects neuronal cells against mutant copper-zinc superoxide dismutase-mediated toxicity. Cell Death Dis. (2014) 5:e1497. doi: 10.1038/cddis.2014.459
173. Wilson ME, Boumaza I, Lacomis D, Bowser R. Cystatin C: a candidate biomarker for amyotrophic lateral sclerosis. PLoS ONE. (2010) 5:e15133. doi: 10.1371/journal.pone.0015133
174. Rossi D, Volanti P, Brambilla L, Colletti T, Spataro R, La Bella V. CSF neurofilament proteins as diagnostic and prognostic biomarkers for amyotrophic lateral sclerosis. J Neurol. (2018) 265:510–21. doi: 10.1007/s00415-017-8730-6
175. Chen X, Chen Y, Wei Q, Ou R, Cao B, Zhao B, et al. Assessment of a multiple biomarker panel for diagnosis of amyotrophic lateral sclerosis. BMC Neurol. (2016) 16:173. doi: 10.1186/s12883-016-0689-x
176. Gonçalves M, De Carvalho M, Peixoto C, Alves P, Barreto C, Oliva A, et al. Phosphoneurofilament heavy chain and vascular endothelial growth factor as cerebrospinal fluid biomarkers for ALS. Amyotroph Lateral Scler Front Degener. (2017) 18:134–6. doi: 10.1080/21678421.2016.1212894
177. Gendron TF, C9ORF72 Neurofilament Study Group LM, Daughrity LM, Heckman MG, Diehl NN, Wuu J, et al. Phosphorylated neurofilament heavy chain: a biomarker of survival for C9ORF72-associated amyotrophic lateral sclerosis. Ann Neurol. (2017) 82:139–46. doi: 10.1002/ana.24980
178. Boylan KB, Glass JD, Crook JE, Yang C, Thomas CS, Desaro P, et al. Phosphorylated neurofilament heavy subunit (pNF-H) in peripheral blood and CSF as a potential prognostic biomarker in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2013) 84:467–72. doi: 10.1136/jnnp-2012-303768
179. Thompson AG, Gray E, Thézénas M-L, Charles PD, Evetts S, Hu MT, et al. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann Neurol. (2018) 83:258–68. doi: 10.1002/ana.25143
180. Hathout Y, Seol H, Han MHJ, Zhang A, Brown KJ, Hoffman EP. Clinical utility of serum biomarkers in Duchenne muscular dystrophy. Clin Proteomics. (2016) 13:9. doi: 10.1186/s12014-016-9109-x
181. Sun X, Zhang H. miR-451 elevation relieves inflammatory pain by suppressing microglial activation-evoked inflammatory response via targeting TLR4. Cell Tissue Res. (2018) doi: 10.1007/s00441-018-2898-7
182. Yao Y, Tsirka SE. Monocyte chemoattractant protein-1 and the blood-brain barrier. Cell Mol Life Sci. (2014) 71:683–97. doi: 10.1007/s00018-013-1459-1
183. Boettger T, Wüst S, Nolte H, Braun T. The miR-206/133b cluster is dispensable for development, survival and regeneration of skeletal muscle. Skelet Muscle. (2014) 4:23. doi: 10.1186/s13395-014-0023-5
184. De Felice B, Guida M, Guida M, Coppola C, De Mieri G, Cotrufo R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene. (2012) 508:35–40. doi: 10.1016/j.gene.2012.07.058
185. Horak M, Novak J, Bienertova-Vasku J. Muscle-specific microRNAs in skeletal muscle development. Dev Biol. (2016) 410:1–13. doi: 10.1016/J.YDBIO.2015.12.013
186. Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, De Carvalho M, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. (2013) 12:310–22. doi: 10.1016/S1474-4422(13)70036-X
187. Valbuena GN, Rizzardini M, Cimini S, Siskos AP, Bendotti C, Cantoni L, et al. Metabolomic analysis reveals increased aerobic glycolysis and amino acid deficit in a cellular model of amyotrophic lateral sclerosis. Mol Neurobiol. (2016) 53:2222–40. doi: 10.1007/s12035-015-9165-7
188. Funalot B, Desport J-C, Sturtz F, Camu W, Couratier P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2009) 10:113–117. doi: 10.1080/17482960802295192
189. Steyn FJ, Ioannides ZA, van Eijk RPA, Heggie S, Thorpe KA, Ceslis A, et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J Neurol Neurosurg Psychiatry. (2018) 89:1016–23. doi: 10.1136/jnnp-2017-317887
190. Abdel-Khalik J, Yutuc E, Crick PJ, Gustafsson J-Å, Warner M, Roman G, et al. Defective cholesterol metabolism in amyotrophic lateral sclerosis. J Lipid Res. (2017) 58:267–78. doi: 10.1194/jlr.P071639
