Neutrophilic and eosinophilic dermatoses associated with hematological malignancy
 Carlo Alberto Maronese1,2†
Carlo Alberto Maronese1,2†  Federica Derlino1†
Federica Derlino1†  Chiara Moltrasio1† Daniele Cattaneo3,4
Chiara Moltrasio1† Daniele Cattaneo3,4  Alessandra Iurlo3
Alessandra Iurlo3  Angelo Valerio Marzano1,2*
Angelo Valerio Marzano1,2*- 1Dermatology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- 2Department of Pathophysiology and Transplantation, Università degli Studi di Milano, Milan, Italy
- 3Hematology Division, Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- 4Department of Oncology and Hemato-Oncology, University of Milan, Milan, Italy
Cutaneous manifestations of hematologic malignancy represent both a clinical challenge for the treating physician and a pathophysiological model for advancing the knowledge on individual neoplasms. Indeed, a growing body of evidence supports the concept of recurrent molecular defects associating with specific clinical features, as best exemplified by VEXAS. Herein neutrophilic and eosinophilic dermatoses of potential interest for both hematologists and dermatologists will be reviewed, including subcorneal pustular dermatosis-type IgA pemphigus, neutrophilic eccrine hidradenitis, Sweet’s syndrome as well as myelodysplasia cutis and VEXAS, pyoderma gangrenosum, eosinophilic annular erythema, eosinophilic dermatosis of hematological malignancy, Wells syndrome and cutaneous involvement in hypereosinophilic syndromes. Possible management approaches are discussed for each, emphasizing scenarios that require treatment of the underlying condition to achieve remission at the skin level.
1 Introduction
Cutaneous manifestations of hematologic malignancies (HMs) represent both a clinical challenge for the treating physician and a pathophysiological model for advancing the knowledge on individual neoplasms. Indeed, a growing body of evidence supports the concept of recurrent molecular defects associated with specific clinical features, as best exemplified by VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) (1). Moreover, acquired somatic mutations in the hematopoietic compartment, other than those documented in true HM, have been documented to fuel a minor proportion of autoinflammatory urticaria, providing yet another example of the complex relationship between hemoproliferative disorders and the skin (2, 3).
While different subsets of cutaneous presentations have been linked to hematologic tumors (4), herein only neutrophilic and eosinophilic dermatoses will be reviewed, due to their special interest for both hematologists and dermatologists.
Neutrophilic dermatoses (ND), also known as neutrophilic diseases, are classified according to their clinico-pathological picture into superficial/epidermal, dermal and deep forms, with a fourth category that encompasses mixed as well as syndromic ND (5). Eosinophilic dermatoses have been classified similarly (6), but it should be underscored that available evidence is relatively more limited for this group. Levels of Evidence are also introduced for each of the discussed entities (7).
2 Neutrophilic dermatoses
2.1 Superficial neutrophilic dermatoses
2.1.1 Subcorneal pustular dermatosis-type IgA pemphigus (level of evidence 4)
IgA pemphigus is a rare neutrophilic acantholytic autoimmune disease, with two subtypes that differ in terms of epidermal immunoglobulin (Ig)A deposition patterns: subcorneal pustular dermatosis (SPD) and intraepidermal neutrophilic IgA dermatosis (IEN) (8).
SPD-type IgA pemphigus is a relapsing, sterile dermatosis clinically characterized by flat, hypopyon pustules, often on a slightly erythematous base. Histologically, it shows subcorneal acantholysis, pustules with intercellular IgA deposits in the upper epidermis and predominant neutrophil infiltration (9). True SPD differs from it for the negativity of immunofluorescence studies (9).
Concomitant lymphoproliferative disorders have been reported in SPD-IgA pemphigus, especially IgA monoclonal gammopathy of uncertain significance (MGUS) with a rate of 9.5% (10). It is speculated that IgA paraproteinemia may affect neutrophil function and migration, however the exact pathogenesis of the relationship with HM is presently unknown.
Cases of IgA multiple myeloma (MM) have also been reported, with six patients described so far (11–15). In four cases, the onset of MM was concomitant with the diagnosis of IgA pemphigus while in the remaining two patients, MM developed 6 and 17 years later, respectively. In most, treatment for MM also improved skin lesions, suggesting that MM treatment should precede standard treatment for IgA pemphigus (15). Indeed, Koga et al., (15) reported daratumumab with lenalidomide plus dexamethasone as an effective therapeutic option for both MM and IgA pemphigus.
HMs less frequently reported as related to true SPD are represented by aplastic anaemia (16), lymphomas (such as CD30+ anaplastic large-cell lymphoma and nodal marginal zone lymphoma) (17) and chronic lymphocytic leukaemia (CLL) (18).
Thorough investigations, with immunofluorescence and whenever possible with immunoblotting, can aid in making the correct diagnosis, differentiating this entity from other immunobullous diseases. It should be kept in mind that, although paraneoplastic pemphigus is commonly associated with HM, HM-associated SPD-type IgA pemphigus is a distinct disease and that the terms should not be used interchangeably (19).
Besides therapy of the underlying HM, which tends to work best also for the associated skin condition, dapsone remains cornerstone in treating SPD and SPD-type IgA pemphigus, starting at a dose of 25 mg, with a target of 50–150 mg/day; the lowest dose necessary to control symptoms should be maintained, and monitoring of hematologic toxicity is mandatory (20). Other therapeutic options include topical and oral corticosteroids (used concurrently or as primary treatment), immunosuppressants such as mycophenolate mofetil and azathioprine, phototherapy, photochemotherapy and acitretin (20). Isolated refractory cases have been managed with tumor necrosis factor (TNF)-α antagonist infliximab and adalimumab, intravenous immunoglobulin or plasmapheresis (10). HM-associated cases, however, are most commonly managed with systemic corticosteroids and dapsone (10–18).
2.1.2 Neutrophilic eccrine hidradenitis (level of evidence 4)
Neutrophilic eccrine hidradenitis (NEH) is a rare, self-limiting ND of unknown aetiology with a characteristic histopathologic pattern, typified by neutrophil-rich infiltrates and necrosis of eccrine sweat glands. The clinical picture is highly heterogenous, manifesting with asymmetric, erythematous-oedematous papules or plaques of variable size, either asymptomatic or pruriginous and painful, closely related to Sweet syndrome (SS) (21). Concordantly, the differential diagnosis of NEH is wide, possibly including SS, erythema multiforme, vasculitis, bacterial (e.g., Pseudomonas) folliculitis and idiopathic eccrine palmoplantar hidradenitis, which is a self-resolving disease observed in childhood.
NEH was initially described in patients with acute myeloid leukaemia (AML) receiving cytarabine (22). Intriguingly, patients with underlying HMs who develop NEH, commonly do so after the first cycle of systemic chemotherapy (23). There is some evidence supporting direct drug toxicity of chemotherapy to the eccrine sweat gland (due to preferential concentration) acting as a trigger for the onset of NEH (24). From a pathophysiologic standpoint, NEH could represent a chemotherapy-induced reactive disorder in the context of an abnormal neutrophil response (25).
Of note, NEH has also been identified in untreated cases of AML and chronic myeloid leukaemia (CML) (22) and, although very rarely, in idiopathic cases (26), the latter being successfully treated with colchicine (26). In such cases, age-appropriate cancer screening has been strongly recommended (21).
Being a self-limiting disease, NEH does not strictly require therapy, but may be managed only with supportive care (27). Moreover, in cases of NEH related to a specific chemotherapeutic agent, dapsone may be useful before drug rechallenge (28).
2.2 Dermal neutrophilic dermatoses
2.2.1 Sweet syndrome and related disorders (level of evidence 3A-4)
SS is a rare ND clinically characterized by the sudden onset of painful, tender, well-demarcated, erythematous papules, plaques and nodules, usually accompanied by fever (>38°C), leukocytosis and elevation of inflammatory markers (Figures 1A,B) (29). Arthralgias, malaise, headache and myalgias may concur. On histology, a dermal infiltrate of mature neutrophils without overt vasculitis is typically observed. Although SS is mostly considered as idiopathic, drug-induced or reactive cases arising in the setting of infectious, inflammatory and neoplastic diseases are recognized. HMs account for 85% of malignancy-associated cases, with AML being the most common associated form. Other entities include myelodysplastic syndromes (MDS), non-Hodgkin lymphomas (NHL), CLL and MM. Cutaneous lesions can present before, after, or simultaneously with malignancies and do not seem to have prognostic implications (30). No clear association has been demonstrated between clinical presentation and the aforementioned settings of occurrence. However, HM-associated SS has been suggested by some to present more frequently in older patients (31), with no sex preference and in those with complete blood count abnormalities (31–33). Also, a lack of arthralgias (31, 33) as well as a more persistent or recurrent course has been described (30).
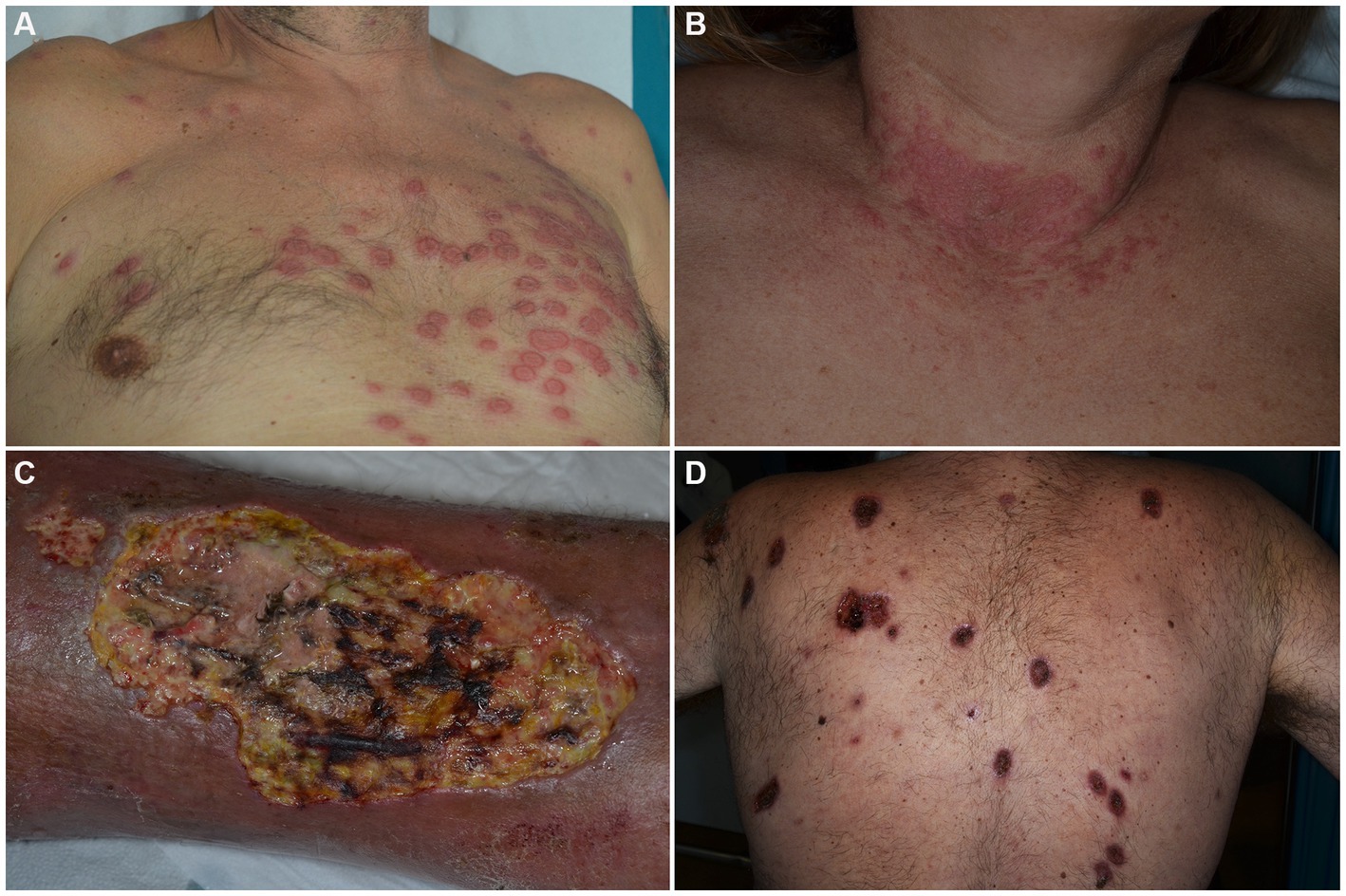
Figure 1. Clinical features of Sweet’s syndrome (A,B) and pyoderma gangrenosum (C,D). Erythematous, infiltrated roundish plaques are observed in the prototypic form among dermal neutrophilic dermatoses (A,B). Ulcerations with typical violaceous, undermined borders are seen in pyoderma gangrenosum, either in its unilesional or multilesional presentation (C,D).
HM-associated forms have been traditionally linked with a more histiocytoid histological appearance, which is defined by the presence of morphologically immature neutrophils that resemble histiocytic elements. While the link between histiocytoid SS and HMs has been questioned (34), a recent systematic review confirmed the association, especially with recurrent cases of histiocytoid SS (35).
