 Minrui Wang1†
Minrui Wang1† Yang Yu
Yang Yu Xiaoqiu Tan
Xiaoqiu Tan Chunxiang Zhang
Chunxiang Zhang- 1Department of Physiology, School of Basic Medical Sciences, Southwest Medical University, Luzhou, Sichuan, China
- 2Department of Pharmaceutical Sciences, School of Pharmacy, Southwest Medical University, Luzhou, Sichuan, China
- 3The Key Laboratory of Medical Electrophysiology of the Ministry of Education, Southwest Medical University, Luzhou, Sichuan, China
- 4Department of Cardiology, the Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, China
Ischemic heart disease (IHD) is a leading cause of disability and death worldwide, with immune regulation playing a crucial role in its pathogenesis. Various immune cells are involved, and as one of the key immune cells residing in the heart, macrophages play an indispensable role in the inflammatory and reparative processes during cardiac ischemia. Exosomes, extracellular vesicles containing lipids, nucleic acids, proteins, and other bioactive molecules, have emerged as important mediators in the regulatory functions of macrophages and hold promise as a novel therapeutic target for IHD. This review summarizes the regulatory mechanisms of different subsets of macrophages and their secreted exosomes during cardiac ischemia over the past five years. It also discusses the current status of clinical research utilizing macrophages and their exosomes, as well as strategies to enhance their therapeutic efficacy through biotechnology. The aim is to provide valuable insights for the treatment of IHD.
1 Introduction
Cardiovascular disease (CVD) is a leading cause of global mortality, with over 18.5 million deaths attributable to CVD in 2019, with ischemic heart disease (IHD) accounting for half of all CVD-related deaths worldwide (1). IHD is characterized by reduced blood flow to the heart, leading to an imbalance between myocardial oxygen supply and demand. Ischemia of the myocardium can progress to ischemia-reperfusion arrhythmias, myocardial infarction (MI), and even heart failure (2).
Due to its complex pathophysiological mechanisms, understanding the specific mechanisms involved in IHD occurrence can contribute to the development of more effective treatment methods aimed at improving patient survival rates (3–6).
The underlying mechanisms of most CVDs involve innate and acquired immune responses. Among them, inflammation is one of the important complications following IHD, such as MI or reperfusion injury, and the inflammatory cascade response plays an important role in myocardial tissue injury, repair, and remodeling, and mastery of the cell-specific signaling mechanisms that mediate the inflammatory response is essential for the treatment of MI (7–11).
Immune cells are involved in microenvironmental changes following the development of IHD, and macrophages are among the most abundant immune cells in the heart (12, 13).
In healthy conditions, cardiac-resident macrophages constitute 6–8% of non-myocardial cells in adult mice (14). After myocardial ischemia, cardiac macrophages undergo marked changes in phenotype and function and are capable of massive expansion through their proliferation and recruitment of monocytes, a behavior implicated in both the injurious and reparative responses of the heart (7, 15, 16). This is primarily evidenced by the rapid apoptosis of resident macrophages within 2 hours and their near depletion within 24 hours following MI (17). Meanwhile, disruption of cardiac homeostasis leads to recruitment and differentiation of Ly6Chi monocytes from the bloodstream into macrophages, replacing resident macrophages and persisting long-term (18–20). Upon ischemic insult, circulating monocytes swiftly transition from a rolling to a flowing state via activation of chemokine ligand 2 (CCL2)/monocyte chemoattractant protein (MCP)-1 signaling (21). This prompts their infiltration into the infarcted area, forming a reservoir of monocytes. Stimulated by factors such as colony-stimulating factor and granulocyte growth factor within macrophage colonies, these monocytes differentiate into mature macrophages, thus constituting the primary source of cardiac macrophages (22).
While resident macrophages are scarce in number, recent research indicates their ability to proliferate following cardiac injury, thereby influencing the subsequent recruitment of monocytes. Following cardiac injury, resident macrophages produce inflammatory and chemotactic factors responsible for clearing and degrading apoptotic cardiomyocytes, impacting cardiac conduction (23–25). These resident macrophages are typically divided into CCR2- and CCR2+ subsets, each with distinct mechanisms and functions (26). Tissue-resident CCR2+ macrophages promote monocyte recruitment through a MYD88-dependent mechanism, leading to MCP release and monocyte mobilization, while CCR2- macrophages inhibit monocyte recruitment (27).
Nevertheless, both in terms of quantity and impact, resident cardiac macrophages are not as influential as circulating monocytes, which play a predominant role in the ischemic heart (28). Circulating monocytes differentiate into M1 type early post-MI and transition to M2 type later (29). Classically activated M1 macrophages primarily engage in phagocytosis, MHC II antigen presentation, and reactive oxygen species production (30–32), whereas M2 macrophages stimulate extracellular matrix production, cell proliferation, and angiogenesis, facilitating tissue remodeling and repair (33–35). In conclusion, the dynamic interplay between resident cardiac macrophages and circulating monocytes plays a pivotal role in the response to MI and subsequent cardiac repair processes. Further investigation into the precise molecular pathways governing macrophage behavior in the injured heart holds promise for the development of targeted strategies to enhance cardiac healing and functional recovery.
Exosomes are extracellular vesicles with a diameter of 40–160 nm that are secreted by cells and serve as important vehicles for paracrine signaling (36). Exosomes play pivotal roles in various macrophage-mediated effects, serving crucial functions in cellular processes. Their biogenesis involves distinct stages, commencing with cellular internalization, where the cell membrane engulfs extracellular material to form vesicles. Subsequent fusion with endosomal compartments generates early endosomes, which mature into intraluminal vesicles (ILVs) within late endosome multivesicular bodies (MVBs). Ultimately, MVBs merge with the cell membrane, releasing ILVs as exosomes (37, 38). Exosomes play a significant role in intracellular and intercellular communication by selectively delivering cargoes such as nucleic acids, proteins, and lipids to target cells and organs. They contribute to important processes including angiogenesis, ventricular remodeling, and immune response regulation following cardiac ischemia (39). The therapeutic benefits of exosomes have been validated in various animal and disease models (40–42). Moreover, the unique physiological properties of exosomes, including their circulatory stability, biocompatibility, low immunogenicity, and low toxicity, make them an excellent carrier for targeted drug delivery, which has been applied in a range of diseases such as cancer and inflammation (43). As typical secretory cells, macrophages are capable of secreting various molecular signaling substances and releasing different types of exosomes in the ischemic microenvironment (44–46). Based on this, this review summarizes the research progress in the regulatory mechanisms and therapeutic effects of different phenotypic macrophages and their exosomes in IHD over the past five years. It evaluates their specific biological functions and provides guidance for the utilization of macrophage-derived exosomes in low-risk, highly targeted therapy for IHD.
2 The role of macrophages in IHD
Due to the non-regenerative nature of adult cardiac cells, prolonged myocardial ischemia often leads to MI, resulting in irreversible loss of cardiomyocytes and even left ventricular remodeling and progressive heart failure (47). Therefore, protecting cardiomyocytes and preventing ventricular remodeling and heart failure are important strategies for treating IHD.
Studies have shown that ventricular remodeling is closely associated with dysregulated immune responses, and monocytes and macrophages are the key effector cells of the immune system (48). During disease onset, a significant accumulation of blood-derived monocytes occurs at the ischemic site of the heart, which subsequently differentiate into macrophages to participate in the immune response (3). In the early phase of MI, the necrosis of a large number of cardiomyocytes triggers an intense inflammatory response, and the number of M1 macrophages will peak at 3–4 days after MI, participating in the removal of damaged cells and phagocytosis of extracellular matrix debris during this inflammatory phase (49). In the following days, the inflammatory phase gradually transitions into the reparative phase, where macrophages shift towards an M2 phenotype. M2 macrophages become predominant from day 5 post-infarction and secrete anti-inflammatory and pro-fibrotic cytokines to facilitate the repair of injured myocardium. It is noteworthy that this transition depends on the timely suppression of the inflammatory response. Prolonged activation of M1 macrophages can lead to extensive cardiomyocyte death, degradation of extracellular matrix, expansion of the infarct area, and adverse ventricular remodeling, ultimately resulting in heart failure (4, 49–54) (Figure 1).
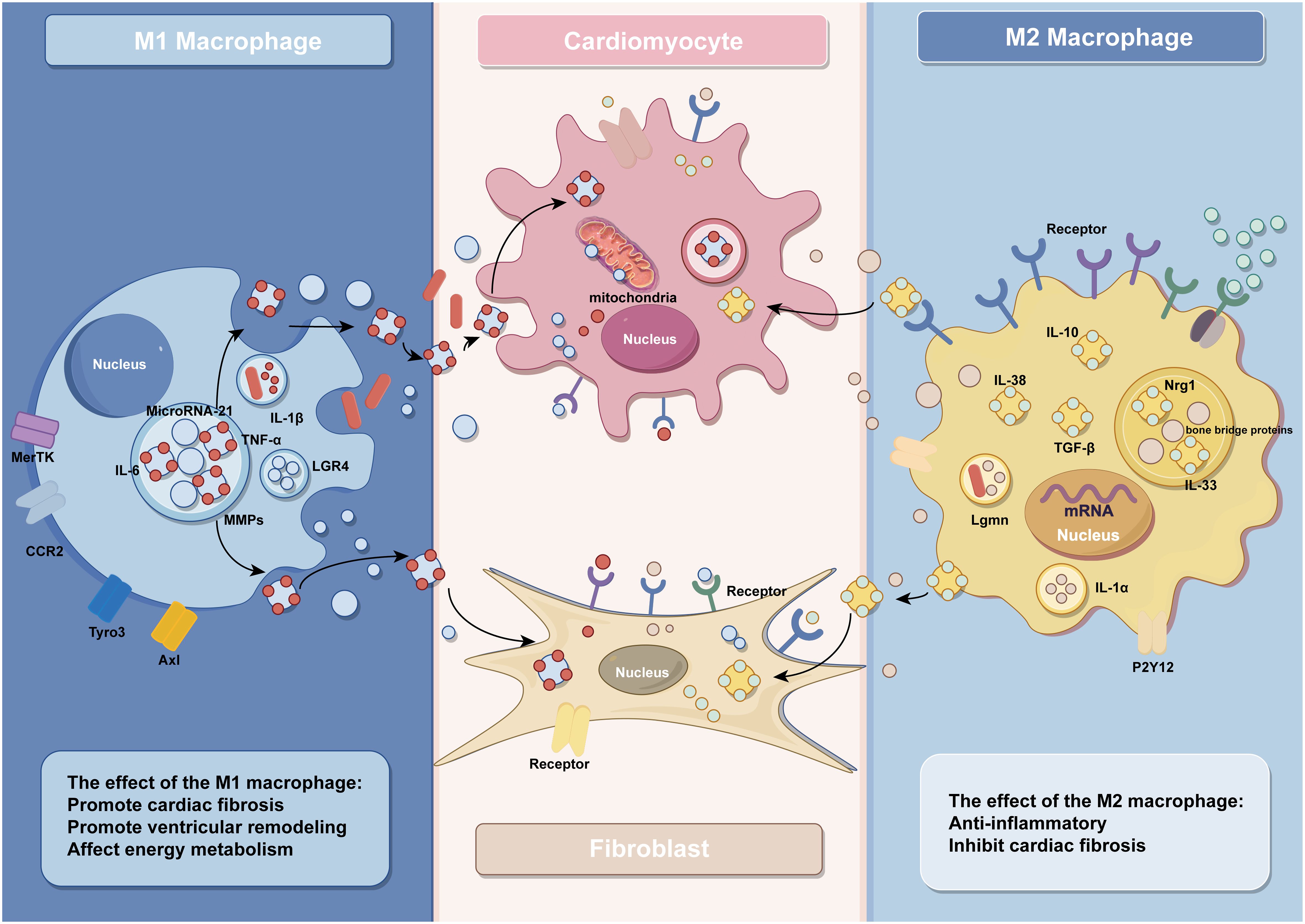
Figure 1 The effect of macrophages and their exosomes in IHD (by Figdraw).
