 Shiya Yao1†
Shiya Yao1† Yuejun Han
Yuejun Han Ketao Jin
Ketao Jin Huanrong Lan
Huanrong Lan- 1Department of Colorectal Surgery, Affiliated Jinhua Hospital, Zhejiang University School of Medicine, Jinhua, Zhejiang, China
- 2Department of Surgical Oncology, Hangzhou Cancer Hospital, Hangzhou, Zhejiang, China
Immunotherapy has made significant advances in the treatment of colorectal cancer (CRC), revolutionizing the therapeutic landscape and highlighting the indispensable role of the tumor immune microenvironment. However, some CRCs have shown poor response to immunotherapy, prompting investigation into the underlying reasons. It has been discovered that certain chemotherapeutic agents possess immune-stimulatory properties, including the induction of immunogenic cell death (ICD), the generation and processing of non-mutated neoantigens (NM-neoAgs), and the B cell follicle-driven T cell response. Based on these findings, the concept of inducing chemotherapy has been introduced, and the combination of inducing chemotherapy and immunotherapy has become a standard treatment option for certain cancers. Clinical trials have confirmed the feasibility and safety of this approach in CRC, offering a promising method for improving the efficacy of immunotherapy. Nevertheless, there are still many challenges and difficulties ahead, and further research is required to optimize its use.
1 Introduction
Colorectal cancer (CRC) is the third most common malignancy globally, after lung cancer, and is the second leading cause of cancer-related deaths in both men and women (1, 2). Despite improved screening for early detection, the global burden of disease and mortality has not significantly decreased (1). Approximately 20% of patients present with metastatic disease at diagnosis, and an additional 25% of patients who present with localized disease will subsequently develop metastases (3). Most patients with metastatic CRC (mCRC) cannot be cured and are managed with palliative systemic therapy, resulting in poor prognosis with a median overall survival (mOS) of approximately 30 months (3). In the past five years, immunotherapy has made a significant impact on the treatment of CRC, with immune checkpoint inhibitors (ICIs) being particularly prominent. The tumor immune microenvironment (TIME) is intimately linked with tumor immunotherapy and represents a key obstacle to successful antitumor immune therapy, potentially limiting its clinical benefit. Numerous studies have demonstrated the efficacy of ICIs in the treatment of microsatellite instability (MSI-H)/mismatch repair-deficient (dMMR) mCRC. However, 95% of mCRC patients are microsatellite stable (MSS)/proficient mismatch repair (pMMR) subtype and are insensitive to immune therapy (4, 5). We will discuss the reasons for poor response to ICIs in this patient population, including defects in antigen presentation and peptide transport, immune evasion, abnormalities in the TIME, low tumor mutation burden, and targeting of apoptotic pathways, among others. Efforts are currently underway to overcome these barriers and improve the sensitivity of immune therapy in this patient population.
It was previously believed that chemotherapy was solely an immunosuppressive agent. However, recent data indicate that chemotherapy drugs can promote immune activation through various pathways, notably via the induction of immunogenic cell death (ICD) mechanisms (6–9). Certain chemotherapy drugs, such as doxorubicin (DOX), paclitaxel (PTX), and oxaliplatin (OXA), can kill tumor cells via ICD, thereby activating innate and adaptive antitumor immune responses. ICD is characterized by the release of danger-associated molecular patterns (DAMPs) and the generation and processing of non-mutated neoantigens (NM-neoAgs) tumor-associated antigens to enhance antigen presentation by promoting dendritic cell (DC) maturation and cytotoxic T lymphocyte (CTL) infiltration (10). This process can reverse the tumor immune suppressive microenvironment and increase the sensitivity of immunotherapy. A series of clinical studies supporting the combination of chemotherapy and ICIs in mCRC is currently ongoing. Based on this, the concept of inducing chemotherapy has been introduced, which involves administering chemotherapy prior to immunotherapy to convert the tumor microenvironment (TME) from “cold” to “hot,” thereby enhancing the response to immunotherapy. Inducing chemotherapy combined with immunotherapy has become part of the standard treatment for certain cancers, with clinical trials confirming its feasibility in CRC. However, many challenges remain in the treatment of mCRC. The fusion of immunotherapy and Single-cell RNA sequencing (scRNA-seq) holds promise for providing truly personalized treatment for an increasing number of mCRC patients, enabling us to achieve personalized treatment strategies (11). Additionally, significant research is needed to optimize this combined treatment approach, including how to optimize dosage regimens, such as dose, timing, and sequence, and biomarker prediction studies. We look forward to further breakthroughs in the future to provide more effective and safer treatment options for cancer patients.
2 Exploring the progress and challenges of immunotherapy in CRC
Immune checkpoint inhibitors (ICIs) have brought new opportunities in cancer treatment (12–15). The programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1) signaling pathway in tumors is a crucial mechanism for evading immune surveillance. The FDA first approved the use of immunotherapy drugs for treating mCRC in 2017 (16–19). Pembrolizumab (an anti-PD-1 monoclonal antibody) has been established as the new standard for first-line treatment in MSI-H/dMMR mCRC (20).