Classic diagnostic criteria for SS include Su and Liu’s set first published in 1986 (36) and then modified by von den Driesch (37) and a separate diagnostic framework for drug-induced SS which was authored by Walker and Cohen (38). The most recent revision by Nofal et al. builds on von den Driesch’s work and distinguishes two constant features (abrupt onset of painful or tender erythematous papules, plaques, or nodules with a dense dermal neutrophilic infiltrate on histology) and a series of variable ones (fever >38°C; atypical skin lesions including hemorrhagic blisters, pustular lesions, cellulitis-like-lesions; presence or absence of leukocytoclastic vasculitis; subcutaneous, histiocytoid, xanthomatoid or cryptococcoid variant on histology; elevated erythrocyte sedimentation rate; elevated C-reactive protein levels; leukocytosis; neutrophilia; anemia), that may help avoid misdiagnosis (39).
With specific regard to MDS-associated forms, a spectrum of so-called myeloid dermatoses has been proposed, including: (i) classic clinicopathologic pictures of SS, (ii) histiocytoid SS and (iii) leukemia cutis, which is distinct, is positioned at the more severe end of the spectrum and is typified by true blast infiltrates in the skin. Representing de facto leukemic progression in the skin, leukemia cutis portends a poor prognosis and presents with either localized or more frequently widespread erythemato-violaceous papules, nodules or masses (40). Recently, this spectrum has expanded to embrace also an intermediate entity, which has been termed myelodysplasia cutis and may be diagnosed upon documentation of dermal infiltrates composed of non-blast myeloid cells clonally related to MDS cells in the bone marrow (41) (Figure 2).
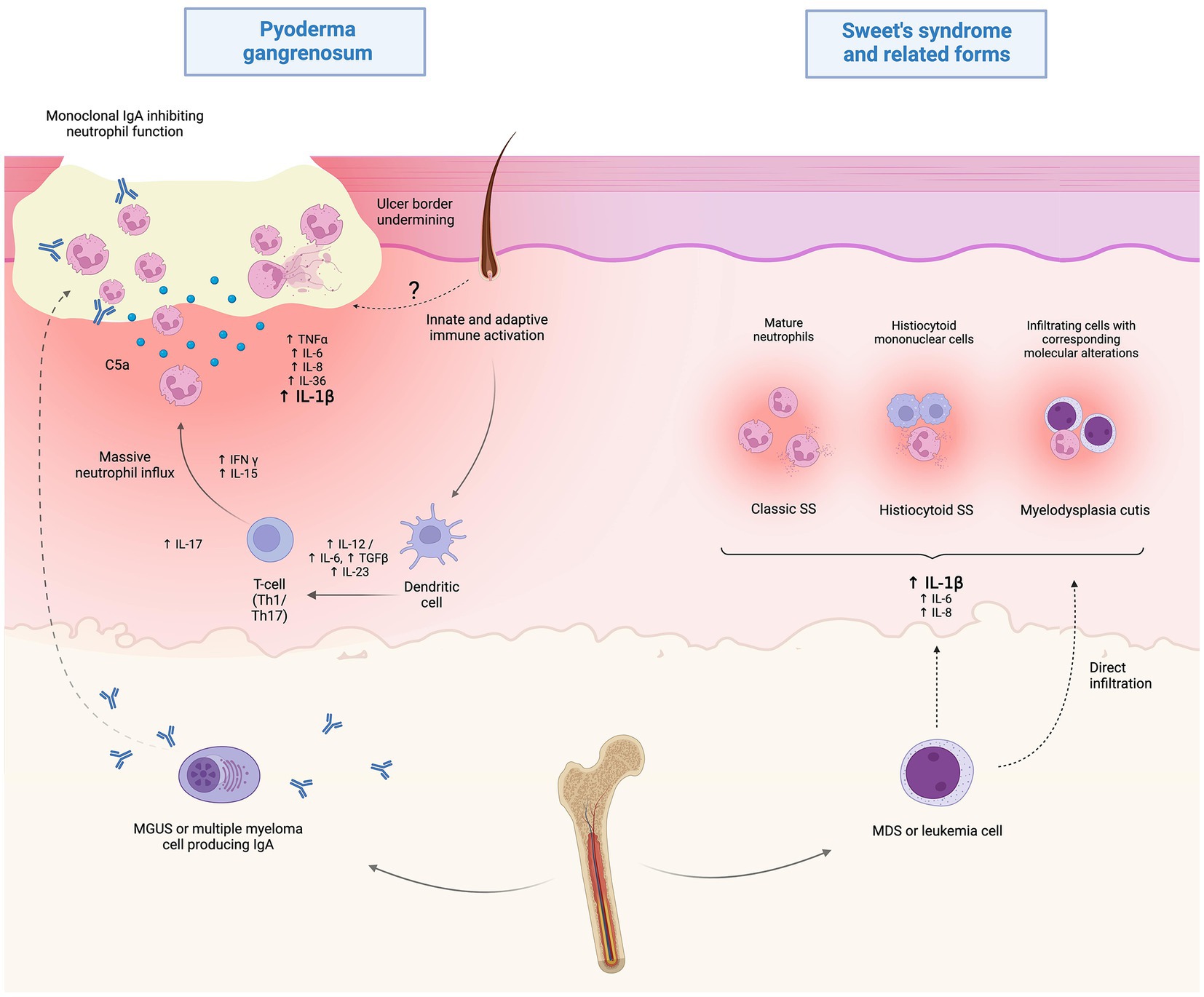
Figure 2. Proposed pathophysiology of hematological malignancy (HM)-associated Sweet’s syndrome (SS) and pyoderma gangrenosum (PG). While the current understanding of HM-associated PG is limited, it is speculated that immunoglobulin A paraproteinemia may play a role by altering neutrophil function in MGUS or multiple myeloma-associated cases. Myelodysplastic or frankly leukemic cells may promote systemic inflammation, resulting in the clinico-pathological picture of SS. Also, direct infiltration of myelodysplastic cells may play a role in the recently disclosed setting of myelodysplasia cutis. Created with BioRender.com.
Clinically, myelodysplasia cutis may present either with features of classic SS, i.e., erythematous, edematous plaques, or with a diffuse papulonodular eruption superimposable to that of leukemia cutis (42). This disconnect highlights the limitations in the current understanding of the pathophysiology of this spectrum and simultaneously underscores the importance of performing thorough investigations in otherwise typical SS cases.
Interestingly, a proportion of SS cases classified as classic or histiocytoid SS (43) as well as myelodysplasia cutis (42) may present in the setting of VEXAS syndrome, a newly recognized, autoinflammatory disease (43).
First described in 2020 in middle-aged men with a series of different, late-onset autoinflammatory manifestations, VEXAS is due to an inactivating acquired mutation in UBA-1, a gene encoding the ubiquitin-activating enzyme E1 (1). Disruption of ubiquitylation leads to altered degradation of proteins and their accumulation as cytoplasmatic vacuoles in an aberrant myeloid precursor, which is thus given a proliferative advantage and determines myeloid-induced inflammation. It is noteworthy that also drug-induced proteasome inhibition leads to SS in some cases (44).
VEXAS syndrome has a high morbidity and may present with fever, cytopenia, vacuoles in myeloid and erythroid precursor cells, bone marrow morphologic dysplasia and cutaneous and/or systemic autoinflammatory manifestations, such as ND (typically SS), vasculitis, relapsing polychondritis and pulmonary sterile neutrophilic infiltrates. It is noteworthy that cutaneous lesions in VEXAS-associated SS may have an arcuate papular morphology in up to a third of cases, which may be a clue to the underlying condition (45, 46). Importantly, it is presently unclear whether VEXAS cases classified as classic SS are true SS or were simply inadequately investigated from a molecular standpoint, lacking demonstration of an identical mutation in both skin and blood.
While dealing with SS in patients with HMs, it should be kept in mind that some cutaneous forms can also be triggered by medications commonly used for the neoplasm itself, like granulocyte colony stimulating factor (G-CSF), all-trans retinoic acid and hypomethylating agents (47).
Concerning possible treatment options, classic SS usually shows an excellent response to systemic steroids, such as prednisone at a dosage of 0.5–1 mg/kg/day, with a slow tapering within 4–8 weeks. Conversely, HM-associated as well as VEXAS-associated SS cases may experience a steroid-refractory, chronic-relapsing course, with the latter representing a possible clue to the underlying condition (30). In such instances, treatment of the neoplastic disorder represents the first choice followed by systemic corticosteroids and/or immunomodulating/immunosuppressive agents. For SS in VEXAS cases, particularly, while methotrexate, cyclosporine, and anti-interleukin (IL)-1 or anti-IL-6 inhibitors tend to result in transient responses, Janus kinase (JAK) inhibitors, e.g., ruxolitinib, are shaping as a more effective approach, leading to dramatic and even durable remissions (48, 49).
Finally, myelodysplasia cutis responds to azacytidine or hypomethylating agents, but hematopoietic stem cell transplantation remains the only curative option for both myelodysplasia cutis and VEXAS (42, 43, 50).
2.2.2 Erythema elevatum diutinum (level of evidence 4)
Erythema elevatum diutinum (EED) is a cutaneous leukocytoclastic vasculitis also classified as a dermal ND, due to its prominent neutrophilic component at histology in the early phases, its peculiar course, and its evolution, that set it apart from other vasculitides (51).
EED initially presents with symmetric, erythemato-violaceus, soft papules and plaques that favor the extensor surfaces of acral body sites and become indurated over time. The clinical evolution reflects the differences in histopathological appearance over time. In the early stages, a dermal infiltrate of polymorphonuclear cells along with fibrin deposition and sometimes SS-like papillary oedema is observed. As the lesions enter the chronic phase, histiocytes become more prevalent, with vascular prominence, lipid deposition, possible spindle cell proliferation and the characteristic finding of progressive concentric perivascular fibrosis (51).
The pathogenesis of EED is still incompletely understood but it is postulated that it originates from chronic antigenic exposure or excess antibody levels. Immune complex deposition occurring in post-capillary venules as a result of infections, hematologic or autoimmune diseases, may then lead to the production of key cytokines, such as IL-8, setting the inflammatory process in motion, similarly to other ND (51).
Several associations have been recorded, including infections, particularly HIV, autoimmune disorders and HMs (52). Among the latter, paraproteinemias (35.3%) hold a prominent role, particularly cases of the IgA-type, but other plasma cell dyscrasias, NHL, MDS, hairy cell leukemia and CLL are also possible (53). Typically, cases last 5–10 years before undergoing self-resolution; however, those associated with IgA paraproteinemia may persist for longer.
Finally, co-occurrence with other ND, such as pyoderma gangrenosum (PG), has also been observed, highlighting the association of HMs with most members of the ND spectrum (54).
2.3 Dermal/hypodermal
2.3.1 Pyoderma gangrenosum (level of evidence 3A-4)
PG is an autoinflammatory polygenic skin disorder classified within the group of deep/hypodermal ND and clinically characterized by rapidly evolving cutaneous ulcers, with undermined borders and peripheral erythema (Figures 1C,D) (55). In addition to the classic ulcerative form which accounts for 85% of cases, PG can occur in other variants such as: bullous, pustular, vegetative, peristomal, genital, infantile and extracutaneous, with individual cases sometimes switching between variants (56). Several mimickers of PG have been identified, the most important being venous ulcers, deep infections, vasculitides (especially ANCA-associated ones) and neoplasms, such as primary cutaneous CD30+ anaplastic large cell lymphoma.
Criteria have been developed to aid in the diagnosis of this entity. The first set of criteria was published by Su et al. in 2004 (57). More recently, criteria for the classic ulcerative variant have been validated by means of a Delphi consensus of international experts, emphasizing the role of histology (58). The third set of criteria, the PARACELSUS score, has been proposed by a German group, particularly for the differential diagnosis with venous leg ulcers. It represents a weighted score incorporating several items (3 points for major criteria: Progressive course of disease, Absence of relevant differential diagnoses, Reddish-violaceous wound border; 2 points for minor ones: Amelioration due to immunosuppressant, Characteristically bizarre ulcer shape, Extreme pain >4 VAS, localized pathergy phenomenon; 1 point for additional ones: Suppurative inflammation in histopathology, Undermined wound margin, Associated systemic disease), whereby a sum of 10 or more strongly supports a diagnosis of PG (59). Among the three, the PARACELSUS score correctly identifies the highest proportion of PG patients (60), but research efforts are ongoing to device newer and better diagnostic criteria, also for clinical trials (61).
An association with systemic diseases is documented in 50% of PG cases (22). HMs, particularly, may be present in up to 10% of patients with PG (62), with AML, CML, MM, MDS, and MGUS being the most frequent associated diagnoses (4).
In a large retrospective cohort study conducted by Ashchyan et al. (63), patient age was shown to influence the risk for certain comorbidities; patients aged 65 years or older had a higher probability of having associated HMs than younger patients. However, other studies have since reported concomitant HMs also in younger PG patients (e.g., average age of 56.6 years), highlighting that a hematologic work-up should not be restricted to patients of a certain age (64).