2.1 The role of M1 macrophages in IHD
2.1.1 M1 macrophages are involved in the pro-inflammatory and fibrotic responses after MI
M1 macrophages in IHD are mainly involved in the inflammatory response of post-infarction myocardial tissue and myocardial tissue fibrosis, thus aggravating cardiac injury (55). As a result of myocardial necrosis, the integrity of ECs and their barrier function are impaired, promoting the release of danger-associated molecular patterns, further activating intercellular crosstalk signaling and releasing a large amount of pro-inflammatory mediators, facilitating polarization of macrophages towards the M1 phenotype (56). Activated M1 macrophages release a significant amount of inflammatory cytokines and growth factors such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), chemokines, etc., which further contribute to the promotion of inflammation and fibrotic responses. For example, angiotensin II (AngII) AT1 receptors are involved in the development of myocardial fibrosis through stimulation of the TNF-α/NF-κB/CD44-triggered κ-signaling pathway (57, 58). Studies have shown that chemokines are key linking factors between myocardial inflammation and fibrosis, such that CC chemokine ligand 2(CCL2) can exert fibrotic effects by recruiting and activating M1 macrophages expressing its receptor CCR2 (59). In addition, C-X-C chemokine receptor 4 (CXCR4) is a vital regulator of macrophage-mediated immune responses, and CXCR4 significantly enhances the expression of chemokine (C-X-C) motif ligand (CXCL3), thereby promoting myofibroblast (MF) differentiation (60). In addition to various cytokines, some microRNAs contained in macrophages regulate the inflammatory and fibrotic responses after MI. For instance, studies conducted by Deepak Ramanujam et al. have demonstrated that microRNA-21 (miR-21) is not only the most abundant microRNA in cardiac macrophages but also a key factor contributing to myocardial tissue fibrosis. M1 macrophages secrete miR-21 in a paracrine manner, targeting cardiac fibroblasts (CFs) and promoting their transition from a quiescent state to MFs (61). After MI, there are significant changes in the protein expression levels within resident macrophages in the heart. Among them, the synthesis and degradation of matrix metalloproteinases play multiple roles in the process of ventricular remodeling. Seven days after infarction, the expression of Mmp14 (MT1-MMP) in macrophages significantly increases. Specific deletion of Mmp14 in mice can significantly alleviate post-MI cardiac dysfunction, reduce fibrosis, and protect the microvascular network in the heart (16). A series of pro-inflammatory reactions induced by M1 macrophages promotes the occurrence of cardiac fibrosis, further leading to impaired cardiac contraction and ejection function, exacerbating the development of heart failure. Therefore, early and rapid intervention and modulation of the secretion of relevant cytokines and gene expression can effectively prevent the progression of IHD.
2.1.2 Involvement of M1 macrophages in ventricular remodeling
Patients with IHD undergoing reperfusion therapy still face the challenges of left ventricular remodeling and heart failure after MI (62). The polarization of macrophages is regulated by multiple proteins, and a reduction in the expression of protective proteins after MI promotes the polarization of macrophages towards the M1 phenotype, exacerbating the inflammatory response and leading to adverse ventricular remodeling. For example, decreased expression of V-set and immunoglobulin domain containing 4(VSIG4), a protein that protects against cardiac injury after ischemia in myocardial tissue, further activates TLR4/NF-κB and accelerates macrophage polarization toward M1 macrophages, which leads to increased apoptosis of cardiomyocytes and aggravates cardiac injury after reperfusion (63). On the other hand, ischemia in the myocardium dramatically increases the expression levels of various proteins that promote M1-type polarization, further exacerbating the development of ventricular remodeling. Among them, Dectin-1, a class of proteins that regulates macrophage differentiation, is highly expressed in the early phase of cardiac ischemia-reperfusion (I/R), and its elevated expression leads to the increased polarization of macrophages toward M1 type and promotes infiltration of Ly-6C+ monocytes and neutrophils, leading to further myocardial injury and ventricular remodeling (64). In addition to this, macrophages in the early stages of MI highly express Lgr4, which leads to inflammatory macrophage activation by promoting cAMP response element binding protein(CREB) mediated c-Fos, Fosl1 and FosB trans activation, further leading to reduced cardiac function, increased myocardial infarct size and poor ventricular remodeling (65). As with Dectin-1 and Lgr4, increased expression of grass bacillus proteinogenic-converting enzyme 9 (PCSK9) in myocardial tissue after acute MI promotes poor myocardial repair by the polarization of M1 macrophages (66–68).
Following MI, macrophages undergo polarization toward M1 phenotype, thereby instigating the inflammatory cascade. Furthermore, certain cytokines released by macrophages impede their differentiation into M2 subtype. Consequently, this cytokine-mediated hindrance compromises the myocardial tissue repair mechanism. For example, the increased expression of YAP and TAZ after MI further increases the secretion of IL-6 by M1 macrophages by interacting with the histone deacetylase 3 (HDAC3)-nuclear receptor co-blocker 1 (NCoR1) blocking complex, thereby decreasing arginase-I (Arg1) expression, further impeding the repair response (69). Timely regulation of protein expression and maintenance of macrophage homeostasis would contribute to modulating the healing process following ischemic injury. For example, research by Wang et al. demonstrated that inhibition of purinergic receptor 2Y12 (P2Y12) in macrophages reduced inflammation and improved reperfusion arrhythmias in a rat I/R model, which served as cardioprotection (70). Relevant references are also presented in Table 1 for easy visualization (Table 1).
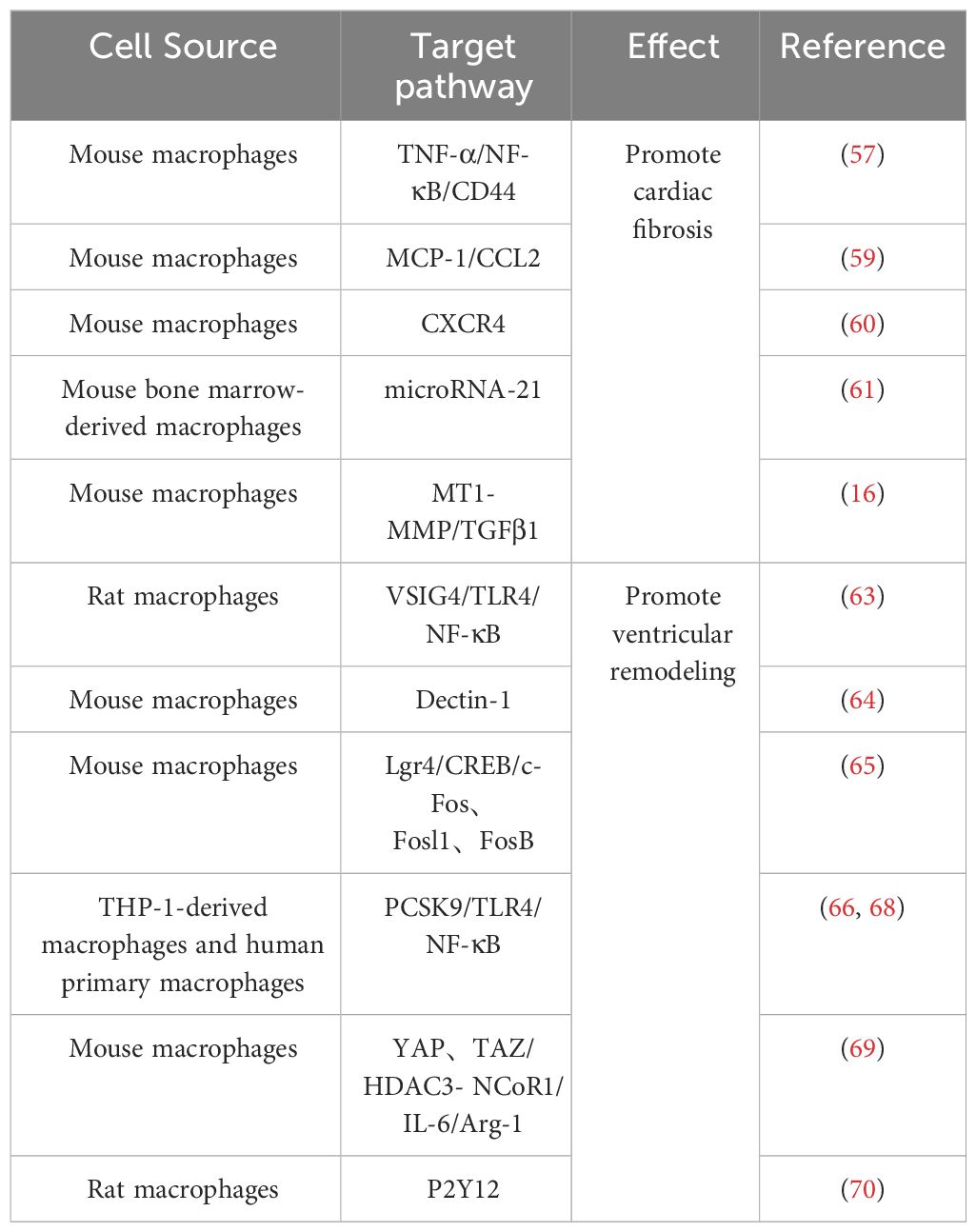
Table 1 Effect of M1 macrophage on IHD.
2.2 The Role of M2 macrophages in IHD
Following early MI, cardiac-resident macrophages become depleted, which promotes adverse cardiac remodeling in the peri-infarct area and severely impairs cardiac function. However, activated M2 macrophages, similar to cardiac-resident macrophages, have the capability to promote tissue repair and regulate the homeostasis of the myocardial microenvironment, thereby exerting crucial cardioprotective functions (Table 2) (25, 78).
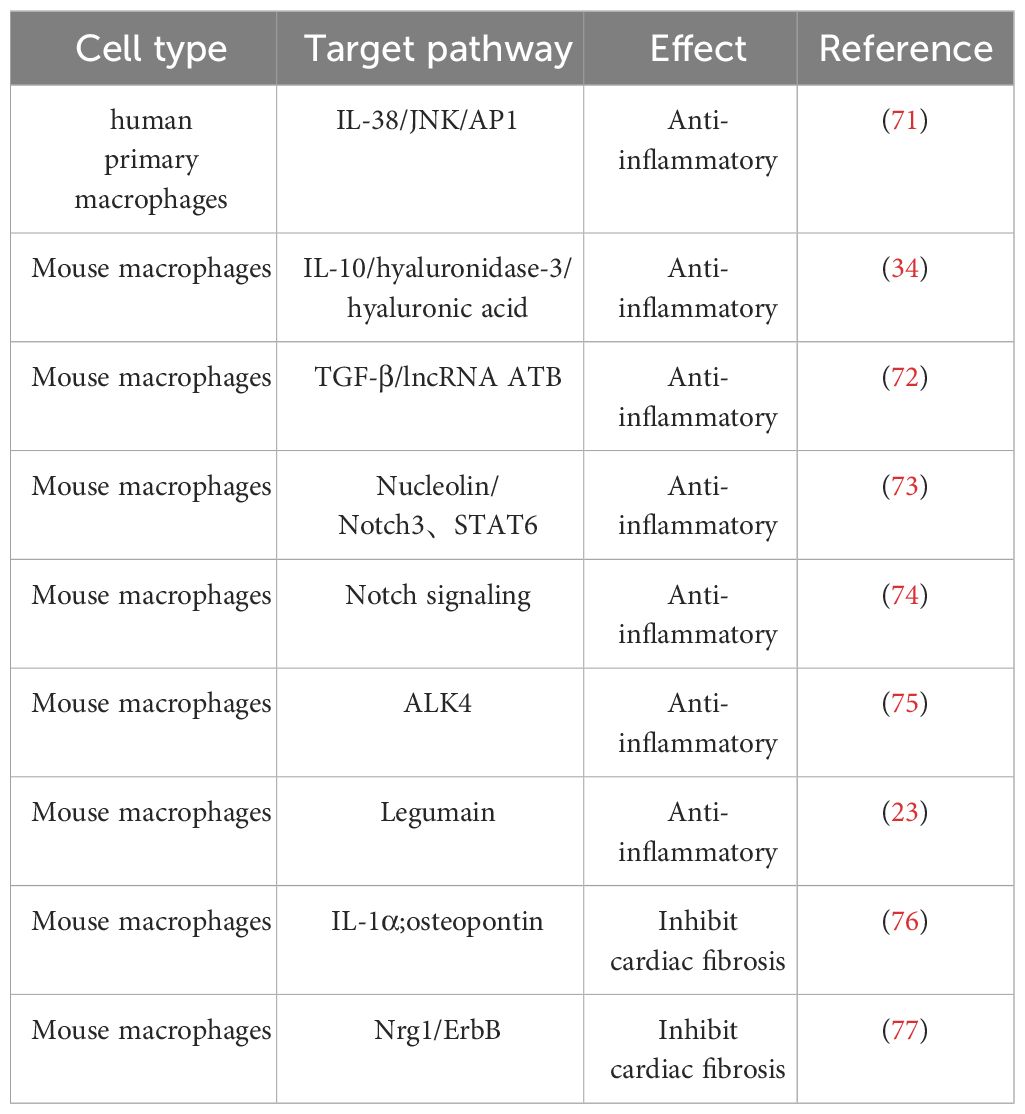
Table 2 Effect of M2 macrophage on IHD.
