 Michael Mauermeir1†
Michael Mauermeir1† Martha Ölke1†Inaya Hayek1
Martha Ölke1†Inaya Hayek1 Jan Schulze-Luehrmann1
Jan Schulze-Luehrmann1 Katja Dettmer2Peter J. Oefner2
Katja Dettmer2Peter J. Oefner2 Christian Berens3
Christian Berens3 Christian Menge3
Christian Menge3 Anja Lührmann1*
Anja Lührmann1*- 1Mikrobiologisches Institut, Universitätsklinikum Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
- 2Institut für Funktionelle Genomik, Universität Regensburg, Regensburg, Germany
- 3Friedrich-Loeffler-Institut, Institut für molekulare Pathogenese, Jena, Germany
Background: Coxiella burnetii is a zoonotic pathogen, infecting humans, livestock, pets, birds and ticks. Domestic ruminants such as cattle, sheep, and goats are the main reservoir and major cause of human infection. Infected ruminants are usually asymptomatic, while in humans infection can cause significant disease. Human and bovine macrophages differ in their permissiveness for C. burnetii strains from different host species and of various genotypes and their subsequent host cell response, but the underlying mechanism(s) at the cellular level are unknown.
Methods: C. burnetii infected primary human and bovine macrophages under normoxic and hypoxic conditions were analyzed for (i) bacterial replication by CFU counts and immunofluorescence; (ii) immune regulators by westernblot and qRT-PCR; cytokines by ELISA; and metabolites by gas chromatography-mass spectrometry (GC-MS).
Results: Here, we confirmed that peripheral blood-derived human macrophages prevent C. burnetii replication under oxygen-limiting conditions. In contrast, oxygen content had no influence on C. burnetii replication in bovine peripheral blood-derived macrophages. In hypoxic infected bovine macrophages, STAT3 is activated, even though HIF1α is stabilized, which otherwise prevents STAT3 activation in human macrophages. In addition, the TNFα mRNA level is higher in hypoxic than normoxic human macrophages, which correlates with increased secretion of TNFα and control of C. burnetii replication. In contrast, oxygen limitation does not impact TNFα mRNA levels in C. burnetii-infected bovine macrophages and secretion of TNFα is blocked. As TNFα is also involved in the control of C. burnetii replication in bovine macrophages, this cytokine is important for cell autonomous control and its absence is partially responsible for the ability of C. burnetii to replicate in hypoxic bovine macrophages. Further unveiling the molecular basis of macrophage-mediated control of C. burnetii replication might be the first step towards the development of host directed intervention measures to mitigate the health burden of this zoonotic agent.
Introduction
Coxiella burnetii is an obligate intracellular pathogen, which causes the zoonotic disease Q fever. In humans, Q fever might be asymptomatic or presents as a mild self-limiting flu-like disease. However, the infection can also progress to an interstitial pneumonia or hepatitis (1). The severity of the primary infection does not predict long-term health consequences, such as post-Q fever fatigue syndrome (QFS) or chronic Q fever (2, 3). QFS occurs in ~20% of patients with symptomatic acute Q fever (4). QFS is defined as fatigue lasting longer than six months in combination with pain, sleeping problems, headache, and concentration issues. Importantly, there is no evidence-based treatment available (5). Approximately 2% of Q fever patients might develop chronic Q fever (6). Endocarditis is the most common manifestation of chronic Q fever, and patients with an underlying valvulopathy are at a higher risk (7). Treatment of chronic Q fever involves administration of doxycycline and hydroxychloroquine for 18-24 months (6).
The primary reservoir of C. burnetii and the major source of human infections are infected livestock (8). Infection in cattle, goats, and sheep is often asymptomatic, but it may also lead to metritis, infertility, abortion, stillbirth or the delivery of weak offspring (9). Infected females shed the pathogen in huge quantities through birthing products and in smaller numbers in urine, feces and milk. However, the route of C. burnetii shedding seems to differ between cattle, sheep, and goats. Thus, shedding in milk is common in cattle, but less widespread in ewes (8). Infection of humans occurs mainly via the respiratory tract, while ingestion of contaminated milk plays a lesser role in transmission of C. burnetii (1, 8). Most human Q fever outbreaks are linked to infected sheep and goats (10), though cattle may also serve as a source for human infections (11). Given a prevalence of ~30% of C. burnetii infection in cattle (12–16), we have to increase our understanding of how C. burnetii adapts to the bovine host in vivo and in vitro.
In vitro studies using bovine cells are rare, but demonstrate that C. burnetii infects and replicates inside bovine epithelial cell lines as well as in primary bovine monocyte-derived macrophages. However, C. burnetii only induces a transient pro-inflammatory cytokine response in macrophages. Reduced levels of pro-inflammatory cytokines correlate with bacterial replication, suggesting that intracellular C. burnetii replication depends on a suitable microenvironment (17–19). This is in agreement with previous findings, showing that TNF inhibits C. burnetii replication (20). Not only the cytokine profile at the site of infection is determinative for bacterial replication, but also the metabolism of the host (21). The availability of the TCA cycle metabolite citrate is essential for C. burnetii replication in human and murine macrophages (22). Under oxygen limiting conditions, the transcription factor hypoxia-inducible factor (HIF) 1α is stabilized (23). HIF1α limits the activation of STAT3, which in turn reduces the intracellular level of citrate and, as a consequence, C. burnetii replication (22). HIF1α does not only influence host cell metabolism, but also inflammation as it controls the expression and secretion of the pro-inflammatory cytokine TNF (24).
In this study, we aimed to investigate the intracellular C. burnetii-containing vacuole (CCV) in bovine cells in more detail, and whether oxygen availability influences host cell metabolism and immune response during C. burnetii infection in bovine macrophages.
Material and methods
Reagents and bacterial strain
Chemicals were purchased from Sigma Aldrich unless indicated otherwise. C. burnetii Nine Mile phase II clone 4 (RSA439), kindly provided by Matteo Bonazzi (CNRS, Montpellier, France), was grown in ACCM-D medium (Sunrise Science Products) at 37°C, 5% CO2 and 2.5% O2 for 7 days.
Bovine peripheral blood derived macrophages
Bovine peripheral blood samples were obtained from healthy heifers of the Holstein-Friesian breed, kept at the Friedrich-Loeffler-Institut, Jena. The animal experiment was reviewed by the Committee on the Ethics of Animal Experiments and the Protection of Animals of the State of Thuringia, Germany, and approved by the competent authority (Permit numbers 22-2684-04-04-102/15, date of permission 17.06.2015, and 22-2684-04-BFI-20-102, date of permission 20.04.2020). Bovine macrophages from peripheral blood were isolated as described (19). Briefly, leukocytes were isolated from citrate whole-blood (5:1 dilution with a 3.8% sodium citrate solution) via centrifugation (2,380 x g, 20 min, room temperature), washed 3 times with PBS/EDTA (0.8% NaCl, 0.02% KH2PO4, 0.02% KCl, 0.142% Na2HPO4 x 2H2O, 0.2% Na-EDTA x 2H2O in ddH2O, pH 7.4) by centrifugation (800 x g, 10 min, room temperature). This was followed by an erythrocyte lysis step in lysis buffer (0.826% NH4Cl, 0.109% NaHCO3, 0.0037% Na2-EDTA x 2H2O in ddH2O) for 5 min. After three more washing steps with PBS/EDTA (300 x g, 10 min), the cells were layered onto Pancoll (Pan Biotech) for density centrifugation (800 x g, 45 min). The mononuclear cell layer was extracted and washed several times with 0.9% NaCl before incubation in Saint-Gobain VueLife® “C” Series Bags (32-C) in Iscove´s modified Dulbecco´s medium (IMDM without phenol red, supplemented with 0.05 µM 2-mercaptoethanol, 20% FCS and antibiotics (1% Penicillin/Streptomycin, 1% Amphotericin B).
After 7 to 9 days of incubation at 37°C and 5% CO2, the cells were re-isolated from VueLife bags, washed several times with cold 0.9% NaCl solution (300 x g, 10 min, room temperature) and resuspended in IMDM medium without phenol red, supplemented with 0.05 µM 2-mercaptoethanol, 2% FCS and antibiotics (1% Penicillin/Streptomycin, 1% Amphotericin B). The cells were seeded into suspension culture plates. After 20-24 h incubation, non-adherent cells were washed away via gentle flushing with medium or sterile 0.9% NaCl solution. Cells were replenished with IMDM medium without phenol red, supplemented with 2-mercaptoethanol, 4% FCS (w/o antibiotics) and incubated at 37°C and 5% CO2 before further processing.
Human peripheral blood derived macrophages
Human peripheral blood samples were obtained from healthy donors at the University Clinic of Erlangen (Ethical Committee Erlangen approval number 111_12B). Peripheral blood mononuclear cells (PBMCs) were harvested from Leukoreduction system chambers using a Pancoll-gradient centrifugation protocol as described earlier (22). In brief, Leukoreduction system chamber content was diluted with PBS (Merck) and layered onto Pancoll (Pan Biotech) for density centrifugation (1,328 x g, 25 min, room temperature). The leukocytes were collected and the remaining erythrocytes lysed via a brief incubation in cold ddH2O. After washing in PBS, PBMCs were positively selected using anti-CD14 beads (Miltenyi Biotec) according to the manufacturer’s protocol. The selected cells were cultivated for seven days in cRPMI (complete RPMI, RPMI 1640 containing 10% FCS, 1% HEPES, 0.5% 2-mercaptoethanol, 1% Penicillin/Streptomycin and 5µl/ml human M-CSF [≙ 50 U/ml, Peprotech]) in cell culture flasks. After seven days, cells were detached with Accutase/PBS (1:4) for 30 min at 37°C and gentle rinsing with PBS, before they were harvested by centrifugation (300 x g, 5 min, room temperature), resuspended in cRPMI w/o antibiotics, and seeded into cell culture plates. The cells were incubated at 37°C and 5% CO2 until further processing.
Infection of cell lines with C. burnetii
Bel-26 and A549 cells were seeded and cultured in DMEM, low glucose and pyruvate (Thermo Fisher), containing 10% FCS and 5% FCS, respectively. The cells were incubated at 37°C, 5% CO2, 21% O2. For infection, C. burnetii was quantified via OD600 measurement (OD600 1 ≙ 109 C. burnetii/ml). The cells were infected at MOI 200. After 2h of infection, cells were washed 3 times with PBS to remove residual bacteria in the medium followed by the addition of fresh medium.
Bovine or human macrophages were infected at MOI 10 and incubated either under normoxic (N – 37°C, 5% CO2, 21% O2) or hypoxic (H – 37°C, 5% CO2, 0.5% O2) conditions.
Quantitation of human and bovine TNFα
Secretion of TNF into cell culture supernatants by human or bovine macrophages either treated with 10µg/ml LPS from E. coli O111:B4 (Sigma) for 4 and 24 h or infected with C. burnetii for 4, 24 and 96 h were measured by ELISA (BD Biosciences or R&D Systems respectively). Additionally, TNFα secretion by bovine macrophages was determined by a bioassay deploying PK-15 cells as previously described (25, 26).
Volume measurement of the CCVs
Z-stack images were taken using the LSM700 (Zeiss) as described before (27). The longest distance of the CCV and the corresponding 90 degree angle were measured with the Zen software (Zeiss). The XYZ dimensions of each CCV were multiplied and plotted as volume with GraphPad Prism 9 (GraphPad software, San Diego, USA).
NAD+/NADH assay
Bovine and human macrophages were seeded at a density of 2x106 cells per well on a 6-well plate. One day after seeding the cells were either left uninfected or were infected with C. burnetii at MOI 10 and incubated under normoxia or hypoxia. At 24 h post-infection, the levels of NAD+ and NADH were measured using the colorimetric NAD/NADH Assay Kit (Abcam) as described in the manufacturer´s protocol.
Phagocytosis assay
Bovine and human macrophages were seeded at a density of 5x105 cells per well on a 24-well plate. One day after seeding, cells were incubated with pHrodo Red E. coli bioparticles in accordance with the instruction (Thermo Fisher Scientific) for 30 min. The cells were washed with PBS, fixed for 20 min with 4% paraformaldehyde (Alfa Aesar) in PBS (Merck) and permeabilized for 2 min with ice-cold methanol. The samples were mounted onto glass slides using ProLong™ Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific) to stain DNA. Sample analysis was performed using a Zeiss LSM 700 confocal microscope.
Measurement of cellular metabolites by GC-MS
Bovine and human macrophages were seeded at a density of 107 cells per 10-cm plate and infected with C. burnetii at MOI 10. Infected cells were pelleted at the time points indicated. Cells were then lysed and extracted for carboxylic acid analysis by GC-MS as previously described in detail (22, 28).