Despite the tremendous potential of immunotherapy in CRC treatment, there are also challenges and limitations. CRC is a common malignancy, with the majority being MSS/pMMR type. Compared to MSI-H/dMMR tumors, MSS/pMMR tumors have poorer response to immunotherapy, primarily due to immune suppression or immune desertification (21–23), characterized by low levels or defects in T-cell infiltration and reduced checkpoint protein expression (5). They generally do not benefit from immune therapies such as PD-1/PD-L1 inhibitors (24, 25), indicating obstacles to the effectiveness of immunotherapy (26). The mechanisms of resistance to immunotherapy in MSS/pMMR CRC are highly complex (Figure 1) (27).
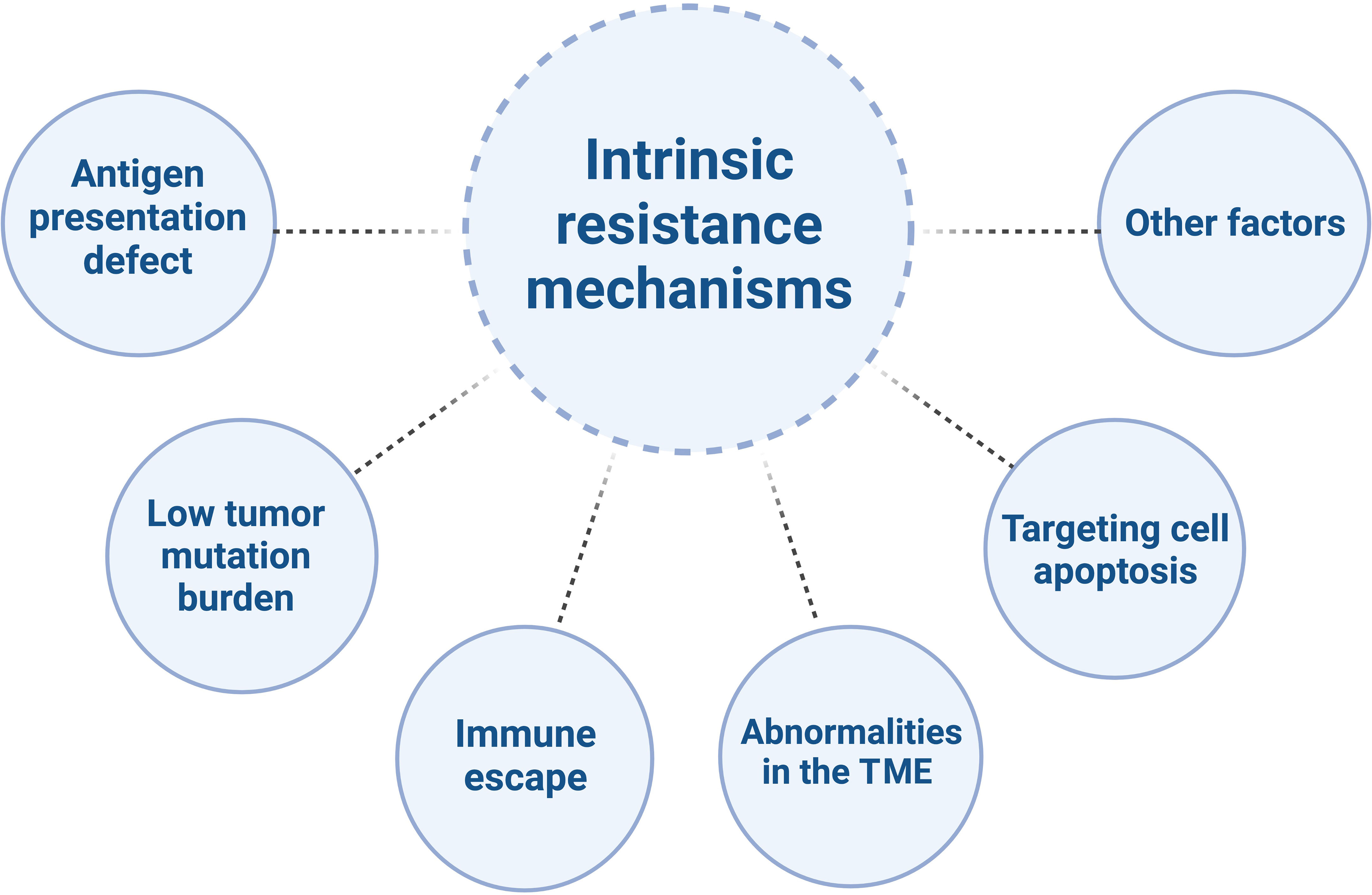
Figure 1 The intrinsic resistance mechanisms of MSS/pMMR mCRC to ICIs.