191. Blasco H, Patin F, Molinier S, Vourc'h P, Le Tilly O, Bakkouche S, et al. A decrease in blood cholesterol after gastrostomy could impact survival in ALS. Eur J Clin Nutr. (2017) 71:1133–5. doi: 10.1038/ejcn.2017.54
192. Vieira AS, Dogini DB, Lopes-Cendes I. Role of non-coding RNAs in non-aging-related neurological disorders. Braz J Med Biol Res. (2018) 51:e7566. doi: 10.1590/1414-431X20187566
193. Wan P, Su W, Zhuo Y. The Role of Long Noncoding RNAs in neurodegenerative diseases. Mol Neurobiol. (2017) 54:2012–21. doi: 10.1007/s12035-016-9793-6
194. Peng W, Wang J, Shan B, Peng Z, Dong Y, Shi W, et al. Diagnostic and prognostic potential of circulating long non-coding RNAs in non small cell lung cancer. Cell Physiol Biochem. (2018) 49:816–27. doi: 10.1159/000493043
195. Bermejo JL, Huang G, Manoochehri M, Mesa KG, Schick M, Silos RG, et al. Long intergenic noncoding RNA 299 methylation in peripheral blood is a biomarker for triple-negative breast cancer. Epigenomics. (2018) 2018:epi-2018-0121. doi: 10.2217/epi-2018-0121
196. Li M, Wang Y-F, Yang X-C, Xu L, Li W-M, Xia K, et al. Circulating long noncoding RNA LIPCAR acts as a novel biomarker in patients with ST-segment elevation myocardial infarction. Med Sci Monit. (2018) 24:5064–70. doi: 10.12659/MSM.909348
197. Nagaraj S, Zoltowska KM, Laskowska-Kaszub K, Wojda U. microRNA diagnostic panel for Alzheimer's disease and epigenetic trade-off between neurodegeneration and cancer. Ageing Res Rev. (2018) 49:125–43. doi: 10.1016/J.ARR.2018.10.008
198. Punga T, Bartoccioni E, Lewandowska M, Damato V, Evoli A, Punga AR. Disease specific enrichment of circulating let-7 family microRNA in MuSK+ myasthenia gravis. J Neuroimmunol. (2016) 292:21–6. doi: 10.1016/j.jneuroim.2016.01.003
199. de Andrade HMT, de Albuquerque M, Avansini SH, de S, Rocha C, Dogini DB, Nucci A, et al. MicroRNAs-424 and 206 are potential prognostic markers in spinal onset amyotrophic lateral sclerosis. J Neurol Sci. (2016) 368:19–24. doi: 10.1016/j.jns.2016.06.046
200. Russell AP, Wada S, Vergani L, Hock MB, Lamon S, Léger B, et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol Dis. (2013) 49:107–17. doi: 10.1016/j.nbd.2012.08.015
201. Si Y, Cui X, Crossman DK, Hao J, Kazamel M, Kwon Y, et al. Muscle microRNA signatures as biomarkers of disease progression in amyotrophic lateral sclerosis. Neurobiol Dis. (2018) 114:85–94. doi: 10.1016/J.NBD.2018.02.009
202. Toivonen JM, Manzano R, Oliván S, Zaragoza P, García-Redondo A, Osta R. MicroRNA-206: a potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS ONE. (2014) 9:e89065. doi: 10.1371/journal.pone.0089065
203. Ranganathan S, Williams E, Ganchev P, Gopalakrishnan V, Lacomis D, Urbinelli L, et al. Proteomic profiling of cerebrospinal fluid identifies biomarkers for amyotrophic lateral sclerosis. J Neurochem. (2005) 95:1461–71. doi: 10.1111/j.1471-4159.2005.03478.x
204. Ryberg H, An J, Darko S, Lustgarten JL, Jaffa M, Gopalakrishnan V, et al. Discovery and verification of amyotrophic lateral sclerosis biomarkers by proteomics. Muscle Nerve. (2010) 42:104–11. doi: 10.1002/mus.21683
205. Pasinetti GM, Ungar LH, Lange DJ, Yemul S, Deng H, Yuan X, et al. Identification of potential CSF biomarkers in ALS. Neurology. (2006) 66:1218–22. doi: 10.1212/01.wnl.0000203129.82104.07
206. Brancia C, Noli B, Boido M, Pilleri R, Boi A, Puddu R, et al. TLQP Peptides in amyotrophic lateral sclerosis: possible blood biomarkers with a neuroprotective role. Neuroscience. (2018) 380:152–63. doi: 10.1016/j.neuroscience.2018.03.023
207. Lunetta C, Lizio A, Maestri E, Sansone VA, Mora G, Miller RG, et al. Serum C-reactive protein as a prognostic biomarker in amyotrophic lateral sclerosis. JAMA Neurol. (2017) 74:660–7. doi: 10.1001/jamaneurol.2016.6179
208. Gaiani A, Martinelli I, Bello L, Querin G, Puthenparampil M, Ruggero S, et al. Diagnostic and Prognostic Biomarkers in amyotrophic lateral sclerosis: neurofilament light chain levels in definite subtypes of disease. JAMA Neurol. (2017) 74:525–32. doi: 10.1001/jamaneurol.2016.5398
