 Jiaqian Zhang
Jiaqian Zhang Hongjiang Liu
Hongjiang Liu Yuehong Chen
Yuehong Chen Huan Liu
Huan Liu Shengxiao Zhang
Shengxiao Zhang Geng Yin
Geng Yin Qibing Xie
Qibing Xie- 1Department of Rheumatology and Immunology, West China Hospital, Sichuan University, Chengdu, China
- 2Department of Rheumatology, The Second Hospital of Shanxi Medical University, Taiyuan, China
- 3Department of General Practice, General Practice Medical Center, West China Hospital, Sichuan University, Chengdu, China
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune condition marked by inflammation of the joints, degradation of the articular cartilage, and bone resorption. Recent studies found the absolute and relative decreases in circulating regulatory T cells (Tregs) in RA patients. Tregs are a unique type of cells exhibiting immunosuppressive functions, known for expressing the Foxp3 gene. They are instrumental in maintaining immunological tolerance and preventing autoimmunity. Increasing the absolute number and/or enhancing the function of Tregs are effective strategies for treating RA. This article reviews the studies on the mechanisms and targeted therapies related to Tregs in RA, with a view to provide better ideas for the treatment of RA.
1 Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterized by symmetric arthritis in distal joints, leading to joint degradation and functional impairment. It often presents with extra-articular manifestations, involving multiple systems, severely impacting overall bodily function and reducing the quality of life for patients (1). Nonsteroidal anti-inflammatory drugs, glucocorticoids, and rheumatoid arthritis medications are commonly used to treat RA to alleviate inflammation and reduce pain. However, the use of these drugs carries the risk of liver, kidney, and gastrointestinal damage (2). The emergence of early diagnosis, novel treatment methods, and effective therapeutic strategies has garnered widespread attention. Currently, the etiology of RA is not fully elucidated, but the immune processes occurring in the synovium and synovial fluid, including synovial cell proliferation and fibrosis, vascular membrane formation, and cartilage and bone erosion, are noteworthy. Naive CD4+ T cells can differentiate into various cell types under antigen-presenting cell stimulation. Dysregulation in the function and/or quantity of these cells can lead to abnormalities in cellular and humoral immunity (3). Regulatory T cells (Tregs) play a crucial role in maintaining immune tolerance and preventing autoimmunity (4, 5). Abnormal quantity and/or functional deficiencies in Tregs have been reported in RA (6–9). Therefore, manipulating and regulating Tregs represent effective therapeutic strategies for many such diseases.
2 Characteristics and functions of Tregs
2.1 Origin and classification of Tregs
Tregs, initially identified in animal models and subsequently in humans, have offered a novel viewpoint on the establishment and preservation of self-tolerance (4). In 1995, Sakaguchi first discovered a CD4+CD25+ T cell subset with autoimmune tolerance capability further named Tregs (10). In 2003, forkhead box protein 3 (Foxp3) was established as a critical transcription factor for Tregs, indispensable for their formation and suppressive functionality. Considering their origin and biological traits, Tregs are classified into two subgroups: thymus-derived regulatory T cells (tTregs), also referred to as natural regulatory T cells (nTregs), and induced regulatory T cells (iTregs), or peripheral regulatory T cells (pTregs). Despite significant differences in origin, gene expression profiles, and biological characteristics between nTregs and iTregs (11), both types are dependent on antigen stimulation and activation in the presence of IL-2 (12). nTregs are strictly controlled by the thymic microenvironment and characteristically express the nuclear transcription factor Foxp3, along with CD25 and CD4 on their cell surface. Induced Tregs, or iTregs, primarily originate from immature CD4+ cells in peripheral lymphoid tissues or from in vitro culture, stimulated by transforming growth factor (TGF)-β and IL-2. The induction of iTregs does not require strong T cell receptor (TCR) stimulation, but rather an appropriate antigen stimulation. Overactivation of downstream TCR signals may interfere with iTreg development. Similarly, strong co-stimulation negatively impacts the generation of iTregs. Conversely, blocking the CD28 co-stimulatory pathway or improving signal transduction through the activation of cytotoxic T lymphocyte-associated protein 4 (CTLA4) and ICOS promotes the development of iTregs (13). Although both nTregs and iTregs express Foxp3, CD25, CTLA-4, and other Treg function-related molecules, the stability of Foxp3 expression in iTregs is much lower than in nTregs. Under certain in vivo conditions, iTregs can be driven to differentiate into effector T cells (14). In nTregs, the DNA in the regulatory T cell-specific demethylated region (TSDR) of the Foxp3 enhancer is demethylated, while in iTregs, the TSDR is only partially demethylated (15). This incomplete demethylation renders iTregs more susceptible to loss of Foxp3 expression and functionality. nTregs represent a stable lineage of cells specific to antigens expressed in the thymus, whereas iTregs constitute a more dynamic population, ensuring tolerance to new antigens encountered in peripheral areas. Collectively, these two populations are vital for maintaining immune tolerance.
2.2 Structure and function of Foxp3
Foxp3, located on the p11.23-13.3 region of the X chromosome, consists of 11 coding exons, 3 non-coding exons, and 104 introns. It serves as the main transcription factor dictating the development and function of Tregs (16). Within the Foxp3 locus, there are three conserved non-coding sequences (CNSs): a promoter (CNS1) and two enhancers (CNS2, CNS3) (17). CNS1 is essential for the induction of peripheral Tregs, and its knockout significantly reduces the number of Tregs in gut-associated lymphoid tissues (18). CNS3 plays a crucial role in the genesis of Tregs, and its deletion leads to a severe reduction in the thymic output of Tregs. CNS2, also known as TSDR, contains a CpG island that is highly demethylated only in functional Tregs. The methylation status of this highly conserved CpG island is pivotal in determining the expression level of Foxp3 and the stability of Tregs, and is deemed the most definitive marker of the Treg lineage (18). Two primary epigenetic mechanisms: DNA methylation and histone modifications play crucial roles in establishing and maintaining Foxp3 expression in Tregs (19, 20). Methylation of the TSDR can lead to chromatin condensation, which decreases the DNA sequence’s accessibility and inhibits the transcription of Foxp3. The partial methylation of TSDR underlies the instability of Foxp3 expression in Tregs. Conversely, the demethylation of TSDR is vital for the stability of Foxp3 expression, promoting transcription in Tregs. Moreover, Foxp3 is also regulated by ubiquitination and phosphorylation processes. Ubiquitination encourages Foxp3 degradation, while the impacts of Foxp3 phosphorylation can vary depending on the modification site. Protein phosphatase 1 acts on Foxp3 at the Ser418 site, removing a phosphate group - a process known as dephosphorylation. On the other hand, PIM1 adds a phosphate group to the Ser422 site of Foxp3, a process known as phosphorylation, which inhibits the function of Foxp3. In a contrasting manner, the lymphocyte-specific protein tyrosine kinase phosphorylates Foxp3 at the Tyr342 site, which augments its function. Therefore, the expression of Foxp3 must be strictly regulated to maintain the homeostasis of T cell-mediated immune responses. Tregs expressing Foxp3 play a significant role in suppressing immune responses (21). Constitutive expression of Foxp3 is a decisive factor in driving immune-suppressive functions (16). Mice with Foxp3 defects develop fatal autoimmune diseases (22), whereas lifelong continuous expression of Foxp3 prevents the occurrence of autoimmunity (23). Tregs with downregulated Foxp3 expression lose their suppressive function and can even produce pro-inflammatory cytokines, such as effector cells (24). Therefore, the stable expression of Foxp3 plays an important role in the balance between autoimmunity and tolerance, as well as the therapeutic effect based on Tregs.
2.3 Immunosuppressive mechanism of Tregs
RA is a systemic, inflammatory autoimmune disease. Aberrant activation of innate immune cells, including dendritic cells (DCs), innate lymphoid cells, and adaptive immune cells, may lead to excessive production of pro-inflammatory cytokines, ultimately resulting in the destruction of bone tissue and cartilage (25).Tregs play an irreplaceable role in maintaining self-tolerance and homeostasis, presenting noticeable differences between healthy individuals and those with autoimmune diseases (26, 27). They regulate the immune response through intercellular interactions or by inhibiting cytokine release and inducing negative regulatory signaling molecules to suppress excessive immunity. The potential mechanisms through which Tregs mediate inhibition are as follows: (Figure 1).
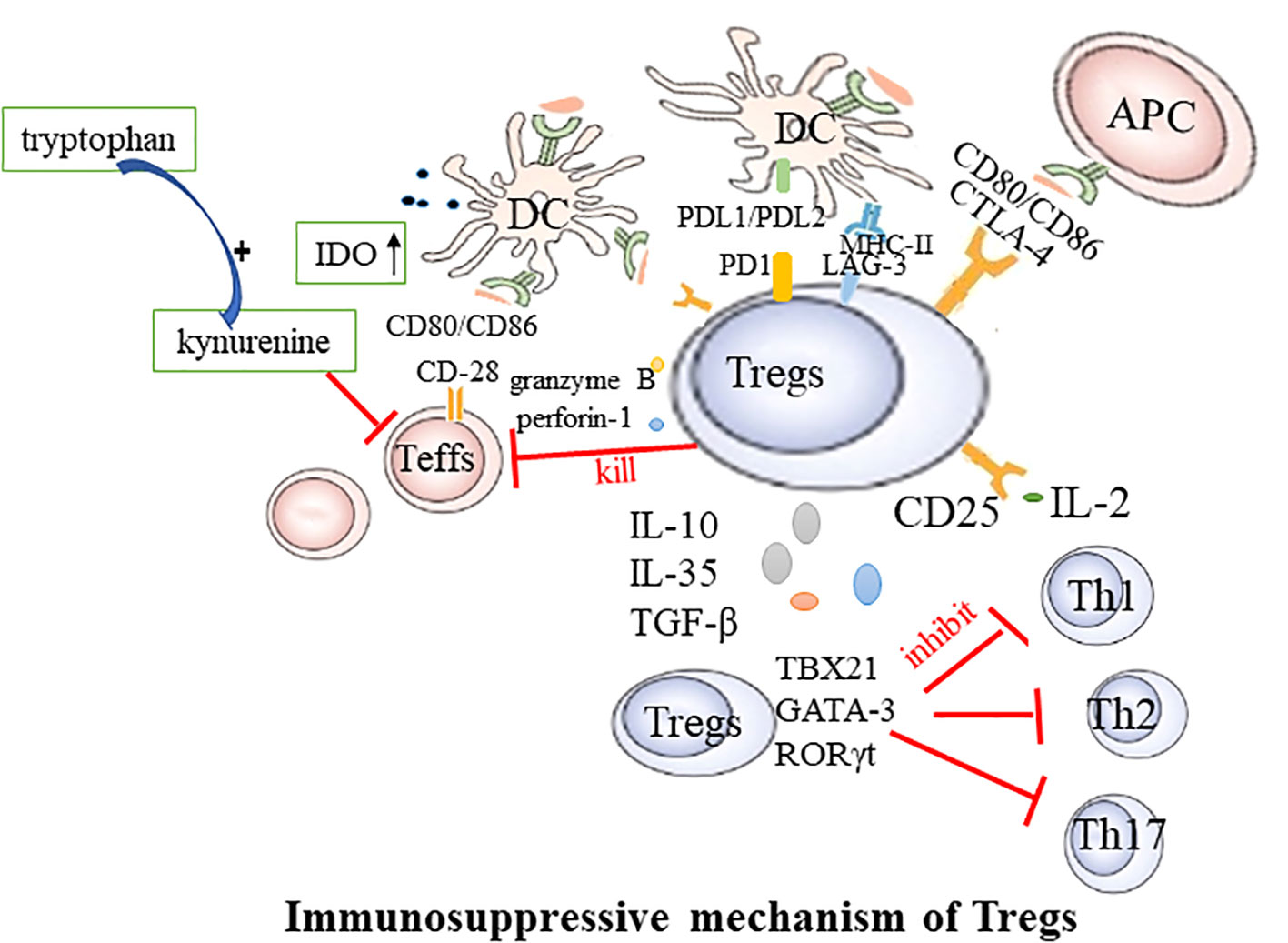
Figure 1 Immunosuppressive mechanism of Tregs. Teffs, effector T cells; DC, dendritic cells; CTLA-4, Cytotoxic T lymphocyte-associated antigen4; LAG-3, Lymphocyte activation gene 3; PD-1, Programmed cell death protein 1; APC, antigen presenting cells; TBX21, T-box transcription factor; Th, T helper cell.
2.3.1 Cell-to-cell contact-dependent
Tregs exercise their suppressive function via direct engagement with DCs or effector T cells (Teffs). This regulation of immune responses is facilitated through a mechanism that relies on cell-to-cell contact and is characterized by the expression of the nuclear transcription factor Foxp3, in conjunction with the elevated presence of CD25, CTLA4, Lymphocyte activation gene 3 (LAG-3), and glucocorticoid-induced TNFR-related protein (GITR) on the cell membrane. CTLA-4, an inhibitory molecule, is abundantly expressed on the Tregs’ surface and is instrumental in their function. Tregs with CTLA-4 deficiency exhibit significantly weakened suppressive effects (28). CTLA-4 curbs T-cell activation by either competitively binding to or downregulating CD80/CD86 present on the surface of DCs (29, 30). Additionally, Tregs can deplete CD80 and CD86 from the surface of antigen-presenting cells (APCs) through a process of endocytosis mediated by CTLA-4, thereby further modulating the stimulatory capacity of APCs (31). CTLA-4 can also induce indoleamine 2,3-dioxygenase (IDO) in dendritic cells. IDO degrades tryptophan into the immunotoxic metabolite kynurenine, inhibiting the proliferation of Teffs. LAG-3 mitigates the costimulatory capacity of dendritic cells (DCs) and the immune response of T cells by binding to MHC-II (32). The surface Programmed Cell Death Protein 1(PD-1), also known as CD279, on Tregs binds to its ligands PD-L1 and PD-L2 on DCs, leading to the inhibition of effector T cell function (33). Additionally, Foxp3+ Tregs expressing the transcription factors TBX21, GATA-3, or retinoic acid receptor-related orphan receptor (ROR) γt can respectively suppress the functions of helper T cells 1 (Th1), 2 (Th2), or 17 (Th17) (34).