Colony forming units
The supernatant of infected cells was removed. The adherent cells were incubated with ice-cold ddH2O for 10 min at room temperature and 30 min at 4°C. Next, the cells were ruptured via repeated pipetting (~50 x), the lysate was transferred to 1.5 ml Eppendorf tubes and pelleted at 21,000 x g for 1 min. The pellets were resuspended in ACCM-D medium and frozen at -20°C until further processing.
The thawed samples were briefly mixed and each sample was plated as triplicates onto ACCM-D agar plates in 10-fold dilution series. Inoculated plates were incubated for 14 days before counting the colony forming units.
RNA
After removal of the cell culture supernatant, 500 µl of TriFast (Peqlab) or RNA-solv (VWR) were added to the dish. Cells were detached via gentle pipetting and transferred to RNAse-free microcentrifuge tubes. The tubes were vortexed for 15 s at maximum speed and stored at -80°C until RNA extraction. RNA extraction was performed according to the manufacturer’s protocol (Peqgold TriFast™ or VWR RNA-Solv®). To maximize RNA yield, 2 µl of Pelletpaint (Merck Millipore) or 1.5 µl Glycoblue coprecipitant (Thermo Fisher) were added to the samples in the isopropanol precipitation step. A switch to RNA-solv and Glycoblue coprecipitant was necessary due to a shortage of supply and discontinuation of TriFast.
qRT-PCR
Reverse transcription of RNA to cDNA was performed using SuperScript™ II Reverse Transcriptase kit with oligo(dT)12-18 primers (Thermo Fisher) according to the manufacturer´s protocol. qRT-PCR was performed using QuantiFast SYBR Green PCR Kit (QIAGEN, 10.3 ng of cDNA template, 500 nM primer, final volume 10 µl) for bovine samples and SYBR-Select Mastermix (Thermo Fisher, 10.3 ng template, 200 nM primer, final volume 10 µl) for human samples. The primer pair sequences of studied genes are listed in Table 1. Analysis was performed using the ΔΔct method. Bovine or human hypoxanthine guanine phosphoribosyl transferase (Hprt1) were used as the respective housekeeping genes.
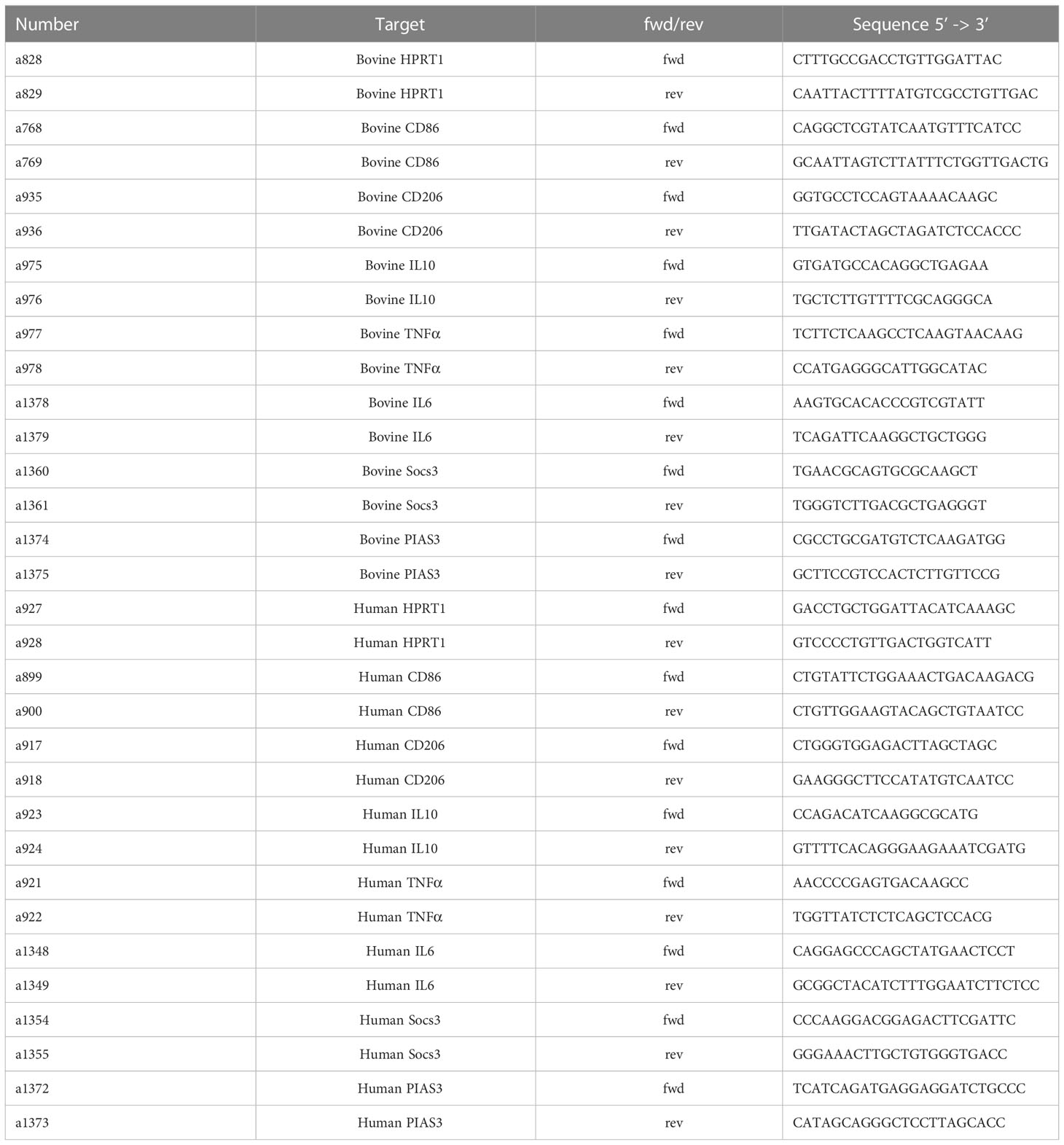
Table 1 Primers used.
Immunoblotting
Proteins were separated by SDS-PAGE Bis-Tris gradient gel (Thermo Fisher) and transferred to a PVDF membrane (Millipore). Proteins were detected with antibodies against HIF1α (Biomol), actin (Merck), pSTAT3 and STAT3 (Cell Signaling), respectively. Detection was performed using horseradish peroxidase conjugated secondary antibodies (Dianova) and a chemiluminescence detection system. Densitometry was performed with ImageJ (NIH).
Indirect immunofluorescence
Cells were seeded on coverslips in 24-well cell plates. One hour prior to fixation, Lysotracker red (Thermo Fisher Scientific) was added (1:1000 final conc.). The cells were fixed with 4% paraformaldehyde (Alfa Aesar) in PBS (Merck) and permeabilized with ice-cold methanol before quenching with PBS containing 5% goat serum and 50mM NH4Cl. Next, the cells were incubated with an anti-C. burnetii antibody (rabbit, in house) and an anti-rabbit antibody labeled with AlexaFluor®488 Dye (Dianova). The samples were mounted onto glass slides using ProLong™ Diamond Antifade Mountant with DAPI (Thermo Fisher) to stain DNA. Sample analysis was performed using a Zeiss Apotome or a Zeiss LSM 700 confocal microscope.
Statistical analysis
The statistical analysis was performed using GraphPad Prism 9. As stated in the figure legends, a one-sample t-test (if datasets are compared to normalized values), a paired or unpaired t-test (for normally distributed datasets), a Mann-Whitney test (for non-normally distributed datasets) or a two-way ANOVA with Tukey’s multiple comparison test were used. A value of p < 0.05 was considered significant.
Results
C. burnetii establishes a replicative phagolysosomal-like compartment in bovine lung epithelial cells
Macrophages are the primary target cells of C. burnetii (1), but lung epithelial cells are the first contact of C. burnetii upon entering the host organism. There is evidence that bovine lung epithelial cells, being less susceptible to C. burnetii infection, do not act as replication sites (17). However, the infection characteristics have not been compared to a C. burnetii-infection in human lung epithelial cells. Thus, we infected the bovine and human lung epithelial cell lines Bel-26 and A549, respectively, with C. burnetii and determined the efficiency of invasion, replication ability, pH and characteristic properties of the C. burnetii-containing vacuole (CCV). First, we analyzed the MOI required to achieve an infection rate of ~70%. While in human A549 cells an MOI of 100 was necessary to reach an infection rate of ~70% at 48 h post-infection, an MOI of 200 was required in bovine Bel-26 cells (data not shown). In both cell lines, C. burnetii was able to establish LysoTracker-positive CCVs (Figure 1A). The pH of the CCVs in these two cell lines did not differ and was slightly acidic with pH values of 5.5 - 5.6 (Figure 1B). At 48 h post-infection, the average number of CCVs per cell was similar with 1.3 in Bel-26 and 1.6 in A549 cells (Figures 1C, D). In contrast to Bel-26 cells, A549 cells harbored an increased percentage of larger CCVs (Figure 1E) and higher bacterial counts (Figure 1F). Thus, at 24 h post-infection A549 cells harbored more than twice as many C. burnetii than Bel-26 cells (Figure 1F). However, C. burnetii replicated similarly well in both cell lines, and multiplied 4 - 5 fold from 24 to 72 h. This data indicated that, notwithstanding the differences detected in invasion efficiency, C. burnetii had established a replicative, LysoTracker-positive compartment in both human and bovine epithelial lung cells and species differences were minor at this level. Whether these pattern reflect the host species or just the respective properties of the cell lines used is unknown. Therefore, we focused on the analysis of the interaction of C. burnetii with primary cells and compared primary peripheral blood derived macrophages from humans and cattle under conditions present at the respiratory mucosal surface (normoxia) and in infected and inflamed tissue (hypoxia).
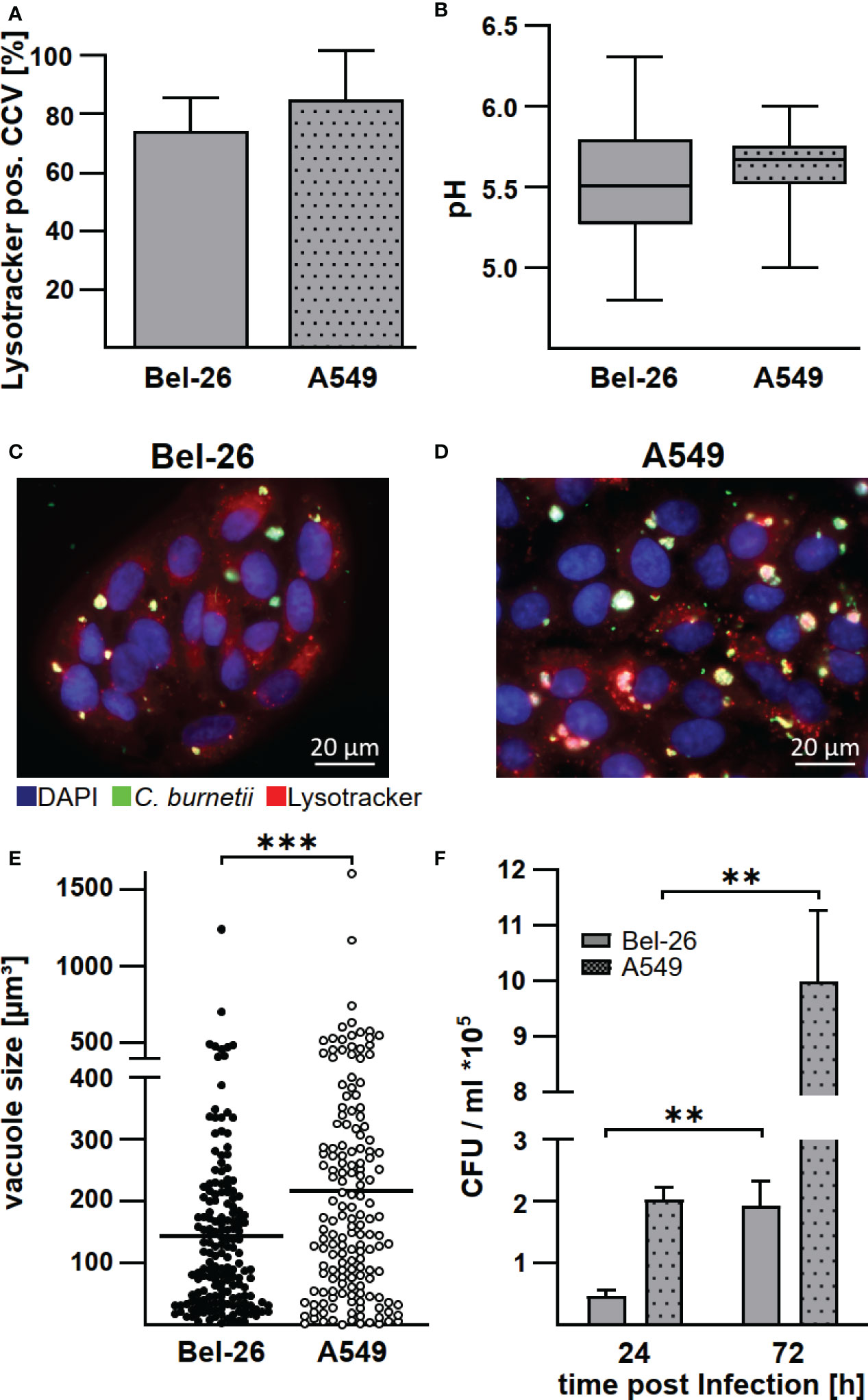
Figure 1 C. burnetii replicate in bovine and human lung epithelial cell lines in a lysosome-like compartment. The bovine lung epithelial cell line Bel-26 and the human lung epithelial cell line A549 were infected with C. burnetii NMII. (A) At 47 h post-infection, 1µM LysoTracker Red DND-99 was added to the culture medium for 1h. The cells were fixed, permeabilized and stained with an anti-C. burnetii antibody and DAPI. The percentage of LysoTracker positive C. burnetii-containing vacuoles (CCVs) of 100 infected cells in each of three independent experiments was determined. Mean ± SD. (B) The pH of the CCVs at 48 h post-infection was determined by calculation of the fluorescence intensity ratios of the dual-wavelength fluorophore LysoSensor Yellow/Blue DND-160. Data represents average values ± SD of 50 infected cells per sample from three independent experiments. (C, D) Representative immunofluorescence micrographs of (C) Bel-26 and (D) A549 cells infected with C. burnetii NMII for 48 h using a ApoTome (Zeiss). (E) The dimension of CCVs at 72 h post-infection was determined from confocal Z-stack images using the the LSM700 microscope and Zen (Zeiss) software. The volume of at least 100 CCVs from two independent experiments are shown. An unpaired t-test was performed. ***p < 0.001. (F) C. burnetii counts of either infected Bel-26 or A549 cells were determined at the time points indicated via counting of colony forming units (CFU). Shown is a representative experiment out of three experiments with similar results, performed with technical triplicates of biological duplicates. Mean ± SD, n = 6, Mann-Whitney test. **p<0.01.
