How do epiphytic and surrounding seawater bacterial communities shift with the development of the Saccharina japonica farmed in the Northern China?
 Ling Cai
Ling Cai Xin Gao1,2
Xin Gao1,2  Mahasweta Saha
Mahasweta Saha Luyang Xiao
Luyang Xiao Gaoge Wang
Gaoge Wang- 1College of Marine Life Science, Ocean University of China, Qingdao, China
- 2Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao, China
- 3Marine Ecology and Biodiversity, Plymouth Marine Laboratory, Prospect Place, Plymouth, United Kingdom
- 4Weihai Changqing Ocean Science & Technology Co., Ltd, Rongcheng, China
Epibacteria of seaweeds play an important role for the development of hosts and are influenced by the planktonic surrounding seawater bacteria. However, to date, the knowledges related to both epiphytic and surrounding seawater bacterial communities associated with northern farmed Saccharina japonica are very limited. In this study, using 16S rRNA gene amplicon sequencing, the shifts of epiphytic and surrounding seawater bacterial communities of the northern farmed S. japonica from mature sporophytes, sporelings (3 time points) to juvenile sporophytes (2 time points) were investigated. The dominant genera of epibacterial communities were Alcanivorax (mature sporophytes and 4-week-old sporelings), Bacillus (7-week-old sporelings and 9-week-old sporelings), Halomonas (4-week-old juvenile sporophytes) and Cobetia (9-week-old juvenile sporophytes). Meanwhile, the Chao1 indexes and beta diversity of epibacterial communities were significantly different with the development of S. japonica (p < 0.05). Furthermore, Alcanivorax, Bacillus and Halomonas were both dominant and core genera, indicating that these taxa may be beneficial to the development of S. japonica. The alpha diversity indexes of both epiphytic and surrounding seawater bacterial communities were significantly different for 9-week-old juvenile sporophytes. Therefore, the epibacterial communities were influenced by both development of S. japonica and the surrounding seawater bacterial communities. This study not only extends the understanding of the bacterial communities associated with the northern farmed S. japonica, but also help to make production management by monitoring the variations in both epiphytic and surrounding seawater bacterial communities.
Introduction
Epibacterial communities associated with seaweeds are essential for the development, metabolic functioning and defense of their hosts (Morrissey et al., 2019; Paix et al., 2021) and are affected by physiological and biochemical characteristics of seaweeds during the development, which may lead to selective enrichment of bacterial colonizers on the surfaces (Florez et al., 2019; Saha and Weinberger, 2019). It is found that the dominant genera of epibacterial communities were various at different developmental stages (Mancuso et al., 2016; Comba González et al., 2021) and alpha diversity indexes increased with the development of seaweeds (Michelou et al., 2013; Weigel and Pfister, 2019; Ihua et al., 2020). These studies indicated that the composition and diversity of epibacterial communities shift with the development of seaweeds. In addition, it has been found that core species play the important role in the structure and function of microbial community (Bonthond et al., 2020; Saha et al., 2020; Phelps et al., 2021). Core species analysis has been applied in studies of epibacterial communities of seaweeds to identify bacteria that may contribute to the normal development of host (Tujula et al., 2010; Han et al., 2021; Phelps et al., 2021). Meanwhile, the planktonic bacteria in the surrounding seawater also play an important role in the succession of epibacterial communities associated with seaweeds (Fahimipour et al., 2017; Lemay et al., 2018a; Cleary and Huang, 2020; Juhmani et al., 2020). So far, there is no consistent different or similar conclusions on the relationship between epiphytic and surrounding seawater bacterial communities. All of these outcomes achieved in the wild seaweeds provide the reference to investigate the shifts of epiphytic and surrounding seawater bacterial communities during the development in the commercially farmed seaweeds.
Saccharina japonica is an importantly farmed seaweed worldwide. China contributed about 90% of yield volume globally (Yan et al., 2022). Compared to wild seaweeds, studies on epiphytic and surrounding seawater bacterial communities of farmed S. japonica are still at infant stage. Usually, the cultivation period for the northern farmed S. japonica in China includes stages of sporelings (from early time of August to mid-October), juvenile sporophytes (from mid-October to next January), adult sporophytes (from February to June) and mature sporophytes with sporangia (from June to early time of August). Using 16S rRNA amplicon sequencing, the epibacterial communities of the northern farmed S. japonica were documented at the stages of sporelings, juvenile sporophytes, adult sporophytes and mature sporophytes. The dominant genera, Shannon indexes and beta diversity of the epibacterial communities exhibited significant succession from mature sporophytes, sporelings and juvenile sporophytes (Han et al., 2021) as well as the adult sporophytes during the harvest season (Zhang et al., 2020b). In addition, it is found that the relative abundance of dominant genera SAR11_clade and Candidatus Actinomarina in the seawater bacterial communities decreased with development of sporelings (Wang et al., 2022). Regarding to the relationship of epiphytic and surrounding seawater bacterial communities during the cultivation developmental period, related research was conducted only at the stage of adult sporophytes during the harvest season. The dominant genera were variable and the alpha diversity indexes were different between epiphytic and surrounding seawater bacterial communities (Zhang, 2017), indicating that the epibacterial communities had their own unique bacterial taxa. So far, there are no comprehensive studies on the succession of both epiphytic and surrounding seawater bacterial communities during the cultivation cycle.
Considering that epibacterial communities play crucial roles during the development of seaweeds and are affected by the surrounding seawater bacterial communities, however, the comprehensive knowledges of epiphytic and surrounding seawater bacterial communities in northern farmed S. japonica are still unknown. Therefore, we proposed the hypothesis: the shifts of epibacterial communities are influenced by both the development stage of farmed S. japonica and the surrounding seawater bacterial communities. Using 16S rRNA gene amplicon sequencing, we first investigated the composition and diversity of the epiphytic and surrounding seawater bacterial communities from mature sporophytes, sporelings to juvenile sporophytes; then we identified the important taxa that contribute to the development of farmed S. japonica by dominant and core genera analysis; finally, we assessed the influences of surrounding seawater bacterial communities on the epibacterial communities. Our results not only enrich the knowledge of microbiota associated with northern farmed S. japonica, but also will help to develop cultivation management from the angle of monitoring the variations in both epiphytic and surrounding seawater bacterial communities.
Materials and methods
Bacterial sampling
In this study, epiphytic and surrounding seawater bacterial samples were collected at six developmental time points from mature sporophytes, sporelings to juvenile sporophytes stages at Qingyutan, Rongcheng, Shandong Province, China (37°10' 2.19" N, 122°34' 54.8" E). Mature sporophytes (MS) with sporangia were collected on 8th August, 2019 and were rinsed with sterile seawater to remove the loose epiphytes on the surface (Figure 1). Epibacteria were sampled by swabbing 50 cm2 of the sporangia surface with sterile cotton swabs near the one-third of mature sporophytes away from the meristematic tissue. The swabs with epibacteria were stored in 50 mL sterile Eppendorf tubes. Usually it takes one month for the zoospores to develop into sporelings (S), which cover the substrate curtains (40 ×115 cm) in the greenhouses. The environmental factors, such as temperature (10 ± 1 °C), salinity (30‰) and light intensity (80 μE m-2 s-1), were controlled in the greenhouses. Method for sampling epibacterial samples of sporelings referred to Han et al. (2021). Epibacteria of 4-week-old sporelings (S1, 0.1-0.2 mm in length), 7-week-old sporelings (S2, 1-5 mm in length) and 9-week-old sporelings (S3, 10-20 mm in length) were sampled by swabbing on the substrate curtains by using sterile cotton swabs on 7th September, 27th September and 11th October 2019, respectively (Figure 1). Swabs with epibacteria were put into 50 mL sterile Eppendorf tubes. Juvenile sporophytes (JS) are referred to those sporelings which were transferred into the sea and cultivated for a period of 1-2 month. Epibacterial samples for both 4-week-old juvenile sporophytes (JS1, 30-40 cm in length) and 9-week-old juvenile sporophytes (JS2, 50-60 cm in length) were collected on 29th November 2019 and 4th January 2020, respectively (Figure 1). The sampling method of epibacteria on juvenile sporophytes were the same as mature sporophytes.