Mutations in antigen presentation-related genes in tumor cells lead to loss of antigens within tumor cells, which makes it difficult for T cells to recognize and attack tumor cells, thereby reducing the sensitivity of tumor cells to immunotherapy. For example, the BRAF V600E mutation has been shown to reduce T cells infiltration into the TME and eliminate neoantigen presentation on cancer cells (28, 29). Inhibiting BRAF signaling has been demonstrated to reduce myeloid-derived suppressor cells, increase the recruitment of tumor-infiltrating lymphocytes, enhance neoantigen presentation on antigen-presenting cells, and collectively enhance anti-tumor immune responses (28, 30–32).
MSS/pMMR CRC tumor cells have a lower tumor mutation burden, which means that the effectiveness of immunotherapy is relatively poor. These types of tumors exhibit a relative deficiency in CD8+ T cell infiltration (33, 34), as well as lower tumor mutation burdens (35–38) and multiple immune antigen defects, leading to tumor immune evasion (39, 40). Immune escape refers to the ability of tumor cells to evade attacks from the immune system through various mechanisms (41). Immune escape mechanisms in MSS/pMMR CRC include the lack of immune stimulatory molecules, overexpression of immune inhibitory molecules, deficiency in major histocompatibility complex class I (MHC I) molecules, and lack of T cell infiltration (42–44). The activating mutations of KRAS, as an upstream regulator of BRAF and a potent activator of MAPK, may play a role in immune escape by impairing interferon-mediated antigen presentation and recruitment of effector T cells to the TME (45, 46). Another preclinical study showed that RAS oncogenes induce immune escape by stabilizing PD-1 RNA and leading to sustained expression of PD-1 (47). Increasing evidence suggests that the MAPK pathway may also be involved in immune exclusion, serving as another biological barrier to the success of immune therapy.
MSS/pMMR CRC is typically characterized by a “cold” or “excluded” TME, meaning that immune cells are unable to infiltrate the tumor or, even if they do, they are unable to exert their cytotoxic effects. Studies have shown that there is a reduced infiltration and activation of T cells in MSS/pMMR CRC, while the levels of immune suppressive cells (such as Tregs and myeloid-derived suppressor cells (MDSCs)) and immune inhibitory molecules (such as IDO1 and transforming growth factor-beta (TGF-β)) are elevated (48–50). This may weaken the anti-tumor response of T cells. For example, the increase in TGF-β is associated with an increase in Tregs, leading to downregulation of anti-tumor immunity. These data also support the role of the TGF-β pathway in downregulating NK cell activity, as NK cells play a role in innate immunity by recognizing cancer cells (51). It is worth noting that TGF-β activation has been observed in CRC liver metastasis, resulting in downregulation of CD4+ and CD8+ T cells (52). Activated B cells were found to be significantly depleted in liver metastases of CRC through scRNA-seq. The inhibitory effect on cancer cells was mediated by the suppression of the Wnt and TGF-β pathways through the SDF-1-CXCR4 axis, which promoted the migration of activated B cells (53).
In MSS/pMMR CRC, inhibition of the tumor cell apoptosis pathway renders the tumor cells insensitive to attacks from the immune system. Aberrant activation of the WNT/β-catenin signaling pathway is frequently observed in MSS CRC but is rare in MSI-H CRC (54, 55). Abnormal activation of the Wnt signaling pathway is associated with T cell exclusion and insufficient infiltration, which may lead to the inhibition of tumor cell apoptosis pathways and consequently result in treatment resistance (25, 54, 56–58). By inhibiting β-catenin pharmacologically, it is possible to increase the number of dendritic cells (DCs), upregulate CCL4, and promote the infiltration of CD8+ T cells in various tumor models, including CRC, thereby reactivating anti-tumor immune responses (59–61). Additionally, the overexpression of members of the Bcl-2 family and the presence of an immune suppressive microenvironment may also lead to the inhibition of tumor cell apoptosis pathways and consequently result in treatment resistance (62, 63). Other factors that may contribute to immunotherapy resistance in CRC include changes in the gut microbiome, which can impact the efficacy of immune therapy and lead to treatment resistance (64).
In summary, immunotherapy has had a profound impact on the traditional treatment of CRC, but it also poses challenges and limitations. Future research needs to further explore the combination of immunotherapy with conventional chemotherapy, overcome immune resistance mechanisms, and understand the influence of the TIME on the efficacy of immune therapy in order to enhance the effectiveness of immunotherapy and advance the treatment of CRC.
3 Mechanism of chemotherapy in promoting immune activation
Recent research in the field of immunotherapy has demonstrated that chemotherapy drugs not only enhance the immunogenicity of cancer cells but also induce immune stimulation by activating effector T cells and inhibiting immune suppressive cells. These exciting findings suggest that the combination of chemotherapy and ICIs may have synergistic anti-cancer effects, providing a promising treatment option for tumor patients who have a poor response to monotherapy with ICIs.