2.2.1 Involvement of M2 macrophages in the anti-inflammatory response after MI
M2 macrophages safeguard the ischemic heart by secreting anti-inflammatory factors, including IL-10, IL-38, and transforming growth factor-β (TGF-β), thereby serving as potent anti-inflammatory agents. Among them, IL-10 can promote the polarization of macrophages to M2 macrophages and improve the cardiac microenvironment through M2 macrophage-dependent hyaluronidase-3/hyaluronic acid degradation mechanism to further subdue the inflammatory response and promote myocardial tissue healing (34). IL-38, a newly discovered member of the IL-1 family, de-activates the c-jun N-terminal kinase/activator protein 1 (JNK/AP1) pathway by binding to interleukin one receptor helper-like protein one and increases IL-36 production, regulates dendritic cell-induced cardiac regulatory T cells, thereby modulating macrophage polarization and improving myocardial post-infarction ventricular remodeling (71). In contrast to IL-10 and IL-38, a recent study has shown that transforming growth factor-β (TGF-β) can reduce cardiomyocyte inflammatory response, oxidative stress, and cell apoptosis through activation of the long non-coding RNA ATB, providing multiple avenues to alleviate cardiac I/R injury (72).
In addition to cytokines, a variety of proteins play important roles in the anti-inflammatory process of M2 macrophages. Legumain, a gene specifically expressed in M2 macrophages, is involved in the post-myocardial infarction (MI) inflammatory response by upregulating IL-10 and TGF-β, while downregulating IL-1β, TNF-α, and IL-6 (23). Similar cardioprotective proteins include nucleolin, which was previously found to significantly attenuate myocardial I/R injury by promoting myocardial angiogenesis and reducing cardiomyocyte apoptosis by Tang et al (79). Furthermore, their subsequent investigation revealed a substantial decrease in nucleolin expression during the early stage of MI, followed by an increase during the later stage. This upregulation of nucleolin, facilitated by the key regulatory factors notch homolog 3 (Notch3) and signal transducer and activator of transcription 6 (STAT6), promotes M2 macrophage polarization, thus contributing to the anti-inflammatory response (80). It is worth noting that the role of the Notch signaling pathway in ischemic myocardium is dual-edged. Activation of the Notch pathway has been shown to suppress ventricular remodeling in MI rats, but overexpression of Notch signaling may have fibrotic effects on cardiac fibrosis (73, 81, 82). Besides, another study has demonstrated that blockade of the Notch signaling pathway promotes M2 polarization of cardiac macrophages and improves cardiac function by inhibiting imbalanced fibrotic remodeling after MI (74).
Promoting the differentiation of macrophages to M2 macrophages by regulating the expression of specific genes in mice has emerged as a target for modulating the anti-inflammatory response. A study by Yuli Yang et al. found that inhibition of ALK4 gene expression in mice significantly inhibited the secretion of inflammatory factors by M1 macrophages while inducing a phenotypic switch from pro-inflammatory M1 macrophages to anti-inflammatory M2 macrophages and ultimately promoting cardiac repair after myocardial injury (75).
2.2.2 Role of M2 macrophages on myocardial fibrosis after MI
M2 macrophages exhibit anti-inflammatory and tissue repair properties by producing high levels of anti-inflammatory cytokines and promoting fibroblast progenitor cell proliferation and differentiation, playing a crucial role in CFs-mediated myocardial repair (50). Analogous to macrophages, these CFs adopt a pro-inflammatory phenotype soon after MI, after which they differentiate into MFs, which secrete anti-inflammatory factors and extracellular matrix proteins to repair and stabilize cardiac tissue (83). M2 macrophages can secrete IL-1α and osteopontin to activate CFs and promote their transformation into MFs, thereby forming more supportive fibrous tissue at the infarct site and repairing the vulnerable ventricular wall of the infarcted heart (76). In addition to promoting CF activation, M2 macrophages inhibit CF senescence and apoptosis by activating the neuroglial protein 1 (Nrg1)/epidermal growth factor receptor (ErbB) pathway (77).
As the exploration of macrophage functions progresses, the classification of M1/M2 appears simplistic and broad. Mosser and Edwards proposed a classification system for macrophage function, categorizing macrophages into classical activation, regulatory, and repair subtypes (84). Further in vitro experiments have subdivided M2 macrophages into four subgroups, including M2a, M2b, M2c, and M2d (85). Among them, M2a macrophages express high levels of fibrogenic factors, contributing to repair during early stages of injury, while M2b macrophages are defined as regulatory cells with potent immunomodulatory and anti-inflammatory effects (85, 86). In vitro experiments by Yue et al. demonstrated that M2a macrophages significantly promoted the proliferation and migration of CFs, the expression of fibrosis-associated proteins, and the differentiation to MFs, whereas M2b macrophages had the exact opposite effect on CFs, with a significant antifibrotic effect (86, 87). Subsequently, they evaluated the effects of M2b macrophage transplantation using a rat I/R model, confirming that M2b macrophages can reduce cardiac fibrosis, improve heart function significantly by inhibiting the mitogen-activated protein kinase (MAPK) signaling pathway, and decreasing the activation of platelet-derived growth factor receptors (PDGFRs) in CFs (88).
In addition to the common regulation of cardiac fibrosis through modulating CFs, M2b macrophages can also regulate the fibrotic process by affecting the lymphatic system. Lymphatic vessels in the heart drain interstitial fluid to maintain cardiac homeostasis, and previous studies have shown that exogenous vascular endothelial growth factor C (VEGFC) can stimulate lymphangiogenesis in the heart, thereby alleviating myocardial edema and fibrosis after MI (89). Recent research has revealed that transplantation of M2b macrophages upregulates the expression of VEGFC and vascular endothelial growth factor receptor 3 (VEGFR3) in the hearts of I/R rats, promoting lymphangiogenesis to reduce myocardial fibrosis and improve cardiac function (90). Taken together, this series of studies demonstrates the potential value of macrophage therapy in heart disease. Further investigations are needed to explore the complexity of macrophage subtypes and differentiation, as well as the interplay between different signaling pathways.
2.3 Macrophage phenotype switching intimately linked to metabolic responses Resource Identification Initiative
In IHD, the phenotypic polarization and metabolic changes of macrophages recruited into circulation and residing in tissues can disrupt the M1/M2 homeostasis, thereby impacting the balance of cardiac inflammatory effects and determining disease regression and prognosis (37, 38). The polarization and activation of macrophages are closely associated with metabolic reprogramming, which is manifested as a bias in energy utilization, thereby altering their inflammatory phenotype (91). Macrophages primarily modulate their inflammatory phenotype through four energy cycles, including: 1) glycolysis, 2) oxidative phosphorylation (OXPHOS), 3) tricarboxylic acid cycle (TCA), and 4) fatty acid oxidation (91, 92). Under normal physiological conditions, macrophages are intimately integrated to tissue and organismal metabolism (93). M1 macrophages rely predominantly on glycolysis, accelerating glucose transport by upregulating the glucose transporter protein GLUT1 to meet the demands of rapid ATP production, whereas M2 macrophages utilize fatty acids as a fuel source for TCA and subsequent OXPHOS (94, 95).
In IHD, changes in cardiac metabolism may disrupt M1/M2 homeostasis, which in turn affects the cardiac inflammatory response and further influences disease regression and prognosis. Studies have shown that ischemia leads to an increased dependence of cells on glycolysis, while hypoxia induces pro-inflammatory gene expression and metabolic reprogramming towards glycolysis (96, 97). Following myocardial tissue injury, macrophages will activate a series of receptor tyrosine kinases such as Tyro3, Axl, and MerTK, which mediate the clearance of apoptotic cells and regulate the production of inflammatory cytokines (98). Among these, cross-signaling between AX1 and TLR4 transduces to glycolytic metabolism and pro-inflammatory IL-1β secretion, leading to an increased inflammatory response within the myocardium, unfavorable ventricular remodeling and impaired contractile function (99). Glycolytic metabolism promotes macrophage-induced fibrosis, whereas inhibition of glycolysis facilitates the restoration of macrophage energy metabolism from glycolysis to normal OXPHOS pathway under a normoxic state, further blocking M1 polarization and thus improving the condition of ischemic cardiomyopathy (100–102). This undoubtedly provides excellent targets for targeted therapies to inhibit inflammation, for example, Zhao et al. used salvianolic acid B to inhibit mammalian target of rapamycin 1 (mtorc1)-induced glycolysis, which reduced myocardial M1 macrophages and increased M2 macrophages in mice at 3 days after I/R and reduced collagen deposition and improved cardiac dysfunction at 7 days after I/R (103). Notably, glycolysis does not have only negative effects. Early endogenous glycolytic reprogramming after MI can promote the transcription of reparative genes by promoting histone demethylation in monocytes, thereby improving cardiac function after MI (104).
Metabolic shifting between glycolysis and mitochondrial OXPHOS is an important mechanism for the transition of macrophages to reparative phenotype, and a timely transition of M1 macrophages to M2 macrophages would contribute to myocardial repair (105). On the first day after MI, macrophages polarize towards the M1 phenotype, exhibiting distinct pro-inflammatory and extracellular matrix degradation characteristics. However, by the third day post-MI, macrophage proliferation and phagocytic capacity increase, accompanied by upregulation of genes associated with mitochondrial function and OXPHOS, indicating metabolic reprogramming (106). In diseased states, this metabolic transition is influenced by multiple factors. For instance, comprehensive metabolomic analysis has indicated that the TCA cycle may be interrupted during the inflammatory process, resulting in selective accumulation of intermediates including succinate, which further impacts oxidative phosphorylation and thus the metabolic switch of macrophages. Additionally, the further oxidation of succinate can drive the generation of a large amount of reactive oxygen species (ROS), exacerbating oxidative damage (100, 101, 107, 108). Recent studies have shown that activated macrophages can produce itaconate to inhibit succinate oxidation and regulate M2 macrophage polarization, indicating that macrophages possess certain metabolic regulation capabilities (109). Moreover, alterations in the cardiac microenvironment exert a profound impact on macrophage metabolism, while the activity of macrophages reciprocally influences the heart. For instance, IL-33 can be released by various cells following necrosis. It not only induces macrophage reprogramming, leading to uncoupling of the mitochondrial respiratory chain and increased production of the mitochondrial-derived metabolite itaconate, thus promoting the resolution of inflammation and initiation of tissue damage repair; but also activates the JAK/STAT signaling pathway to induce M2 macrophage polarization, thereby impeding the progression of cardiac fibrosis and improving cardiac systolic and diastolic function (110, 111). Indeed, Liu et al. demonstrated that in vitro induced M2 macrophages could be transplanted into hearts of heart failure mice models, confirmed that M2 macrophages can transfer mitochondria to damaged cardiomyocytes, which promote cell survival under stress conditions and alleviate cardiac fibrosis and cardiomyocyte apoptosis (112).
Currently, there have been relevant studies on treating diseases by regulating the polarization state and functions of macrophages at the metabolic level, showing promising therapeutic benefits (95). For example, Chen et al. demonstrated that supplementing with ω-alkynyl arachidonic acid can inhibit glycolysis and promote M2 macrophage polarization, leading to reduced infarct size, prevention of the development of left ventricular dysfunction, and improved clinical outcomes in a mouse model of MI (113). Further investigation will contribute to unraveling the intricate interplay between macrophage energy metabolism and the cardiac microenvironment and deepen our understanding of this dynamic change, advancing the development of relevant therapeutic strategies.