According to a recent systematic review on HM-associated PG (64), MDS (24.4%) and IgA-type MGUS (22.1%) represent the two most frequent associations. As both disorders can progress to overt malignancies, such as AML - reported in 11.5% of cases - and MM, respectively, it is important to identify patients with MGUS or MDS early and monitor them closely for disease progression, so that appropriate treatment can be started promptly (64). Indeed, the same authors pointed out that in most cases the diagnosis of MDS and MGUS preceded the onset of PG, whereas it was made concurrently in patients with AML. Of note, while ulcerative PG accounted for the majority of HM-associated PG, the bullous variant, which is usually characterized by a very severe and rapidly progressing picture, has been recorded in up to half of cases associated with AML (64). From a pathogenetic standpoint, while paraproteinemia-associated cases are speculated to result from an IgA-mediated impairment of neutrophil function and/or altered chemotaxis, little is known about the exact mechanisms underlying MDS-associated forms.
PG management encompasses a variety of systemic, topical and wound care options, which can be chosen and combined based on disease extent, inflammatory versus non inflammatory phase and comorbidities (55). In a proportion of cases, comorbidity-directed therapies aimed to control the associated HM also result in PG remission, proving indirect insights into the disease pathogenesis. Although chemotherapy alone led to healing of HM-associated PG in just 7.5% of reported cases (64), figures may be different with newer treatment approaches for HM.
In different case reports, PG in the setting of MDS has been successfully treated with a combination of systemic corticosteroids, immunosuppressive and immunomodulators agents (64–66).
Thalidomide, particularly, by modulating the release of inflammatory mediators like TNF-α and inhibiting the chemotaxis of monocytes and leucocytes and phagocytosis by neutrophils (67), has been shown to be very effective for both MDS and PG (68–71).
Similarly, MGUS-associated PG cases experienced partial and/or complete remission following MGUS-directed treatments, including autologous peripheral blood stem cell transplantation (72) and, particularly, after the administration of novel oral agents.
In detail, dramatic responses to bortezomib, a proteasome inhibitor, have been reported in PG cases with IgA MGUS (73–75); and smoldering MM (75). It is particularly noteworthy that switch to bortezomib resulted in prompt healing of either giant (74) or anti-TNF-α refractory PG (63) in just 1 month. Similarly, ixazomib, a next generation oral proteasome inhibitor, was reported to be effective in a case of PG with concurrent IgA smoldering MM. Strikingly, a complete response was achieved in just a few weeks since initiation, paralleling the resolution of MGUS (76). Indeed, an aggressive management of HM aimed at the resolution of concomitant IgA MGUS has been advocated for to achieve PG remission (77).
Overall, the development of PG in cases of HMs may confer a worse prognosis to the underlying disease and an appropriate haematological and clinical work-up should be mandatory. Also, careful consideration should be given prior to prescribing cyclosporine and TNF-α inhibitors in patients with HMs associated to PG, to avoid immunosuppression (78).
3 Eosinophilic dermatoses
3.1 Eosinophilic annular erythema (level of evidence 4)
Eosinophilic annular erythema (EAE) is a rare, superficial eosinophilic dermatosis (ED) that manifests with severely pruritic, large annular plaques hallmarked by intense peripheral erythema and central pigmentation. It was originally described in children (hence the name annular erythema of infancy) but can occur also in adults. Previously classified as a superficial variant of Wells syndrome (WS), EAE is an autonomous entity with a wide range of disease associations. The diagnosis is reached via clinicopathologic correlation, with several clues allowing to differentiate it from WS. Aside from the predominance of annular lesions, in EAE the inflammatory infiltrate is mainly perivascular (not deep dermal as in WS) and no flame figures are observed (at least in typical cases) (79). In a nation-wide multicenter French study, 4/18 (22.22%) patients had a concomitant HM, including polycythemia vera (n = 2) and B-cell lymphoma (n = 2). However, a lower rate was reported in the literature (80). While the peculiar cutaneous picture of EAE should prompt the consideration of screening for underlying malignancy, especially in the elderly, the strength of the relationship with HMs is subject to debate. Also, some cases of EAE may represent figurate variants of eosinophilic dermatosis of hematologic malignancy (EDHM) (81).
3.2 Eosinophilic dermatosis of hematologic malignancy (level of evidence 4)
EDHM represents a chronic-relapsing pruritic disorder occurring primarily in patients suffering from B-cell neoplasms, particularly CLL in which 6–8% of patients may be affected (accounting for up to 77% of EDHM cases), but also NHL (such as mantle cell lymphoma), acute leukemias, MM/MGUS and even T-cell lymphomas (82).
Now classified within the spectrum/group of ED (6, 82–84), the nomenclature of EDHM has undergone several changes since its original description, passing from exaggerated delayed hypersensitivity to mosquito bite in CLL (85) to insect bite-like reaction in patients with HMs (86) to ED of myeloproliferative neoplasms (MPN) (87) to EDHM (88). Two alternative names, i.e., T-cell papulosis associated with B-cell malignancy (89) and hematologic-related malignancy-induced eosinophilic dermatosis (He Remained) (90), have then been proposed, emphasizing its morphological features and the pathophysiological relationship with HMs, respectively (82). Finally, the all-comprehensive definition of polymorphic eruption of HMs has been proposed to embrace the full spectrum of EDHM clinicopathological nuances as well as its follicular variants (4).
The typical picture of EDHM consists of recurrent papules, papulo-vesicles, plaques, nodules, urticarial lesions and tense bullae, distributed to the limbs, trunk, and head and neck area in decreasing order of frequency (Figures 3A–C). Exceptionally, eyelid involvement has also been observed (91).
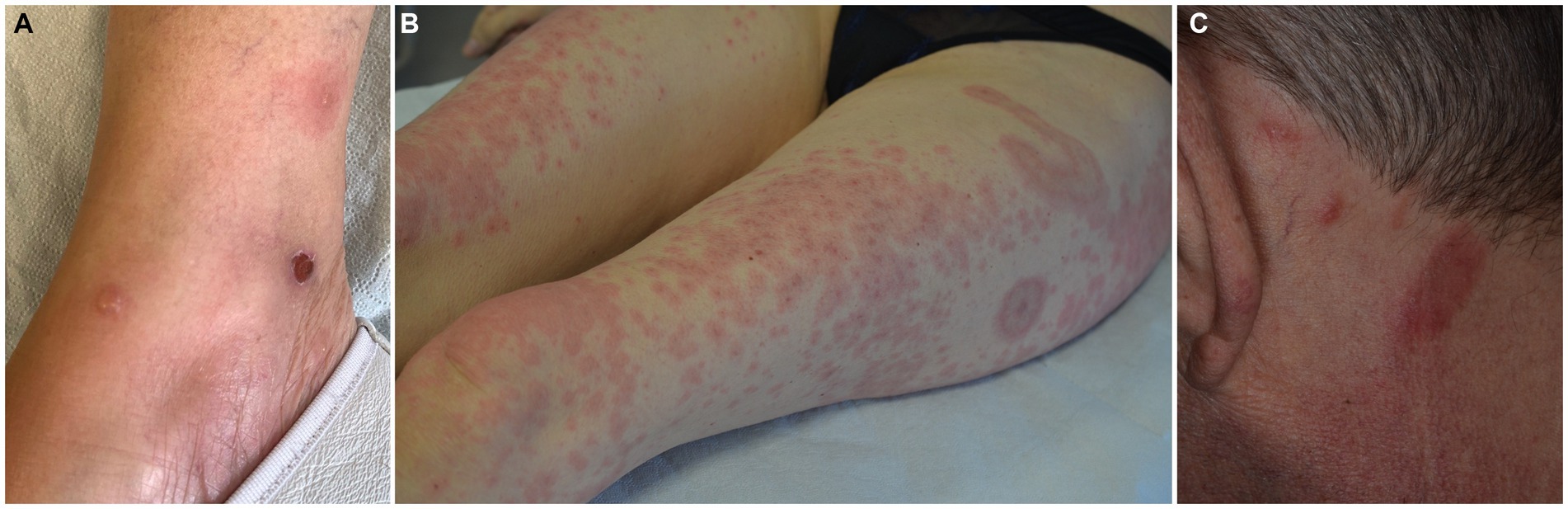
Figure 3. Spectrum of clinical manifestations in eosinophilic dermatosis of hematological malignancy, including pemphigoid-like (A), Wells syndrome-like (B) and eosinophilic pustular folliculitis (C) presentations.
Intense itching is reported, and secondary excoriation is often noted.
It is noteworthy that pemphigoid-like tense bullae have been described in up to a third of cases (84, 92) highlighting the importance of performing immunofluorescence studies. A third, smaller subset of patients presents with eosinophilic cellulitis-like forms (4).
While evidence on seasonal variation is controversial (89, 92–94), it has progressively become apparent that a substantial proportion of cases occurs independently of actual insect bites.
From a pathophysiological perspective, EDHM is T helper 2 (Th2)-skewed process, showing a reduced FOXP3/CD4 ratio (indicating a relative deficiency in the regulatory T -TReg- compartment) and significant IL-4, IL-31 and eotaxin-1 overexpression in the skin. Notably, serum IL-4 is also significantly elevated (84). Skin presence of the same neoplastic B-cell clone as in extracutaneous tissues was demonstrated in 13/15 (86.6%) cases associated with CLL, leading to speculation on a possible direct role in EDHM pathogenesis (89, 95, 96). One unifying hypothesis would have an inconspicuous proportion of neoplastic B-cell clones infiltrating the skin and orchestrating a shift toward type 2 immunity, with subsequent eosinophil chemotaxis (84) (Figure 4). While cases with non-eosinophil-rich histologic pictures have been described (89), it is important to underscore that these can relapse showing typical EDHM histology and viceversa (4).
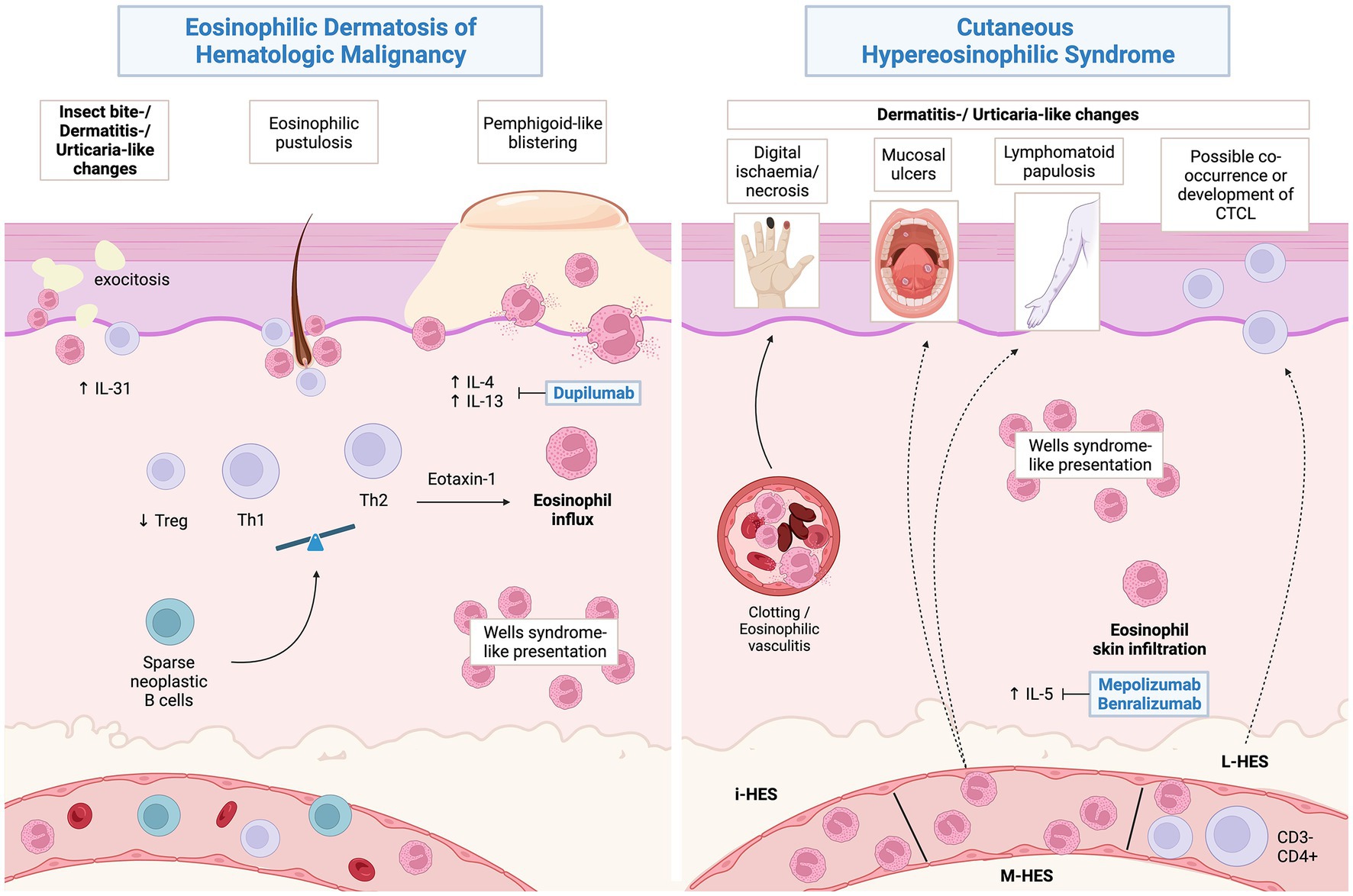
Figure 4. Proposed pathophysiology of eosinophilic dermatosis of hematological malignancy and possible associations of hypereosinophilic syndromes. Created with BioRender.com.