2.3.2 Cell contact-independent
Tregs wield a potent immunosuppressive influence on the proliferation and cytokine production of effector T cells. By depleting and generating key cytokines, Tregs regulate immune responses and the maintenance of immune homeostasis. The high level of CD25 expression on the surface of Tregs contributes to the massive consumption of IL-2. This cytokine is required for Teffs proliferation and activation. Therefore, Tregs may competitively deplete this growth factor from pathogenic Teffs, thereby indirectly inhibiting their activity (35). The suppressive effects of Tregs may also occur through the secretion of soluble immunosuppressive cytokines, including IL-10, TGF-β, and IL-35 (36). These cytokines further contribute to the immunomodulatory effects of Tregs. For example, B-lymphocyte-induced maturation protein 1 promotes the differentiation of Tregs, upregulates IL-10, and inhibits CD4+ T cell-induced activation of fibroblast-like synoviocytes (37). In mouse models of collagen-induced arthritis and inflammatory bowel disease, Tregs lacking IL-35 are unable to control disease progression, highlighting the importance of this cytokine in immune regulation (38). Serum IL-35 levels are reduced in rheumatoid arthritis (RA) patients, demonstrating potential as a biomarker for active RA (39). Tregs can kill responsive T cells through the secretion of granzyme B and perforin-1 (40). In addition to their local effects, Tregs can “sense” cytokines or chemokines in the environment and migrate to sites of inflammation. During the active immune response, stimulation of TCR and cytokines guide Tregs to migrate to the site of inflammation. Once there, they suppress the inflammatory response, limit tissue damage, and promote resolution of inflammation (41).
3 Characteristics of Tregs in RA
Under normal circumstances, Tregs regulate immune tolerance status. Studies have shown that the occurrence and development of RA are related to various changes in anti-inflammatory Tregs and their counter-regulatory effects. Abnormal number and functional defects of Tregs play a relevant role in the pathogenesis and development of RA (7, 9, 42).
3.1 Deficiency of Tregs in RA
A deficiency of Tregs has been observed in both RA animal models and patients. In collagen-induced arthritis (CIA), a commonly used model for RA that involves immunizing mice with heterologous CII, an abnormal number of Tregs has been shown to influence the disease. In CIA mice, increased Treg counts were closely associated with reduced joint swelling, lower arthritis scores, reduced cartilage surface damage, and reduced joint cavity desmoplasia (43). Transfer of CD4+CD25+ cells into immunized mice during induction of antigen-induced arthritis resulted in reduced arthritis severity. Effective depletion of Tregs at any point in the life of a healthy individual triggers a rapid autoimmune and inflammatory response, resulting in multi-organ autoimmunity (44). The role of Tregs in pathogenesis is not fully understood, and the number of Tregs reported over the years to be increased, decreased, or unchanged in synovial fluid and peripheral blood is controversial (Table 1). The controversy surrounding the reported fluctuations in regulatory T cells (Tregs) numbers in the synovial fluid and peripheral blood of rheumatoid arthritis (RA) patients can be attributed to several factors. Variations in the local immune microenvironment within the joint may contribute to differences observed in Tregs numbers. The heterogeneous nature of the RA patient population, including diverse disease progressions, clinical manifestations, and treatment responses, may lead to inconsistent findings across studies. Methodological disparities, such as differences in detection methods, criteria for defining Tregs and different statistical counting methods (absolute value or percentage of Tregs), further contribute to the discrepancies. Therapeutic interventions, ranging from immunosuppressive agents to anti-inflammatory medications, may impact Tregs numbers and function in RA patients. Additionally, the variability in sample collection timing and individual differences, including genetic variations, play roles in shaping the observed differences in Tregs numbers among RA patients. Therefore, a comprehensive understanding of Tregs’ role in RA necessitates consideration of study design, sample sources, detection methods, and other influencing factors. Although the proportion of Tregs in the peripheral blood of RA patients is reported differently, most studies consistently report an abundance of Tregs in SF. The low frequency of Tregs in the peripheral blood of RA patients may be due to these cells migrating to local inflammatory tissues such as synovial fluid by expressing specific chemokine receptors. Some investigations of the absolute number of lymphocyte subsets in RA patients and its clinical significance found that the absolute number of Treg subsets can better reflect the true state of RA patients, making it more clinically significant (64, 67). In patients with rheumatic diseases, the proportion of Tregs varies according to the cell identification method used. A more stringent definition of Tregs is needed when assessing the status of such patients.
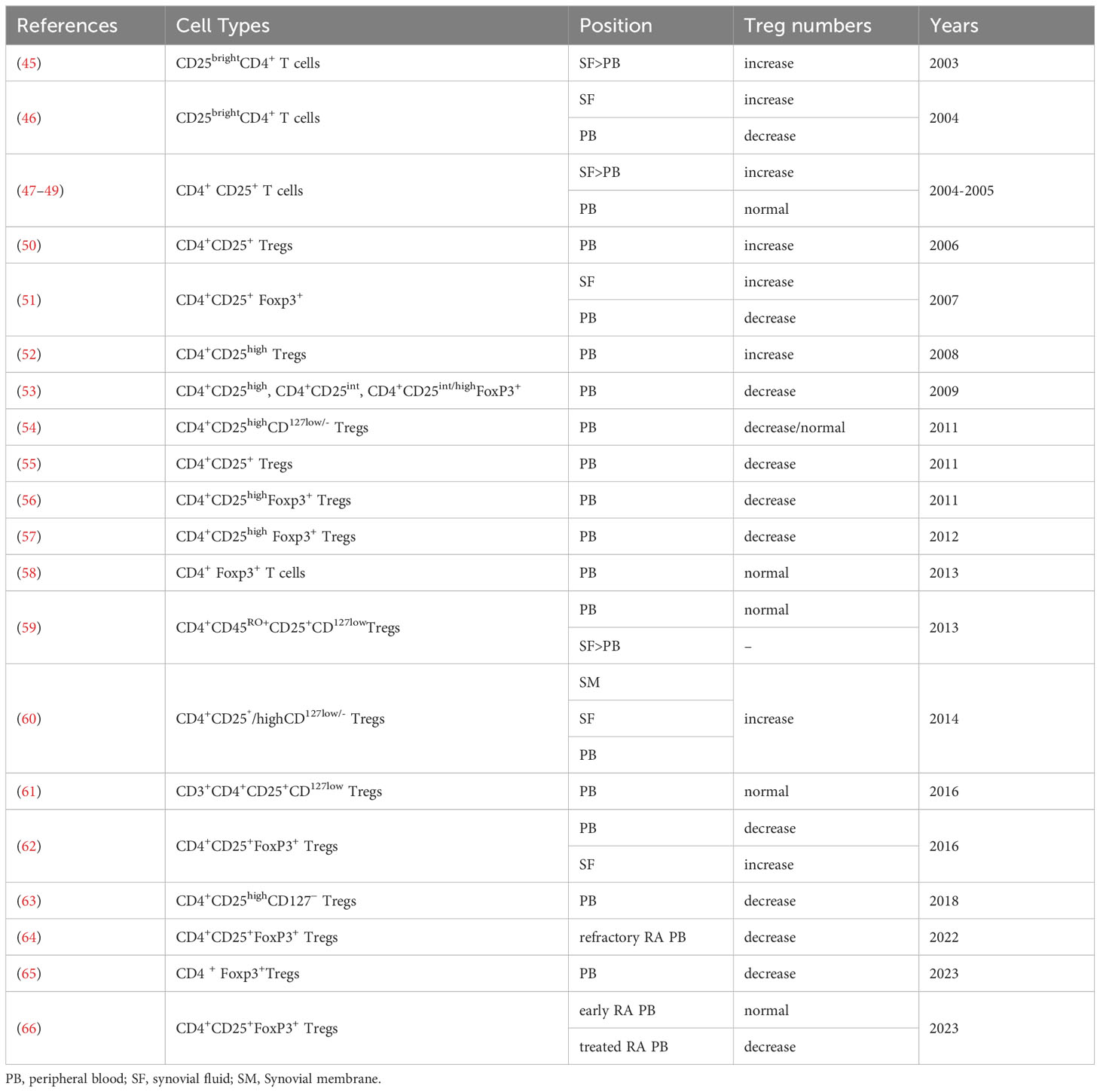
Table 1 Treg numbers in RA patients.
3.2 Malfunction of Tregs in RA
In the CIA model of Sprague Dawley rats, there is a significant decrease in the expression level of Foxp3, a key transcription factor in Tregs. This finding further underscores the importance of Foxp3 in maintaining immune homeostasis and preventing autoimmune diseases (68). During the pathological process of RA, the levels of pro-inflammatory cytokines in the serum, such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), are significantly elevated. These factors play a crucial role in the initiation and progression of the disease (69). Through complex signaling pathways, these factors impact the stability of Foxp3 in Tregs, altering their functionality and reducing their efficacy in suppressing immune responses. At the gene transcription level, IL-6 promotes DNA methylation at the CpG site of the upstream enhancer in nTreg, a process that weakens the transcriptional activity of the Foxp3 gene. At the protein level, IL-6 activates the enzyme PIM1, which specifically phosphorylates the S422 site of Foxp3. This process negatively regulates the chromatin binding activity of Foxp3, thereby inhibiting the function of Tregs (70). On the other hand, IL-1β can downregulate Foxp3 expression induced by TGF-β, while TNF-α activates protein phosphatase 1 to dephosphorylate the Ser418 site of Foxp3. Both mechanisms could potentially reduce the suppressive function of Tregs (71, 72). Moreover, inflammatory cytokines can regulate the expression of the Foxp3 transcription factor directly through nuclear factor-κB, thereby affecting the development and function of Tregs (73).
Inflammatory cytokines, beyond their direct regulation of Foxp3, wield an indirect influence on the functionality of Tregs by modulating the behavior of other cellular entities. For instance, a constellation of inflammatory cytokines, including but not limited to IL-1β, IL-6, and TNF-α, orchestrate a complex interplay that indirectly governs the proliferation of Tregs. This regulation occurs through the modulation of DCs, pivotal players in our immune response. The intricate dance of cell signaling also involves IL-21, a cytokine found in conventional T cells. It indirectly maintains the balance - or homeostasis - of Tregs by curtailing the availability of another cytokine, IL-2 (74). Adding another layer to this complex picture, Sun et al. (75) has traced the functional impairments seen in Tregs in the context of RA back to a reduction in the expression of a particular protein - T-cell immunoglobulin and mucin-domain containing-3 (62). In a separate but related discovery, Wang L. and his team found that exosomes derived from RA can inhibit the induction of Tregs. They achieve this by transferring a specific microRNA, miR-17, which subsequently suppresses the differentiation of Tregs by targeting the expression of TGFBR II, a crucial receptor in this process (76). In the battlefield of RA, Tregs are often unable to halt the onslaught of abnormal immune responses and the relentless progression of the disease. The question remains: is this inability linked to an early aging process in Tregs, which could lead to alterations in their phenotype and function? This is still an elusive piece of the puzzle that researchers are striving to understand (77).
3.3 Imbalance of Tregs/Teffs in RA
In RA patients, the number and function of peripheral Tregs are defective, leading to excessive activation of Teffs and promoting the onset of RA. This phenomenon is particularly evident during the active phase of RA (57, 68). The expression of Th17 cells, Th1, and Th17-related cytokines, and RORγt in peripheral blood of patients with active RA is increased, and the expression of Tregs and Foxp3 is decreased (78). The etiology and pathogenesis of RA remain unclear, but T cell dysfunction, particularly the abnormal activation of helper CD4+T cells, is a primary driver in the occurrence and development of RA (79). These T cells differentiate into different subsets: Th1, Th2, Th17, and Tregs, under the regulation of specific transcription factors (80).
The imbalance of Th1/Th2 cells plays a crucial role in the occurrence and development of RA (81). Th1 is primarily characterized by the secretion of high-level γ interferon, and also secrete granulocyte-macrophage colony-stimulating factor, IL-2, TNF-α, etc, to mediate cellular immunity. The characteristic cytokines of Th2 is IL-4, which also secretes IL-5、IL-6、 IL-10、IL-13, etc., to mediate humoral immunity (82, 83). Studies have shown that the joint inflammation in RA is dominated by Th1 (84). The Th1/Th2 ratio increased during RA activity (85), and improving the Th1/Th2 balance positively influences RA therapy. Th1 and Th2 are also derived from CD4+T cells induced to differentiate under specific cytokine conditions. Therefore, Tregs-targeted RA therapy may also influence Th1/Th2 balance. The imbalance between anti-inflammatory Tregs and pro-inflammatory Th17 accelerates RA progress (55, 86). Tregs primarily maintain self-tolerance and further suppress autoimmunity, while proinflammatory Th17 induces and propagates inflammation. Studies have found that the number and function of Th17 and Tregs in the peripheral blood of RA patients dramatically change, and the Th17/Tregs ratio increases (66, 87–89). During the pathogenesis of RA, Th17, as a pro-inflammatory helper T cell subset, can induce osteoblast nuclear factor-κB receptor activator ligand (RANKL) expression by upregulating osteoclast formation, leading to bone erosion (90). The initial differentiation of Th17 depends on TGF-β and IL-6, and IL-23 promotes functional maturation of Th17 (90). Interleukin-6-triggered Janus kinase (JAK) 2-STAT3 pathway is required for Th17 development. The STAT3 signaling pathway directly regulates RORγt (91), the master molecule of Th17. IL-17A is a characteristic cytokine of Th17 that is abundantly expressed in arthritic joints. It can directly stimulate RANKL expression and mediate cartilage proteoglycan loss and bone resorption, leading to the destruction of articular cartilage and bone (92). In addition, IL-17A is a potent instigator in the inflammatory response, capable of inducing pro-inflammatory cytokines such as TNF-α and IL-6. This leads to a cascade of inflammation and cell infiltration into local joints, contributing to the hallmark symptoms of arthritis (93, 94). These pro-inflammatory cytokines, including TNF-α and IL-6, along with other inflammatory mediators like IL-17, play a pivotal role in the pathogenesis of RA. They act in additive or synergistic ways, amplifying the inflammatory response and driving the progression of RA (93). This suggests a heightened immune response, with the body’s own defenses turned against itself, leading to the chronic inflammation and joint damage characteristic of RA. In active RA patients, there is an observed elevation in the levels of Th17 cells and IL-17A in peripheral blood. Tregs and Th17 antagonize each other in RA, forming a dynamic balance. The imbalance of this immune balance is a significant cause of RA (66, 95, 96) Therefore, increasing peripheral blood Tregs in RA patients, reducing Th17 or IL-17A levels, and improving the Treg/Th17 cell imbalance may be key to RA treatment (Figure 2).
