 Yannick S. Rakké
Yannick S. Rakké Sonja I. Buschow
Sonja I. Buschow Jan N. M. IJzermans1
Jan N. M. IJzermans1 Dave Sprengers
Dave Sprengers- 1Department of Surgery, Erasmus MC-Transplant Institute, University Medical Center, Rotterdam, Netherlands
- 2Department of Gastroenterology and Hepatology, Erasmus MC-Cancer Institute-University Medical Center, Rotterdam, Netherlands
Hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA) are the first and second most common primary liver cancer (PLC). For decades, systemic therapies consisting of tyrosine kinase inhibitors (TKIs) or chemotherapy have formed the cornerstone of treating advanced-stage HCC and CCA, respectively. More recently, immunotherapy using immune checkpoint inhibition (ICI) has shown anti-tumour reactivity in some patients. The combination regimen of anti-PD-L1 and anti-VEGF antibodies has been approved as new first-line treatment of advanced-stage HCC. Furthermore, gemcibatine plus cisplatin (GEMCIS) with an anti-PD-L1 antibody is awaiting global approval for the treatment of advanced-stage CCA. As effective anti-tumour reactivity using ICI is achieved in a minor subset of both HCC and CCA patients only, alternative immune strategies to sensitise the tumour microenvironment of PLC are waited for. Here we discuss immune checkpoint stimulation (ICS) as additional tool to enhance anti-tumour reactivity. Up-to-date information on the clinical application of ICS in onco-immunology is provided. This review provides a rationale of the application of next-generation ICS either alone or in combination regimen to potentially enhance anti-tumour reactivity in PLC patients.
1 Introduction
Primary liver cancer (PLC), including hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA), is the third leading cause of cancer-related death ranking sixth in incidence worldwide (1). Its incidence is expected to increase in Western society (2, 3). Early- or intermediate-stage HCC can be treated successfully using ablative therapy, surgical resection, or liver transplantation while early-stage CCA can be treated using surgical resection only. Moreover, the majority of PLC patients get diagnosed at advanced-stage disease leaving them to no other option than systemic therapies including immune checkpoint inhibitors (ICI) and multi-tyrosine kinase inhibitors (TKIs; e.g., sorafenib, regorafenib) for HCC and chemotherapy for CCA (e.g., cisplatin/gemcitabine, FOLFIRINOX) (4, 5). So far, these remaining treatment options for advanced-stage PLC have only shown modest survival benefits and more effective treatment approaches are urgently needed. Next to ICI, engaging immune (co-)stimulatory molecules (i.e., immune checkpoint stimulation (ICS) in the tumour microenvironment alone or combined with other immune enhancing therapies represents a promising novel opportunity. In this review, we first summarise the current knowledge on ICI and its pitfalls in the hepatic tumour immune microenvironment (TIME). Then we provide an overview of insights gained from (pre)clinical studies regarding the interactions between co-stimulatory molecules and their ligands expressed on different T-cell subsets, antigen presenting cells and other cell types in the context of the hepatic TIME. Lastly, we highlight the opportunities to enhance and support currently applied ICI and more targeted immune therapies using ICS in PLC.
2 Immune checkpoint inhibition fails to induce anti-tumour immunity in the majority of primary liver cancer patients
ICI is often applied in the form of antibodies that interfere with binding of co-inhibitory receptors (i.e., cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and programmed cell death-1 (PD-1)) and their cognate ligands (CD80/86 and programmed cell death ligand-1 (PD-L1), resp.). ICIs have been shown to effectively enhance pre-existing anti-tumour immune-responses among multiple immune-active cancer types such as melanoma, non-small cell lung cancer, and renal cell carcinoma (6–8).Accordingly, anti-PD1 antagonistic antibodies (pembrolizumab and nivolumab, resp.) prolonged survival in advanced-stage HCC patients that did not respond well to TKIs only. However, clinical efficacy of anti-PD1-mediated ICI was modest and failed to sustain its benefit when compared to TKIs directly upon randomisation among TKI-naive HCC patients (9, 10). Combination regimen of various ICIs or chemotherapy with ICI appeared to be more successful. Anti-PD-L1 antagonistic antibodies (atezolizumab) with anti-vascular endothelial growth factor (anti-VEGF; bevacizumab) improved overall and progression-free survival (OS and PFS) outcomes compared to the TKI sorafenib (11, 12). Similarly, sintilimumab (anti-PD-1) plus IBI305 (bevacizumab biosimilar) improved survival rates compared to sorafenib in Chinese patients with unresectable, hepatitis B virus (HBV)- associated HCC (13). Moreover, durvalumab (anti-PD-L1) in combination with the anti-CTLA-4 agonistic antibody tremelimumab improved OS compared to sorafenib (14). As atezolizumab-bevacizumab (atezo-bev) and durvalumab-tremelimumab (durva-trem) were proven to successfully induce clinical anti-tumour efficacy, both regimens have been approved by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) to treat unresectable HCC patients in the first line of care. Also, in refractory or recurrent CCA, a combination regimen of ICI with chemotherapy was superior to chemotherapy alone. In the TOPAZ-1 trial, durvalumab with gemcibatine plus cisplatin (GEMCIS) improved both OS and PFS compared to chemotherapy with placebo (15). Based on these data durva-GEMCIS has been granted FDA-approval as first-line standard-of-care in advanced-stage CCA.
Still, even though these immunotherapy combination regimens have proven to successfully induce anti-tumour immunity objective responses could only be confirmed in 20-27% of HCC patients and about 27% of CCA patients (11, 13–15). This data underlines the complexity of the hepatic tumour immune microenvironment (TIME) potentially explaining inter-patient variation regarding clinical efficacy. Establishment of a suppressive TIME may enable tumour cells to evade and restrain efficient anti-tumour immunity (16). Compared to adjacent tissue compartments, the hepatic TIME has been shown to be enriched specifically for immune suppressive cells (e.g., regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumour-associated macrophages (TAMs)), rather than immune effector cells (17–19). Furthermore, in HCC and CCA, tumours can be either inflamed or non-inflamed, but the latter is the dominant phenotype in both settings indicating tumour-immunity may also often be poorly developed (20, 21) Indeed, the anti-tumour adaptive immunity in PLC has been shown to be hampered by impaired T cell priming and local dysfunction or exhaustion of tumour-infiltrating lymphocytes (TILs) (22, 23).
The notion that combination regimens have proven to be clinically more effective compared to monotherapy in PLC suggests that successful reinvigoration of anti-tumour immunity in PLC might need a multi-factorial approach. Whereas ICI primarily intents to enhance existing cytotoxic CD8 T cell (CTL)-immune effector function, PLC-directed immunotherapies may require inhibition of immune suppressive cells as well. With respect to the latter, bevacizumab has been described to inhibit MDSCs, TAMs, and Tregs and these effects could partly explain its enhanced anti-tumour activity in the atezo-bev regimen (24) (Figure 1). Likewise, combination regimes of ICI with ICS using either agonistic antibodies or ligands targeting costimulatory molecules, could enhance T cell-mediated anti-tumour immunity. These ICS may act through depletion of immune suppressive cells as well as by enhancing the priming and activation of immune effector cells. Thereby, ICS might provide an interesting alternative or supportive immune therapeutic approach in PLC (25). Recent early phase I clinical trials have reported on the clinical safety and efficacy of ICS used alone or in combination regimes in various advanced solid tumours (26–30). To now develop more effective combination regimes based on ICI and ICS for the treatment of PLC, a comprehensive overview of the different stimulatory checkpoint mechanisms that might support intra-tumoural T-cell responses and their interplay with immune inhibitory checkpoint mechanisms in PLC is required and this we aim to supply with this review.
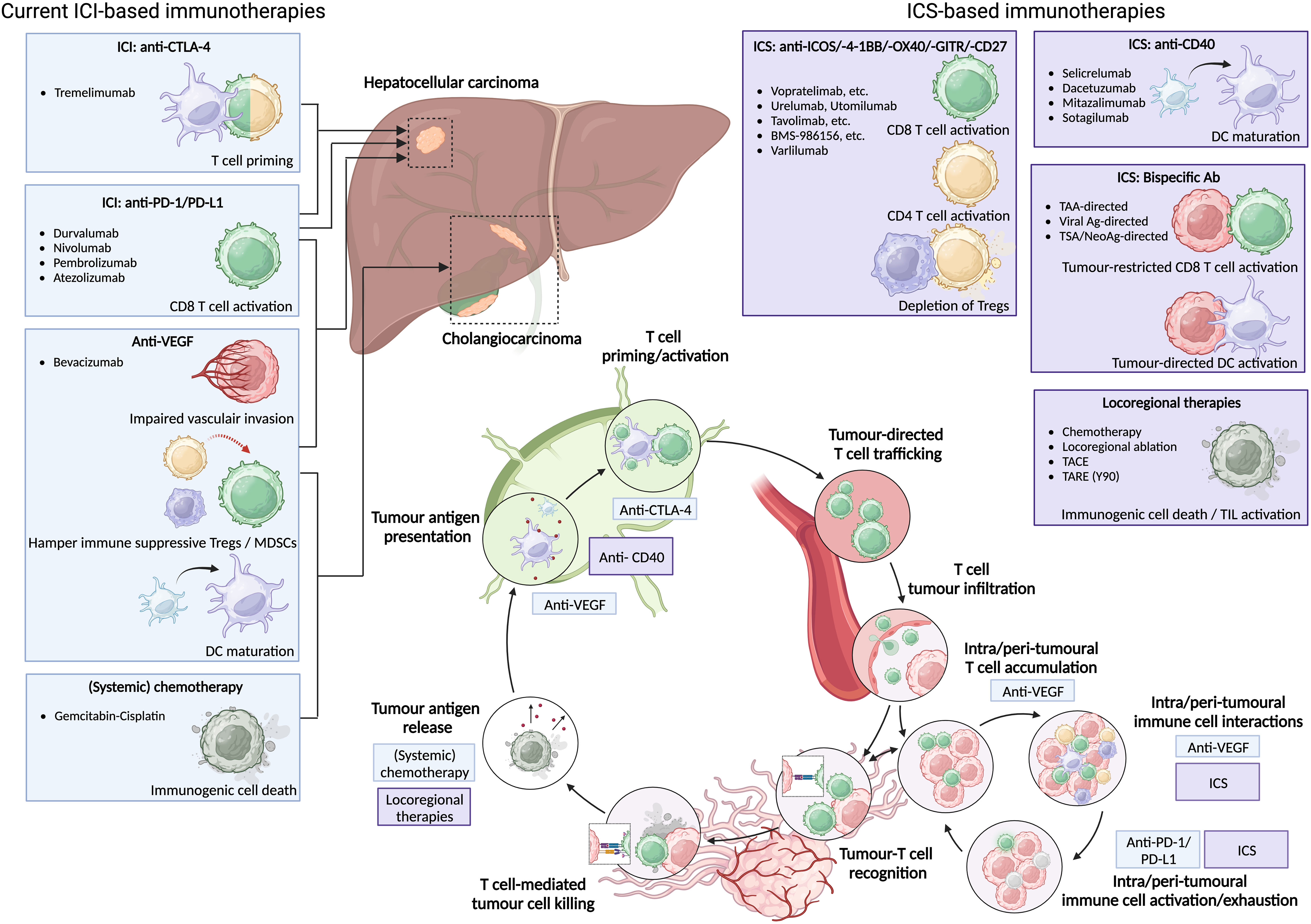
Figure 1 Current ICI-based immunotherapies for HCC and (i)CCA can be supported by ICS-based immunotherapies to enhance anti-tumour immunity. Current ICI-based immunotherapies for HCC consist of anti-PD-1/PD-L1 monotherapy, anti-CTLA-4 plus anti-PD-1/PD-L1, anti-PD-L1 plus anti-VEGF; or anti-VEGF plus Gem-Cis for CCA. Novel ICS-based immunotherapies can either provide additional CD4 and CD8 T cell activation, induce depletion of immunosuppressive Tregs, or induce DC maturation. Moreover, ICS-based bispecific antibodies might induce tumour-directed DC activation as well as tumour-restricted CD8 T cell activation and subsequent tumour cell killing via tumour-associated antigen (TAA), viral antigen (viral Ag), or tumour-specific/neoantigen (TSA/neoAg). All can be enhanced by additional release of tumour antigen via locoregional therapies.
3 Co-stimulatory immune checkpoints are widely expressed among liver-resident innate and adaptive immune subsets
Stimulatory immune checkpoint interactions are crucial for effective T cell activation. In 1987, CD28 was the first co-stimulatory receptor that was demonstrated to enhance T cell receptor (TCR) signaling, thereby laying the foundation for the three-signal model of T cell activation that requires both TCR and co-stimulatory signaling as well as cytokines for full T cell activation and differentiation (31). Following recognition of the cognate peptide-MHC complex by the TCR, co-signaling receptors co-localise with TCR molecules at the immunological synapse. In contrast to co-inhibitory immune checkpoint interactions that provide feedback inhibitory signals to activated CTL in the effector phase, co-stimulatory interactions rather are important during antigen presentation, priming, and subsequent T cell activation or differentiation (25, 32) (Table 1). Co-stimulatory receptors are divided into two distinct groups: immunoglobulin superfamily (IgSF) members and tumour necrosis factor receptor superfamily (TNFRSF) members, and these subclasses are in turn divided according to protein structure and function (25).
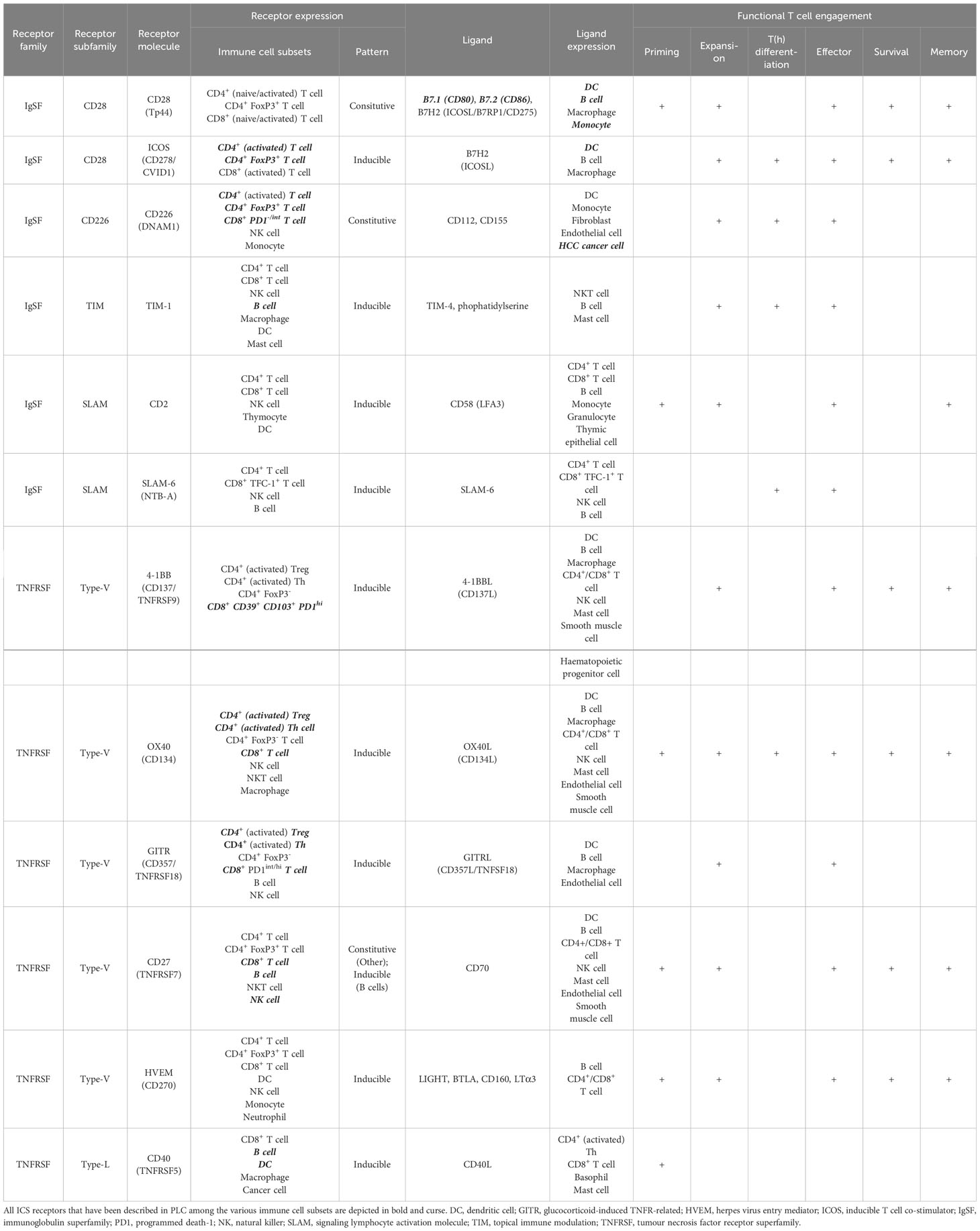
Table 1 Immune co-stimulatory receptors as divided by IgSF and TNFRSF members are expressed among different immune cell subsets playing various roles in functional T cell engagement.
3.1 IgSF structure, expression, and ligands
The IgSF comprises various cell surface and soluble proteins. Its members share structural features with immunoglobulins, including an Ig-domain. IgSF members include cell surface antigen receptors, cell adhesion molecules, cytokine receptors, and co-inhibitory or -stimulatory signaling receptors. The human co-stimulatory IgSF members consists of 12 receptors and 16 ligands that are represented in the CD28, CD226, TIM, and CD2/SLAM receptor subfamilies (25). We here highlight the most relevant subfamily members.
