 Bristy Basak
Bristy Basak Sachiko Akashi-Takamura
Sachiko Akashi-Takamura- Department of Microbiology and Immunology, School of Medicine, Aichi Medical University, Nagakute, Aichi, Japan
Lipopolysaccharide (LPS) induces potent cell activation via Toll-like receptor 4/myeloid differentiation protein 2 (TLR4/MD-2), often leading to septic death and cytokine storm. TLR4 signaling is diverted to the classical acute innate immune, inflammation-driving pathway in conjunction with the classical NF-κB pivot of MyD88, leading to epigenetic linkage shifts in nuclear pro-inflammatory transcription and chromatin structure-function; in addition, TLR4 signaling to the TIR domain-containing adapter-induced IFN-β (TRIF) apparatus and to nuclear pivots that signal the association of interferons alpha and beta (IFN-α and IFN-β) with acute inflammation, often coupled with oxidants favor inhibition or resistance to tissue injury. Although the immune response to LPS, which causes sepsis, has been clarified in this manner, there are still many current gaps in sepsis immunology to reduce mortality. Recently, selective agonists and inhibitors of LPS signals have been reported, and there are scattered reports on LPS tolerance and control of sepsis development. In particular, IRF3 signaling has been reported to be involved not only in sepsis but also in increased pathogen clearance associated with changes in the gut microbiota. Here, we summarize the LPS recognition system, main findings related to the IRF3, and finally immunological gaps in sepsis.
1 Introduction
Septic shock is characterized by a dangerous degree of hypotension, high fever, tachycardia and tachypnea due to severe sepsis (1) by various mechanisms, including the direct effects of bacterial toxins, such as endotoxin. Severe sepsis causes circulatory, cellular, and metabolic disturbances, as a result, organs are unable to receive adequate blood supply, resulting in dysfunction (2). In spite of careful treatments, the mortality rate in sepsis is still high (3). Immune-modulating therapy is the main treatment of septic shock, and extracorporeal blood purification remains contentious (2). Furthermore, it is unclear how the various innate immune pathways relate to induction or regulation of sepsis, because there are still many current gaps in sepsis immunology to reduce mortality.
Interferon regulatory factor 3 (IRF3) is a transcription regulator in many cell types, and key to rapid antiviral immune responses (4). After microbial infection, IRF3 is rapidly phosphorylated by kinases that are part of several pattern recognition receptors (PRRs) pathways such as TLR3 (5), TLR4 (6), retinoic acid–inducible gene I (RIG-I) (7), melanoma differentiation-associated protein 5 (MDA5) (8), and stimulator of IFN genes (STING) (9). After phosphorylation, IRF3 dimerizes and localizes to the nucleus, then it works as an integrating transcription factor for promoters of type I IFNs, several cytokines, as well as several antiviral IFN-stimulated genes (ISGs) (4). There are many reviews of IRF3 function during viral infection, but few recent reviews of IRF3 function in response to endotoxin. Here, we firstly review the mechanism of LPS recognition, secondly present results from IRF3-deficient mice, IRF3 inhibitors, and molecules that affect IRF3 in endotoxic sepsis, and outline the effects of IRF3 on bacterial clearance. Finally, we provide an overview of current gaps in sepsis immunology.
2 LPS recognition and response mechanism
LPS, also known as endotoxin, is a glycan-based pathogen-associated molecular pattern (PAMP) found on the cell surface of gram-negative bacteria that is composed of three domains: an amphipathic glycophospholipid (lipid A) component, hydrophilic polysaccharides in the core, and an O-antigen consisting of repeated units of common hexose sugar (10). Lipid A is a major inducer of the endotoxic properties of LPS. The induction of inflammatory responses by LPS starts with its binding to LPS binding protein (LBP), a 60 KD acute phase protein synthesized preliminary in the liver (11, 12). LPS aggregates in aqueous environments owing to its amphiphilic nature (13). LBP disrupts aggregation prior to its transfer to a cluster of differentiation 14 (CD14), which can be found either in a soluble form or linked to the cell surface by glycosylphosphatidylinositol anchor (14, 15).
2.1 TLR4 signaling diverting into the MyD88 classic NF-κB activation
The signaling responses are mainly supplied by TLR4 which is non-covalently linked to the soluble protein MD-2 (sMD-2) and secreted as a large disulfide-bound multimeric glycoprotein. CD14 immediately leaves LPS-LBP complexes after LPS is transferred to MD-2 (13). The association of the monomers LPS and MD-2 with the ectodomain of TLR4 triggers a series of consecutive events, including the dimerization of TLR4/MD-2 (16). This association triggers the formation of a hexametric ligand-receptor complex consisting of two copies of the TLR4/MD-2/LPS homodimer, an active complex. This results in the assembly of the intracellular Toll interleukin-1 receptor (TIR) domain, which further recruits downstream adaptor proteins that mediate the signaling cascade. TLR4 transduces the signals via two distinct pathways: MyD88 and TRIF dependent.