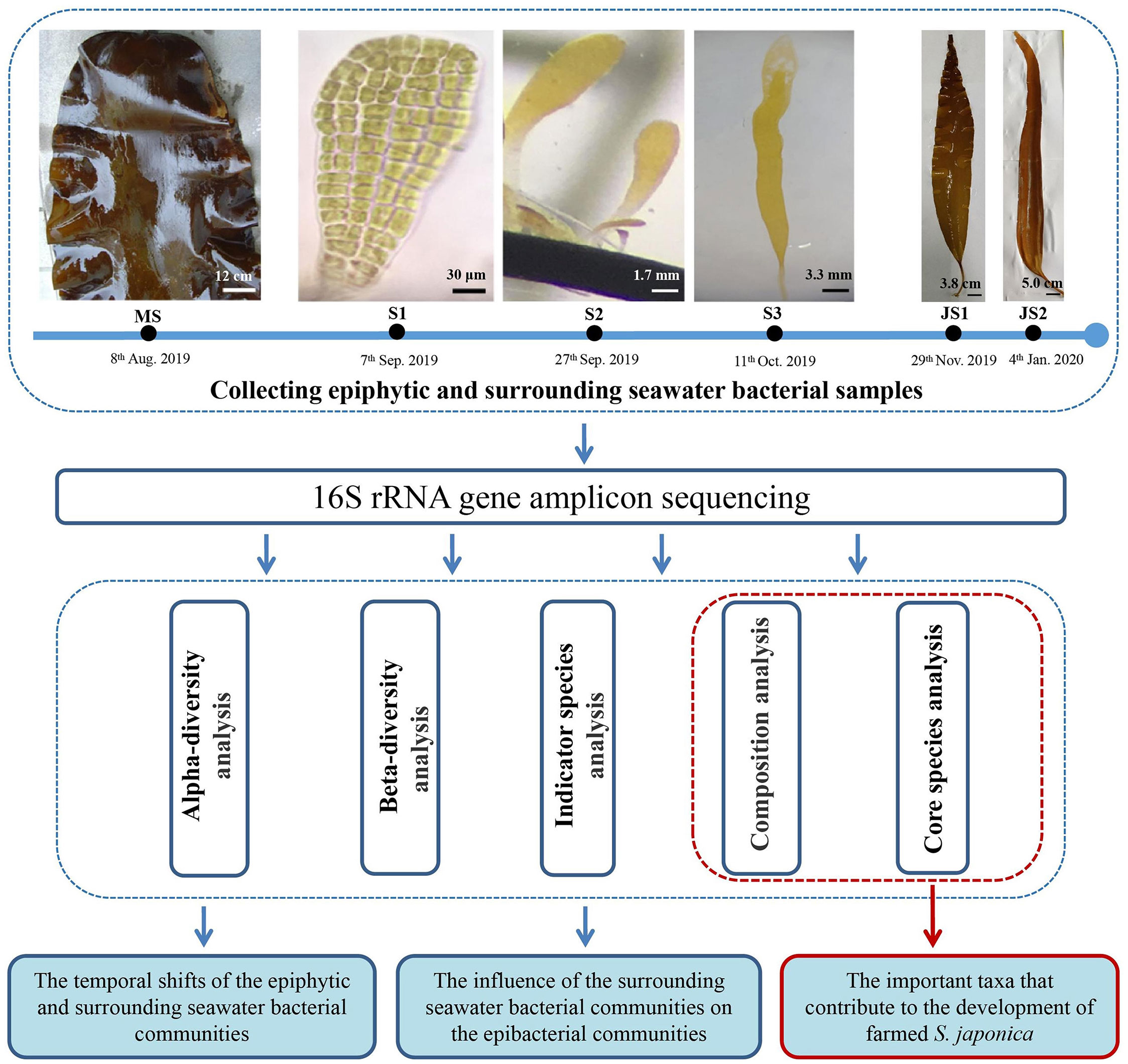
Figure 1 Experimental design for the shifts of epiphytic and surrounding seawater bacterial communities of northern farmed S. japonica. MS: mature sporophyte; S1: the 4-week-old sporeling; S2: 7-week-old sporeling; S3: 9-week-old sporeling; JS1: 4-week-old juvenile sporophyte; JS2: 9-week-old juvenile sporophyte. Bars, MS: 12 cm; S1: 30 μm, S2: 1.7 mm; S3: 3.3 mm; JS1: 3.8 cm; JS2: 5.0 cm.
Regarding to the collection of seawater bacterial samples, 2.5 L surrounding seawater were collected by using sterile bottles at each developmental time points. Seawater samples were firstly filtered through the membrane of 3 μm pore size to wash off the larger biotic or abiotic particles, and then were filtered through a membrane of 0.22 μm pore size (Whatman, UK). These membranes with seawater bacteria were put into 50 mL sterile Eppendorf tubes. All bacterial samples were kept frozen at -80 °C until DNA was extracted. Six replicates for both epibacterial and seawater bacterial samples were designed for each developmental time points, respectively.
DNA extraction, amplification, and 16S rRNA gene amplicon sequencing
We used HiPure Soil DNA kits (Magen, Guangzhou, China) to extract DNA of bacteria following the manufacturer’s instructions. The V3-V4 hypervariable regions of the bacterial 16S rRNA gene were amplified by PCR (95°C for 2 min, followed by 27 cycles at 98°C for 10 s, 62°C for 30 s, 68°C for 30 s and a final extension at 68°C for 10 min) with the primers 341F (5′- CCTACGGGNGGCWGCAG-3′) and 806R (5′- GGACTACHVGGGTATCTAAT -3′). PCR reactions were performed in 50 μL mixture, containing 10 × Buffer KOD (5 μL), dNTPs (5 μL, 2 mM), primers 341F and 806R (1.5 μL of each, 10 μM), KOD DNA Polymerase (Toyobo, Japan) and 100 ng template DNA.
PCR amplicons were purified from 2% agarose gels using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA). Quantification was performed using ABI StepOnePlus Real-Time PCR System (Life Technologies, Foster City, USA). The 2 × 250 bp paired-end reads were generated with Guangzhou Genedenovo Biotechnology (China) on the Illumina Novaseq6000 platform (Guo et al., 2017).
Analysis of illumina sequencing data
The DADA2 (version 1.14) was used to denoise and remove low-quality reads (Callahan et al., 2016). Then paired end denoised reads were merged as tags with a minimum overlap of 12 bp. Chimera sequences were identified and were deleted by UCHIME algorithm (Edgar et al., 2011). The high-quality tags were clustered into ASVs (amplicon sequence variants) with 100% similarity. The confidence threshold values ranged from 0.8 to 1.0. All ASV sequences were classified into organisms by a naive Bayesian model using RDP classifier 2.2 (Wang, 2007) based on SILVA database (version 132) (Pruesse et al., 2007). Because there were much higher standard errors in some groups, we removed the three replicates with outliers. Thus, finally, only three replicates close to the average value were selected for the analysis in this study. The sequences of chloroplast, mitochondria and archaea origin were removed from the dataset.
Core species analysis
Genera present in all replicates of epiphytic or surrounding seawater bacterial communities were selected and defined as the core genera. Core genus analysis of both epiphytic and surrounding seawater bacterial communities were performed in R project Venn Diagram package 1.6.16 (Chen and Boutros, 2011). Further, top core genera were identified by screening the core genera with relative abundance more than 1.0%, and was visualized in R project ggplot2 package 2.2.1 (Wickham, 2011).
Statistical analysis
The stacked bar plot of the communities composition was visualized in R project ggplot2 package 2.2.1 (Wickham, 2011). The Chao1, Pielou’s and Shannon diversity indices were calculated in QIIME (version 1.9.1) and the comparison between samples were calculated by Welch’s test. Non-metric multidimensional scaling (NMDS) and Permutational multivariate analysis of variance (PERMANOVA) were calculated based on unweighted unifrac dissimilarities in R project Vegan package 2.5.3.