2.4 Macrophage phenotype switching intimately linked to microenvironment
Macrophage polarization plays a pivotal role in host defense and tissue repair, with its regulation extending beyond metabolic cues to encompass various microenvironmental factors such as cytokines, cell receptors, and microRNAs. Cytokines, notably, are key determinants in the transition from M1 to M2 macrophage phenotypes (114). Exemplifying this, IL-4 orchestrates the shift from M1 to M2 via JAK1/STAT6 signaling (115). Additionally, interferon regulatory factors (IRFs) exert critical control over macrophage phenotypic polarization, transition, and function. While IRF-1, IRF-5, and IRF-8 contribute to pro-inflammatory M1 formation, IRF-3 and IRF-4 govern M2 polarization (116). Furthermore, insulin-like growth factor 2 mRNA-binding protein 2 (IGF2BP2) mediates the conversion of M1 to M2 macrophages through an m6A-dependent mechanism targeting scleroderma-related protein 1 (117). Insulin has also been found to modulate macrophage polarization by activating the PI3K/Akt/Rac-1 and PPAR-γ signaling pathways (118). Cell surface receptor proteins also participate in M1 to M2 transition; for instance, dual blockade of TLR4 and TNFR1 promotes M1 to M2 polarization (119). MicroRNAs, like miR-126, play a pivotal role in shifting macrophages from pro-inflammatory M1 to anti-inflammatory M2 phenotype by downregulating VEGFA and KLF4 expression (120). In summary, macrophage polarization is intricately regulated by diverse factors, collectively shaping cellular function and phenotypic transitions.
2.5 M1/M2 macrophages phenotype correlates with cardiac rupture post-MI
Following MI, the heightened inflammatory response poses a significant risk of cardiac rupture (121, 122). Notably, clinical evidence underscores a direct correlation between the intensity of inflammation and the incidence of cardiac rupture in MI patients (123, 124). The risk of cardiac rupture post-MI is primarily associated with neutrophils, M1 macrophages, M2 macrophages, myeloperoxidase (MPO), and matrix metalloproteinases (MMPs), especially the infiltration of macrophages (125, 126). In the aftermath of MI, macrophages exhibit a biphasic activation pattern: pro-inflammatory M1 macrophages peak within the initial 3 days, while pro-fibrotic/repairing M2 macrophages reach their zenith around day 7 post-MI (127). Building upon this understanding, recent investigations have sought to elucidate the distinct roles played by different macrophage subtypes in modulating the risk of cardiac rupture. One study found that administration of the natural tetrapeptide Acetyl-Ser-Asp-Lys-Pro (Ac-SDKP) reduced the number of M1 macrophages in cardiac tissue post-MI, thereby significantly decreasing the incidence of cardiac rupture (128). Another study promoted apoptosis of M1 macrophages by knocking out apoptosis inhibitor of macrophage (AIM), consequently reducing the occurrence of cardiac rupture post-MI (129). Moreover, MMP-28 has garnered attention for its ability to augment the activation of M2 macrophages, thereby exerting a protective effect against cardiac rupture following MI (130).Furthermore, studies focusing on macrophage-specific Lgr4 deletion have unveiled a compelling mechanistic link, wherein ablation of this receptor culminates in a discernible shift in macrophage subtype composition within the infarcted milieu, characterized by a decrease in M1 macrophages juxtaposed with an augmentation in M2 macrophages, ultimately translating into a lowered incidence of cardiac rupture (65). Collectively, these findings underscore the pivotal role played by the delicate balance between M1 and M2 macrophages in dictating the susceptibility to cardiac rupture post-MI.
In summary, a comprehensive understanding of the intricate immunological landscape post-MI, with a specific focus on the nuanced modulation of macrophage subpopulations, holds immense promise in delineating novel therapeutic strategies aimed at mitigating the risk of cardiac rupture. These insights not only deepen our appreciation of the pathophysiological underpinnings of post-MI complications but also pave the way for the development of targeted interventions with translational potential, heralding a new dawn in the realm of cardiovascular medicine.
3 The role of different phenotypic macrophage-derived exosomes in IHD
Exosomes carry a cargo of proteins, RNA, DNA, lipids, and metabolites (such as amino acids, ATP, and acylamide) from the cell surface and interior. The types and levels of exosome cargo are influenced by donor cells, microenvironment, or physiological conditions. Through endocytosis, direct membrane fusion, or binding to cell surface receptors, exosomes can effectively target and deliver these biomolecules carrying important information to recipient cells, playing multifaceted roles in altering cell phenotypes, regulating gene expression, controlling the recruitment of inflammatory cells, etc., and participating in the pathogenesis and development of IHD (36, 131–133). Exosomes mediate signal exchanges between immune cells and cardiomyocytes. Studying the exosome cargo and their functions not only provides insight into the communication between cells in healthy and diseased states but also lays a foundation for the clinical application of exosomes. (Table 3).
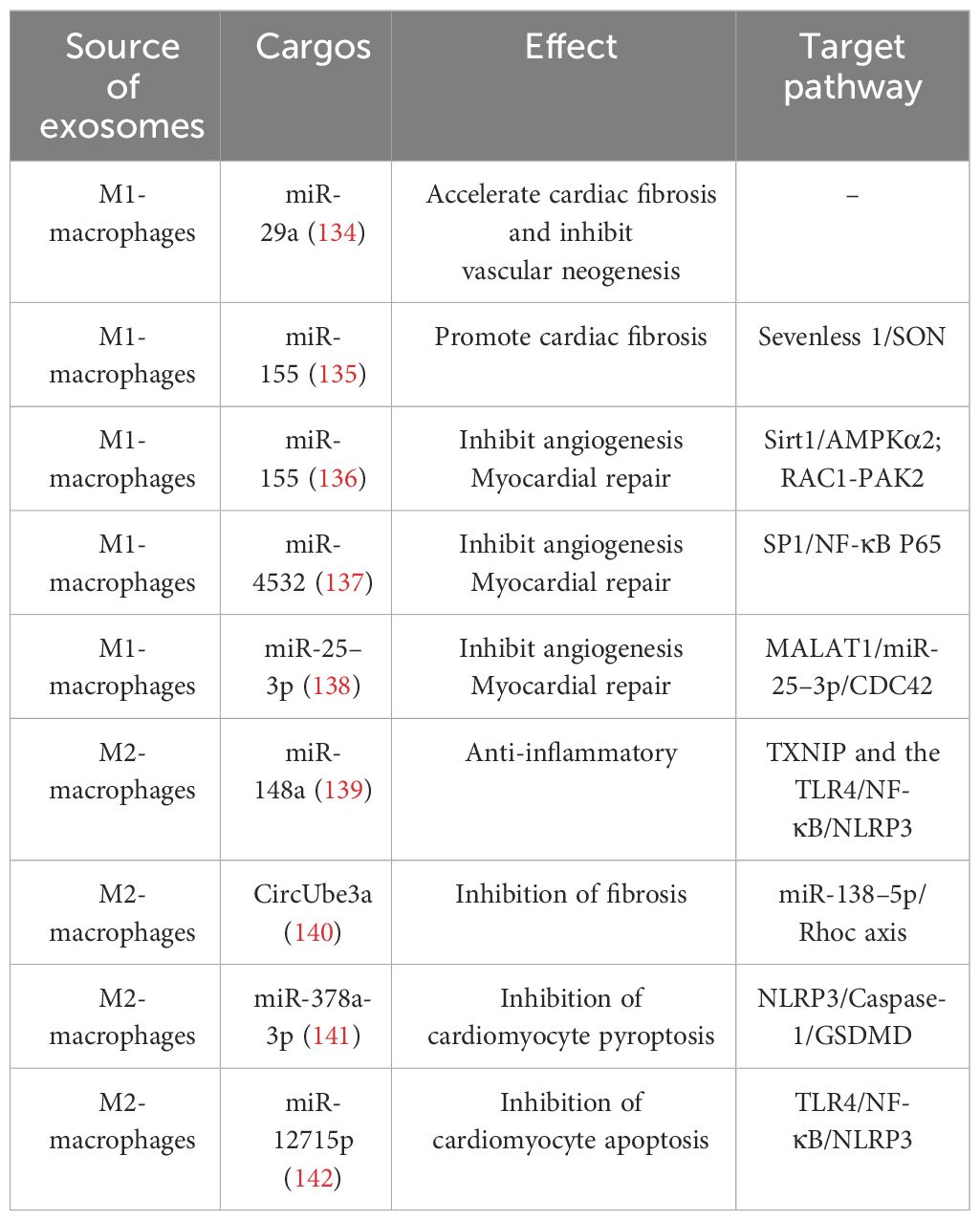
Table 3 Effect of macrophage-derived exosomes on IHD.
3.1 Role of M1 macrophage-derived exosomes in IHD
Previous studies have primarily focused on the role of M1 macrophage-derived exosomes in promoting inflammatory responses; in fact, it also plays an important crosstalk role in mediating between macrophages and cardiac cells. In response to hypoxia/reoxygenation stimulation, miR-29a in exosomes secreted by activated M1 macrophages mediated cardiomyocyte pyroptosis (134). Through paracrine effects, M1 macrophage-secreted exosomes are well-targeted to CFs and ECs to accelerate cardiac fibrosis and inhibit vascular neogenesis. Exosomes derived from M1 macrophages exhibit high expression of miRNA-155, which acts as a paracrine regulatory factor for CF proliferation and inflammation. Through exosome-mediated targeting of CFs, miRNA-155 can down-regulate the expression of Son of Sevenless gene (Sos1) to inhibit fibroblast proliferation, and decrease the expression of the anti-inflammatory gene Suppressor of Cytokine Signaling 1 (Socs1) to accelerate the inflammatory response of CFs (135). As a typical multifunctional miRNA, miRNA-155 can simultaneously target multiple molecular nodes. When transferred to ECs through exosomes derived from M1 macrophages, miRNA-155 can inhibit the Sirtuin 1 (Sirt1)/protein kinase AMP-activated catalytic subunit alpha 2 (AMPKα2)-endothelial nitric oxide synthase and Rac family small GTPase 1 (RAC1)-p21 (RAC1)-activated kinase 2 (PAK2) signaling pathways, thereby reducing the angiogenic capacity of ECs and impairing cardiac healing (136). M1 macrophage-derived exosomes can also exacerbate ECs injury by targeting the transport of miR-4532 and activating the SP1 and NF-κB P65 signaling pathways (137). Additionally, the highly expressed lncRNA Metastasis-Associated Lung Adenocarcinoma Transcript 1 (MALAT1) in exosomes secreted by M1 macrophages can competitively bind with miR-25–3p in ECs, promoting the expression of Cell Division Cycle Protein 42 (CDC42), which in turn activates the Mitogen-Activated Protein Kinase (MEK)/Extracellular Signal-Regulated Kinases (ERK) pathway and inhibits angiogenesis and myocardial regeneration (138).
3.2 Role of M2 macrophage-derived exosomes in IHD
M2 macrophage-derived exosomes account for an essential part of the therapeutic role played by M2 macrophages. They carry miR-148a that inhibits the expression of thioredoxin-interacting protein (TXNIP) and inactivates the TLR4/NF-κB/NLRP3 inflammasome signaling pathway, thus playing a cardioprotective role (139). They also mediate CF proliferation, migration, and MF transformation by transferring CircUbe3a to recipient cells targeting the miR-138–5p/Rhoc axis (140). Concurrently, exosomes derived from M2 macrophages can regulate the death program of cardiomyocytes through various mechanisms, with numerous miRNAs playing a role. Concurrently, exosomes derived from M2 macrophages can regulate the death program of cardiomyocytes through various mechanisms, with numerous miRNAs playing a role. Specifically, miR-1271–5p reduces the apoptosis of cardiomyocytes by downregulating the expression of SOX6, whereas miR-378a-3p mitigates cardiomyocyte pyroptosis by inhibiting the expression of ELAVL1, thus destabilizing the NLRP3 inflammasome and subsequently blocking the activation of the NLRP3/Caspase-1/GSDMD pathway (141, 142). In summary, exosomes derived from M2 macrophages can mitigate the damage caused by IHD and improve the prognosis of the disease through various pathways, such as alleviating inflammatory injury, reducing cardiomyocyte death, and promoting cardiac repair. Therefore, they hold promise as potential sources for therapeutic exosomes.