Studies have shown that chemotherapy drugs may exert their effects through immune stimulation mechanisms (65). For example, anthracycline drugs can induce immunogenic cell death (ICD) and directly block immune inhibitory pathways in the TIME, leading to the release of neoantigens (NM-neoAgs) from cancer cells (6, 66–72). ICD is characterized by the presentation of dying cancer cells to antigen-presenting cells (APCs) in the form of danger-associated molecular patterns (DAMPs), which act as lymphoma adjuvant-like signals (73). Specifically, during the early stages of apoptosis, ICD induces the exposure of calreticulin (CRT) on the cell surface and releases extracellular ATP by upregulating autophagy during the detachment phase of apoptosis. It also promotes the release of high mobility group box 1 (HMGB1) during the secondary necrosis phase of cell death (71, 72, 74–82). CRT, ATP, and HMGB1 bind to their respective receptors (CD91 receptor, purinergic P2Y2 or P2X7 receptors, and TLR4) expressed on DCs, triggering their entry into the tumor tissue and upregulating antigen uptake processes. The resulting mature DCs (mDCs) present antigens and trigger a series of further immune responses. New antigens and danger signal molecules are released, forming a “cancer immune cycle” (83) (Figure 2). Overall, the ability of DCs to capture and present antigens is enhanced, and their ability to process NM-neoAgs and recruit CD4+ T cells/CD8+ T cells for an enhanced adaptive immune response in the tumor is more effective, thereby enhancing the immune system’s ability to clear cancer cells (66). Additionally, pro-inflammatory cytokines such as TNF, IL-6, and IL-1β, which are detected during and after ICD induction, can increase MHC I expression on APCs, promote T cell differentiation, and activate NK cells (85). Activated DCs (such as IL-12) and other innate immune cells (such as cytokines produced by IFN-α/β) enhance NK cell function, leading to the secretion of IFN-γ and TNF (86).
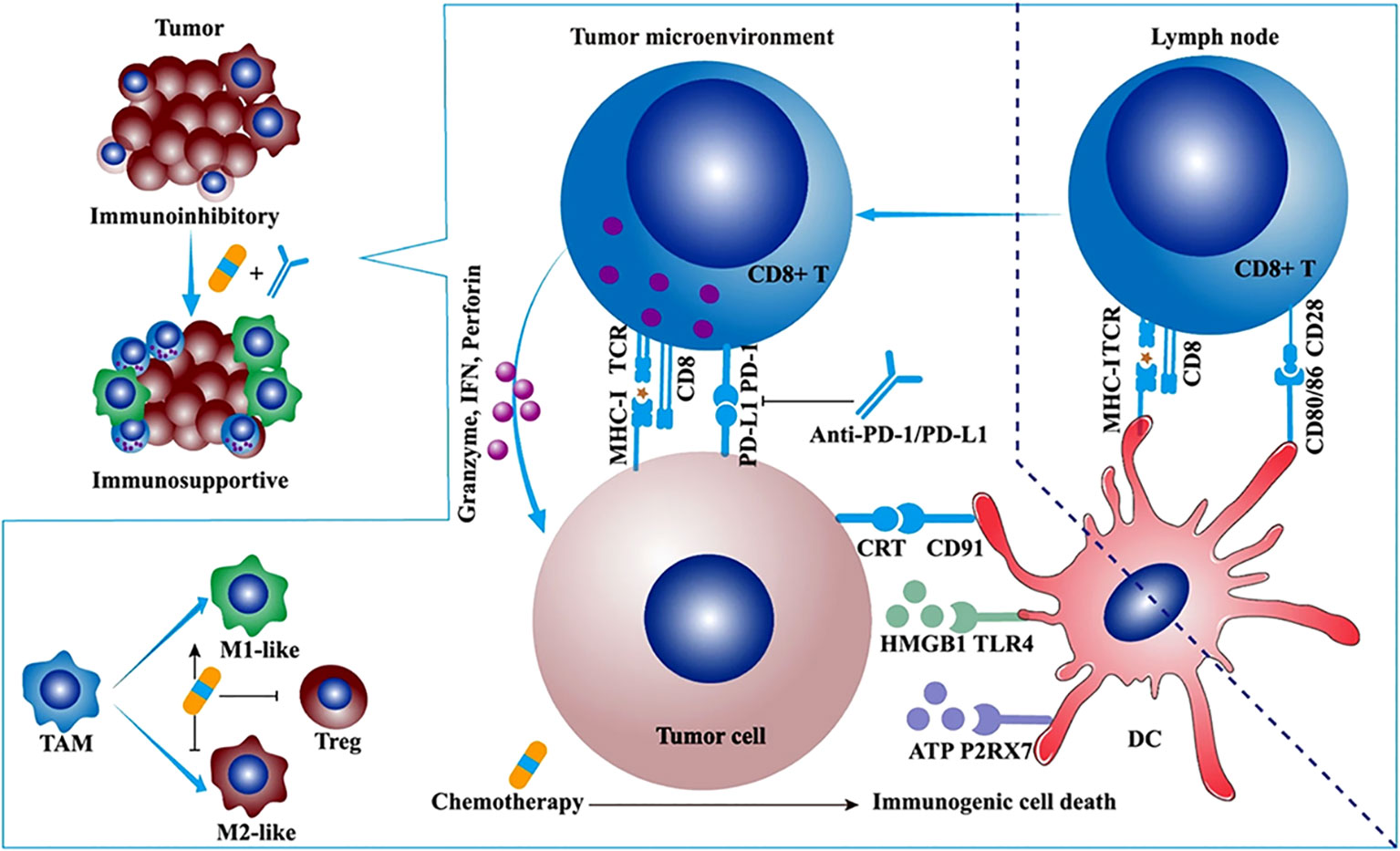