The MyD88-dependent pathway commences on the plasma membrane, whereas the TRIF-mediated signaling begins after internalization of the TLR4/MD-2/LPS complex into endosomes (17). The MyD88-dependent pathway is indispensable for the production of inflammatory cytokines (18, 19). After TIRAP (also known as MAL) is recruited to the TLR4, MyD88 triggers the formation of a complex by binding to serine/threonine kinases, interleukin-1 receptor-associated kinases 2 and 4 (IRAK2 and IRAK4) (20, 21). Then, TNF receptor-associated factor 6 (TRAF6), an ubiquitin E3 ligase, is recruited and NF-κB is activated, then proinflammatory cytokines are produced (Figure 1, priming step).
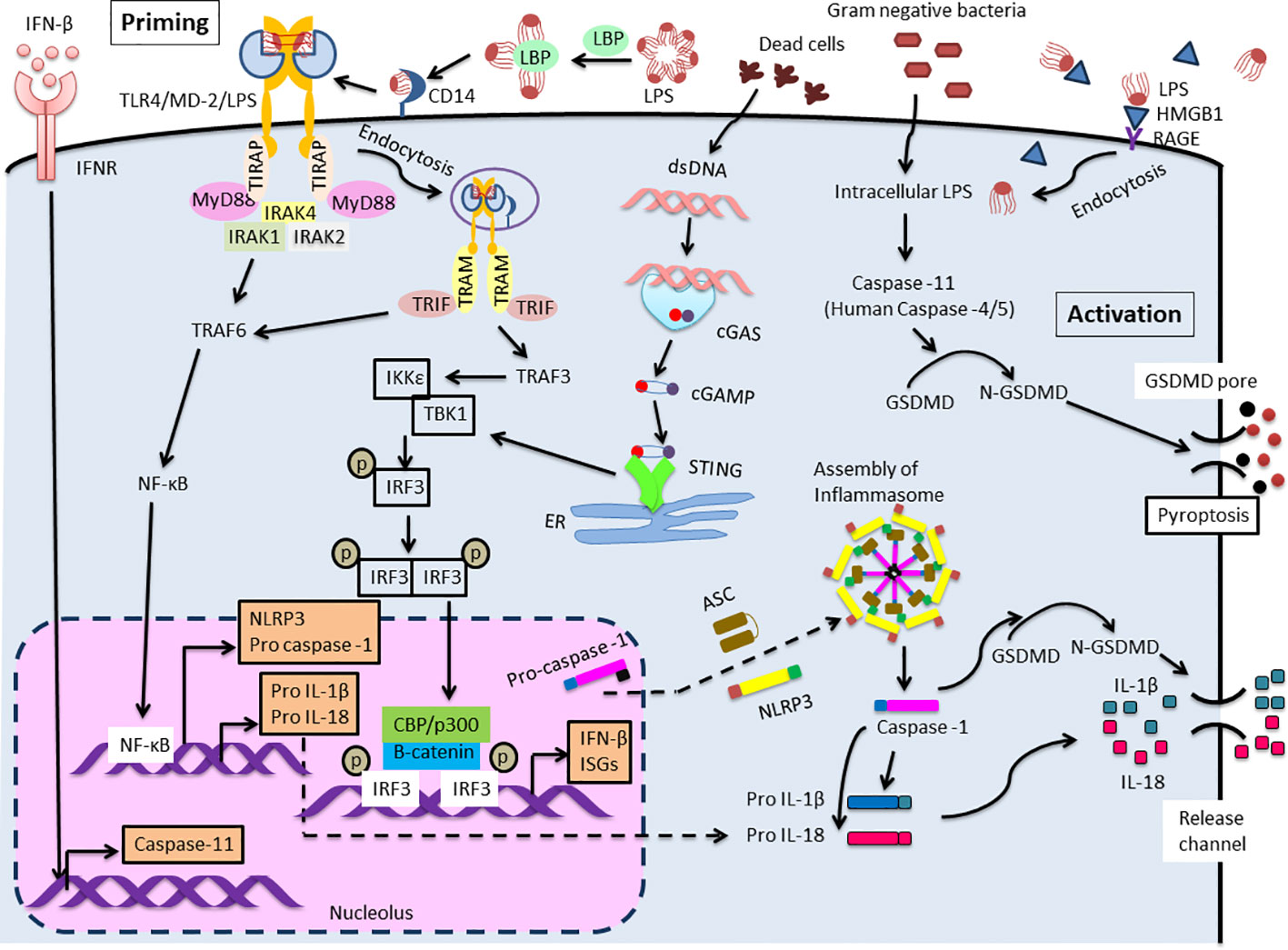
Figure 1 Extra- and Intra-cellular LPS recognition mechanism. TLR4 mediated NLRP3 (canonical) inflammasome signaling cascade consists of NLRP3, ASC and caspase-1 which leads to maturation and release of IL-1β and IL-18. Two distinct pathways are involved to trigger this signaling cascade. The first one is priming initiated with the recognition of extracellular LPS by TLR4. This activates NF-κB leading to pro IL-1β and pro IL-18 and up-regulates the expression of NLRP3 and pro-caspase-1. Expression of caspase-11 is also elevated by IFN-β/IFNR signaling. IRF3 and STING function as primary stimulator for IFN-β production. Another one is activation induced in response to intracellular LPS which is recognized by caspase-11 (Human: caspase-4/5). This results in the formation of non-canonical inflammasome complex and then activation, that is followed by pyroptosis through GSDMD cleavage. Meanwhile, the activation of IL-1β and IL-18 during the process of pyroptosis is mediated by processing of precursors of these cytokines by caspase-1.