Eosinophilic (pustular) folliculitis and acneiform follicular mucinosis may also be part of the EDHM spectrum, demonstrating superimposable histological features (4, 97). A series focusing on folliculotropic forms reported a higher proportion of head and neck involvement (89). Indeed, a recent study evaluating cutaneous hypersensitivity reactions in a cohort consisting of 501 patients with CLL/small lymphocytic lymphoma (SLL) showed a predominately folliculocentric CD4+ T-cell infiltrate in 5/17 cases, all occurring in the head and neck area (94). In some rare instances, the infiltrates may also involve the subcutaneous tissue, with a clinicopathologic picture of eosinophilic panniculitis (98).
Due to its protean manifestations, EDHM poses a significant challenge in diagnostic terms, with possible differential diagnoses including bullous pemphigoid, true insect-bite reaction, eczema, urticaria, urticarial vasculitis (99), leukemia cutis and disseminated Herpes zoster (100). Mycosis fungoides, especially the follicular variant, is sometimes listed as a possible differential diagnosis of eosinophilic folliculitis/folliculotropic EDHM.
Originally, Byrd et al. proposed a set of criteria to support the diagnosis of EDHM including: (i) pruritic, papular, nodular, and/or vesiculobullous eruption that is resistant to conservative management; (ii) a superficial and deep eosinophil-rich dermal lymphohistiocytic infiltrate on histology; (iii) exclusion of other causes of tissue eosinophilia; and (iv) pre-existing diagnosis of HM (87). However, said criteria fail to incorporate all the nuances in the EDHM spectrum (4).
It should be underscored that EDHM holds no prognostic relevance with respect to the underlying HM (93); however, it may benefit from treatments targeted at the associated HM. In the past, systemic and topical corticosteroids, dapsone, other nonspecific immunosuppressant/immunomodulating agents (e.g., doxycycline) as well as UVA1 light phototherapy have proved effective at controlling the cutaneous picture (92, 93). While the condition is responsive to a variety of options, relapses are frequent (92) and long-term maintenance may be required. Recently, dupilumab an anti-IL4Rα monoclonal antibody (mAb) targeting type 2 inflammation demonstrated high effectiveness in a good proportion of EDHM cases (5/9) (101–104), paving the way for pathogenesis-driven therapy of this challenging condition.
3.3 Wells syndrome (level of evidence 3A-4)
WS is a dermal eosinophilic dermatosis, mainly observed in adults and hallmarked by a benign, yet sometimes recurrent clinical course (105). WS classically presents with urticarial erythematous-edematous plaques, but sometimes also with more infiltrated lesions, vesicles, or blisters. An annular configuration with central blanching and a purplish border may be observed. Generally, complete remission of a flare occurs within 4 to 8 weeks from onset, but a more prolonged course is possible, up to several years (105, 106).
Many clinical varieties of WS have been identified, including plaque-type (most common in children), annular granuloma–like (most commonly seen in adults), urticaria-like, papulovesicular, bullous, papulonodular and fixed drug eruption–like (106).
From a histopathological point of view, three stages are described. At the first stage of the disease, only dermal edema along with eosinophilic infiltrates is documented. A second (sub-acute) phase then ensues, being characterized by the presence in the mid-deep dermis of the so-called “flame-figures,” i.e., structures made of degenerated collagen fibers and eosinophils at the center with a dense infiltrate of histiocytes admixed with numerous eosinophils at the periphery. With time, as lesions enter the third phase, only granulomatous changes with histiocytes and giant cells around the “flame figures” are seen. Vasculitic changes are not observed in WS. It is important to underscore that while characteristic, “flame-figures” are not pathognomonic of WS, possibly appearing also in other forms of the eosinophilic dermatosis spectrum (105).
Two sets of diagnostic criteria have been proposed by Caputo et al. and Heelan et al., in 2006 and 2013, respectively, (106, 107). The most recent one requires two out of four major criteria (documentation of any of the previously reported clinical variants; relapsing, remitting course; no evidence of systemic disease; eosinophilic infiltrates with no vasculitis on histology) alongside at least one out of four minor criteria (flame figures; granulomatous changes on histology; peripheral eosinophilia not persistent and not greater than >1,500/μl; presence of a triggering factor) (107).
Whether distinguishing Wells syndrome (WS) from EDHM or cutaneous hypereosinophilic syndrome (cHES) (108) in hematologic patients is possible is still matter of debate (109).
Indeed, a proportion of cases originally reported as WS in patients with B-cell malignancies (mantle cell lymphoma, CLL) may have been clinically consistent with EDHM (110–112). Vice versa, clinically typical WS, with large erythematous, oedematous areas, has been recognized in some cases of CLL (113, 114). Co-occurrence of WS with EDHM in patients with B-cell neoplasms credits the idea of one spectrum of disorders, with different nuances in clinical expressivity (113, 115, 116).
Eosinophilic cellulitis may also be a presenting feature of idiopathic HES (108, 117, 118), rarely showing a more severe, necrotic evolution (119).
Recurrent eosinophilic cellulitis has been reported - albeit anecdotally - also in myeloid/lymphoid neoplasms with eosinophilia harbouring the FIP1L1-PDGFRA fusion transcript (120) or t(5,12)(q33;p13) translocation (121).
It is important to underline that WS-like cutaneous features may be present both in patients with EDHM and in those with HES, peripheral blood eosinophilia being the major distinguishing feature between the two scenarios.
Classic treatment options for WS include topical and systemic corticosteroids tapered over the course of several weeks, as well as cyclosporine or dapsone either as steroid-sparing agents or as add-on for refractory cases (122). Interestingly, newer drugs capable of selectively targeting type 2 immunity, such as anti-IL-4/13 (123, 124), anti-IL-5 (125, 126), anti-IL-5R (127) and JAK2 inhibitors (128) have been reported to be effective in isolated cases of WS, similarly to both EDHM and HES. Dapsone and biologics are of particular interest as they appear to control cutaneous manifestations without acting as immunosuppressants.
Cases of WS associated with HM have been traditionally managed with systemic corticosteroids, usually with rapid responses (113, 115, 116).
3.4 Hypereosinophilic syndrome and cutaneous hypereosinophilic syndrome (level of evidence 3A-4)
HES is a condition defined by the presence of peripheral blood hypereosinophilia (≥1.5 × 109/L) in association with tissue/organ damage. According to the recent international consensus classification (129), the following etiologic scenarios are recognized: (i) secondary/reactive HES (eosinophils are reactive and non-clonal) including lymphocyte variant-HES (L-HES); (ii) primary HES (associated with a hematopoietic neoplasms); (iii) idiopathic HES. The hematologic neoplasms associated with the second scenario include: myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions (M/LN-eo-TK); eosinophilia associated with other myeloid neoplasms, e.g., CML or AML with inv. (16); and chronic eosinophilic leukemia, not otherwise specified (CEL, NOS). In contrast, the term of “idiopathic HE” or “HE of unknown significance” (HEus) is used to describe persistent hypereosinophilia (≥6 months) without associated organ/tissue damage (129).
Concerning skin manifestations of HES, while a typical picture consisting of eczema- or urticaria-like features can broadly be defined, some nuances are characteristic of each form and will be discussed separately. Hopefully, a better categorization of cHES is to be expected as our molecular understanding of HES becomes progressively more detailed.
Lymphocyte-variant HES is linked to clonal circulating Th2 CD4 T cells (>0.5% T cells), most commonly with a defective CD3− CD4+ immunophenotype and shows the greatest frequency (79%) of cutaneous involvement among HES forms (130). In line with its Th2 polarization, L-HES commonly presents urticaria/angioedema, pruritus and eczematous lesions. Unusual manifestations include subcutaneous nodules and palmoplantar haemorrhagic blisters (131). Of note, although a certain degree of overlap exists between EDHM and cHES (e.g., nonspecific erythematous eruptions, pruritus, urticaria, eosinophilic cellulitis), true, pemphigoid-like blistering is only rarely observed in HES (131).
As both nodal and primary cutaneous peripheral T-cell lymphomas may develop during the course of L-HES, close follow-up may be advised. Of note, cHES may also initially masque a concomitant cutaneous lymphoma. In a large series, where skin T-cell clonality was available for eight cases, a dominant clone identical to the blood T-cell clone was found in seven, of whom two had cHES alone and five had concurrent primary cutaneous T-cell lymphoma (132).
Primary HES, formerly identified as myeloid HES comprising also CEL, NOS, affects the skin in 23.1–25% of cases (130), with more robust data available for the predominant subgroup harbouring the FIP1L1–PDGFRA fusion gene (32%) which results in a constitutively activated platelet-derived growth factor receptor-alpha (PDGFRA) (133).
Besides the rare occurrence of eosinophilic cellulitis, M/LN-eo-TK with the FIP1L1–PDGFRA fusion gene usually presents with transient erythematous (eczema-like) eruptions (121), pruritus, urticaria and dermographism (130).
Lymphomatoid papulosis, cutaneous nodules, purpura and mouth ulcers have been reported as well (133, 134). Indeed, chronic recurrent ulcerations affecting the oral or genital mucosae are a distinctive, incapacitating manifestation of HES and may represent the first sign of the condition (135–138). Mucosal ulcers have been linked particularly with the FIP1L1–PDGFRA fusion gene-positive form (137, 139, 140), occurring in 8/151 of these patients in a large series (133); however, they have been reported also in idiopathic HES (141, 142) and albeit anecdotally in L-HES (143).
Lymphomatoid papulosis is another characteristic association of M/LN-eo-TK with the FIP1L1–PDGFRA fusion gene, co-occurring in approximately 7% (11/151) of cases (133, 144).
Recently, Kitayama et al. demonstrated direct skin infiltration by neoplastic, FIP1L1–PDGFRA fusion gene-positive eosinophils in a patient with M/LN-eo-TK harbouring the mentioned translocation and presenting with a pruritic erythematous maculo-papular rash, dermographism and slight hyperpigmentation (145). The authors also reported an increase in dermal mast cells (145), which may be in keeping with pre-clinical evidence showing synergism between FIP1L1–PDGFRA fusion gene and the Stem Cell Factor/c-Kit pathway, thus promoting mast cell activation and survival (146).
Of note, a complex relationship links the abovementioned MPN and systemic mastocytosis (SM), one that may have had diagnostic repercussions on a proportion of previous reports (145, 147). From a hematologic perspective, it is imperative to differentiate M/LN-eo-TK presenting with a mast cell proliferation (no KIT D816V mutation) from SM with or without an associated myeloid neoplasm (129, 148). Conversely, from a dermatological perspective, the presence of an abundant mast cell component in the skin may hypothetically explain some features of the cutaneous picture such as dermographism and residual hyperpigmentation (145), the latter possibly serving as a clue to it.
Cutaneous involvement in idiopathic HES occurs in approximately a third of cases (130). According to a review on 32 individual patients, it presents as a pruritic and sometimes painful, erythematous-oedematous, papular eruption mainly on the extremities but also on the trunk. Other possible yet nonspecific skin changes include urticaria/angioedema, telangiectasia, palmar erythema or lichenification, cutaneous atrophy, superficial venous thrombophlebitis, hyperpigmentation (149), erythroderma (150) and eosinophilic cellulitis (117, 119).
A peculiar and possibly underrecognized subset of patients with idiopathic HES presents with necrotizing eosinophilic vasculitis, either as single organ vasculitis or with multisystem involvement. Similarly to idiopathic HES, the diagnosis of HES-associated vasculitis requires the exclusion of secondary causes, particularly eosinophilic granulomatosis with polyangiitis (EGPA) (151). HES-associated vasculitis, also known as idiopathic eosinophilic vasculitis, affects the skin in approximately half of cases, manifesting clinically with a picture of pruritic papular or frankly urticarial lesions, followed - in decreasing order of frequency - by purpuric papules, livedo, angioedema and even skin ulcers or digital necrosis (including splinter haemorrhages and nail fold infarcts), sometimes preceded by Raynaud’s phenomenon (151–159). Lesions predominate on the extremities, but the trunk and the head and neck area may also be affected, such as in patients with temporal arteritis (151, 160).
From a practical perspective, the demonstration of a clinicopathologic picture of eosinophilic vasculitis, including the characteristic digital ulcers, may hypothetically favor the diagnosis of idiopathic HES over L-HES or forms associated with myeloid/lymphoid neoplasia (161).
Importantly, cHES with non-eosinophil rich infiltrates on histology has also been observed. Among such rare instances, cases of interstitial granulomatous dermatitis are of particular interest (133, 162, 163). Indeed, the presence of a granulomatous infiltrate in the skin manifesting clinically with roundish erythematous plaques showing characteristic central umbilication and xanthomization may be a distinctive feature of the newly defined M/LN-eo-TK with t(9,12)(q22;p13) ETV6::SYK (164–166).
Treatment of HES is challenging and variant-specific approaches are needed to obtain optimal results.