Indicator species were those genera which influence structure differences in both epiphytic or surrounding seawater microbial communities. Indicator species were analyzed by calculating the indicator value (IndVal) in labdsv package (version 2.0-1) of R project and performing cross-validation test (Roberts, 2016) based on the abundance and occurrence frequency at genus level. Genera with IndVal ≥ 0.7 and p value ≤ 0.05 was selected as indicator genus (Glasl et al., 2019).
Results
The compositional variation of epiphytic and surrounding seawater bacterial communities
There were 18 epibacterial and 18 seawater bacterial samples were analyzed from mature sporophytes, sporelings to juvenile sporophytes of northern farmed S. japonica (Figure 1). A total of 4,653,122 paired-end raw reads and 4,499,030 clean reads were obtained after filtering low quality reads and removing the mitochondria, chloroplasts and eukaryotes sequences. The 24,821 amplicon sequence variants (ASVs) and were assigned across 36 samples. Among the samples, 14,035 ASVs were clustered in the epibacterial (EB) group, which much higher than those in the seawater (SW) group (13,406 ASVs). Only 10.6% ASVs were shared between epiphytic and seawater bacterial communities (Supplementary Figure 1). ASVs were classified into 41 phyla, 111 classes, 247 orders, 372 families and 958 genera. The rarefaction curves showed good diversity coverage (> 99.2%), indicating the sequencing amount was sufficient (Supplementary Figure 2).
Temporal shifts of epibacterial communities associated with northern farmed S. japonica were analyzed from mature sporophytes to juvenile sporophytes (Figure 2). In the epibacterial communities, Proteobacteria (mean relative abundance: 49.1% to 71.1%) was the most dominant phylum at each time point (Figure 2A). At the genus level, Alcanivorax was the most dominant genus at both MS-EB (20.4%) and S1-EB (5.5%). Bacillus was the most dominant at S2-EB (19.9%) and S3-EB (17.0%). Halomonas and Cobetia were the most dominant genera at JS1-EB (12.1%) and JS2-EB (15.5%), respectively (Figure 2B). Regarding to the surrounding seawater bacterial communities, the most dominant phylum was Firmicutes at MS-SW (85.8%) and S3-SW (33.5%). Actinobacteria was the most dominant phylum at S1-SW (51.5%). Meanwhile, Proteobacteria was the most dominant phylum at S2-SW(42.2%), JS1-SW(71.8%) and JS2-SW (52.5%). From mature sporophytes, sporelings to juvenile sporophytes, the relative abundance of Firmicutes decreased from 85.8% to 24.6%, and Proteobacteria increased from 6.5% to 62.1% (Figure 2A). At genus level, the most dominant genera were Virgibacillus at MS-SW (30.8%), Rubritalea at S2-SW (29.2%), Stenotrophomonas at JS1-SW (22.0%) and Cobetia at JS2-SW (25.2%). Oceanobacillus was the most dominant genus at both S1-SW (4.4%) and S3-SW (19.6%) (Figure 2B).
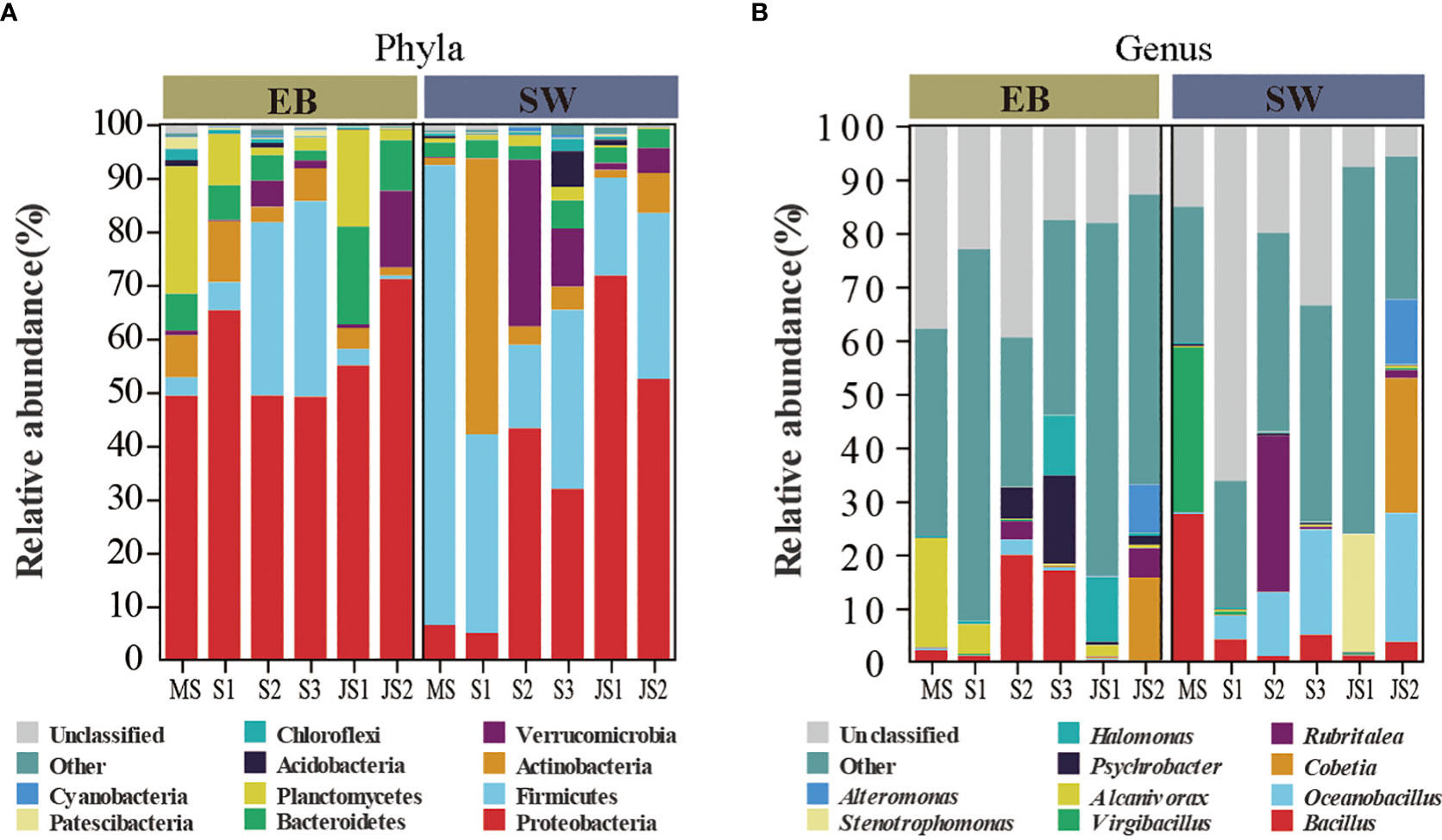
Figure 2 The composition of epiphytic and surrounding seawater bacterial communities. MS: mature sporophyte; S1: the 4-week-old sporeling; S2: 7-week-old sporeling; S3: 9-week-old sporeling; JS1: 4-week-old juvenile sporophyte; JS2: 9-week-old juvenile sporophyte. Top 10 ASVs with relative abundance in both epiphytic and seawater bacterial communities at (A) phylum and (B) genus level.