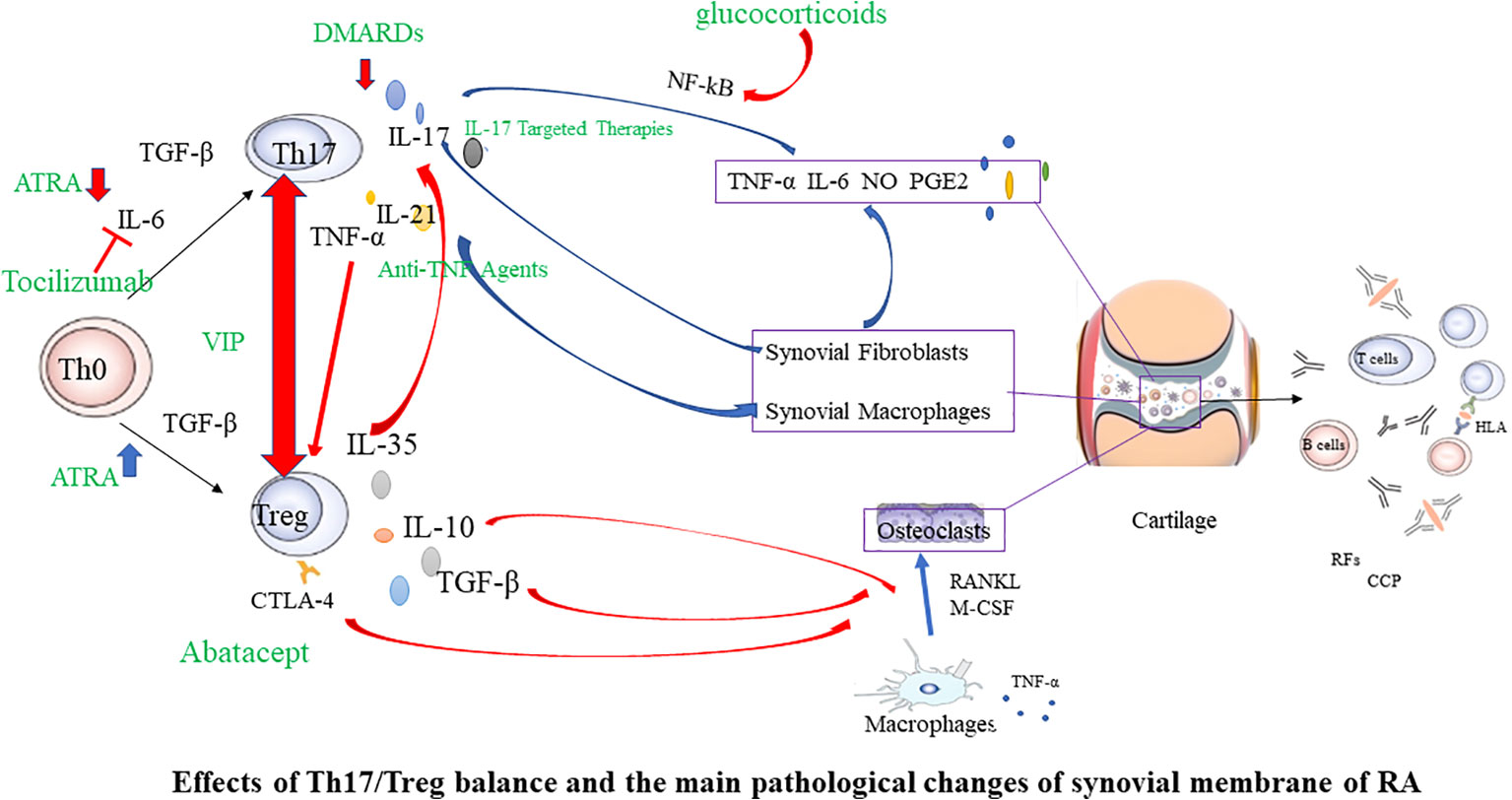
Figure 2 Effects of Th17/Treg balance and the main pathological changes of synovial membrane of RA. In the initial Th0 cells, in the presence of TGF-β, they differentiate into Tregs. However, when IL-6 is present simultaneously, it induces the expression of RORγt, promoting the differentiation into Th17 cells. Tregs have the ability to downregulate the expression of IL-17, thereby inhibiting the differentiation of Th17 cells. Conversely, inhibiting the production of Th17 cells helps promote the development of Tregs. In the joints, synovial cells and chondrocytes are the major local cell populations affected by rheumatoid arthritis. Synovial cells can be classified as fibroblast-like and macrophage-like synoviocytes. In affected joints, synovium swells due to infiltration by fibroblast-like synoviocytes, various T cell subsets, macrophages, and B cells. Interactions among these cells lead to the excessive production of numerous cytokines, sustaining synovial inflammation and joint damage. Red arrows indicate inhibition, and blue arrows indicate promotion. Green font marks drugs. RANKL, nuclear factor-κB ligand; M-CSF, macrophage colony-stimulating factor; TNF, tumor necrosis factor; IL, interleukin; TGF, transforming growth factor; RF, rheumatoid factor; CCP, cyclic citrullinated peptide; CTLA-4, Cytotoxic T lymphocyte-associated antigen4; Th17, T helper cell type 17; DMARDs, disease modifying antirheumatic drugs; ATRA, All-trans retinoic acid; VIP, Vasoactive intestinal peptide.
4 Tregs-targeted therapies in RA
The search for new, safe, and effective drugs specifically activating Tregs is a significant aspect of cellular immunotherapy. In humans, disease-modifying antirheumatic drugs (DMARDs) are the primary therapeutic agents for RA, reducing synovitis and systemic inflammation, and improving function. Biological agents are employed when arthritis is uncontrolled or when DMARDs induce toxic effects. The therapeutic efficacy of biologics, such as monoclonal antibodies used to neutralize inflammatory cytokines or block cytokine receptors, has been established in halting the progression of rheumatic diseases. However, the risk of infection and high costs limit the prescription of these biological agents. Given the powerful immunosuppressive ability of Tregs, immunotherapy targeting Tregs has become a major direction for the research of autoimmune diseases, including RA. This specialized treatment strives to reinstate physiological self-tolerance by boosting the inherent suppression of pathogenic autoreactive T cells by Tregs, circumventing the side effects typically associated with prolonged immunosuppression or biotherapy. Recently, clinical trials have been initiated to evaluate therapies focused on fostering the expansion of Tregs in autoimmune diseases. In the following sections, we will summarize Treg-targeted therapies in RA (Figures 2, 3).
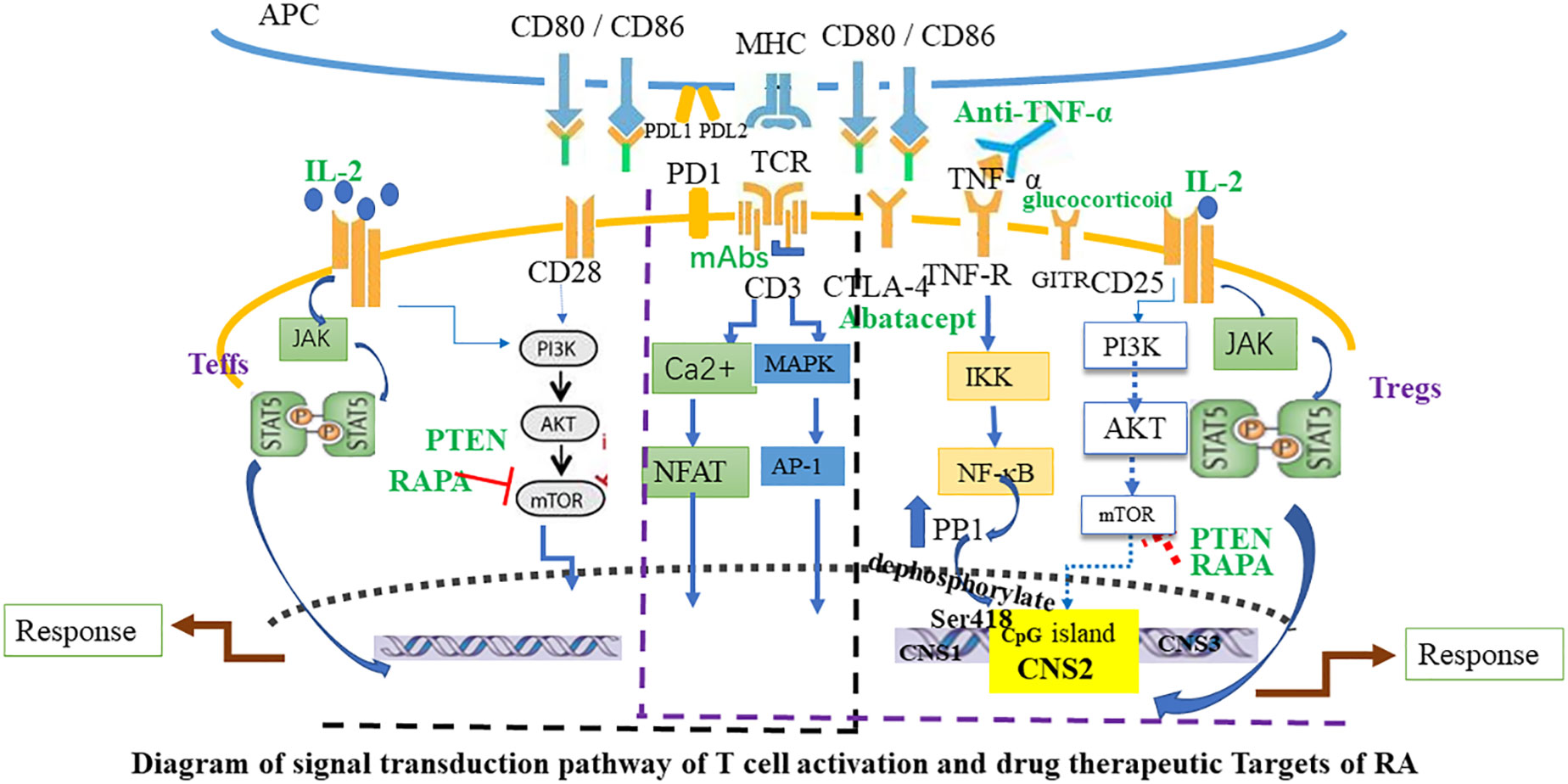
Figure 3 Diagram of signal transduction pathway of T cell activation and drug therapeutic Targets of RA. The activation of Teffs and Tregs is mediated by combined signaling through the TCR and co-stimulatory molecules such as CD28, and through the interleukin-2 receptor (IL-2R). Black font marks molecules expressed by T cells and APC. Green font marks drugs. Blue arrows indicate signaling pathways and red T-bars point to targets of immunosuppressive drugs. APC, antigen-presenting cell; Teffs, effector T cells; JAK, Janus kinase; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; mAbs, CD3 monoclonal antibodies; CTLA-4, Cytotoxic T lymphocyte-associated antigen 4; PD-1, Programmed cell death protein 1; GITR, glucocorticoid-induced TNFR-related protein; AP-1, activator protein 1; PP1, protein phosphatase 1; PTEN, Phosphatase and tensin homolog; RAPA, Rapamycin.
4.1 Glucocorticoids
Glucocorticoids, a frequently used class of drugs, can enhance the presence of Tregs by upregulating Foxp3 expression (97). These drugs can interact with specific receptors present in T cells, leading to a disruption in the communication between TCR signal transduction and the pathways that follow (98). The interaction of glucocorticoids with the exterior of T cells can obstruct the vital interaction between T cells and APCs, a process crucial for an effective immune response. This interaction also downregulates the rolling and adhesion of leukocytes, a key step in immune cell migration, and disrupts the cytoskeleton of T cells, effectively inhibiting their migratory capabilities (99). Moreover, glucocorticoids can shift the balance within the T cell population, favoring the predominance of Th2 cells and Tregs, both of which play crucial roles in immune regulation (100). Notably, patients with autoimmune and atopic disorders, who are treated with glucocorticoids, have demonstrated an increased proportion of Tregs within their T cell subsets (101). Glucocorticoids can also spur the generation of pTregs by boosting the expression of the glucocorticoid-induced leucine zipper (GILZ). This protein further stimulates TGF-β signaling and Foxp3 expression (102). Interestingly, while GILZ appears to be a potent catalyst for Foxp3+ expression, the absence of GILZ does not entirely obstruct Foxp3+ expression in Tregs. This observation implies that glucocorticoids may enhance Tregs through several, concurrent mechanisms (103). Lilla et al. (104) found that regulatory T cells were less sensitive to glucocorticoid-induced apoptosis than CD4+ T cells and better understood the changes in GC-induced apoptosis-related proteins. Studies have reported that dexamethasone given in the absence of Tregs completely loses its ability to control inflammation, and the Tregs themselves lack glucocorticoid receptors, resulting in rapid loss of therapeutic ability. Glucocorticoids Anti-inflammatory effects are mediated by Foxp3 + T cell regulation through an MR-342-dependent mechanism (105). In conclusion, glucocorticoids could potentially amplify the prevalence and activity of Tregs. Moreover, they can shape a favorable immune environment for Tregs by adjusting local cytokine expression.
4.2 DMARDs
In terms of therapeutic strategies, studies have evaluated the immune responses in mice immunized with ovalbumin and treated with Methotrexate (MTX), Cyclophosphamide (CTX), or a combination of both. Observations have shown that combination therapy of MTX and CTX, as opposed to monotherapy with either drug, induces a bias towards Tregs and inhibits Th17 by interfering with the maturation and antigen-presenting abilities of DCs (106). Upon interaction with this compound, there is a prolonged upregulation of Foxp3, TGF-β, and IL-10 in CD4+ cells, an augmentation of Treg suppressive function, and a reduction in IL-17 mRNA (107). In the context of hydroxychloroquine, when this compound is introduced to peripheral blood mononuclear cells from RA patients in vitro, there is a decrease in the secretion of IL-17, IL-6, and IL-22 in the culture supernatant (108). Treating early, untreated RA patients with methotrexate can increase the proportion and absolute number of Tregs with high levels of activation markers, indicating an enhancement of their functional capabilities (109). A study evaluated the effect of MTX treatment on the percentage and absolute number of CD4+Foxp3+ Tregs in the peripheral blood of untreated patients with early-stage RA and found that increases and phenotypic changes in Treg cells were associated with MTX treatment closely (109). MTX-loaded nanoparticles reduced the severity of experimental arthritis models in mice and reciprocally regulated Th17 and Tregs in vivo. Nanoparticles are promising therapeutics due to their ability to deliver and release drugs (110). Patients battling autoimmune diseases often necessitate lifelong immunotherapy, a regimen that can result in severe adverse reactions and medicinal side effects. Initiating drug treatment at an early stage can effectively impede the disease’s progression and decrease the rate of advancement. Given the numerous reports on the regenerative capabilities of Tregs, the most effective therapeutic strategy is to induce self-tolerance prior to the onset of significant tissue damage.