Figure 2 The synergistic antitumor efficacies and mechanisms of α-PD-1/PD-L1 in combination with chemotherapy, radiotherapy, or angiogenesis inhibitor. Chemotherapy synergizes with α-PD-1/PD-L1. Some cytotoxic chemotherapeutic drugs could induce immunogenic cell death and stimulate antitumor immune response. Immunogenic cell death (ICD) is featured with some upregulated damage-associated molecular patterns (DAMPs) such as calreticulin (CRT), ATP, and high-mobility group box 1 (HMGB1). The ATP-P2RX7, CRT-CD91, and HMGB1-TLR4 pathways facilitate the antigen capture and presentation of DC, ultimately motivating adaptive antitumor immune response. Apart from ICD, low-dose chemotherapy depletes regulatory T cells (Tregs) and promotes the repolarization of tumor-associated macrophage (TAM) from M2-like to M1-like phenotype (84).
The TIME is a key barrier to anti-tumor immunity and may limit the clinical efficacy of immunotherapy (87, 88). Immune cells not only act as “gatekeepers” of the tumor but can also have positive or negative effects on tumor growth and metastasis (89, 90). For example, Tregs and MDSCs can suppress the immune response of T cells and natural killer cells (NK cells), providing favorable conditions for cancer invasion, inhibiting anti-tumor immune responses, and promoting metastasis (91, 92). TAMs can exhibit anti-tumor properties (93). When activated, TAMs can promote the proliferation and activation of anti-tumor T cells, thereby inhibiting tumor growth and metastasis (93).Although the number and function of NK cells may be suppressed, recent research has shown that certain drugs can enhance the function of NK cells and play a significant role in tumor treatment (94). Studies have shown that chemotherapy can eliminate specific cells, such as Tregs and MDSCs (95, 96), which have immunosuppressive characteristics. This can transform non-inflammatory tumors (referred to as “cold tumors”) into tumors rich in cytotoxic cells (referred to as “hot tumors”) (97), especially when used at doses below the maximum tolerated dose. Recent research indicates that anti-PD-1 therapy can reshape the tumor immune microenvironment based on chemotherapy-induced changes, providing new insights for improving the effectiveness of combination immunotherapy and chemotherapy (98).
Furthermore, some studies suggest that chemotherapy-induced DNA damage fragments in the cell nucleus may actively translocate to the cytoplasm to prevent their erroneous insertion into the genomic DNA. This process activates the innate immune cGAS-STING (cyclic GMP-AMP synthase/stimulator of interferon genes) pathway, leading to an immune-rich microenvironment and triggering innate immune responses (Figure 3A) (99–102).