C. burnetii replicated under hypoxia only in bovine, but not in human macrophages
Established protocols for generating differentiated macrophages from human and bovine blood (19, 22) differ in procedure and cultivation media. To ensure comparability, we first analyzed basic functions of the differentiated macrophages. Bovine and human macrophages secreted TNFα after LPS stimulation, although with different kinetics (Figures 2A, B), and phagocytosed pHrhodo Red E.coli similarly well (Figures 2C, D). This data indicate that the bovine and human primary macrophage cultures used herein display a similar level of differentiation.
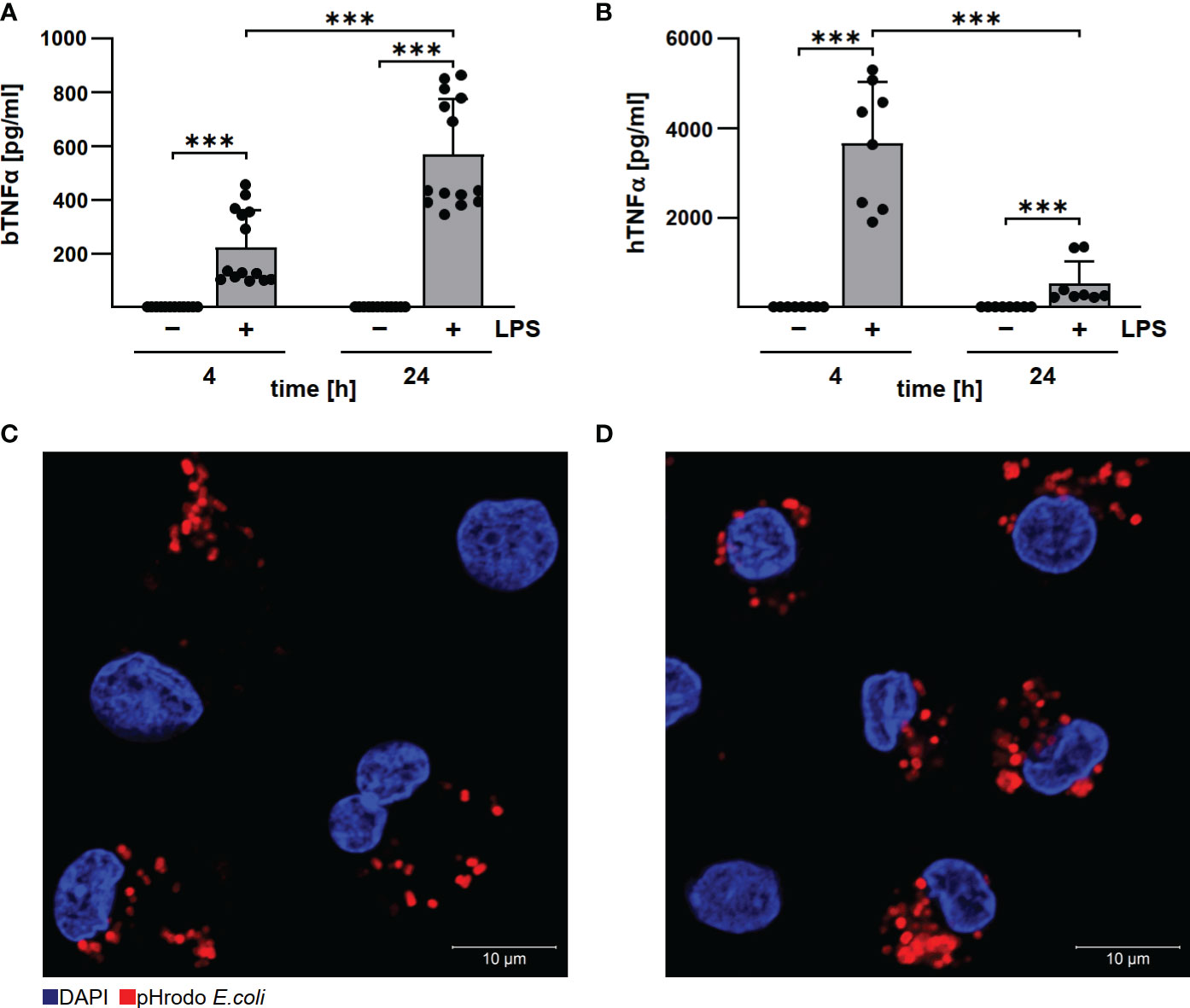
Figure 2 Human and bovine macrophages react to LPS and have phagocytic capacity. Bovine (A) and human (B) macrophages were stimulated with 10 µg/ml LPS for the time periods indicated. Cells were incubated for 4 and 24 h under normoxia. TNFα levels were analyzed by ELISA. Mean ± SD, n = 3, two-way ANOVA with Tukey’s multiple comparisons test. ***p<0.001. (C, D) Bovine and human macrophages were incubated with pHrodo Red E. coli bioparticles for 30 min. The cells were stained with DAPI. Representative immunofluorescence micrographs of (C) bovine and (D) human macrophages using a confocal laser scanning microscope (LSM700) are shown.
Since we had discovered recently, that C. burnetii replicated only in the presence of oxygen in human and murine primary macrophages (22), we analyzed the influence of oxygen availability on the ability of C. burnetii to replicate intracellularly in bovine macrophages. In contrast to human macrophages, C. burnetii replicated in bovine macrophages under both normoxic and hypoxic conditions. This was demonstrated by immunofluorescence imaging of infected cells (Figure 3A) and by colony forming unit (CFU) analysis (Figure 3B), implying that the host cell species might be decisive for C. burnetii replication under hypoxic conditions.
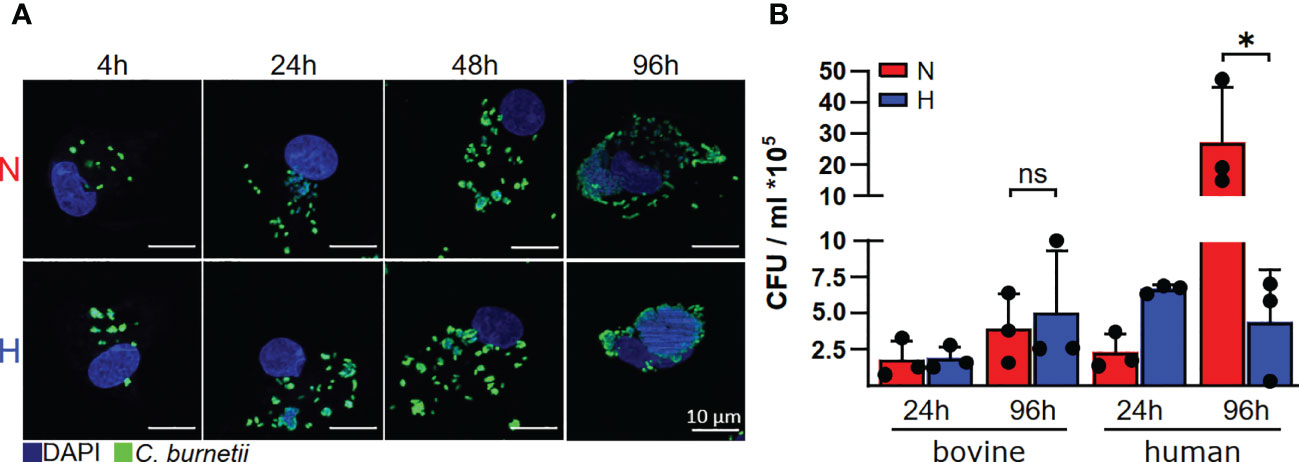
Figure 3 C. burnetii replication is inhibited by human, but not by bovine macrophages. (A) Bovine macrophages were infected with C. burnetii NMII for the time periods indicated under normoxic (N) or hypoxic (H) conditions. The cells were fixed and stained with DAPI and an anti-C. burnetii antibody. Representative immunofluorescence micrographs from three independent experiments with similar results are shown. (B) Human macrophages from three different donors and three independent bovine macrophage preparations from two bovine donor animals were infected with C. burnetii NMII for 24 and 96 hours under normoxic (N) or hypoxic (H) conditions. Bacterial counts were determined by counting colony forming units (CFU). Unpaired t-test or Mann-Whitney test, n=3. *p<0.05, ns=p>0.05.
C. burnetii-induced modulation of the HIF1α-STAT3 axis differs between human and bovine macrophages
HIF1α is the key transcription factor allowing the cell to adapt and to react to shifts in oxygen content (29). Under ample oxygen availability, proline residues 402 and 564 of HIF1α are hydroxylated by prolyl hydroxylases (PHD), which are cellular oxygen sensors. Hydroxylated HIF1α is recognized by the ubiquitin ligase Von Hippel-Lindau protein (pVHL), which marks HIF1α for proteasomal degradation (30, 31). When oxygen is missing, the PHDs are disabled, which leads to stabilization of HIF1α and, as a consequence, to the expression of genes involved in the adaption to hypoxia, metabolic processes and immune system regulation (23, 32). HIF1α also prevents STAT3 activation, thereby interfering with C. burnetii replication inside hypoxic human macrophages (22). Therefore, we analyzed the HIF1α protein levels in bovine macrophages infected with C. burnetii under normoxic and hypoxic conditions. Hypoxia induced stabilization of HIF1α, which was not further enhanced by C. burnetii infection (Figures 4A, B). This is in contrast to our observation in human and murine macrophages, where the infection resulted in augmented HIF1α levels, which impaired the activation of STAT3, resulting in inhibition of C. burnetii replication (22). Interestingly, C. burnetii infection of bovine macrophages resulted in phosphorylation of STAT3 independent of the available oxygen concentration (Figures 4C, D). This data suggests that the activation of STAT3 under hypoxia seems to be distinct in different C. burnetii host-species. In human and murine macrophages, hypoxia leads to stabilization of HIF1α, to inhibition of STAT3 activation, and inability of C. burnetii to replicate (22). In hypoxic bovine macrophages, in contrast, STAT3 was activated and C. burnetii multiplied.