3.1.1 CD28 receptor subfamily: CD28
CD28 (alias: Tp44) is a co-stimulatory molecule that was first described in 1987 (31, 33). Generally, CD28 is expressed constitutively on CD4+ and CD8+ T cells, including those in the TIME. Expression has been demonstrated among bone marrow stromal cells and other immune subsets such as plasma cells, neutrophils, and eosinophils (34). CD28-directed ligands belong to the B7 family of which CD80 (alias: B7-1) and CD86 (alias: B7-2) demonstrate the highest binding affinity (Kd: 4uM and 15-40uM, resp.) (25, 35, 36). CD80 and 86 are upregulated on APCs upon activation and maturation following immune stress responses. CD28 signaling induces activation of NFAT, mTOR, ERK and NFkB lowering the threshold for TCR signaling and subsequent T cell activation and proliferation, survival and effector function (Figure 2). However, in the TIME, CD80/86-mediated CD28 signaling may be hampered due to competitive binding of PD-L-1 and CTLA-4 to their respect receptors (37). Both PD-1 and CTLA-4 signaling impair CD28 signaling as intracellular domains of activated or PD-L-1 bound PD-1 bind to the CD28 cytoplasmic tail with high affinity. Moreover, CD28-mediated T cell co-stimulation has been shown to be crucial for PD-1 therapy in cancer patients as loss of CD28 via T cell exhaustion was shown to correlate to clinical irresponsiveness towards PD-1 ICI (37).
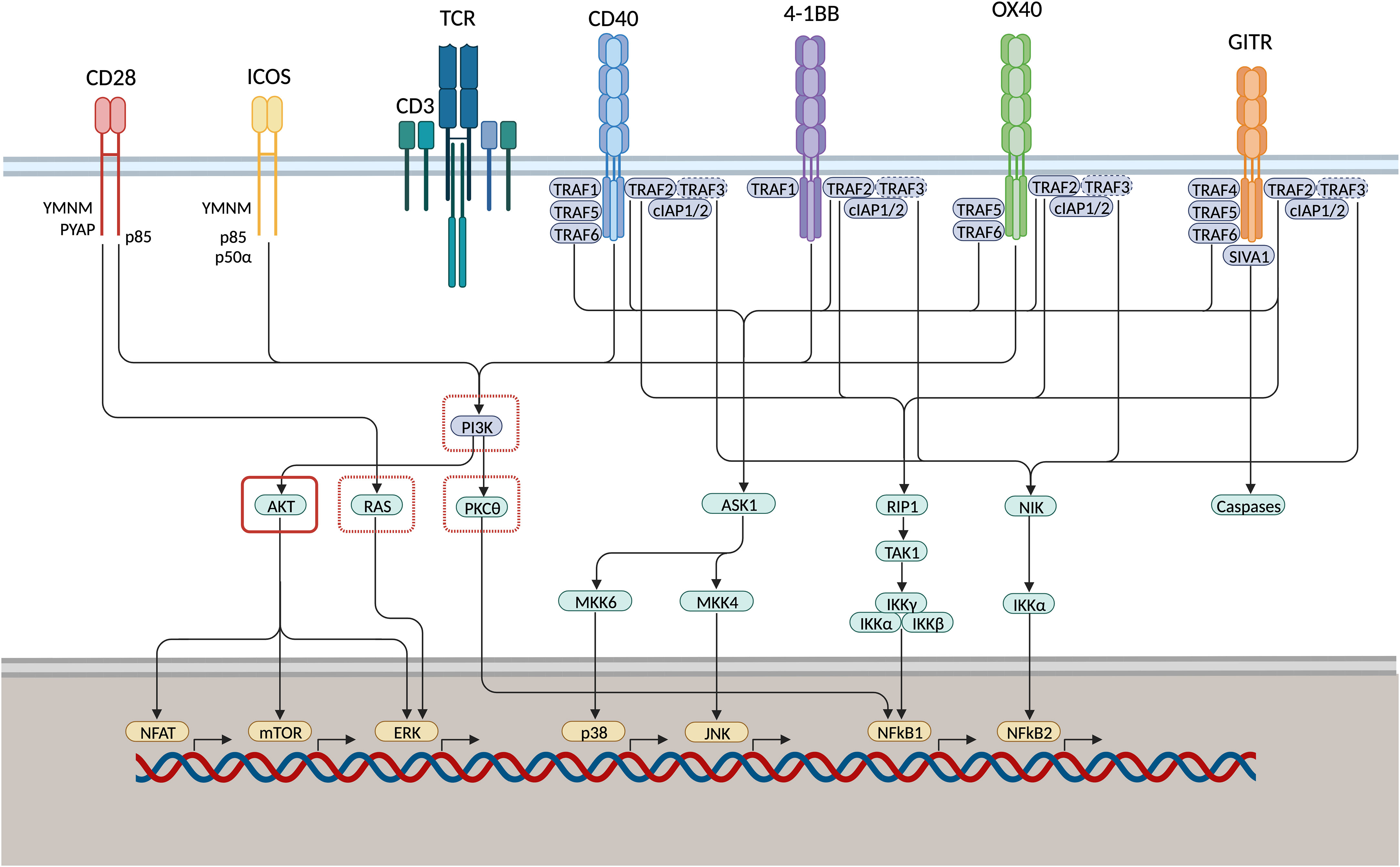
Figure 2 IgSF and TNFRSF members induce T cell proliferation, survival, and effector function via shared co-stimulatory signaling pathways. CD28 and inducible T-cell co-stimulator (ICOS) associate with phosphoinositide 3-kinase (PI3K) through their YMNM/PYAP- or YMFM-motif, respectively. TNF receptor monomers multimerise into trimeric ligand-receptor complexes that engage TNF receptor-associated factor (TRAF) adaptor proteins. Upon ligation, GITR associates with TRAF2/4/5/6, 4-1BB associates with TRAF1/2, OX40 associates with TRAF2/5/6, CD40 associates with TRAF1/2/5/6. p38 and JNK are activated subsequent to TRAF2 and -5-mediated regulation of the MAPK pathways. NfkB is induced via the canonical and non-canonical signaling cascade. NFkB1 is induced through activation of kinase RIP1, TAK1 and IKK complexes mediated via TRAF2 or TRAF5 association. Moreover, TRAF2 engages with TRAF3 via cIAP1/2, thereby inducing TRAF3 degradation. As TRAF3 mediates NIK degradation under natural conditions, NFkB2 activation is induced downstream of phosphorylation of the inhibitory kappa B kinase-alpha (IKK) as a result of NF-kB-inducing kinase (NIK) stabilisation. GITR associates with pro-apoptosis factor SIVA1, activating downstream caspases. This might function as a negative feedback loop to pro-survival signaling cascades vai Bcl-xL. PI3K, RAS, and PKC𝜃 are inhibited through PD-1 signaling (red dashed square) directly or via recruitment of protein tyrosine phosphatase SHP1. AKT can be inhibited through PD-1 and CTLA-4 signaling (red solid square) via recruitment of SHP1 or protein phosphatase 2A (PP2A), respectively. Therefore, co-stimulatory signaling pathways might be enhanced through concomitant application of both ICS and ICB.
In HCC, CD28 has been described to be expressed on TILs albeit at lower levels compared to PBMC-derived T cell (38). This might be explained by the fact that TIL fractions are enriched for central and effector memory T cells (Tcm and TEMRA, resp.) rather than naïve CD8 T cells (Tn). Alternatively, diminished CD28 may be a reflection of T cell exhaustion (37). Among CD3+ TILs, Hsu and colleagues have demonstrated high levels of PD1/CD28-coexpression (39). Furthermore, CD28 is upregulated on CD8+ PD1hi HCC TILs compared to the PD1- and PD1int compartments thereby potentially delineating tumour-reactive TILs (40). In the TIME multiple cell types can deliver CD28 stimulatory signals as CD80 and CD86 are expressed on intra-tumoural B cell, BDCA1+ myeloid DC (mDC), and CD14+ monocytes in both HCC and CCA (18). HCC tumour cells themselves, however, have demonstrated relatively low B7 family member expression (41, 42).
As CD28 has been described to be expressed constitutively on naive T cells, agonistic targeting of CD28 is hard to restrict to the TIME or to circulating tumour-specific T cells. This was illustrated by a first-in-human clinical trial of a CD28 agonistic antibody where all healthy volunteers developed life-threatening immune-related adverse events (irAE) as a direct result of a cytokine release syndrome (43). Direct CD28 stimulation of TILs does, however, did show potential in vitro, reinvigorating anti-tumour CD8 TIL function and metabolic activity (44). Alternatively, CD28 stimulation might rather be established indirectly through antibody-mediated blockade of anti-PD-L-1 and CTLA-4 allowing increased binding of CD80/86 to CD28 and/or by lifting of inhibitory signaling via PD-1 and CTLA-4. Especially in HCC the former might contribute to the effect of ICI therapies targeting PD-L-1 and CTLA-4, since HCC tumour cells of early-relapsed disease demonstrated enhanced interaction of PD-L-1 and CTLA-4 with CD80/86 compared to primary HCC tumour cells (45). In these cases anti-PD-1/CTLA-4 blockade might enhance CD28 signaling and thereby could support T cell activation by local antigen presentation, thus improving tumour immune surveillance.
3.1.2 CD28 receptor subfamily: ICOS
Engagement of CD28 will trigger most T cells. To specifically target only already activated cells using ICS, one might focus on activation-induced co-stimulatory receptors such as inducible T-cell co-stimulator (ICOS; aliases: CD278, CVID1) (46) (Figure 2). Only a small fraction of resting memory T cells shows expression of ICOS at low levels (25). ICOS expression has been demonstrated to be expressed at intermediate levels in immune-active tumour-infiltrating Th1 cells (47). Furthermore, both in colorectal cancer (CRC) and non-small cell lung cancer (NSCLC), Duhen and colleagues demonstrated tumour-reactive CD4+ Th or follicular T helper (Tfh) TILs co-express PD1 and ICOS (48). Important to consider when targeting ICOS is that in the TIME ICOS may also be highly expressed on FoxP3+ Tregs, identifying highly immune suppressive activated Tregs among ICOShi fractions (47, 49). Hence, stimulating ICOS may also augment immune suppression via Tregs. The ligand for ICOS is B7H2 (aliases: ICOSLG, B7RP1, CD275) (50). ICOS and CD28 share the B7H2 ligand, although ICOS binds to B7H2 with significantly higher affinity. B7H2 is expressed constitutively on mature APCs (e.g., B cells, macrophages, and DCs) (50). Moreover, B7H2 is largely expressed on somatic cells such as tumour cells including HCC, under local control of TNFα (51, 52). Similar to CD28, agonistic ICOS engagement triggers the activation of multiple pathways that support antigen specific T cell activation (Figure 2).
Upregulation of ICOSL on HCC tumour-derived plasmacytoid DCs (pDCs) was hypothesised to activate type 1 regulatory T (Tr1) cells only (53). However, in HCC TILs, ICOS is upregulated both on Tregs as well as on CD4+ TILs that demonstrated features of recent activation and displayed increased proliferative capacity as well (54–57). Therefore, ICOS signaling in HCC might both enhance and hamper local tumour control by liver-resident immune cells. In contrast, CCA-derived intra-tumoural Tregs demonstrated high expression of ICOS but not effector Th or CTL (19).
Consistent with these expression patterns, in preclinical studies, ICOS signaling either promoted pro-tumour responses (via Tregs) or anti-tumour responses (via Th1, Tfh, or CTL) (47). Hence, agonistic ICOS mAbs alone will most likely not achieve any anti-tumour activity due to predominant action on Treg subsets. Interestingly, in patients treated with anti-CTLA-4 mAb, ICOS appears to be upregulated on effector T cells (58). Moreover, ICOS knock-out mice do not respond well to anti-CLTA-4, suggesting a significant role for ICOS-signaling on effector T cell-mediated anti-tumour activity particularly when Tregs are blocked (59). Concordantly, in a murine tumour model, concomitant stimulation of ICOS and blockade of CTLA-4 has proven to elicit potent synergistic anti-tumour responses, pleading for clinical exploration of combination regimen of ICOS stimulation with ICI, especially anti-CTLA-4 antibodies (60, 61). Future research may focus on antibody engineering to enhance the potential of ICOShi Treg depletion via antibody-dependent cytotoxicity and also the timing of Treg depletion with respect to ICOS stimulation. On another note, ICOS stimulation has recently been combined with PD-1 blockade. Yap and colleagues reported on a phase 1/2 trial on vopratelimab (humanised IgG1 agonistic ICOS antibody; alias: JTX-2011) combined or not with nivolumab in refractory advanced-stage solid tumours, including 2 CCA patients in the monotherapy arm (62). Though vopratelimab alone or in combination with nivolumab was tolerated well, objective response rates (ORRs) were only 1.4% and 2.3% respectively.
3.1.3 CD226, TIM, and CD2/SLAM IgSF subfamily members as potential targets for ICS
CD226 (alias: DNAM1, TLiSA1) is a constitutively expressed co-stimulatory receptor that was discovered first in 1985 (63). Being expressed mostly by effector T cells, Tregs, and NK cells, CD226 regulates immune activity via interplay with its ligands CD155 and CD112.
At the tumour site CD226 expression tends to be diminished due to PD-1 and TIGIT signaling, local regulation through TGF-ß, and proteasomal cleavage (64). Given the significant immunostimulatory role of CD226 via VAV1, agonistic CD226-targeting mAbs could potentially serve as promising anti-tumour regimen (65, 66). However, CD226 plays a significant role in blood platelet adhesion and activation as well, potentially complicating its clinical application. T cell Ig and ITIM domain (TIGIT) competes with CD226 for binding CD155 (Kd 114-119nM vs. 1-3nM) and CD112 (Kd 0.31-8.97uM vs. not measureable) (67). In HCC-derived CTL, Th, and Treg TILs the TIGIT/CD226 ratio appeared to be upregulated (68). Therefore, enhancing CD226 signaling specifically on TILs might be achieved indirectly using selective blockade of TIGIT. Indeed, low PD1 expressing CD8+ T cells reacted to anti-PD-1 and anti-TIGIT combination regimen in a CD226-dependent manner in vitro whereas these CTLs did not respond to anti-PD-1 alone. Concordantly, in the setting of NSCLC, Banta, et al. demonstrated the importance of CD226 expression for optimal anti-tumour CD8+ T cell responses in the context of therapeutic ICI via PD-1 or TIGIT (69).
TIM-1 is a co-stimulatory receptor that is part of the IgSF TIM subfamily. TIM-1 ligands are TIM-4 and phophatidylserine. TIM-1 expression is induced upon activation of T cells, NK cells, B cells, macrophages, DCs, and mast cells (70). In a mouse transplant model, agonistic TIM-1 mAb have shown to stimulate effector T cell function and deprogram Tregs (71). In HCC, TIM-1 expressing B cells have been demonstrated to delineate immune suppressive subsets. TIM-1 targeting might therefore have a stimulatory effect by deprogramming Bregs as well (72). Stimulating effector T cells and hampering regulatory subsets, TIM-1-mediated ICS might be a potential candidate for enhancing anti-tumour activity (73). However, recently TIM-1 expression by cervical cancer cells was associated with cancer proliferation and migration indicating harmful effects may also arise. The expression of TIM-1 on liver tumour cells is unknown, hence more studies are required before agonistic targeting of TIM-1 can be applied in the clinic (74).
SLAMF6 (alias: NTB-A) is expressed on B, NK, and T cells as well. Remarkably, SLAMF6 expression is strongly correlated to the expression of T cell factor 1 (TCF-1) (75). Both are described to delineate progenitor exhausted CD8+ T cells from terminally-exhausted CTLs, making SLAMF6-mediated ICS of great interest in rescuing tumour-specific CTLs (76). SLAMF6 has neither been characterised in HCC nor in CCA. However, in HCC progenitor exhausted CD8+ T cells have been demonstrated ex vivo by PD1int, TCF-1+, TOXlo expression, providing a potential reservoir for SLAMF6-mediated ICS to reinvigorate anti-tumour immunity (68).
3.2 TNFRSF structure, expression, and ligands
The human TNF-super-family (TNFSF) consists of 29 receptors and 19 ligands. Upon ligation, TNF receptor monomers multimerise into trimeric ligand-receptor complexes that engage TNF receptor-associated factor (TRAF) adaptor proteins (Figure 2). TNF-/TNF-super-family members are divided into four separate categories of which the type-V (divergent) and type-L (conventional) play a prominent role in stimulatory T cell co-signalling (25). To date, 4-1BB, OX40, GITR, CD27, and CD40 are considered appropriate candidates for therapeutic immunomodulation.
3.2.1 Type V TNFSF receptor subfamily: 4-1BB
The activation-induced co-stimulatory molecule 4-1BB (aliases: TNFRSF9, CD137, ILA) was first described in 1989 (77). 4-1BB is transiently expressed upon TCR engagement on TILs, including activated CD8+ T cells (78–80), memory and regulatory CD4+ T cells (81), follicular CD4+ T (Tfh) cells (82), but also on NK cells (83). Additionally, 4-1BB expression by TILs is enhanced partly under hypoxic conditions through hypoxia-inducible factor 1-α (79). 4-1BB binds uniquely to its 4-1BB ligand (4-1BBL; aliases: TNFSF9, CD137L). 4-1BBL is expressed on antigen presenting cells such as dendritic cells (DCs), B lymphocytes, and macrophages (84, 85). Furthermore, upon inflammation, non-immunological human cells (e.g., smooth muscle cells, endothelial cells, hematopoietic stem cells) demonstrate expression of 4-1BBLas well, suggesting a role for these cells in effector T cell enhancement (86, 87). Besides a membrane-bound form (m4-1BB), 4-1BB also exists in a soluble form (s4-1BB) that results from alternative splicing (88). Although initially s4-1BB was observed in patients with autoimmune disease, increased levels have been demonstrated in haematological malignancies as well (89). Moreover, s4-1BB is hypothesised to function as a cancer immune escape mechanism by competing with m4-1BB for binding to 4-1BBL, thereby hampering intratumoural 4-1BB T cell costimulation (90).
In HCC and intrahepatic CCA, 4-1BB has been described to be exclusively expressed by CD4+ and CD8+ TILs (91, 92). Interestingly, Kim and colleagues have shown that 4-1BB delineates a distinct activation status among exhausted PD1hi CD8+ HCC-derived TILs (92). Compared to 4-1BB- TILs, 4-1BB+ PD1hi CD8+ T cells expressed higher levels of TCF-1, CD28, and T-bet/Eomes, indicating a greater potential for TIL reinvigoration. Accordingly, co-stimulation of CD8+ TILs using a humanised IgG4 4-1BB mAb further reinvigorated anti-PD-1-mediated CD8 function in vitro. As 4-1BB+ PD1hi CD8+ TILs are hypothesised to be tumour-reactive T cells, 4-1BB costimulation using agonistic antibodies may be promising anti-tumour strategy in HCC patients (92). Care should be taken, however, as 4-1BB has also been demonstrated to be expressed highly on Tregs in colorectal cancer-derived liver metastasis (CRLM), as well as on TIL-derived Tregs in HCC (93, 94). Nevertheless, pre-clinical mouse studies have revealed a potential of dual anti-tumour activities of 4-1BB mAb by which both Treg depletion and CD8 T cell promotion can be achieved by smartly exploiting antibody isotypes and FcγR-availability (95).