4.3 Low-dose interleukin-2
Interleukin-2 (IL-2) is crucial for Treg-mediated tolerance and it plays a pivotal role in the generation, expansion, survival, and function of Tregs (111, 112). IL-2 receptor signaling can facilitate the expansion and differentiation of T cells and activate a series of signaling pathways in T cells, including MAPK, PI3K, STAT5, and so on (113). Among them, the STAT5 signaling pathway is an important pathway for Tregs (114). STAT5 activation can directly regulate the expression of Foxp3 and inhibit the methylation of CpG islands in the CNS2 region by binding to Foxp3 promoter and CNS2 region. Simultaneously, Tregs intrinsically express high-affinity CD25 (IL-2 receptor alpha chain) along with unique IL-2-inducing signals and downstream genes (115). They exhibit a preferential response to low doses of IL-2 in comparison to other immune cells (116). The administration of low-dose IL-2 has been significantly effective in reducing disease activity (117). Thus, IL-2 helps tip the immune balance toward regulation rather than inflammation. Research by Scott N. Furlan et al. (118) found that compared to infusing Tregs alone with rapamycin, adding IL-2 in vivo to rapamycin supports the logarithmic growth of Tregs and effectively increases the number of Tregs in peripheral blood compartments. Therefore, low-dose IL-2, which is well-tolerated, can induce Tregs and mediate clinical improvements in autoimmune and inflammatory diseases. Employing exogenous low-dose IL-2 to selectively stimulate Tregs appears advantageous for the body, and it holds substantial potential for the treatment of immune diseases. Currently, the development of human anti-IL-2 monoclonal antibodies is underway (119, 120). Scientists have engineered a specific IL-2 monoclonal antibody (mAb), JES6-1, that selectively proliferates Tregs and demonstrates superior disease management in a mouse model of colitis, in comparison to a non-covalently linked complex of IL-2 and JES6-1 (121). In a prospective, open-label, Phase I to IIa study in patients with RA, low-dose IL-2 was shown to be well tolerated and effective, activating and expanding specific Tregs without activating effector T cells, thereby increasing The Treg/Teff ratio (122). Low-dose IL-2 therapy has been tried to treat a variety of autoimmune diseases and is a promising approach for the treatment of RA.
4.4 Rapamycin and all-trans retinoic acid
Initially discovered as a powerful antifungal agent derived from Streptomyces hygroscopicus, Rapamycin (RAPA) was subsequently acknowledged for its significant immunosuppressive qualities. It accomplishes this immunosuppression by inhibiting the serine/threonine kinase mTOR, a downstream effector in the phosphatidylinositol 3-kinase (PI3K) and Akt signaling pathway (123, 124). mTOR exerts its effect through two unique complexes, specifically mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Rapamycin exhibits a stronger inhibitory effect on mTORC1 and only suppresses mTORC2 after prolonged exposure. These mTOR complexes play a crucial role in mediating a myriad of cellular activities. The differentiation of Th1 and Th17 cells from CD4+ T cells is directed by mTORC1, while mTORC2 plays an indispensable role in the differentiation of Th2 cells (125). Interestingly, Tregs exhibit resistance to the growth-inhibitory effects mediated by rapamycin. Studies have indicated that rapamycin promotes the proliferation and survival of Tregs, while simultaneously inhibiting the growth, migration, and cytokine production of Th1 and Th17 cells (126–128). Initially, Bruyn et al. (129) found that the clinical efficacy of methotrexate combined with rapamycin analog everolimus in the treatment of RA was much better than that of methotrexate alone. Subsequently, more and more studies have shown that rapamycin is safe and effective in the treatment of RA. The original research group evaluated the effect of rapamycin on CD4 + CD25 + Foxp3 + Tregs in RA patients, suggesting that rapamycin can promote the proliferation of Tregs in low-activity RA patients, thereby balancing Th17/Tregs. Balance of cell ratios increases the likelihood of clinical remission in RA and reduces the risk of disease recurrence (130). Wen et al. (128) found that rapamycin immunomodulatory therapy can not only effectively reverse the reduced Tregs and suppress effector T cells, but also alleviate the clinical symptoms of patients with active RA and reduce the application of immunosuppression. It can be seen that the use of rapamycin can not only control clinical symptoms, but also greatly reduce the dosage of hormones and immunosuppressants in patients. The PI3K-Akt-mTOR signaling axis tightly governs the development, homeostasis, and functionality of Tregs (131). However, due to the widespread expression of the PI3K-Akt-mTOR signaling pathway, the effects of rapamycin can be observed in a variety of immune and non-immune cells. It’s still unclear whether rapamycin selectively inhibits the proliferation of non-Tregs, thereby indirectly promoting the expansion of Foxp3+ Tregs (132). Despite a significant increase in Treg levels, rapamycin leads to a transient deterioration of β-cell function in patients with Type 1 Diabetes Mellitus (T1DM) (133). Hence, the negative impacts of this approach in clinical practice cannot be overlooked, and its application in autoimmune diseases requires further investigation. Comparative studies have shown that both rapamycin and all-trans retinoic acid (ATRA) have similar promoting and stabilizing effects during the expansion process of Tregs (134), and under inflammatory conditions, ATRA exhibits superior stability in nTregs compared to rapamycin (135). ATRA enhances the differentiation and stability of iTregs without altering DNA methylation (136). ATRA significantly activates the ERK1/2 signaling pathway, promoting Foxp3 expression (136). DNA methylation at the Foxp3 gene locus plays an important role in the expression and maintenance of Foxp3 in subpopulations (137). In the presence of inflammatory factors, ATRA can inhibit the methylation of the Foxp3 gene in nTregs (135). This may be one of the molecular mechanisms by which ATRA plays a role in diseases of the immune system. Therefore, ATRA can be used as monotherapy or adjuvant therapy for autoimmune diseases.
4.5 Biological agents
4.5.1 Anti-TNF agents
In the past 10 years, more and more studies have emphasized the impact of biological agents on Tregs in RA patients. Ehrenstein et al. initially observed that infliximab (a chimeric monoclonal antibody against TNF) treatment increased the proportion of circulating CD4 + Foxp3+ cells and restored the inhibitory activity of CD25high Tregs (138). Studies suggest that the expression of Foxp3 in CD4+ lymphocytes escalates in patients treated with etanercept, a fusion protein that functions as a TNF inhibitor (139). Patients treated with TNF-α blockers had a higher proportion of Tregs than patients treated with methotrexate (MTX) (140). Anti-TNF-α treatment increases the Tregs/effector T cell ratio, indicating that it can restore the suppressive function of Tregs (57, 141). Furthermore, Tregs isolated from RA patients who have responded favorably to adalimumab monotherapy seem to display more pronounced suppressive activity (142, 143).However, some studies did not observe any difference in the percentage of Tregs before and after treatment with the human monoclonal antibody adalimumab (50, 144). Some have shown that blocking TNF affects the number and function of Tregs in mouse arthritis models and RA patients (145, 146). Recent studies by Tseng et al. found that TNFR2 is required for Treg development and function under homeostatic and inflammatory conditions. They further verified that TNFR2 is required to prevent Foxp3 promoter DNA methylation, emphasizing the importance of TNFR2. TNFR2 signaling plays an important role in the maintenance of Tregs stability, and TNFR2 deficiency results in a Th17-like phenotype (147). Valencia et al. found that TNF is mediated by TNFRII, which is constitutively expressed by Tregs, and that downregulation of expression results in a reduction of Foxp3 mRNA, directly impairing the inhibitory activity of RA CD25high Tregs (148). The expansion and activation of TNFRII+ Tregs may be one of the mechanisms by which anti-TNF drugs control RA inflammation (149). A recent investigation also demonstrated that TNF-α modulates the balance between Tregs and pathogenic Th17 and Th1 cells in the synovium of RA patients via Foxp3 dephosphorylation (72, 148). In RA patients, TNF blocker treatment restored the suppressive activity of Tregs, which was associated with reduced protein phosphatase 1 (PP1) expression and increased Foxp3 phosphorylation in Tregs. The critical phosphorylation site Ser418 in the c-terminal DNA-binding domain of Foxp3 is a critical site for the inhibitory function of Tregs. In RA-derived Tregs, TNF-α amplifies the expression of protein phosphatase 1 (PP1) via the IKK-NF-κB pathway, which results in the dephosphorylation of the crucial Ser418 site of Foxp3. This action subsequently leads to the inactivation of Foxp3, causing damage (150). Interestingly, animal model studies have shown that ntregs require the presence of TNFα to function effectively, but iTregs do not require this stimulation (151). Therefore, TNF-α is a major driver of inflammation in RA patients, and TNF blockers are an effective treatment for RA.
4.5.2 CTLA-4-immunologlobulin-G1
The fusion protein linked with the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4-Ig), which tactically adjusts the costimulatory signaling between CD28 and CD80/86, appears to function as a biological DMARDs. Notably, this substance has a dual role: it not only engages with T cells, but also has a significant interaction with other cell populations that play a critical role in the development and progression of RA (152). The first description of CTLA-4 abnormalities in RA-deficient Tregs dates back to 2008, when RA patients had lower CTLA-4 levels and higher internalization rates in Tregs than healthy subjects (153). Considering that the forced expression of CTLA-4 on Tregs in the context of RA can rejuvenate their inhibitory function, and conversely, the blockade of CTLA-4 on healthy Tregs can impair their functionality, researchers theorize that CTLA-4 present on RA Tregs could be a prospective therapeutic target for direct manipulation within these cells. The contribution of CTLA4-Ig in the treatment of RA extends beyond merely curtailing T cell activation. One of the protective mechanisms of CTLA4-Ig is to directly inhibit the formation of osteoclasts. In fact, CTLA-4 expressed by normal Tregs binds to CD80/CD86 on dendritic cells, inducing the activation of IDO, and leading to the apoptosis of osteoclast precursors (154). Although anti-CTLA‐4 antibodies have been used in autoimmune therapy, their underlying mechanisms have not been fully elucidated. One study showed that CTLA4-Ig inhibited CIA by modifying CIA mouse DCs and converting them into tolerant DCs, thereby increasing the number of CD4+CD25+Foxp3+ Tregs. This may be a new immune regulatory mechanism of CTLA4-Ig (155). CTLA-4-Ig, in conjunction with TCR ligation, possesses the added capacity to transform naive CD4+CD25- T cells into Foxp3+Tregs and amplify their population (156). Abatacept is a biological treatment that employs the extracellular domain of the CTLA-4 protein, fused with the Fc portion of IgG1, to safely disrupt the costimulatory signal of APC (157). RA patients who carry the CTLA-4 G polymorphism demonstrated a superior EULAR response and a reduced disease activity rate following treatment with abatacept (158). In the European Union, the use of abatacept has been granted approval for patients with highly active and progressive RA who have not previously undergone treatment with methotrexate (159). However, the exact mechanism of the drug’s efficacy and its effects on the cells that most commonly utilize the CTLA-4 protein (tregs) is unclear and may be conflicting. It has been reported that abatacept has a very complex effect on Treg population number and suppressive function (157). For instance, administering abatacept for RA seems to revive, at least partially, the impaired suppressive action of circulating Tregs (160, 161). However, this finding has not been corroborated by in vitro studies involving Tregs from synovial fluid (162). In addition, one study has shown that CTLA-4Ig treatment significantly reduced the proportions of Tregs (163).
4.5.3 IL-6 receptor blocker
IL-6 is a key cytokine determining whether naive T lymphocytes differentiate into Tregs or Th17 cells (164). This understanding has inspired us to delve deeper into the effect of inhibiting IL-6 on the Treg/Th17 cell balance in autoimmune diseases like RA. Evidence from experimental RA models suggests that early intervention with anti-IL-6 receptor antibodies can decrease the prevalence of circulating Th17 cells, thereby mitigating clinical symptoms (165). Consistent with these studies, treatment of early-stage RA with the IL-6 receptor blocker tocilizumab for 3 months resulted in a similar reduction in the number of Th17 (166). This reduction was demonstrated in another study that counted Th17 percentages at 4 months after tocilizumab treatment (57). Other studies did not observe any difference in Th17 percentage 6 months after treatment (167, 168). IL-6 is related to the plasticity of Tregs. Data from RA patients treated with tocilizumab show an increase in Treg numbers after treatment, consistent with a good clinical response (57, 168, 169). IL-6 is a major driver of increased inflammation in RA patients, and tocilizumab has been shown to treat RA patients and alleviate their disease (170–172).
4.6 Treg-based cellular therapies
In recent years, cell therapy centered around Tregs has emerged as a novel target for treating RA, garnering widespread attention (173–177). One strategy for Tregs therapy in RA involves isolating Tregs from patients and then expanding them in vitro. The goal is to enhance immune suppression by increasing the number of Tregs, thereby preventing excessive immune responses leading to arthritis inflammation. Studies have confirmed that adoptively transferred Tregs in the CIA model quickly appear in synovial tissue after injection, blocking T-cell proliferation and type II collagen-specific proliferation, significantly reducing disease severity and slowing disease progression (178). Transfer of Tregs into T-cell-deficient mice has been shown to increase bone volume (179). While various Tregs populations exist in peripheral blood with predictable functional and phenotypic differences based on cell surface markers (180), their clinical application is limited due to their scarcity, lack of reactivity, and unclear specificity. Successful Tregs transfer therapy for RA patients requires inputting a sufficient number of cells and effectively expanding antigen-specific Tregs without losing their specificity or function. Researchers are exploring methods to enhance the specificity of Tregs through genetic engineering, aiming to improve their inhibitory effect on specific autoantigens. This can be achieved by introducing specific antigen receptors (such as TCRs (181, 182) or chimeric antigen receptors (CARs) (183–185), allowing Tregs to more precisely target and suppress inflammation in the joint area. The adoptive transfer of these antigen-specific Tregs has been shown to be stable in vivo and reverse CIA progression by inhibiting CD4+ T cell proliferation and the key inflammatory cytokine TNF-α (186). Compared to non-specific TCR transduction or polyclonal Tregs, TCR Tregs exhibit enhanced inhibitory functions, making them more capable of restoring dominant immune tolerance and completely alleviating targeted autoimmune diseases (187). CAR Tregs have stronger signaling capabilities than TCR Tregs (188). Recently, Safari and colleagues (189) revealed the potential of the clustered regularly interspaced short palindromic repeat (CRISPR) genome editing technique combined with the CRISPR-associated (Cas) 9 system to modify Tregs (189).