4 The therapeutic potential of macrophages and their exosomes in IHD
Macrophages and their exosomes play important roles in the progression of IHD at different stages. M1 macrophages primarily mediate early inflammatory responses, while M2 macrophages regulate the cardiac repair process. As crucial mediators of intercellular communication, exosomes have significant roles in modulating immune responses and facilitating communication between macrophages, cardiac cells, and the microenvironment. Further exploration of the functions and specific mechanisms of macrophages and their exosomes holds promise for uncovering their potential therapeutic applications.
In recent years, regenerative medicine has been widely applied in the treatment of IHD. The efficacy of stem cell therapy, particularly mesenchymal stem cell (MSC) therapy, has been confirmed by numerous preclinical and clinical studies (143, 144). There is also abundant preclinical evidence supporting the use of exosome therapy in animal models (145). Macrophages, due to their specific functional roles in the pathological process of IHD, often serve as target cells in related research. For instance, the studies conducted by Deng et al. and Xu et al. explored the therapeutic mechanisms of MSC-derived exosomes in the treatment of MI, although they act through different signaling pathways, both studies demonstrated that they improved cardiac injury by modulating macrophage phenotypic polarization (146, 147). There have also been studies on directly transplanting macrophages for the treatment of IHD. As mentioned earlier, transplantation of M2 macrophages has shown beneficial effects in reducing cardiac fibrosis and improving heart function in both I/R rats and heart failure mice models (88, 112). Macrophages have the advantage of an innate ability to migrate and settle into damaged tissue, which contributes to the functional implantation of transplanted cells in the damaged heart (78). However, cell therapy itself faces challenges such as poor recruitment and survival rates after transplantation into ischemic hearts. Additionally, macrophage transplantation presents difficulties in maintaining activated macrophages and has lower clinical feasibility (148). Pretreatment of macrophages may be able to partially address these issues. Chen et al. pre-treated bone marrow-derived macrophages with a sodium-dependent glucose transporter 2 inhibitor (SGLT2i) before transplantation into a mouse model of MI. This resulted in the suppression of inflammation, reduction of myocardial cell apoptosis, and promotion of the transformation of native cardiac macrophages into the M2 phenotype, which contributed to the reduction of adverse ventricular remodeling after MI (149). Similarly, Podaru et al. stimulated bone marrow-derived monocytes with macrophage colony-stimulating factor (M-CSF) and IL-4 to induce their differentiation into M2 macrophages before transplantation, resulting in significant improvement in cardiac function and structure in MI mice. The highlight of this study is that the generated M2 macrophages not only enhance the reparative secretion profile of endogenous reparative macrophages but also possess good stability, maintaining an M2-like phenotype even in the inflammatory environment after MI (150). Overall, macrophage transplantation for the treatment of IHD has accumulated some preclinical research data. However, further optimization of cell delivery routes and studies involving the use of human cells are needed before entering clinical trials. In comparison, a more convenient alternative may be the direct delivery of exosomes that carry the therapeutic functions of parent cells. For macrophages, blocking the transport of M1 macrophage-derived exosomes and promoting targeted delivery of M2 macrophage-derived exosomes, or promoting macrophage transformation from the M1 to M2 phenotype through the uptake of exogenous exosomes can all contribute to the repair of damaged myocardial tissue.
Researchers have further developed and optimized exosomes based on their characteristics. In terms of the therapeutic benefits of exosomes themselves, although exosomes exhibit good targeting ability, they have low persistence, and the use of biologics that can prolong their duration of action would enhance their efficacy. Biomaterials, such as hydrogels, have been widely employed in the delivery of exosomes due to their excellent biocompatibility, stability, and mechanical properties, which effectively extend the duration of action and even enhance therapeutic effects (151). For instance, Zou et al. constructed a composite system called Gel@Exo by combining conductive hydrogel with umbilical cord MSC-derived exosomes to improve their therapeutic effects on MI. This composite system offers advantages such as controllable gel kinetics, injectability, conductivity matching with natural myocardium, adaptability to heartbeats, softness, dynamic stability, and good cellular compatibility. It significantly improves the retention time of exosomes in the heart and optimizes their therapeutic effects (152). Furthermore, engineering modifications of exosomes can enhance their stability, bioactivity, and target binding capability at both cellular and tissue-specific levels, thereby further improving their therapeutic efficacy for diseases (153, 154). Techniques such as pre-treating parent cells and incorporating self-assembling peptides into the exosomes membrane allow the generated exosomes to better cope with the complex physiological environment of ischemic hearts (155, 156).
5 M1/M2 macrophages and their exosomes in clinical application
Clinical trials of macrophages and their exosomes for the treatment of cardiovascular diseases are still in the early stages, but preliminary results have shown promise. In 2013, Perin et al. (NCT00824005) significantly improved the left ventricular ejection fraction (LVEF) of patients with chronic ischemic heart disease by administering non-expanded autologous bone marrow macrophages via transendocardial injections. This trial demonstrated the potential positive outcomes of ex vivo expansion of macrophages for cardiac repair in patients with chronic ischemic heart disease (157). Further investigations revealed a total of 11 completed clinical studies on cell therapy using macrophages, focusing on conditions such as cardiomyopathy, arterial diseases, tumors, and other ailments. Among them, three trials (NCT01670981, NCT01020968, and NCT00765518) involved intramyocardial injection of Ixmyelocel-T, which contained a mixture of macrophages, granulocytes, monocytes, mixed myeloid progenitor cells, lymphocytes, and mesenchymal stem/stromal cells, to treat heart failure due to ischemic dilated cardiomyopathy. These studies showed a reduction in major adverse cardiovascular events and improvement in symptoms, attributing the effectiveness to the M2 macrophages in Ixmyelocel-T. These M2 macrophages were found to be effective in removing apoptotic cells, limiting tissue damage, and promoting wound healing, confirming the efficacy of macrophage therapy (158, 159).Additionally, two studies on autologous M2 macrophage therapy in children with severe cerebral palsy and patients with non-acute stroke confirmed the clinical efficacy of M2 macrophages in promoting neurological recovery without serious adverse events or cellular rejection (160, 161), further demonstrating the safety and efficacy of this approach.
As for exosome-related clinical trials, research has focused on exosomes secreted by plasma, mesenchymal stem cells, and human induced pluripotent stem cells for conditions like cancer, myocardial infarction, and decompensated cirrhosis. However, there have been no studies on exosomes secreted by M1 and M2 macrophages for the treatment of ischemic heart disease (IHD) yet. Nonetheless, a clinical trial investigating the phenotype of macrophages and their exosomes in type 2 diabetic patients who develop myocardial infarction (NCT02768935) suggested that the treatment of IHD with macrophages and their exosomes was progressing toward clinical therapy.
Overall, while there are fewer clinical trials involving macrophages and their exosomes in IHD, the available data have laid the initial groundwork for the development of new therapeutic strategies. As more studies are conducted, we can expect to gather more clinical evidence regarding the potential use of macrophages and their exosomes in the treatment of cardiovascular diseases.
6 Perspective
From the perspective of the delivery characteristics of exosomes, as endogenous extracellular vesicles, they possess excellent blood stability, substance transport properties, and high targeting ability, making them promising candidates as nanocarriers for drug delivery (162–165). This has the potential to overcome the limitations associated with traditional treatments for CVDs, such as low bioavailability, poor retention, inadequate targeting, and complex drug resistance. By utilizing methods such as ultrasound, transfection, incubation, transgenesis, freeze-thaw cycles, and heat shock, drugs can be loaded into exosomes for delivery, aiming to achieve therapeutic effects and improve prognosis (162). The study by Gao et al. utilized macrophage-derived exosomes as a nanoplatform for loading the anti-inflammatory drug methylprednisolone acetate, enhancing its anti-inflammatory and antioxidant effects, rescuing the viability of an in vitro inflammatory cardiomyocyte model, and providing preliminary evidence for the effectiveness of macrophage-derived exosomes as a drug carrier for cardiac immunotherapy (166). On the other hand, due to their abundant cell sources and diverse cargo, exosomes exhibit complex functional heterogeneity, which presents tremendous potential in clinical applications, including disease diagnosis. For instance, several studies have indicated that non-coding RNAs, such as miR-183, miR-21, and miR-208a, found in circulating extracellular vesicles, are closely associated with myocardial ischemic injury and hold promise as future biomarkers for IHD (167–169).
7 Conclusion
In summary, macrophages and their exosomes actively participate in and regulate the progression of IHD, potentially serving as key therapeutic tools for IHD treatment in the future. Additionally, ongoing optimization of cell therapy and advanced engineering techniques for exosomes are driving the development of related therapeutic strategies. Further exploration of the functions and mechanisms of macrophages and their exosomes will contribute to a deeper understanding of the pathophysiological mechanisms underlying IHD and provide new insights for its treatment.