The temporal shifts in diversity of epiphytic and surrounding seawater bacterial communities
There were significant differences in Chao 1 indexes among six developmental time points (p < 0.05). Except S1-EB, the Chao 1 (species richness estimates) indexes of epibacterial communities exhibited increasing trend with the development with northern farmed S. japonica (Figure 3A). Regarding to Pielou’s evenness and Shannon indexes, there were no significant shifts with the development of S. japonica (Figures 3B, C; p > 0.05). In surrounding seawater bacterial communities, Chao 1 indexes were significantly different with development of S. japonica (Figure 3D, p < 0.05). The Pielou’s evenness was not significantly different (Figure 3E, p > 0.05), but the Shannon indexes exhibited significant differences (Figure 3F, p < 0.05). At the same developmental time point, the Chao 1 indexes (Table 1, p < 0.01) and Pielou’s evenness (Table 1, p < 0.05) of epiphytic and seawater bacterial communities were significantly different at MS and JS2. While, the Shannon indexes of epiphytic and surrounding seawater bacterial communities exhibited significant differences at JS2 (Table 1, p < 0.05).
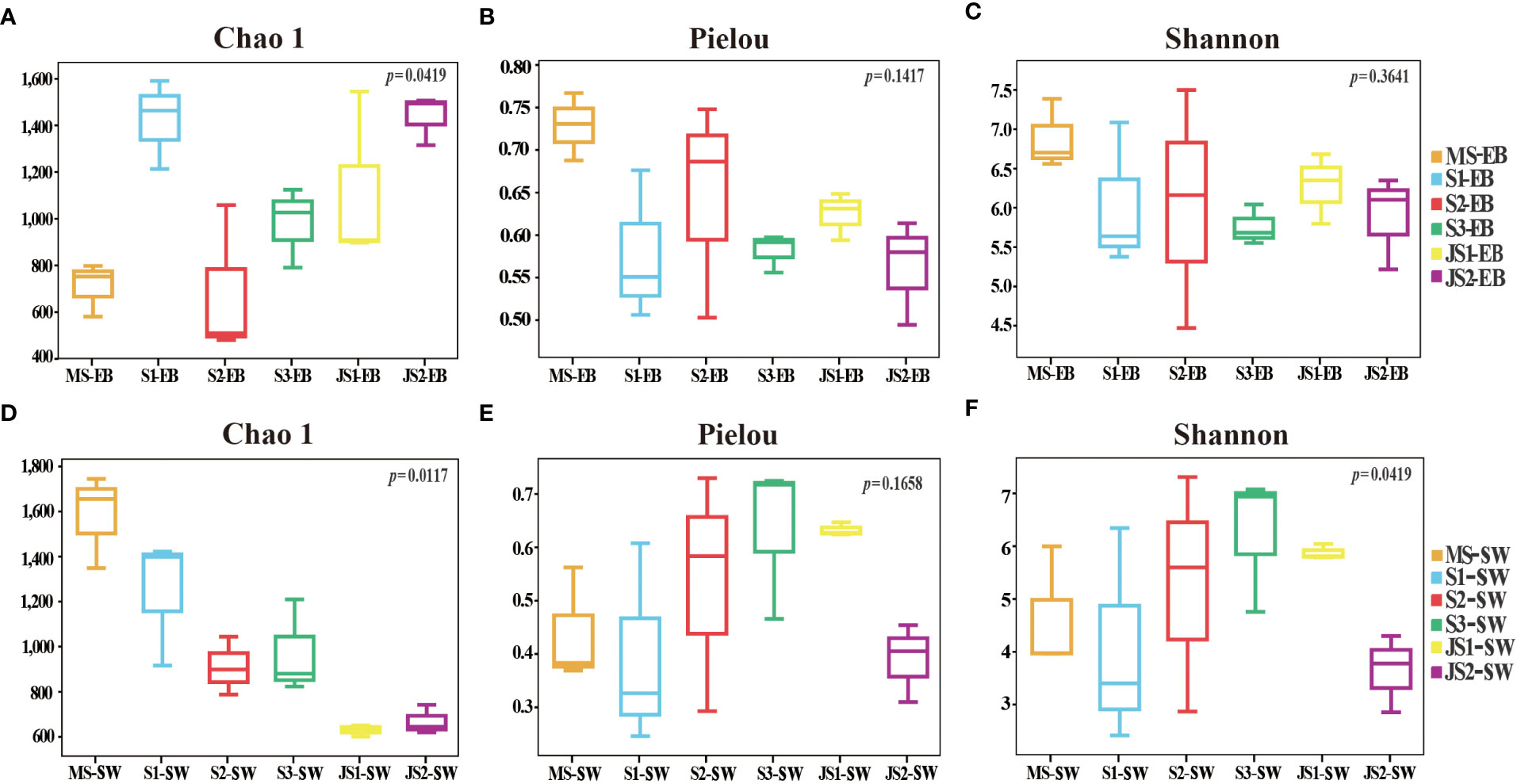
Figure 3 Alpha diversity of the epiphytic and surrounding seawater bacterial communities. (A, D): Chao1 species richness; (B, E): Pielou’s evenness; (C, F): Shannon diversity indexes.
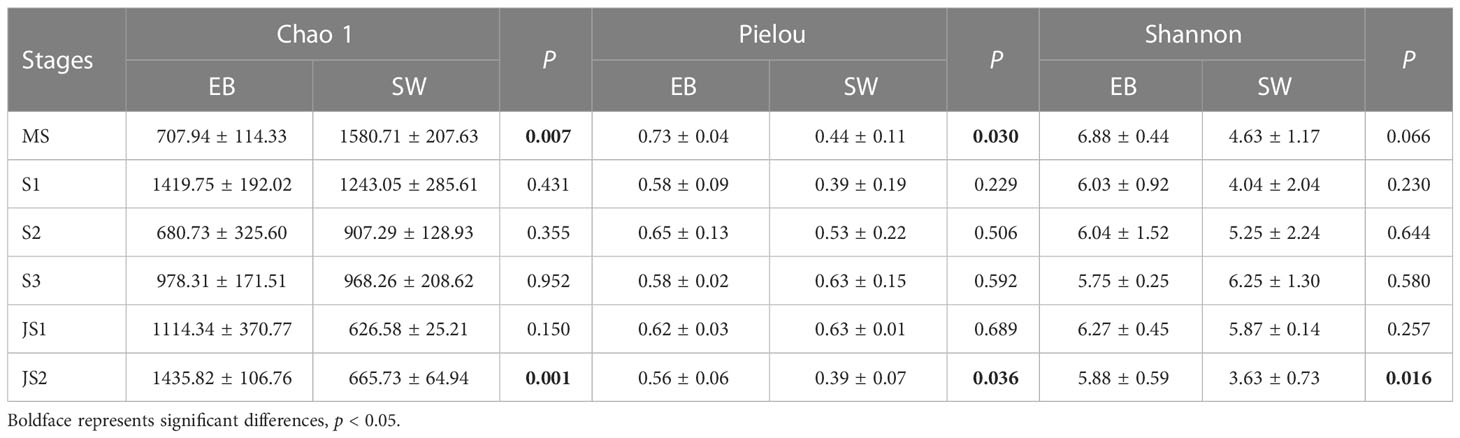
Table 1 Alpha diversity of both epiphytic and surrounding seawater bacterial communities at six developmental time points.
The beta diversity of epibacterial communities displayed significant differences at six developmental time points (Figure 4A). PERMANOVA showed that developmental time explained 40.8% of epibacterial communities variation (PERMANOVA: F value = 1.66, R2 = 0.41, P = 0.001, Table 2). Meanwhile, there were significant differences in the beta diversity of seawater bacterial communities at the different time points (PERMANOVA: F value = 1.89, R2 = 0.44, P = 0.001, Table 2; Figure 4B). Moreover, there was no significant difference between the structure of the epiphytic and the surrounding seawater bacterial communities. (PERMANOVA: F value = 1.2, R2 = 0.03, P = 0.058, Table 2; Supplementary Figure 3). The structure of epiphytic and surrounding seawater bacterial communities were also not significantly different at the same developmental time point by pairwise comparisons (Supplementary Table 1, p > 0.05).