In another realm, research is exploring the combination of Tregs therapy with other immune-modulating therapies to enhance efficacy. This may involve the co-application of Tregs with immunomodulatory drugs or other cell therapies, regulating the immune system at multiple levels to achieve a more comprehensive treatment outcome. An ongoing phase I clinical trial (NCT02772679) is investigating the combined administration of IL-2 and polyclonal Tregs. As mentioned earlier, low-dose IL-2 treatment itself has the effect of expanding Tregs in vivo. The combined administration of polyclonal Tregs with low-dose IL-2 is expected to enhance the number and function of Tregs after infusion. Immunosuppressive drugs commonly used to treat autoimmune diseases have been shown to dose-dependently reduce Treg proliferation and survival (190, 191). Combining Tregs with other cell therapy methods may produce synergistic effects in the treatment of autoimmune diseases, and such approaches are yet to be explored (192). Adoptive transfer of Tregs, whether used in treating autoimmune diseases or employing a strategy of increasing Tregs, aims not only to correct the balance between Tregs and Teff but also faces challenges. One challenge is maintaining the stability and functionality of Tregs during in vitro expansion (193, 194). Research on how to isolate phenotypically stable, highly active, and functionally suppressed Tregs and designing strategies to increase their numbers while maintaining their characteristics is crucial for developing Treg-based therapies for autoimmune diseases. Additionally, factors such as individual differences and potential adverse reactions during the treatment process need to be carefully considered. Therefore, while Tregs therapy brings new therapeutic prospects for RA patients, further in-depth research and clinical trials are needed to validate its safety and effectiveness.
4.7 Others
Phosphatase and tensin homolog (PTEN), a tumor suppressor and a phosphatase specific to the 3’ position of phosphatidylinositol 3,4,5-triphosphate (195), has a pivotal role in cell metabolism and motility (196). Research indicates that PTEN exhibits therapeutic potential in CIA rats and has associations with RA (197, 198). PTEN can enhance the stability of Tregs, while its absence can trigger autoinflammatory conditions (199, 200). Systemic administration of PTEN attenuated arthritis severity in CIA mice, and an increased expression of PTEN curtailed T-cell activation and ameliorated the imbalance between Th17 cells and Tregs (200). PTEN operates as an intracellular phosphatase, putting the brakes on the PI3K-Akt-mTOR signaling pathway. Excessive stimulation of this pathway, a scenario observed in PTEN-deficient Tregs, can incite Treg instability due to the hyperactivation of mTORC2 (199, 201). A hallmark of synovial fibroblast activation in RA patients is the downregulation of PTEN expression (202). These studies have shown that PTEN plays a key role in the occurrence and development of immune responses, and it may be used to modify Tregs function. Overexpression of PTEN can inhibit the severity of CIA, thereby reducing the inflammatory response and osteoclast generation. Significantly, the absence of p53 is linked with a reduction in PTEN gene expression, which in turn triggers the phosphorylation of STAT3, thereby exacerbating autoimmune arthritis. Consequently, this discovery implies that PTEN could potentially be harnessed to alter the function of Tregs (200). In addition, studies have shown that TGF-β, vasoactive intestinal peptide (VIP) and immunoglobulin D (Immunoglobulin D, IgD)-Fc-Ig play an important role in targeting Tregs to restore immune tolerance in RA. TGF-β is an important growth factor that promotes Th17 and Treg differentiation. In the context of experimental arthritis, while the local suppression of TGF-β1 signaling can enhance the Th17/Treg equilibrium, it does not yield improvements in joint pathology (203). VIP is a neuropeptide with anti-inflammatory effects and has therapeutic potential in a variety of immune diseases. It is involved in maintaining immune tolerance through a novel mechanism that induces the generation of Tregs (204). IgD-fc-ig selectively interferes with the interaction between IgD and IgDR, putting a halt to the abnormal proliferation and activation of T cells provoked by IgD. It corrects the imbalance between Th17 and Tregs, lessens the production of inflammatory cytokines (205). The composition of the gut microbiota in patients with RA undergoes changes, potentially exacerbating immune dysregulation (206, 207). Fecal microbiota transplantation, involving the introduction of specific bacteria, may become an adjunctive approach in treating RA. Short-chain fatty acids, produced by certain gut bacteria, have demonstrated anti-inflammatory effects (208).They can upregulate anti-inflammatory genes in dendritic cells, enhance histone acetylation of the Foxp3 locus, and increase the stability of the Foxp3 transcription factor (209). Other molecules produced by bacteria, such as Bifidobacterium’s polysaccharide A, can induce the generation of Tregs (210). Due to safety concerns, fecal transplantation therapy has not yet been approved clinically. However, once this therapy becomes viable, it could be considered as a potential Treg-promoting strategy, synergizing with Tregs-based therapies or other methods promoting Tregs to enhance their therapeutic efficacy.
5 Summary
The maintenance of self-immune tolerance is greatly reliant on Tregs. A decline in the number and/or function of Tregs in RA patients indicates that abnormalities in Tregs are intimately associated with the progression of RA. The transfer of Tregs and preservation of Tregs functions present promising strategies to mitigate joint inflammation in RA patients. CAR-Tregs could potentially serve as a useful tool for treating, or even curing, autoimmune diseases. However, the regulatory mechanisms of Tregs are not yet fully comprehended and warrant further investigation. There is a pressing need to bolster research in clinical investigations and related animal experiments to explore the immune mechanism of Tregs in the pathogenesis of RA. It is anticipated that the scope for future treatment of RA will expand considerably.
Author contributions
JZ: Writing – original draft. HJL: Writing – review & editing. YC: Funding acquisition, Writing – review & editing. HL: Writing – review & editing. SZ: Writing – review & editing. QX: Funding acquisition, Writing – review & editing. GY: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No.82201985), and Key Research and Development (R&D) Projects of Sichuan Province (2021YFS0164).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
RA, Rheumatoid arthritis; Foxp3, forehead box P3; Tregs, regulatory T cells; TCR, T cell receptor; CTLA4, cytotoxic T lymphocyte-associated protein 4; TSDR, regulatory T cell-specific demethylation region; CNSs, conserved non-coding sequences; DCs, dendritic cells; Teffs, effector T cells; IDO, indoleamine 2,3-dioxygenase; LAG-3, Lymphocyte activation gene 3; PD-1, Programmed Cell Death Protein 1; APCs, antigen presenting cells; ROR γt, retinoic acid receptor-associated orphan receptor γt; Th1, T helper cell type 1; CIA, collagen-induced arthritis; TNF, tumor necrosis factor; IL, interleukin; RANKL, nuclear factor-κB ligand; TGF, transforming growth factor; JAK, Janus kinase; DMARDs, disease modifying antirheumatic drugs; GILZ, glucocorticoid-induced leucine zipper; MTX, methotrexate; CTX, cyclophosphamide; RAPA, Rapamycin; PI3K, phosphatidylinositol 3-kinase; ATRA, All-trans retinoic acid; PTEN, Phosphatase and tensin homolog; VIP, Vasoactive intestinal peptide; IgD, Immunoglobulin D; CARs, chimeric antigen receptors; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats
References
1. Scherer HU, Haupl T, Burmester GR. The etiology of rheumatoid arthritis. J Autoimmun (2020) 110:102400. doi: 10.1016/j.jaut.2019.102400
2. Wu D, Luo Y, Li T, Zhao X, Lv T, Fang G, et al. Systemic complications of rheumatoid arthritis: Focus on pathogenesis and treatment. Front Immunol (2022) 13:1051082. doi: 10.3389/fimmu.2022.1051082
3. Jang S, Kwon EJ, Lee JJ. Rheumatoid arthritis: pathogenic roles of diverse immune cells. Int J Mol Sci (2022) 23(2):905. doi: 10.3390/ijms23020905
4. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10(7):490–500. doi: 10.1038/nri2785
5. Tao JH, Cheng M, Tang JP, Liu Q, Pan F, Li XP. Foxp3, regulatory T cell, and autoimmune diseases. Inflammation (2017) 40(1):328–39. doi: 10.1007/s10753-016-0470-8
6. Shen MY, Jiang BP, Zhang MF, Wang X, Zhu H, Gu ZN, et al. MicroRNA-143-3p ameliorates collagen-induced arthritis by polarizing naive CD4(+) T cells into Treg cells. J Clin Lab Anal (2023) 37(5):e24845. doi: 10.1002/jcla.24845
7. Yan S, Kotschenreuther K, Deng S, Kofler DM. Regulatory T cells in rheumatoid arthritis: functions, development, regulation, and therapeutic potential. Cell Mol Life Sci (2022) 79(10):533. doi: 10.1007/s00018-022-04563-0
8. Jiang Q, Yang G, Liu Q, Wang S, Cui D. Function and role of regulatory T cells in rheumatoid arthritis. Front Immunol (2021) 12:626193. doi: 10.3389/fimmu.2021.626193
9. Li X, Xu H, Huang J, Luo D, Lv S, Lu X, et al. Dysfunctions, molecular mechanisms, and therapeutic strategies of regulatory T cells in rheumatoid arthritis. Front Pharmacol (2021) 12:716081. doi: 10.3389/fphar.2021.716081
10. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
11. Haribhai D, Lin W, Edwards B, Ziegelbauer J, Salzman NH, Carlson MR, et al. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J Immunol (Baltimore Md: 1950) (2009) 182(6):3461–8. doi: 10.4049/jimmunol.0802535
12. Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity (2008) 28(1):100–11. doi: 10.1016/j.immuni.2007.11.021
13. Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol (2005) 6(2):152–62. doi: 10.1038/ni1160
14. Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of foxp3 expression in regulatory T cells by DNA methylation. J Immunol. (2009) 182(1):259–73. doi: 10.4049/jimmunol.182.1.259
15. Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity (2011) 35(1):109–22. doi: 10.1016/j.immuni.2011.03.029
16. d’Hennezel E, Ben-Shoshan M, Ochs HD, Torgerson TR, Russell LJ, Lejtenyi C, et al. FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. N Engl J Med (2009) 361(17):1710–3. doi: 10.1056/NEJMc0907093
17. Mantel PY, Ouaked N, Ruckert B, Karagiannidis C, Welz R, Blaser K, et al. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol (Baltimore Md: 1950) (2006) 176(6):3593–602. doi: 10.4049/jimmunol.176.6.3593
18. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463(7282):808–12. doi: 10.1038/nature08750
19. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet (2013) 14(3):204–20. doi: 10.1038/nrg3354
20. Kitagawa Y, Ohkura N, Sakaguchi S. Epigenetic control of thymic Treg-cell development. Eur J Immunol (2015) 45(1):11–6. doi: 10.1002/eji.201444577
21. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122
22. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol (2003) 4(4):337–42. doi: 10.1038/ni909
23. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol (2007) 8(2):191–7. doi: 10.1038/ni1428
24. O’Connor RA, Leech MD, Suffner J, Hammerling GJ, Anderton SM. Myelin-reactive, TGF-beta-induced regulatory T cells can be programmed to develop Th1-like effector function but remain less proinflammatory than myelin-reactive Th1 effectors and can suppress pathogenic T cell clonal expansion in vivo. J Immunol (Baltimore Md: 1950) (2010) 185(12):7235–43. doi: 10.4049/jimmunol.1001551
25. Chen Z, Bozec A, Ramming A, Schett G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat Rev Rheumatol (2019) 15(1):9–17. doi: 10.1038/s41584-018-0109-2
26. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol (2018) 19(7):665–73. doi: 10.1038/s41590-018-0120-4
27. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
28. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
29. Bourque J, Hawiger D. Immunomodulatory bonds of the partnership between dendritic cells and T cells. Crit Rev Immunol (2018) 38(5):379–401. doi: 10.1615/CritRevImmunol.2018026790
30. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U.S.A. (2021) 118(30):e2023739118. doi: 10.1073/pnas.2023739118
31. Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol (2009) 21(10):1105–11. doi: 10.1093/intimm/dxp095
32. Maruhashi T, Okazaki IM, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol (2018) 19(12):1415–26. doi: 10.1038/s41590-018-0217-9
33. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi: 10.1038/nri3405
34. Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol (2013) 13(6):461–7. doi: 10.1038/nri3464
35. Busse D, de la Rosa M, Hobiger K, Thurley K, Flossdorf M, Scheffold A, et al. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc Natl Acad Sci United States America (2010) 107(7):3058–63. doi: 10.1073/pnas.0812851107
36. Collison LW, Vignali DA. In vitro Treg suppression assays. Methods Mol Biol (2011) 707:21–37. doi: 10.1007/978-1-61737-979-6_2
37. Meng Q, Wen Z, Meng W, Bian H, Gu H, Zuo R, et al. et al: Blimp1 suppressed CD4(+) T cells-induced activation of fibroblast-like synoviocytes by upregulating IL-10 via the rho pathway. Environ Toxicol (2023) 38(1):146–58. doi: 10.1002/tox.23672
38. Thiolat A, Denys A, Petit M, Biton J, Lemeiter D, Herve R, et al. Interleukin-35 gene therapy exacerbates experimental rheumatoid arthritis in mice. Cytokine (2014) 69(1):87–93. doi: 10.1016/j.cyto.2014.05.015
39. Li Y, Jie Y, Wang X, Lu J. Serum IL-35 is decreased in overweight patients with rheumatoid arthritis: its correlation with Th1/Th2/Th17-related cytokines. BMC Immunol (2021) 22(1):42. doi: 10.1186/s12865-020-00395-4
40. Frank MJ, Reagan PM, Bartlett NL, Gordon LI, Friedberg JW, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discovery (2018) 8(10):1258–69. doi: 10.1158/2159-8290.CD-18-0743
41. Yuan X, Dee MJ, Altman NH, Malek TR. IL-2Rbeta-dependent signaling and CD103 functionally cooperate to maintain tolerance in the gut mucosa. J Immunol (Baltimore Md: 1950) (2015) 194(3):1334–46. doi: 10.4049/jimmunol.1400955
42. Wang L, Wang Y, Liu C, He J, He X, Zhang X, et al. Treg-targeted efficient-inducible platform for collagen-induced arthritis treatment. Mater Today Bio (2023) 19:100557. doi: 10.1016/j.mtbio.2023.100557
43. Chen L, Wu X, Zhong J, Li D. L161982 alleviates collagen-induced arthritis in mice by increasing Treg cells and down-regulating Interleukin-17 and monocyte-chemoattractant protein-1 levels. BMC Musculoskelet Disord (2017) 18(1):462. doi: 10.1186/s12891-017-1819-3
44. Liston A, Dooley J, Yshii L. Brain-resident regulatory T cells and their role in health and disease. Immunol Lett (2022) 248:26–30. doi: 10.1016/j.imlet.2022.06.005
45. Cao D, Malmstrom V, Baecher-Allan C, Hafler D, Klareskog L, Trollmo C. Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol (2003) 33(1):215–23. doi: 10.1002/immu.200390024
46. Cao D, van Vollenhoven R, Klareskog L, Trollmo C, Malmstrom V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther (2004) 6(4):R335–346. doi: 10.1186/ar1192
47. Mottonen M, Heikkinen J, Mustonen L, Isomaki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol (2005) 140(2):360–7. doi: 10.1111/j.1365-2249.2005.02754.x
48. Liu MF, Wang CR, Fung LL, Lin LH, Tsai CN. The presence of cytokine-suppressive CD4+CD25+ T cells in the peripheral blood and synovial fluid of patients with rheumatoid arthritis. Scand J Immunol (2005) 62(3):312–7. doi: 10.1111/j.1365-3083.2005.01656.x
49. van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum (2004) 50(9):2775–85. doi: 10.1002/art.20499
50. Dombrecht EJ, Aerts NE, Schuerwegh AJ, Hagendorens MM, Ebo DG, Van Offel JF, et al. Influence of anti-tumor necrosis factor therapy (Adalimumab) on regulatory T cells and dendritic cells in rheumatoid arthritis. Clin Exp Rheumatol (2006) 24(1):31–7.