Author contributions
MW: Visualization, Writing – original draft, Writing – review & editing. CL: Writing – original draft, Writing – review & editing. YL: Writing – original draft, Writing – review & editing. YJ: Visualization, Writing – review & editing. YY: Writing – review & editing. XT: Writing – review & editing. CZ: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82030007, 2020), Science and Technology Project of Sichuan Province [23NSFSC1345, 2022YFS0614, 2022YFS0578, 2023NSFSC0620].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
2. Jensen RV, Hjortbak MV, Bøtker HE. Ischemic heart disease: an update. Semin Nucl Med. (2020) 50:195–207. doi: 10.1053/j.semnuclmed.2020.02.007
3. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. (2020) 116:1101–12. doi: 10.1093/cvr/cvz336
4. Zhang Z, Xu Y, Cao C, Wang B, Guo J, Qin Z, et al. Exosomes as a messager to regulate the crosstalk between macrophages and cardiomyocytes under hypoxia conditions. J Cell Mol Med. (2022) 26:1486–500. doi: 10.1111/jcmm.17162
5. Gimbrone M, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
6. Saleh M, Ambrose JA. Understanding myocardial infarction. F1000Research. (2018) 7:F1000 Faculty Rev-1378. doi: 10.12688/f1000research
7. Benjamin E, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: A report from the American heart association. Circulation. (2019) 139:e56–e528. doi: 10.1161/CIR.0000000000000659
8. Bayer AL, Alcaide P. MyD88: At the heart of inflammatory signaling and cardiovascular disease. J Mol Cell Cardiol. (2021) 161:75–85. doi: 10.1016/j.yjmcc.2021.08.001
9. Cai S, Zhao M, Zhou B, Yoshii A, Bugg D, Villet O, et al. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J Clin Invest. (2023) 133(4):e159498. doi: 10.1172/JCI159498
10. Akhmerov A, Parimon T. Extracellular vesicles, inflammation, and cardiovascular disease. Cells. (2022) 11(14):2229. doi: 10.3390/cells11142229
11. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. (2016) 119:91–112. doi: 10.1161/CIRCRESAHA.116.303577
12. Kologrivova I, Shtatolkina M, Suslova T, Ryabov V. Cells of the immune system in cardiac remodeling: main players in resolution of inflammation and repair after myocardial infarction. Front Immunol. (2021) 12:664457. doi: 10.3389/fimmu.2021.664457
13. Ong SB, Hernández-Reséndiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, et al. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. (2018) 186:73–87. doi: 10.1016/j.pharmthera.2018.01.001
14. Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res. (2014) 115:284–95. doi: 10.1161/CIRCRESAHA.115.303567
15. Grune J, Yamazoe M, Nahrendorf M. Electroimmunology and cardiac arrhythmia. Nat Rev Cardiol. (2021) 18:547–64. doi: 10.1038/s41569-021-00520-9
16. Alonso-Herranz L, Sahún-Español Á, Paredes A, Gonzalo P, Gkontra P, Núñez V, et al. Macrophages promote endothelial-to-mesenchymal transition via MT1-MMP/TGFβ1 after myocardial infarction. Elife. (2020) 9:e57920. doi: 10.7554/eLife.57920
17. Bonaventura A, Montecucco F, Dallegri F. Cellular recruitment in myocardial ischaemia/reperfusion injury. Eur J Clin Invest. (2016) 46:590–601. doi: 10.1111/eci.12633
18. Wu Q, Yao Q, Hu T, Yu J, Jiang K, Wan Y, et al. Dapagliflozin protects against chronic heart failure in mice by inhibiting macrophage-mediated inflammation, independent of SGLT2. Cell Rep Med. (2023) 4:101334. doi: 10.1016/j.xcrm.2023.101334
19. Davies LC, Rosas M, Jenkins SJ, Liao CT, Scurr MJ, Brombacher F, et al. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat Commun. (2013) 4:1886. doi: 10.1038/ncomms2877
20. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
21. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. (2014) 40:91–104. doi: 10.1016/j.immuni.2013.11.019
22. Jung K, Kim P, Leuschner F, Gorbatov R, Kim JK, Ueno T, et al. Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ Res. (2013) 112:891–9. doi: 10.1161/CIRCRESAHA.111.300484
23. Jia D, Chen S, Bai P, Luo C, Liu J, Sun A, et al. Cardiac resident macrophage-derived legumain improves cardiac repair by promoting clearance and degradation of apoptotic cardiomyocytes after myocardial infarction. Circulation. (2022) 145:1542–56. doi: 10.1161/CIRCULATIONAHA.121.057549
24. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell. (2017) 169:510–522.e20. doi: 10.1016/j.cell.2017.03.050
25. Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. (2019) 20:29–39. doi: 10.1038/s41590-018-0272-2
26. Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. (2018) 24:1234–45. doi: 10.1038/s41591-018-0059-x
27. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, et al. Tissue resident CCR2- and CCR2+ Cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. (2019) 124:263–78. doi: 10.1161/CIRCRESAHA.118.314028
28. Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. (2014) 114:1611–22. doi: 10.1161/CIRCRESAHA.114.303204
29. Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res. (2014) 102:240–8. doi: 10.1093/cvr/cvu025
30. Li Z, Liu X, Zhang X, Zhang W, Gong M, Qin X, et al. TRIM21 aggravates cardiac injury after myocardial infarction by promoting M1 macrophage polarization. Front Immunol. (2022) 13:1053171. doi: 10.3389/fimmu.2022.1053171
31. Jiang S, Wang R, Han L, Kuerban K, Ye L, Pan S, et al. Activation of autophagy reverses gemcitabine-induced immune inhibition of RAW264.7 macrophages by promoting TNF-α, IL-6 and MHC-II expression. Immunol Res. (2021) 69:352–62. doi: 10.1007/s12026-021-09210-7
32. Rendra E, Riabov V, Mossel DM, Sevastyanova T, Harmsen MC, Kzhyshkowska J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology. (2019) 224:242–53. doi: 10.1016/j.imbio.2018.11.010
33. Chen Y, Wu G, Li M, Hesse M, Ma Y, Chen W, et al. LDHA-mediated metabolic reprogramming promoted cardiomyocyte proliferation by alleviating ROS and inducing M2 macrophage polarization. Redox Biol. (2022) 56:102446. doi: 10.1016/j.redox.2022.102446
34. Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, et al. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. (2017) 112:33. doi: 10.1007/s00395-017-0622-5
35. Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol. (2018) 19:327–41. doi: 10.1038/s41590-018-0064-8
36. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. (2020) 367(6478):eaau6977. doi: 10.1126/science.aau6977
37. Lee YJ, Shin KJ, Jang HJ, Ryu JS, Lee CY, Yoon JH, et al. GPR143 controls ESCRT-dependent exosome biogenesis and promotes cancer metastasis. Dev Cell. (2023) 58:320–334.e8. doi: 10.1016/j.devcel.2023.01.006
38. Marie PP, Fan SJ, Mason J, Wells A, Mendes CC, Wainwright SM, et al. Accessory ESCRT-III proteins are conserved and selective regulators of Rab11a-exosome formation. J Extracell Vesicles. (2023) 12:e12311. doi: 10.1002/jev2.12311
39. Liu Y, Wang M, Yu Y, Li C, Zhang C. Advances in the study of exosomes derived from mesenchymal stem cells and cardiac cells for the treatment of myocardial infarction. Cell Commun Signal. (2023) 21:202. doi: 10.1186/s12964-023-01227-9
40. Lai JJ, Chau ZL, Chen SY, Hill JJ, Korpany KV, Liang NW, et al. Exosome processing and characterization approaches for research and technology development. Adv Sci (Weinh). (2022) 9:e2103222. doi: 10.1002/advs.202103222
41. Shao H, Im H, Castro CM, Breakefield X, Weissleder R, Lee H. New technologies for analysis of extracellular vesicles. Chem Rev. (2018) 118:1917–50. doi: 10.1021/acs.chemrev.7b00534
42. Akbar N, Azzimato V, Choudhury RP, Aouadi M. Extracellular vesicles in metabolic disease. Diabetologia. (2019) 62:2179–87. doi: 10.1007/s00125-019-05014-5
43. Vader P, Mol EA, Pasterkamp G, Schiffelers RM. Extracellular vesicles for drug delivery. Adv Drug Delivery Rev. (2016) 106:148–56. doi: 10.1016/j.addr.2016.02.006
44. Elsharkasy OM, Nordin JZ, Hagey DW, de Jong OG, Schiffelers RM, Andaloussi SE, et al. Extracellular vesicles as drug delivery systems: Why and how? Adv Drug Delivery Rev. (2020) 159:332–43. doi: 10.1016/j.addr.2020.04.004
45. Wang Z, Zhu H, Shi H, Zhao H, Gao R, Weng X, et al. Exosomes derived from M1 macrophages aggravate neointimal hyperplasia following carotid artery injuries in mice through miR-222/CDKN1B/CDKN1C pathway. Cell Death Dis. (2019) 10:422. doi: 10.1038/s41419-019-1667-1
46. Sherman CD, Lodha S, Sahoo S. EV cargo sorting in therapeutic development for cardiovascular disease. Cells. (2021) 10(6):1500. doi: 10.3390/cells10061500
47. Wu X, Iroegbu CD, Peng J, Guo J, Yang J, Fan C. Cell death and exosomes regulation after myocardial infarction and ischemia-reperfusion. Front Cell Dev Biol. (2021) 9:673677. doi: 10.3389/fcell.2021.673677
48. Bejjani AT, Saab SA, Muhieddine DH, Habeichi NJ, Booz GW, Zouein FA. Spatiotemporal dynamics of immune cells in early left ventricular remodeling after acute myocardial infarction in mice. J Cardiovasc Pharmacol. (2020) 75:112–22. doi: 10.1097/FJC.0000000000000777
49. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. (2014) 11:255–65. doi: 10.1038/nrcardio.2014.28
50. Li Z, Ding Y, Peng Y, Yu J, Pan C, Cai Y, et al. Effects of IL-38 on macrophages and myocardial ischemic injury. Front Immunol. (2022) 13:894002. doi: 10.3389/fimmu.2022.894002
51. Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. (2016) 7:12260. doi: 10.1038/ncomms12260
52. Chen M, Li X, Wang S, Yu L, Tang J, Zhou S. The role of cardiac macrophage and cytokines on ventricular arrhythmias. Front Physiol. (2020) 11:1113. doi: 10.3389/fphys.2020.01113
53. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U.S.A. (2014) 111:16029–34. doi: 10.1073/pnas.1406508111
54. Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest. (2013) 43:986–95. doi: 10.1111/eci.12118
55. Kim Y, Nurakhayev S, Nurkesh A, Zharkinbekov Z, Saparov A. Macrophage polarization in cardiac tissue repair following myocardial infarction. Int J Mol Sci. (2021) 22(5):2715. doi: 10.3390/ijms22052715
56. Xiong YY, Gong ZT, Tang RJ, Yang YJ. The pivotal roles of exosomes derived from endogenous immune cells and exogenous stem cells in myocardial repair after acute myocardial infarction. Theranostics. (2021) 11:1046–58. doi: 10.7150/thno.53326
57. Yang LW, Qin DZ, James E, McKallip RJ, Wang NP, Zhang WW, et al. CD44 deficiency in mice protects the heart against angiotensin ii-induced cardiac fibrosis. Shock. (2019) 51:372–80. doi: 10.1097/SHK.0000000000001132
58. Frangogiannis NG. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. (2019) 65:70–99. doi: 10.1016/j.mam.2018.07.001
59. Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. (2007) 115:584–92. doi: 10.1161/CIRCULATIONAHA.106.646091
60. Zhang N, Ma Q, You Y, Xia X, Xie C, Huang Y, et al. CXCR4-dependent macrophage-to-fibroblast signaling contributes to cardiac diastolic dysfunction in heart failure with preserved ejection fraction. Int J Biol Sci. (2022) 18:1271–87. doi: 10.7150/ijbs.65802
61. Ramanujam D, Schön AP, Beck C, Vaccarello P, Felician G, Dueck A, et al. MicroRNA-21-dependent macrophage-to-fibroblast signaling determines the cardiac response to pressure overload. Circulation. (2021) 143:1513–25. doi: 10.1161/CIRCULATIONAHA.120.050682
62. Han D, Kang SH, Yoon CH, Youn TJ, Chae IH. Attenuation of ischemia-reperfusion injury by intracoronary chelating agent administration. Sci Rep. (2022) 12:2050. doi: 10.1038/s41598-022-05479-2
63. Wang Y, Ding J, Song H, Teng Y, Fang X. VSIG4 regulates macrophages polarization and alleviates inflammation through activating PI3K/AKT and inhibiting TLR4/NF-κB pathway in myocardial ischemia-reperfusion injury rats. Physiol Int. (2022). doi: 10.1556/2060.2022.00055
64. Fan Q, Tao R, Zhang H, Xie H, Lu L, Wang T, et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation. (2019) 139:663–78. doi: 10.1161/CIRCULATIONAHA.118.036044