Figure 4 The beta diversity and indicator species of the epiphytic and the surrounding seawater bacterial communities at six developmental time points. Non-metric multidimensional scaling (NMDS) plot on amplicon sequence variants (ASV) of (A) epiphytic and (B) surrounding seawater bacterial communities based on the unweighted Unifrac, the circle represented EB group and the triangle represents SW group. The indicator species of (C) epiphytic and (D) surrounding seawater bacterial communities at six developmental time points, the horizontal and the vertical axes indicating the developmental time points and indicator genera, respectively. The bubble size representing the IndVal of the genus, and the bubble color represents the six developmental time points. MS: mature sporophyte; S1: the 4-week-old sporeling; S2: 7-week-old sporeling; S3: 9-week-old sporeling; JS1: 4-week-old juvenile sporophyte; JS2: 9-week-old juvenile sporophyte.
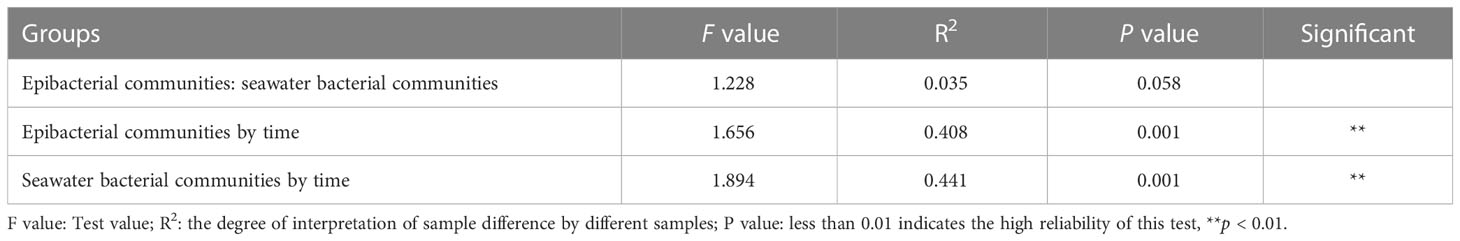
Table 2 Permutational multivariate analysis of variance (PERMANOVA) with adonis based on unweighted unifrac dissimilarities of epiphytic and surrounding seawater bacterial communities by the six developmental time points.
In order to further explore the structure differences of epiphytic or surrounding seawater bacterial communities at six developmental time points, indicator species were analyzed based on IndVal ≥ 0.7 and p value ≤ 0.05 at genus level. The indicator species of MS-EB were Gracilimonas, Vibrio, C1-B045. Thalassospira was indicator species at the S1-EB. Whereas, Pseudomonas Rhodopirellula and Salegentibacter were the indicator species at JS1-EB. There were no indicator species at S2-EB, S3-EB and JS2-EB (Figure 4C). In addition, bubble plot showed different indicator species at different developmental time point to reflect the structure of temporal differences in surrounding seawater bacterial communities (Figure 4D). Virgibacillus and Sediminibacillus were indicator species at the MS-SW. Rubritalea was indicator species at the S2-SW. Acinetobacter, Brevundimonas and Pseudomonas were indicator species at the JS1-SW.
The core genera of epiphytic and surrounding seawater bacterial communities
In this study, core genera were referred to those that were present in all samples at the developmental time points, regardless of their relative abundance. Overall, there were 48 core genera in the epibacterial communities (Supplementary Figure 4A). Among these core genera, the mean relative abundance greater than 1.0% were referred to top core genera. The top core genera included Bacillus, Alcanivorax, Halomonas, Psychrobacter, Blastopirellula, Erythrobacter, Rubritalea, Pseudomonas, SM1A02, Muricauda and Gimesia (Table 3). From MS to S3, the relative abundance of Bacillus in the epibacterial communities gradually increased (ranging from 0.1% to 17.0%) (Supplementary Figure 5). There were 29 core genera (Supplementary Figure 4B) and 7 top core genera (Table 3) in the seawater bacterial communities. Among these top core genera, the relative abundance of Bacillus (27.2%), Virgibacillus (31.9%) and Sediminibacillus (15.8%) were higher at MS-SW. While, the relative abundance of Bradyrhizobium (4.6%) and Rubritalea (23.4%) was higher at both S2-SW (Supplementary Figure 5). Acinetobacter (10.4%) and Pseudomonas (10.4%) was higher at both JS1-SW. Bacillus, Rubritalea and Pseudomonas were top core genera between epiphytic and surrounding seawater bacterial communities. From MS to S2, the relative abundance of Bacillus in the surrounding seawater bacterial communities gradually decreased, which was the opposite of the epibacterial communities (Supplementary Figure 5). The top core genera Rubritalea between epiphytic and surrounding seawater bacterial communities had the highest relative abundance at S2, and the highest relative abundance of Pseudomonas at JS1. The relative abundance of Rubritalea and Pseudomonas were higher in the surrounding seawater bacterial communities at S2 and JS1 (Table 3).
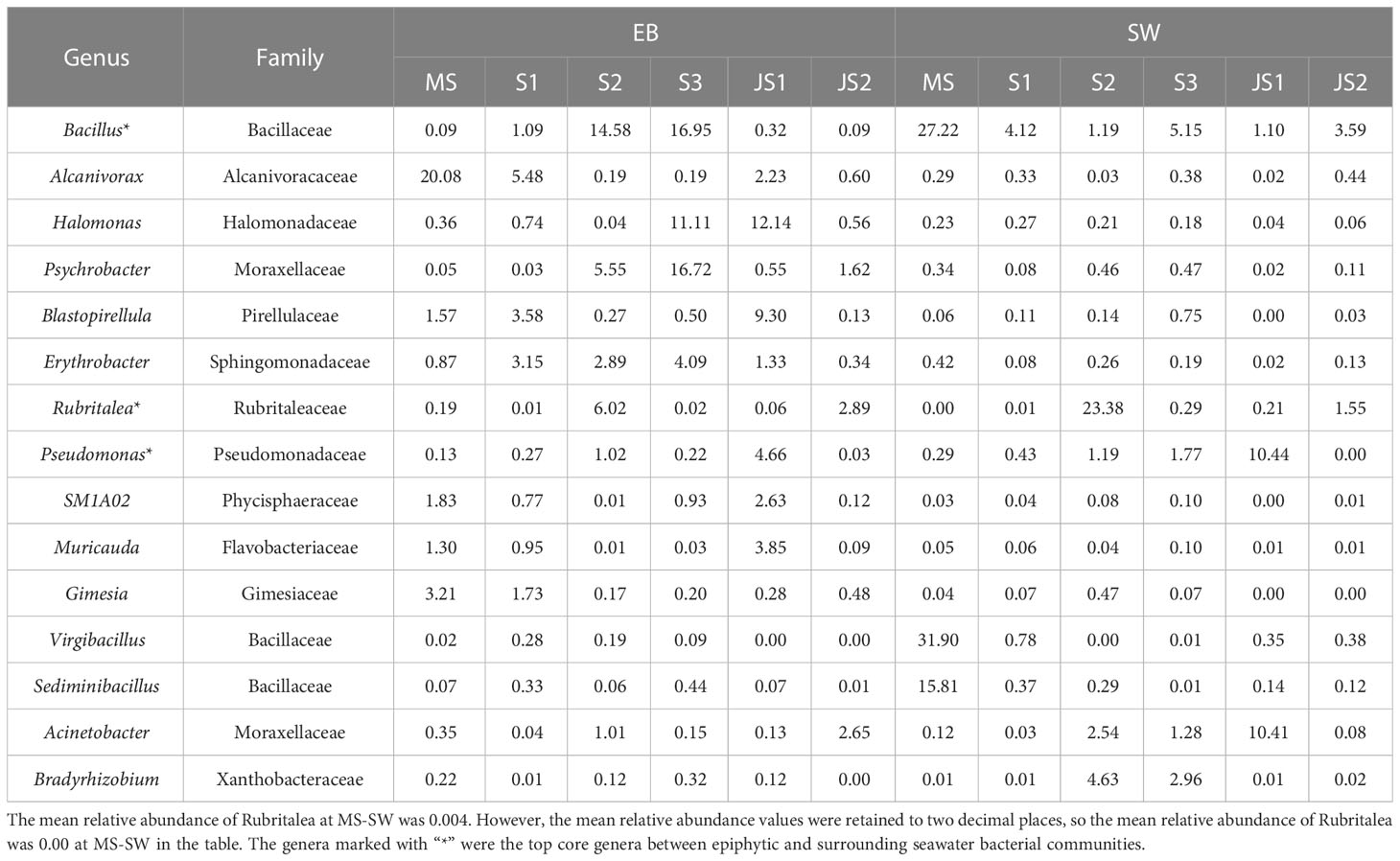
Table 3 Top core genera with their mean relative abundances of epiphytic and surrounding seawater bacterial communities at six developmental time points.