209. Mitchell RM, Simmons Z, Beard JL, Stephens HE, Connor JR. Plasma biomarkers associated with ALS and their relationship to iron homeostasis. Muscle Nerve. (2010) 42:95–103. doi: 10.1002/mus.21625
210. Simpson EP, Henry YK, Henkel JS, Smith RG, Appel SH. Increased lipid peroxidation in sera of ALS patients: a potential biomarker of disease burden. Neurology. (2004) 62:1758–65. doi: 10.1212/WNL.62.10.1758
211. Nardo G, Pozzi S, Pignataro M, Lauranzano E, Spano G, Garbelli S, et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE. (2011) 6:e25545. doi: 10.1371/journal.pone.0025545
212. Filareti M, Luotti S, Pasetto L, Pignataro M, Paolella K, Messina P, et al. Decreased levels of foldase and chaperone proteins are associated with an early-onset amyotrophic lateral sclerosis. Front Mol Neurosci. (2017) 10:99. doi: 10.3389/fnmol.2017.00099
213. Chen Y, Wei Q, Chen X, Li C, Cao B, Ou R, et al. Aberration of miRNAs expression in leukocytes from sporadic amyotrophic lateral sclerosis. Front Mol Neurosci. (2016) 9:69. doi: 10.3389/fnmol.2016.00069
214. De Felice B, Annunziata A, Fiorentino G, Borra M, Biffali E, Coppola C, et al. miR-338-3p is over-expressed in blood, CFS, serum and spinal cord from sporadic amyotrophic lateral sclerosis patients. Neurogenetics. (2014) 15:243–53. doi: 10.1007/s10048-014-0420-2
215. Vrabec K, Boštjanč;ič; E, Koritnik B, Leonardis L, Dolenc Grošelj L, Zidar J, et al. Differential expression of several miRNAs and the host genes AATK and DNM2 in leukocytes of sporadic ALS patients. Front Mol Neurosci. (2018) 11:106. doi: 10.3389/fnmol.2018.00106
216. Waller R, Goodall EF, Milo M, Cooper-Knock J, Da Costa M, Hobson E, et al. Serum miRNAs miR-206, 143-3p and 374b-5p as potential biomarkers for amyotrophic lateral sclerosis (ALS). Neurobiol Aging. (2017) 55:123–31. doi: 10.1016/j.neurobiolaging.2017.03.027
217. Tasca E, Pegoraro V, Merico A, Angelini C. Circulating microRNAs as biomarkers of muscle differentiation and atrophy in ALS. Clin Neuropathol. (2016) 35:22–30. doi: 10.5414/NP300889
218. Raheja R, Regev K, Healy BC, Mazzola MA, Beynon V, Von Glehn F, et al. Correlating serum micrornas and clinical parameters in amyotrophic lateral sclerosis. Muscle Nerve. (2018) 58:261–9. doi: 10.1002/mus.26106
219. Blasco H, Corcia P, Moreau C, Veau S, Fournier C, Vourc'h P, et al. 1H-NMR-based metabolomic profiling of CSF in early amyotrophic lateral sclerosis. PLoS ONE. (2010) 5:e13223. doi: 10.1371/journal.pone.0013223
220. Lawton KA, Cudkowicz ME, Brown MV, Alexander D, Caffrey R, Wulff JE, et al. Biochemical alterations associated with ALS. Amyotroph Lateral Scler. (2012) 13:110–8. doi: 10.3109/17482968.2011.619197
221. Ihara Y, Nobukuni K, Takata H, Hayabara T. Oxidative stress and metal content in blood and cerebrospinal fluid of amyotrophic lateral sclerosis patients with and without a Cu, Zn-superoxide dismutase mutation. Neurol Res. (2005) 27:105–8. doi: 10.1179/016164105X18430
222. Wuolikainen A, Andersen PM, Moritz T, Marklund SL, Antti H. ALS patients with mutations in the SOD1 gene have an unique metabolomic profile in the cerebrospinal fluid compared with ALS patients without mutations. Mol Genet Metab. (2012) 105:472–8. doi: 10.1016/j.ymgme.2011.11.201
223. Kumar A, Bala L, Kalita J, Misra UK, Singh RL, Khetrapal CL, et al. Metabolomic analysis of serum by (1) H NMR spectroscopy in amyotrophic lateral sclerosis. Clin Chim Acta. (2010) 411:563–7. doi: 10.1016/j.cca.2010.01.016
224. Lawton KA, Brown MV, Alexander D, Li Z, Wulff JE, Lawson R, et al. Plasma metabolomic biomarker panel to distinguish patients with amyotrophic lateral sclerosis from disease mimics. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15:362–70. doi: 10.3109/21678421.2014.908311
225. Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, et al. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. (1990) 28:18–25. doi: 10.1002/ana.410280106
226. Iwasaki Y, Ikeda K, Kinoshita M. Plasma amino acid levels in patients with amyotrophic lateral sclerosis. J Neurol Sci. (1992) 107:219–22.