65. Huang CK, Dai D, Xie H, Zhu Z, Hu J, Su M, et al. Lgr4 governs a pro-inflammatory program in macrophages to antagonize post-infarction cardiac repair. Circ Res. (2020) 127:953–73. doi: 10.1161/CIRCRESAHA.119.315807
66. Ricci C, Ruscica M, Camera M, Rossetti L, Macchi C, Colciago A, et al. PCSK9 induces a pro-inflammatory response in macrophages. Sci Rep. (2018) 8:2267. doi: 10.1038/s41598-018-20425-x
67. Wang F, Li M, Zhang A, Li H, Jiang C, Guo J. PCSK9 modulates macrophage polarization-mediated ventricular remodeling after myocardial infarction. J Immunol Res. (2022) 2022:7685796. doi: 10.1155/2022/7685796
68. Tang ZH, Peng J, Ren Z, Yang J, Li TT, Li TH, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis. (2017) 262:113–22. doi: 10.1016/j.atherosclerosis.2017.04.023
69. Mia MM, Cibi DM, Abdul Ghani SAB, Song W, Tee N, Ghosh S, et al. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PloS Biol. (2020) 18:e3000941. doi: 10.1371/journal.pbio.3000941
70. Wang L, Li N, Wang F, Cui L. P2Y12 inhibition in macrophages reduces ventricular arrhythmias in rats after myocardial ischemia-reperfusion. Adv Clin Exp Med. (2021) 30:413–20. doi: 10.17219/acem/133139
71. Wei Y, Xing J, Su X, Li X, Yan X, Zhao J, et al. IL-38 attenuates myocardial ischemia-reperfusion injury by inhibiting macrophage inflammation. Immun Inflammation Dis. (2023) 11:e898. doi: 10.1002/iid3.898
72. Lin H, Xu WS, Liu XW, Wang Z, Yan J, Zhang T. Macrophages induce the expression of lncRNA ATB via the secretion of TGF-β to relieve ischemia-reperfusion injury in cardiomyocytes. Exp Ther Med. (2021) 22:910. doi: 10.3892/etm
73. Yin J, Hu H, Li X, Xue M, Cheng W, Wang Y, et al. Inhibition of Notch signaling pathway attenuates sympathetic hyperinnervation together with the augmentation of M2 macrophages in rats post-myocardial infarction. Am J Physiol Cell Physiol. (2016) 310:C41–53. doi: 10.1152/ajpcell.00163.2015
74. Li Z, Nie M, Yu L, Tao D, Wang Q, He Y, et al. Blockade of the notch signaling pathway promotes M2 macrophage polarization to suppress cardiac fibrosis remodeling in mice with myocardial infarction. Front Cardiovasc Med. (2021) 8:639476. doi: 10.3389/fcvm.2021.639476
75. Yang Y, Wang Q, Cai X, Wei Z, Hou J, Fei Y, et al. Activin receptor-like kinase 4 haplodeficiency alleviates the cardiac inflammation and pacing-induced ventricular arrhythmias after myocardial infarction. Aging (Albany NY). (2021) 13:17473–88. doi: 10.18632/aging.v13i13
76. Shiraishi M, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. (2016) 126:2151–66. doi: 10.1172/JCI85782
77. Shiraishi M, Yamaguchi A, Suzuki K. Nrg1/ErbB signaling-mediated regulation of fibrosis after myocardial infarction. FASEB J. (2022) 36:e22150. doi: 10.1096/fj.202101428RR
78. Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res. (2018) 191:15–28. doi: 10.1016/j.trsl.2017.10.001
79. Tong Z, Tang Y, Jiang B, Wu Y, Liu Y, Li Y, et al. Phosphorylation of nucleolin is indispensable to upregulate miR-21 and inhibit apoptosis in cardiomyocytes. J Cell Physiol. (2019) 234:4044–53. doi: 10.1002/jcp.27191
80. Tang Y, Lin X, Chen C, Tong Z, Sun H, Li Y, et al. Nucleolin improves heart function during recovery from myocardial infarction by modulating macrophage polarization. J Cardiovasc Pharmacol Ther. (2021) 26:386–95. doi: 10.1177/1074248421989570
81. Kavian N, Servettaz A, Weill B, Batteux F. New insights into the mechanism of notch signalling in fibrosis. Open Rheumatol J. (2012) 6:96–102. doi: 10.2174/1874312901206010096
82. Liu Y, Wang H, Wang X, Xie G. MiR-29b inhibits ventricular remodeling by activating notch signaling pathway in the rat myocardial infarction model. Heart Surg Forum. (2019) 22:E019–e023. doi: 10.1532/hsf.2079
83. Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. (2014) 70:74–82. doi: 10.1016/j.yjmcc.2013.11.015
84. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
85. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. (2014) 262:153–66. doi: 10.1111/imr.12218
86. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol. (2019) 106:345–58. doi: 10.1002/JLB.3RU1018-378RR
87. Yue Y, Huang S, Wang L, Wu Z, Liang M, Li H, et al. M2b macrophages regulate cardiac fibroblast activation and alleviate cardiac fibrosis after reperfusion injury. Circ J. (2020) 84:626–35. doi: 10.1253/circj.CJ-19-0959
88. Yue Y, Huang S, Li H, Li W, Hou J, Luo L, et al. M2b macrophages protect against myocardial remodeling after ischemia/reperfusion injury by regulating kinase activation of platelet-derived growth factor receptor of cardiac fibroblast. Ann Transl Med. (2020) 8:1409. doi: 10.21037/atm
89. Henri O, Pouehe C, Houssari M, Galas L, Nicol L, Edwards-Lévy F, et al. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation. (2016) 133:1484–97;discussion 1497. doi: 10.1161/CIRCULATIONAHA.115.020143
90. Wang C, Yue Y, Huang S, Wang K, Yang X, Chen J, et al. M2b macrophages stimulate lymphangiogenesis to reduce myocardial fibrosis after myocardial ischaemia/reperfusion injury. Pharm Biol. (2022) 60:384–93. doi: 10.1080/13880209.2022.2033798
91. Sun X, Li Y, Deng Q, Hu Y, Dong J, Wang W, et al. Macrophage polarization, metabolic reprogramming, and inflammatory effects in ischemic heart disease. Front Immunol. (2022) 13:934040. doi: 10.3389/fimmu.2022.934040
92. Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. (2018) 19:526–37. doi: 10.1038/s41590-018-0113-3
93. Nicolás-Ávila JA, Pena-Couso L, Muñoz-Cánoves P, Hidalgo A. Macrophages, metabolism and heterophagy in the heart. Circ Res. (2022) 130:418–31. doi: 10.1161/CIRCRESAHA.121.319812
94. Mouton AJ, Li X, Hall ME, Hall JE. Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ Res. (2020) 126:789–806. doi: 10.1161/CIRCRESAHA.119.312321
95. Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. (2017) 8:289. doi: 10.3389/fimmu.2017.00289
96. Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U.S.A. (2017) 114:580–5. doi: 10.1073/pnas.1614035114
97. Aarup A, Pedersen TX, Junker N, Christoffersen C, Bartels ED, Madsen M, et al. Hypoxia-inducible factor-1α Expression in macrophages promotes development of atherosclerosis. Arterioscler Thromb Vasc Biol. (2016) 36:1782–90. doi: 10.1161/ATVBAHA.116.307830
98. Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. (2007) 131:1124–36. doi: 10.1016/j.cell.2007.10.034
99. DeBerge M, Glinton K, Subramanian M, Wilsbacher LD, Rothlin CV, Tabas I, et al. Macrophage AXL receptor tyrosine kinase inflames the heart after reperfused myocardial infarction. J Clin Invest. (2021) 131(6):e139576. doi: 10.1172/JCI139576
100. Zhang S, Bories G, Lantz C, Emmons R, Becker A, Liu E, et al. Immunometabolism of phagocytes and relationships to cardiac repair. Front Cardiovasc Med. (2019) 6:42. doi: 10.3389/fcvm.2019.00042
101. Thorp EB. Macrophage metabolic signaling during ischemic injury and cardiac repair. Immunometabolism. (2021) 3(2):e210018. doi: 10.20900/immunometab20210018
102. Xie N, Cui H, Ge J, Banerjee S, Guo S, Dubey S, et al. Metabolic characterization and RNA profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L834–l844. doi: 10.1152/ajplung.00235.2017
103. Zhao M, Li F, Jian Y, Wang X, Yang H, Wang J, et al. Salvianolic acid B regulates macrophage polarization in ischemic/reperfused hearts by inhibiting mTORC1-induced glycolysis. Eur J Pharmacol. (2020) 871:172916. doi: 10.1016/j.ejphar.2020.172916
104. Wang N, Wang W, Wang X, Mang G, Chen J, Yan X, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ Res. (2022) 131:893–908. doi: 10.1161/CIRCRESAHA.122.320488
105. Mouton AJ, Flynn ER, Moak SP, Aitken NM, Omoto ACM, Li X, et al. Dimethyl fumarate preserves left ventricular infarct integrity following myocardial infarction via modulation of cardiac macrophage and fibroblast oxidative metabolism. J Mol Cell Cardiol. (2021) 158:38–48. doi: 10.1016/j.yjmcc.2021.05.008
106. Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, et al. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol. (2018) 113:26. doi: 10.1007/s00395-018-0686-x
107. Yakupova EI, Maleev GV, Krivtsov AV, Plotnikov EY. Macrophage polarization in hypoxia and ischemia/reperfusion: Insights into the role of energetic metabolism. Exp Biol Med (Maywood). (2022) 247:958–71. doi: 10.1177/15353702221080130
108. Zhao P, Zhou W, Zhang Y, Li J, Zhao Y, Pan L, et al. Aminooxyacetic acid attenuates post-infarct cardiac dysfunction by balancing macrophage polarization through modulating macrophage metabolism in mice. J Cell Mol Med. (2020) 24:2593–609. doi: 10.1111/jcmm.14972
109. O’Neill LAJ, Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol. (2019) 19:273–81. doi: 10.1038/s41577-019-0128-5
110. Faas M, Ipseiz N, Ackermann J, Culemann S, Grüneboom A, Schröder F, et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity. (2021) 54:2531–2546.e5. doi: 10.1016/j.immuni.2021.09.010
111. Li J, Shen D, Tang J, Wang Y, Wang B, Xiao Y, et al. IL33 attenuates ventricular remodeling after myocardial infarction through inducing alternatively activated macrophages ethical standards statement. Eur J Pharmacol. (2019) 854:307–19. doi: 10.1016/j.ejphar.2019.04.046
112. Liu Y, Wu M, Zhong C, Xu B, Kang L. M2-like macrophages transplantation protects against the doxorubicin-induced heart failure via mitochondrial transfer. Biomater Res. (2022) 26:14. doi: 10.1186/s40824-022-00260-y
113. Cheng Y, Feng Y, Xia Z, Li X, Rong J. ω-Alkynyl arachidonic acid promotes anti-inflammatory macrophage M2 polarization against acute myocardial infarction via regulating the cross-talk between PKM2, HIF-1α and iNOS. Biochim Biophys Acta Mol Cell Biol Lipids. (2017) 1862:1595–605. doi: 10.1016/j.bbalip.2017.09.009
114. Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol. (2015) 53:676–88. doi: 10.1165/rcmb.2015-0012OC
115. He Y, Gao Y, Zhang Q, Zhou G, Cao F, Yao S. IL-4 switches microglia/macrophage M1/M2 polarization and alleviates neurological damage by modulating the JAK1/STAT6 pathway following ICH. Neuroscience. (2020) 437:161–71. doi: 10.1016/j.neuroscience.2020.03.008
116. Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology. (2018) 223:101–11. doi: 10.1016/j.imbio.2017.10.005
117. Wang X, Ji Y, Feng P, Liu R, Li G, Zheng J, et al. The m6A reader IGF2BP2 regulates macrophage phenotypic activation and inflammatory diseases by stabilizing TSC1 and PPARγ. Adv Sci (Weinh). (2021) 8:2100209. doi: 10.1002/advs.202100209
118. Yu T, Gao M, Yang P, Liu D, Wang D, Song F, et al. Insulin promotes macrophage phenotype transition through PI3K/Akt and PPAR-γ signaling during diabetic wound healing. J Cell Physiol. (2019) 234:4217–31. doi: 10.1002/jcp.27185
119. Sawoo R, Dey R, Ghosh R, Bishayi B. TLR4 and TNFR1 blockade dampen M1 macrophage activation and shifts them towards an M2 phenotype. Immunol Res. (2021) 69:334–51. doi: 10.1007/s12026-021-09209-0
120. Shou X, Wang Y, Jiang Q, Chen J, Liu Q. miR-126 promotes M1 to M2 macrophage phenotype switching via VEGFA and KLF4. PeerJ. (2023) 11:e15180. doi: 10.7717/peerj.15180
121. Gao XM, White DA, Dart AM, Du XJ. Post-infarct cardiac rupture: recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther. (2012) 134:156–79. doi: 10.1016/j.pharmthera.2011.12.010
122. Yang Y, Ma Y, Han W, Li J, Xiang Y, Liu F, et al. Age-related differences in postinfarct left ventricular rupture and remodeling. Am J Physiol Heart Circ Physiol. (2008) 294:H1815–22. doi: 10.1152/ajpheart.00831.2007