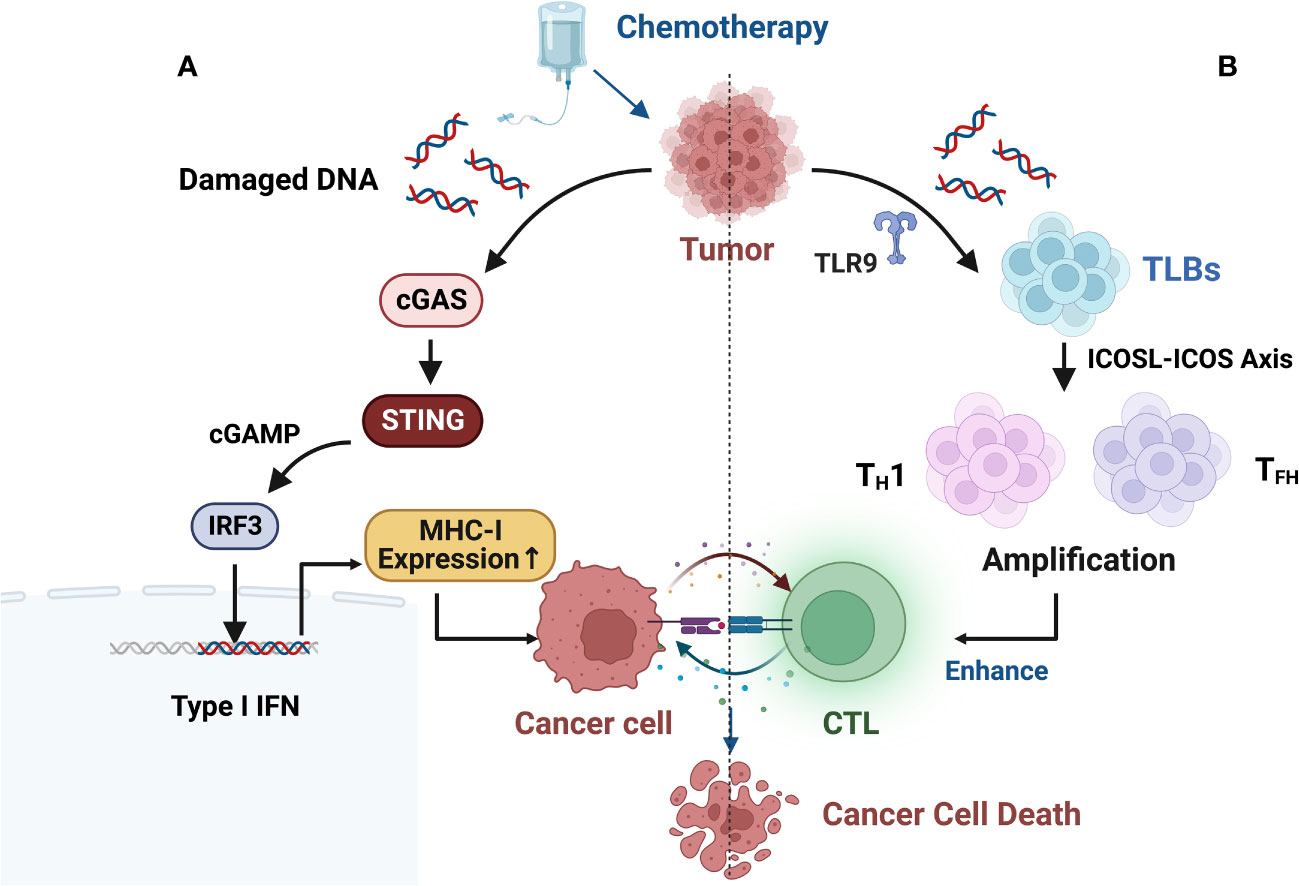
Figure 3 (A) Chemotherapy-induced DNA damage fragments in the nucleus may be actively exported to the cytoplasm to prevent their erroneous insertion into genomic DNA, thereby activating the innate immune cGAS-STING (cyclic GMP-AMP synthase/stimulator of interferon genes) pathway, resulting in an immune-rich microenvironment and triggering innate immune responses. (B) B cell-centered anti-tumor immune network. B-cell-centered anti-tumor immune network. Gemcitabine plus cisplatin (GP) chemotherapy activates an anti-tumor immune response dominated by a type of innate immune B cell (ILB). GP-mediated release of tumor cell DNA fragments can induce an ILB subset located in the third lymphoid structure induced by chemotherapy via Toll-like receptor 9 (TLR9) signaling. Subsequently, ILB promotes the expansion of type 1 helper T cells (TH1) and follicular helper T cells (TFH) via the ICOSL-ICOS signaling axis, thereby facilitating the cytotoxicity of CD8+ T lymphocytes (CTLs). Meanwhile, ILB can also activate the STING-IFN-I pathway of tumor cells, upregulating the expression of MHC I on tumor cells, forming a positive feedback loop.
Recent research has revealed new mechanisms of chemotherapy-induced immune modulation. Effective chemotherapy can induce B-cell-centered effector T-cell responses, suggesting that chemotherapy holds promising potential as a combination therapy with ICIs. This can be achieved by upregulating MHC I expression to directly modulate tumor immunogenicity and by enhancing the efficacy and quantity of CD8+ T cells through the enhanced interaction between endogenous-like B cells (ILBs) and effector helper T cells (TH cells) (Figure 3B). ILBs enhance TFH and TH1 cells via the ICOSL-ICOS axis. The inducible T-cell co-stimulatory (ICOS) pathway is another important pathway in tumor immunotherapy. ICOS is a co-stimulatory receptor expressed on activated T cells and is crucial for their survival and function. The ICOS pathway plays a critical role in balancing effector T cells and Tregs, and its dysregulation has been associated with the development and progression of various types of cancer (103). Lu et al. (104) also found that chemotherapy induces complement signaling pathways, enhancing the anti-tumor properties of B cells. Overall, these data suggest that in addition to therapeutic interventions targeting the restoration of conventional DC function, chemotherapy interventions can reshape the plasticity of B cells and establish an anti-tumor environment. Similar findings and breakthroughs are hoped to be achieved in CRC.