227. Gray E, Larkin JR, Claridge TDW, Talbot K, Sibson NR, Turner MR. The longitudinal cerebrospinal fluid metabolomic profile of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:456–63. doi: 10.3109/21678421.2015.1053490
228. Wuolikainen A, Jonsson P, Ahnlund M, Antti H, Marklund SL, Moritz T, et al. Multi-platform mass spectrometry analysis of the CSF and plasma metabolomes of rigorously matched amyotrophic lateral sclerosis, Parkinson's disease and control subjects. Mol Biosyst. (2016) 12:1287–98. doi: 10.1039/c5mb00711a
229. Dupuis L, Corcia P, Fergani A, Gonzalez De Aguilar J-L, Bonnefont-Rousselot D, Bittar R, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. (2008) 70:1004–9. doi: 10.1212/01.wnl.0000285080.70324.27
230. Valentino F, Bivona G, Butera D, Paladino P, Fazzari M, Piccoli T, et al. Elevated cerebrospinal fluid and plasma homocysteine levels in ALS. Eur J Neurol. (2010) 17:84–9. doi: 10.1111/j.1468-1331.2009.02752.x
231. Levin J, Bötzel K, Giese A, Vogeser M, Lorenzl S. Elevated levels of methylmalonate and homocysteine in Parkinson's disease, progressive supranuclear palsy and amyotrophic lateral sclerosis. Dement Geriatr Cogn Disord. (2010) 29:553–9. doi: 10.1159/000314841
232. Zoccolella S, Simone IL, Lamberti P, Samarelli V, Tortelli R, Serlenga L, et al. Elevated plasma homocysteine levels in patients with amyotrophic lateral sclerosis. Neurology. (2008) 70:222–5. doi: 10.1212/01.wnl.0000297193.53986.6f
233. Cieslarova Z, Lopes FS, do Lago CL, França MC, Colnaghi Simionato AV. Capillary electrophoresis tandem mass spectrometry determination of glutamic acid and homocysteine's metabolites: potential biomarkers of amyotrophic lateral sclerosis. Talanta. (2017) 170:63–8. doi: 10.1016/J.TALANTA.2017.03.103
234. Brettschneider J, Lehmensiek V, Mogel H, Pfeifle M, Dorst J, Hendrich C, et al. Proteome analysis reveals candidate markers of disease progression in amyotrophic lateral sclerosis (ALS). Neurosci Lett. (2010) 468:23–7. doi: 10.1016/j.neulet.2009.10.053
235. Zhou J-Y, Afjehi-Sadat L, Asress S, Duong DM, Cudkowicz M, Glass JD, et al. Galectin-3 is a candidate biomarker for amyotrophic lateral sclerosis: discovery by a proteomics approach. J Proteome Res. (2010) 9:5133–41. doi: 10.1021/pr100409r
236. Liu J, Gao L, Zang D. Elevated levels of IFN-γ in CSF and serum of patients with amyotrophic lateral sclerosis. PLoS ONE. (2015) 10:e0136937. doi: 10.1371/journal.pone.0136937
237. Gao L, Zhou S, Cai H, Gong Z, Zang D. VEGF levels in CSF and serum in mild ALS patients. J Neurol Sci. (2014) 346:216–20. doi: 10.1016/j.jns.2014.08.031
238. Lind A-L, Wu D, Freyhult E, Bodolea C, Ekegren T, Larsson A, et al. A multiplex protein panel applied to cerebrospinal fluid reveals three new biomarker candidates in ALS but none in neuropathic pain patients. PLoS ONE. (2016) 11:e0149821. doi: 10.1371/journal.pone.0149821
239. Kano O, Tanaka K, Kanno T, Iwasaki Y, Ikeda J-E. Neuronal apoptosis inhibitory protein is implicated in amyotrophic lateral sclerosis symptoms. Sci Rep. (2018) 8:6. doi: 10.1038/s41598-017-18627-w
240. Lima C, Pinto S, Napoleão P, Pronto-Laborinho AC, Barros MA, Freitas T, et al. Identification of erythrocyte biomarkers in amyotrophic lateral sclerosis. Clin Hemorheol Microcirc. (2016) 63:423–37. doi: 10.3233/CH-162066
241. Edri-Brami M, Rosental B, Hayoun D, Welt M, Rosen H, Wirguin I, et al. Glycans in sera of amyotrophic lateral sclerosis patients and their role in killing neuronal cells. PLoS ONE. (2012) 7:e35772. doi: 10.1371/journal.pone.0035772
242. Otto M, Bahn E, Wiltfang J, Boekhoff I, Beuche W. Decrease of S100 beta protein in serum of patients with amyotrophic lateral sclerosis. Neurosci Lett. (1998) 240:171–3.