51. Jiao Z, Wang W, Jia R, Li J, You H, Chen L, et al. Accumulation of FoxP3-expressing CD4+CD25+ T cells with distinct chemokine receptors in synovial fluid of patients with active rheumatoid arthritis. Scand J Rheumatol (2007) 36(6):428–33. doi: 10.1080/03009740701482800
52. Han GM, O’Neil-Andersen NJ, Zurier RB, Lawrence DA. CD4+CD25high T cell numbers are enriched in the peripheral blood of patients with rheumatoid arthritis. Cell Immunol (2008) 253(1-2):92–101. doi: 10.1016/j.cellimm.2008.05.007
53. Sempere-Ortells JM, Perez-Garcia V, Marin-Alberca G, Peris-Pertusa A, Benito JM, Marco FM, et al. Quantification and phenotype of regulatory T cells in rheumatoid arthritis according to disease activity score-28. Autoimmunity (2009) 42(8):636–45. doi: 10.3109/08916930903061491
54. Kawashiri SY, Kawakami A, Okada A, Koga T, Tamai M, Yamasaki S, et al. CD4+CD25(high)CD127(low/-) Treg cell frequency from peripheral blood correlates with disease activity in patients with rheumatoid arthritis. J Rheumatol (2011) 38(12):2517–21. doi: 10.3899/jrheum.110283
55. Niu Q, Cai B, Huang ZC, Shi YY, Wang LL. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol Int (2012) 32(9):2731–6. doi: 10.1007/s00296-011-1984-x
56. Lina C, Conghua W, Nan L, Ping Z. Combined treatment of etanercept and MTX reverses Th1/Th2, Th17/Treg imbalance in patients with rheumatoid arthritis. J Clin Immunol (2011) 31(4):596–605. doi: 10.1007/s10875-011-9542-6
57. Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, et al. Brief report: inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum (2012) 64(8):2499–503. doi: 10.1002/art.34477
58. Ji L, Geng Y, Zhou W, Zhang Z. A study on relationship among apoptosis rates, number of peripheral T cell subtypes and disease activity in rheumatoid arthritis. Int J Rheum Dis (2016) 19(2):167–71. doi: 10.1111/1756-185X.12211
59. Walter GJ, Evans HG, Menon B, Gullick NJ, Kirkham BW, Cope AP, et al. Interaction with activated monocytes enhances cytokine expression and suppressive activity of human CD4+CD45ro+CD25+CD127(low) regulatory T cells. Arthritis Rheum (2013) 65(3):627–38. doi: 10.1002/art.37832
60. Moradi B, Schnatzer P, Hagmann S, Rosshirt N, Gotterbarm T, Kretzer JP, et al. CD4(+)CD25(+)/highCD127low/(-) regulatory T cells are enriched in rheumatoid arthritis and osteoarthritis joints–analysis of frequency and phenotype in synovial membrane, synovial fluid and peripheral blood. Arthritis Res Ther (2014) 16(2):R97. doi: 10.1186/ar4545
61. Walter GJ, Fleskens V, Frederiksen KS, Rajasekhar M, Menon B, Gerwien JG, et al. Phenotypic, functional, and gene expression profiling of peripheral CD45RA+ and CD45RO+ CD4+CD25+CD127(low) treg cells in patients with chronic rheumatoid arthritis. Arthritis Rheumatol (2016) 68(1):103–16. doi: 10.1002/art.39408
62. Morita T, Shima Y, Wing JB, Sakaguchi S, Ogata A, Kumanogoh A. The proportion of regulatory T cells in patients with rheumatoid arthritis: A meta-analysis. PloS One (2016) 11(9):e0162306. doi: 10.1371/journal.pone.0162306
63. Zhang X, Zhang X, Zhuang L, Xu C, Li T, Zhang G, et al. Decreased regulatory T-cell frequency and interleukin-35 levels in patients with rheumatoid arthritis. Exp Ther Med (2018) 16(6):5366–72. doi: 10.3892/etm.2018.6885
64. Wang J, Zhang SX, Chang JS, Cheng T, Jiang XJ, Su QY, et al. Low-dose IL-2 improved clinical symptoms by restoring reduced regulatory T cells in patients with refractory rheumatoid arthritis: A randomized controlled trial. Front Immunol (2022) 13:947341. doi: 10.3389/fimmu.2022.947341
65. Mulhearn B, Marshall L, Sutcliffe M, Hannes SK, Fonseka C, Hussell T, et al. Automated clustering reveals CD4(+) T cell subset imbalances in rheumatoid arthritis. Front Immunol (2023) 14:1094872. doi: 10.3389/fimmu.2023.1094872
66. Su R, Li B, Wu R, Xie Y, Gao A, Gao C, et al. Stratified distribution of Th17 and Treg cells in patients with multi-stage rheumatoid arthritis. Arthritis Res Ther (2023) 25(1):55. doi: 10.1186/s13075-023-03041-7
67. Zhang SX, Wang J, Wang CH, Jia RH, Yan M, Hu FY, et al. Low-dose IL-2 therapy limits the reduction in absolute numbers of circulating regulatory T cells in rheumatoid arthritis. Ther Adv Musculoskelet Dis (2021) 13:1759720X211011370. doi: 10.1177/1759720X211011370
68. Liu J, Hong X, Lin D, Luo X, Zhu M, Mo H. Artesunate influences Th17/Treg lymphocyte balance by modulating Treg apoptosis and Th17 proliferation in a murine model of rheumatoid arthritis. Exp Ther Med (2017) 13(5):2267–73. doi: 10.3892/etm.2017.4232
69. Petrelli F, Mariani FM, Alunno A, Puxeddu I. Pathogenesis of rheumatoid arthritis: one year in review 2022. Clin Exp Rheumatol (2022) 40(3):475–82. doi: 10.55563/clinexprheumatol/l9lyen
70. Li Z, Lin F, Zhuo C, Deng G, Chen Z, Yin S, et al. et al: PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J Biol Chem (2014) 289(39):26872–81. doi: 10.1074/jbc.M114.586651
71. Ikeda S, Saijo S, Murayama MA, Shimizu K, Akitsu A, Iwakura Y. Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGF-beta-induced Foxp3 expression. J Immunol (Baltimore Md: 1950) (2014) 192(4):1449–58. doi: 10.4049/jimmunol.1300387
72. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. et al: Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med (2013) 19(3):322–8. doi: 10.1038/nm.3085
73. Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity (2009) 31(6):921–31. doi: 10.1016/j.immuni.2009.09.022
74. Attridge K, Wang CJ, Wardzinski L, Kenefeck R, Chamberlain JL, Manzotti C, et al. IL-21 inhibits T cell IL-2 production and impairs Treg homeostasis. Blood (2012) 119(20):4656–64. doi: 10.1182/blood-2011-10-388546
75. Sun H, Gao W, Pan W, Zhang Q, Wang G, Feng D, et al. Tim3(+) foxp3 (+) treg cells are potent inhibitors of effector T cells and are suppressed in rheumatoid arthritis. Inflammation (2017) 40(4):1342–50. doi: 10.1007/s10753-017-0577-6
76. Wang L, Wang C, Jia X, Yu J. Circulating exosomal miR-17 inhibits the induction of regulatory T cells via suppressing TGFBR II expression in rheumatoid arthritis. Cell Physiol Biochem (2018) 50(5):1754–63. doi: 10.1159/000494793
77. Fessler J, Raicht A, Husic R, Ficjan A, Schwarz C, Duftner C, et al. Novel senescent regulatory T-cell subset with impaired suppressive function in rheumatoid arthritis. Front Immunol (2017) 8:300. doi: 10.3389/fimmu.2017.00300
78. Wang W, Shao S, Jiao Z, Guo M, Xu H, Wang S. The Th17/Treg imbalance and cytokine environment in peripheral blood of patients with rheumatoid arthritis. Rheumatol Int (2012) 32(4):887–93. doi: 10.1007/s00296-010-1710-0
79. Aterido A, Palacio C, Marsal S, Avila G, Julia A. Novel insights into the regulatory architecture of CD4+ T cells in rheumatoid arthritis. PloS One (2014) 9(6):e100690. doi: 10.1371/journal.pone.0100690
80. Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity (2009) 30(5):646–55. doi: 10.1016/j.immuni.2009.05.001
81. Guo Y, Xing E, Song H, Feng G, Liang X, An G, et al. Therapeutic effect of dioscin on collagen-induced arthritis through reduction of Th1/Th2. Int Immunopharmacol (2016) 39:79–83. doi: 10.1016/j.intimp.2016.06.029
82. Srivastava RK, Dar HY, Mishra PK. Immunoporosis: immunology of osteoporosis-role of T cells. Front Immunol (2018) 9:657. doi: 10.3389/fimmu.2018.00657
83. Lubberts E. Role of T lymphocytes in the development of rheumatoid arthritis. Implications for treatment. Curr Pharm Des (2015) 21(2):142–6. doi: 10.2174/1381612820666140825122247
84. Zhou L, Wang J, Li J, Li T, Chen Y, June RR, et al. 1,25-Dihydroxyvitamin D3 ameliorates collagen-induced arthritis via suppression of Th17 cells through miR-124 mediated inhibition of IL-6 signaling. Front Immunol (2019) 10:178. doi: 10.3389/fimmu.2019.00178
85. Luan J, Zhang K, Yang P, Zhang Y, Feng F, Zhu YM, et al. The combination of FK506 and an anti-CD147 mAb exerts potential therapeutic effects on a mouse model of collagen-induced arthritis. Mol Immunol (2018) 101:1–9. doi: 10.1016/j.molimm.2018.05.013
86. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20(1):62–8. doi: 10.1038/nm.3432
87. Gizinski AM, Fox DA. T cell subsets and their role in the pathogenesis of rheumatic disease. Curr Opin Rheumatol (2014) 26(2):204–10. doi: 10.1097/BOR.0000000000000036
88. Alunno A, Manetti M, Caterbi S, Ibba-Manneschi L, Bistoni O, Bartoloni E, et al. Altered immunoregulation in rheumatoid arthritis: the role of regulatory T cells and proinflammatory Th17 cells and therapeutic implications. Mediators Inflammation (2015) 2015:751793. doi: 10.1155/2015/751793
89. Gautam S, Kumar R, Kumar U, Kumar S, Luthra K, Dada R. Yoga maintains Th17/Treg cell homeostasis and reduces the rate of T cell aging in rheumatoid arthritis: a randomized controlled trial. Sci Rep (2023) 13(1):14924. doi: 10.1038/s41598-023-42231-w
90. Kikuta J, Wada Y, Kowada T, Wang Z, Sun-Wada GH, Nishiyama I, et al. et al: Dynamic visualization of RANKL and Th17-mediated osteoclast function. J Clin Invest (2013) 123(2):866–73. doi: 10.1172/JCI65054
91. Zuniga LA, Jain R, Haines C, Cua DJ. Th17 cell development: from the cradle to the grave. Immunol Rev (2013) 252(1):78–88. doi: 10.1111/imr.12036
92. Schinocca C, Rizzo C, Fasano S, Grasso G, La Barbera L, Ciccia F, et al. Role of the IL-23/IL-17 pathway in rheumatic diseases: an overview. Front Immunol (2021) 12:637829. doi: 10.3389/fimmu.2021.637829
93. Kondo N, Kuroda T, Kobayashi D. Cytokine networks in the pathogenesis of rheumatoid arthritis. Int J Mol Sci (2021) 22(20):10922. doi: 10.3390/ijms222010922
94. Koper-Lenkiewicz OM, Sutkowska K, Wawrusiewicz-Kurylonek N, Kowalewska E, Matowicka-Karna J. Proinflammatory cytokines (IL-1, -6, -8, -15, -17, -18, -23, TNF-alpha) single nucleotide polymorphisms in rheumatoid arthritis-A literature review. Int J Mol Sci (2022) 23(4):2106. doi: 10.3390/ijms23042106
95. Paradowska-Gorycka A, Wajda A, Romanowska-Prochnicka K, Walczuk E, Kuca-Warnawin E, Kmiolek T, et al. Th17/treg-related transcriptional factor expression and cytokine profile in patients with rheumatoid arthritis. Front Immunol (2020) 11:572858. doi: 10.3389/fimmu.2020.572858
96. Wang T, Rui J, Shan W, Xue F, Feng D, Dong L, et al. Imbalance of Th17, Treg, and helper innate lymphoid cell in the peripheral blood of patients with rheumatoid arthritis. Clin Rheumatol (2022) 41(12):3837–49. doi: 10.1007/s10067-022-06315-8
97. Karagiannidis C, Akdis M, Holopainen P, Woolley NJ, Hense G, Ruckert B, et al. Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J Allergy Clin Immunol (2004) 114(6):1425–33. doi: 10.1016/j.jaci.2004.07.014
98. Lowenberg M, Verhaar AP, van den Brink GR, Hommes DW. Glucocorticoid signaling: a nongenomic mechanism for T-cell immunosuppression. Trends Mol Med (2007) 13(4):158–63. doi: 10.1016/j.molmed.2007.02.001
99. Muller N, Fischer HJ, Tischner D, van den Brandt J, Reichardt HM. Glucocorticoids induce effector T cell depolarization via ERM proteins, thereby impeding migration and APC conjugation. J Immunol (Baltimore Md: 1950) (2013) 190(8):4360–70. doi: 10.4049/jimmunol.1201520
100. Mahata B, Zhang X, Kolodziejczyk AA, Proserpio V, Haim-Vilmovsky L, Taylor AE, et al. Single-cell RNA sequencing reveals T helper cells synthesizing steroids de novo to contribute to immune homeostasis. Cell Rep (2014) 7(4):1130–42. doi: 10.1016/j.celrep.2014.04.011
101. Suarez A, Lopez P, Gomez J, Gutierrez C. Enrichment of CD4+ CD25high T cell population in patients with systemic lupus erythematosus treated with glucocorticoids. Ann rheumatic Dis (2006) 65(11):1512–7. doi: 10.1136/ard.2005.049924
102. Bereshchenko O, Coppo M, Bruscoli S, Biagioli M, Cimino M, Frammartino T, et al. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep (2014) 7(2):464–75. doi: 10.1016/j.celrep.2014.03.004
103. Libert C, Dejager L. How steroids steer T cells. Cell Rep (2014) 7(4):938–9. doi: 10.1016/j.celrep.2014.04.041
104. Prenek L, Litvai T, Balazs N, Kugyelka R, Boldizsar F, Najbauer J, et al. Regulatory T cells are less sensitive to glucocorticoid hormone induced apoptosis than CD4(+) T cells. Apoptosis (2020) 25(9-10):715–29. doi: 10.1007/s10495-020-01629-x
105. Kim D, Nguyen QT, Lee J, Lee SH, Janocha A, Kim S, et al. Anti-inflammatory roles of glucocorticoids are mediated by Foxp3(+) regulatory T cells via a miR-342-dependent mechanism. Immunity (2020) 53(3):581–596 e585. doi: 10.1016/j.immuni.2020.07.002
106. Yu X, Wang C, Luo J, Zhao X, Wang L, Li X. Combination with methotrexate and cyclophosphamide attenuated maturation of dendritic cells: inducing Treg skewing and Th17 suppression in vivo. Clin Dev Immunol (2013) 2013:238035. doi: 10.1155/2013/238035