2.2 TLR4 signaling diverting into the TRIF activation
A lack of MyD88 in cells might lead to the MyD88-independent pathway, initiating the late phase of the TLR4 signaling pathway (22). Additionally, CD14 mediates the internalization of the TLR4/MD-2/LPS complex into endosomes (23). Once MyD88 and TIRAP are discharged from the plasma membrane, TLR4 is triggered to harbor into the endosomes where it binds to TRIF. TRAM, also known as TICAM-2, facilitates the interaction between TRIF and TLR4 (24). Then, the E3 ubiquitin ligase TRAF3 is recruited and subsequently activates the noncanonical IKK kinases: TANK-binding kinase 1 (TBK1) and IKKϵ. TRAF3 involves in both MyD88 and the TRIF-mediated pathway, regulating both cytokine and IFN production (25). In addition, abolishing degradative (not self) TRAF3 ubiquitination inhibited all proinflammatory cytokines without affecting the IFN response. The consensus motif of TRIF is phosphorylated by TBK1 and causes recruitment of interferon regulatory factor3 (IRF3) (26). IRF3 is phosphorylated by TBK1, activated, dimerized, and translocated to the nucleus (27). IRF3 deficient mice cannot induce type 1 IFN production especially IFNβ (28). (Figure 1).
2.3 Caspase-11 activity and pyroptosis
It has recently become clear that LPS is not only recognized extracellularly via TLR4/MD-2, but also recognized intracellularly. Kayagaki et al. reported that intracellular LPS-activated caspase-11 causes cell death called pyroptosis (29). Pyroptosis is an inflammation-induced programmed cell death that involves rapid disruption of cellular membranes and the release of damage-associated molecular patterns (DAMPs). Pyroptosis is induced by caspase-1 or mouse caspase-11 (human caspase-4/5) cleaves gasdermin D (GSDMD), a pore-forming protein that is usually exists in the auto-suppressed state (30). Recently, the “tag-team” LPS recognition system, consisting of both TLR4 and caspase-11, has come to be understood as a regulator of the response to harmful infections. Extracellular LPS primarily stimulates TLR4/MD-2, followed by inducing a priming signal for the expression of inflammasome components. Intracellular LPS induces caspase-11-dependent inflammasome activation in the cytoplasm (31) (Figure 1, activation step). Intracellular molecules, such as caspase, or GSDMD, will be clinical target of shock therapy in the future. Further, blood LPS, and blood HMGB1, that is an important endotoxin delivery DAMPs protein and mediates caspase-11-dependent lethality in sepsis (32), may be also target for the clinical therapy.
3 IRF3 function in sepsis
Next, we focus on the role of IRF3 in sepsis, because IRF3 is not only an important molecule in the sepsis mortality but is also associated with important roles in microbiome.
3.1 IRF3 is an important gene for IFN production and cell death
IRFs are a family of transcription regulators for a wide range of genes associated with immune responses (33). Although IRF3 is expressed in almost all cells, it is localized in the cytoplasm as an inactive form. However, upon viral or duplex RNA stimulation, a specific serine residue at the C-terminus is phosphorylated, and IRF3 homodimerization was induced, then IRF3 dimer was transferred into the nucleus. After binding of β-catenin to IRF3, IRF3 also forms a complex with p300/CBP, and acquires DNA binding ability, thereby converting into the active form, subsequently increase IFN-β expression (34–37) (Figure 1). Further, the activation of IRF3 occurs not only through TLRs-TRIF axis, but also thorough intracellular receptors such as (RLRs)(RIG-I/MDA5)– mitochondrial antiviral signaling protein (MAVS) (38, 39) and the cytosolic DNA receptor cyclic GMP-AMP (cGAMP) synthase (cGAS)/STING axis (40). STING is a stimulator of IFN genes and the signaling pathway is activated not only by viral DNA but also by autologous genomic DNA and mitochondrial DNA through the activation of the cGAMP, then induces type I interferon, thereby relating to the promoting autophagy (41) and various kinds of cell death (e.g., apoptosis, necroptosis, pyroptosis, ferroptosis, mitotic cell death, and immunogenic cell death) (42).
3.2 The importance of IRF3 in endotoxin shock
IRF3 KO mice are resistant to LPS-induced endotoxin shock (28). Because these mice were found another null mutation in the Bcl2l12 gene, Taniguchi et al. further showed that selective IRF3 deficiency in myeloid cells reduced the serum levels of type I IFN and increased the survival rate in the endotoxin-induced shock (43). Cecal ligation and puncture (CLP) model induces peritonitis by puncturing the distal part of the ligated cecum to induce leakage of feces, and this model is considered to be more clinically similar to sepsis. In the CLP model, disease score, mortality, hypothermia, and bacterial load are all reduced in IRF3 KO mice, compared to their WT counterparts (44, 45). Furthermore, in adoptive transfer experiments by using IRF3 KO bone marrow chimera, little protection from sepsis was shown, whereas chimeras with IRF3-KO stroma showed a substantial degree of protection. This result shows that IRF3 in the stroma principally enhances sepsis pathogenesis (45). From above, IRF3 functions as an immune protector through IFN production; however, IRF3 activation augments sepsis mortality.