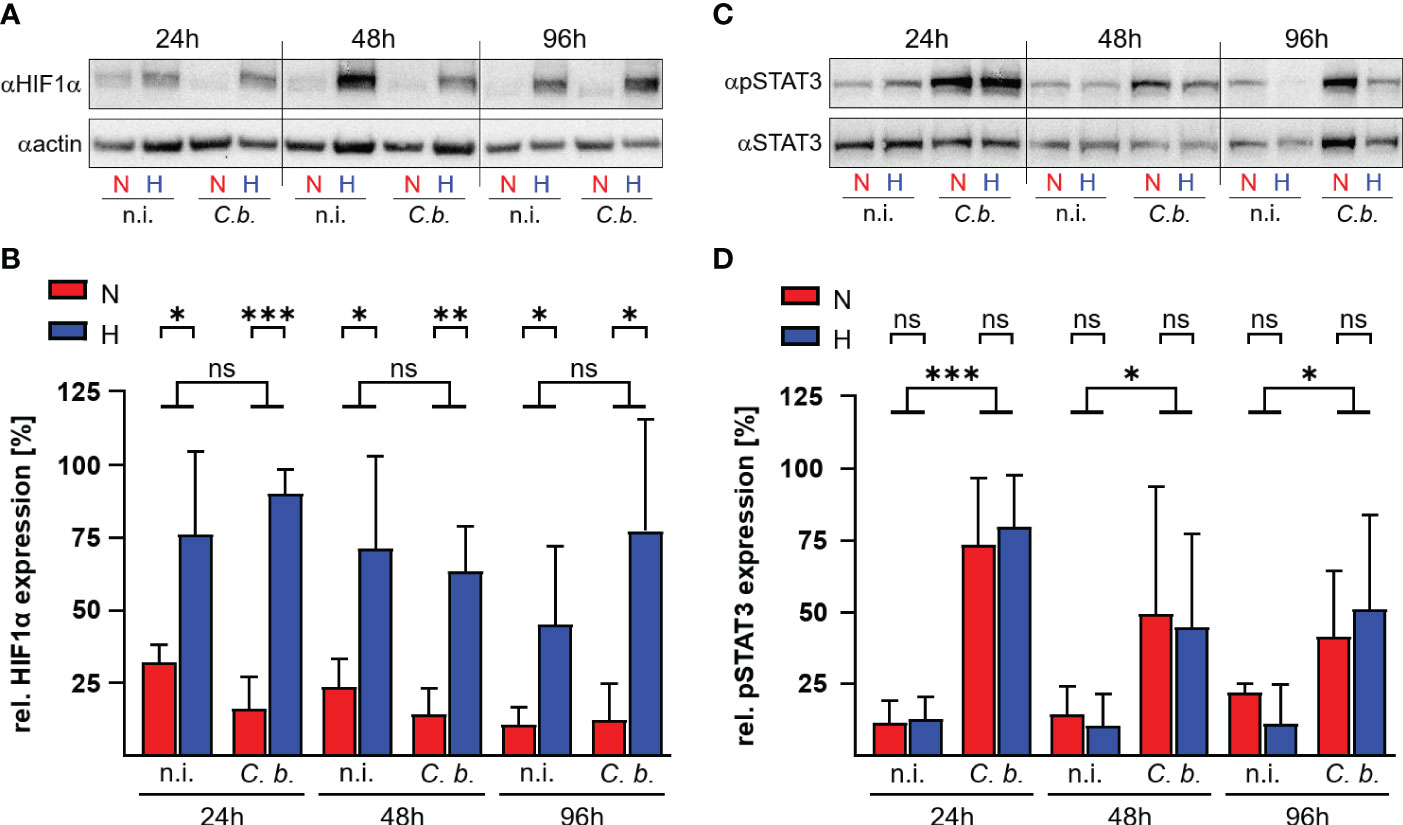
Figure 4 C. burnetii infection induces STAT3 activation in hypoxic bovine macrophages. Bovine monocyte derived macrophages were infected (C. b.) or not infected (n.i.) with C. burnetii NMII for the time periods indicated and kept under normoxic (N) or hypoxic (H) conditions. (A, B) The samples were subjected to immunoblot analysis using antibodies against HIF1α or actin. (A) One representative immunoblot of three independent experiments with similar results is shown. (B) The ratio of HIF1α to actin expression was calculated using ImageJ. Mean ± SD, n = 3, student´s t-test. ***p<0.001, **p<0.01, *p<0.05, ns>0.05. (C, D) The samples were subjected to immunoblot analysis using antibodies against phospho-STAT3 (pSTAT3) or STAT3. (C) One representative immunoblot of three independent experiments is shown. (D) The ratio of pSTAT3 to STAT3 expression was calculated from the immunoblots using ImageJ. Mean ± SD, n = 3, unpaired t-test. ***p<0.001, *p<0.05, ns>0.05.
STAT3 regulators differ in their expression level in C. burnetii-infected bovine and human macrophages
To learn why STAT3 is activated in C. burnetii-infected bovine macrophages, but not in C. burnetii-infected human macrophages under hypoxia, we analyzed the mRNA levels of IL-6, a known STAT3 inducer, and SOCS3 and PIAS3, two known STAT3 inhibitors (33). We detected differences in the expression levels of IL-6 and PIAS3 during C. burnetii-infection between bovine and human macrophages (Figure 5). In bovine macrophages, oxygen availability neither influenced the expression level of IL-6 nor that of the at 24 and 96 h post-infection. In contrast, the IL-6 expression level was increased in human macrophages under hypoxia at 24 and 96 h post-infection and the PIAS3 expression level was increased at 24 h post-infection under normoxia. However, these expression patterns only partially explain the STAT3 activation levels described above (Figures 4C, D). The observed pattern of STAT3 inducer and inhibitor expression apparently is not compliant with mitigated STAT3 activation in hypoxic human macrophages. Interestingly, we observed an oxygen-independent expression of the STAT3 activator IL-6, while the STAT3 inhibitors PIAS3 and SOCS3 were strongly reduced in C. burnetii-infected bovine macrophage (Figure 5), which might favor activation of STAT3 under both, normoxic and hypoxic conditions in bovine macrophages.
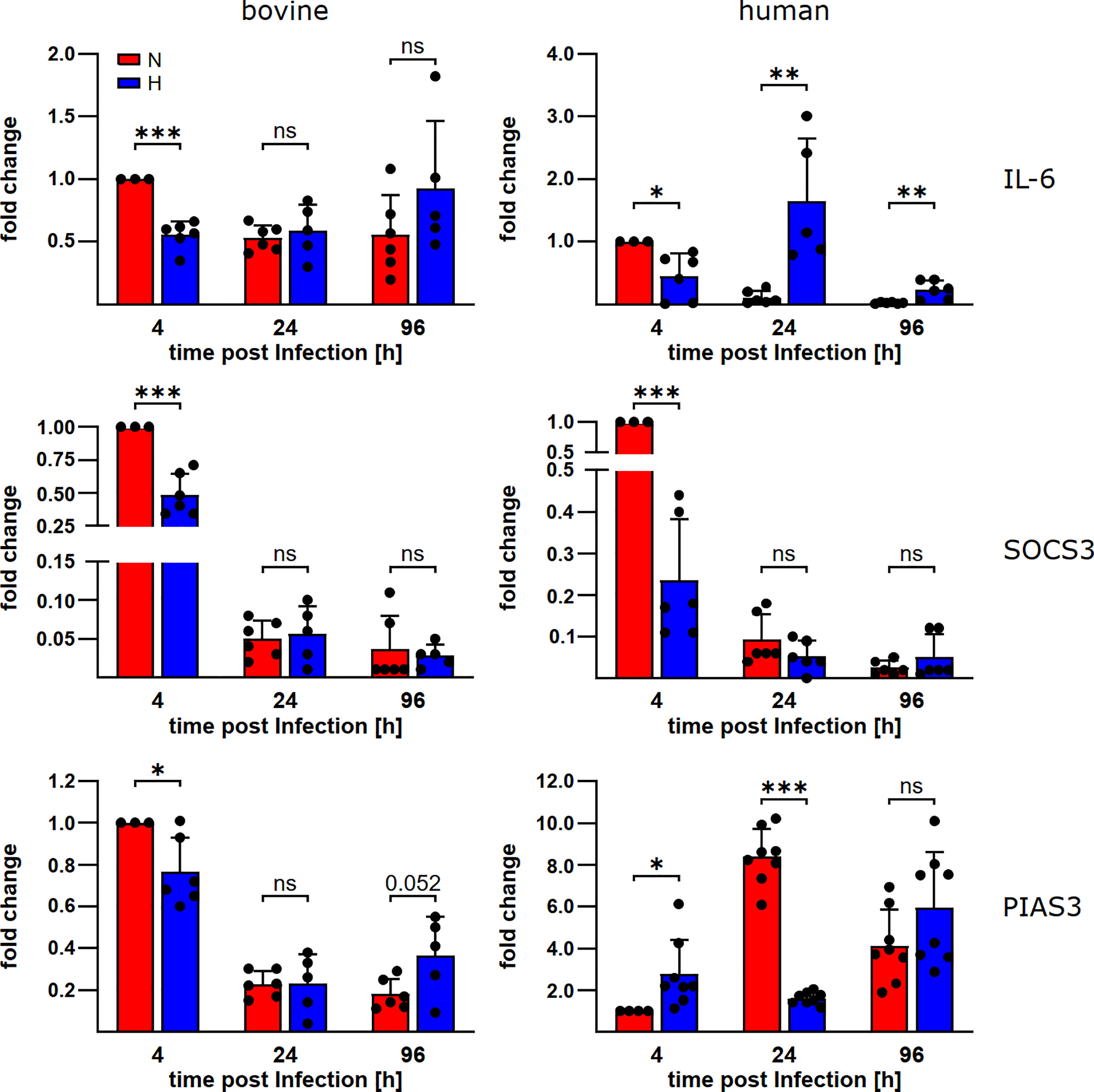
Figure 5 C. burnetii-infection influences gene expression of STAT3 regulators. Gene expression was determined for the genes encoding IL-6, SOCS3 and PIAS3 in C. burnetii NMII infected normoxic and hypoxic bovine (left) and human (right) macrophages at the time points indicated. The data from three to four independent experiments are shown as mean ± SD of 2^-ΔΔCT values (using human or bovine HPRT as a calibrator). Fold changes are shown relative to the 4 h time point under N. One-sample t-test, unpaired t-test or Mann-Whitney test, n=5-6 (bovine) and n=6-8 (human) ***p<0.001, **p<0.01, *p<0.05, ns=p>0.05.
C. burnetii-induced modulation of the level of TCA metabolites differs between human and bovine macrophages
In human and murine macrophages, hypoxia leads to a limitation of citrate, which results in the inability of C. burnetii to replicate (22). Thus, we assessed the carboxylic acid metabolism in C. burnetii-infected hypoxic bovine macrophages to determine its potential role in the ability of C. burnetii to replicate. The levels of pyruvate and lactate were generally higher in infected hypoxic than normoxic human and bovine macrophages, but only the level of lactate showed a significant increase in hypoxic human and bovine macrophages over the time course of infection (Figures 6A, B). Under hypoxic conditions, we observed reduced citrate levels in C. burnetii-infected human macrophages (Figure 6C), similar to previous results in C. burnetii-infected murine macrophages (22). Although we did not observe citrate level reduction in C. burnetii-infected hypoxic bovine macrophages at 24 h post-infection, it was similarly reduced under hypoxia as in human macrophages at 96 h post-infection (Figure 6C). This indicates that the level of citrate under hypoxia might not be responsible for the ability of C. burnetii to replicate in hypoxic bovine macrophages. Furthermore, C. burnetii-infected human macrophages hardly produced itaconate under all conditions tested (Figure 6D), which is in line with previous observations (34). In contrast, infected bovine macrophages produced significant amounts of itaconate, with higher levels under hypoxic conditions. These levels decreased during the infection under both conditions (Figure 6D). Itaconate is derived from citrate, can prevent growth of several bacteria and has immune-regulatory function (34–36). As itaconate also inhibits growth of C. burnetii (37), the increased level of itaconate in bovine macrophages contrasts with the ability of C. burnetii to replicate intracellularly. Another metabolite, whose intracellular concentration was significantly increased under hypoxic conditions in bovine, but not human C. burnetii-infected macrophages was 2-hydroxyglutarate (2HG) (Figure 7A). An acidic intracellular environment resulting from oxygen limitation is believed to favor the reduction of glutamine-derived α-ketoglutarate to 2HG by lactate dehydrogenase A and malate dehydrogenase, with the increase in 2HG varying substantially among different cell types from about 2- to 25-fold (38). Production of intracellular 2HG consumes NADH, thereby alleviating hypoxia-induced reductive stress, which is defined as an excess accumulation of reducing equivalents brought about mostly by suppressed oxidation of NADH by respiratory complex I under hypoxia. In line, we found the NADH-to-NAD+ ratio, to be lower in hypoxic bovine macrophages, but not in hypoxic human macrophages at 24 h post-infection (Figure 7B).