Care should be taken though as in 2008, high doses of urelumab (alias: BMS-663513), a fully humanised IgG4 mAb targeting 4-1BB, led to two hepatotoxicity-related deaths. Lower dosage regimens, however, appeared to be safe in later studies in hematological cancer patients and were accompanied by CR/PR of 0%/6%, 6%/6%, and 17%/0% in DLBCL, FL, and other B cell lymphomas, respectively (26) (96) In addition, Utomilumab (alias: PF-05082566), a fully humanised IgG2 mAb, has been studied as a single agent, engaging 4-1BB to mediate T cell ICS. It was well-tolerated safety profile among 55 patients (MCL, CRC, GC, PDAC, (N)SCLC, CCA, BC, Lymphoma, Sarcoma, etc.) (97). When combined with pembrolizumab, no dose-limiting toxicities were observed and 6 out of 23 patients demonstrated CR or PR (2 CR: RCC, SCLC; 4 PR: TC, RCC, NSCLC, and HNSCC) (98). Interestingly, clinical activity correlated with increased levels of peripheral activated memory/effector CD8+ T cells. Also, other combination regimen of utomilumab with mogamulizumab (anti-CCR4) or avelumab (anti-PD-L1) appeared to be safe, but anti-tumour activity remained relatively small (99, 100). Based on these experiences a very diverse landscape of second-generation 4-1BB agonists has recently been developed, with many entering clinical Phase 1 and 2 trials (101).
3.2.2 Type V TNFSF receptor subfamily: OX40
OX40 (aliases: TNFRSF4, CD134) is a co-stimulatory molecule that was discovered in 1987 by Paterson and colleagues (102). OX40 expression can be induced following TCR cross-linking on activated CD4+ and CD8+ T cells, and Tregs. Moreover, it is induced upon the activation of NK cells, NKT cells, and neutrophils (103–105). OX40 is overexpressed on T cells upon sustained TCR stimulation in the presence of CD28-mediated costimulation as well as IL2 (106). In vitro, OX40 gets upregulated on CD4+ and CD8+ TILs when exposed to autologous tumour cells (107). Accordingly, OX40 appears to be expressed at higher levels in TILs or tumour-draining lymph nodes when compared to PBMC-derived immune cells in melanoma, ductal mamma carcinoma, and head and neck cancer (108–112). OX40 ligand (OX40L; alias: TNFSF4, gp34) functions as the unique ligand for the OX40 receptor (113). OX40L is expressed transiently on antigen presenting cells such as DCs, B lymphocytes, and macrophages upon ligation of certain pattern recognition or cytokine receptors (e.g., TLR2, TLR4, TLR9, CD40, TSLPR, IL18R) (114–117). Moreover, T cells have been demonstrated to upregulate OX40L themselves as a result of TCR crosslinking upon T-T cell interactions, giving rise to sustained CD4+ T cell longevity (118). Innate-derived immune cells such as NK cells and type 2/3 innate lymphoid cells (ILC) express OX40L when triggered by NKG2D or alarmin molecules, respectively (119, 120). As reported for 4-1BBL, non-immunological cells (endothelial cells, smooth muscle cells) can express OX40L under inflammatory conditions as well (121, 122). Though, the soluble variant of OX40L (sOX40L) is increased in some types of cancer, it cannot oligomerise and hence sOX40L does not properly stimulate OX40 (123).
In HCC, OX40 was found enriched in the TIME (124–127). Expression was enhanced specifically among Treg and CD4+ activated helper T cell (aTh) TIL subsets (124, 126). Ligation of OX40 markedly increased CD4+ T cell expansion in vitro (126). CD8+ TIL fractions, in contrast, showed only modest expression levels of OX40 (126). However, OX40 was largely co-expressed with exhaustion marker PD1 on CD8+ T cells from HCV-related HCC patients, suggesting prior antigen specific activation and thus hinting to pre-existing in situ anti-tumour reactivity (124). Moreover, OX40 correlated to higher expression of other immune-activation markers such as: CD68, TIM-3, and LAG3 (125). Though, OX40 expression is overall associated with anti-tumour immunity, in HCC it has also been correlated to more aggressive disease (i.e., displaying increased alpha feto-protein (AFP) and vascular invasion) and to impaired survival (125). In contrast, in CCA, increased expression of OX40 on PBMC-derived Th and CD8+ T cell was rather correlated to improved recurrence-free survival, hinting to a potential anti-tumour effect (128). Despite the association of OX40 to more aggressive disease in HCC, Treg, Th, and CTL in HCC-derived TILs can be skewed to the pro-inflammatory state upon multimerisation of OX40 using a hexameric OX40 ligand or bead-bound or Fc-engineered OX40 antibody in vitro. From these experiments it was suggested that FcγR(IIB)-mediated antibody multimerisation, to allow for OX40 trimerisation, is critical to effectively induce OX40-mediated anti-tumour immunity (126). Clinical trials on OX40 stimulation so far, however, have all applied traditional agonistic mAb with insufficient FcγR(IIB) affinities.
In a first attempt, a therapeutic agonistic mouse mAb to OX40 demonstrated tumour regression in 12 out of 30 late-stage cancer patients (129). Interestingly, in this study regression and SD were observed among patients with CCA. However, induction of human anti-mouse antibodies made re-administration of the agent impossible. More recently, Tavolimab (alias: MEDI0562), a humanised agonistic OX40 IgG1 antibody, was deemed safe in advanced-stage solid tumours but PR was observed in only 2 out of 50 patients, not comprising the single HCC patient included (130). Nonetheless, a marked increase in proliferation of peripheral CD4+ and CD8+ memory T cells was observed. Moreover, intra-tumoural FoxP3+ T cells decreased. Also, another fully humanised agonistic OX40 IgG1 antibody (BMS-986178) was shown to be safe in metastatic solid tumours (131). Clinically meaningful anti-tumour efficacy was not observed among any of the HCC patients included in this trial. Similar safety profiles on OX40 monotherapy have been observed in other recent clinical trials studying ICAGN01949 (fully humanised agonistic OX40 IgG1 mAb), GSK3174998 (fully humanised agonistic IgG1 mAb), or ivuxolimab (alias: PF-04518600; fully humanised agonistic IgG2 mAb) in advanced solid tumours and ivuxolimab in AML, respectively (132–135). Interestingly, one HCC patient, a non-responder to prior sorafenib treatment, demonstrated long-term tumour regression on ivuxolimab (132). A 30mg flat dose of ivuxolimab is currently evaluated in an expansion trial for efficacy, safety, and pharmacodynamics in HCC patients specifically. In patients treated with ICAGN01949 also one patient with metastatic CCA receiving 700mg demonstrated PR as best response with a largest decrease in tumour size of 41.9% from baseline (133). OX40 ligation may also hold promise in the neoadjuvant setting as was suggested by a recent study applying tavolimab pre-operably to HNSCC patients. In most patients enhanced immune activation in peripheral CD4+ and CD8+ T cells was observed and 4 out of 17 patients displayed expansion of putative tumour reactive CD103+CD39+CD8+ TILs. Importantly, in contrast to immune non-responsive patients, none of these patients developed recurrent disease (29).
Because of the modest efficacy of OX-40-mediated ICS, combination regimens have been clinically tested as well. BMS-986178 alone did not show any objective response in advanced-stage solid tumours. In combination with nivolumab and/or ipilimumab (anti-CTLA-4), ORR ranged from 0 to 13%, but did not surpass expected ORRs for anti-PD-1/CTLA-4 monotherapies (28). A second trial studying the combination of anti-OX40 antibodies with ICI (tavolimab vs. tavolimab with durvalumab (anti-PD-L1) or tremelimumab (anti-CTLA-4)) again demonstrated no improved clinical activity compared to monotherapy ICI (136). Combination therapies of multiple ICS strategies involving OX40-engagement (i.e., ivuxolimab with utolimumab and GSK3174998 with GSK1795091) do show anti-tumour immune reactivity. These trials, however, lacked a monotherapy arm (137, 138). Taken together, a clear clinical benefit using agonistic mAb to OX40 has not yet been demonstrated but there is remaining clinical potential for future FcγR(IIB)-binding antibodies.
3.2.3 Type V TNFSF receptor subfamily: GITR
Glucocorticoid-induced TNFR-related protein (GITR) (aliases: TNFRSF18, CD357, AITR) is a co-stimulatory molecule that was firstly described in 1997 (139). Tregs demonstrate constitutive high expression levels of GITR, whereas naive and memory TILs have lower expression levels (140, 141). Upon T cell activation via CD28 signalling, GITR expression can be enhanced rapidly in both Treg and effector TILs (140). GITR is expressed transiently at low to intermediate levels in B cells, and innate lymphocyte subsets such as macrophages, NK cells, and NKT cells as well (142–144). GITR binds uniquely to GITR ligand (GITRL; aliases: TNFSF18, CD357L) (145). GITRL is expressed on activated APCs such as macrophages, DCs, and B cells (143, 146, 147). GITRL has been demonstrated to be transiently upregulated upon TLR4 activation (148). Moreover, GITRL is expressed by endothelial cells (145, 149). It is therefore hypothesised to play a role in mediating leukocyte adhesion and migration.
In HCC, TIL-derived (CD4+CD25+ or CD4+FoxP3+) Tregs have enhanced GITR expression compared to Tregs in adjacent tissues or PBMC (150) (55). In vitro, co-culture of HCC-derived CD4+CD25- Th cells and CD4+CD25+ Tregs showed that GITR ligation prevented hypo-responsiveness of effector CD4+ T cells, suggesting GITR engagement either hampered Tregs or stimulated Th cells directly (55). Even though GITR is most abundant on activated CD4+ Tregs, it is also detected on CD4+ Th and CD8+ effector TILs (151–153). Interestingly, these TILs demonstrated co-expression of GITR with other activation induced checkpoint inhibitors and stimulators such as CTLA-4, PD1, and 4-1BB offering opportunity for combination therapies (151, 152). When GITR ligation was combined with CTLA-4-mediated ICI in vitro on HCC-derived TILs, immunosuppression by tumour-derived Tregs was abrogated completely (151). in addition, HCC-derived CD8+ TILs were functionally enhanced when GITR ligation was combined with PD-1 blockade, further paving the way for combination therapies (92, 152). Similar results were obtained for intra-hepatic cholangiocarcinoma (iCCA) where CD4+ Tregs also demonstrated higher GITR expression compared to CD4+ Th, and CD8+ effector cells (19). Additionally, in vitro GITRL enhanced proliferation of both pre-stimulated CD4+ and CD8+ TILs compared to anti-PD-1- or -CTLA-4-mediated ICI. Interestingly, in CCA GITRL is downregulated on mDCs and monocytes in TILs when compared to adjacent liver tissues (19). Thereby, potentially hampering stimulation of anti-tumour TIL activity. Similar to CD4+ T cells, CD8+ TILs largely co-expressed GITR with other checkpoint molecules underscoring the potential of combination-therapies in both CCA and HCC.
Clinical activity of GITR-mediated ICS using TRX-518, a humanised agonistic GITR glycosylated IgG1 mAb, was evaluated in 43 patients with refractory solid tumours including 1 HCC, 1 fibrolamellar, and 1 CCA patient (30). TRX-518 was safe and depleted peripheral Tregs. Nevertheless, patients developed neither PR nor CR. CD8+ T cell exhaustion was hypothesised to cause clinical inactivity, pleading for combinatorial approaches using PD-1-mediated ICI. Similarly, MK-1248, a humanised agonistic GITR IgG4 antibody had no clinical effect as monotherapy for patients with solid cancers, including 1 HCC patient (154). However, when combined with pembrolizumab 1 and 2 out of 17 patients developed CR and PR, respectively (HNSCC, melanoma and cancer e.c.i.). In contrast, in an interim-analysis of a phase 2 trial on advanced solid tumours, no additional clinical activity of GITR engagement using BMS-986156 (humanised agonistic GITR IgG1 mAb) to nivolumab was demonstrated (2 and 19 out of 252 CR and PR, resp.) (27) Still, 5 out of the 12 HCC patients in the combination arm experienced radiological disease control of which four had SD and one had PR. In ICI-naïve but not ICI experienced melanoma patients, MK-4166 (humanised agonistic GITR IgG1 mAb), significantly increased the ORR (5 and 3 out of 13 CR and PR, resp.). Moreover, combination of MK-4166 with pembrolizumab did not result in enhanced clinical activity as was observed in the single HCC patient that got doublet therapy (155). In accordance, GWN323, a humanised agonistic GITR IgG1 mAb, appeared to be safe, but showed minimal clinical activity as monotherapy and modest clinical benefit in combination with spartalizumab (anti-PD-1 mAb) in both advanced solid tumours as well as lymphomas (156). In conclusion, GITR appears to be an attractive target for ICS in vitro. In light of the importance of multimerisation of TNFRSF like OX40 the focus should maybe be shifted towards novel strategies to deliver adequate GITR-mediated ICS such as multimerisation approaches, to potentially reproduce advantageous results in vivo as well.
3.2.4 Type V TNFSF receptor subfamily: CD27
In contrast to other TNFRSF members, CD27 (alias: TNFRSF7) is expressed constitutively on naive and effector T cells. This suggests that CD27 may act earlier upon activation in priming of T cells. Though CD27 gets downregulated on effector phenotypes, it is still expressed at moderate levels by central- and effector-memory T cells. NK cells also express CD27, albeit at lower levels in activated subsets. Moreover, CD27 is expressed by germinal centre and memory B cells (157, 158). It ligates specifically to CD70 (alias: TNFSF7, CD27L) that is expressed upon activation of DCs, B cells, and T cells.
In HCC tumours CD27 was found expressed on tumour-infiltrating B and T cell but the majority of CD27+ TILs were CD3+ T cells (159, 160). Compared to healthy controls, circulating CD27+CD19+ B cells in HCC appeared to be decreased especially with disease progression (161). Accordingly, expression of CD27 on T and B cells has been shown to be associated positively with patient survival (159, 162). CD27 has been demonstrated to be co-expressed with CD38 in HCC tumours, potentially delineating NK cells (159). Concordantly, a fraction of HCC-derived liver-resident NK cells expressed CD27. Additionally, NK cell CD27 was downregulated upon increased tumour burden which corresponded with impaired cytotoxic capacity in vitro, suggesting CD27 expression may associate with prognosis (163).
Besides T cell priming and effector differentiation, continuous ligation of CD27 is considered to play a significant role in the induction of T cell exhaustion as well as Treg survival. Nevertheless, CD27 agonistic antibodies exhibit anti-tumour functionality in both pre-clinical in vitro and in vivo models by enhancing CD27-mediated ICS and depletion of Tregs via ADCC (164, 165). Agonistic targeting of CD27 to stimulate anti-tumour T cell reactivity has been studied on a smaller scale when compared to other ICS-mediated T cell engagement. Treatment of 25 (Melanoma, CRC, OC, PC, RCC, NSCLC) and 31 patients (melanoma, RCC), with varlilumab (alias: CDX-1127), a fully humanised agonistic CD27 IgG1 mAb, in a phase 1 dose-escalation and -expansion trial, respectively was well tolerated (166). Only 1 patient experienced dose-limiting toxicity (grade 3 asymptomatic hyponatremia). Generally, pro-inflammatory immune activation was observed as characterised by increased terminally differentiated effector memory CD8+ T cells, HLA-DR expression on CD4+ T cells, and INFγ responses to recall antigens. One out of 15 RCC patients in the expansion cohort demonstrated a PR. Similarly, in hematologic cancers, varlilumab was tolerated well up to the maximum tested dose (167). 30 and 4 patients were tested in a dose-expansion and -escalation design, respectively of which 1 out of 4 Non-Hodgkin’s Lymphoma (NHL) patients developed CR. Both trials have shown modest clinical activity, pleading for the importance of combination therapies. Already assessed was the combination of varlilumab with nivolumab that could be applied safely in patients with several advanced solid tumours, not involving PLC (168). Though the combination regimen was not compared to nivolumab only, the ORR was not greater than expected for anti-PD-1 monotherapy. Results from a trial (NCT03396445) testing clinical efficacy of another fully humanised agonistic CD27 antibody (MK-5890) in a cross-over design comparing MK-5890 monotherapy to the combination with pembrolizumab (anti-PD-L1) are pending. To our knowledge, no data on treatment of PLC with CD27 targeting Abs are available yet.
3.2.5 Type L receptor subfamily: CD40
CD40 (aliases: TNFRSF5, Bp50) is a co-stimulatory receptor that was discovered in 1986 (169). CD40 is constitutively expressed on APCs (e.g., macrophages, DCs, B cells) (170, 171). Furthermore, CD40 expression can be enhanced on fibroblasts as well as epithelial cells upon exposure to interferons (IFN) and tumour necrosis factor (TNF) (171, 172). In contrast to all other TFNRSF members, the orientation of CD40/CD40L on the DC:T cell synapse is inverted, with the receptor and ligand being expressed on the APC and T cell respectively, indicating a role in T cell priming rather than effector functionalities. CD40 ligand (CD40L; aliases: TNFSF5, CD154) serves as the sole ligand of the CD40 receptor (173). CD40L is primarily expressed by activated T cells and is upregulated upon TCR signalling (174). Under pro-inflammatory conditions, other immune cells that express CD40L are activated B cells, NK cells, mast cells, and basophils (175–178). CD40L expressing CD4+ T cells mainly interact with B cells in germinal centres and are therefore defined as Tfh cells (179). However, in immune oncology, CD40-mediated ICS is largely focused on the “licensing” of DCs allowing them to promote anti-tumour T cell activation and the skewing of macrophages (180). Generally, upon receptor crosslinking by CD40L, APCs upregulate major histocompatibility complex (MHC) molecules and co-stimulatory IgSF or TNFRSF ligands (e.g., CD70) (181). Moreover, CD40-activation induces secretion of cytokines that are crucial for CD8+ T cell activation as well as Th1 polarisation (e.g., IL12). By accomplishing enhanced antigen presentation and pro-inflammatory T cell support, CD40 activation potentiates anti-tumour immunity (181).