107. Pericolini E, Gabrielli E, Alunno A, Bartoloni Bocci E, Perito S, Chow SK, et al. Functional improvement of regulatory T cells from rheumatoid arthritis subjects induced by capsular polysaccharide glucuronoxylomannogalactan. PloS One (2014) 9(10):e111163. doi: 10.1371/journal.pone.0111163
108. Silva JC, Mariz HA, Rocha LF Jr., Oliveira PS, Dantas AT, Duarte AL, et al. Hydroxychloroquine decreases Th17-related cytokines in systemic lupus erythematosus and rheumatoid arthritis patients. Clinics (Sao Paulo Brazil) (2013) 68(6):766–71. doi: 10.6061/clinics/2013(06)07
109. Avdeeva A, Rubtsov Y, Dyikanov D, Popkova T, Nasonov E. Regulatory T cells in patients with early untreated rheumatoid arthritis: Phenotypic changes in the course of methotrexate treatment. Biochimie (2020) 174:9–17. doi: 10.1016/j.biochi.2020.03.014
110. Park JS, Lee D, Yang S, Jeong HY, Na HS, Cho KH, et al. Methotrexate-loaded nanoparticles ameliorate experimental model of autoimmune arthritis by regulating the balance of interleukin-17-producing T cells and regulatory T cells. J Transl Med (2022) 20(1):85. doi: 10.1186/s12967-022-03267-0
111. Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Ciullini Mannurita S, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol (2013) 146(3):248–61. doi: 10.1016/j.clim.2013.01.004
112. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108(5):1571–9. doi: 10.1182/blood-2006-02-004747
113. Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol (2011) 12(6):551–9. doi: 10.1038/ni.2030
114. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in T(reg) cell function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540
115. Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity (2009) 30(2):204–17. doi: 10.1016/j.immuni.2008.11.014
116. Yu A, Snowhite I, Vendrame F, Rosenzwajg M, Klatzmann D, Pugliese A, et al. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes (2015) 64(6):2172–83. doi: 10.2337/db14-1322
117. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med (2016) 22(9):991–3. doi: 10.1038/nm.4148
118. Furlan SN, Singh K, Lopez C, Tkachev V, Hunt DJ, Hibbard J, et al. IL-2 enhances ex vivo-expanded regulatory T-cell persistence after adoptive transfer. Blood Adv (2020) 4(8):1594–605. doi: 10.1182/bloodadvances.2019001248
119. Raffin C, Vo LT, Bluestone JA. T(reg) cell-based therapies: challenges and perspectives. Nat Rev Immunol (2020) 20(3):158–72. doi: 10.1038/s41577-019-0232-6
120. Wu R, Li N, Zhao X, Ding T, Xue H, Gao C, et al. Low-dose Interleukin-2: Biology and therapeutic prospects in rheumatoid arthritis. Autoimmun Rev (2020) 19(10):102645. doi: 10.1016/j.autrev.2020.102645
121. Spangler JB, Trotta E, Tomala J, Peck A, Young TA, Savvides CS, et al. Engineering a single-agent cytokine/antibody fusion that selectively expands regulatory T cells for autoimmune disease therapy. J Immunol (Baltimore Md: 1950) (2018) 201(7):2094–106. doi: 10.4049/jimmunol.1800578
122. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis (2019) 78(2):209–17. doi: 10.1136/annrheumdis-2018-214229
123. Singh K, Sun S, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic. IV. Mechanism of action. J antibiot (1979) 32(6):630–45. doi: 10.7164/antibiotics.32.630
124. Garber K. Targeting mTOR: something old, something new. J Natl Cancer Instit (2009) 101(5):288–90. doi: 10.1093/jnci/djp034
125. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12(4):295–303. doi: 10.1038/ni.2005
126. Romano M, Sen M, Scotta C, Alhabbab RY, Rico-Armada A, Lechler RI, et al. Isolation and expansion of thymus-derived regulatory T cells for use in pediatric heart transplant patients. Eur J Immunol (2021) 51(8):2086–92. doi: 10.1002/eji.202048949
127. Kishimoto TK, Fournier M, Michaud A, Rizzo G, Roy C, Capela T, et al. Rapamycin nanoparticles increase the therapeutic window of engineered interleukin-2 and drive expansion of antigen-specific regulatory T cells for protection against autoimmune disease. J Autoimmun (2023) 140:103125. doi: 10.1016/j.jaut.2023.103125
128. Wen HY, Wang J, Zhang SX, Luo J, Zhao XC, Zhang C, et al. Low-dose sirolimus immunoregulation therapy in patients with active rheumatoid arthritis: A 24-week follow-up of the randomized, open-label, parallel-controlled trial. J Immunol Res (2019) 2019:7684352. doi: 10.1155/2019/7684352
129. Bruyn GA, Tate G, Caeiro F, Maldonado-Cocco J, Westhovens R, Tannenbaum H, et al. Everolimus in patients with rheumatoid arthritis receiving concomitant methotrexate: a 3-month, double-blind, randomised, placebo-controlled, parallel-group, proof-of-concept study. Ann rheumatic Dis (2008) 67(8):1090–5. doi: 10.1136/ard.2007.078808
130. Niu HQ, Li ZH, Zhao WP, Zhao XC, Zhang C, Luo J, et al. Sirolimus selectively increases circulating Treg cell numbers and restores the Th17/Treg balance in rheumatoid arthritis patients with low disease activity or in DAS28 remission who previously received conventional disease-modifying anti-rheumatic drugs. Clin Exp Rheumatol (2019). 38(1):58–66. doi: 10.1136/annrheumdis-2019-eular.2936
131. Spence A, Klementowicz JE, Bluestone JA, Tang Q. Targeting Treg signaling for the treatment of autoimmune diseases. Curr Opin Immunol (2015) 37:11–20. doi: 10.1016/j.coi.2015.09.002
132. Hill JA, Hall JA, Sun CM, Cai Q, Ghyselinck N, Chambon P, et al. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity (2008) 29(5):758–70. doi: 10.1016/j.immuni.2008.09.018
133. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes (2012) 61(9):2340–8. doi: 10.2337/db12-0049
134. Scotta C, Esposito M, Fazekasova H, Fanelli G, Edozie FC, Ali N, et al. Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4(+)CD25(+)FOXP3(+) T regulatory cell subpopulations. Haematologica (2013) 98(8):1291–9. doi: 10.3324/haematol.2012.074088
135. Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, et al. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci United States America (2014) 111(33):E3432–3440. doi: 10.1073/pnas.1408780111
136. Lu L, Ma J, Li Z, Lan Q, Chen M, Liu Y, et al. All-trans retinoic acid promotes TGF-beta-induced Tregs via histone modification but not DNA demethylation on Foxp3 gene locus. PloS One (2011) 6(9):e24590. doi: 10.1371/journal.pone.0024590
137. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PloS Biol (2007) 5(2):e38. doi: 10.1371/journal.pbio.0050038
138. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med (2004) 200(3):277–85. doi: 10.1084/jem.20040165
139. Huang Z, Yang B, Shi Y, Cai B, Li Y, Feng W, et al. Anti-TNF-alpha therapy improves Treg and suppresses Teff in patients with rheumatoid arthritis. Cell Immunol (2012) 279(1):25–9. doi: 10.1016/j.cellimm.2012.09.001
140. Farid E, Mumtaz M, Hajji F, Ebrahim RA, Abdulla H, Tabbara K. T regulatory cells in rheumatoid arthritis with reference to anti-citrullinated peptide antibody and TNF-alpha inhibitor therapy. Egypt J Immunol (2020) 27(1):55–63.
141. Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med (2007) 204(1):33–9. doi: 10.1084/jem.20061531
142. McGovern JL, Nguyen DX, Notley CA, Mauri C, Isenberg DA, Ehrenstein MR. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis rheuma (2012) 64(10):3129–38. doi: 10.1002/art.34565
143. Vigna-Perez M, Abud-Mendoza C, Portillo-Salazar H, Alvarado-Sanchez B, Cuevas-Orta E, Moreno-Valdes R, et al. Immune effects of therapy with Adalimumab in patients with rheumatoid arthritis. Clin Exp Immunol (2005) 141(2):372–80. doi: 10.1111/j.1365-2249.2005.02859.x
144. Blache C, Lequerre T, Roucheux A, Beutheu S, Dedreux I, Jacquot S, et al. Number and phenotype of rheumatoid arthritis patients’ CD4+CD25hi regulatory T cells are not affected by adalimumab or etanercept. Rheumatol (Oxford England) (2011) 50(10):1814–22. doi: 10.1093/rheumatology/ker183
145. McCann FE, Perocheau DP, Ruspi G, Blazek K, Davies ML, Feldmann M, et al. Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol (2014) 66(10):2728–38. doi: 10.1002/art.38755
146. Nguyen DX, Ehrenstein MR. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J Exp Med (2016) 213(7):1241–53. doi: 10.1084/jem.20151255
147. Tseng WY, Huang YS, Clanchy F, McNamee K, Perocheau D, Ogbechi J, et al. TNF receptor 2 signaling prevents DNA methylation at the Foxp3 promoter and prevents pathogenic conversion of regulatory T cells. Proc Natl Acad Sci U.S.A. (2019) 116(43):21666–72. doi: 10.1073/pnas.1909687116
148. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood (2006) 108(1):253–61. doi: 10.1182/blood-2005-11-4567
149. Santinon F, Batignes M, Mebrek ML, Biton J, Clavel G, Herve R, et al. Involvement of tumor necrosis factor receptor type II in foxP3 stability and as a marker of treg cells specifically expanded by anti-tumor necrosis factor treatments in rheumatoid arthritis. Arthritis Rheumatol (Hoboken NJ) (2020) 72(4):576–87. doi: 10.1002/art.41134
150. Bystrom J, Clanchy FI, Taher TE, Mangat P, Jawad AS, Williams RO, et al. TNFα in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine (2018) 101:4–13. doi: 10.1016/j.cyto.2016.09.001
151. Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, et al. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J Immunol (Baltimore Md: 1950) (2011) 186(12):6779–87. doi: 10.4049/jimmunol.1003868
152. Cutolo M, Sulli A, Paolino S, Pizzorni C. CTLA-4 blockade in the treatment of rheumatoid arthritis: an update. Expert Rev Clin Immunol (2016) 12(4):417–25. doi: 10.1586/1744666X.2016.1133295
153. Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci United States America (2008) 105(49):19396–401. doi: 10.1073/pnas.0806855105
154. Bernard NJ. Rheumatoid arthritis: Who knows why regulatory T cells are defective in RA … IDO. Nat Rev Rheumatol (2014) 10(7):381. doi: 10.1038/nrrheum.2014.96
155. Ko HJ, Cho ML, Lee SY, Oh HJ, Heo YJ, Moon YM, et al. CTLA4-Ig modifies dendritic cells from mice with collagen-induced arthritis to increase the CD4+CD25+Foxp3+ regulatory T cell population. J Autoimmun (2010) 34(2):111–20. doi: 10.1016/j.jaut.2009.07.006
156. Razmara M, Hilliard B, Ziarani AK, Chen YH, Tykocinski ML. CTLA-4 x Ig converts naive CD4+CD25- T cells into CD4+CD25+ regulatory T cells. Int Immunol (2008) 20(4):471–83. doi: 10.1093/intimm/dxn007
157. Langdon K, Haleagrahara N. Regulatory T-cell dynamics with abatacept treatment in rheumatoid arthritis. Int Rev Immunol (2018) 37(4):206–14. doi: 10.1080/08830185.2018.1465943
158. Marquez Pete N, Maldonado Montoro MDM, Perez Ramirez C, Sanchez Martin A, Martinez de la Plata JE, Martinez Martinez F, et al. Impact of single-nucleotide polymorphisms of CTLA-4, CD80 and CD86 on the effectiveness of abatacept in patients with rheumatoid arthritis. J Pers Med (2020) 10(4):220. doi: 10.3390/jpm10040220
159. Blair HA, Deeks ED. Abatacept: A review in rheumatoid arthritis. Drugs (2017) 77(11):1221–33. doi: 10.1007/s40265-017-0775-4
160. Picchianti Diamanti A, Rosado MM, Scarsella M, Germano V, Giorda E, Cascioli S, et al. Abatacept (cytotoxic T lymphocyte antigen 4-immunoglobulin) improves B cell function and regulatory T cell inhibitory capacity in rheumatoid arthritis patients non-responding to anti-tumour necrosis factor-alpha agents. Clin Exp Immunol (2014) 177(3):630–40. doi: 10.1111/cei.12367
161. Alvarez-Quiroga C, Abud-Mendoza C, Doniz-Padilla L, Juarez-Reyes A, Monsivais-Urenda A, Baranda L, et al. CTLA-4-Ig therapy diminishes the frequency but enhances the function of Treg cells in patients with rheumatoid arthritis. J Clin Immunol (2011) 31(4):588–95. doi: 10.1007/s10875-011-9527-5
162. Pieper J, Herrath J, Raghavan S, Muhammad K, Vollenhoven R, Malmstrom V. CTLA4-Ig (abatacept) therapy modulates T cell effector functions in autoantibody-positive rheumatoid arthritis patients. BMC Immunol (2013) 14:34. doi: 10.1186/1471-2172-14-34
163. Aldridge J, Andersson K, Gjertsson I, Hultgard Ekwall AK, Hallstrom M, van Vollenhoven R, et al. Blood PD-1+TFh and CTLA-4+CD4+ T cells predict remission after CTLA-4Ig treatment in early rheumatoid arthritis. Rheumatol (Oxford) (2022) 61(3):1233–42. doi: 10.1093/rheumatology/keab454
164. Hagenstein J, Melderis S, Nosko A, Warkotsch MT, Richter JV, Ramcke T, et al. A novel role for IL-6 receptor classic signaling: induction of RORgammat(+)Foxp3(+) tregs with enhanced suppressive capacity. J Am Soc Nephrol (2019) 30(8):1439–53. doi: 10.1681/ASN.2019020118
165. Fujimoto M, Serada S, Mihara M, Uchiyama Y, Yoshida H, Koike N, et al. Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis rheuma (2008) 58(12):3710–9. doi: 10.1002/art.24126
166. Guggino G, Giardina AR, Raimondo S, Giardina G, Sireci G, Dieli F, et al. Targeting IL-6 signalling in early rheumatoid arthritis is followed by Th1 and Th17 suppression and Th2 expansion. Clin Exp Rheumatol (2014) 32(1):77–81.