While corticosteroids represent a first-line option across HES variants, primary forms associated with a HM tend to respond inadequately. Among the latter, presence of specific fusion transcripts, i.e., FIP1L1–PDGFRA, serves as predictor a good clinical and hematologic response to imatinib 100–400 mg die (167). M/LN-eo-TK with other fusion genes or CEL, NOS may also benefit from the same approach (usually requiring higher dosages) or from next generation tyrosine kinase inhibitors; however, treatment-tailoring based on the underlying neoplasm, possibly with stem-cell transplantation, is required to achieve blood and skin remission (129, 130). L-HES is less responsive than the idiopathic variant to systemic corticosteroids and may require additional lines of treatment. Among the latter, pegylated interferon alpha 2a is regarded an effective and well-tolerated option, opposing the Th2 polarization of the condition (168). Regarding biologics, mepolizumab, an anti-IL-5 mAb recently approved for HES without an identifiable secondary non hematologic cause, resulted in good control of most HES manifestations; however, reported skin outcomes are conflicting (169).
In a phase II trial, patients treated with benralizumab, an anti-IL-5R mAb, also demonstrated good clinical and hematological responses. However, among patients with complete individual descriptive data, only 2/4 with L-HES had sustained cutaneous response while the other half showed quick loss of response; those with idiopathic HES generally had substantial (2/3) or at least partial (1/3) improvements of their respective cutaneous pictures (170). This is consistent with two subsequent reports on idiopathic HES (171, 172). Isolated observations also support the effectiveness of dupilumab in idiopathic HES, however larger studies are needed to define its placing, if any (173, 174).
4 Conclusion
Neutrophilic and eosinophilic dermatoses associated with HM represent a heterogeneous group of skin conditions, which may either parallel the course of the underlying hematological disorder (PG, SS) or be independent from it (e.g., EDHM). Several entities belonging to both neutrophilic and eosinophilic dermatoses have recently been subject to provisional reclassification, thanks to a better overall understanding of their molecular aspects. EDHM now incorporates also eosinophilic pustular folliculitis as well as pemphigoid-like and Wells-like presentations. Conversely, several different scenarios have been distinguished in addition to classic SS, i.e., histiocytoid SS, VEXAS-related SS and the newly defined setting of myelodysplasia cutis. Better definition of the molecular characteristics of these dermatoses has also led to promising premises for pathogenesis-driven treatments, as for dupilumab in EDHM; however, mechanisms linking each HM to specific cutaneous phenotypes are still incompletely understood and warrant further research.
Author contributions
CMa: Supervision, Writing – review & editing, Conceptualization, Investigation, Writing – original draft. FD: Investigation, Writing – original draft, Writing – review & editing. CMo: Investigation, Writing – original draft, Writing – review & editing, Supervision. DC: Writing – review & editing. AI: Writing – review & editing. AM: Writing – review & editing, Supervision, Validation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was partially supported by the Italian Ministry of Health (Ricerca Corrente 2023), Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan (Italy).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Beck, DB, Ferrada, MA, Sikora, KA, Ombrello, AK, Collins, JC, Pei, W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
2. Assrawi, E, Louvrier, C, Lepelletier, C, Georgin-Lavialle, S, Bouaziz, JD, Awad, F, et al. Somatic mosaic NLRP3 mutations and Inflammasome activation in late-onset chronic Urticaria. J Invest Dermatol. (2020) 140:791–798.e2. doi: 10.1016/j.jid.2019.06.153
3. De Langhe, E, Van Loo, S, Malengier-Devlies, B, Metzemaekers, M, Staels, F, Vandenhaute, J, et al. TET2-driver and NLRC4-passenger variants in adult-onset autoinflammation. N Engl J Med. (2023) 388:1626–9. doi: 10.1056/NEJMc2212928
4. Maglie, R, Genovese, G, Solimani, F, Guglielmo, A, Pileri, A, Portelli, F, et al. Immune-mediated dermatoses in patients with Haematological malignancies: a comprehensive review. Am J Clin Dermatol. (2020) 21:833–54. doi: 10.1007/s40257-020-00553-9
5. Marzano, AV, Borghi, A, Wallach, D, and Cugno, M. A comprehensive review of neutrophilic diseases. Clin Rev Allergy Immunol. (2018) 54:114–30. doi: 10.1007/s12016-017-8621-8
6. Marzano, AV, and Genovese, G. Eosinophilic dermatoses: recognition and management. Am J Clin Dermatol. (2020) 21:525–39. doi: 10.1007/s40257-020-00520-4
7. Centre for Evidence-Based Medicine. Oxford Centre for Evidence-Based Medicine: Levels of Evidence; (2009). Available at: https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009 (Accessed October 31, 2023)
8. Düker, I, Schaller, J, Rose, C, Zillikens, D, Hashimoto, T, and Kunze, J. Subcorneal pustular dermatosis-type IgA pemphigus with autoantibodies to desmocollins 1, 2, and 3. Arch Dermatol. (2009) 145:1159–62. doi: 10.1001/archdermatol.2009.224
9. Cardones, A, and Hall, R III. Bullous diseases of the skin and mucous membranes. Clin. Immunol. (2019) 2019:857–70. doi: 10.1016/B978-0-7020-6896-6.00063-6
10. Kridin, K, Patel, PM, Jones, VA, Cordova, A, and Amber, KT. IgA pemphigus: a systematic review. J Am Acad Dermatol. (2020) 82:1386–92. doi: 10.1016/j.jaad.2019.11.059
11. Bernard, P, Amici, JM, Bedane, C, Catanzano, G, and Bonnetblanc, JM. Dermatose neutrophilique à IgA intra-épidermique associée à un myélome à IgA [Intra-epidermal neutrophilic IgA dermatosis associated with IgA myeloma]. Ann Dermatol Venereol. (1990) 117:890–2.
12. Takata, M, Inaoki, M, Shodo, M, Hirone, T, and Kaya, H. Subcorneal pustular dermatosis associated with IgA myeloma and intraepidermal IgA deposits. Dermatology. (1994) 189 Suppl 1:111–4. doi: 10.1159/000246947
13. Szturz, P, Adam, Z, Klincová, M, Feit, J, Krejčí, M, Pour, L, et al. Multiple myeloma associated IgA pemphigus: treatment with bortezomib- and lenalidomide-based regimen. Clin Lymphoma Myeloma Leuk. (2011) 11:517–20. doi: 10.1016/j.clml.2011.06.014
14. Lee, N, Lockwood, SJ, Richardson, P, Paba-Prada, C, and Saavedra, AP. IgA multiple myeloma in a patient with an IgG pemphigus foliaceus-like exanthem. Int J Dermatol. (2017) 56:1058–60. doi: 10.1111/ijd.13721
15. Koga, H, Tsutsumi, M, Teye, K, Ishii, N, Yamaguchi, M, Nagafuji, K, et al. Subcorneal pustular dermatosis-type IgA pemphigus associated with multiple myeloma: a case report and literature review. J Dermatol. (2023) 50:234–8. doi: 10.1111/1346-8138.16516
16. Park, BS, Cho, KH, Eun, HC, and Youn, JI. Subcorneal pustular dermatosis in a patient with aplastic anemia. J Am Acad Dermatol. (1998) 39:287–9. doi: 10.1016/s0190-9622(98)70093-3
17. Ratnarathorn, M, and Newman, J. Subcorneal pustular dermatosis (Sneddon-Wilkinson disease) occurring in association with nodal marginal zone lymphoma: a case report. Dermatol Online J. (2008) 14:6. doi: 10.5070/D336N564F4
18. Brown, SJ, Barrett, PD, Hendrick, A, and Langtry, JA. Subcorneal pustular dermatosis in association with chronic lymphocytic leukaemia. Acta Derm Venereol. (2003) 83:306–7. doi: 10.1080/00015550310016661
19. Antiga, E, Bech, R, Maglie, R, Genovese, G, Borradori, L, Bockle, B, et al. S2k guidelines on the management of paraneoplastic pemphigus/paraneoplastic autoimmune multiorgan syndrome initiated by the European academy of dermatology and venereology (EADV). J Eur Acad Dermatol Venereol. (2023) 37:1118–34. doi: 10.1111/jdv.18931
20. Cheng, S, Edmonds, E, Ben-Gashir, M, and Yu, RC. Subcorneal pustular dermatosis: 50 years on. Clin Exp Dermatol. (2008) 33:229–33. doi: 10.1111/j.1365-2230.2008.02706.x
21. Crane, JS. Neutrophilic eccrine hidradenitis In: StatPearls. Treasure Island (FL): StatPearls Publishing (2023)
22. de Souza, PK, Amorim, RO, Sousa, LS, and Batista, MD. Dermatological manifestations of hematologic neoplasms. Part II: nonspecific skin lesions/paraneoplastic diseases. An Bras Dermatol. (2023) 98:141–58. doi: 10.1016/j.abd.2022.08.005
23. Pierson, JC, Helm, TN, Taylor, JS, Elston, DM, and Tuthill, RJ. Neutrophilic eccrine hidradenitis heralding the onset of acute myelogenous leukemia. Arch Dermatol. (1993) 129:791–2. doi: 10.1001/archderm.1993.01680270135026
24. Li, AW, Yin, ES, Stahl, M, Kim, TK, Panse, G, Zeidan, AM, et al. The skin as a window to the blood: cutaneous manifestations of myeloid malignancies. Blood Rev. (2017) 31:370–88. doi: 10.1016/j.blre.2017.07.003
25. Saada, V, Aractingi, S, Leblond, V, Marinho, E, Frances, C, and Chosidow, O. Hidradénite eccrine neutrophilique associée à une rechute de leucémie aiguë myéloblastique [Neutrophilic eccrine hidradenitis associated with relapse of acute myeloblastic leukemia]. Ann Dermatol Venereol. (1998) 125:420–2.
26. Belot, V, Perrinaud, A, Corven, C, de Muret, A, Lorette, G, and Machet, L. Hidradénite eccrine neutrophilique idiopathique de l’adulte d’évolution prolongée traitée par colchicine [Adult idiopathic neutrophilic eccrine hidradenitis treated with colchicine]. Presse Med. (2006) 35:1475–8. doi: 10.1016/s0755-4982(06)74837-0
27. Cohen, PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. (2007) 46:106–11. doi: 10.1111/j.1365-4632.2006.02605.x
28. Copaescu, AM, Castilloux, JF, Chababi-Atallah, M, Sinave, C, and Bertrand, J. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. (2013) 5:340–6. doi: 10.1159/000356229
29. Joshi, TP, Friske, SK, Hsiou, DA, and Duvic, M. New practical aspects of sweet syndrome. Am J Clin Dermatol. (2022) 23:301–18. doi: 10.1007/s40257-022-00673-4
30. Orfaly, VE, Shakshouk, H, Heath, M, Hamilton, A, and Ortega-Loayza, AG. Sweet syndrome: a review of published Cases. Dermatology. (2023) 239:664–9. doi: 10.1159/000530519
31. Jung, EH, Park, JH, Hwan Kim, K, Kim, JS, Sil Choi, I, Byun, JM, et al. Characteristics of sweet syndrome in patients with or without malignancy. Ann Hematol. (2022) 101:1499–508. doi: 10.1007/s00277-022-04850-7
32. Zheng, S, Li, S, Tang, S, Pan, Y, Ding, Y, Qiao, J, et al. Insights into the characteristics of sweet syndrome in patients with and without hematologic malignancy. Front Med (Lausanne). (2020) 7:20. doi: 10.3389/fmed.2020.00020
33. Nelson, CA, Noe, MH, McMahon, CM, Gowda, A, Wu, B, Ashchyan, HJ, et al. Sweet syndrome in patients with and without malignancy: a retrospective analysis of 83 patients from a tertiary academic referral center. J Am Acad Dermatol. (2018) 78:303–309.e4. doi: 10.1016/j.jaad.2017.09.013
34. Alegría-Landa, V, Rodríguez-Pinilla, SM, Santos-Briz, A, Rodríguez-Peralto, JL, Alegre, V, Cerroni, L, et al. Clinicopathologic, Immunohistochemical, and molecular features of Histiocytoid sweet syndrome. JAMA Dermatol. (2017) 153:651–9. doi: 10.1001/jamadermatol.2016.6092