In conclusion, the mechanisms of immune modulation activated by chemotherapy provide new insights for the clinical treatment of combination immunotherapy. Further research is needed to explore the combined strategies of chemotherapy and immunotherapy in order to achieve better treatment outcomes and wider clinical applications.
4 Cytotoxic chemotherapy drugs that promote anti-tumor immunity
Chemotherapy drugs were initially designed to directly inhibit or kill malignant cells to achieve their therapeutic effect. Recently, some frontline drugs have been found to further promote anti-tumor immunity by increasing tumor immunogenicity, improving T cells infiltration, or depleting immune-suppressive populations (Figure 4). There is increasing evidence that chemotherapy triggers complex immune events (105–109), which is due to the ability of drugs to induce ICD in tumor cells and to directly modulate immune cells. Some chemotherapy drugs have been shown to exert immune-stimulatory effects by inhibiting immune-suppressive cells and/or activating effector cells, or by increasing immunogenicity and T cells infiltration (110–112). Chemotherapy drugs that promote anti-tumor immunity can be classified into several categories based on their mechanisms (Table 1).
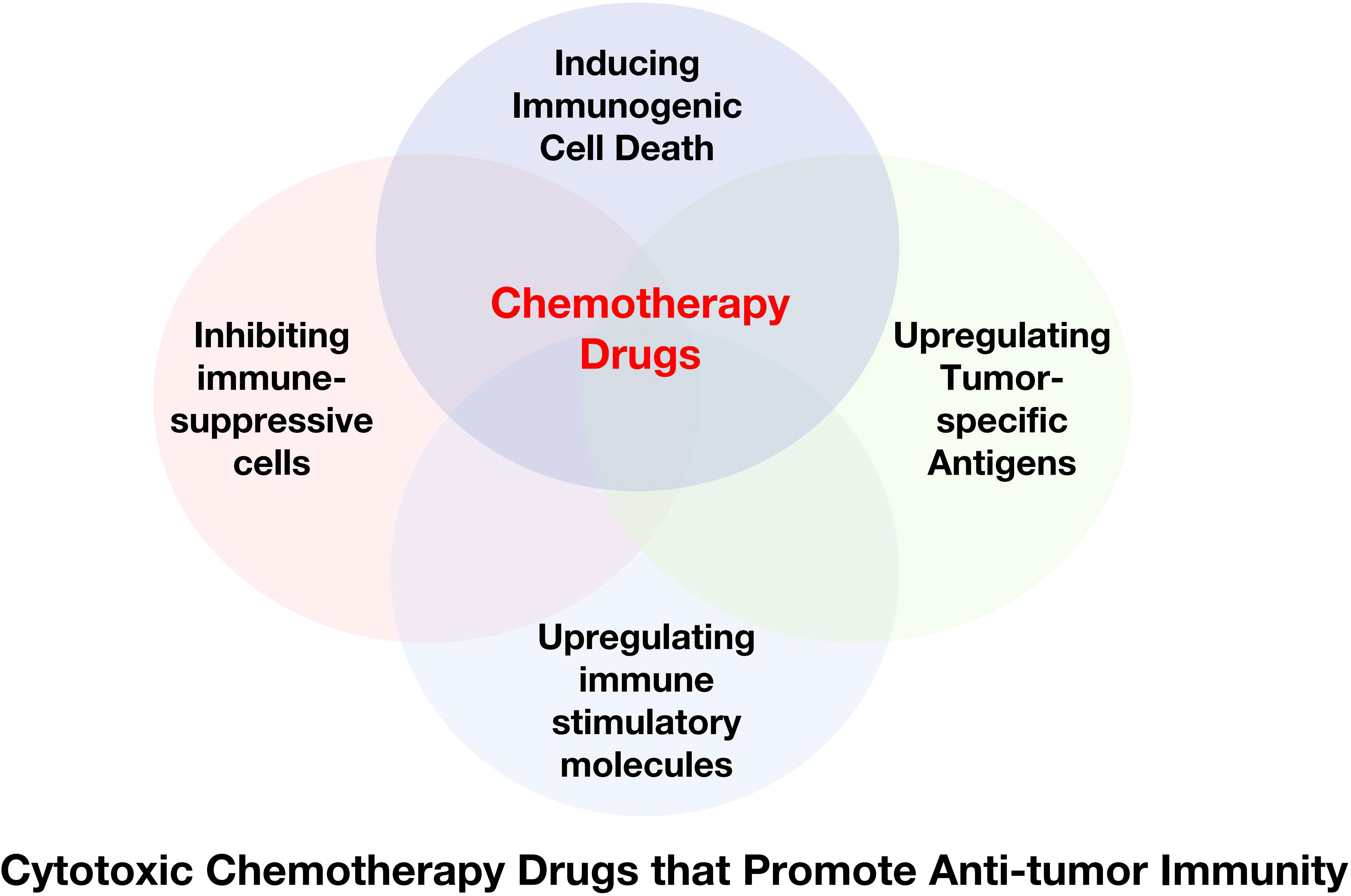
Figure 4 Cytotoxic Chemotherapy Drugs that Promote Anti-tumor Immunity. Some chemotherapy drugs have been shown to exert immune-stimulating effects by inducing immunogenic cell death, inhibiting immune suppressor cells, and/or activating effector cells, or upregulating tumor-specific antigens.