243. Häggmark A, Mikus M, Mohsenchian A, Hong M-G, Forsström B, Gajewska B, et al. Plasma profiling reveals three proteins associated to amyotrophic lateral sclerosis. Ann Clin Transl Neurol. (2014) 1:544–53. doi: 10.1002/acn3.83
244. Williams SM, Khan G, Harris BT, Ravits J, Sierks MR. TDP-43 protein variants as biomarkers in amyotrophic lateral sclerosis. BMC Neurosci. (2017) 18:20. doi: 10.1186/s12868-017-0334-7
245. Freischmidt A, Müller K, Ludolph AC, Weishaupt JH. Systemic dysregulation of TDP-43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol Commun. (2013) 1:42. doi: 10.1186/2051-5960-1-42
246. Benigni M, Ricci C, Jones AR, Giannini F, Al-Chalabi A, Battistini S. Identification of miRNAs as potential biomarkers in cerebrospinal fluid from amyotrophic lateral sclerosis patients. Neuromolecular Med. (2016) 18:551–60. doi: 10.1007/s12017-016-8396-8
247. Waller R, Wyles M, Heath PR, Kazoka M, Wollff H, Shaw PJ, et al. Small RNA sequencing of sporadic amyotrophic lateral sclerosis cerebrospinal fluid reveals differentially expressed miRNAs related to neural and glial activity. Front Neurosci. (2017) 11:731. doi: 10.3389/fnins.2017.00731
248. Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. (2012) 122:3063–87. doi: 10.1172/JCI62636
249. Freischmidt A, Müller K, Zondler L, Weydt P, Volk AE, BoŽič; AL, et al. Serum microRNAs in patients with genetic amyotrophic lateral sclerosis and pre-manifest mutation carriers. Brain. (2014) 137:2938–50. doi: 10.1093/brain/awu249
250. Freischmidt A, Müller K, Zondler L, Weydt P, Mayer B, von Arnim CAF, et al. Serum microRNAs in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. (2015) 36:2660.e15-2660.e20. doi: 10.1016/j.neurobiolaging.2015.06.003
251. Matamala JM, Arias-Carrasco R, Sanchez C, Uhrig M, Bargsted L, Matus S, et al. Genome-wide circulating microRNA expression profiling reveals potential biomarkers for amyotrophic lateral sclerosis. Neurobiol Aging. (2018) 64:123–38. doi: 10.1016/j.neurobiolaging.2017.12.020
252. Takahashi I, Hama Y, Matsushima M, Hirotani M, Kano T, Hohzen H, et al. Identification of plasma microRNAs as a biomarker of sporadic amyotrophic lateral sclerosis. Mol Brain. (2015) 8:67. doi: 10.1186/s13041-015-0161-7
253. Saris CG, Horvath S, van Vught PW, van Es MA, Blauw HM, Fuller TF, et al. Weighted gene co-expression network analysis of the peripheral blood from Amyotrophic Lateral Sclerosis patients. BMC Genomics. (2009) 10:405. doi: 10.1186/1471-2164-10-405
254. Mougeot J-LC, Li Z, Price AE, Wright FA, Brooks BR. Microarray analysis of peripheral blood lymphocytes from ALS patients and the SAFE detection of the KEGG ALS pathway. BMC Med Genomics. (2011) 4:74. doi: 10.1186/1755-8794-4-74
255. Zhao W, Beers DR, Hooten KG, Sieglaff DH, Zhang A, Kalyana-Sundaram S, et al. Characterization of gene expression phenotype in amyotrophic lateral sclerosis monocytes. JAMA Neurol. (2017) 74:677. doi: 10.1001/jamaneurol.2017.0357
256. Nachmany H, Wald S, Abekasis M, Bulvik S, Weil M. Two potential biomarkers identified in mesenchymal stem cells and leukocytes of patients with sporadic amyotrophic lateral sclerosis. Dis Markers. (2012) 32:211–20. doi: 10.3233/DMA-2011-0885
257. Lilo E, Wald-Altman S, Solmesky LJ, Ben Yaakov K, Gershoni-Emek N, Bulvik S, et al. Characterization of human sporadic ALS biomarkers in the familial ALS transgenic mSOD1(G93A) mouse model. Hum Mol Genet. (2013) 22:4720–5. doi: 10.1093/hmg/ddt325