35. Haber, R, Feghali, J, and El Gemayel, M. Risk of malignancy in histiocytoid sweet syndrome: a systematic review and reappraisal. J Am Acad Dermatol. (2020) 83:661–3. doi: 10.1016/j.jaad.2020.02.048
37. von den Driesch, P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. (1994) 31:535–56. doi: 10.1016/s0190-9622(94)70215-2
38. Walker, DC, and Cohen, PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. (1996) 34:918–23. doi: 10.1016/s0190-9622(96)90080-8
39. Nofal, A, Abdelmaksoud, A, Amer, H, Nofal, E, Yosef, A, Gharib, K, et al. Sweet’s syndrome: diagnostic criteria revisited. J Dtsch Dermatol Ges. (2017) 15:1081–8. doi: 10.1111/ddg.13350
40. Yook, HJ, Son, JH, Kim, YH, Han, JH, Lee, JH, Park, YM, et al. Leukaemia cutis: clinical features and outcomes of 56 patients. Acta Derm Venereol. (2022) 102:adv00647. doi: 10.2340/actadv.v102.1123
41. Calvo, KR. Skin in the game: the emergence of myelodysplasia cutis. Blood. (2022) 139:1132–4. doi: 10.1182/blood.2021014788
42. Delaleu, J, Kim, R, Zhao, LP, de Masson, A, Vignon-Pennamen, MD, Cassius, C, et al. Clinical, pathological, and molecular features of myelodysplasia cutis. Blood. (2022) 139:1251–3. doi: 10.1182/blood.2021013967
43. Sterling, D, Duncan, ME, Philippidou, M, Salisbury, JR, Kulasekararaj, AG, and Basu, TN. VEXAS syndrome (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) for the dermatologist. J Am Acad Dermatol. (2022) 89:1209–14. doi: 10.1016/j.jaad.2022.01.042
44. Kim, JS, Roh, HS, Lee, JW, Lee, MW, and Yu, HJ. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. (2012) 51:1491–3. doi: 10.1111/j.1365-4632.2011.05141.x
45. Zakine, È, Papageorgiou, L, Bourguiba, R, Mekinian, A, Terrier, B, Kosmider, O, et al. Clinical and pathological features of cutaneous manifestations in VEXAS syndrome: a multicenter retrospective study of 59 cases. J Am Acad Dermatol. (2023) 88:917–20. doi: 10.1016/j.jaad.2022.10.052
46. Gurnari, C, Mannion, P, Pandit, I, Pagliuca, S, Voso, MT, Maciejewski, JP, et al. UBA1 screening in sweet syndrome with hematological neoplasms reveals a novel association between VEXAS and chronic Myelomonocytic leukemia. Hema. (2022) 6:e775. doi: 10.1097/HS9.0000000000000775
47. Maller, B, Bigness, A, Moiño, D, and Greene, J. Sweet’s syndrome associated with hematological malignancies. Leuk Res. (2020) 99:106461. doi: 10.1016/j.leukres.2020.106461
48. Heiblig, M, Ferrada, MA, Koster, MJ, Barba, T, Gerfaud-Valentin, M, Mékinian, A, et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: a retrospective multicenter study. Blood. (2022) 140:927–31. doi: 10.1182/blood.2022016642
49. Boyadzhieva, Z, Ruffer, N, Kötter, I, and Krusche, M. How to treat VEXAS syndrome: a systematic review on effectiveness and safety of current treatment strategies. Rheumatology (Oxford). (2023) 62:3518–25. doi: 10.1093/rheumatology/kead240
50. Patel, N, Dulau-Florea, A, and Calvo, KR. Characteristic bone marrow findings in patients with UBA1 somatic mutations and VEXAS syndrome. Semin Hematol. (2021) 58:204–11. doi: 10.1053/j.seminhematol.2021.10.007
51. Momen, SE, Jorizzo, J, and Al-Niaimi, F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. (2014) 28:1594–602. doi: 10.1111/jdv.12566
52. Doktor, V, Hadi, A, Hadi, A, Phelps, R, and Goodheart, H. Erythema elevatum diutinum: a case report and review of literature. Int J Dermatol. (2019) 58:408–15. doi: 10.1111/ijd.14169
53. Sandhu, JK, Albrecht, J, Agnihotri, G, and Tsoukas, MM. Erythema elevatum et diutinum as a systemic disease. Clin Dermatol. (2019) 37:679–83. doi: 10.1016/j.clindermatol.2019.07.028
54. Vide, J, Brás, G, Guimarães, JE, and Mota, A. Pyoderma gangrenosum and erythema elevatum diutinum associated with a high-risk myelodysplastic syndrome: case report. Hematol Transfus Cell Ther. (2018) 40:192–5. doi: 10.1016/j.htct.2017.11.005
55. Maronese, CA, Pimentel, MA, Li, MM, Genovese, G, Ortega-Loayza, AG, and Marzano, AV. Pyoderma Gangrenosum: an updated literature review on established and emerging pharmacological treatments. Am J Clin Dermatol. (2022) 23:615–34. doi: 10.1007/s40257-022-00699-8
56. Maverakis, E, Marzano, AV, Le, ST, Callen, JP, Brüggen, MC, Guenova, E, et al. Pyoderma gangrenosum. Nat Rev Dis Primers. (2020) 6:81. doi: 10.1038/s41572-020-0213-x
57. Su, WP, Davis, MD, Weenig, RH, Powell, FC, and Perry, HO. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. (2004) 43:790–800. doi: 10.1111/j.1365-4632.2004.02128.x
58. Maverakis, E, Ma, C, Shinkai, K, Fiorentino, D, Callen, JP, Wollina, U, et al. Diagnostic criteria of ulcerative pyoderma Gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. (2018) 154:461–6. doi: 10.1001/jamadermatol.2017.5980
59. Jockenhöfer, F, Wollina, U, Salva, KA, Benson, S, and Dissemond, J. The PARACELSUS score: a novel diagnostic tool for pyoderma gangrenosum. Br J Dermatol. (2019) 180:615–20. doi: 10.1111/bjd.16401
60. Haag, C, Hansen, T, Hajar, T, Latour, E, Keller, J, Shinkai, K, et al. Comparison of three diagnostic frameworks for pyoderma Gangrenosum. J Invest Dermatol. (2021) 141:59–63. doi: 10.1016/j.jid.2020.04.019
61. Rick, J, Gould, LJ, Marzano, AV, Garg, A, Chen, D, Oakes, DL, et al. The “understanding pyoderma Gangrenosum, review and assessment of disease effects (UPGRADE)” project: a protocol for the development of the core outcome domain set for trials in pyoderma gangrenosum. Arch Dermatol Res. (2023) 315:983–8. doi: 10.1007/s00403-022-02424-1
62. Honigman, A, and Kelly, RI. Pyoderma gangrenosum: a review of patient’s demographics, disease and treatment in 118 patients. Australas J Dermatol. (2022) 63:267–9. doi: 10.1111/ajd.13840
63. Ashchyan, HJ, Butler, DC, Nelson, CA, Noe, MH, Tsiaras, WG, Lockwood, SJ, et al. The Association of age with Clinical Presentation and Comorbidities of pyoderma Gangrenosum. JAMA Dermatol. (2018) 154:409–13. doi: 10.1001/jamadermatol.2017.5978
64. Montagnon, CM, Fracica, EA, Patel, AA, Camilleri, MJ, Murad, MH, Dingli, D, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. (2020) 82:1346–59. doi: 10.1016/j.jaad.2019.09.032
65. Tamaki, K, Nakazawa, T, Mamehara, A, Tsuji, G, Saigo, K, Kawano, S, et al. Successful treatment of pyoderma gangrenosum associated with myelodysplastic syndrome using high-dose intravenous immunoglobulin. Intern Med. (2008) 47:2077–81. doi: 10.2169/internalmedicine.47.1280
66. Yamauchi, T, Ishida, K, Iwashima, Y, Ikegaya, S, Kawai, Y, Wakahara, M, et al. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome. J Infect Chemother. (2003) 9:268–71. doi: 10.1007/s10156-003-0254-6
67. Wu, JJ, Huang, DB, Pang, KR, Hsu, S, and Tyring, SK. Thalidomide: dermatological indications, mechanisms of action and side-effects. Br J Dermatol. (2005) 153:254–73. doi: 10.1111/j.1365-2133.2005.06747.x
68. Koca, E, Duman, AE, Cetiner, D, Buyukasik, Y, Haznedaroglu, IC, Uner, A, et al. Successful treatment of myelodysplastic syndrome-induced pyoderma gangrenosum. Neth J Med. (2006) 11:105–7. doi: 10.1080/10245330600574177
69. Federman, GL, and Federman, DG. Recalcitrant pyoderma gangrenosum treated with thalidomide. Mayo Clin Proc. (2000) 75:842–4. doi: 10.4065/75.8.842
70. Hecker, MS, and Lebwohl, MG. Recalcitrant pyoderma gangrenosum: treatment with thalidomide. J Am Acad Dermatol. (1998) 38:490–1. doi: 10.1016/s0190-9622(98)70513-4
71. Rustin, MH, Gilkes, JJ, and Robinson, TW. Pyoderma gangrenosum associated with Behçet’s disease: treatment with thalidomide. J Am Acad Dermatol. (1990) 23:941–4. doi: 10.1016/s0190-9622(08)80705-0
72. Kyle, RA, and Rajkumar, SV. Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. Curr Hematol Malig Rep. (2010) 5:62–9. doi: 10.1007/s11899-010-0047-9
73. Velasco-Tamariz, V, Carreño-Tarragona, G, Tous-Romero, F, Gil-de la Cruz, E, Martín-Clavero, E, and Rivera-Díaz, R. Dramatic resolution of disseminated pyoderma gangrenosum associated with monoclonal gammopathy after therapy with bortezomib and dexamethasone. Int Wound J. (2017) 14:1382–4. doi: 10.1111/iwj.12746
74. Bostan, E, Günaydın, SD, Karaduman, A, Evans, SE, Elçin, G, Gülseren, D, et al. Excellent response to bortezomib in a patient with widespread ulcerative pyoderma gangrenosum accompanied by pulmonary involvement and IgA monoclonal gammopathy. Int Wound J. (2019) 16:1052–4. doi: 10.1111/iwj.13135
75. Nahi, H, Afram, G, Brauner, H, Talme, T, and Kuzmina, N. Pyoderma gangrenosum with plasma cell dyscrasia should be subject for anti-myeloma treatment. Int J Dermatol. (2021) 60:e271–3. doi: 10.1111/ijd.15504
76. Cespedes, DA, Gallitano, SM, and Bhutani, D. Ixazomib as treatment for pyoderma gangrenosum associated with IgA smoldering multiple myeloma. Ann Hematol. (2022) 101:441–2. doi: 10.1007/s00277-021-04524-w
77. Machan, A, Azendour, H, Frikh, R, Hjira, N, and Boui, M. The dilemma of treating pyoderma gangrenosum associated with monoclonal gammopathy of undetermined significance. Dermatol Online J. (2020) 26:13030/qt1bk7d8hj. doi: 10.5070/D3265048791
78. Dissemond, J, Marzano, AV, Hampton, PJ, and Ortega-Loayza, AG. Pyoderma Gangrenosum: treatment options. Drugs. (2023) 83:1255–67. doi: 10.1007/s40265-023-01931-3
79. Heras, MO, Muñoz, NP, Sancho, MI, and Millet, PU. Eosinophilic annular erythema in adults: report of two cases and review of the literature. An Bras Dermatol. (2017) 92:65–8. doi: 10.1590/abd1806-4841.20176373
80. Chastagner, M, Shourik, J, Jachiet, M, Battistella, M, Lefevre, G, Gibier, JB, et al. Treatment of eosinophilic annular erythema: retrospective multicenter study and literature review. Ann Dermatol Venereol. (2022) 149:123–7. doi: 10.1016/j.annder.2021.07.007
81. Miljković, J, and Bartenjev, I. Hypereosinophilic dermatitis-like erythema annulare centrifugum in a patient with chronic lymphocytic leukaemia. J Eur Acad Dermatol Venereol. (2005) 19:228–31. doi: 10.1111/j.1468-3083.2005.01052.x
82. Cohen, PR. What’s in a name? A new nomenclature has been proposed for eosinophilic dermatosis of hematologic malignancy (EDHM): hematologic-related malignancy-induced eosinophilic dermatosis (he remained). Dermatol Online J. (2020) 26:13030/qt1zd6p6z2. doi: 10.5070/D3266049329
83. Leiferman, KM, and Peters, MS. Eosinophil-related disease and the skin. J Allergy Clin Immunol Pract. (2018) 6:1462–1482.e6. doi: 10.1016/j.jaip.2018.06.002
84. Maglie, R, Ugolini, F, De Logu, F, Nassini, R, Simi, S, Nardiello, P, et al. Overexpression of helper T cell type 2-related molecules in the skin of patients with eosinophilic dermatosis of hematologic malignancy. J Am Acad Dermatol. (2022) 87:761–70. doi: 10.1016/j.jaad.2021.07.007
85. Weed, RI. Exaggerated delayed hypersensitivity to mosquito bites in chronic lymphocytic leukemia. Blood. (1965) 26:257–68. doi: 10.1182/blood.V26.3.257.257
86. Barzilai, A, Shpiro, D, Goldberg, I, Yacob-Hirsch, Y, Diaz-Cascajo, C, Meytes, D, et al. Insect bite-like reaction in patients with hematologic malignant neoplasms. Arch Dermatol. (1999) 135:1503–7. doi: 10.1001/archderm.135.12.1503
87. Byrd, JA, Scherschun, L, Cahffins, ML, and Fivenson, DP. Eosinophilic dermatosis of myeloproliferative disease: characterization of a unique eruption in patients with hematologic disorders. Arch Dermatol. (2001) 137:1378–80.