167. Thiolat A, Semerano L, Pers YM, Biton J, Lemeiter D, Portales P, et al. Interleukin-6 receptor blockade enhances CD39+ regulatory T cell development in rheumatoid arthritis and in experimental arthritis. Arthritis Rheumatol (2014) 66(2):273–83. doi: 10.1002/art.38246
168. Pesce B, Soto L, Sabugo F, Wurmann P, Cuchacovich M, Lopez MN, et al. Effect of interleukin-6 receptor blockade on the balance between regulatory T cells and T helper type 17 cells in rheumatoid arthritis patients. Clin Exp Immunol (2013) 171(3):237–42. doi: 10.1111/cei.12017
169. Kikuchi J, Hashizume M, Kaneko Y, Yoshimoto K, Nishina N, Takeuchi T. Peripheral blood CD4(+)CD25(+)CD127(low) regulatory T cells are significantly increased by tocilizumab treatment in patients with rheumatoid arthritis: increase in regulatory T cells correlates with clinical response. Arthritis Res Ther (2015) 17(1):10. doi: 10.1186/s13075-015-0526-4
170. Benson R, Zhao SS, Goodson N, Abernethy R, Mewar D, Barnes T. Biologic monotherapy in the biologic naive patient with rheumatoid arthritis (RA): results from an observational study. Rheumatol Int (2020) 40(7):1045–9. doi: 10.1007/s00296-020-04531-6
171. Rivellese F, Surace AEA, Goldmann K, Sciacca E, Cubuk C, Giorli G, et al. Rituximab versus tocilizumab in rheumatoid arthritis: synovial biopsy-based biomarker analysis of the phase 4 R4RA randomized trial. Nat Med (2022) 28(6):1256–68. doi: 10.1038/s41591-022-01789-0
172. Harshan S, Dey P, Raghunathan S. Altered transcriptional regulation of glycolysis in circulating CD8(+) T cells of rheumatoid arthritis patients. Genes (Basel) (2022) 13(7):1216. doi: 10.3390/genes13071216
173. Scheinecker C, Goschl L, Bonelli M. Treg cells in health and autoimmune diseases: New insights from single cell analysis. J Autoimmun (2020) 110:102376. doi: 10.1016/j.jaut.2019.102376
174. Moghaddam MZ, Mousavi MJ, Ghotloo S. Cell-based therapies for the treatment of rheumatoid arthritis. Immun Inflammation Dis (2023) 11(11):e1091. doi: 10.1002/iid3.1091
175. Eggenhuizen PJ, Ng BH, Ooi JD. Treg enhancing therapies to treat autoimmune diseases. Int J Mol Sci (2020) 21(19):7015. doi: 10.3390/ijms21197015
176. Liu R, Zhao P, Tan W, Zhang M. Cell therapies for refractory rheumatoid arthritis. Clin Exp Rheumatol (2018) 36(5):911–9.
177. Debreova M, Culenova M, Smolinska V, Nicodemou A, Csobonyeiova M, Danisovic L. Rheumatoid arthritis: From synovium biology to cell-based therapy. Cytotherapy (2022) 24(4):365–75. doi: 10.1016/j.jcyt.2021.10.003
178. Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ, van Ewijk W, van Laar JM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum (2005) 52(7):2212–21. doi: 10.1002/art.21195
179. Zaiss MM, Sarter K, Hess A, Engelke K, Bohm C, Nimmerjahn F, et al. Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis rheuma (2010) 62(8):2328–38. doi: 10.1002/art.27535
180. Klein S, Kretz CC, Krammer PH, Kuhn A. CD127(low/-) and FoxP3(+) expression levels characterize different regulatory T-cell populations in human peripheral blood. J Invest Dermatol (2010) 130(2):492–9. doi: 10.1038/jid.2009.313
181. Ochi T, Fujiwara H, Okamoto S, An J, Nagai K, Shirakata T, et al. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood (2011) 118(6):1495–503. doi: 10.1182/blood-2011-02-337089
182. Legut M, Dolton G, Mian AA, Ottmann OG, Sewell AK. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood (2018) 131(3):311–22. doi: 10.1182/blood-2017-05-787598
183. Cook PJ, Yang SJ, Uenishi GI, Grimm A, West SE, Wang LJ, et al. A chemically inducible IL-2 receptor signaling complex allows for effective in vitro and in vivo selection of engineered CD4+ T cells. Mol Ther (2023) 31(8):2472–88. doi: 10.1016/j.ymthe.2023.04.021
184. Li YJ, Chen Z. Cell-based therapies for rheumatoid arthritis: opportunities and challenges. Ther Adv Musculoskelet Dis (2022) 14:1759720X221100294. doi: 10.1177/1759720X221100294
185. Buc M. Mechanisms of action of regulatory lymphocytes and a possibility to use them in the treatment of autoimmune diseases and GvH reactions. Bratisl Lek Listy (2020) 121(1):3–7. doi: 10.4149/BLL_2020_001
186. Sun G, Hou Y, Gong W, Liu S, Li J, Yuan Y, et al. Adoptive induced antigen-specific treg cells reverse inflammation in collagen-induced arthritis mouse model. Inflammation (2018) 41(2):485–95. doi: 10.1007/s10753-017-0704-4
187. Yeh WI, Seay HR, Newby B, Posgai AL, Moniz FB, Michels A, et al. Avidity and bystander suppressive capacity of human regulatory T cells expressing de novo autoreactive T-cell receptors in type 1 diabetes. Front Immunol (2017) 8:1313. doi: 10.3389/fimmu.2017.01313
188. Harris DT, Kranz DM. Adoptive T cell therapies: A comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci (2016) 37(3):220–30. doi: 10.1016/j.tips.2015.11.004
189. Safari F, Farajnia S, Arya M, Zarredar H, Nasrolahi A. CRISPR and personalized Treg therapy: new insights into the treatment of rheumatoid arthritis. J Immunopharmacol immunotoxicol (2018) 40(3):201–11. doi: 10.1080/08923973.2018.1437625
190. Scotta C, Fanelli G, Hoong SJ, Romano M, Lamperti EN, Sukthankar M, et al. Impact of immunosuppressive drugs on the therapeutic efficacy of ex vivo expanded human regulatory T cells. Haematologica (2016) 101(1):91–100. doi: 10.3324/haematol.2015.128934
191. Bonnefoy F, Daoui A, Valmary-Degano S, Toussirot E, Saas P, Perruche S. Apoptotic cell infusion treats ongoing collagen-induced arthritis, even in the presence of methotrexate, and is synergic with anti-TNF therapy. Arthritis Res Ther (2016) 18(1):184. doi: 10.1186/s13075-016-1084-0
192. Zielinski M, Zalinska M, Iwaszkiewicz-Grzes D, Gliwinski M, Hennig M, Jazwinska-Curyllo A, et al. Combined therapy with CD4(+) CD25highCD127(-) T regulatory cells and anti-CD20 antibody in recent-onset type 1 diabetes is superior to monotherapy: Randomized phase I/II trial. Diabetes Obes Metab (2022) 24(8):1534–43. doi: 10.1111/dom.14723
193. Petrillo MG, Ronchetti S, Ricci E, Alunno A, Gerli R, Nocentini G, et al. GITR+ regulatory T cells in the treatment of autoimmune diseases. Autoimmun Rev (2015) 14(2):117–26. doi: 10.1016/j.autrev.2014.10.011
194. Miyara M, Ito Y, Sakaguchi S. TREG-cell therapies for autoimmune rheumatic diseases. Nat Rev Rheumatol (2014) 10(9):543–51. doi: 10.1038/nrrheum.2014.105
195. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene (2005) 24(50):7455–64. doi: 10.1038/sj.onc.1209085
196. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol (2012) 13(5):283–96. doi: 10.1038/nrm3330
197. Wang CR, Shiau AL, Chen SY, Lin LL, Tai MH, Shieh GS, et al. Amelioration of collagen-induced arthritis in rats by adenovirus-mediated PTEN gene transfer. Arthritis rheuma (2008) 58(6):1650–6. doi: 10.1002/art.23517
198. Sun S, Steinberg BM. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J Gen Virol (2002) 83(Pt 7):1651–8. doi: 10.1099/0022-1317-83-7-1651
199. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol (2015) 16(2):178–87. doi: 10.1038/ni.3076
200. Lee SH, Park JS, Byun JK, Jhun J, Jung K, Seo HB, et al. PTEN ameliorates autoimmune arthritis through down-regulating STAT3 activation with reciprocal balance of Th17 and Tregs. Sci Rep (2016) 6:34617. doi: 10.1038/srep34617
201. Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol (2015) 16(2):188–96. doi: 10.1038/ni.3077
202. Pap T, Franz JK, Hummel KM, Jeisy E, Gay R, Gay S. Activation of synovial fibroblasts in rheumatoid arthritis: lack of Expression of the tumour suppressor PTEN at sites of invasive growth and destruction. Arthritis Res (2000) 2(1):59–64. doi: 10.1186/ar69
203. Aarts J, van Caam A, Chen X, Marijnissen RJ, Helsen MM, Walgreen B, et al. Local inhibition of TGF-beta1 signaling improves Th17/Treg balance but not joint pathology during experimental arthritis. Sci Rep (2022) 12(1):3182. doi: 10.1038/s41598-022-07075-w
204. Gonzalez-Rey E, Delgado M. Vasoactive intestinal peptide and regulatory T-cell induction: a new mechanism and therapeutic potential for immune homeostasis. Trends Mol Med (2007) 13(6):241–51. doi: 10.1016/j.molmed.2007.04.003
205. Zhang J, Hu X, Dong X, Chen W, Zhang L, Chang Y, et al. Regulation of T cell activities in rheumatoid arthritis by the novel fusion protein igD-fc-ig. Front Immunol (2020) 11:755. doi: 10.3389/fimmu.2020.00755
206. Zhao T, Wei Y, Zhu Y, Xie Z, Hai Q, Li Z, et al. Gut microbiota and rheumatoid arthritis: From pathogenesis to novel therapeutic opportunities. Front Immunol (2022) 13:1007165. doi: 10.3389/fimmu.2022.1007165
207. Jiang ZM, Zeng SL, Huang TQ, Lin Y, Wang FF, Gao XJ, et al. Sinomenine ameliorates rheumatoid arthritis by modulating tryptophan metabolism and activating aryl hydrocarbon receptor via gut microbiota regulation. Sci Bull (Beijing) (2023) 68(14):1540–55. doi: 10.1016/j.scib.2023.06.027
208. Kim CH. Complex regulatory effects of gut microbial short-chain fatty acids on immune tolerance and autoimmunity. Cell Mol Immunol (2023) 20(4):341–50. doi: 10.1038/s41423-023-00987-1
209. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
Keywords: regulatory T cells, rheumatoid arthritis, therapy, Foxp3, Th17
Citation: Zhang J, Liu H, Chen Y, Liu H, Zhang S, Yin G and Xie Q (2024) Augmenting regulatory T cells: new therapeutic strategy for rheumatoid arthritis. Front. Immunol. 15:1312919. doi: 10.3389/fimmu.2024.1312919
Received: 10 October 2023; Accepted: 08 January 2024;
Published: 23 January 2024.
Edited by:
Su-Jin Moon, Catholic University of Korea, Republic of KoreaReviewed by:
Maciej Zieliński, University of Gdańsk, PolandFarooq Riaz, Chinese Academy of Sciences (CAS), China
Copyright © 2024 Zhang, Liu, Chen, Liu, Zhang, Yin and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geng Yin, yingeng1975@163.com; Qibing Xie, xieqibing1971@163.com