In HCC, CD40 is expressed on tumour infiltrating B cells and DCs. Total CD40 tumour expression correlated positively to improved survival, highlighting a significant role in anti-tumour immunity (159). Also in CCA, intra-tumoural CD40 expression was demonstrated to be an independent predictor for improved survival (182). However, in situ, the priming of anti-cancer immunity by APCs may be hampered in HCC, since intra-tumoural activated DCs showed less expression of CD40 when compared to adjacent tissues in the majority of patients, especially in those with tumour suppressor gene mutations (183, 184). Therefore, impaired DC maturation in HCC and CCA may prove to be a critical feature of tumour escape that offers therapeutic opportunity. Encouragingly, HCC PBMC-derived B cells transfected with HCC total RNA were able to induce cytotoxic T cell responses ex vivo upon activation with CD40L (185). Furthermore, CD40L-actived B cells of HCC patients were able to induce in vitro CD4+ and CD8+ responses in autologous TIL fractions to tumour-associated antigens (glypican-3, MAGE-C2) (18). These in vitro studies indicate that in HCC anti-tumour T cells can be induced when tumour antigens are effectively presented by well matured APC. As such there is potential for CD40 agonistic drugs in PLC. In four different CCA mouse models, treatment with an agonistic CD40 antibody alone achieved moderate anti-tumour immunity only (182). However, when combined with anti-PD-1 ICI, anti-tumour effects enhanced significantly by the induction of CD8+ T cell responses via activation of DCs and macrophages. Interestingly, anti-tumour activities were abrogated upon macrophage depletion, thus pointing out a critical role of myeloid cells for both CD40-mediated immunotherapy as well as effective anti-PD-1 therapy in CCA. These data highlight the possibility of targeting CD40 to facilitate T cell priming of non-inflammed tumours and support the combination of agonistic CD40-antibodies with T-cell targeting immunotherapy.
In 2007, selicrelumab (aliases: CP-870,893; RO7009789), a fully humanised agonistic CD40 IgG2 mAb, elicited a partial response in 4 out of 29 patients with advanced solid tumours. PR was observed in metastatic melanoma patients, and 1 out of 2 CCA patients experienced regression of a large hepatic metastasis (186). Even though, 55% of all study subjects developed grade 1-2 cytokine release syndrome (CRS) the safety profile was deemed acceptable. Similarly, in hematological cancer (B cell NHL), CD40 engagement via dacetuzumab (humanised agonistic CD40 IgG1 mAb; alias: SGN-40) was tolerated well amongst patients. Objective anti-tumour responses were reported in only a subset of patients (6 out of 50; 1 CR, 5 PR) (187). Other phase I and II trials on dacetuzumab in patients with multiple myeloma (MM) and diffuse large B-cell lymphoma (DLBCL), or on ChiLob7/4 (chimeric agonistic CD40 IgG1 mAb) in DLBCL patients have shown a similar safety profile as well as modest clinical activity (188–190). Recently, mitazalimumab, a fully humanised agonistic CD40 IgG1 (alias: ADC1013) has been shown to encompass a manageable safety profile among 95 patients with advanced solid tumours, unfortunately not including any PLC patients. Only 1 out of 95 patients (RCC) experienced partial response (191). These data have pleaded for identifying patients that are sensitive to CD40 engagement and again argue for combining CD40 agonistic antibodies with other regimen to enhance clinical activity. To enhance polarisation of macrophages to the pro-inflammatory M1 phenotype, sotigalimab (humised IgG1 agonistic CD40 antibody; alias: APX005M) was combined with inhibition of CSFR1 via cabiralizumab and co-administered with nivolumab (anti-PD-1) in patients with anti-PD-1/PD-L1-resistant melanoma, RCC, and NSCLC (192). Though the triplet therapy was tolerated reasonably and patient’s pharmacodynamics analysis suggested enhanced pro-inflammatory state, PR was reached in 1 out of 26 patients only. Lastly, in the phase 2 PRINCE trial studying nivolumab/chemotherapy, sotigalimab/chemotherapy, and nivolumab/sotigalimab/chemotherapy in metastatic PDAC patients, modest increase in OS has been observed in the nivolumab and sotigagalimab arms versus historical controls (193). However, only the nivolumab/chemotherapy arm met the primary study endpoint. Interestingly, analysis on PR-patients in the sotigalimab arm revealed that mainly genetic signatures related to CD4+ T cells, B cells, and DC subsets tend to predict for improved OS, in line with the envisioned mechanism of CD40 stimulation. Therefore, since CD40-mediated ICS induces T cell priming, agonistic CD40 antibodies should rather be used in combination regimen that offer tumour specific antigens as T cell targets or alleviate T cell suppression, like vaccination approaches or ICI respectively. Recent studies also suggests that we may not be looking at the right compartment to observe an immunomodulatory effect of the treatment. We may need to focus on tumour-draining lymph nodes for markers that predict response to therapy (194).
4 Enhancing ICS-mediated anti-tumour activity in PLC
4.1 Enhancing ICS-mediated anti-tumour activity in PLC requires a different approach compared to ICI
To date, the majority of phase 1 and 2 clinical trials on agonistic mAbs targeting immune co-stimulatory receptors as monotherapy or in combination with ICI have consistently shown no to modest anti-tumour activity only (Table 2). Though current regimens are tolerated well, none so far demonstrate enhanced clinical efficacy. Strategies to enhance ICS-mediated functionality either at low dosages or at higher dosages in a more tumour-restricted manner could aid to unlock the full therapeutic potential of ICS-mediated immunotherapy to enhance pre-existing in situ anti-tumour immunity. For PLC, the diverse repertoire of liver-resident immune cells and the expression of relating Fcγ receptors (FcγRs) on these cells provides unique opportunities to deliver enhanced on-target ICS.
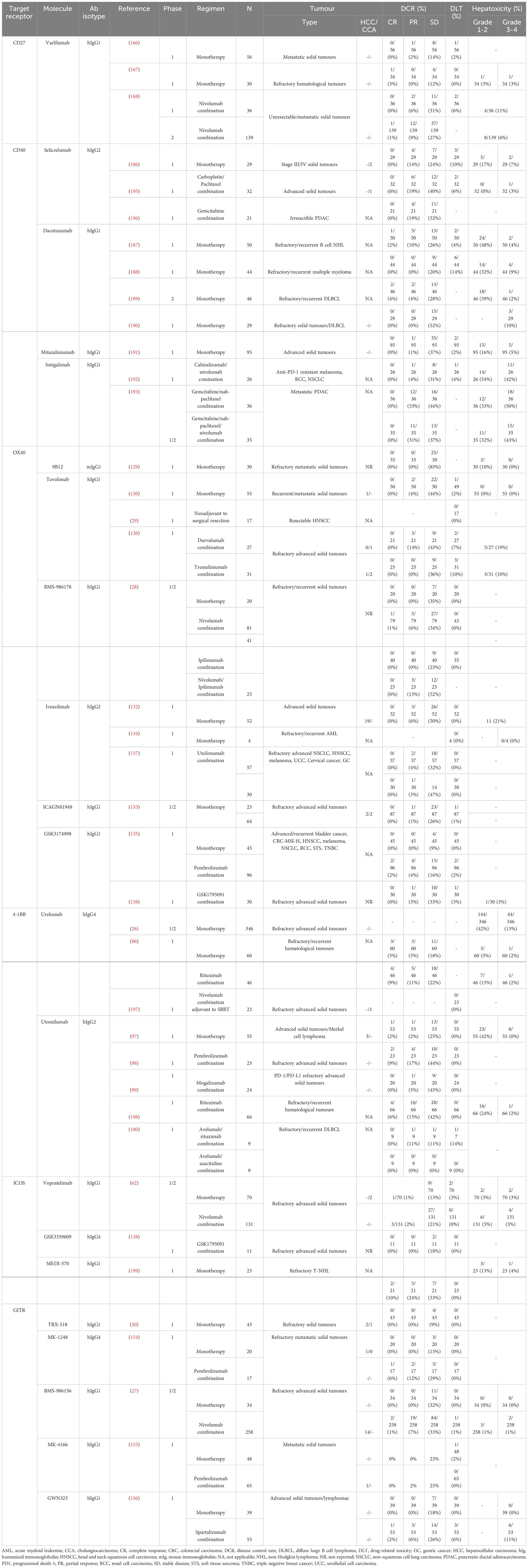
Table 2 Immune co-stimulatory receptors are targeted using various antibodies in phase 1/2 clinical trials among various types of cancer.
4.1.1 Translating principal differences of agonistic ICS Abs compared to antagonistic ICI Abs
Up to now, much clinical experience on anti-cancer immunotherapy has been obtained from large cohort studies. However, these studies have primarily focused on ICI using antagonising mAb. In contrast, high affinity towards cognate epitopes as well as ligand competition are of less importance to agonistic ICS approaches and this needs to be considered when developing such strategies.
4.1.2 Receptor super clustering is essential for downstream signaling of ICS
By nature, CD28-, ICOS-, and TNFRSF-mediated ICS signaling requires clustering or higher-order oligomerisation of costimulatory receptors: receptor super clustering (200). For example, soluble TNFRSF ligands demonstrate reduced agonistic capacities compared to membrane bound forms. A key factor for optimal ICS following agonistic binding is the potential of the mAb for multivalent binding and induction of receptor super clustering (201). Natural IgSF ligands (e.g., B7H1, ICOSL) configure as homodimers that account for bivalent engagement of separate singular IgSF receptors. Moreover, natural TNF ligands mostly present as tertiary confirmations that engage with cognate receptors in receptor-trimer complexes (i.e., 3:3 configuration). Receptor complex multimerisation has been demonstrated to greatly enhance co-stimulatory receptor activation. Preclinical and clinical trials have proven the synergistic effect of receptor clustering through the application of receptor ligands as well as multivalent ligands compared to monomeric signaling (202–204). Multivalent agonistic aptamers directed to OX40 and 4-1BB have shown to induce greater TNFRSF activation (205, 206). In vitro activation of HCC-derived TILs was enhanced upon exposure to multimeric OX40L and GITRL, whereas monomeric Abs did not or hardly stimulate CD4+ and CD8+ T cell proliferation, respectively (126, 152). Concordantly, whenever OX40 mAbs were coupled to beads, creating multimeric anti-OX40 IgG2, TIL expansion increased. These results highlight the significant role of TNFRSF multimerisation for activating TILs in HCC. Consequently, current agonistic ICS mAb may establish suboptimal receptor super clustering and this should be optimised to enhance ICS-mediated T cell activation.
4.1.3 Enhancement of FcγRIIB affinity increases the agonist potential of mAb through multimerisation
Natural IgG is capable of bridging multiple receptors via the two connected antigen-binding fragments (Fab) that induce monomer-monomer or oligomer interactions albeit insufficient to support proper ICS (207). Agonistic mAb should either actively drive receptor oligomerisation, stabilise self-assembled receptor oligomers, or bridge between pre-existing receptor-trimers (Figure 3). Receptor-trimer bridging can be achieved using FcγR-mediated receptor crosslinking. FcγRs bind the constant crystallizable fragment (Fc) domain of IgGs, and depending of the subtypes this binding induces activating or inhibitory signals regulating the function of various immune subsets (208). In human, six different FcγRs have been described of which 5 immune-activating (high affinity immunoglobulin-γ FcRI, FcγRI; low affinity immunoglobulin- γ FcRIIa, FcγRIIA; low affinity immunoglobulin-γ FcRIIc, FcγRIIC; low affinity immunoglobulin-γ FcRIIIa, FcγIIIA; low affinity immunoglobulin-γ FcRIIIb, FcγIIIB) and 1 inhibitory variant (FcγRIIB) (208). The inhibitory FcγRIIB is expressed on APC and myeloid subsets (DC, macrophage, B cell, NK cell, etc.) and uniquely functions as a scaffold crosslinking IgGs which can be exploited to crosslink also agonistic mAb (208).
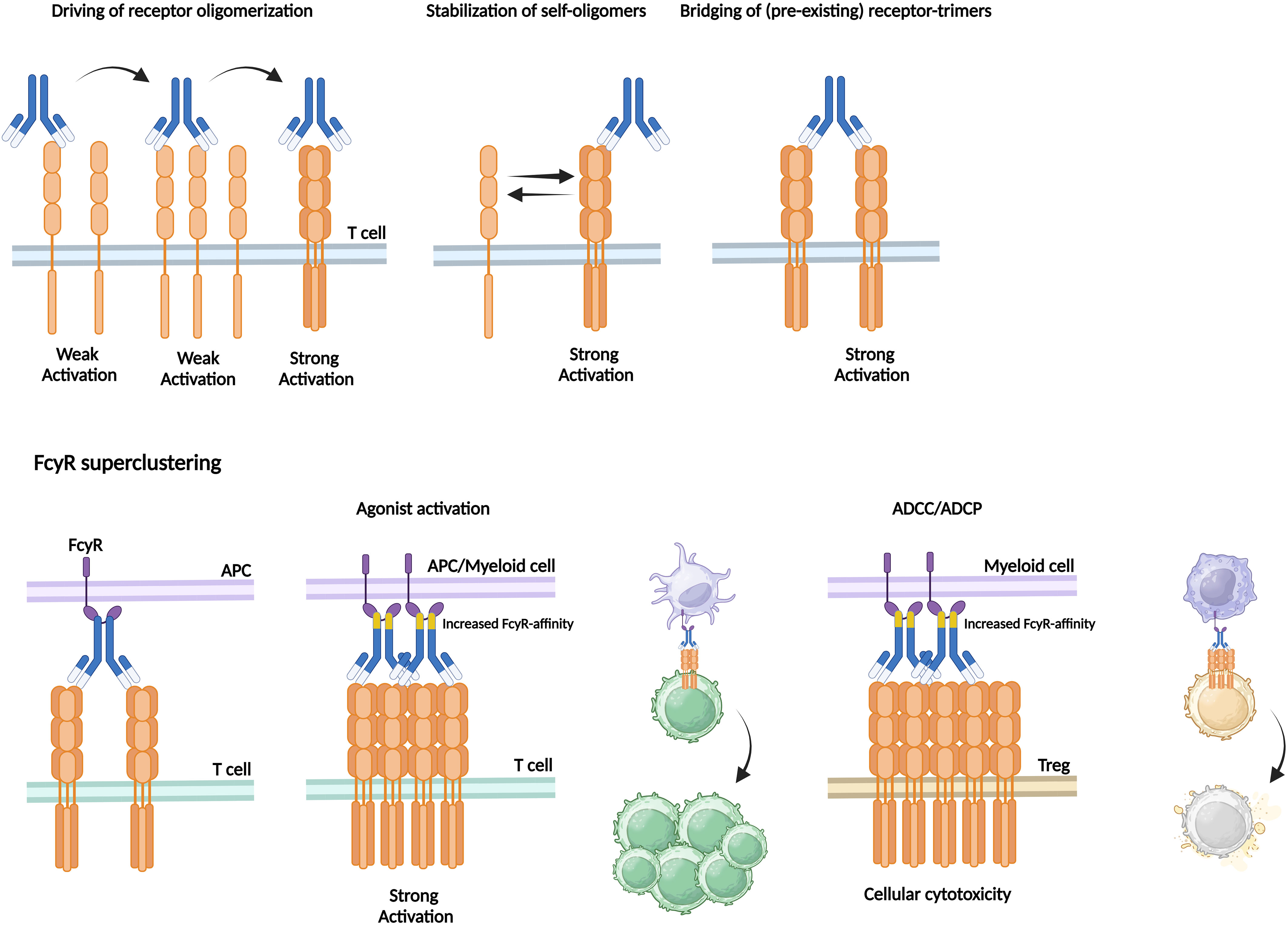
Figure 3 Agonistic mAb require receptor oligomerisation as can be enhanced by increased FcγR-affinity. To induce strong activation, agonistic mAb should either actively drive receptor oligomerisation, stabilise self-assembled receptor oligomers, or bridge between pre-existing receptor-trimers. Receptor-trimer bridging is achieved via FcγR-mediated receptor crosslinking. FcγR-expressing APCs bind to the Fc-region of the antibody that is bound to the target receptor expressed by T cells. Increased FcγR-affinity can enhance the extent of FcγR superclustering thereby inducing strong T cell activation followed by proliferation or enhanced ADCC/ADCP-mediated cellular cytotoxicity of for in stance immunosuppressive Tregs.
In HCC-derived TILs FcγRIIB is widely expressed, facilitating the FcγRIIB-dependent effect of any agonistic ICS mAb. The majority of intra-tumoural B cells and cDCs express FcγRIIB and to a smaller extent it is expressed on monocytes and NK cells (126). Binding to inhibitory FcγRIIB has been widely demonstrated to be of utmost importance for agonistic mAbs to drive potent CD28-, CD40-, OX40-, and 4-1BB-mediated ICS via receptor trimerisation (209–211). After mAb engage with their cognate TNFRSF epitope their Fc domains are captured by FcγRIIB-expressing APCs or myeloid cells forming a scaffold facilitating TNFRSF clustering and activation in trans. Accordingly, Campos-Carrascosa and colleagues treated HCC-derived TILs with an Fc-engineered αOX40 human IgG1 antibody, termed αOX40_v12, that contained six mutations leading to increased affinity to FcγRIIB (E223D, G237D, H268D, P271G, Y296D, A330R). When compared to the native agonistic αOX40 human IgG1 antibody αOX40_v12 appeared to improve in vitro TIL expansion and functionality. These novel generation Fc-engineered agonistic mAbs are currently widely studied (126, 212, 213).
4.1.4 Enhancement of activating FcγR engagement potentiates ADCC/ADCP-mediated anti-tumour activity of ICS agonistic Abs
In contrast to current ICI that relies mostly on direct T cell activation, agonistic ICS mAbs may also stimulate CD8+ effector T cells indirectly by the depletion of immunosuppressive cells (e.g., Treg). Antibodies binding to activating FcγRs expressed by NK cells, macrophages, or granulocytes, can trigger cell-mediated cytotoxic effector functions such as antibody-dependent cellular cytotoxicity (ADCC) and phagocytosis (ADCP) (214, 215). Upon mAb-antigen recognition, target cells are opsonised by antibodies, recognised by activating FcγR expressing cells and subsequently directly killed or phagocytosed by these cells. In human, FcγR3, expressed on NK cells and monocytes, is considered the primary activating receptor driving ADCC (216).