Discussion
Compared to the wild seaweeds, epimicrobial communities of farmed seaweeds are still in infancy. This study investigated the temporal shifts of epiphytic and surrounding seawater bacterial communities at six developmental time points of commercially northern farmed S. japonica by 16S rRNA amplicon sequencing. These results are consistent with our hypothesis. First, the composition of epibacterial communities shifted with the development of S. japonica. Moreover, the Chao1 indexes and beta diversity of epibacterial communities were significantly different with the development of S. japonica (p < 0.05). Meanwhile, we identified the important bacteria that contribute to the normal development of farmed S. japonica by analyzing the dominant and core genera of epibacterial communities. Furthermore, in contrast to the surrounding seawater bacterial communities, the composition and structure of the epibacterial communities were influenced by both host development and the surrounding seawater bacterial communities. To our knowledge, this is the first characterization of temporal shifts and the differences between epiphytic and surrounding seawater bacterial communities in the commercially northern farmed S. japonica.
Dominant and core genera of the epibacterial communities
It was widely accepted that dominant taxa in microbial communities of seaweeds were species- specific at genus level (Bengtsson and Øvreås, 2010; Mancuso et al., 2016; Lemay et al., 2018b, Lemay et al., 2021a; Guo et al., 2022). In this study, the dominant genera with the highest relative abundance were Alcanivorax (MS-EB and S1-EB), Bacillus (S2-EB and S3-EB), Halomonas (JS1-EB) and Cobetia (JS2-EB) (Figure 2B). Previous studies had shown that Flavobacterium, Yoonia- Loktanella and Planctomyces were the most dominant genera for brown alga Sargassum horneri (Mei et al., 2019), Sargassum thunbergii (Guo et al., 2022) and the farmed Caulerpa lentillifera (Pang et al., 2022), respectively. This difference indicated that dominant genera were seaweeds specific (Roth-Schulze et al., 2016; Korlević et al., 2021). What’s interesting, in this study, the dominant genera Alcanivorax, Bacillus and Halomonas were also the core genera. Alcanivorax is a degrading bacterium for marine hydrocarbon pollutants (Olivera et al., 2009), which may render its seaweed hosts to decontaminate the hydrocarbon pollutants and contribute to the healthy development. Bacillus, especially Bacillus subtilis, has strong antibacterial activity against the pathogens Aeromonas hydrophila, Vibrio vulnificus and Vibrio parahaemolyticus of the brown alga Sargassum myriocystum (Chakraborty et al., 2017). Moreover, Halomonas has been found at the wide range of pH and temperatures as well as at almost any range of salinity, which enables Halomonas to colonize the variety of habitats (Kim et al., 2013). Halomonas is also beneficial to the morphogenesis and development of seaweeds, which excretes a specific regulator like cytokinins that enhanced cell division (Spoerner et al., 2012). For example, Halomonas sp. Z3 has been reported to promote the development and increase the number of individuals of the brown alga Ectocarpus sp. (Tapia et al., 2016). Based on the relative abundance advantage and beneficial effects, we speculate that Alcanivorax, Bacillus and Halomonas may play beneficial roles during the development of northern farmed S. japonica. However, their effects on the seaweed development still remain to be further verified by isolating the pure bacterial strains.
Temporal shifts in diversity of the epibacterial communities
The temporal shifts of diversity with the development of seaweeds was one of the evidences of microbial communities shifts (Weigel and Pfister, 2019). Previous studies indicate that the alpha diversity gradually increased with the development of Laminaria saccharina (Staufenberger et al., 2008), Cystoseira compressa (Mancuso et al., 2016) and the Sargassum muticum (Serebryakova et al., 2018). Consistent with these previous researches, we found the Chao 1 indexes of epibacterial communities increased and was significantly different with the development of northern farmed S. japonica (p < 0.05). Meanwhile, beta diversity revealed significant differences in the epibacterial communities among different developmental stages (Figure 4A, p < 0.01). The epiphytic bacterial communities exhibited a significant increase in species diversity as well as significant differences in community structure, but the Pielou’s evenness was stable during the development of northern farmed S. japonica (Figure 3B). This suggested that the epibacterial communities continued to recruit new bacteria to assemble the communities with the development of S. japonica, rather than based on the increase in the abundance of already colonized bacteria. In addition, it is reported that different metabolites (e.g., enzymes and phenolics) can be secreted by the seaweeds at different development time points (Collén and Davison, 2001; Malik et al., 2020; Han et al., 2021; Lemay et al., 2021a), which can attract the attachment of certain bacteria (Zheng et al., 2005) and thus have the unique microbial composition and structure at different developmental time points.
Epibacterial communities of S. japonica influenced by the surrounding seawater bacterial communities
Studies have shown that epibacterial communities of seaweeds were specific and were influenced by the surrounding seawater bacterial communities (Fahimipour et al., 2017; Lemay et al., 2018a; Cleary and Huang, 2020; Juhmani et al., 2020). It was found that the dominant bacteria of Macrocystis pyrifera (Michelou et al., 2013), Thalassia testudinum and Syringodium filliforme (Ugarelli et al., 2018) were shared between the epiphytic and surrounding seawater communities at the phylum level. Consistent with these previous investigations, we observed that Proteobacteria was the dominant phyla in both epiphytic and surrounding seawater bacterial communities. Moreover, the top core genera in the epibacterial communities were all present in the surrounding seawater bacterial communities, which is similar to the results in Nereocystis luetkeana and M. pyrifera (Weigel and Pfister, 2019). The top core genera Rubritalea and Pseudomonas between epiphytic and surrounding seawater bacterial communities had the highest relative abundance at the same developmental time point, and their relative abundance were higher in the surrounding seawater bacterial communities. Meanwhile, the structure of the epiphytic and surrounding seawater bacterial communities did not differ significantly. These results suggested that the surrounding seawater bacterial communities influenced the epibacterial communities. It could be explained by the fact that the surface of S. japonica was constantly in contact with the diversified free-living bacteria in the surrounding seawater and most of the epibacteria are recruited from the surrounding seawater (Mancuso et al., 2016; Weigel and Pfister, 2019). It is worth noting that microbial communities of seaweeds have been shown to be influenced by a variety of factors, including host species, geography and environment, and so on (Hollants et al., 2013; Morrissey et al., 2019). Therefore, the factors influencing the shifts of epibacterial communities of the farmed S. japonica will need to be explored more fully and deeply in the future.
It has been found that the epibacterial communities associated with seaweeds are also influenced by the seaweeds themselves (Weigel and Pfister, 2019; James et al., 2020; Lemay et al., 2021b). In this study, the alpha diversity of the epibacterial communities, including Chao1, Pielou’s evenness and the Shannon indexes, were significantly higher than those of surrounding seawater bacterial communities at JS2 (p < 0.05). The reason for the higher alpha diversity at JS2 maybe due to the faster growth of S. japoninca. Zimmerman and Kremer (1986) and Zhang et al. (2020a) reported that the concentrations of mannitol and laminarin on S. japonica reduced when it grew fast. Therefore, we speculate that the decreased concentrations of mannitol and laminarin on S. japonica at JS2 may attract or inhibit bacterial attachment and then led to a higher alpha diversity of the epibacterial communities. This indicated that the epibacterial communities associated with S. japonica were also influenced by the seaweeds themselves. Our results were consistent with previous findings obtained from Ecklonia radiata (Marzinelli et al., 2015), Cystoseira compressa (Mancuso et al., 2016) and Caulerpa taxifolia (Morrissey et al., 2019). This suggested that the epibacterial communities were more affected by the host itself than the surrounding seawater bacterial communities at the faster growth stage of S. japonica.