258. Wuolikainen A, Moritz T, Marklund SL, Antti H, Andersen PM. Disease-related changes in the cerebrospinal fluid metabolome in amyotrophic lateral sclerosis detected by GC/TOFMS. PLoS ONE. (2011) 6:e17947. doi: 10.1371/journal.pone.0017947
259. Spreux-Varoquaux O, Bensimon G, Lacomblez L, Salachas F, Pradat PF, Le Forestier N, et al. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J Neurol Sci. (2002) 193:73–8. doi: 10.1016/S0022-510X(01)00661-X
260. Dupuis L, Spreux-Varoquaux O, Bensimon G, Jullien P, Lacomblez L, Salachas F, et al. Platelet serotonin level predicts survival in amyotrophic lateral sclerosis. PLoS ONE. (2010) 5:e13346. doi: 10.1371/journal.pone.0013346
261. Simone IL, Ruggieri M, Tortelli R, Ceci E, D'Errico E, Leo A, et al. Serum N-acetylaspartate level in amyotrophic lateral sclerosis. Arch Neurol. (2011) 68:1308. doi: 10.1001/archneurol.2011.217
262. Keizman D, Ish-Shalom M, Berliner S, Maimon N, Vered Y, Artamonov I.et al. Low uric acid levels in serum of patients with ALS: further evidence for oxidative stress? J Neurol Sci. (2009) 285:95–9. doi: 10.1016/j.jns.2009.06.002
263. Dorst J, Kühnlein P, Hendrich C, Kassubek J, Sperfeld AD, Ludolph AC. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J Neurol. (2011) 258:613–7. doi: 10.1007/s00415-010-5805-z
264. Dedic SIK, Stevic Z, Dedic V, Stojanovic VR, Milicev M, Lavrnic D. Is hyperlipidemia correlated with longer survival in patients with amyotrophic lateral sclerosis? Neurol Res. (2012) 34:576–80. doi: 10.1179/1743132812Y.0000000049
265. Rozen S, Cudkowicz ME, Bogdanov M, Matson WR, Kristal BS, Beecher C, et al. Metabolomic analysis and signatures in motor neuron disease. Metabolomics. (2005) 1:101–8. doi: 10.1007/s11306-005-4810-1
266. Patin F, Corcia P, Vourc'h P, Nadal-Desbarats L, Baranek T, Goossens J-F, et al. Omics to explore amyotrophic lateral sclerosis evolution: the central role of arginine and proline metabolism. Mol Neurobiol. (2017) 54:5361–74. doi: 10.1007/s12035-016-0078-x
267. Conti A, Riva N, Pesca M, Iannaccone S, Cannistraci CV, Corbo M, et al. Increased expression of Myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim Biophys Acta. (2014) 1842:99–106. doi: 10.1016/J.BBADIS.2013.10.013
268. Yin F, Ye F, Tan L, Liu K, Xuan Z, Zhang J, et al. Alterations of signaling pathways in muscle tissues of patients with amyotrophic lateral sclerosis. Muscle Nerve. (2012) 46:856–60. doi: 10.1002/mus.23411
269. Elf K, Shevchenko G, Nygren I, Larsson L, Bergquist J, Askmark H, et al. Alterations in muscle proteome of patients diagnosed with amyotrophic lateral sclerosis. J Proteomics. (2014) 108:55–64. doi: 10.1016/j.jprot.2014.05.004
270. Narayan M, Seeley KW, Jinwal UK. Identification of Apo B48 and other novel biomarkers in amyotrophic lateral sclerosis patient fibroblasts. Biomark Med. (2016) 10:453–462. doi: 10.2217/bmm-2016-0025
271. Paré B, Touzel-Deschênes L, Lamontagne R, Lamarre M-S, Scott F-D, Khuong HT, et al. Early detection of structural abnormalities and cytoplasmic accumulation of TDP-43 in tissue-engineered skins derived from ALS patients. Acta Neuropathol Commun. (2015) 3:5. doi: 10.1186/s40478-014-0181-z
272. Mendonça DMF, Chimelli L, Martinez AMB. Quantitative evidence for neurofilament heavy subunit aggregation in motor neurons of spinal cords of patients with amyotrophic lateral sclerosis. Brazilian J Med Biol Res. (2005) 38:925–33.