Whereas inhibitory FcγR2B engagement stimulates downstream signaling through receptor multimerisation, activating FcγR binding facilitates ADCC and ADCP activities of agonist ICS mAbs (157). Notably, engagement by FcγR2B reduces antibody availability for activating FcγRs (217). Therefore, IgG isotype selection is critical for the design of ICS mAb therapies. IgG1 has highest affinity to all activating FcγRs, whereas IgG2 and IgG4 only bind moderately to FcγRII and FcγR1 (IgG4 only) (216). mAbs with so called high activating: inhibitory (A:I) ratios tend to have greater cell-mediated cytotoxic effector functions, but lower agonistic activity (218). To establish effective anti-tumour immunity through ICS mAbs (esp. of the IgG1 isotype), the relative effect of immune cell activation versus immune cell depletion is likely determined by the tumour’s immune context.
As the TIME of PLC is highly enriched by immunosuppressive Tregs that hamper cytotoxic T cell-mediated anti-tumour immunity, greater ADCC effects might be desired rather than direct immune cell activation. Both HCC- and CCA-derived tumour infiltrating Tregs express high levels of co-stimulatory IgSF (ICOS) and TNFRSF (4-1BB, OX40, GITR) members (18, 55, 91, 92, 124, 126). As such, agonistic ICS mAbs of the IgG1 isotype might be of particular interest in depleting immune suppressive Tregs through ADCC mediated by liver resident Kupffer cells and NK cells that express relative high levels of FcγRIII (219, 220). Similar to discussed Fc-engineering strategies to increase binding to FcγRIIB, approaches to specifically enhance binding of the Fc domain to activating FcγRs could be employed to potentiate ADCC/ADCP-mediated anti-tumour activity of ICS agonistic Abs. Full anti-tumour immunity is however likely not to be expected from depletion of immunosuppressive subsets alone. Among various solid tumours in human, including HCC and CCA, Treg depletion by GITR agonism did increase Teff: Treg ratios, but was not sufficient to activate cytolytic cells due to persistent in situ TIL exhaustion (30). Accordingly, cytotoxic T cell re-invigoration in PLC might be supported by depletion of Tregs or any other immunosuppressing subsets but should likely be supported by ICS-mediated T cell activation and ICI in the appropriate dosing-regimen as well.
4.1.5 Agonist ICS mAbs require different dosing-regimen compared to ICI mAbs
In contrast to conventional ICI mAbs, ICS demonstrate a variable dose-response relationship. Classical mAbs primarily achieve clinical activity through receptor antagonism or ADCC, ADCP, and complement-dependent cytotoxicity (CDC). These modes of action establish optimal functionality at binding saturation of the cognate receptors. At peak receptor occupancy, increased dosage concentration will not induce any additional effect thereby reaching a plateau (201). In contrast, preclinical in vitro studies on ICS have shown clinical activity to decrease after a peak at specific concentrations has been reached (221, 222). Thus, agonistic ICS mAbs act in a bell-shaped dose-response rather than the classical sigmoidal dose-response functionality.
This mechanism might be partly attributed to the mAbs’ stoichiometric binding properties to the cognate receptors (201, 222). Theoretically, formation of maximal receptor superclustering should be achieved upon bisected molar concentrations of antibody to receptor thereby providing optimal bridging between the both. If antibody or receptor abundance exceed each other, inadequate antibody-receptor bridging would be established leading to isolated complexes with a 2:1 or 1:2 stoichiometry, respectively. At the dose-optimum, effective ICS-mediated T cell activation can be solely established directly via maximal receptor multimerisation. Therefore, optimal dosing strategies may require adaptations based on biomarkers (e.g., T cell proliferation or activation) to carefully monitor these dynamics.
Secondly, optimal activity at a certain dose-optimum might be explained by dynamical T cell functionality as well. Prolonged T cell activation through chronic antigen exposure via ICS-supported TCR signaling drives immune cell exhaustion leading to downregulation and activation-induced cell death. Whereas ICI mAbs disinhibit T cells that have been primed already, high dose agonistic ICS mAb concentrations could facilitate substantial T cell priming and overstimulation favouring subsequent T cell exhaustion. Combination regimen incorporating ICI might be a logical consideration to revert T cell exhaustion. However, albeit administered concomitantly rather than sequentially, various clinical trials failed to show any additional effect of ICS/ICI combination therapy over ICS monotherapies (27, 28, 135, 136, 154, 168). Therefore, better understanding of the process of T cell differentiation; in particular on the relation between priming, activation and exhaustion, is crucial. Effective combination regimen should preferentially first apply ICS agonists to enable tumour-specific T cell activation possibly even towards exhaustion. As activation and exhaustion are characterised by enhanced co-inhibitory receptor expression, this approach should then be followed by ICI mAbs to prevent suppression via co-inhibitory receptor ligands in the TIME (215, 223).
Other than ICI that bind to broadly expressed co-inhibitory receptors, co-stimulatory receptor expression might be relatively low on effector TILs, particularly on the cytotoxic immune compartments. As mentioned previously, co-stimulatory receptors are expressed fairly transiently on activated CD4+ and CD8+ T cells. However, HCC- and CCA-derived TILs demonstrate relatively lower expression of ICOS, OX40, and GITR on CD8+ T and (a)Th cells when compared to (a)Tregs (18, 126, 152). and thus it might be required to re-prime TILs prior to administration of any agonistic ICS Ab (combination) regimen. Strikingly, the pre-activation status of TILs correlated positively to ex vivo response rates upon OX40-mediated ICS (126). Several strategies could be of use here. i.e., toll-like receptor (TLR) agonists, vaccination, radiation or low dose metronomic chemotherapy to achieve immunogenic cell death. In HCC, previous locoregional therapies demonstrate to enhance Ki67 expression in HCC-derived TILs (126, 224). Thus, these data support the concept of priming the TIME prior to ICS-mediated anti-tumour immunity in PLC.
4.2 Tumour-targeted delivery of ICS through bispecific antibody approaches can reduce off-target (hepatotoxic) effects
Although, agonistic ICS mAbs aim to enhance anti-tumour functionality, they have been demonstrated to cause treatment-related immune-mediated adverse events. To some extent, prior priming of immune cells using locoregional therapies might direct immune activation towards the TIME. However, these effects may extend to non-tumourous surrounding tissues as well, potentially contributing to severe organ damage. Therefore, more tumour-restricted delivery of immune activation may be warranted by obligate bispecific antibodies (bsAbs).
Engineered IgG-like bsAbs have a single IgG incorporating two Fab arms that have distinct antigen specificities and can therefore be directed to distinct receptors (225). Physical linkage of two binding specificities warrants a dependency that can be either spatial (in-trans) or temporal (in-cis) (226). In-trans binding redirects effector T cell cytotoxicity to specifically eliminate target cells by linking T cells with tumour cells to form an immune synapse via a T-cell and tumour-binding domain. Similarly, ICS agonists functionality can be directed to the TIME using tumour-restricted antigen thereby increasing therapeutic efficacy and minimising any off-target ICS activities. Moreover, application of an ICS binding domain will not only attract T cells to the tumour site, but co-stimulatory receptor expressing NK cells as well, potentially leading to NK cell mediated toxicity. In-cis binding bi-specific antibodies co-targeting 2 receptors on the same cells may be used to restrict ICS activation to tumour-reactive T cells. Binding to distinct receptors would allow to simultaneously block two pathways (antagonist-antagonist pairing; e.g., PD-1 and CTLA-4) or pair antagonist to agonist (e.g., PD-1 to 4-1BB) and agonist to agonist (e.g., OX40 to 4-1BB). Though dual antagonist pairing (i.e., PD-1 x LAG3 and PD-1 x TIM3) seems to robustly enhance anti-tumour activity in the preclinical setting, heterodimerisation of different co-stimulatory receptors has to be studied in more detail (227–232). Intriguingly, some TNFRSF members have been demonstrated to signal as mixed oligomers (233). As TNFRSF generally engage shared downstream TRAF adaptor proteins, simultaneous receptor binding of bsAbs to distinct co-stimulatory receptors might allow for downstream ICS signaling that is equally effective as homodimerisation of individual receptors, albeit in a more tumour-specific manner.
In PLC, bsAbs have great potential in tumour-restricted delivery of ICS-mediated activation. Incorporation of a binding epitope directed to either tumour-associated antigen (TAA) or tumour-specific neoantigen (neoAg) might direct tumour cell targeting. Tumour-associated antigens (TAA) feature non-mutated amino acid sequences that are enriched within cancer cells, but may be presented by HLA on the surface of non-malignant cells as well. In HCC, TAA compromise oncofetal and cancer-germline antigens such as glypican 3 (GPC3) and melanoma-associated gene C1 (MAGE-C1) (234, 235). In CCA patients, TAA have been described in small cohorts only (236) and evidence on TAA-mediated oncogenicity and systemic TAA-reactive immune responses remains limited (128). However, in metastasised CCA patients Löffler and colleagues reported efficient tumour immune cell infiltration upon TAA-peptide vaccination in CCA metastatic lesions, suggesting the pre-existence of a TAA-reactive immune cell repertoire in these patients (237). bsAbs that target oncofetal protein GPC3 and 4-1BB are already in preclinical development (PRS-342) and might be promising candidates in directing and stimulating recently activated 4-1BB+ TILs to HCC tumour tissues. Lastly, dual receptor engagement might restrict ICS to tumour reactive HCC-derived TILs. As these subsets were described to be delineated by the expression of 4-1BB and PD1, bsAbs targeting both receptors could potentially specifically enhance anti-tumour immune activities (92).
5 Future application of ICS-mediated agonistic Abs in PLC management and conclusions
Though current immunotherapies for PLC have clearly shown to have some clinical effects, ICI-mediated antagonist antibodies reach clinical anti-tumour efficacy in a minor subset of advanced-stage HCC and CCA patients only (12, 15). Multi-facetted approaches addressing cytotoxic as well as immunosuppressive elements of the PLC TIME seem to improve anti-tumour immune activation (238). Therefore, attention and expectations have shifted towards combination treatments incorporating anti-PD-1, -PD-L1, and -CTLA-4 mAbs rather than single-agent ICI-regimen. In PLC, co-stimulatory receptors are widely expressed among immune regulatory and activatory cell subsets, thereby potentially facilitating reinvigoration of in situ anti-tumour activity in a dual manner.
When applied properly, ICS-mediated immune activation might hold great promise in enhancing the pool of HCC and CCA ‘responders’ at various stages of disease. As the HCC- and CCA-derived TIME are highly enriched for immunosuppressive cell subsets, single-agent ICS-regimen should primarily aim for the enhancement of A:I ratios and subsequent ADCC and ADCP functionalities. Elimination of Treg function appears crucial as the suppressive capacity of Tregs is potentially enhanced upon current anti-PD-L1 ICI regimen (239). Intra-tumoural Treg removal might allow more efficient activation of cytotoxic CD8+ T cells, either via direct ICS alone or in combination with in situ (ICS-supported) vaccination followed by previous ICI approaches.
Specifically in PLC, the use of new generation ICS-mediated Abs should be investigated to enhance binding to activating FcyRs to promote ADCC/ADCP. Given FcyRIIB-mediated inhibition of ADCC, minimal to no engagement to inhibitory FcyRs should be strived at. Alternative approaches to enhance co-stimulatory receptor multimerisation could still consist of biAbs or state-of-the-art Fc-coupled fusion proteins (240). Moreover, adequate dosing regimen should be determined for patients taking in situ TIL activation status into account. Application of such optimised ICS-mediated single- or combination-regimen might potentially make the HCC and CCA-derived TIME more susceptible to immunotherapies in advanced-stage disease.
Author contributions
YR: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing. SB: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing. JI: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing. DS: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors thank N. Delleman for her help in creating the illustrations. All schematic illustrations were created with BioRender.com.
Conflict of interest
SB has received grants from Pfizer, Merus, ISA Therapeutics BV. DS has received grants from Pfizer, Merus, ISA Therapeutics BV.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology (2021) 73:4–13. doi: 10.1002/hep.31288
3. Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol (2020) 17:557–88. doi: 10.1038/s41575-020-0310-z
4. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers (2021) 7:6. doi: 10.1038/s41572-020-00240-3
5. Valle JW, Kelley RK, Nervi B, Oh D-Y, Zhu AX. Biliary tract cancer. Lancet (2021) 397:428–44. doi: 10.1016/S0140-6736(21)00153-7
6. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet (2021) 398:1002–14. doi: 10.1016/S0140-6736(21)01206-X
7. Vansteenkiste J, Wauters E, Reymen B, Ackermann CJ, Peters S, De Ruysscher D. Current status of immune checkpoint inhibition in early-stage NSCLC. Ann Oncol (2019) 30:1244–53. doi: 10.1093/annonc/mdz175
8. Braun DA, Bakouny Z, Hirsch L, Flippot R, Van Allen EM, Wu CJ, et al. Beyond conventional immune-checkpoint inhibition — novel immunotherapies for renal cell carcinoma. Nat Rev Clin Oncol (2021) 18:199–214. doi: 10.1038/s41571-020-00455-z
9. Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: A randomized, double-blind, phase III trial. J Clin Oncol (2020) 38:193–202. doi: 10.1200/JCO.19.01307
10. Yau T, Park J-W, Finn RS, Cheng A-L, Mathurin P, Edeline J, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol (2022) 23:77–90. doi: 10.1016/S1470-2045(21)00604-5
11. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. New Engl J Med (2020) 382:1894–905. doi: 10.1056/NEJMOA1915745/SUPPL_FILE/NEJMOA1915745_DATA-SHARING.PDF
12. Cheng AL, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol (2022) 76:862–73. doi: 10.1016/j.jhep.2021.11.030
13. Ren Z, Xu J, Bai Y, Xu A, Cang S, Du C, et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2–3 study. Lancet Oncol (2021) 22:977–90. doi: 10.1016/S1470-2045(21)00252-7
14. Abou-Alfa GK, Lau G, Kudo M, Chan SL, Kelley RK, Furuse J, et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid (2022) 1(8). doi: 10.1056/EVIDoa2100070
15. Oh D-Y, Ruth He A, Qin S, Chen L-T, Okusaka T, Vogel A, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evidence (2022) 1(8). doi: 10.1056/EVIDoa2200015
16. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
17. Leone V, Ali A, Weber A, Tschaharganeh DF, Heikenwalder M. Liver inflammation and hepatobiliary cancers. Trends Cancer (2021) 7:606–23. doi: 10.1016/j.trecan.2021.01.012
18. Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology (2017) 153:1107–1119.e10. doi: 10.1053/j.gastro.2017.06.017
19. Zhou G, Sprengers D, Mancham S, Erkens R, Boor PPC, van Beek AA, et al. Reduction of immunosuppressive tumor microenvironment in cholangiocarcinoma by ex vivo targeting immune checkpoint molecules. J Hepatol (2019) 71:753–62. doi: 10.1016/j.jhep.2019.05.026
20. Montironi C, Castet F, Haber PK, Pinyol R, Torres-Martin M, Torrens L, et al. Inflamed and non-inflamed classes of HCC: a revised immunogenomic classification. Gut (2023) 72:129–40. doi: 10.1136/gutjnl-2021-325918
21. Loeuillard E, Conboy CB, Gores GJ, Rizvi S. Immunobiology of cholangiocarcinoma. JHEP Rep (2019) 1:297–311. doi: 10.1016/j.jhepr.2019.06.003
22. Philip M, Schietinger A. CD8+ T cell differentiation and dysfunction in cancer. Nat Rev Immunol (2022) 22:209–23. doi: 10.1038/s41577-021-00574-3
23. Barsch M, Salié H, Schlaak AE, Zhang Z, Hess M, Mayer LS, et al. T-cell exhaustion and residency dynamics inform clinical outcomes in hepatocellular carcinoma. J Hepatol (2022) 77:397–409. doi: 10.1016/j.jhep.2022.02.032
24. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol (2018) 15:325–40. doi: 10.1038/nrclinonc.2018.29
25. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13:227–42. doi: 10.1038/nri3405
26. Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res (2017) 23:1929–36. doi: 10.1158/1078-0432.CCR-16-1272
27. Heinhuis KM, Carlino M, Joerger M, Di Nicola M, Meniawy T, Rottey S, et al. Safety, tolerability, and potential clinical activity of a glucocorticoid-induced TNF receptor–related protein agonist alone or in combination with nivolumab for patients with advanced solid tumors: A phase 1/2a dose-escalation and cohort-expansion clinical trial. JAMA Oncol (2020) 6:100–7. doi: 10.1001/JAMAONCOL.2019.3848
28. Gutierrez M, Moreno V, Heinhuis KM, Olszanski AJ, Spreafico A, Ong M, et al. OX40 agonist BMS-986178 alone or in combination with nivolumab and/or ipilimumab in patients with advanced solid tumors. Clin Cancer Res (2021) 27:460–72. doi: 10.1158/1078-0432.CCR-20-1830
29. Duhen R, Ballesteros-Merino C, Frye AK, Tran E, Rajamanickam V, Chang S-C, et al. Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells. Nat Commun (2021) 12:1047. doi: 10.1038/s41467-021-21383-1
30. Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational design of anti-GITR-based combination immunotherapy. Nat Med (2019) 25:759–66. doi: 10.1038/s41591-019-0420-8
31. June CH, Ledbetter JA, Gillespie MM, Lindsten T, Thompson CB. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol (1987) 7:4472. doi: 10.1128/MCB.7.12.4472
32. Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J Autoimmun (2018) 95:77–99. doi: 10.1016/J.JAUT.2018.08.007
33. Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol (1991) 147:2461–6. doi: 10.4049/JIMMUNOL.147.8.2461
34. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity (2016) 44:973–88. doi: 10.1016/J.IMMUNI.2016.04.020
35. Yao S, Zhu Y, Zhu G, Augustine M, Zheng L, Goode DJ, et al. B7-H2 is a costimulatory ligand for CD28 in human. Immunity (2011) 34:729–40. doi: 10.1016/j.immuni.2011.03.014
36. Sansom DM. CD28, CTLA-4 and their ligands: who does what and to whom? Immunology (2000) 101:169–77. doi: 10.1046/j.1365-2567.2000.00121.x
37. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Sci (1979) (2017) 355:1423–7. doi: 10.1126/science.aaf0683
38. Maki A, Matsuda M, Asakawa M, Kono H, Fujii H, Matsumoto Y. Decreased expression of CD28 coincides with the down-modulation of CD3ζ and augmentation of caspase-3 activity in T cells from hepatocellular carcinoma-bearing patients and hepatitis C virus-infected patients. J Gastroenterol Hepatol (2004) 19:1348–56. doi: 10.1111/J.1440-1746.2004.03455.X
39. Hsu PN, Yang TC, Kao JT, Cheng KS, Lee YJ, Wang YM, et al. Increased PD-1 and decreased CD28 expression in chronic hepatitis B patients with advanced hepatocellular carcinoma. Liver Int (2010) 30:1379–86. doi: 10.1111/J.1478-3231.2010.02323.X
40. Ma J, Zheng B, Goswami S, Meng L, Zhang D, Cao C, et al. PD1Hi CD8+ T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J Immunother Cancer (2019) 7:331. doi: 10.1186/S40425-019-0814-7
41. Tatsumi T, Takehara T, Katayama K, Mochizuki K, Yamamoto M, Kanto T, et al. Expression of costimulatory molecules B7-1 (CD80) and B7-2 (CD86) on human hepatocellular carcinoma. Hepatology (1997) 25:1108–14. doi: 10.1002/HEP.510250511
42. Chan RCF, Xie Y. CD80 transfected human hepatocellular carcinoma cells activate cytotoxic T lymphocytes to target HCC cells with shared tumor antigens. Oncol Rep (2004) 12:435–42. doi: 10.3892/OR.12.2.435/HTML
43. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. New Engl J Med (2006) 355:1018–28. doi: 10.1056/NEJMoa063842
44. Beckermann KE, Hongo R, Ye X, Young K, Carbonell K, Healey DCC, et al. CD28 costimulation drives tumor-infiltrating T cell glycolysis to promote inflammation. JCI Insight (2020) 5(16):e138729. doi: 10.1172/jci.insight.138729
45. Sun Y, Wu L, Zhong Y, Zhou K, Hou Y, Wang Z, et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell (2021) 184:404–421.e16. doi: 10.1016/j.cell.2020.11.041
46. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397:263–6. doi: 10.1038/16717
47. Solinas C, Gu-Trantien C, Willard-Gallo K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open (2020) 5:544. doi: 10.1136/esmoopen-2019-000544
48. Duhen R, Fesneau O, Samson KA, Frye AK, Beymer M, Rajamanickam V, et al. PD-1 and ICOS coexpression identifies tumor-reactive CD4+ T cells in human solid tumors. J Clin Invest (2022) 132(12):e156821. doi: 10.1172/JCI156821
49. Strauss L, Bergmann C, Szczepanski M.J, Lang S, Kirkwood JM, Whiteside TL, et al. Expression of ICOS on human melanoma-infiltrating CD4+CD25highFoxp3+ T regulatory cells: implications and impact on tumor-mediated immune suppression. J Immunol (2008) 180:2967–80. doi: 10.4049/JIMMUNOL.180.5.2967
50. Yoshinaga SK, Whorlskey JS, Khare SD, Sarmiento U, Guo J, Horan T, et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature (1999) 402:827–32. doi: 10.1038/45582
51. Swallow MM, Wallin JJ, Sha WC. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFα. Immunity (1999) 11:423–32. doi: 10.1016/S1074-7613(00)80117-X
52. Zheng Y, Liao N, Wu Y, Gao J, Li Z, Liu W, et al. High expression of B7-H2 or B7-H3 is associated with poor prognosis in hepatocellular carcinoma. Mol Med Rep (2019) 19(5):4315–25. doi: 10.3892/mmr.2019.10080
53. Pedroza-Gonzalez A, Zhou G, Vargas-Mendez E, Boor PP, Mancham S, Verhoef C, et al. Tumor-infiltrating plasmacytoid dendritic cells promote immunosuppression by Tr1 cells in human liver tumors. Oncoimmunology (2015) 4(6):e1008355. doi: 10.1080/2162402X.2015.1008355/SUPPL_FILE/KONI_A_1008355_SM3671.ZIP
54. Tu JF, Ding YH, Ying XH, Wu FZ, Zhou XM, Zhang DK, et al. Regulatory T cells, especially ICOS+ FOXP3+ regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep (2016) 6:1–8. doi: 10.1038/srep35056
55. Pedroza-Gonzalez A, Verhoef C, Ijzermans JNM, Peppelenbosch MP, Kwekkeboom J, Verheij J, et al. Activated tumor-infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology (2013) 57:183–94. doi: 10.1002/HEP.26013
56. Di Blasi D, Boldanova T, Mori L, Terracciano L, Heim MH, De Libero G. Unique T-cell populations define immune-inflamed hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol (2020) 9:195–218. doi: 10.1016/J.JCMGH.2019.08.004
57. Lu LC, Deantonio C, Palu CC, Lee YH, Mitchell LS, Cowan M, et al. ICOS-positive regulatory T cells in hepatocellular carcinoma: the perspective from digital pathology analysis. Oncology (2022) 100:419–28. doi: 10.1159/000525239
58. Ng Tang D, Shen Y, Sun J, Wen S, Wolchok JD, Yuan J, et al. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol Res (2013) 1:229–34. doi: 10.1158/2326-6066.CIR-13-0020
59. Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti–CTLA-4 therapy. Cancer Res (2011) 71:5445–54. doi: 10.1158/0008-5472.CAN-11-1138
60. Soldevilla MM, Villanueva H, Meraviglia-Crivelli D, Menon AP, Ruiz M, Cebollero J, et al. ICOS costimulation at the tumor site in combination with CTLA-4 blockade therapy elicits strong tumor immunity. Mol Ther (2019) 27:1878–91. doi: 10.1016/j.ymthe.2019.07.013
61. Shi LZ, Goswami S, Fu T, Guan B, Chen J, Xiong L, et al. Blockade of CTLA-4 and PD-1 enhances adoptive T-cell therapy efficacy in an ICOS-mediated manner. Cancer Immunol Res (2019) 7:1803–12. doi: 10.1158/2326-6066.CIR-18-0873
62. Yap TA, Gainor JF, Callahan MK, Falchook GS, Pachynski RK, LoRusso P, et al. First-in-human phase I/II ICONIC trial of the ICOS agonist vopratelimab alone and with nivolumab: ICOS-high CD4 T-cell populations and predictors of response. Clin Cancer Res (2022) 28:3695–708. doi: 10.1158/1078-0432.CCR-21-4256
63. Burns GF, Triglia T, Werkmeister JA, Begley CG, Boyd AW. TLiSA1, a human T lineage-specific activation antigen involved in the differentiation of cytotoxic T lymphocytes and anomalous killer cells from their precursors. J Exp Med (1985) 161:1063–78. doi: 10.1084/jem.161.5.1063
64. Conner M, Hance KW, Yadavilli S, Smothers J, Waight JD. Emergence of the CD226 axis in cancer immunotherapy. Front Immunol (2022) 13:914406. doi: 10.3389/fimmu.2022.914406
65. Gaud G, Roncagalli R, Chaoui K, Bernard I, Familiades J, Colacios C, et al. The costimulatory molecule CD226 signals through VAV1 to amplify TCR signals and promote IL-17 production by CD4 + T cells. Sci Signal (2018) 11(538):eaar3083. doi: 10.1126/scisignal.aar3083
66. Chiang EY, Mellman I. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. J Immunother Cancer (2022) 10:e004711. doi: 10.1136/jitc-2022-004711
67. Chauvin J-M, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer (2020) 8:e000957. doi: 10.1136/jitc-2020-000957
68. Ge Z, Zhou G, Campos Carrascosa L, Gausvik E, Boor PPC, Noordam L, et al. TIGIT and PD1 Co-blockade Restores ex vivo Functions of Human Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Cell Mol Gastroenterol Hepatol (2021) 12:443–64. doi: 10.1016/j.jcmgh.2021.03.003
69. Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O’Gorman WE, et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity (2022) 55:512–526.e9. doi: 10.1016/j.immuni.2022.02.005
70. Jeong S, Park S-H. Co-stimulatory receptors in cancers and their implications for cancer immunotherapy. Immune Netw (2020) 20(1):e3. doi: 10.4110/in.2020.20.e3
71. Degauque N, Mariat C, Kenny J, Zhang D, Gao W, Vu MD, et al. Immunostimulatory Tim-1–specific antibody deprograms Tregs and prevents transplant tolerance in mice. J Clin Invest (2008) 118:735–41. doi: 10.1172/JCI32562
72. Xue H, Lin F, Tan H, Zhu Z-Q, Zhang Z-Y, Zhao L. Overrepresentation of IL-10-expressing B cells suppresses cytotoxic CD4+ T cell activity in HBV-induced hepatocellular carcinoma. PloS One (2016) 11:e0154815. doi: 10.1371/journal.pone.0154815
73. Xiao Y, Motomura S, Deyev V, Podack ER. TNF superfamily member 13, APRIL, inhibits allergic lung inflammation. Eur J Immunol (2011) 41:164–71. doi: 10.1002/eji.201040436
74. Chen L, Qing J, Xiao Y, Huang X, Chi Y, Chen Z. TIM-1 promotes proliferation and metastasis, and inhibits apoptosis, in cervical cancer through the PI3K/AKT/p53 pathway. BMC Cancer (2022) 22:370. doi: 10.1186/s12885-022-09386-7
75. Gartshteyn Y, Askanase AD, Mor A. SLAM associated protein signaling in T cells: tilting the balance toward autoimmunity. Front Immunol (2021) 12:654839. doi: 10.3389/fimmu.2021.654839
76. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
77. Kwon BS, Weissman SM. cDNA sequences of two inducible T-cell genes. Proc Natl Acad Sci (1989) 86:1963–7. doi: 10.1073/PNAS.86.6.1963
78. Hernandez-Chacon JA, Li Y, Wu RC, Bernatchez C, Wang Y, Weber JS, et al. Co-stimulation through the CD137/4-1BB pathway protects human melanoma tumor-infiltrating lymphocytes from activation-induced cell death and enhances anti-tumor effector function. J Immunother (2011) 34:236. doi: 10.1097/CJI.0B013E318209E7EC
79. Palazón A, Martínez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, et al. The HIF-1α hypoxia response in tumor-infi ltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discovery (2012) 2:608–23. doi: 10.1158/2159-8290.CD-11-0314/42917/P/THE-HIF-1-HYPOXIA-RESPONSE-IN-TUMOR-INFILTRATING-T
80. Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, et al. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res (2014) 20:44–55. doi: 10.1158/1078-0432.CCR-13-0945/85960/AM/CD137-ACCURATELY-IDENTIFIES-AND-ENRICHES-FOR
81. Freeman ZT, Nirschl TR, Hovelson DH, Johnston RJ, Engelhardt JJ, Selby MJ, et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J Clin Invest (2020) 130:1405–16. doi: 10.1172/JCI128672
82. Alfaro C, Echeveste JI, Rodriguez-Ruiz ME, Solorzano JL, Perez-Gracia JL, Idoate MA, et al. Functional expression of CD137 (4-1BB) on T helper follicular cells. Oncoimmunology (2015) 4:12. doi: 10.1080/2162402X.2015.1054597/SUPPL_FILE/KONI_A_1054597_SM9418.ZIP
83. Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol (1998) 190:167–72. doi: 10.1006/CIMM.1998.1396
84. Alderson MR, Smith CA, Tough TW, Davis-Smith T, Armitage RJ, Falk B, et al. Moslecular and biological characterization of human 4-1BB and its ligands. Eur J Immunol (1994) 24:2219–27. doi: 10.1002/EJI.1830240943
85. Pollok KE, Kim Y -J, Hurtado J, Zhou Z, Kim KK, Kwon BS. 4-1BB T-cell antigen binds to mature B cells and macrophages, and costimulates anti-μ-primed splenic B cells. Eur J Immunol (1994) 24:367–74. doi: 10.1002/EJI.1830240215
86. Seko Y, Sugishita K, Sato O, Takagi A, Tada Y, Matsuo H, et al. Expression of costimulatory molecules (4-1BBL and fas) and major histocompatibility class I chain-related A (MICA) in aortic tissue with takayasu’s arteritis. J Vasc Res (2004) 41:84–90. doi: 10.1159/000076437
87. Lee SW, Park Y, So T, Kwon BS, Cheroutre H, Mittler RS, et al. Identification of regulatory functions for 4-1BB and 4-1BBL in myelopoiesis and the development of dendritic cells. Nat Immunol (2008) 9:917–26. doi: 10.1038/ni.1632
88. Michel J, Langstein J, Hofstädter F, Schwarz H. A soluble form of CD137 (ILA/4-1BB), a member of the TNF receptor family, is released by activated lymphocytes and is detectable in sera of patients with rheumatoid arthritis. Eur J Immunol (1998) 28(1):290–5. doi: 10.1002/(SICI)1521-4141(199801)28:01
89. Salih HR, Schmetzer HM, Burke C, Starling GC, Dunn R, Pelka-Fleischer R, et al. Soluble CD137 (4-1BB) ligand is released following leukocyte activation and is found in sera of patients with hematological Malignancies. J Immunol (2001) 167:4059–66. doi: 10.4049/JIMMUNOL.167.7.4059
90. Labiano S, Palazón A, Bolaños E, Azpilikueta A, Sánchez-Paulete AR, Morales-Kastresana A, et al. Hypoxia-induced soluble CD137 in Malignant cells blocks CD137L-costimulation as an immune escape mechanism. Oncoimmunology (2016) 5(1):e1062967. doi: 10.1080/2162402X.2015.1062967/SUPPL_FILE/KONI_A_1062967_SM8002.ZIP
91. Wan YLe, Zheng SS, Zhao ZC, Li MW, Jia CK, Zhang H. Expression of co-stimulator 4-1BB molecule in hepatocellular carcinoma and adjacent non-tumor liver tissue, and its possible role in tumor immunity. World J Gastroenterol (2004) 10:195–9. doi: 10.3748/wjg.v10.i2.195
92. Kim HD, Park S, Jeong S, Lee YJ, Lee H, Kim CG, et al. 4-1BB delineates distinct activation status of exhausted tumor-infiltrating CD8+ T cells in hepatocellular carcinoma. Hepatology (2020) 71:955–71. doi: 10.1002/HEP.30881
93. Rakké YS, Campos Carrascosa L, van Beek AA, de Ruiter V, van Gemerden RS, Doukas M, et al. GITR ligation improves anti-PD1-mediated restoration of human MMR-proficient colorectal carcinoma tumor-derived T cells. Cell Mol Gastroenterol Hepatol (2023) 15:77–97. doi: 10.1016/j.jcmgh.2022.09.007
94. Desai J, Noordam L, Boor P, Bouzid R, Mittag D, Tacken P, et al. 883 MCLA-145, an anti CD137×PD-L1 bispecific antibody, induces T cell activation and proliferation in ex vivo models of hepatocellular carcinoma. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2022-SITC2022.0883
95. Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, et al. Antibodies to costimulatory receptor 4-1BB enhance anti-tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity (2018) 49:958–970.e7. doi: 10.1016/j.immuni.2018.09.014
96. Timmerman J, Herbaux C, Ribrag V, Zelenetz AD, Houot R, Neelapu SS, et al. Urelumab alone or in combination with rituximab in patients with relapsed or refractory B-cell lymphoma. Am J Hematol (2020) 95:510–20. doi: 10.1002/ajh.25757
97. Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, et al. Phase i study of single-agent utomilumab (PF-05082566), a 4-1bb/cd137 agonist, in patients with advanced cancer. Clin Cancer Res (2018) 24:1816–23. doi: 10.1158/1078-0432.CCR-17-1922/356969/P/PHASE-I-STUDY-OF-SINGLE-AGENT-UTOMILUMAB-PF
98. Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, et al. Phase Ib study of utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in combination with pembrolizumab (MK-3475) in patients with advanced solid tumors. Clin Cancer Res (2017) 23:5349–57. doi: 10.1158/1078-0432.CCR-17-1243/116631/AM/PHASE-IB-STUDY-OF-UTOMILUMAB-PF-05082566-A-4-1BB
99. Cohen EEW, Pishvaian MJ, Shepard DR, Wang D, Weiss J, Johnson ML, et al. A phase Ib study of utomilumab (PF-05082566) in combination with mogamulizumab in patients with advanced solid tumors. J Immunother Cancer (2019) 7:342. doi: 10.1186/s40425-019-0815-6
100. Hawkes EA, Phillips T, Budde LE, Santoro A, Saba NS, Roncolato F, et al. Avelumab in combination regimens for relapsed/refractory DLBCL: results from the phase ib JAVELIN DLBCL study. Target Oncol (2021) 16:761–71. doi: 10.1007/S11523-021-00849-8/FIGURES/3
101. Claus C, Ferrara-Koller C, Klein C. The emerging landscape of novel 4-1BB (CD137) agonistic drugs for cancer immunotherapy. MAbs (2023) 15:1. doi: 10.1080/19420862.2023.2167189
102. Paterson D, Jefferies W, Green J, Brandon M, Corthesy P, Puklavec M, et al. Antigens of activated rat T lymphocytes including a molecule of 50,000 Mr detected only on CD4 positive T blasts. Mol Immunol (1987) 24:1281–90. doi: 10.1016/0161-5890(87)90122-2
103. Mallett S, Fossum S, Barclay AN. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes–a molecule related to nerve growth factor receptor. EMBO J (1990) 9:1063–8. doi: 10.1002/J.1460-2075.1990.TB08211.X
104. Hendriks J, Xiao Y, Rossen JWA, van der Sluijs KF, Sugamura K, Ishii N, et al. During viral infection of the respiratory tract, CD27, 4-1BB, and OX40 collectively determine formation of CD8+ Memory T cells and their capacity for secondary expansion. J Immunol (2005) 175:1665–76. doi: 10.4049/jimmunol.175.3.1665
105. Turaj AH, Cox KL, Penfold CA, French RR, Mockridge CI, Willoughby JE, et al. Augmentation of CD134 (OX40)-dependent NK anti-tumour activity is dependent on antibody cross-linking. Sci Rep (2018) 8:1–11. doi: 10.1038/s41598-018-20656-y
106. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes bcl-xL and bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity (2001) 15:445–55. doi: 10.1016/S1074-7613(01)00191-1
107. Peng W, Williams LJ, Xu C, Melendez B, McKenzie JA, Chen Y, et al. Anti-OX40 antibody directly enhances the function of tumor-reactive CD8þ T cells and synergizes with PI3Kb inhibition in PTEN loss melanoma. Clin Cancer Res (2019) 25:6406–16. doi: 10.1158/1078-0432.CCR-19-1259/75393/AM/ANTI-OX40-ANTIBODY-DIRECTLY-ENHANCES-THE-FUNCTION
108. Vetto JT, Lum S, Morris A, Sicotte M, Davis J, Lemon M, et al. Presence of the T-cell activation marker OX-40 on tumor infiltrating lymphocytes and draining lymph node cells from patients with melanoma and head and neck cancers. Am J Surg (1997) 174:258–65. doi: 10.1016/S0002-9610(97)00139-6
109. Sarff MC, Edwards D, Dhungel B, Wegmann KW, Corless C, Weinberg AD, et al. OX40 (CD134) expression in sentinel lymph nodes correlates with prognostic features of primary melanomas. Am J Surg (2008) 195:621–5. doi: 10.1016/j.amjsurg.2007.12.036
110. Montler R, Bell RB, Thalhofer C, Leidner R, Feng Z, Fox BA, et al. OX40, PD-1 and CTLA-4 are selectively expressed on tumor-infiltrating T cells in head and neck cancer. Clin Transl Immunol (2016) 5:e70. doi: 10.1038/CTI.2016.16
111. Ramstad T, Lawnicki L, Vetto J, Weinberg A. Immunohistochemical analysis of primary breast tumors and tumor-draining lymph nodes by means of the T-cell costimulatory molecule OX-40. Am J Surg (2000) 179:400–6. doi: 10.1016/S0002-9610(00)00361-5
112. Xie F, Wang Q, Chen Y, Gu Y, Mao H, Zeng W, et al. Costimulatory molecule OX40/OX40L expression in ductal carcinoma in situ and invasive ductal carcinoma of breast: An immunohistochemistry-based pilot study. Pathol Res Pract (2010) 206:735–9. doi: 10.1016/J.PRP.2010.05.016
113. Baum PR, Gayle RB, Ramsdell F, Srinivasan S, Sorensen RA, Watson ML, et al. Molecular characterization of murine and human OX40/OX40 ligand systems: identification of a human OX40 ligand as the HTLV-1-regulated protein gp34. EMBO J (1994) 13:3992–4001. doi: 10.1002/J.1460-2075.1994.TB06715.X
114. Ohshima Y, Tanaka Y, Tozawa H, Takahashi Y, Maliszewski C, Delespesse G. Expression and function of OX40 ligand on human dendritic cells. J Immunol (1997) 159:3838–48.