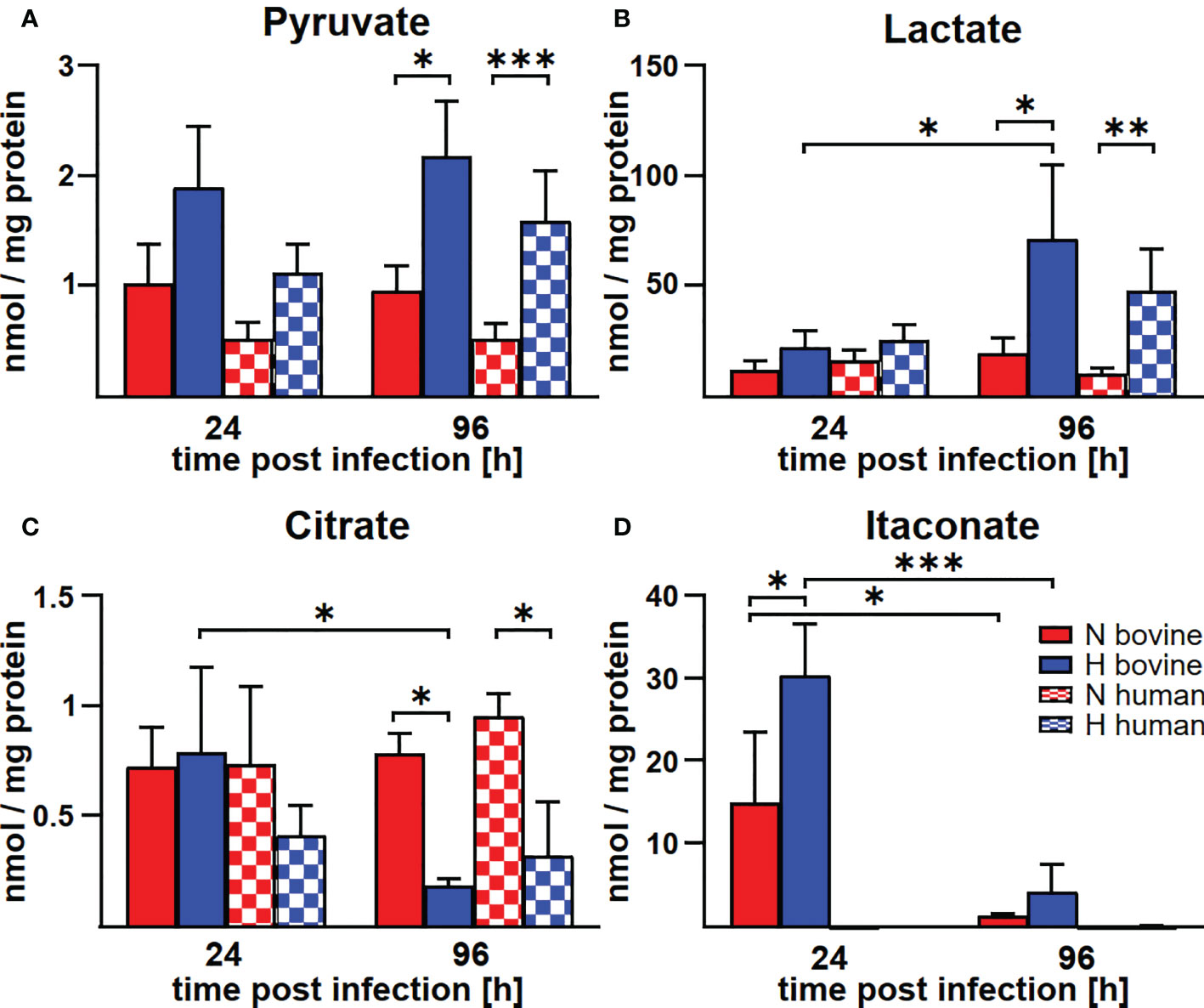
Figure 6 Hypoxia alters metabolite concentrations in C. burnetii-infected human and bovine macrophages similarly, with the exception of itaconate. (A-D) Human and bovine macrophages were infected with C. burnetii NMII for the time periods indicated under normoxia (N) or hypoxia (H). Carboxylic acids were analyzed by GC-MS. The amounts of (A) pyruvate, (B) lactate, (C) citrate, and (D) itaconate respectively, are shown in nmol/mg protein from three to four independent experiments. Mean ± SD, n = 3-4, two-way ANOVA with Tukey’s multiple comparison test. ***p<0.001, **p<0.01, *p<0.05.
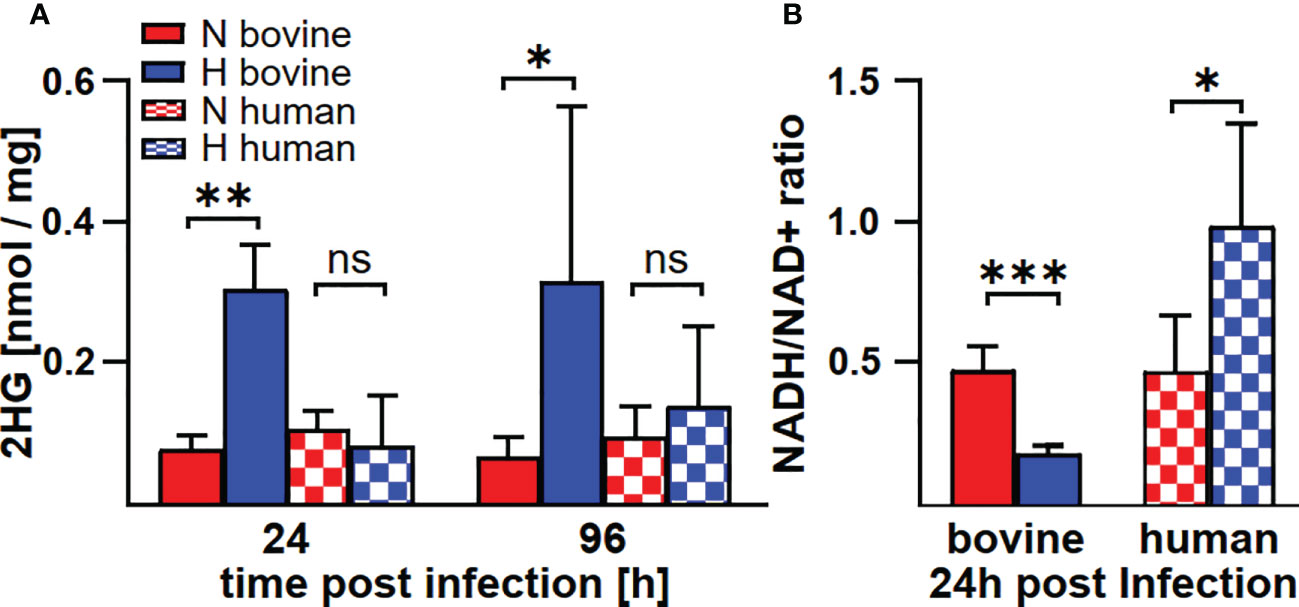
Figure 7 C. burnetii infection induces an increase of 2HG and a reduction of the NADH/NAD+ ratio only in hypoxic macrophages. Human and bovine macrophages were infected with C. burnetii NMII for the time periods indicated under normoxia (N) or hypoxia (H). (A) The amounts of 2HG (2-hydroxyglutarate) were analyzed by GC-MS and are shown in nmol/mg protein for three to four independent experiments. Unpaired t-test, n=3-4. **p<0.01, *p<0.05, ns=p>0.05. (B) NADH and NAD+ were analyzed in pmol using a colorimetric NAD+/NADH assay. The NADH/NAD+ ratio was calculated from 2-3 independent experiments performed in duplicate or triplicate. Mean ± SD, unpaired t-test. ***p<0.001, **p<0.01, *p<0.05.
Neither a C. burnetii infection nor the level of available oxygen affected the intracellular levels of the Krebs cycle intermediates succinate, fumarate, and malate in both human and bovine macrophages (Table 2).
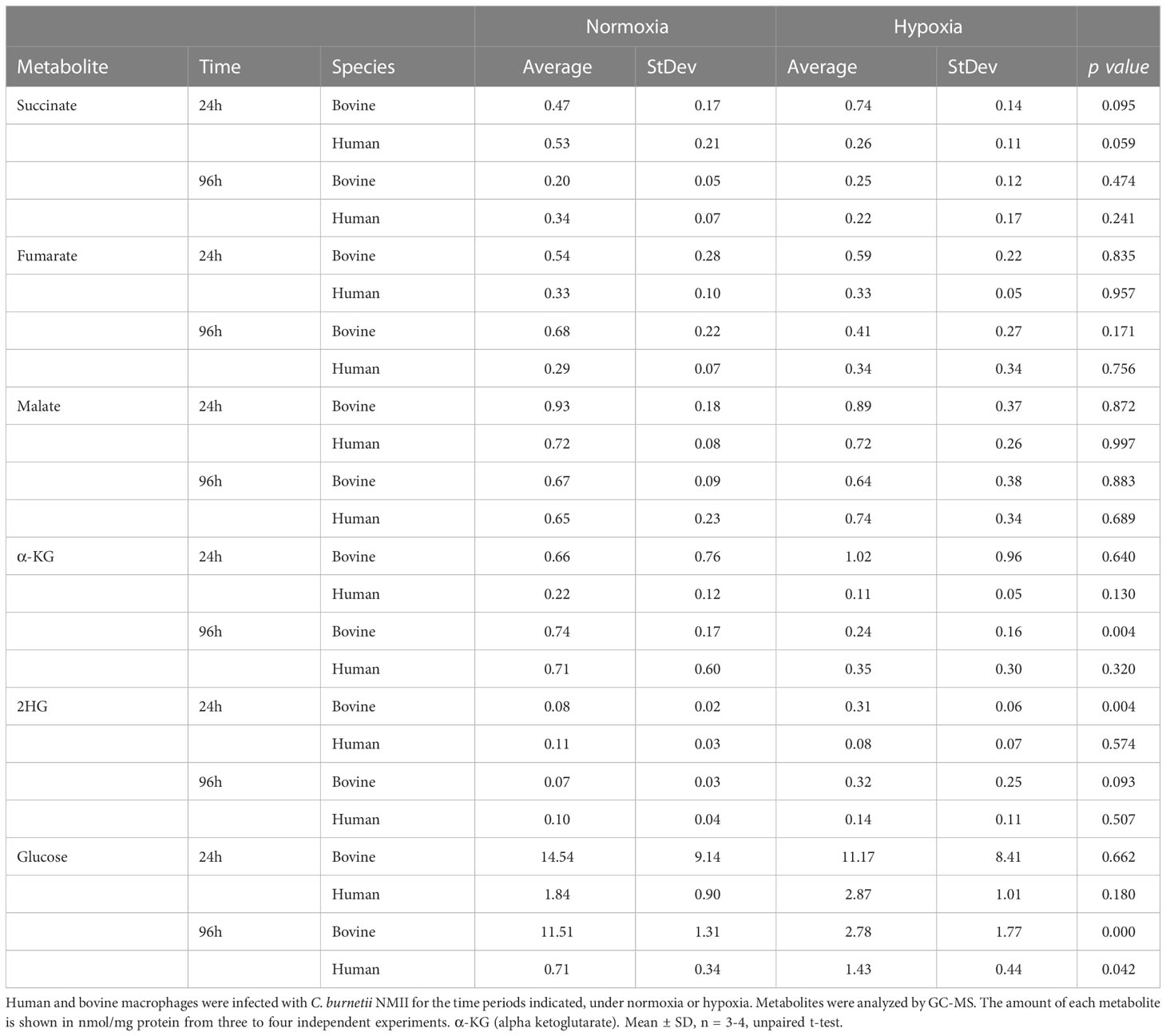
Table 2 Krebs cycle intermediates under normoxia and hypoxia.
C. burnetii-induced upregulation of TNFα expression was stabilized only under hypoxia in human macrophages
To investigate why C. burnetii replication was supported in hypoxic bovine macrophages, but not in hypoxic human macrophages, we analyzed the respective macrophage polarization, as the infection with C. burnetii stimulates an atypical M2 phenotype in human macrophages (39). Indeed, a similar polarization of murine macrophages towards an M2 phenotype is important for the permissiveness of these host cells to C. burnetii replication (40). Thus, we evaluated the expression of two M1 genes (TNFα and CD86) and two M2 genes (IL-10 and CD206) at different time points post-infection under normoxic and hypoxic conditions in human and bovine macrophages. In human macrophages, we observed that the infection induced a strong upregulation of TNFα and a slight upregulation of IL-10 at 4 h post-infection independent of the availability of oxygen (Figure 8). Over the time-course of the infection, the induction of the genes analyzed decreased gradually. Importantly, the levels of TNFα mRNA at 24 and 96 h post-infection were significantly higher in infected human macrophages kept under hypoxia. C. burnetii-infected bovine macrophages were also characterized by an upregulation of TNFα and IL-10 at early time-points post-infection. Like in infected human macrophages, the upregulation of the genes analyzed decreased over time. In contrast to the situation in human cells, we did not observe a difference in TNFα induction between infected hypoxic and normoxic bovine macrophages (Figure 8). As TNFα is a cytokine known to restrict C. burnetii replication (41), the difference in TNFα expression might explain why C. burnetii is capable of replicating in hypoxic bovine macrophages, but not in hypoxic human macrophages.