3.3 Effect of IRF3 inhibitors on endotoxin shock
Several specific inhibitors of IRF3 have been reported. We focused on those that have been reported to inhibit endotoxic shock. Piceatannol (trans-3,3′,4,5′-tetrahydroxystilbene) is one of the stilbene derivative found in many plants such as red grapes. Piceatannol is known as a phytoalexin to protect the plant against fungal infection, and exhibits antioxidant and anti-inflammatory effects (46, 47). Piceatannol was previously reported as a Syk-specific tyrosine kinase inhibitor (48). Recently, the inhibitory effect on IRF3 is also known in more detail. In the nucleus, IRF3 binds to interferon stimulated response elements (ISREs) in the promoters of a subset of ISGs. Piceatannol prevents LPS-induced ISG induction by inhibiting the signaling cascade leading to IRF3 binding to the ISRE. Thus, piceatannol inhibits LPS-induced activation of IRF3, prevents the production of cytokines, and reduces septic shock after LPS challenge in animals (49). Although the reduction in mortality was limited (from >70% to < 35%), the same protective effect was also recognized using tetramethoxystilbene (TMS), a methylated form of piceatannol, instead of piceatannol. This result supports the notion that the protective effect might be related to the tyrosine kinase inhibitor, not the antioxidative properties of piceatannol (49).
Fucoxanthin (FX), which is a carotenoid derived from brown algae, binds to IRF3 and inhibits the phosphorylation of IRF3, then reduces pro-inflammatory cytokine production, thereby decreasing mortality in a mouse CLP-induced sepsis model (50). Further, FX also affects microbiota composition in intestine, that influences the development of sepsis. FX significantly reduced the bacterial load in the abdominal cavity in CLP-induced sepsis mice model via and increased the short-chain fatty acids (SCFAs) levels such as acetic and propionic acids, which showed negative correlations with the expression levels of inflammatory factors, in their intestines (51). FX alters the microbial diversity and promotes SCFAs production, in CLP-induced sepsis mice model, thus reshaping gut homeostasis (51).
The importance of IRF3 activation in the induction of septic shock is also confirmed by the results of epidermal growth factor receptor (EGFR) inhibitors (52). The binding of LPS to TLR4 and EGF to EGFR, respectively, both on the plasma membrane led to endocytosis. EGFR is important for the activation of PI3K/AKT pathway which is required for β-catenin activation and IRF-driven gene expression of TLR4 signaling. Further, the kinase activity of EGFR is needed for the activation both of IRF3 and its co-activator, β-catenin. If EGFR activity is inhibited, LPS-induced induction of IFN and ISGs is spoiled, then septic shock response is impaired. Indeed, the EGFR kinase activity inhibitor (gefitinib)-treated mice were protected from LPS-induced septic shock by the selective block of the IRF-driven genes in TLR4 signaling (52). These results demonstrate the selective regulation of TLR4 signaling by direct and indirect IRF3 inhibitors and emphasize the potential use of these inhibitors to treat sepsis.
3.4 Intracellular molecules and IRF3 activation
Accumulating evidence suggests that inflammation, cardiomyocyte apoptosis, and pyroptosis are involved in developing sepsis and sepsis-induced cardiomyopathy (SIC). IRF3 is important for sepsis-induced cardiac injury (53). In wild type mice and cardiomyocytes, LPS-induced cardiac injury stimulates STING through cGAS -cGAMP, which further mobilizes TBK1, then IRF3 is phosphorylated. Phosphorylated IRF3 subsequently translocated into nucleus and increased the expression of NOD-like receptor protein 3 (NLRP3). STING knockout attenuates LPS-induced cardiac injury by preventing NLRP3-mediated inflammation, apoptosis, and pyroptosis. This study suggests that STING-IRF3 might be a therapeutic target for SIC (53).
Furthermore, caspase-11 activation can also be affected by other proteins (54). The caspase-11 inflammasome in macrophages is negatively regulated by the zinc (Zn2+)-regulating protein, metallothionein 3 (MT3). In challenge with intracellular lipopolysaccharide, macrophages increased MT3 expression, which reduced the activation of caspase-11, caspase-1 and IL-1β. It is considered that MT3 increased intra-macrophage Zn2+ to downmodulate the TRIF-IRF3-STAT1 axis, a prerequisite for caspase-11 effector function. MT3 suppresses the activation of the caspase-11 inflammasome in vivo, whereas caspase-11 and MT3 synergistically impair antibacterial immunity (54). Thus, the inflammasome activation signal is influenced by various factors.
3.5 The relationship between intestinal microbiota and IRF3
As mentioned above, the CLP model is mainly used because it closely mimics the clinical progression of sepsis in humans (44). In the CLP model, bacteria, fungi, and metabolites migrate into the abdominal cavity, leading to abdominal infections and systemic sepsis. The gut microbiome plays an important role for keeping the body homeostasis, pathogen defense, food digestion and absorption, and immune system regulation (55). Preclinical studies show that microbiome-dependent metabolic pathways are able to drive immunological response against invading pathogens. Microbiome is also related to the onset of sepsis (56).