115. Stüber E, Neurath M, Calderhead D, Perry Fell H, Strober W. Cross-linking of OX40 ligand, a member of the TNF/NGF cytokine family, induces proliferation and differentiation in murine splenic B cells. Immunity (1995) 2:507–21. doi: 10.1016/1074-7613(95)90031-4
116. Ito T, Wang Y-H, Duramad O, Hori T, Delespesse GJ, Watanabe N, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med (2005) 202:1213–23. doi: 10.1084/jem.20051135
117. Maxwell JR, Yadav R, Rossi RJ, Ruby CE, Weinberg AD, Aguila HL, et al. IL-18 bridges innate and adaptive immunity through IFN-γ and the CD134 pathway. J Immunol (2006) 177:234–45. doi: 10.4049/JIMMUNOL.177.1.234
118. Soroosh P, Ine S, Sugamura K, Ishii N. OX40-OX40 ligand interaction through T cell-T cell contact contributes to CD4 T cell longevity. J Immunol (2006) 176:5975–87. doi: 10.4049/JIMMUNOL.176.10.5975
119. Zingoni A, Sornasse T, Cocks BG, Tanaka Y, Santoni A, Lanier LL. Cross-talk between activated human NK cells and CD4+ T cells via OX40-OX40 ligand interactions. J Immunol (2004) 173:3716–24. doi: 10.4049/JIMMUNOL.173.6.3716
120. Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity (2018) 48:1195–1207.e6. doi: 10.1016/j.immuni.2018.05.003
121. Imura A, Hori T, Imada K, Ishikawa T, Tanaka Y, Maeda M, et al. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J Exp Med (1996) 183:2185–95. doi: 10.1084/JEM.183.5.2185
122. Burgess JK, Blake AE, Boustany S, Johnson PRA, Armour CL, Black JL, et al. CD40 and OX40 ligand are increased on stimulated asthmatic airway smooth muscle. J Allergy Clin Immunol (2005) 115:302–8. doi: 10.1016/j.jaci.2004.11.004
123. Müller N, Wyzgol A, Münkel S, Pfizenmaier K, Wajant H. Activity of soluble OX40 ligand is enhanced by oligomerization and cell surface immobilization. FEBS J (2008) 275:2296–304. doi: 10.1111/J.1742-4658.2008.06382.X
124. Piconese S, Timperi E, Pacella I, Schinzari V, Tripodo C, Rossi M, et al. Human OX40 tunes the function of regulatory T cells in tumor and nontumor areas of hepatitis C virus–infected liver tissue. Hepatology (2014) 60:1494–507. doi: 10.1002/HEP.27188
125. Xie K, Xu L, Wu H, Liao H, Luo L, Liao M, et al. OX40 expression in hepatocellular carcinoma is associated with a distinct immune microenvironment, specific mutation signature, and poor prognosis. Oncoimmunology (2018) 7(4):e1404214. doi: 10.1080/2162402X.2017.1404214/SUPPL_FILE/KONI_A_1404214_SM8526.DOCX
126. Campos Carrascosa L, van Beek AA, de Ruiter V, Doukas M, Wei J, Fisher TS, et al. FcγRIIB engagement drives agonistic activity of Fc-engineered αOX40 antibody to stimulate human tumor-infiltrating T cells. J Immunother Cancer (2020) 8:e000816. doi: 10.1136/jitc-2020-000816
127. Du P, Wang Z, Geng J, Wang Y. Expression and clinical significance of OX40 and OX40L mRNA in hepatocellular carcinoma. Bull Exp Biol Med (2021) 170:485–8. doi: 10.1007/S10517-021-05093-8/METRICS
128. Kida A, Mizukoshi E, Kido H, Toyama T, Terashima T, Arai K, et al. The characteristics of the immune cell profiles in peripheral blood in cholangiocarcinoma patients. Hepatol Int (2021) 15:695–706. doi: 10.1007/S12072-021-10177-8/METRICS
129. Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res (2013) 73:7189–98. doi: 10.1158/0008-5472.CAN-12-4174/650906/AM/OX40-IS-A-POTENT-IMMUNE-STIMULATING-TARGET-IN-LATE
130. Glisson BS, Leidner RS, Ferris RL, Powderly J, Rizvi NA, Keam B, et al. Safety and clinical activity of MEDI0562, a humanized OX40 agonist monoclonal antibody, in adult patients with advanced solid tumors. Clin Cancer Res (2020) 26:5358–67. doi: 10.1158/1078-0432.CCR-19-3070/76962/AM/SAFETY-AND-CLINICAL-ACTIVITY-OF-MEDI0562-A
131. Gutierrez M, Moreno V, Heinhuis KM, Olszanski AJ, Spreafico A, Ong M, et al. OX40 agonist BMS-986178 alone or in combination with nivolumab and/or ipilimumab in patients with advanced solid tumors. Clin Cancer Res (2021) 27:460–72. doi: 10.1158/1078-0432.CCR-20-1830/77539/AM/OX40-AGONIST-BMS-986178-ALONE-OR-IN-COMBINATION
132. Diab A, Hamid O, Thompson JA, Ros W, Eskens FALM, Doi T, et al. Open-label, dose-escalation study of the OX40 agonist ivuxolimab in patients with locally advanced or metastatic cancers. Clin Cancer Res (2022) 28:71–83. doi: 10.1158/1078-0432.CCR-21-0845/673892/AM/A-PHASE-I-OPEN-LABEL-DOSE-ESCALATION-STUDY-OF-THE
133. Davis EJ, Martin-Liberal J, Kristeleit R, Cho DC, Blagden SP, Berthold D, et al. First-in-human phase I/II, open-label study of the anti-OX40 agonist INCAGN01949 in patients with advanced solid tumors. J Immunother Cancer (2022) 10:e004235. doi: 10.1136/JITC-2021-004235
134. Short NJ, Borthakur G, Pemmaraju N, Dinardo CD, Kadia TM, Jabbour E, et al. A multi-arm phase Ib/II study designed for rapid, parallel evaluation of novel immunotherapy combinations in relapsed/refractory acute myeloid leukemia. Leuk Lymphoma (2022) 63(9):2161–70. doi: 10.1080/10428194.2022.2062345
135. Postel-Vinay S, Lam VK, Ros W, Bauer TM, Hansen AR, Cho DC, et al. First-in-human phase I study of the OX40 agonist GSK3174998 with or without pembrolizumab in patients with selected advanced solid tumors (ENGAGE-1). J Immunother Cancer (2023) 11:e005301. doi: 10.1136/JITC-2022-005301
136. Goldman JW, Piha-Paul SA, Curti B, Pedersen KS, Bauer TM, Groenland SL, et al. Safety and tolerability of MEDI0562, an OX40 agonist mAb, in combination with durvalumab or tremelimumab in adult patients with advanced solid tumors. Clin Cancer Res (2022) 28:3709–19. doi: 10.1158/1078-0432.CCR-21-3016/704886/AM/SAFETY-AND-TOLERABILITY-OF-MEDI0562-AN-OX40
137. Hamid O, Chiappori AA, Thompson JA, Doi T, Hu-Lieskovan S, Eskens FALM, et al. First-in-human study of an OX40 (ivuxolimab) and 4-1BB (utomilumab) agonistic antibody combination in patients with advanced solid tumors. J Immunother Cancer (2022) 10:e005471. doi: 10.1136/jitc-2022-005471
138. Steeghs N, Hansen AR, Hanna GJ, Garralda E, Park H, Strauss J, et al. Manufacturing-dependent change in biological activity of the TLR4 agonist GSK1795091 and implications for lipid A analog development. Clin Transl Sci (2022) 15:2625–39. doi: 10.1111/CTS.13387
139. Nocentini G, Giunchi L, Ronchetti S, Krausz LT, Bartoli A, Moraca R, et al. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc Natl Acad Sci U.S.A (1997) 94:6216–21. doi: 10.1073/PNAS.94.12.6216/ASSET/1A12F622-26EF-4356-A11D-458EC20E7948/ASSETS/GRAPHIC/PQ1173812006.JPEG
140. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol (2002) 3:135–42. doi: 10.1038/ni759
141. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4+CD25+ Immunoregulatory T Cells: Gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity (2002) 16:311–23. doi: 10.1016/S1074-7613(02)00280-7
142. Teodorovic LS, Riccardi C, Torres RM, Pelanda R. Murine B cell development and antibody responses to model antigens are not impaired in the absence of the TNF receptor GITR. PloS One (2012) 7:e31632. doi: 10.1371/JOURNAL.PONE.0031632
143. Hanabuchi S, Watanabe N, Wang YH, Wang YH, Ito T, Shaw J, et al. Human plasmacytoid predendritic cells activate NK cells through glucocorticoid-induced tumor necrosis factor receptor-ligand (GITRL). Blood (2006) 107:3617–23. doi: 10.1182/BLOOD-2005-08-3419
144. Kim HJ, Kim HY, Kim BK, Kim S, Chung DH. Engagement of glucocorticoid-induced TNF receptor costimulates NKT cell activation In Vitro and In Vivo. J Immunol (2006) 176:3507–15. doi: 10.4049/JIMMUNOL.176.6.3507
145. Kwon B, Yu KY, Ni J, Yu GL, Jang IK, Kim YJ, et al. Identification of a novel activation-inducible protein of the tumor necrosis factor receptor superfamily and its ligand. J Biol Chem (1999) 274:6056–61. doi: 10.1074/JBC.274.10.6056
146. Yu KY, Kim HS, Song SY, Min SS, Jeong JJ, Youn BS. Identification of a ligand for glucocorticoid-induced tumor necrosis factor receptor constitutively expressed in dendritic cells. Biochem Biophys Res Commun (2003) 310:433–8. doi: 10.1016/J.BBRC.2003.09.024
147. Cardona ID, Goleva E, Ou LS, Leung DYM. Staphylococcal enterotoxin B inhibits regulatory T cells by inducing glucocorticoid-induced TNF receptor-related protein ligand on monocytes. J Allergy Clin Immunol (2006) 117:688–95. doi: 10.1016/j.jaci.2005.11.037
148. Suvas S, Kim B, Sarangi PP, Tone M, Waldmann H, Rouse BT. In vivo kinetics of GITR and GITR ligand expression and their functional significance in regulating viral immunopathology. J Virol (2005) 79:11935–42. doi: 10.1128/JVI.79.18.11935-11942.2005/ASSET/37C12AD7-837D-4C12-926B-92665C1D2282/ASSETS/GRAPHIC/ZJV0180567910006.JPEG
149. Nardelli B, Zaritskaya L, McAuliffe W, Ni Y, Lincoln C, Yun HC, et al. Osteostat/tumor necrosis factor superfamily 18 inhibits osteoclastogenesis and is selectively expressed by vascular endothelial cells. Endocrinology (2006) 147:70–8. doi: 10.1210/EN.2005-0518
150. Ormandy L, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res (2005) 65:2457–64. doi: 10.1158/0008-5472.CAN-04-3232
151. Pedroza-Gonzalez A, Zhou G, Singh SP, Boor PPC, Pan Q, Grunhagen D, et al. GITR engagement in combination with CTLA-4 blockade completely abrogates immunosuppression mediated by human liver tumor-derived regulatory T cells ex vivo. Oncoimmunology (2015) 4(12):e1051297. doi: 10.1080/2162402X.2015.1051297
152. van Beek AA, Zhou G, Doukas M, Boor PPC, Noordam L, Mancham S, et al. GITR ligation enhances functionality of tumor-infiltrating T cells in hepatocellular carcinoma. Int J Cancer (2019) 145:1111–24. doi: 10.1002/IJC.32181
153. Vence L, Bucktrout SL, Curbelo IF, Blando J, Smith BM, Mahne AE, et al. Characterization and comparison of GITR expression in solid tumors. Clin Cancer Res (2019) 25:6501–10. doi: 10.1158/1078-0432.CCR-19-0289/75138/AM/CHARACTERIZATION-AND-COMPARISON-OF-GITR-EXPRESSION
154. Geva R, Voskoboynik M, Dobrenkov K, Mayawala K, Gwo J, Wnek R, et al. First-in-human phase 1 study of MK-1248, an anti–glucocorticoid-induced tumor necrosis factor receptor agonist monoclonal antibody, as monotherapy or with pembrolizumab in patients with advanced solid tumors. Cancer (2020) 126:4926–35. doi: 10.1002/CNCR.33133
155. Papadopoulos KP, Autio K, Golan T, Dobrenkov K, Chartash E, Chen Q, et al. Phase I study of MK-4166, an anti-human glucocorticoid-induced tnf receptor antibody, alone or with pembrolizumab in advanced solid tumors. Clin Cancer Res (2021) 27:1904–11. doi: 10.1158/1078-0432.CCR-20-2886/78568/AM/PHASE-I-STUDY-OF-MK-4166-AN-ANTI-HUMAN
156. Piha-Paul SA, Geva R, Tan TJ, Lim DWT, Hierro C, Doi T, et al. First-in-human phase I/Ib open-label dose-escalation study of GWN323 (anti-GITR) as a single agent and in combination with spartalizumab (anti-PD-1) in patients with advanced solid tumors and lymphomas. J Immunother Cancer (2021) 9:e002863. doi: 10.1136/JITC-2021-002863
157. Wajant H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ (2015) 22:1727–41. doi: 10.1038/cdd.2015.109
158. Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood (2018) 131:39–48. doi: 10.1182/blood-2017-07-741025
159. Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut (2017) 66:342–51. doi: 10.1136/gutjnl-2015-310814
160. Unitt E, Rushbrook SM, Marshall A, Davies S, Gibbs P, Morris LS, et al. Compromised lymphocytes infiltrate hepatocellular carcinoma: The role of T-regulatory cells. Hepatology (2005) 41:722–30. doi: 10.1002/hep.20644
161. Wang X, Wang L, Ji F, Zhu J, Ayana D, Fang X. Decreased CD27 on B lymphocytes in patients with primary hepatocellular carcinoma. J Int Med Res (2012) 40:307–16. doi: 10.1177/147323001204000131
162. Tian M-X, Liu W-R, Wang H, Zhou Y-F, Jin L, Jiang X-F, et al. Tissue-infiltrating lymphocytes signature predicts survival in patients with early/intermediate stage hepatocellular carcinoma. BMC Med (2019) 17:106. doi: 10.1186/s12916-019-1341-6
163. Zhang Q-F, Yin W-W, Xia Y, Yi Y-Y, He Q-F, Wang X, et al. Liver-infiltrating CD11b–CD27– NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell Mol Immunol (2017) 14:819–29. doi: 10.1038/cmi.2016.28
164. Wasiuk A, Testa J, Weidlick J, Sisson C, Vitale L, Widger J, et al. CD27-mediated regulatory T cell depletion and effector T cell costimulation both contribute to antitumor efficacy. J Immunol (2017) 199:4110–23. doi: 10.4049/jimmunol.1700606
165. Ramakrishna V, Sundarapandiyan K, Zhao B, Bylesjo M, Marsh HC, Keler T. Characterization of the human T cell response to in vitro CD27 costimulation with varlilumab. J Immunother Cancer (2015) 3:37. doi: 10.1186/s40425-015-0080-2
166. Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, in patients with advanced solid tumors. J Clin Oncol (2017) 35:2028–36. doi: 10.1200/JCO.2016.70.1508
167. Ansell SM, Flinn I, Taylor MH, Sikic BI, Brody J, Nemunaitis J, et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, for hematologic Malignancies. Blood Adv (2020) 4:1917–26. doi: 10.1182/BLOODADVANCES.2019001079
168. Sanborn RE, Pishvaian MJ, Callahan MK, Weise A, Sikic BI, Rahma O, et al. Safety, tolerability and efficacy of agonist anti-CD27 antibody (varlilumab) administered in combination with anti-PD-1 (nivolumab) in advanced solid tumors. J Immunother Cancer (2022) 10:e005147–e005147. doi: 10.1136/JITC-2022-005147
169. Clark EA, Ledbetter JA. Activation of human B cells mediated through two distinct cell surface differentiation antigens, Bp35 and Bp50. Proc Natl Acad Sci (1986) 83:4494–8. doi: 10.1073/PNAS.83.12.4494
170. Alderson MR, Armitage RJ, Tough TW, Strockbine L, Fanslow WC, Spriggs MK. CD40 expression by human monocytes: regulation by cytokines and activation of monocytes by the ligand for CD40. J Exp Med (1993) 178:669–74. doi: 10.1084/JEM.178.2.669
171. Banchereau J, Dubois B, Fayette J, Burdin N, Briere F, Miossec P, et al. Functional CD40 antigen on B cells, dendritic cells and fibroblasts. Adv Exp Med Biol (1995) 378:79–83. doi: 10.1007/978-1-4615-1971-3_16/COVER
172. Karmann K, Hughes CCW, Schechner J, Fanslow WC, Pober JS. CD40 on human endothelial cells: inducibility by cytokines and functional regulation of adhesion molecule expression. Proc Natl Acad Sci (1995) 92:4342–6. doi: 10.1073/PNAS.92.10.4342
173. Lederman S, Yellin MJ, Krichevsky A, Belko J, Lee JJ, Chess L. Identification of a novel surface protein on activated CD4+ T cells that induces contact-dependent B cell differentiation (help). J Exp Med (1992) 175:1091–101. doi: 10.1084/JEM.175.4.1091
174. Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci (1992) 89:6550–4. doi: 10.1073/PNAS.89.14.6550
175. Bolduc A, Long E, Stapler D, Cascalho M, Tsubata T, Koni PA, et al. Constitutive CD40L expression on B cells prematurely terminates germinal center response and leads to augmented plasma cell production in T cell areas. J Immunol (2010) 185:220–30. doi: 10.4049/JIMMUNOL.0901689
176. Carbone E, Ruggiero G, Terrazzano G, Palomba C, Manzo C, Fontana S, et al. A new mechanism of NK cell cytotoxicity activation: the CD40–CD40 ligand interaction. J Exp Med (1997) 185:2053. doi: 10.1084/JEM.185.12.2053
177. Palma AM, Hanes MR, Marshall JS. Mast cell modulation of B cell responses: an under-appreciated partnership in host defence. Front Immunol (2021) 12:718499/BIBTEX. doi: 10.3389/FIMMU.2021.718499/BIBTEX
178. Yanagihara Y, Kajiwara K, Basaki Y, Ikizawa K, Ebisawa M, Ra C, et al. Cultured basophils but not cultured mast cells induce human IgE synthesis in B cells after immunologic stimulation. Clin Exp Immunol (2001) 111:136–43. doi: 10.1046/J.1365-2249.1998.00474.X
179. Deenick EK, Ma CS. The regulation and role of T follicular helper cells in immunity. Immunology (2011) 134:361–7. doi: 10.1111/J.1365-2567.2011.03487.X
180. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol (2018) 18:635–47. doi: 10.1038/s41577-018-0044-0
181. Vonderheide RH. CD40 agonist antibodies in cancer immunotherapy. Annu Rev Med (2020) 71:47–58. doi: 10.1146/annurev-med-062518-045435
182. Diggs LP, Ruf B, Ma C, Heinrich B, Cui L, Zhang Q, et al. CD40-mediated immune cell activation enhances response to anti-PD-1 in murine intrahepatic cholangiocarcinoma. J Hepatol (2021) 74:1145–54. doi: 10.1016/j.jhep.2020.11.037
183. Zekri A-RN, El Deeb S, Bahnassy AA, Badr AM, Abdellateif MS, Esmat G, et al. Role of relevant immune-modulators and cytokines in hepatocellular carcinoma and premalignant hepatic lesions. World J Gastroenterol (2018) 24:1228–38. doi: 10.3748/wjg.v24.i11.1228
184. Liu Z, Liu L, Guo CG, Yu S, Meng L, Zhou X, et al. Tumor suppressor gene mutations correlate with prognosis and immunotherapy benefit in hepatocellular carcinoma. Int Immunopharmacol (2021) 101:108340. doi: 10.1016/J.INTIMP.2021.108340
185. Shen S, Xu Z, Qian X, Ding Y, Yu L, Liu B. Autogeneic rna-electroporated CD40-ligand activated b-cells from hepatocellular carcinoma patients induce CD8+ T-cell responses ex vivo. Exp Oncol (2007) 29:137–43.
186. Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol (2007) 25:876–83. doi: 10.1200/JCO.2006.08.3311
187. Advani R, Forero-Torres A, Furman RR, Rosenblatt JD, Younes A, Ren H, et al. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. J Clin Oncol (2009) 27:4371–7. doi: 10.1200/JCO.2008.21.3017
188. Hussein M, Berenson JR, Niesvizky R, Munshi N, Matous J, Sobecks R, et al. A phase I multidose study of dacetuzumab (SGN-40; humanized anti-CD40 monoclonal antibody) in patients with multiple myeloma. Haematologica (2010) 95:845–8. doi: 10.3324/HAEMATOL.2009.008003
189. De Vos S, Forero-Torres A, Ansell SM, Kahl B, Cheson BD, Bartlett NL, et al. A phase II study of dacetuzumab (SGN-40) in patients with relapsed diffuse large B-cell lymphoma (DLBCL) and correlative analyses of patient-specific factors. J Hematol Oncol (2014) 7:1–9. doi: 10.1186/1756-8722-7-44/TABLES/6
190. Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, et al. Clinical and biological effects of an agonist anti-CD40 antibody a cancer research UK phase I study. Clin Cancer Res (2015) 21:1321–8. doi: 10.1158/1078-0432.CCR-14-2355/175429/AM/CLINICAL-AND-BIOLOGICAL-EFFECTS-OF-AN-AGONIST-ANTI
191. Moreno V, Perets R, Peretz-Yablonski T, Fourneau N, Girgis S, Guo Y, et al. A phase 1 study of intravenous mitazalimab, a CD40 agonistic monoclonal antibody, in patients with advanced solid tumors. Invest New Drugs (2022) 41:93–104. doi: 10.1007/S10637-022-01319-2/METRICS
192. Weiss SA, Djureinovic D, Jessel S, Krykbaeva I, Zhang L, Jilaveanu L, et al. A phase i study of APX005M and cabiralizumab with or without nivolumab in patients with melanoma, kidney cancer, or non-small cell lung cancer resistant to anti-PD-1/PD-L1. Clin Cancer Res (2021) 27:4757–67. doi: 10.1158/1078-0432.CCR-21-0903/673905/AM/A-PHASE-I-STUDY-OF-APX005M-AND-CABIRALIZUMAB-WITH
193. Padrón LJ, Maurer DM, O’Hara MH, O’Reilly EM, Wolff RA, Wainberg ZA, et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat Med 2022 28:6 (2022) 28:1167–77. doi: 10.1038/s41591-022-01829-9
194. Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, et al. The PD-1/PD-L1-checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell (2020) 38:685–700.e8. doi: 10.1016/j.ccell.2020.09.001
195. Vonderheide, et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. doi: 10.4161/onci.23033
196. Beatty, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. doi: 10.1126/science.1198443
197. Foster, et al. Phase I study of stereotactic body radiotherapy plus nivolumab and urelumab or cabiralizumab in advanced solid tumors. doi: 10.1158/1078-0432.CCR-21-0810
198. Gopal, et al. First-in-human study of utomilumab, a 4-1BB/CD137 agonist, in combination with rituximab in patients with follicular and other CD20+ non-hodgkin lymphomas. doi: 10.1158/1078-0432.CCR-19-2973
199. Chavez, et al. Targeting the inducible T-cell costimulator (ICOS) in patients with relapsed/refractory T-follicular helper phenotype peripheral T-cell and angioimmunoblastic T-cell lymphoma. doi: 10.1158/1078-0432.CCR-22-2955
200. Kucka K, Wajant H. Receptor oligomerization and its relevance for signaling by receptors of the tumor necrosis factor receptor superfamily. Front Cell Dev Biol (2021) 8:615141/BIBTEX. doi: 10.3389/FCELL.2020.615141/BIBTEX
201. Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discovery (2018) 17:509–27. doi: 10.1038/nrd.2018.75
202. Wyzgol A, Müller N, Fick A, Munkel S, Grigoleit GU, Pfizenmaier K, et al. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J Immunol (2009) 183:1851–61. doi: 10.4049/jimmunol.0802597
203. Chin SM, Kimberlin CR, Roe-Zurz Z, Zhang P, Xu A, Liao-Chan S, et al. Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat Commun (2018) 9:1–13. doi: 10.1038/s41467-018-07136-7
204. Richards DM, Marschall V, Billian-Frey K, Heinonen K, Merz C, Redondo Müller M, et al. HERA-GITRL activates T cells and promotes anti-tumor efficacy independent of FcγR-binding functionality. J Immunother Cancer (2019) 7:191. doi: 10.1186/S40425-019-0671-4
205. McNamara JO, Kolonias D, Pastor F, Mittler RS, Chen L, Giangrande PH, et al. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J Clin Invest (2008) 118:376–86. doi: 10.1172/JCI33365
206. Dollins CM, Nair S, Boczkowski D, Lee J, Layzer JM, Gilboa E, et al. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem Biol (2008) 15:675–82. doi: 10.1016/J.CHEMBIOL.2008.05.016
207. Nelson AD, Hoffmann MM, Parks CA, Dasari S, Schrum AG, Gil D. IgG Fab fragments forming bivalent complexes by a conformational mechanism that is reversible by osmolytes. J Biol Chem (2012) 287:42936–50. doi: 10.1074/jbc.M112.410217
208. Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol (2020) 20:633–43. doi: 10.1038/s41577-020-00410-0
209. Li F, Ravetch JV. Inhibitory Fcγ receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Sci (1979) (2011) 333:1030–4. doi: 10.1126/SCIENCE.1206954/SUPPL_FILE/LI.SOM.PDF
210. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med (2013) 210:1685–93. doi: 10.1084/JEM.20130573
211. Qi X, Li F, Wu Y, Cheng C, Han P, Wang J, et al. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcγR affinity. Nat Commun (2019) 10:2141. doi: 10.1038/s41467-019-10088-1
212. Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res (2013) 73:7189–98. doi: 10.1158/0008-5472.CAN-12-4174/650906/AM/OX40-IS-A-POTENT-IMMUNE-STIMULATING-TARGET-IN-LATE
213. Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, et al. Antibodies to costimulatory receptor 4-1BB enhance anti-tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity (2018) 49:958–970.e7. doi: 10.1016/J.IMMUNI.2018.09.014
214. Stewart R, Hammond SA, Oberst M, Wilkinson RW. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J Immunother Cancer (2014) 2:29. doi: 10.1186/s40425-014-0029-x
215. Kraehenbuehl L, Weng C-H, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol (2022) 19:37–50. doi: 10.1038/s41571-021-00552-7
216. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol (2014) 5:520. doi: 10.3389/fimmu.2014.00520
217. Simpson AP, Roghanian A, Oldham RJ, Chan HTC, Penfold CA, Kim HJ, et al. FcγRIIB controls antibody-mediated target cell depletion by ITIM-independent mechanisms. Cell Rep (2022) 40:111099. doi: 10.1016/j.celrep.2022.111099
218. Beers SA, Glennie MJ, White AL. Influence of immunoglobulin isotype on therapeutic antibody function. Blood (2016) 127:1097–101. doi: 10.1182/blood-2015-09-625343
219. Sajid M, Liu L, Sun C. The dynamic role of NK cells in liver cancers: role in HCC and HBV associated HCC and its therapeutic implications. Front Immunol (2022) 13:887186. doi: 10.3389/fimmu.2022.887186
220. Bruggeman CW, Houtzager J, Dierdorp B, Kers J, Pals ST, Lutter R, et al. Tissue-specific expression of IgG receptors by human macrophages ex vivo. PloS One (2019) 14:e0223264. doi: 10.1371/journal.pone.0223264
221. Waibler Z, Sender LY, Kamp C, Müller-Berghaus J, Liedert B, Schneider CK, et al. Toward experimental assessment of receptor occupancy: TGN1412 revisited. J Allergy Clin Immunol (2008) 122:890–2. doi: 10.1016/j.jaci.2008.07.049
222. Liu H, Wu W, Sun G, Chia T, Cao L, Liu X, et al. Optimal target saturation of ligand-blocking anti-GITR antibody IBI37G5 dictates FcγR-independent GITR agonism and antitumor activity. Cell Rep Med (2022) 3(6):100660. doi: 10.1016/j.xcrm.2022.100660
223. Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, et al. Timing of PD-1 blockade is critical to effective combination immunotherapy with anti-OX40. Clin Cancer Res (2017) 23:6165–77. doi: 10.1158/1078-0432.CCR-16-2677
224. Noordam L, de Beijer MTA, Mancham S, Vogler I, Boor PPC, de Ruiter V, et al. Systemic T-cell and humoral responses against cancer testis antigens in hepatocellular carcinoma patients. Oncoimmunology (2022) 11(1):2131096. doi: 10.1080/2162402X.2022.2131096
225. Suurs FV, Lub-de Hooge MN, de Vries EGE, de Groot DJA. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther (2019) 201:103–19. doi: 10.1016/J.PHARMTHERA.2019.04.006
226. Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery (2019) 18:585–608. doi: 10.1038/s41573-019-0028-1
227. Jeong S, Park E, Kim H-D, Sung E, Kim H, Jeon J, et al. Novel anti-4-1BB×PD-L1 bispecific antibody augments anti-tumor immunity through tumor-directed T-cell activation and checkpoint blockade. J Immunother Cancer (2021) 9:e002428. doi: 10.1136/jitc-2021-002428
228. Peper-Gabriel JK, Pavlidou M, Pattarini L, Morales-Kastresana A, Jaquin TJ, Gallou C, et al. The PD-L1/4-1BB bispecific antibody–anticalin fusion protein PRS-344/S095012 elicits strong T-cell stimulation in a tumor-localized manner. Clin Cancer Res (2022) 28:3387–99. doi: 10.1158/1078-0432.CCR-21-2762
229. Fromm G, de Silva S, Johannes K, Patel A, Hornblower JC, Schreiber TH. Agonist redirected checkpoint, PD1-Fc-OX40L, for cancer immunotherapy. J Immunother Cancer (2018) 6:149. doi: 10.1186/s40425-018-0454-3
230. Chan S, Belmar N, Ho S, Rogers B, Stickler M, Graham M, et al. An anti-PD-1–GITR-L bispecific agonist induces GITR clustering-mediated T cell activation for cancer immunotherapy. Nat Cancer (2022) 3:337–54. doi: 10.1038/s43018-022-00334-9
231. Clancy-Thompson E, Perry T, Pryts S, Jaiswal A, Oganesyan V, van DN, et al. 461 Generation of AZD7789, a novel PD-1 and TIM-3 targeting bispecific antibody, which binds to a differentiated epitope of TIM-3. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2022-SITC2022.0461
232. Shi N, Zhou Y, Liu Y, Zhang R, Jiang X, Ren C, et al. PD-1/LAG-3 bispecific antibody potentiates T cell activation and increases antitumor efficacy. Front Immunol (2022) 13:1047610. doi: 10.3389/fimmu.2022.1047610
233. Smulski CR, Decossas M, Chekkat N, Beyrath J, Willen L, Guichard G, et al. Hetero-oligomerization between the TNF receptor superfamily members CD40, Fas and TRAILR2 modulate CD40 signalling. Cell Death Dis (2017) 8:e2601–1. doi: 10.1038/cddis.2017.22
234. Sideras K, Bots SJ, Biermann K, Sprengers D, Polak WG, IJzermans JNM, et al. Tumour antigen expression in hepatocellular carcinoma in a low-endemic western area. Br J Cancer (2015) 112:1911–20. doi: 10.1038/bjc.2015.92
235. Noordam L, Ge Z, Özturk H, Doukas M, Mancham S, Boor PPC, et al. Expression of cancer testis antigens in tumor-adjacent normal liver is associated with post-resection recurrence of hepatocellular carcinoma. Cancers (Basel) (2021) 13:2499. doi: 10.3390/cancers13102499
236. Zhou J, Li Y, Chen S, Deng A. Expression and prognostic significance of cancer-testis antigens (CTA) in intrahepatic cholagiocarcinoma. J Exp Clin Cancer Res (2011) 30:2. doi: 10.1186/1756-9966-30-2
237. Löffler MW, Chandran PA, Laske K, Schroeder C, Bonzheim I, Walzer M, et al. Personalized peptide vaccine-induced immune response associated with long-term survival of a metastatic cholangiocarcinoma patient. J Hepatol (2016) 65:849–55. doi: 10.1016/j.jhep.2016.06.027
238. Rimassa L, Finn RS, Sangro B. Combination immunotherapy for hepatocellular carcinoma. J Hepatol (2023) 79:506–15. doi: 10.1016/j.jhep.2023.03.003
239. van Gulijk M, van Krimpen A, Schetters S, Eterman M, van Elsas M, Mankor J, et al. PD-L1 checkpoint blockade promotes regulatory T cell activity that underlies therapy resistance. Sci Immunol (2023) 8(83):eabn6173. doi: 10.1126/sciimmunol.abn6173
Keywords: immune checkpoint stimulation, immunotherapy, hepatocellular carcinoma, cholangiocarcinoma, immunoglobulin superfamily, tumour necrosis factor receptor superfamily, receptor super clustering, bispecific antibody
Citation: Rakké YS, Buschow SI, IJzermans JNM and Sprengers D (2024) Engaging stimulatory immune checkpoint interactions in the tumour immune microenvironment of primary liver cancers – how to push the gas after having released the brake. Front. Immunol. 15:1357333. doi: 10.3389/fimmu.2024.1357333
Received: 17 December 2023; Accepted: 31 January 2024;
Published: 19 February 2024.
Edited by:
Jesse Haramati, University of Guadalajara, MexicoReviewed by:
Karim Amrane, Morlaix Hospital, FranceSuresh Kalathil, University at Buffalo, United States
Copyright © 2024 Rakké, Buschow, IJzermans and Sprengers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dave Sprengers, d.sprengers@erasmusmc.nl