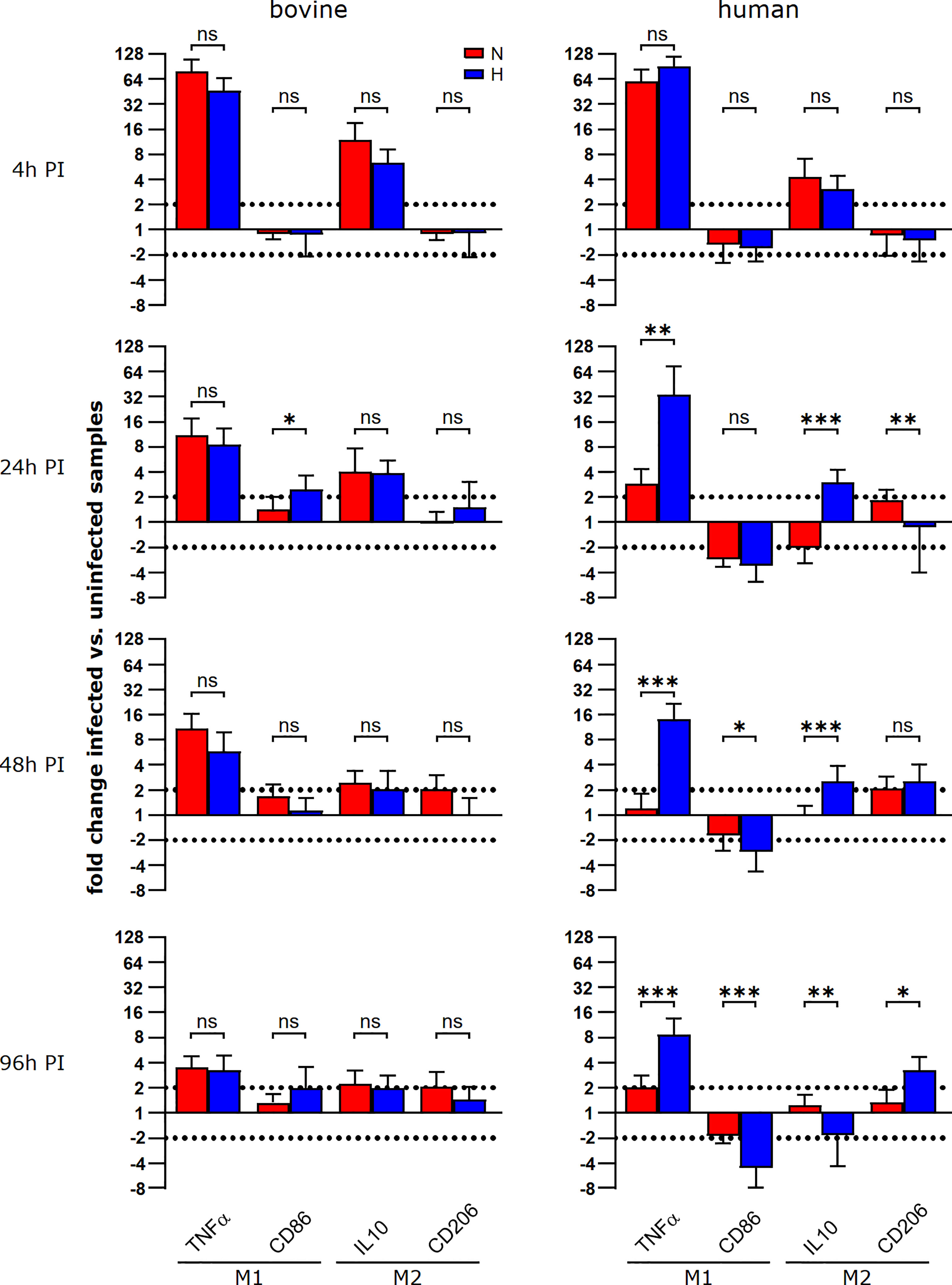
Figure 8 During the course of a C. burnetii infection, TNFα expression is only maintained in hypoxic human macrophages. Gene expression was determined for M1/M2 polarization marker genes in C. burnetii NMII infected human or bovine macrophages at the time points indicated. Data are shown as mean ± SD of ΔΔCT values (using uninfected samples as a calibrator), Mann-Whitney test, n=5-8 (bovine) and n=6-10 (human) ***p<0.001, **p<0.01, *p<0.05, ns=p>0.05.
Neither normoxic nor hypoxic C. burnetii-infected bovine macrophages secrete TNF
Levels of transcription of cytokine genes and of secretion of the encoded proteins do not necessarily correlate (42). In addition, not only the expression, but also the release of TNFα is controlled by HIF1α (24). In the supernatant of C. burnetii infected human macrophages, we observed the highest amount of TNFα at 24 h post-infection. The level of TNFα decreased over time. At later time points post-infection, the amount of TNFα in the supernatant was higher for infected hypoxic than normoxic human macrophages (Figure 9A), confirming that the cytokine levels correlated with the mRNA levels (Figure 8). Although bovine macrophages proved capable of secreting bTNFα after LPS stimulation (Figure 2A), we could not detect bTNFα in the supernatant of C. burnetii-infected bovine macrophages under all conditions tested using either a bTNFα-specific ELISA (Figure 9A) or a TNFα bioassay (not shown).
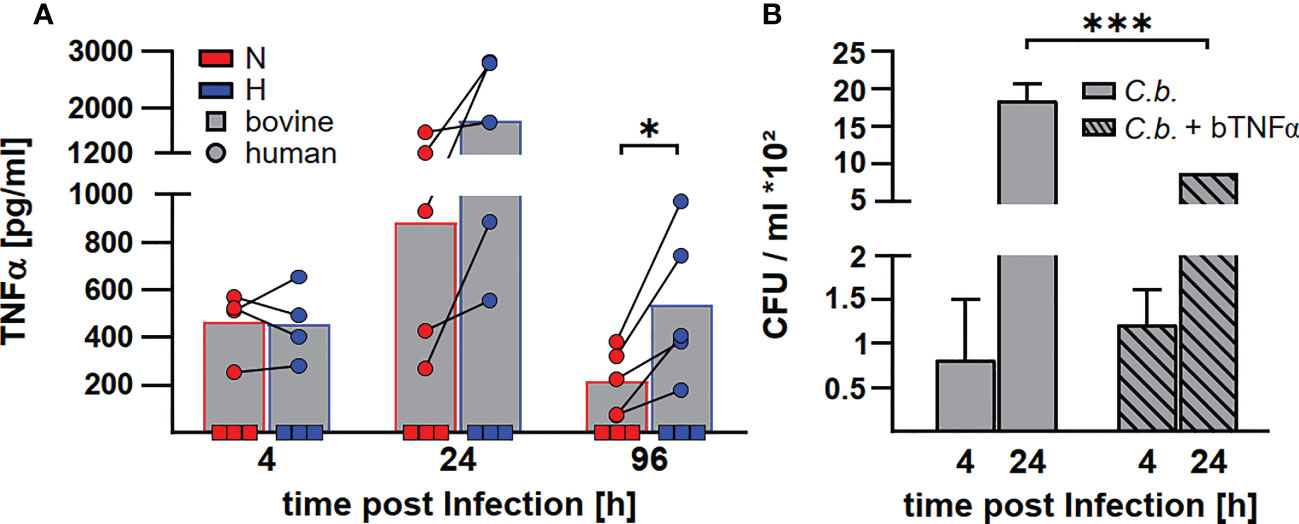
Figure 9 C. burnetii replication in infected bovine macrophages correlates with the lack of TNFα secretion. (A) Human macrophages from four different donors (circle) and three independent bovine macrophage preparations from two bovine donor animals (square) were infected with C. burnetii NMII for 4, 24 and 96 h under normoxia (N) or hypoxia (H). TNFα levels were analyzed by ELISA. Mean ± SD, paired t-test. *p<0.05. (B) Three independent bovine macrophage preparations from two bovine donor animals were infected with C. burnetii NMII and either left untreated or treated with 10ng/ml bTNFα. Cells were incubated for 4 and 24 h under normoxia. Bacterial counts were determined by counting colony forming units (CFU). Shown is a representative experiment performed with technical triplicates out of two experiments with similar results. Mean ± SD, unpaired t-test. ***p<0.01, *p<0.05.
TNF is involved in the control of C. burnetii replication in bovine macrophages
As the lack of TNFα secretion might contribute to the lack of control of C. burnetii replication in hypoxic bovine macrophages (41), we treated C. burnetii-infected bovine macrophages with bTNFα and analyzed CFU counts at different time points post-infection under normoxic conditions. Addition of bTNFα to C. burnetii-infected bovine macrophages resulted in lower bacterial counts (Figure 9B), suggesting that the lack of TNFα in C. burnetii infected bovine macrophages is important for the inability to prevent bacterial replication under hypoxic conditions.
Discussion
Once C. burnetii is internalized, the C. burnetii-containing vacuole (CCV) matures to an acidic, phagolysosomal-like parasitophorous vacuole (27, 43). During the course of the cellular infection, early phagolysosomes harbor a single bacterial cell. Sequentially, they fuse to a single large parasitophorous vacuole containing multiple C. burnetii cells (44). Mature CCVs have a pH of 4.7 to 5.2, which stimulates the activation of bacterial metabolism and the assimilation of essential nutrients (45–47). C. burnetii requires a functional Dot/Icm type IV secretion system (T4SS) for establishing the replicative CCV (48, 49). In addition to pH tolerance, a dismutase-catalase system promotes intracellular survival of C. burnetii. It eliminates reactive oxygen species generated by the bacteria themselves but also, most importantly, those generated by the host cell (50). Consequently, the infectious process might differ at multiple points/steps that are decisive for host differences in C. burnetii pathogenesis.
Previous studies have shown that species differences do exist down to the cellular level, however, genetic background and/or molecular basis have not been uncovered (19). Hypoxia plays a major role in the host - C. burnetii interaction. Therefore, we analyzed the interaction of human and bovine monocyte-derived macrophages with C. burnetii under normoxic and hypoxic conditions. The following reasons prompted us to analyze the influence of hypoxia during the course of a C. burnetii infection: i) the oxygen level is low at the site of infection in an acute colitis model (51); ii) murine macrophages prevent C. burnetii replication under hypoxia; iii) C. burnetii might enter a state of persistence under hypoxia (22); and iv) the course of a C. burnetii infection differs in humans and cattle (1). Our results indicate that the host-pathogen interaction differs also at the cellular level. Thus, C. burnetii replication is controlled in hypoxic human monocyte-derived macrophages, but not in bovine monocyte-derived macrophages (Figure 3). This difference might be of biological importance. In hypoxic human macrophages, C. burnetii might enter a stage of persistence, similar to the situation in hypoxic murine macrophages (22). Thus, hypoxia and/or HIF1α prevent bacterial replication without elimination of the pathogen (52), which might allow reoccurring or chronic infections. In the case of C. burnetii, it might support and/or allow Q fever to reach a chronic state in humans. Interestingly, infection with C. burnetii is common in cattle, but clinical disease is rare (53). Infertility, abortion and mastitis have been reported (54). However, there are conflicting reports whether seropositive cows have better or worse reproduction (54, 55). Whether the difference in controlling C. burnetii infection under hypoxia might result in different clinical outcomes can only be speculated about. Importantly, the lack of control of C. burnetii replication by hypoxic bovine macrophages might be mediated by STAT3 activation (Figure 4), as C. burnetii replication in murine macrophages under normoxia depends on the presence of STAT3. In addition, expression of a constitutively active STAT3 in murine macrophages also allows replication under hypoxia (22). The observed constant expression of the STAT3 activator IL-6 along with strongly limited expression of the STAT3 inhibitors PIAS3 and SOCS3 in C. burnetii-infected bovine macrophage (Figure 5) could favor activation of STAT3 and might explain the deficit of bovine macrophages to control C. burnetii replication under oxygen-limited conditions. However, the pathway(s) leading to STAT3 activation in C. burnetii-infected hypoxic bovine macrophages and how this impacts C. burnetii replication has to be analyzed in more detail.