Recently, the relationship between intestinal microbiota and IRF3 has received much attention. Although the ligands are unknown, IRF3 is activated in macrophage cultures with live or sonicated commensal bacteria (44). Also, IRF3 enhances pathogen clearance by restoring host immunity. Fecal microbiota transplantation (FMT) rescued mice from lethal infection due to a pathogen community, isolated from a sepsis patient by restoring systemic immunity in an IRF3-dependent manner (57). Pathogenic community infection reduces fecal butyrate levels, which normalizes IRF3 levels. FMT, by providing operational taxonomic units (OTUs) such as Bacteroides, which can produce butyrate, and increase the expression level of IRF3, then protects against systemic pathogen community infections (57). In sepsis models, such as CLP, IRF3 functions as an enhancer of sepsis. In contrast, in pathogenic community infections, IRF3 restores the protective immunity in sepsis.
4 Important gaps in sepsis immunology
Even with the above mechanisms of LPS recognition as a trigger for sepsis, a variety of events, as shown in Table 1, actually play a significant role in the pathogenesis of sepsis.

Table 1 Important gaps in sepsis immunology (No mark: known, *: unknown).
Sepsis is a highly heterogeneous syndrome, which contains not only excessive inflammation but also immunosuppressive events. This makes it difficult to understand the status of the host response and to consider the suitable treatment strategy. Flow cytometry readouts such as monocyte HLA-DR and PD-L1 expression may be useful for the monitoring marker (58), however highly sensitive biomarker to determine the severity of sepsis even in mixture immunological situation is needed.
Nitric oxide (NO) and reactive oxygen species (ROS) play important roles in nonspecific infection defense mechanisms. The excess NO produced by inducible NO synthase (iNOS) induction reacts with ROS at the site of infection to form chemically reactive active NO species such as peroxynitrite. It exhibits strong antibacterial activity, and plays an important role in innate immunity and infection defense functions by phagocytic cells such as macrophages. On the other hand, NO and ROS damage host cells and tissues and cause oxidative stress. For the reason, the function of NO and ROS are considered as a “double-edged sword” (60).
Nutrition and cell metabolism also highly affects immunity. As shown in Table 1, both malnutrition and overnutrition are negatively affect immunity. Zinc, one of the micronutrients, is important for the modulatory effect in sepsis inflammation. As mentioned above, Zinc also acts the indirect inhibitor on TRIF-IRF3 activation signals (54). Metabolic state is also strongly related to the inflammasome signaling and lethality.
“Epigenetics” is a regulatory mechanism of gene expression without changes in DNA sequence, and consists of DNA methylation, histone modifications, and non-coding RNAs (77). Host inflammatory response through MyD88-dependent pathway is also characterized by epigenetic modifications in key regulatory genes, such as TNF. In human monocytic cell lines, stimulation with LPS decreased methylation of the TNF promoter, which in turn displaced nucleosomes from the NF-κB binding site, then NF-κB was able to bind to the TNF promoter, resulting in upregulation of TNF transcription (77, 81). Epigenetic mechanisms are likely central to the pathogenesis of sepsis because it associates with the host-pathogen interaction (histone modification), the pathogenesis of the early pro-inflammatory response (DNA methylation), and the establishment of endotoxin tolerance (DNA methylation) (77).
Microbiota is also important for immunity. The gut microbiome has a protective role in sepsis by keeping the gut barrier, regulating leukocyte function, and modulating immunity (82). Recently, it was reported that microbiome-dependent metabolic pathways can drive distinct immune response to pathogens (82). Other study showed that sepsis patient is characterized by a loss of diversity, lower number of the key commensal genera, and overgrowth of pathogens (82, 83). Further, Probiotics and Fecal transplantation is useful for the recolonization of the gut and for decreasing sepsis incidence and mortality (84).
5 Conclusion
IRF3 activation is important for the mortality both in challenged with endotoxin-induced shock and in CLP model. Recent clinical targets for endotoxic shock therapy are becoming intracellular molecules and intracellularly incorporated LPS. However, IRF3 is also important and may be one of the therapeutic targets because it affects not only the sepsis induction but also the pathogen clearance via the restoration of host immunity.
One of the features that distinguishes sepsis from uncomplicated infections is the dysregulation of the host response. Another is that at the clinical diagnosis of sepsis, not only an inflammatory response but also an immunosuppression has already occurred. Thus, immunodeficiency indirectly contributes to the perpetuation of organ failure. These two characteristics are also responsible for the difficulty of sepsis treatment (59). Further, as shown in Table 1, there are still gaps to be addressed. Addressing these gaps should help to better understand the physiopathology of sepsis and provide translational opportunities to improve its prevention, diagnosis, and treatment.
Author contributions
BB: Writing – original draft. SA: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by 2023 Grants for Young Researchers for Education and Research in Aichi Medical University.