273. Kovanda A, Leonardis L, Zidar J, Koritnik B, Dolenc-Groselj L, Ristic Kovacic S, et al. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci Rep. (2018) 8:5609. doi: 10.1038/s41598-018-23139-2
274. Campos-Melo D, Droppelmann CA, He Z, Volkening K, Strong MJ. Altered microRNA expression profile in amyotrophic lateral sclerosis: a role in the regulation of NFL mRNA levels. Mol Brain. (2013) 6:26. doi: 10.1186/1756-6606-6-26
275. Koval ED, Shaner C, Zhang P, du Maine X, Fischer K, Tay J, et al. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet. (2013) 22:4127–35. doi: 10.1093/hmg/ddt261
276. Ishtiaq M, Campos-Melo D, Volkening K, Strong MJ. Analysis of novel NEFL mRNA targeting microRNAs in amyotrophic lateral sclerosis. PLoS ONE. (2014) 9:e85653. doi: 10.1371/journal.pone.0085653
277. Emde A, Eitan C, Liou L-L, Libby RT, Rivkin N, Magen I, et al. Dysregulated miRNA biogenesis downstream of cellular stress and ALS-causing mutations: a new mechanism for ALS. EMBO J. (2015) 34:2633–2651. doi: 10.15252/embj.201490493
278. Si Y, Cui X, Kim S, Wians R, Sorge R, Oh SJ, et al. Smads as muscle biomarkers in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. (2014) 1:778–87. doi: 10.1002/acn3.117
279. Shtilbans A, Choi S-G, Fowkes ME, Khitrov G, Shahbazi M, Ting J, et al. Differential gene expression in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2011) 12:250–6. doi: 10.3109/17482968.2011.560946
280. Bernardini C, Censi F, Lattanzi W, Barba M, Calcagnini G, Giuliani A, et al. Mitochondrial network genes in the skeletal muscle of amyotrophic lateral sclerosis patients. PLoS ONE. (2013) 8:e57739. doi: 10.1371/journal.pone.0057739
281. Si Y, Kim S, Cui X, Zheng L, Oh SJ, Anderson T, et al. Transforming growth factor beta (TGF-β) Is a muscle biomarker of disease progression in ALS and correlates with smad expression. PLoS ONE. (2015) 10:e0138425. doi: 10.1371/journal.pone.0138425
282. Dupuis L, Gonzalez de Aguilar J-L, di Scala F, Rene F, de Tapia M, Pradat P-F, et al. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol Dis. (2002) 10:358–365. doi: 10.1006/NBDI.2002.0522
283. Malaspina A, Kaushik N, De Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem. (2008) 77:132–45. doi: 10.1046/j.1471-4159.2001.00231.x
284. Jiang Y-M, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, Terao S, et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol. (2005) 57:236–51. doi: 10.1002/ana.20379
285. Ishigaki S, Niwa J, Ando Y, Yoshihara T, Sawada K, Doyu M, et al. Differentially expressed genes in sporadic amyotrophic lateral sclerosis spinal cords-screening by molecular indexing and subsequent cDNA microarray analysis. FEBS Lett. (2002) 531:354–8. doi: 10.1016/S0014-5793(02)03546-9
286. Dangond F, Hwang D, Camelo S, Pasinelli P, Frosch MP, Stephanopoulos G, et al. Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol Genomics. (2004) 16:229–39. doi: 10.1152/physiolgenomics.00087.2001
287. Wang X-S, Simmons Z, Liu W, Boyer PJ, Connor JR. Differential expression of genes in amyotrophic lateral sclerosis revealed by profiling the post mortem cortex. Amyotroph Lateral Scler. (2006) 7:201–10. doi: 10.1080/17482960600947689
288. Lederer CW, Torrisi A, Pantelidou M, Santama N, Cavallaro S. Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genomics. (2007) 8:26. doi: 10.1186/1471-2164-8-26
289. Offen D, Barhum Y, Melamed E, Embacher N, Schindler C, Ransmayr G. Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant. J Mol Neurosci. (2009) 38:85–93. doi: 10.1007/s12031-007-9004-z
290. Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H, et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE. (2010) 5:e9872. doi: 10.1371/journal.pone.0009872
291. Riva N, Clarelli F, Domi T, Cerri F, Gallia F, Trimarco A, et al. Unraveling gene expression profiles in peripheral motor nerve from amyotrophic lateral sclerosis patients: insights into pathogenesis. Sci Rep. (2016) 6:39297. doi: 10.1038/srep39297
292. Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem. (2002) 80:616–25. doi: 10.1046/j.0022-3042.2001.00731.x
293. Jones AP, Gunawardena WJ, Coutinho CM, Gatt JA, Shaw IC, Mitchell JD. Preliminary results of proton magnetic resonance spectroscopy in motor neurone disease (amytrophic lateral sclerosis). J Neurol Sci. (1995) 129(Suppl.):85–9.