The transcription factor STAT3 has key roles in inflammation and immunity (33). It suppresses signal transduction mediated by TLRs (56) and has anti-inflammatory function in mice and in humans (33). In addition, STAT3 also affects cellular metabolism. Thus, STAT3 increases the expression of Indy, a citrate transporter, and of citrate synthase, thereby elevating the intracellular level of the TCA metabolite citrate (57, 58). Indeed, higher citrate levels were only observed in C. burnetii-infected normoxic, but not hypoxic human macrophages (Figure 6C), suggesting that the increased level of STAT3 correlates with increased citrate levels (Figures 4, 6C). However, we were unable to observe this correlation in bovine macrophages at 96 h post-infection (Figures 4, 6C). Whether this might point to a difference in regulation of host cell metabolites between bovine and human macrophages has to be clarified. The significant difference between the cellular levels of itaconate supports such an assumption. Itaconate is produced by IRG1 from cis-aconitate, which is produced by ACO2 from citrate (59). We did not detect itaconate in C. burnetii-infected human macrophages (Figure 6D), in agreement with previous observations (34). In contrast, normoxic murine macrophages infected with C. burnetii and Legionella pneumophila produced high amounts of itaconate, which was reduced under hypoxic conditions (22). C. burnetii infected bovine macrophages also produced itaconate, but, in contrast to murine macrophages, it was elevated under hypoxic conditions (Figure 6D). As itaconate impedes bacterial replication (36), it is unlikely that the increased itaconate level in infected hypoxic bovine macrophages accounts for C. burnetii replication. However, itaconate also has an immune-regulatory function. Thus, treatment of LPS-stimulated murine macrophages with itaconate or its derivatives reduced the production of the pro-inflammatory cytokines IL-1β and IL-6, but not of TNF (35, 60). As TNFα is essential in restricting C. burnetii in murine macrophages, increased itaconate levels might not permit C. burnetii replication. However, we observed a host species-specific difference in TNFα mRNA levels during infection. In infected human macrophages, TNFα expression was always higher under hypoxic conditions at all time-points tested, while in infected bovine macrophages we did not observe an influence of the oxygen level on the TNFα mRNA level (Figure 8). Importantly, bovine macrophages were herein found not to secrete TNFα in the context of C. burnetii infection, despite of infection-induced mRNA synthesis (Figure 8 and Figure 9) and the ability to secrete TNFα after LPS stimulation (Figure 2). Thus, the ability of C. burnetii to replicate in hypoxic bovine macrophages might be mediated by a block of TNFα secretion. TNF trafficking and release to the extracellular milieu is well studied (61). After translation in the ER as type II membrane precursor pro-TNF, it is rapidly delivered to the Golgi, from where it traffics to the cell surface. The transport of TNF from the trans-Golgi network (TGN) depends on golgin and golgin-245 (62). TNF-loaded vesicles from the TGN fuse with recycling endosomes (RE), which depends on the SNARE proteins Stx6, Stx7 and Vti1b (63, 64). Transport of TNF from the RE to the cell surface is facilitated by Stx4 and VAMP-3 (63, 65). To be released as a soluble cytokine at the cell surface, pro-TNF has to be cleaved by the TNF-converting enzyme (TACE, ADAM17) (66). The block of TNFα secretion by C. burnetii-infected bovine macrophages may occur at any of these different steps. However, it might also be possible that C. burnetii activates or stabilizes synaptotagmin 11, which inhibits cytokine secretion (67). Further research will be required to elucidate at which point TNFα secretion is blocked and, more importantly, how.
Taken together, our results indicate that C. burnetii activates distinct signaling cascades in human and bovine macrophages, leading to different levels of control of bacterial replication. The lack of control of C. burnetii by hypoxic bovine macrophages is associated with STAT3 activation and a block of TNFα secretion (Figure 10). Addition of TNFα enables the bovine macrophages to at least partially restrict bacterial replication (Figure 9B), supporting the importance of this cytokine for cell autonomous control of C. burnetii replication. It can only be speculated how this difference in controlling C. burnetii replication might contribute to disease severity. At first glance, it is counterintuitive that the lack of control might result in the development of less severe disease. However, if C. burnetii is controlled, but not eliminated, this might lead to persistence. Chronic Q fever develops months to years after primary infection (68), indicating that bacterial persistence is an important step in disease development. In contrast, constant low-level bacterial replication might activate the innate and adaptive immune systems, allowing better control of the pathogen in vivo and consequently prevention of disease.
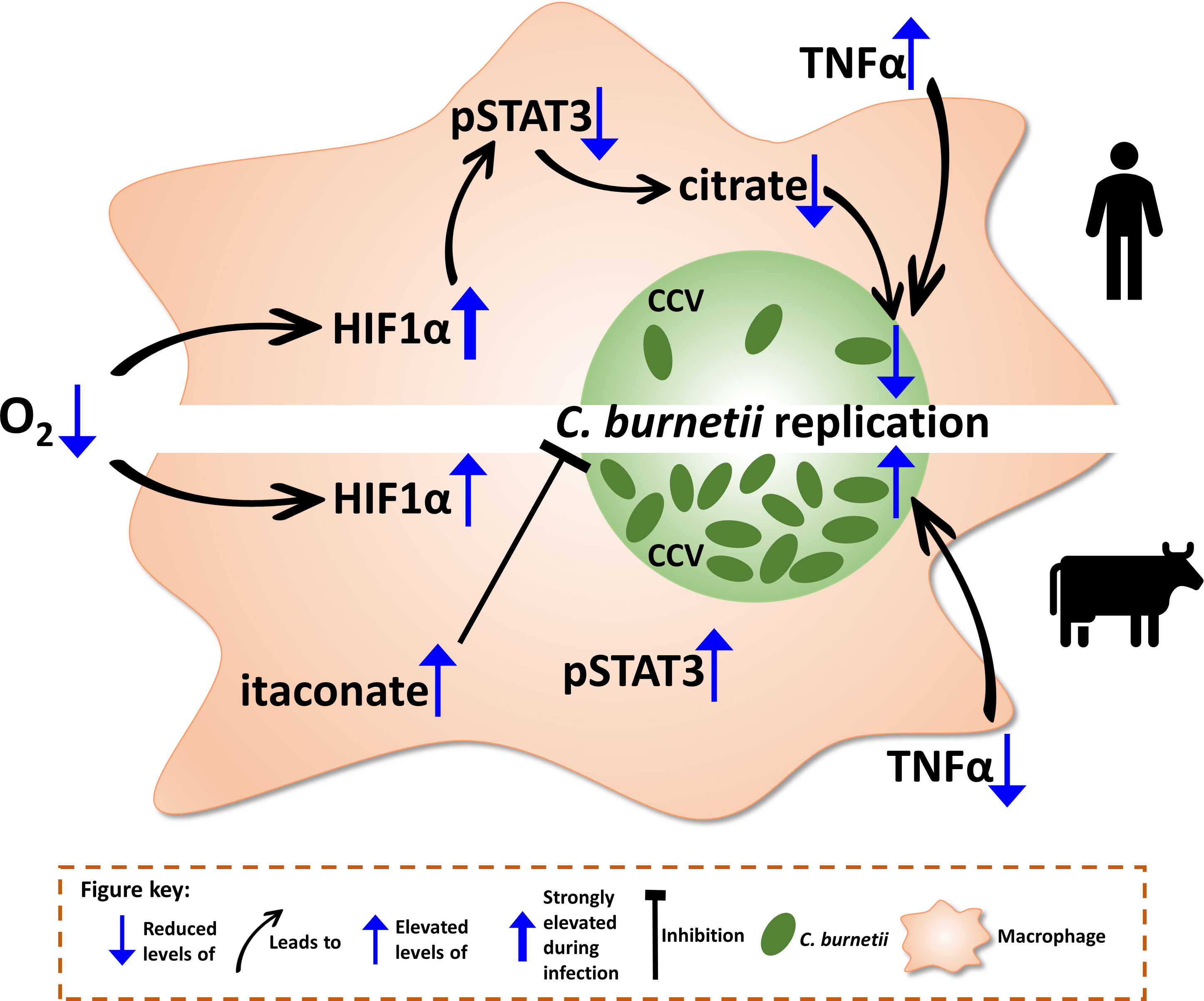
Figure 10 Schematic model of the consequences of hypoxia on bovine and human macrophages infected with C. burnetii. When oxygen (O2) levels drop, the transcription factor hypoxia-inducible factor 1-alpha (HIF1α) is stabilized. While in human macrophages (hMΦ), a C. burnetii infection leads to an elevation of HIF1α stabilization, HIF1α levels in bovine macrophages (bMΦ) are not altered upon infection. In infected hMΦ, HIF1α prevents activation of signal transducer and activator of transcription 3 (STAT3) by phosphorylation, which in turn causes a decrease in intracellular citrate levels, leading to the impediment of C. burnetii replication. However, this impairment of STAT3 activation by HIF1α is not observed in bMΦ causing phosphorylation of STAT3. Furthermore, the tumor necrosis factor alpha (TNFα) secretion is increased in hypoxic hMΦ correlating with the control of C. burnetii replication. In contrast, TNFα secretion is blocked in bMΦ, eliminating the factor of control over C. burnetii. Thus, C. burnetii is able to replicate. Finally, itaconate levels are increased in bMΦ, which have been shown to inhibit C. burnetii replication.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by Ethical Committee Erlangen approval number 111_12B. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. The animal study was reviewed and approved by Permit numbers 22-2684-04-04-102/15 and 22-2684-04-BFI-20-102.
Author contributions
AL conceived, designed and supervised the study. AL and CM provided resources. MM, MÖ, IH, JS-L, and KD performed the experiments. MM, IH, JS-L, KD, PO, CB, CM and AL analyzed the data. MM, IH, CB, PO, CM and AL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through the Collaborative Research Initiative 1181 (CRC1181) project A06 and by the Bundesministerium für Bildung und Forschung (BMBF) under the project number 01KI1726A/01KI2008A of “Q-GAPS” as part of the research network zoonotic infectious diseases to AL. KD was supported by the Bavarian Ministry of Science and the Arts within the Bavarian Research Network bayresq.net. IH was funded by the Bavarian Equal Opportunities Sponsorship – Realisierung von Chancengleichheit von Frauen in Forschung und Lehre (FFL) – Realization Equal Opportunities for Women in Research and Teaching.