123. Nahrendorf M, Aikawa E, Figueiredo JL, Stangenberg L, van den Borne SW, Blankesteijn WM, et al. Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular remodelling: implications for FXIII therapy and antithrombin use in myocardial infarction. Eur Heart J. (2008) 29:445–54. doi: 10.1093/eurheartj/ehm558
124. Kameda K, Matsunaga T, Abe N, Fujiwara T, Hanada H, Fukui K, et al. Increased pericardial fluid level of matrix metalloproteinase-9 activity in patients with acute myocardial infarction: possible role in the development of cardiac rupture. Circ J. (2006) 70:673–8. doi: 10.1253/circj.70.673
125. Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest. (2005) 115:599–609. doi: 10.1172/JCI22304
126. Fang L, Gao XM, Moore XL, Kiriazis H, Su Y, Ming Z, et al. Differences in inflammation, MMP activation and collagen damage account for gender difference in murine cardiac rupture following myocardial infarction. J Mol Cell Cardiol. (2007) 43:535–44. doi: 10.1016/j.yjmcc.2007.06.011
127. Liu W, Zhang X, Zhao M, Zhang X, Chi J, Liu Y, et al. Activation in M1 but not M2 Macrophages Contributes to Cardiac Remodeling after Myocardial Infarction in Rats: a Critical Role of the Calcium Sensing Receptor/NRLP3 Inflammasome. Cell Physiol Biochem. (2015) 35:2483–500. doi: 10.1159/000374048
128. Nakagawa P, Romero CA, Jiang X, D'Ambrosio M, Bordcoch G, Peterson EL, et al. Ac-SDKP decreases mortality and cardiac rupture after acute myocardial infarction. PloS One. (2018) 13:e0190300. doi: 10.1371/journal.pone.0190300
129. Ishikawa S, Noma T, Fu HY, Matsuzaki T, Ishizawa M, Ishikawa K, et al. Apoptosis inhibitor of macrophage depletion decreased M1 macrophage accumulation and the incidence of cardiac rupture after myocardial infarction in mice. PloS One. (2017) 12:e0187894. doi: 10.1371/journal.pone.0187894
130. Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, et al. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res. (2013) 112:675–88. doi: 10.1161/CIRCRESAHA.111.300502
131. Ohayon L, Zhang X, Dutta P. The role of extracellular vesicles in regulating local and systemic inflammation in cardiovascular disease. Pharmacol Res. (2021) 170:105692. doi: 10.1016/j.phrs.2021.105692
132. Vinaiphat A, Sze SK. Advances in extracellular vesicles analysis. Adv Clin Chem. (2020) 97:73–116. doi: 10.1016/bs.acc.2019.12.003
133. Gonda A, Kabagwira J, Senthil GN, Wall NR. Internalization of exosomes through receptor-mediated endocytosis. Mol Cancer Res. (2019) 17:337–47. doi: 10.1158/1541-7786.MCR-18-0891
134. Wang Y, Qiu Z, Yuan J, Li C, Zhao R, Liu W, et al. Hypoxia-reoxygenation induces macrophage polarization and causes the release of exosomal miR-29a to mediate cardiomyocyte pyroptosis. In Vitro. Cell Dev Biol Anim. (2021) 57:30–41. doi: 10.1007/s11626-020-00524-8
135. Wang C, Zhang C, Liu L, A X, Chen B, Li Y, et al. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol Ther. (2017) 25:192–204. doi: 10.1016/j.ymthe.2016.09.001
136. Liu S, Chen J, Shi J, Zhou W, Wang L, Fang W, et al. M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res Cardiol. (2020) 115:22. doi: 10.1007/s00395-020-0781-7
137. Liu P, Wang S, Wang G, Zhao M, Du F, Li K, et al. Macrophage-derived exosomal miR-4532 promotes endothelial cells injury by targeting SP1 and NF-κB P65 signalling activation. J Cell Mol Med. (2022) 26:5165–80. doi: 10.1111/jcmm.17541
138. Chen B, Luo L, Wei X, Gong D, Li Z, Li S, et al. M1 bone marrow-derived macrophage-derived extracellular vesicles inhibit angiogenesis and myocardial regeneration following myocardial infarction via the MALAT1/microRNA-25–3p/CDC42 axis. Oxid Med Cell Longev 2021. (2021) p:9959746. doi: 10.1155/2021/9959746
139. Dai Y, Wang S, Chang S, Ren D, Shali S, Li C, et al. M2 macrophage-derived exosomes carry microRNA-148a to alleviate myocardial ischemia/reperfusion injury via inhibiting TXNIP and the TLR4/NF-κB/NLRP3 inflammasome signaling pathway. J Mol Cell Cardiol. (2020) 142:65–79. doi: 10.1016/j.yjmcc.2020.02.007
140. Wang Y, Li C, Zhao R, Qiu Z, Shen C, Wang Z, et al. CircUbe3a from M2 macrophage-derived small extracellular vesicles mediates myocardial fibrosis after acute myocardial infarction. Theranostics. (2021) 11:6315–33. doi: 10.7150/thno.52843
141. Yuan W, Liang X, Liu Y, Wang H. Mechanism of miR-378a-3p enriched in M2 macrophage-derived extracellular vesicles in cardiomyocyte pyroptosis after MI. Hypertens Res. (2022) 45:650–64. doi: 10.1038/s41440-022-00851-1
142. Hu H, Qi L, Ren C, Yan S. M2 macrophage-derived exosomes regulate myocardial ischemia-reperfusion and pyroptosis via ROS/NLRP3 pathway. Heart Surg Forum. (2022) 25:E698–e708. doi: 10.1532/hsf.4919
143. Arjmand B, Abedi M, Arabi M, Alavi-Moghadam S, Rezaei-Tavirani M, Hadavandkhani M, et al. Regenerative medicine for the treatment of ischemic heart disease; status and future perspectives. Front Cell Dev Biol. (2021) 9:704903. doi: 10.3389/fcell.2021.704903
144. Golpanian S, Wolf A, Hatzistergos KE, Hare JM. Rebuilding the damaged heart: mesenchymal stem cells, cell-based therapy, and engineered heart tissue. Physiol Rev. (2016) 96:1127–68. doi: 10.1152/physrev.00019.2015
145. Zheng YL, Wang WD, Cai PY, Zheng F, Zhou YF, Li MM, et al. Stem cell-derived exosomes in the treatment of acute myocardial infarction in preclinical animal models: a meta-analysis of randomized controlled trials. Stem Cell Res Ther. (2022) 13:151. doi: 10.1186/s13287-022-02833-z
146. Xu R, Zhang F, Chai R, Zhou W, Hu M, Liu B, et al. Exosomes derived from pro-inflammatory bone marrow-derived mesenchymal stem cells reduce inflammation and myocardial injury via mediating macrophage polarization. J Cell Mol Med. (2019) 23:7617–31. doi: 10.1111/jcmm.14635
147. Deng S, Zhou X, Ge Z, Song Y, Wang H, Liu X, et al. Exosomes from adipose-derived mesenchymal stem cells ameliorate cardiac damage after myocardial infarction by activating S1P/SK1/S1PR1 signaling and promoting macrophage M2 polarization. Int J Biochem Cell Biol. (2019) 114:105564. doi: 10.1016/j.biocel.2019.105564
148. Huang P, Wang L, Li Q, Xu J, Xu J, Xiong Y, et al. Combinatorial treatment of acute myocardial infarction using stem cells and their derived exosomes resulted in improved heart performance. Stem Cell Res Ther. (2019) 10:300. doi: 10.1186/s13287-019-1353-3
149. Chen R, Zhang Y, Zhou H, Hu Y, Chen Y. SGLT2 inhibitor-pretreated macrophage transplantation improves adverse ventricular remodeling after acute myocardial infarction. J Cardiovasc Pharmacol. (2023) 82:287–97. doi: 10.1097/FJC.0000000000001466
150. Podaru MN, Fields L, Kainuma S, Ichihara Y, Hussain M, Ito T, et al. Reparative macrophage transplantation for myocardial repair: a refinement of bone marrow mononuclear cell-based therapy. Basic Res Cardiol. (2019) 114:34. doi: 10.1007/s00395-019-0742-1
151. Tariq U, Gupta M, Pathak S, Patil R, Dohare A, Misra SK. Role of biomaterials in cardiac repair and regeneration: therapeutic intervention for myocardial infarction. ACS Biomater Sci Eng. (2022) 8:3271–98. doi: 10.1021/acsbiomaterials.2c00454
152. Zou Y, Li L, Li Y, Chen S, Xie X, Jin X, et al. Restoring Cardiac Functions after Myocardial Infarction-Ischemia/Reperfusion via an Exosome Anchoring Conductive Hydrogel. ACS Appl Mater Interfaces. (2021) 13:56892–908. doi: 10.1021/acsami.1c16481
153. de Abreu RC, Fernandes H, da Costa Martins PA, Sahoo S, Emanueli C, Ferreira L. Native and bioengineered extracellular vesicles for cardiovascular therapeutics. Nat Rev Cardiol. (2020) 17:685–97. doi: 10.1038/s41569-020-0389-5
154. Xu J, Wang J, Chen Y, Hou Y, Hu J, Wang G. Recent advances of natural and bioengineered extracellular vesicles and their application in vascular regeneration. Regener Biomater. (2022) 9:rbac064. doi: 10.1093/rb/rbac064
155. Wang X, Chen Y, Zhao Z, Meng Q, Yu Y, Sun J, et al. Engineered exosomes with ischemic myocardium-targeting peptide for targeted therapy in myocardial infarction. J Am Heart Assoc. (2018) 7:e008737. doi: 10.1161/JAHA.118.008737
156. Liu HY, Yu LF, Zhou TG, Wang YD, Sun DH, Chen HR, et al. Lipopolysaccharide-stimulated bone marrow mesenchymal stem cells-derived exosomes inhibit H2O2-induced cardiomyocyte inflammation and oxidative stress via regulating miR-181a-5p/ATF2 axis. Eur Rev Med Pharmacol Sci. (2020) 24:10069–77. doi: 10.26355/eurrev_202010_23224
157. Perin EC, Willerson JT, Pepine CJ, Henry TD, Ellis SG, Zhao DX, et al. Effect of transendocardial delivery of autologous bone marrow mononuclear cells on functional capacity, left ventricular function, and perfusion in chronic heart failure: the FOCUS-CCTRN trial. Jama. (2012) 307:1717–26. doi: 10.1001/jama.2012.418
158. Ledford KJ, Zeigler F, Bartel RL. Ixmyelocel-T, an expanded multicellular therapy, contains a unique population of M2-like macrophages. Stem Cell Res Ther. (2013) 4:134. doi: 10.1186/scrt345
159. Henry TD, Traverse JH, Hammon BL, East CA, Bruckner B, Remmers AE, et al. Safety and efficacy of ixmyelocel-T: an expanded, autologous multi-cellular therapy, in dilated cardiomyopathy. Circ Res. (2014) 115:730–7. doi: 10.1161/CIRCRESAHA.115.304554
160. Chernykh ER, Kafanova MY, Shevela EY, Sirota SI, Adonina EI, Sakhno LV, et al. Clinical experience with autologous M2 macrophages in children with severe cerebral palsy. Cell Transplant. (2014) 23 Suppl 1:S97–104. doi: 10.3727/096368914X684925
161. Chernykh ER, Shevela EY, Starostina NM, Morozov SA, Davydova MN, Menyaeva EV, et al. Safety and therapeutic potential of M2 macrophages in stroke treatment. Cell Transplant. (2016) 25:1461–71. doi: 10.3727/096368915X690279
162. Xi XM, Xia SJ, Lu R. Drug loading techniques for exosome-based drug delivery systems. Pharmazie. (2021) 76:61–7. doi: 10.1691/ph.2021.0128
163. Kang T, Atukorala I, Mathivanan S. Biogenesis of extracellular vesicles. Subcell Biochem. (2021) 97:19–43. doi: 10.1007/978-3-030-67171-6_2
164. Lai J, Huang C, Guo Y, Rao L. Engineered extracellular vesicles and their mimics in cardiovascular diseases. J Control Release. (2022) 347:27–43. doi: 10.1016/j.jconrel.2022.04.046
165. Wu R, Gao W, Yao K, Ge J. Roles of exosomes derived from immune cells in cardiovascular diseases. Front Immunol. (2019) 10:648. doi: 10.3389/fimmu.2019.00648
166. Gao H, Wang S, Liu Z, Hirvonen JT, Santos A H. Mycophenolic acid-loaded naïve macrophage-derived extracellular vesicles rescue cardiac myoblast after inflammatory injury. ACS Appl Bio Mater. (2023) 6:4269–76. doi: 10.1021/acsabm.3c00475
167. Zhao X, Jia Y, Chen H, Yao H, Guo W. Plasma-derived exosomal miR-183 associates with protein kinase activity and may serve as a novel predictive biomarker of myocardial ischemic injury. Exp Ther Med. (2019) 18:179–87. doi: 10.3892/etm
168. Ling H, Guo Z, Shi Y, Zhang L, Song C. Serum exosomal microRNA-21, microRNA-126, and PTEN are novel biomarkers for diagnosis of acute coronary syndrome. Front Physiol. (2020) 11:654. doi: 10.3389/fphys.2020.00654
Keywords: ischemic heart disease, immune cells, macrophage, extracellular vesicles, exosomes
Citation: Wang M, Li C, Liu Y, Jin Y, Yu Y, Tan X and Zhang C (2024) The effect of macrophages and their exosomes in ischemic heart disease. Front. Immunol. 15:1402468. doi: 10.3389/fimmu.2024.1402468
Received: 17 March 2024; Accepted: 29 April 2024;
Published: 10 May 2024.
Edited by:
Guo-Chang Fan, University of Cincinnati, United StatesReviewed by:
Bingcai Qi, Capital Medical University, ChinaDongze Qin, Albert Einstein College of Medicine, United States
Kobina Essandoh, University of Michigan, United States
Copyright © 2024 Wang, Li, Liu, Jin, Yu, Tan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqiu Tan, tanxiaoqiu@swmu.edu.cn; Chunxiang Zhang, zhangchunxiangKZL@163.com
†These authors have contributed equally to this work