88. Farber, MJ, La Forgia, S, Sahu, J, and Lee, JB. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. (2012) 39:690–5. doi: 10.1111/j.1600-0560.2012.01906.x
89. Visseaux, L, Durlach, A, Barete, S, Beylot-Barry, M, Bonnet, N, Chassine, A, et al. T-cell papulosis associated with B-cell malignancy: a distinctive clinicopathologic entity. J Eur Acad Dermatol Venereol. (2018) 32:1469–75. doi: 10.1111/jdv.14805
90. Cohen, PR. Hematologic-related malignancy-induced eosinophilic dermatosis (he Remainded): an eosinophilic dermatosis predominantly associated with chronic lymphocytic leukemia. J Am Acad Dermatol. (2020) 82:e13–4. doi: 10.1016/j.jaad.2019.08.062
91. Ho, TC, Compton, L, Sheinbein, D, and Custer, PL. Eosinophilic dermatosis of malignancy involving the eyelid. Ophthalmic Plast Reconstr Surg. (2021) 37:e196–8. doi: 10.1097/IOP.0000000000002004
92. Grandi, V, Maglie, R, Antiga, E, Vannucchi, M, Delfino, C, Lastrucci, I, et al. Eosinophilic dermatosis of hematologic malignancy: a retrospective cohort of 37 patients from an Italian center. J Am Acad Dermatol. (2019) 81:246–9. doi: 10.1016/j.jaad.2018.11.048
93. Bairey, O, Goldschmidt, N, Ruchlemer, R, Tadmor, T, Rahimi-Levene, N, Yuklea, M, et al. Insect-bite-like reaction in patients with chronic lymphocytic leukemia: a study from the Israeli chronic lymphocytic leukemia study group. Eur J Haematol. (2012) 89:491–6. doi: 10.1111/ejh.12015
94. Abbott, J, Corean, J, Snyder, AM, Florell, SR, Miles, R, Stephens, D, et al. Folliculocentric lymphocytic hypersensitivity reactions in CLL/SLL patients: a unique clinicopathologic entity amongst non-specific hypersensitivity reactions. Skin Health Dis. (2023) 3:e208. doi: 10.1002/ski2.208
95. Mitteldorf, C, Tronnier, M, Merz, H, Haenssle, HA, Bertsch, HP, Schön, MP, et al. Insect bite-like reactions in a patient with B-cell chronic lymphocytic leukaemia: fluorescence in situ hybridization analysis revealed neoplastic B cells within the skin infiltrate. Br J Dermatol. (2012) 167:944–6. doi: 10.1111/j.1365-2133.2012.10966.x
96. Meiss, F, Technau-Hafsi, K, Kern, JS, and May, AM. Eosinophilic dermatosis of hematologic malignancy: correlation of molecular characteristics of skin lesions and extracutaneous manifestations of hematologic malignancy. J Cutan Pathol. (2019) 46:175–81. doi: 10.1111/cup.13389
97. Bailey, CAR, Laurain, DA, Sheinbein, DM, Jones, HA, Compton, LA, and Rosman, IS. Eosinophilic folliculitis, eosinophilic dermatosis of hematologic malignancy and acneiform follicular mucinosis: two case reports and a review of the literature highlighting the spectrum of histopathology. J Cutan Pathol. (2021) 48:439–50. doi: 10.1111/cup.13932
98. Rodríguez-Lojo, R, Almagro, M, Piñeyro, F, Pérez-Varela, L, Fernández-Jorge, B, Del Pozo, J, et al. Eosinophilic panniculitis and insect bite-like eruption in a patient with chronic lymphocytic leukaemia: a spectrum of the same entity. Dermatol Res Pract. (2010) 2010:263827. doi: 10.1155/2010/263827
99. Marzano, AV, Maronese, CA, Genovese, G, Ferrucci, S, Moltrasio, C, Asero, R, et al. Urticarial vasculitis: clinical and laboratory findings with a particular emphasis on differential diagnosis. J Allergy Clin Immunol. (2022) 149:1137–49. doi: 10.1016/j.jaci.2022.02.007
100. Bari, O, and Cohen, PR. Eosinophilic dermatosis of hematologic malignancy mimicking varicella zoster infection: report in a woman with chronic lymphocytic leukemia and review of the literature. Dermatol Pract Concept. (2017) 7:6–15. doi: 10.5826/dpc.0703a02
101. Maglie, R, Ugolini, F, De Logu, F, Simi, S, Senatore, S, Montefusco, F, et al. Dupilumab for the treatment of recalcitrant eosinophilic dermatosis of haematologic malignancy. J Eur Acad Dermatol Venereol. (2021) 35:e501–3. doi: 10.1111/jdv.17232
102. Sibaud, V, Brun, A, Meyer, N, Oberic, L, Lamant, L, and Ysebaert, L. Efficacy of dupilumab in eosinophilic dermatosis of haematologic malignancy (EDHM) needs to be confirmed. J Eur Acad Dermatol Venereol. (2022) 36:e213–5. doi: 10.1111/jdv.17748
103. Jin, A, Pousti, BT, Savage, KT, Mollanazar, NK, Lee, JB, and Hsu, S. Eosinophilic dermatosis of hematologic malignancy responding to dupilumab in a patient with chronic lymphocytic leukemia. JAAD Case Rep. (2019) 5:815–7. doi: 10.1016/j.jdcr.2019.07.026
104. Goyal, A, Lofgreen, S, Mariash, E, Bershow, A, and Gaddis, KJ. Targeted inhibition of IL-4/13 with dupilumab is an effective treatment for eosinophilic dermatosis of hematologic malignancy. Dermatol Ther. (2020) 33:e13725. doi: 10.1111/dth.13725
105. Weins, AB, Biedermann, T, Weiss, T, and Weiss, JM. Wells syndrome. J Dtsch Dermatol Ges. (2016) 14:989–93. doi: 10.1111/ddg.13132
106. Caputo, R, Marzano, AV, Vezzoli, P, and Lunardon, L. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. (2006) 142:1157–61. doi: 10.1001/archderm.142.9.1157
107. Heelan, K, Ryan, JF, Shear, NH, and Egan, CA. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. (2013) 7:113–20. doi: 10.3315/jdcr.2013.1157
108. Smith, SM, Kiracofe, EA, Clark, LN, and Gru, AA. Idiopathic Hypereosinophilic syndrome with cutaneous manifestations and flame figures: a Spectrum of eosinophilic dermatoses whose features overlap with Wells’ syndrome. Am J Dermatopathol. (2015) 37:910–4. doi: 10.1097/DAD.0000000000000279
109. Qiao, J, Sun, CE, Zhu, W, Zhu, D, and Fang, H. Flame figures associated with eosinophilic dermatosis of hematologic malignancy: is it possible to distinguish the condition from eosinophilic cellulitis in patients with hematoproliferative disease? Int J Clin Exp Pathol. (2013) 6:1683–7.
110. Zeeli, T, Feinmesser, M, Segal, R, and David, M. Insect-bite-like Wells’ syndrome in association with mantle-zone lymphoma. Br J Dermatol. (2006) 155:614–6. doi: 10.1111/j.1365-2133.2006.07345.x
111. Šajn, M, Luzar, B, and Zver, S. Wells’ syndrome possibly caused by hematologic malignancy, influenza vaccination or ibrutinib: a case report. World J Clin Cases. (2022) 10:10997–1003. doi: 10.12998/wjcc.v10.i30.10997
112. Gambichler, T, Othlinghaus, N, Rotterdam, S, Altmeyer, P, and Stücker, M. Impetiginized Wells’ syndrome in a patient with chronic lymphocytic leukaemia. Clin Exp Dermatol. (2009) 34:e274–5. doi: 10.1111/j.1365-2230.2008.03192.x
113. Misago, N, Okwa, T, Tanaka, M, and Narisawa, Y. Eosinophilic cellulitis (Wells’ syndrome) and an insect bite-like reaction in a patient with non-Hodgkin B cell lymphoma. Eur J Dermatol. (2011) 21:422–3. doi: 10.1684/ejd.2011.1296
114. Prabhsimranjot, S, Makardhwaj, SS, Perez, A, Grossman, K, Benasher, D, and Astrow, A. Steroid dependent recalcitrant Well’s syndrome in a patient with chronic lymphocytic leukemia (CLL) responding to hydroxychloroquine (HCQS). Blood. (2016) 128:5579. doi: 10.1182/blood.V128.22.5579.5579
115. Shin, D, and Kim, DY. Chronic relapsing eosinophilic cellulitis associated, although independent in severity, with chronic lymphocytic leukemia. J Eur Acad Dermatol Venereol. (2016) 30:159–61. doi: 10.1111/jdv.12652
116. Lorente-Lavirgen, AI, López-López, R, Baquero-Sánchez, E, Pulpillo-Ruiz, A, De Zulueta-Dorado, T, and Conejo-Mir, J. Pruritic nodules and plaques on the arms with blisters in a patient with chronic lymphocytic leukemia. Int J Dermatol. (2014) 53:277–9. doi: 10.1111/ijd.12257
117. Fujii, K, Tanabe, H, Kanno, Y, Konishi, K, and Ohgou, N. Eosinophilic cellulitis as a cutaneous manifestation of idiopathic hypereosinophilic syndrome. J Am Acad Dermatol. (2003) 49:1174–7. doi: 10.1016/s0190-9622(03)00466-3
118. Bogenrieder, T, Griese, DP, Schiffner, R, Büttner, R, Riegger, GA, Hohenleutner, U, et al. Wells’ syndrome associated with idiopathic hypereosinophilic syndrome. Br J Dermatol. (1997) 137:978–82. doi: 10.1046/j.1365-2133.1997.19972078.x
119. Lachance, M, Bernard, J, Lavoie, A, Gagné, É, and Grenier, PO. Idiopathic hypereosinophilic syndrome with eosinophilic cellulitis-like cutaneous involvement treated with mepolizumab and dapsone. JAAD Case Rep. (2022) 22:11–3. doi: 10.1016/j.jdcr.2022.01.022
120. Davis, RF, Dusanjh, P, Majid, A, Fletcher, A, Wardlaw, A, Siebert, R, et al. Eosinophilic cellulitis as a presenting feature of chronic eosinophilic leukaemia, secondary to a deletion on chromosome 4q12 creating the FIP1L1-PDGFRA fusion gene. Br J Dermatol. (2006) 155:1087–9. doi: 10.1111/j.1365-2133.2006.07490.x
121. Smith, KJ, Jacobson, E, Hamza, S, and Skelton, H. Unexplained hypereosinophilia and the need for cytogenetic and molecular genetic analyses. Arch Dermatol. (2004) 140:584–8. doi: 10.1001/archderm.140.5.584
122. Räßler, F, Lukács, J, and Elsner, P. Treatment of eosinophilic cellulitis (Wells syndrome) - a systematic review. J Eur Acad Dermatol Venereol. (2016) 30:1465–79. doi: 10.1111/jdv.13706
123. Traidl, S, Angela, Y, Kapp, A, Suhling, H, Schacht, V, and Werfel, T. Dupilumab in eosinophilic cellulitis (Wells’ syndrome) - case report of a potential new treatment option. J Dtsch Dermatol Ges. (2021) 19:1653–5. doi: 10.1111/ddg.14598
124. Kirven, RM, and Plotner, AN. Wells syndrome successfully treated with dupilumab. Int J Dermatol. (2023) 62:e454–5. doi: 10.1111/ijd.16501
125. Terhorst-Molawi, D, Altrichter, S, Röwert, J, Magerl, M, Zuberbier, T, Maurer, M, et al. Effective treatment with mepolizumab in a patient with refractory Wells syndrome. J Dtsch Dermatol Ges. (2020) 18:737–9. doi: 10.1111/ddg.14151
126. Herout, S, Bauer, WM, Schuster, C, and Stingl, G. Eosinophilic cellulitis (Wells syndrome) successfully treated with mepolizumab. JAAD Case Rep. (2018) 4:548–50. Published 2018 Jun 6. doi: 10.1016/j.jdcr.2018.02.011
127. Blomberg, M, Winther, C, Høyrup, S, and Skov, L. Treatment of widespread eosinophilic cellulitis (Wells’ syndrome) with Benralizumab. Acta Derm Venereol. (2020) 100:adv00332. doi: 10.2340/00015555-3697
128. Morot, J, Del Duca, E, Chastagner, M, Fernandes, M, Estrada, Y, Lefevre, MA, et al. Hyperactivation of the JAK2/STAT5 signaling pathway and evaluation of Baricitinib treatment among patients with eosinophilic cellulitis. JAMA Dermatol. (2023) 159:820–9. doi: 10.1001/jamadermatol.2023.1651
129. Wang, SA, Orazi, A, Gotlib, J, Reiter, A, Tzankov, A, Hasserjian, RP, et al. The international consensus classification of eosinophilic disorders and systemic mastocytosis. Am J Hematol. (2023) 98:1286–306. doi: 10.1002/ajh.26966
130. Requena, G, van den Bosch, J, Akuthota, P, Kovalszki, A, Steinfeld, J, Kwon, N, et al. Clinical profile and treatment in Hypereosinophilic syndrome variants: a pragmatic review. J Allergy Clin Immunol Pract. (2022) 10:2125–34. doi: 10.1016/j.jaip.2022.03.034
131. Rodríguez-Lomba, E, Molina-López, I, Parra-Blanco, V, and Suárez-Fernández, R. Recurrent hemorrhagic blisters: an atypical presentation of lymphocytic hypereosinophilic syndrome with cutaneous manifestations. J Dtsch Dermatol Ges. (2020) 18:487–9. doi: 10.1111/ddg.14083
132. Laurent, C, Lefèvre, G, Kahn, JE, Staumont-Salle, D, Felten, R, Puget, M, et al. Cutaneous manifestations of lymphoid-variant hypereosinophilic syndrome. Br J Dermatol. (2022) 187:1011–3. doi: 10.1111/bjd.21782
133. Rohmer, J, Couteau-Chardon, A, Trichereau, J, Panel, K, Gesquiere, C, Ben Abdelali, R, et al. Epidemiology, clinical picture and long-term outcomes of FIP1L1-PDGFRA-positive myeloid neoplasm with eosinophilia: data from 151 patients. Am J Hematol. (2020) 95:1314–23. doi: 10.1002/ajh.25945
134. Legrand, F, Renneville, A, MacIntyre, E, Mastrilli, S, Ackermann, F, Cayuela, JM, et al. The Spectrum of FIP1L1-PDGFRA-associated chronic eosinophilic leukemia: new insights based on a survey of 44 Cases. Medicine (Baltimore). (2013) 92:e1–9. doi: 10.1097/MD.0b013e3182a71eba
135. Leiferman, KM, O’Duffy, JD, Perry, HO, Greipp, PR, Giuliani, ER, and Gleich, GJ. Recurrent incapacitating mucosal ulcerations. A prodrome of the hypereosinophilic syndrome. JAMA. (1982) 247:1018–20. doi: 10.1001/jama.1982.03320320054032
136. Barouky, R, Bencharif, L, Badet, F, Salles, G, Vital Durand, D, and Rousset, H. Mucosal ulcerations revealing primitive hypereosinophilic syndrome. Eur J Dermatol. (2003) 13:207–8.