Conclusions
Using 16S rRNA gene amplicon sequencing, we investigated the shifts of epiphytic and surrounding seawater bacterial communities of northern farmed S. japonica from mature to juvenile sporophyte stages. Our results indicated that the dominant genera of epibacterial communities shifted with the development of S. japonica. Moreover, the Chao1 indexes and beta diversity of epibacterial communities were significantly different among the six developmental time points. Combining analysis of the dominant and core genera, Alcanivorax, Bacillus and Halomonas may contribute to the development of northern farmed S. japonica. Finally, the epibacterial communities were influenced by both S. japonica itself and the surrounding seawater bacterial communities. These findings provide novel insights into the bacterial communities associated with northern farmed S. japonica.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA903939.
Author contributions
GW conceived, designed the experiment and revised the manuscript. LCa analyzed the sequencing data and wrote the manuscript. XG involved in the analysis of sequencing data. MS participated in sequencing analysis and helped to improve the English writing. YH collected samples of Saccharina japonica. LCh and LX provided the samples of S. japonica. All authors contributed to the article and approved the submitted version.
Funding
This study was sponsored by the National Natural Science Foundation of China (42076106; 41576158), Sino-German Center for Research Promotion (GZ1357), and National Key R & D Program of China (2018YFD0900305).
Acknowledgments
We are grateful to Guangzhou Genedenovo Biotechnology Co. Ltd for their kindly technical support.
Conflict of interest
Author LCh and LX are employed by Weihai Changqing Ocean Science & Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1117926/full#supplementary-material
References
Bengtsson M. M., Øvreås L. (2010). Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea. BMC Microbiol. 10, 261. doi: 10.1186/1471-2180-10-261
Bonthond G., Bayer T., Krueger-Hadfield S. A., Barboza F. R., Nakaoka M., Valero M., et al. (2020). How do microbiota associated with an invasive seaweed vary across scales? Mol. Ecol. 29, 2094–2108. doi: 10.1111/mec.15470
Callahan B. J., Mcmurdie P. J., Rosen M. J. (2016). DADA2: high-resolution sample inference from illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chakraborty K., Thilakan B., Chakraborty R. D., Raola V. K., Joy M. (2017). O-Heterocyclic derivatives with antibacterial properties from marine bacterium Bacillus subtilis associated with seaweed, Sargassum myriocystum. Appl. Microbiol. Biotechnol. 101, 569–583. doi: 10.1007/s00253-016-7810-3
Chen H., Boutros P. C. (2011). VennDiagram: a package for the generation of highly- customizable Venn and Euler diagrams in r. BMC Bioinf. 12, 35. doi: 10.1186/1471-2105-12-35
Cleary D. F., Huang Y. M. (2020). A comparison of the prokaryotic communities associated with seven seaweed species, sediment, and seawater from the penghu archipelago, Taiwan. Mar. Biol. Res. 16, 744–761. doi: 10.1080/17451000.2020.1859119
Collén J., Davison I. R. (2001). Seasonality and thermal acclimation of reactive oxygen metabolism in Fucus vesiculosus (Phaeophyceae). J. Phycol. 37, 474–481. doi: 10.1046/j.1529-8817.2001.037004474.x
Comba González N. B., Niño Corredor A. N., López Kleine L., Montoya Castaño D. (2021). Temporal changes of the epiphytic bacteria community from the marine macroalga Ulva lactuca (Santa Marta, Colombian-Caribbean). Curr. Microbiol. 78, 534–543. doi: 10.1007/s00284-020-02302-x
Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fahimipour A. K., Kardish M. R., Lang J. M., Green J. L., Eisen J. A., Stachowicz J. J. (2017). Global-scale structure of the eelgrass microbiome. Appl. Environ. Microbiol. 83, e03391–e03316. doi: 10.1128/AEM.03391-16
Florez J. Z., Camus C., Hengst M. B., Marchant F., Buschmann A. H. (2019). Structure of the epiphytic bacterial communities of Macrocystis pyrifera in localities with contrasting nitrogen concentrations and temperature. Algal. Res. 44, 101706. doi: 10.1016/j.algal.2019.101706
Glasl B., Bourne D. G., Frade P. R., Thomas T., Schaffelke B., Webster N. S. (2019). Microbial indicators of environmental perturbations in coral reef ecosystems. Microbiome. 7, 94. doi: 10.1186/s40168-019-0705-7
Guo Z., Wang L., Jiang Z., Liang Z. (2022). Comparison studies of epiphytic microbial communities on four macroalgae and their rocky substrates. Mar. Pollut. Bull. 176, 113435. doi: 10.1016/j.marpolbul.2022.113435
Guo M. J., Wu F. H., Hao G., Qi Q., Rong L., Li N. (2017). Bacillus subtilis Improves immunity and disease resistance in rabbits. Front. Immunol. 8. doi: 10.3389/fimmu.2017.00354
Han Q., Zhang X., Chang L., Xiao L., Ahmad R., Saha M., et al. (2021). Dynamic shift of the epibacterial communities on commercially cultivated Saccharina japonica from mature sporophytes to sporelings and juvenile sporophytes. J. Appl. Phycol. 33, 1171–1179. doi: 10.1007/s10811-020-02329-4
Hollants J., Leliaert F., De Clerck O., Willems A. (2013). What we can learn from sushi: a review on seaweed-bacterial associations. FEMS Microbiol. Ecol. 83, 1–16. doi: 10.1111/j.1574-6941.2012.01446.x
Ihua M. W., FitzGerald J. A., Guihéneuf F., Jackson S. A., Claesson M. J., Stengel D. B., et al. (2020). Diversity of bacteria populations associated with different thallus regions of the brown alga Laminaria digitata. PloS One 15, e0242675. doi: 10.1371/journal.pone
James A. K., English C. J., Nidzieko N. J., Carlson C. A., Wilbanks E. G. (2020). Giant kelp microbiome altered in the presence of epiphytes. Limnol. Oceanogr. 5, 354–362. doi: 10.1002/lol2.10157
Juhmani A. S., Vezzi A., Wahsha M., Buosi A., Pascale F., Schiavon R., et al. (2020). Diversity and dynamics of seaweed associated microbial communities inhabiting the lagoon of venice. Microorganisms. 8, 1657. doi: 10.3390/microorganisms8111657
Kim K. K., Lee J. S., Stevens D. A. (2013). Microbiology and epidemiology of Halomonas species. Future Microbiol. 8, 1559–1573. doi: 10.2217/fmb.13.108
Korlević M., Markovski M., Zhao Z., Herndl G. J., Najdek M. (2021). Seasonal dynamics of epiphytic microbial communities on marine macrophyte surfaces. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.671342
Lemay M. A., Chen M. Y., Mazel F., Hind K. R., Starko S., Keeling P. J., et al. (2021b). Morphological complexity affects the diversity of marine microbiomes. ISME J. 15, 1372–1386. doi: 10.1038/s41396-020-00856-z
Lemay M. A., Davis K. M., Martone P. T., Parfrey L. W. (2021a). Kelp-associated microbiota are structured by host anatomy1. J. Phycol. 57, 1119–1130. doi: 10.1111/jpy.13169
Lemay M. A., Martone P. T., Hind K. R., Lindstrom S. C., Wegener Parfrey L. (2018b). Alternate life history phases of a common seaweed have distinct microbial surface communities. Mol. Ecol. 27, 3555–3568. doi: 10.1111/mec.14815
Lemay M. A., Martone P. T., Keeling P. J., Burt J. M., Krumhansl K. A., Sanders R. D., et al. (2018a). Sympatric kelp species share a large portion of their surface bacterial communities. Environ. Microbiol. 20, 658–670. doi: 10.1111/1462-2920.13993
Malik S. A. A., Bedoux G., Maldonado J. Q. G., Freile-Pelegrín Y., Robledo D., Bourgougnon N. (2020). Defence on surface: macroalgae and their surface-associated microbiome. Adv. Bot. Res. 95, 327–368. doi: 10.1016/bs.abr.2019.11.009