Acknowledgments
We thank labor members (Tatsuya Yamazaki, Susumu Tomono, and Masanori Inui) for suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest (1992) 101:1644–55. doi: 10.1378/chest.101.6.1644
2. Kellum JA, Ronco C. The role of endotoxin in septic shock. Crit Care (2023) 27:400. doi: 10.1186/s13054-023-04690-5
3. Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med (2008) 36:296–327. doi: 10.1097/01.CCM.0000298158.12101.41
4. Au WC, Moore PA, Lowther W, Juang YT, Pitha. PM. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferoninduced genes. Proc Natl Acad Sci USA (1995) 92:11657–61. doi: 10.1073/pnas.92.25.11657
5. Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, et al. IRF3 mediates a TLR3/ TLR4-specific antiviral gene program. Immunity (2002) 17:251–63. doi: 10.1016/S1074-7613(02)00390-4
6. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med (2003) 198:1043–55. doi: 10.1084/jem.20031023
7. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol (2004) 5:730–7. doi: 10.1038/ni1087
8. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5- mediated type I interferon induction. Nat Immunol (2005) 6:981–8. doi: 10.1038/ni1243
9. Tanaka Y, Chen. ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal (2012) 5:ra20. doi: 10.1126/scisignal.2002521
10. Caroff M, Novikov A. Lipopolysaccharides: structure, function and bacterial identification. Oilseeds fats Crops Lipids (2020) 27:31. doi: 10.1051/ocl/2020025
11. Schumann RR, Kirschning CJ, Unbehaun A, Aberle HP, Knope HP, Lamping N, et al. The lipopolysaccharide-binding protein is a secretory class 1 acute-phase protein whose gene is transcriptionally activated by APRF/STAT/3 and other cytokine-inducible nuclear proteins. Mol Cell Biol (1996) 16:3490–503. doi: 10.1128/MCB.16.7.3490
12. Tobias PS, Soldau K, Ulevitch RJ. Isolation of a lipopolysaccharide-binding acute phase reactant from rabbit serum. J Exp Med (1986) 164:777–93. doi: 10.1084/jem.164.3.777
13. Ryu JK, Kim SJ, Rah SH, Kang JI, Jung HE, Lee D, et al. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for efficient LPS recognition and transfer. Immunity (2017) 46:38–50. doi: 10.1016/j.immuni.2016.11.007
14. Brandenburg K, Seydel U. Conformation and supramolecular structure of lipid A. Adv Exp Med Biol (2010) 667:25–38. doi: 10.1007/978-1-4419-1603-7_3
15. Jerala R. Structural biology of the LPS recognition. Int J Med Microbiol (2007) 297(5):353–63. doi: 10.1016/j.ijmm.2007.04.001
16. Zamyatina A, Heine H. Lipopolysaccharide recognition in the crossroads of TLR4 and caspase-4/11 mediated inflammatory pathways. Front Immunol (2020) 11:585146. doi: 10.3389/fimmu.2020.585146
17. Guven-Maiorov E, Keskin O, Gursoy A, VanWaes C, Chen Z, Tsai CJ, et al. The architecture of the TIR domain signalosome in the Toll-like receptor-4 signaling pathway. Sci Rep (2015) 5(1):13128. doi: 10.1038/srep13128
18. Chen Z, Shao Z, Mei S, Yan Z, Ding X, Billiar T, et al. Sepsis upregulates CD14 expression in a MyD88-Dependent and Trif-Independent pathway. Shock (2018) 49:82–9. doi: 10.1097/SHK.0000000000000913
19. Reynoso M, Hobbs S, Kolb AL, Matheny JRW, Roberts BM. MyD88 and not TRIF knockout is sufficient to abolish LPS-induced inflammatory responses in bone-derived macrophages. FEBS Lett (2023) 597(9):1225–32. doi: 10.1002/1873-3468.14616
20. Moncrieffe MC, Bollschweiler D, Li B, Penczek PA, Hopkins L, Bryant CE, et al. MyD88 death-Domain oligomerization determines myddosome architecture: implications for toll-like receptor signaling. Structure (2020) 28:281–89.e283. doi: 10.1016/j.str.2020.01.003
21. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature (2010) 465(7300):885–90. doi: 10.1038/nature09121
22. Yang DJ, Wang XD, Fu XY, Lu HM, Zhou ZG, Liu Y. MyD88 deficiency aggravates the severity of acute pancreatitis by promoting MyD88-independent TRIF pathway-mediated necrosis. Ann Transl Med (2022) 10:1214. doi: 10.21037/atm-22-5134
23. Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell (2011) 147(4):868–80. doi: 10.1016/j.cell.2011.09.051
24. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol (2014) 14(8):546–58. doi: 10.1038/nri3713
25. Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of TRAF3 ubiquitination selectively activate type I interferon and proinflammatory cytokine expression. Nat Immunol (2010) 11(1):70. doi: 10.1038/ni.1819
26. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
27. Jefferies CA. Regulating IRFs in IFN driven disease. Front Immunol (2019) 10:325. doi: 10.3389/fimmu.2019.00325
28. Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, et al. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun (2003) 306:860–6. doi: 10.1016/S0006-291X(03)01049-0
29. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341:1246–9. doi: 10.1126/science.1240248
30. Kopitar-Jerala N. The role of interferons in inflammation and inflammasome activation. Front Immunol (2017) 8:873. doi: 10.3389/fimmu.2017.00873