294. Gredal O, Rosenbaum S, Topp S, Karlsborg M, Strange P, Werdelin L. Quantification of brain metabolites in amyotrophic lateral sclerosis by localized proton magnetic resonance spectroscopy. Neurology. (1997) 48:878–81.
295. Cwik VA, Hanstock CC, Allen PS, Martin WR. Estimation of brainstem neuronal loss in amyotrophic lateral sclerosis with in vivo proton magnetic resonance spectroscopy. Neurology. (1998) 50:72–7.
296. Ikeda K, Iwasaki Y, Kinoshita M, Ichijo M, Fujii H, Matsuoka Y, et al. Quantification of brain metabolites in ALS by localized proton magnetic spectroscopy. Neurology. (1998) 50:576–7.
297. Carew JD, Nair G, Pineda-Alonso N, Usher S, Hu X, Benatar M. Magnetic resonance spectroscopy of the cervical cord in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2011) 12:185–191. doi: 10.3109/17482968.2010.515223
298. Ikeda K, Murata K, Kawase Y, Kawabe K, Kano O, Yoshii Y, et al. Relationship between cervical cord 1 H-magnetic resonance spectroscopy and clinoco-electromyographic profile in amyotrophic lateral sclerosis. Muscle Nerve. (2013) 47:61–7. doi: 10.1002/mus.23467
299. Cistaro A, Pagani M, Montuschi A, Calvo A, Moglia C, Canosa A, et al. The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur J Nucl Med Mol Imaging. (2014) 41:844–52. doi: 10.1007/s00259-013-2667-5
300. Cheong I, Marjanska M, Deelchand DK, Eberly LE, Walk D, Öz G. Ultra-high field proton MR spectroscopy in early-stage amyotrophic lateral sclerosis. Neurochem Res. (2017) 42:1833–44. doi: 10.1007/s11064-017-2248-2
301. Foerster BR, Pomper MG, Callaghan BC, Petrou M, Edden RAE, Mohamed MA, et al. An imbalance between excitatory and inhibitory neurotransmitters in amyotrophic lateral sclerosis revealed by use of 3-T proton magnetic resonance spectroscopy. JAMA Neurol. (2013) 70:1009–16. doi: 10.1001/jamaneurol.2013.234
302. Unrath A, Ludolph AC, Kassubek J. Brain metabolites in definite amyotrophic lateral sclerosis. A longitudinal proton magnetic resonance spectroscopy study. J Neurol. (2007) 254:1099–106. doi: 10.1007/s00415-006-0495-2
Keywords: circulating biomarkers, ALS, patients stratification, multi-system biomarkers, motor neuron disease
Citation: Vijayakumar UG, Milla V, Cynthia Stafford MY, Bjourson AJ, Duddy W and Duguez SM-R (2019) A Systematic Review of Suggested Molecular Strata, Biomarkers and Their Tissue Sources in ALS. Front. Neurol. 10:400. doi: 10.3389/fneur.2019.00400
Received: 29 November 2018; Accepted: 02 April 2019;
Published: 14 May 2019.
Edited by:
Pierre-francois Pradat, Hôpitaux Universitaires Pitié Salpêtrière, FranceReviewed by:
Valentina Bonetto, Istituto Di Ricerche Farmacologiche Mario Negri, ItalyKim A. Staats, University of Southern California, United States
Copyright © 2019 Vijayakumar, Milla, Cynthia Stafford, Bjourson, Duddy and Duguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie Duguez, s.duguez@ulster.ac.uk
†These authors have contributed equally to this work