Acknowledgments
The authors sincerely thank Norman Müller and Beate Burkert (Friedrich-Loeffler-Institut, Institute for Molecular Pathogenesis) for their excellent technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Eldin C, Melenotte C, Mediannikov O, Ghigo E, Million M, Edouard S, et al. From Q fever to Coxiella burnetii infection: a paradigm change. Clin Microbiol Rev (2017) 30:115–90. doi: 10.1128/CMR.00045-16
3. Morroy G, Keijmel SP, Delsing CE, Bleijenberg G, Langendam M, Timen A, et al. Fatigue following acute Q-fever: A systematic literature review. PloS One (2016) 11:e0155884. doi: 10.1371/journal.pone.0155884
4. Ayres JG, Flint N, Smith EG, Tunnicliffe WS, Fletcher TJ, Hammond K, et al. Post-infection fatigue syndrome following Q fever. QJM Monthly J Assoc Phys (1998) 91:105–23. doi: 10.1093/qjmed/91.2.105
5. Bronner MB, Haagsma JA, Dontje ML, Barmentloo L, Kouwenberg R, Olde Loohuis AGM, et al. Long-term impact of a Q-fever outbreak: An evaluation of health symptoms, health-related quality of life, participation and health care satisfaction after ten years. J Psychosom Res (2020) 139:110258. doi: 10.1016/j.jpsychores.2020.110258
6. Kampschreur LM, Wegdam-Blans MC, Wever PC, Renders NH, Delsing CE, Sprong T, et al. Chronic Q fever diagnosis- consensus guideline versus expert opinion. Emerg Infect Dis (2015) 21:1183–8. doi: 10.3201/eid2107.130955
7. Million M, Walter G, Thuny F, Habib G, Raoult D. Evolution from acute Q fever to endocarditis is associated with underlying valvulopathy and age and can be prevented by prolonged antibiotic treatment. Clin Infect Dis (2013) 57:836–44. doi: 10.1093/cid/cit419
8. Rodolakis A. Q fever in dairy animals. Ann New York Acad Sci (2009) 1166:90–3. doi: 10.1111/j.1749-6632.2009.04511.x
9. Van den Brom R, van Engelen E, Roest HI, van der Hoek W, Vellema P. Coxiella burnetii infections in sheep or goats: an opinionated review. Vet Microbiol (2015) 181:119–29. doi: 10.1016/j.vetmic.2015.07.011
10. Georgiev M, Afonso A, Neubauer H, Needham H, Thiery R, Rodolakis A, et al. Q fever in humans and farm animals in four European countries 1982 to 2010. Euro Surveill (2013) 18:20407–19. doi: 10.2807/ese.18.08.20407-en
11. Pouquet M, Bareille N, Guatteo R, Moret L, Beaudeau F. Coxiella burnetii infection in humans: to what extent do cattle in infected areas free from small ruminants play a role? Epidemiol Infect (2020) 148:e232. doi: 10.1017/S0950268820001880
12. Guatteo R, Seegers H, Taurel AF, Joly A, Beaudeau F. Prevalence of Coxiella burnetii infection in domestic ruminants: a critical review. Vet Microbiol (2011) 149:1–16. doi: 10.1016/j.vetmic.2010.10.007
13. Gache K, Rousset E, Perrin JB, D.E. Cremoux R, Hosteing S, Jourdain E, et al. Estimation of the frequency of Q fever in sheep, goat and cattle herds in France: results of a 3-year study of the seroprevalence of Q fever and excretion level of Coxiella burnetii in abortive episodes. Epidemiol Infect (2017) 145:3131–42. doi: 10.1017/S0950268817002308
14. Velasova M, Damaso A, Prakashbabu BC, Gibbons J, Wheelhouse N, Longbottom D, et al. Herd-level prevalence of selected endemic infectious diseases of dairy cows in great Britain. J Dairy Sci (2017) 100:9215–33. doi: 10.3168/jds.2016-11863
15. Dobos A, Kreizinger Z, Kovacs AB, Gyuranecz M. Prevalence of Coxiella burnetii in central and Eastern European dairy herds. Comp Immunol Microbiol Infect Dis (2020) 72:101489. doi: 10.1016/j.cimid.2020.101489
16. Szymanska-Czerwinska M, Jodelko A, Zareba-Marchewka K, Niemczuk K. Shedding and genetic diversity of Coxiella burnetii in polish dairy cattle. PloS One (2019) 14:e0210244. doi: 10.1371/journal.pone.0210244
17. Sobotta K, Bonkowski K, Liebler-Tenorio E, Germon P, Rainard P, Hambruch N, et al. Permissiveness of bovine epithelial cells from lung, intestine, placenta and udder for infection with Coxiella burnetii. Vet Res (2017) 48:23. doi: 10.1186/s13567-017-0430-9
18. Sobotta K, Hillarius K, Jimenez PH, Kerner K, Heydel C, Menge C. Interaction of Coxiella burnetii strains of different sources and genotypes with bovine and human monocyte-derived macrophages. Front Cell Infect Microbiol (2017) 7:543. doi: 10.3389/fcimb.2017.00543
19. Sobotta K, Hillarius K, Mager M, Kerner K, Heydel C, Menge C. Coxiella burnetii infects primary bovine macrophages and limits their host cell response. Infect Immun (2016) 84:1722–34. doi: 10.1128/IAI.01208-15
20. Ghigo E, Capo C, Raoult D, Mege JL. Interleukin-10 stimulates Coxiella burnetii replication in human monocytes through tumor necrosis factor down-modulation: role in microbicidal defect of Q fever. Infect Immun (2001) 69:2345–52. doi: 10.1128/IAI.69.4.2345-2352.2001
21. Hayek I, Schatz V, Bogdan C, Jantsch J, Lührmann A. Mechanisms controlling bacterial infection in myeloid cells under hypoxic conditions. Cell Mol Life Sci (2021) 78:1887–907. doi: 10.1007/s00018-020-03684-8
22. Hayek I, Fischer F, Schulze-Luehrmann J, Dettmer K, Sobotta K, Schatz V, et al. Limitation of TCA cycle intermediates represents an oxygen-independent nutritional antibacterial effector mechanism of macrophages. Cell Rep (2019) 26:3502–3510.e3506. doi: 10.1016/j.celrep.2019.02.103
23. Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol Cell (2008) 30:393–402. doi: 10.1016/j.molcel.2008.04.009
24. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol (2009) 9:609–17. doi: 10.1038/nri2607
25. Pauli U, Bertoni G, Duerr M, Peterhans E. A bioassay for the detection of tumor necrosis factor from eight different species: evaluation of neutralization rates of a monoclonal antibody against human TNF-alpha. J Immunol Methods (1994) 171:263–5. doi: 10.1016/0022-1759(94)90047-7
26. Borrmann E, Möbius P, Diller R, Köhler H. Divergent cytokine responses of macrophages to Mycobacterium avium subsp. paratuberculosis strains of types II and III in a standardized in vitro model. Vet Microbiol (2011) 152:101–11. doi: 10.1016/j.vetmic.2011.04.002
27. Schulze-Luehrmann J, Eckart RA, Ölke M, Saftig P, Liebler-Tenorio E, Lührmann A. LAMP proteins account for the maturation delay during the establishment of the Coxiella burnetii-containing vacuole. Cell Microbiol (2016) 18:181–94. doi: 10.1111/cmi.12494
28. Dettmer K, Nurnberger N, Kaspar H, Gruber MA, Almstetter MF, Oefner PJ. Metabolite extraction from adherently growing mammalian cells for metabolomics studies: optimization of harvesting and extraction protocols. Anal Bioanal Chem (2011) 399:1127–39. doi: 10.1007/s00216-010-4425-x
29. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U.S.A. (1995) 92:5510–4. doi: 10.1073/pnas.92.12.5510
30. Greer SN, Metcalf JL, Wang Y, Ohh M. The updated biology of hypoxia-inducible factor. EMBO J (2012) 31:2448–60. doi: 10.1038/emboj.2012.125
31. Bardos JI, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim Biophys Acta (2005) 1755:107–20. doi: 10.1016/j.bbcan.2005.05.001
32. Corcoran SE, O'Neill LA. HIF1alpha and metabolic reprogramming in inflammation. J Clin Invest (2016) 126:3699–707. doi: 10.1172/JCI84431
33. Hillmer EJ, Zhang H, Li HS, Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev (2016) 31:1–15. doi: 10.1016/j.cytogfr.2016.05.001
34. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U.S.A. (2013) 110:7820–5. doi: 10.1073/pnas.1218599110
35. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature (2018) 556:113–7. doi: 10.1038/nature25986
36. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs modify the proteome of Legionella-containing vacuoles and restrict infection via IRG1-derived itaconic acid. PloS Pathog (2016) 12:e1005408. doi: 10.1371/journal.ppat.1005408
37. Kohl L, Siddique M, Bodendorfer B, Berger R, Preikschat A, Daniel C, et al. Macrophages inhibit Coxiella burnetii by the ACOD1-itaconate pathway for containment of Q fever. EMBO Mol Med (2022):e15931. doi: 10.15252/emmm.202215931
38. Du X, Hu H. The roles of 2-hydroxyglutarate. Front Cell Dev Biol (2021) 9:651317. doi: 10.3389/fcell.2021.651317
39. Benoit M, Barbarat B, Bernard A, Olive D, Mege JL. Coxiella burnetii, the agent of Q fever, stimulates an atypical M2 activation program in human macrophages. Eur J Immunol (2008) 38:1065–70. doi: 10.1002/eji.200738067
40. Fernandes TD, Cunha LD, Ribeiro JM, Massis LM, Lima-Junior DS, Newton HJ, et al. Murine alveolar macrophages are highly susceptible to replication of Coxiella burnetii phase II in vitro. Infect Immun (2016) 84:2439–48. doi: 10.1128/IAI.00411-16
41. Bradley WP, Boyer MA, Nguyen HT, Birdwell LD, Yu J, Ribeiro JM, et al. Primary role for toll-like receptor-driven tumor necrosis factor rather than cytosolic immune detection in restricting Coxiella burnetii phase II replication within mouse macrophages. Infect Immun (2016) 84:998–1015. doi: 10.1128/IAI.01536-15
42. Shebl FM, Pinto LA, Garcia-Pineres A, Lempicki R, Williams M, Harro C, et al. Comparison of mRNA and protein measures of cytokines following vaccination with human papillomavirus-16 L1 virus-like particles. Cancer Epidemiol Biomarkers Prev (2010) 19:978–81. doi: 10.1158/1055-9965.EPI-10-0064
43. Lührmann A, Newton HJ, Bonazzi M. Beginning to understand the role of the type IV secretion system effector proteins in Coxiella burnetii pathogenesis. Curr Topics Microbiol Immunol (2017) 413:243–68. doi: 10.1007/978-3-319-75241-9_10
44. Norlander L. Q fever epidemiology and pathogenesis. Microbes Infect (2000) 2:417–24. doi: 10.1016/S1286-4579(00)00325-7
45. Hackstadt T. Estimation of the cytoplasmic pH of Coxiella burnetii and effect of substrate oxidation on proton motive force. J Bacteriol (1983) 154:591–7. doi: 10.1128/jb.154.2.591-597.1983
46. Hackstadt T, Williams JC. pH dependence of the Coxiella burnetii glutamate transport system. J Bacteriol (1983) 154:598–603. doi: 10.1128/jb.154.2.598-603.1983
47. Howe D, Mallavia LP. Coxiella burnetii exhibits morphological change and delays phagolysosomal fusion after internalization by J774A.1 cells. Infect Immun (2000) 68:3815–21. doi: 10.1128/IAI.68.7.3815-3821.2000
48. Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, et al. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio (2011) 2:e00175–00111. doi: 10.1128/mBio.00175-11
49. Carey KL, Newton HJ, Lührmann A, Roy CR. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PloS Pathog (2011) 7:e1002056. doi: 10.1371/journal.ppat.1002056
50. Baca OG, Li YP, Kumar H. Survival of the Q fever agent Coxiella burnetii in the phagolysosome. Trends Microbiol (1994) 2:476–80. doi: 10.1016/0966-842X(94)90651-3
51. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity (2014) 40:66–77. doi: 10.1016/j.immuni.2013.11.020
52. Schaffer K, Taylor CT. The impact of hypoxia on bacterial infection. FEBS J (2015) 282:2260–6. doi: 10.1111/febs.13270
53. Pexara A, Solomakos N, Govaris A. Q fever and seroprevalence of Coxiella burnetii in domestic ruminants. Vet Ital (2018) 54:265–79. doi: 12834/Vetlt.1113.6046.3
54. Arricau-Bouvery N, Rodolakis A. Is Q fever an emerging or re-emerging zoonosis? Vet Res (2005) 36:327–49. doi: 10.1051/vetres:2005010
55. Lopez-Gatius F, Almeria S, Garcia-Ispierto I. Serological screening for Coxiella burnetii infection and related reproductive performance in high producing dairy cows. Res Vet Sci (2012) 93:67–73. doi: 10.1016/j.rvsc.2011.07.017
56. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity (1999) 10:39–49. doi: 10.1016/S1074-7613(00)80005-9
57. von Loeffelholz C, Lieske S, Neuschafer-Rube F, Willmes DM, Raschzok N, Sauer IM, et al. The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology (2017) 66:616–30. doi: 10.1002/hep.29089
58. MacPherson S, Horkoff M, Gravel C, Hoffmann T, Zuber J, Lum JJ. STAT3 regulation of citrate synthase is essential during the initiation of lymphocyte cell growth. Cell Rep (2017) 19:910–8. doi: 10.1016/j.celrep.2017.04.012
59. Williams NC, O'Neill LAJ. A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol (2018) 9:141. doi: 10.3389/fimmu.2018.00141
60. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
61. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol (2014) 5:491. doi: 10.3389/fimmu.2014.00491
62. Lieu ZZ, Lock JG, Hammond LA, La Gruta NL, Stow JL, Gleeson PA. A trans-golgi network golgin is required for the regulated secretion of TNF in activated macrophages in vivo. Proc Natl Acad Sci U.S.A. (2008) 105:3351–6. doi: 10.1073/pnas.0800137105
63. Murray RZ, Kay JG, Sangermani DG, Stow JL. A role for the phagosome in cytokine secretion. Science (2005) 310:1492–5. doi: 10.1126/science.1120225
64. Murray RZ, Wylie FG, Khromykh T, Hume DA, Stow JL. Syntaxin 6 and Vti1b form a novel SNARE complex, which is up-regulated in activated macrophages to facilitate exocytosis of tumor necrosis factor-alpha. J Biol Chem (2005) 280:10478–83. doi: 10.1074/jbc.M414420200
65. Pagan JK, Wylie FG, Joseph S, Widberg C, Bryant NJ, James DE, et al. The t-SNARE syntaxin 4 is regulated during macrophage activation to function in membrane traffic and cytokine secretion. Curr Biol CB (2003) 13:156–60. doi: 10.1016/S0960-9822(03)00006-X
66. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature (1997) 385:729–33. doi: 10.1038/385729a0
67. Arango Duque G, Fukuda M, Descoteaux A. Synaptotagmin XI regulates phagocytosis and cytokine secretion in macrophages. J Immunol (2013) 190:1737–45. doi: 10.4049/jimmunol.1202500
Keywords: Coxiella burnetii, bovine macrophages, normoxia, hypoxia, HIF1α, STAT3, citrate, TNF
Citation: Mauermeir M, Ölke M, Hayek I, Schulze-Luehrmann J, Dettmer K, Oefner PJ, Berens C, Menge C and Lührmann A (2023) Bovine blood derived macrophages are unable to control Coxiella burnetii replication under hypoxic conditions. Front. Immunol. 14:960927. doi: 10.3389/fimmu.2023.960927
Received: 03 June 2022; Accepted: 16 January 2023;
Published: 30 January 2023.
Edited by:
Ulrich Emil Schaible, Research Center Borstel (LG), GermanyReviewed by:
Lis Noelia Velasquez, University Medical Center Hamburg-Eppendorf, GermanyAnders Omsland, Washington State University, United States
Alan G. Goodman, Washington State University, United States
Copyright © 2023 Mauermeir, Ölke, Hayek, Schulze-Luehrmann, Dettmer, Oefner, Berens, Menge and Lührmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anja Lührmann, anja.luehrmann@uk-erlangen.de
†These authors have contributed equally to this work