137. Leiferman, KM, and Gleich, GJ. Hypereosinophilic syndrome: case presentation and update. J Allergy Clin Immunol. (2004) 113:50–8. doi: 10.1016/j.jaci.2003.10.051
138. Curto-Barredo, L, Segura, S, Ishii, N, Hashimoto, T, Mascaró, JM Jr, Espinet, B, et al. Pemphigus-like hypereosinophilic syndrome with FIP1L1-PDGFRA fusion gene: a challenging and uncommon clinical presentation. J Dermatol. (2019) 46:531–4. doi: 10.1111/1346-8138.14888
139. Tsuda, K, Tanimoto, T, Hayakawa, K, and Komatsu, T. Oral mucosal manifestations of chronic eosinophilic leukaemia with FIP1L1-PDGFRα. BMJ Case Rep. (2016) 2016:bcr2015213266. doi: 10.1136/bcr-2015-213266
140. Isvy-Joubert, A, Hickman, G, Flageul, B, Battistella, M, Bagot, M, Galicier, L, et al. Syndrome hyperéosinophilique d’origine myéloproliférative révélé par des ulcérations muqueuses bipolaires [Myeloproliferative hypereosinophilic syndrome revealed by bipolar mucosal ulcerations]. Ann Dermatol Venereol. (2012) 139:636–40. doi: 10.1016/j.annder.2012.05.008
141. Butterfield, JH, and Gleich, GJ. Interferon-alpha treatment of six patients with the idiopathic hypereosinophilic syndrome. Ann Intern Med. (1994) 121:648–53. doi: 10.7326/0003-4819-121-9-199411010-00003
142. Billon, C, Gautier, C, Villaret, E, Ducos, MH, Martin, JC, and Geniaux, M. Ulcérations muqueuses isolées révélatrices d’un syndrome hyperéosinophilique idiopathique [Isolated mucosal ulcers disclosing idiopathic hypereosinophilic syndrome]. Ann Dermatol Venereol. (1997) 124:248–50.
143. d’Elbée, JM, Parrens, M, Mercié, P, Longy Boursier, M, Dieval, C, de Mascarel, A, et al. Hypereosinophilic syndrome - lymphocytic variant transforming into peripheral T-cell lymphoma with severe oral manifestations. Oral Surg Oral Med Oral Pathol Oral Radiol. (2013) 116:e185–90. doi: 10.1016/j.oooo.2013.03.017
144. Bellani, V, Croci, GA, Bucelli, C, Maronese, CA, Alberti, S, Iurlo, A, et al. Lymphomatoid papulosis associated with myeloid neoplasm with eosinophilia and FIP1L1::PDGFRA rearrangement: successful imatinib treatment in two cases. J Dermatol. (2023) 50:1330–4. doi: 10.1111/1346-8138.16836
145. Kitayama, S, Makino, T, Yoto, A, Mori, S, Furukawa, F, Torai, R, et al. Detection of FIP1L1-PDGFRA fusion gene-positive cells in the skin lesion of a patient with hypereosinophilic syndrome. Clin Exp Dermatol. (2023) 48:364–7. doi: 10.1093/ced/llac112
146. Yamada, Y, Sanchez-Aguilera, A, Brandt, EB, McBride, M, Al-Moamen, NJ, Finkelman, FD, et al. FIP1L1/PDGFRalpha synergizes with SCF to induce systemic mastocytosis in a murine model of chronic eosinophilic leukemia/hypereosinophilic syndrome. Blood. (2008) 112:2500–7. doi: 10.1182/blood-2007-11-126268
147. Wiednig, M, Beham-Schmid, C, Kranzelbinder, B, and Aberer, E. Clonal mast cell proliferation in pruriginous skin in hypereosinophilic syndrome. Dermatology. (2013) 227:67–71. doi: 10.1159/000351807
148. Tefferi, A, Pardanani, A, and Li, CY. Hypereosinophilic syndrome with elevated serum tryptase versus systemic mast cell disease associated with eosinophilia: 2 distinct entities? Blood. (2003) 102:3073–4. doi: 10.1182/blood-2003-07-2274
149. Inayat, F, O’Neill, SS, Zafar, F, Marupudi, S, and Vasim, I. Idiopathic hypereosinophilic syndrome with cutaneous involvement: a comparative review of 32 cases. BMJ Case Rep. (2018) 11:bcr2018227137. doi: 10.1136/bcr-2018-227137
150. Merlotto, MR, Cantadori, LO, Sakabe, D, and Miot, HA. Case for diagnosis. Erythroderma as manifestation of hypereosinophilic syndrome. An Bras Dermatol. (2018) 93:451–3. doi: 10.1590/abd1806-4841.20187419
151. Lefèvre, G, Leurs, A, Gibier, JB, Copin, MC, Staumont-Sallé, D, Dezoteux, F, et al. Idiopathic eosinophilic vasculitis: another side of hypereosinophilic syndrome? A comprehensive analysis of 117 cases in asthma-free patients. J Allergy Clin Immunol Pract. (2020) 8:1329–1340.e3. doi: 10.1016/j.jaip.2019.12.011
152. Oppliger, R, Gay-Crosier, F, Dayer, E, and Hauser, C. Digital necrosis in a patient with hypereosinophilic syndrome in the absence of cutaneous eosinophilic vasculitis. Br J Dermatol. (2001) 144:1087–9. doi: 10.1046/j.1365-2133.2001.04207.x
153. Narayan, S, Ezughah, F, Standen, GR, Pawade, J, and Kennedy, CT. Idiopathic hypereosinophilic syndrome associated with cutaneous infarction and deep venous thrombosis. Br J Dermatol. (2003) 148:817–20. doi: 10.1046/j.1365-2133.2003.05309.x
154. Hamada, T, Kimura, Y, Hayashi, S, Nakama, T, and Hashimoto, T. Hypereosinophilic syndrome with peripheral circulatory insufficiency and cutaneous microthrombi. Arch Dermatol. (2007) 143:799–813. doi: 10.1001/archderm.143.6.812
155. Alzayer, H, and Hasan, MA. Hypereosinophilic vasculitis: a case report. Medicine (Baltimore). (2019) 98:e15392. doi: 10.1097/MD.0000000000015392
156. Ito, K, Hara, H, Okada, T, and Terui, T. Hypereosinophilic syndrome with various skin lesions and juvenile temporal arteritis. Clin Exp Dermatol. (2009) 34:e192–5. doi: 10.1111/j.1365-2230.2008.03008.x
157. Jang, KA, Lim, YS, Choi, JH, Sung, KJ, Moon, KC, and Koh, JK. Hypereosinophilic syndrome presenting as cutaneous necrotizing eosinophilic vasculitis and Raynaud’s phenomenon complicated by digital gangrene. Br J Dermatol. (2000) 143:641–4. doi: 10.1111/j.1365-2133.2000.03726.x
158. Serrano-Santiago, SM, Colon-Donate, M, Rivera-Franceschini, C, Caceres-Perkins, W, and Nazario-Jimenez, S. Digital ischemia as a presenting feature of Hypereosinophilic syndrome-associated Vasculitis. J Allergy Clin Immunol Pract. (2023) 11:2572–3. doi: 10.1016/j.jaip.2023.05.010
159. Kanno, T, Fukumoto, T, and Hamada, T. Eosinophilic vasculitis as a manifestation of idiopathic hypereosinophilic syndrome. J Dermatol. (2022) 49:e140–1. doi: 10.1111/1346-8138.16279
160. Ishii, K, Mizuuchi, T, Yamamoto, Y, Mori, H, Tago, M, Kato, E, et al. Development of eosinophilic temporal arteritis and digital ischemia in a patient with hypereosinophilic syndrome. Intern Med. (2020) 59:1323–30. doi: 10.2169/internalmedicine.3707-19
161. Aractingi, S, Bachmeyer, C, Pautier, P, Barboteu, M, Meignin, V, Daniel, MT, et al. Necrotic cutaneous lesions induced by hypereosinophilic syndrome secondary to a T-cell lymphoma. J Am Acad Dermatol. (2002) 46:S133–6. doi: 10.1067/mjd.2002.107233
162. Cases-Merida, S, Lorente-Lavirgen, A, and Pérez-Gil, A. Interstitial granulomatous dermatitis due to a rare myeloproliferative neoplasia. Indian J Dermatol. (2018) 63:264–7. doi: 10.4103/ijd.IJD_432_17
163. Zayas, J, Peters, MS, Butterfield, JH, Pongdee, T, and Sokumbi, O. Clinical and histopathological features of hypereosinophilic syndrome with cutaneous involvement: the Mayo Clinic experience. J Cutan Pathol. (2023) 50:455–65. doi: 10.1111/cup.14410
164. Beauverd, Y, Cortés, B, Masouye, I, and Chalandon, Y. Complete remission of disseminated granulomatous dermatitis related to chronic eosinophilic leukemia following allogeneic stem cell transplantation. Leuk Lymphoma. (2017) 58:470–2. doi: 10.1080/10428194.2016.1190965
165. Lierman, E, Smits, S, Debackere, K, André, M, Michaux, L, and Vandenberghe, P. T(9,12)(q22;p13) ETV6::SYK: a new recurrent cytogenetic aberration and tyrosine kinase gene fusion in myeloid or lymphoid neoplasms associated with eosinophilia. Br J Haematol. (2023) 200:665–8. doi: 10.1111/bjh.18569
166. Debois, D, Marot, L, Andre, M, and Dachelet, C. Thalidomide as an effective treatment for adult multiple xanthogranuloma. JAAD Case Rep. (2018) 4:896–8. doi: 10.1016/j.jdcr.2018.05.005
167. Iurlo, A, and Cattaneo, D. Biologic therapies for hypereosinophilic disorders: from tyrosine kinase inhibitors to monoclonal antibodies. Towards an increasingly customized management? Blood Rev. (2023) 58:101014. doi: 10.1016/j.blre.2022.101014
168. Choi, C, Moller, D, Tan, J, Dou, C, Peterson, EA, Medvedev, N, et al. Pegylated interferon alpha 2a is an effective and well-tolerated treatment option for lymphocyte-variant hypereosinophilic syndrome. Br J Haematol. (2020) 188:e68–72. doi: 10.1111/bjh.16332
169. Roufosse, F, Butterfield, J, Steinfeld, J, Bentley, JH, von Maltzahn, R, Kwon, N, et al. Mepolizumab therapy improves the most bothersome symptoms in patients with hypereosinophilic syndrome. Front Med (Lausanne). (2023) 10:1035250. doi: 10.3389/fmed.2023.1035250
170. Kuang, FL, Legrand, F, Makiya, M, Ware, J, Wetzler, L, Brown, T, et al. Benralizumab for PDGFRA-negative Hypereosinophilic syndrome. N Engl J Med. (2019) 380:1336–46. doi: 10.1056/NEJMoa1812185
171. Alen Coutinho, I, Regateiro, FS, Loureiro, C, and Todo-Bom, A. Benralizumab in severe and refractory PDGFRA-negative Hypereosinophilic syndrome. J Clin Immunol. (2021) 41:688–90. doi: 10.1007/s10875-020-00946-9
172. Fujii, K, Takahashi, H, Hayakawa, N, and Iwasaki, Y. Idiopathic hypereosinophilic syndrome in remission with benralizumab treatment after relapse with mepolizumab. Respirol Case Rep. (2020) 8:e00665. doi: 10.1002/rcr2.665
173. Jiang, X, Ye, J, Wu, X, Zhu, J, Chen, S, and Cheng, H. A case of complete recovery in a hypereosinophilic dermatitis patient with dupilumab. Inflamm Res. (2023) 72:875–8. doi: 10.1007/s00011-023-01715-1
Keywords: neutrophilic, eosinophilic, dermatoses, pyoderma, sweet
Citation: Maronese CA, Derlino F, Moltrasio C, Cattaneo D, Iurlo A and Marzano AV (2024) Neutrophilic and eosinophilic dermatoses associated with hematological malignancy. Front. Med. 10:1324258. doi: 10.3389/fmed.2023.1324258
Edited by:
Thibault Comont, Centre Hospitalier Universitaire de Toulouse, FranceReviewed by:
Fabio Del Duca, Sapienza University of Rome, ItalyAlvise Sernicola, University of Padua, Italy
Copyright © 2024 Maronese, Derlino, Moltrasio, Cattaneo, Iurlo and Marzano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelo Valerio Marzano, angelo.marzano@unimi.it
†These authors have contributed equally to this work and share first authorship