31. Stowe I, Lee B, Kayagaki N. Caspase-11: arming the guards against bacterial infection. Immunol Rev (2015) 265:75–84. doi: 10.1111/imr.12292
32. Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, et al. The endotoxin delivery protein HMGB1 mediates Caspase-11-Dependent lethality in sepsis. Immunity (2018) 49:740–53.e747. doi: 10.1016/j.immuni.2018.08.016
33. Nguyen H, Hiscott J, Pitha PM. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev (1997) 8:293–312. doi: 10.1016/S1359-6101(97)00019-1
34. Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol (1999) 19:2465–74. doi: 10.1128/MCB.19.4.2465
35. Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell (1998) 1:507–18. doi: 10.1016/S1097-2765(00)80051-9
36. Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J (1998) 17:1087–95. doi: 10.1093/emboj/17.4.1087
37. Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol (1998) 18:2986–96. doi: 10.1128/MCB.18.5.2986
38. Sasai M, Shingai M, Funami K, Yoneyama M, Fujita T, Matsumoto M, et al. NAK-associated protein 1 participates in both the TLR3 and the cytoplasmic pathways in type I IFN induction. J Immunol (2006) 177:8676–83. doi: 10.4049/jimmunol.177.12.8676
39. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
40. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
41. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
42. Zhang R, Kang R, Tang D. The STING1 network regulates autophagy and cell death. Signal Transduct Target Ther (2021) 6:208. doi: 10.1038/s41392-021-00613-4
43. Yanai H, Chiba S, Hangai S, Kometani K, Inoue A, Kimura Y, et al. Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc Natl Acad Sci USA (2018) 115:5253–8. doi: 10.1073/pnas.1803936115
44. Walker WE, Bozzi AT, Goldstein DR. IRF3 contributes to sepsis pathogenesis in the mouse cecal ligation and puncture model. J Leukoc Biol (2012) 92:1261–8. doi: 10.1189/jlb.0312138
45. Heipertz EL, Harper J, Goswami DG, Lopez CA, Nellikappallil J, Zamora R, et al. IRF3 signaling within the mouse stroma influences sepsis pathogenesis. J Immunol (2021) 206:398–409. doi: 10.4049/jimmunol.1900217
46. Waffo Teguo P, Fauconneau B, Deffieux G, Huguet F, Vercauteren J, Merillon JM. Isolation, identification, and antioxidant activity of three stilbene glucosides newly extracted from vitis vinifera cell cultures. J Nat Prod (1998) 61:655–7. doi: 10.1021/np9704819
47. Kageura T, Matsuda H, Morikawa T, Toguchida I, Harima S, Oda M, et al. Inhibitors from rhubarb on lipopolysaccharide-induced nitric oxide production in macrophages: structural requirements of stilbenes for the activity. Bioorg Med Chem (2001) 9:1887–93. doi: 10.1016/S0968-0896(01)00093-1
48. Oliver JM, Burg DL, Wilson BS, McLaughlin JL, Geahlen RL. Inhibition of mast cell Fc epsilon R1-mediated signaling and effector function by the Syk-selective inhibitor, piceatannol. J Biol Chem (1994) 269:29697–703. doi: 10.1016/S0021-9258(18)43936-1
49. Dang O, Navarro L, David M. Inhibition of lipopolysaccharide-induced interferon regulatory factor 3 activation and protection from septic shock by hydroxystilbenes. Shock (2004) 21:470–5. doi: 10.1097/00024382-200405000-00012
50. Su J, Guan B, Chen K, Feng Z, Guo K, Wang X, et al. Fucoxanthin attenuates inflammation via interferon regulatory factor 3 (IRF3) to improve sepsis. J Agric Food Chem (2023) 71:12497–510. doi: 10.1021/acs.jafc.3c03247
51. Su J, Guan B, Su Q, Hu S, Wu S, Tong Z, et al. Fucoxanthin ameliorates sepsis via modulating microbiota by targeting IRF3 activation. Int J Mol Sci (2023) 24. doi: 10.3390/ijms241813803
52. Chattopadhyay S, Veleeparambil M, Poddar D, Abdulkhalek S, Bandyopadhyay SK, Fensterl V, et al. EGFR kinase activity is required for TLR4 signaling and the septic shock response. EMBO Rep (2015) 16:1535–47. doi: 10.15252/embr.201540337
53. Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W, et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol (2019) 24:101215. doi: 10.1016/j.redox.2019.101215
54. Chowdhury D, Gardner JC, Satpati A, Nookala S, Mukundan S, Porollo A, et al. Metallothionein 3-Zinc axis suppresses caspase-11 inflammasome activation and impairs antibacterial immunity. Front Immunol (2021) 12:755961. doi: 10.3389/fimmu.2021.755961
55. Elmassry MM, Zayed A, Farag MA. Gut homeostasis and microbiota under attack: impact of the different types of food contaminants on gut health. Crit Rev Food Sci Nutr (2022) 62:738–63. doi: 10.1080/10408398.2020.1828263
56. Pickard JM, Zeng MY, Caruso R, Nunez G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev (2017) 279:70–89. doi: 10.1111/imr.12567
57. Kim SM, DeFazio JR, Hyoju SK, Sangani K, Keskey R, Krezalek MA, et al. Fecal microbiota transplant rescues mice from human pathogen mediated sepsis by restoring systemic immunity. Nat Commun (2020) 11:2354. doi: 10.1038/s41467-020-15545-w
58. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol (2017) 17:407–20. doi: 10.1038/nri.2017.36