Mancuso F. P., D'hondt S., Willems A., Airoldi L., De Clerck O. (2016). Diversity and temporal dynamics of the epiphytic bacterial communities associated with the canopy-forming seaweed Cystoseira compressa (Esper) gerloff and nizamuddin. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00476
Marzinelli E. M., Campbell A. H., Zozaya Valdes E., Vergés A., Nielsen S., Wernberg T., et al. (2015). Continental-scale variation in seaweed host-associated bacterial communities is a function of host condition, not geography. Environ. Microbiol. 17, 4078–4088. doi: 10.1111/1462-2920.12972
Mei X., Wu C., Zhao J., Yan T., Jiang P. (2019). Community structure of bacteria associated with drifting Sargassum horneri, the causative species of golden tide in the yellow Sea. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.01192
Michelou V. K., Caporaso J. G., Knight R., Palumbi S. R., Harder T. (2013). The ecology of microbial communities associated with Macrocystis pyrifera. PloS One 8, e67480. doi: 10.1371/journal.pone.0067480
Morrissey K. L., Çavaş L., Willems A., De Clerck O. (2019). Disentangling the influence of environment, host specificity and thallus differentiation on bacterial communities in siphonous green seaweeds. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00717
Olivera N. L., Nievas M. L., Lozada M., Del Prado G., Dionisi H. M., Siñeriz F. (2009). Isolation and characterization of biosurfactant-producing Alcanivorax strains: hydrocarbon accession strategies and alkane hydroxylase gene analysis. Res. Microbiol. 160, 19–26. doi: 10.1016/j.resmic.2008.09.011
Paix B., Layglon N., Le Poupon C., D'Onofrio S., Misson B., Garnier C., et al. (2021). Integration of spatio-temporal variations of surface metabolomes and epibacterial communities highlights the importance of copper stress as a major factor shaping host-microbiota interactions within a Mediterranean seaweed holobiont. Microbiome. 9, 201. doi: 10.1186/s40168-021-01124-8
Pang M., Huang Z., Lv L., Li X., Jin G. (2022). Seasonal succession of bacterial communities in cultured Caulerpa lentillifera detected by high-throughput sequencing. Open Life Sci. 17, 10–21. doi: 10.1515/biol-2022-0001
Phelps C. M., McMahon K., Bissett A., Bernasconi R., Steinberg P. D., Thomas T., et al. (2021). The surface bacterial community of an Australian kelp shows cross-continental variation and relative stability within regions. FEMS Microbiol. Ecol. 97, fiab089. doi: 10.1093/femsec/fiab089
Pruesse E., Quast C., Knittel K., Fuchs B. M., Ludwig W., Peplies J. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Roberts D. W. (2016) Labdsv: ordination and multivariate analysis for ecology. Available at: http://cran.r-project.org/package=labdsv.
Roth-Schulze A. J., Zozaya-Valdés E., Steinberg P. D., Thomas T. (2016). Partitioning of functional and taxonomic diversity in surface associated microbial communities. Environ. Microbiol. 18, 4391–4402. doi: 10.1111/1462-2920.13325
Saha M., Ferguson R. M. W., Dove S., Künzel S., Meichssner R., Neulinger S. C., et al. (2020). Salinity and time can alter epibacterial communities of an invasive seaweed. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02870
Saha M., Weinberger F. (2019). Microbial “gardening” by a seaweed holobiont: surface metabolites attract protective and deter pathogenic epibacterial settlement. J. Ecol. 107, 2255–2265. doi: 10.1111/1365-2745.13193
Serebryakova A., Aires T., Viard F., Serrão E. A., Engelen A. H. (2018). Summer shifts of bacterial communities associated with the invasive brown seaweed Sargassum muticum are location and tissue dependent. PloS One 13, e0206734. doi: 10.1371/journal.pone.0206734
Spoerner M., Wichard T., Bachhuber T., Stratmann J., Oertel W. (2012). Growth and thallus morphogenesis of Ulva mutabilis (Chlorophyta) depends on a combination of two bacterial species excreting regulatory factors. J. Phycol. 48, 1433–1447. doi: 10.1111/j.1529-8817.2012.01231.x
Staufenberger T., Thiel V., Wiese J., Imhoff J. F. (2008). Phylogenetic analysis of bacteria associated with Laminaria saccharina. FEMS Microbiol. Ecol. 64, 65–77. doi: 10.1111/j.1574-6941.2008.00445.x
Tapia J. E., González B., Goulitquer S., Potin P., Correa J. A. (2016). Microbiota influences morphology and reproduction of the brown alga Ectocarpus sp. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00197
Tujula N. A., Crocetti G. R., Burke C., Thomas T., Holmström C., Kjelleberg S. (2010). Variability and abundance of the epiphytic bacterial community associated with a green marine Ulvacean alga. ISME J. 4, 301–311. doi: 10.1038/ismej.2009.107
Ugarelli K., Laas P., Stingl U. (2018). The microbial communities of leaves and roots associated with turtle grass (Thalassia testudinum) and manatee grass (Syringodium filliforme) are distinct from seawater and sediment communities, but are similar between species and sampling sites. Microorganisms. 7, 4. doi: 10.3390/microorganisms7010004
Wang Q. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang S., Yan Y., Qian H., Li J., Liu T., Mo Z. (2022). Dynamics of planktonic microbial community associated with Saccharina japonica seedling. J. Mar. Sci. Eng. 10, 726. doi: 10.3390/jmse10060726
Weigel B. L., Pfister C. A. (2019). Successional dynamics and seascape-level patterns of microbial communities on the canopy-forming kelps Nereocystis luetkeana and Macrocystis pyrifera. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00346
Yan Y., Wang S., Li J., Liu F., Mo Z. (2022). Alterations in epiphytic bacterial communities during the occurrence of green rot disease in Saccharina japonica seedlings. J. Mar. Sci. Eng. 10, 730. doi: 10.3390/jmse10060730
Zhang R. (2017). Dynamics of the bacterial communities associated with commercially cultivated saccharina japonica and the surrounding seawater during the harvest season (Qingdao: Ocean University of China).
Zhang L., Cao Z., Liang G., Li X., Wu H., Yang G. (2020a). Comparative transcriptome analysis reveals candidate genes related to structural and storage carbohydrate biosynthesis in kelp Saccharina japonica (Laminariales, phaeophyceae). J. Phycol. 56, 1168–1183. doi: 10.1111/jpy.13016
Zhang R., Chang L., Xiao L., Zhang X., Han Q., Li N., et al. (2020b). Diversity of the epiphytic bacterial communities associated with commercially cultivated healthy and diseased Saccharina japonica during the harvest season. J. Appl. Phycol. 32, 2071–2080. doi: 10.1007/s10811-019-02025-y
Zheng L., Han X. T., Chen H. M., Lin W., Yan X. J. (2005). Marine bacteria associated with marine macroorganisms: the potential antimicrobial resources. Ann. Microbiol. 55, 119–124.
Keywords: epibacterial communities, surrounding seawater bacterial communities, Saccharina japonica, temporal shift, 16S rRNA gene amplicon sequencing, seaweeds
Citation: Cai L, Gao X, Saha M, Han Y, Chang L, Xiao L and Wang G (2023) How do epiphytic and surrounding seawater bacterial communities shift with the development of the Saccharina japonica farmed in the Northern China? Front. Mar. Sci. 10:1117926. doi: 10.3389/fmars.2023.1117926
Received: 07 December 2022; Accepted: 20 April 2023;
Published: 04 May 2023.
Edited by:
Ana Rotter, National Institute of Biology (NIB), SloveniaCopyright © 2023 Cai, Gao, Saha, Han, Chang, Xiao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaoge Wang, wgaoge@ouc.edu.cn