59. Rubio I, Osuchowski MF, Shankar-Hari M, Skirecki T, Winkler MS, Lachmann G, et al. Current gaps in sepsis immunology: new opportunities for translational research. Lancet Infect Dis (2019) 19:e422–36. doi: 10.1016/S1473-3099(19)30567-5
60. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol (2014) 24:R453–462. doi: 10.1016/j.cub.2014.03.034
61. Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem (2012) 287:36617–22. doi: 10.1074/jbc.M112.407130
62. Gupta SC, Hevia D, Patchva S, Park B, Koh W, Aggarwal BB. Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal (2012) 16:1295–322. doi: 10.1089/ars.2011.4414
63. Grijalba MT, Vercesi AE, Schreier S. Ca2+-induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2+-stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry (1999) 38:13279–87. doi: 10.1021/bi9828674
64. Durand A, Duburcq T, Dekeyser T, Neviere R, Howsam M, Favory R, et al. Involvement of mitochondrial disorders in septic cardiomyopathy. Oxid Med Cell Longev (2017) 2017:4076348. doi: 10.1155/2017/4076348
65. Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA (2014) 111:15514–9. doi: 10.1073/pnas.1414859111
66. Zhang Z, Han N, Shen Y. S100A12 promotes inflammation and cell apoptosis in sepsis-induced ARDS via activation of NLRP3 in fl ammasome signaling. Mol Immunol (2020) 122:38–48. doi: 10.1016/j.molimm.2020.03.022
67. Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res (2013) 23:201–12. doi: 10.1038/cr.2013.6
68. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496:238–42. doi: 10.1038/nature11986
69. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell (2016) 167:457–70.e413. doi: 10.1016/j.cell.2016.08.064
70. Zhang YY, Ning BT. Signaling pathways and intervention therapies in sepsis. Signal Transduct Target Ther (2021) 6:407. doi: 10.1038/s41392-021-00816-9
71. Fitting C, Dhawan S, Cavaillon JM. Compartmentalization of tolerance to endotoxin. J Infect Dis (2004) 189:1295–303. doi: 10.1086/382657
72. Suzuki T, Shimizu T, Szalay L, Choudhry MA, Rue LW, Bland KI, et al. Androstenediol ameliorates alterations in immune cells cytokine production capacity in a two-hit model of trauma-hemorrhage and sepsis. Cytokine (2006) 34:76–84. doi: 10.1016/j.cyto.2006.04.007
73. Becker JU, Theodosis C, Jacob ST, Wira CR, Groce NE. Surviving sepsis in low-income and middle-income countries: new directions for care and research. Lancet Infect Dis (2009) 9:577–82. doi: 10.1016/S1473-3099(09)70135-5
74. Souffriau J, Libert C. Mechanistic insights into the protective impact of zinc on sepsis. Cytokine Growth Factor Rev (2018) 39:92–101. doi: 10.1016/j.cytogfr.2017.12.002
75. Kempker JA, Panwar B, Judd SE, Jenny NS, Wang HE, Gutierrez OM. Plasma 25-Hydroxyvitamin D and the longitudinal risk of sepsis in the REGARDS cohort. Clin Infect Dis (2019) 68:1926–31. doi: 10.1093/cid/ciy794
76. Casaer MP, Mesotten D, Hermans G, Wouters PJ, Schetz M, Meyfroidt G, et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med (2011) 365:506–17. doi: 10.1056/NEJMoa1102662
77. Binnie A, Tsang JLY, Hu P, Carrasqueiro G, Castelo-Branco P, Dos Santos CC. Epigenetics of sepsis. Crit Care Med (2020) 48:745–56. doi: 10.1097/CCM.0000000000004247
78. Prescott HC, Dickson RP, Rogers MA, Langa KM, Iwashyna TJ. Hospitalization type and subsequent severe sepsis. Am J Respir Crit Care Med (2015) 192:581–8. doi: 10.1164/rccm.201503-0483OC
79. Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJ, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut (2016) 65:575–83. doi: 10.1136/gutjnl-2015-309728
80. Karmarkar D, Rock KL. Microbiota signalling through MyD88 is necessary for a systemic neutrophilic inflammatory response. Immunology (2013) 140:483–92. doi: 10.1111/imm.12159
81. El Gazzar M, Liu T, Yoza BK, McCall CE. Dynamic and selective nucleosome repositioning during endotoxin tolerance. J Biol Chem (2010) 285:1259–71. doi: 10.1074/jbc.M109.067330
82. Haak BW, Wiersinga WJ. The role of the gut microbiota in sepsis. Lancet Gastroenterol Hepatol (2017) 2:135–43. doi: 10.1016/S2468-1253(16)30119-4
83. Lankelma JM, van Vught LA, Belzer C, Schultz MJ, van der Poll T, de Vos WM, et al. Critically ill patients demonstrate large interpersonal variation in intestinal microbiota dysregulation: a pilot study. Intensive Care Med (2017) 43:59–68. doi: 10.1007/s00134-016-4613-z
Keywords: lipopolysaccharide (LPS), TLR4 signaling, MyD88-depending pathway, IRF3 signaling, sepsis control
Citation: Basak B and Akashi-Takamura S (2024) IRF3 function and immunological gaps in sepsis. Front. Immunol. 15:1336813. doi: 10.3389/fimmu.2024.1336813
Received: 11 November 2023; Accepted: 22 January 2024;
Published: 05 February 2024.
Edited by:
Richard Hotchkiss, Washington University in St. Louis, United StatesReviewed by:
Charles E. McCall, Wake Forest Baptist Medical Center, United StatesCopyright © 2024 Basak and Akashi-Takamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sachiko Akashi-Takamura, sachiko@aichi-med-u.ac.jp