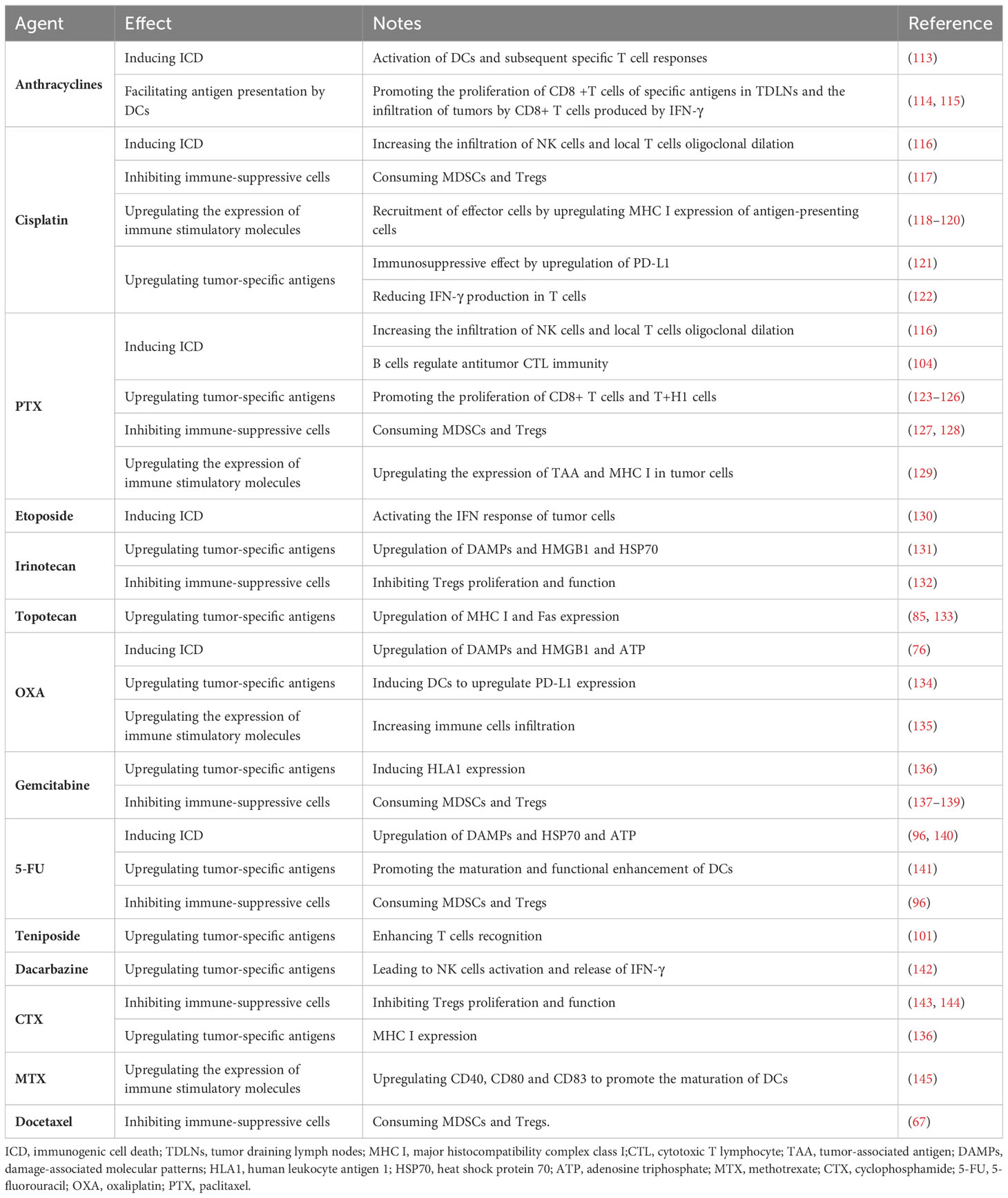
Table 1 Immunological effects of conventional antitumor agents.
4.1 Inducing ICD
ICD is a form of apoptosis that can induce an effective anti-tumor immune response. Drugs that induce ICD include anthracycline chemotherapy drugs, OXA, and PTX (146–149). As mentioned earlier, these drugs induce an anti-tumor immune response by activating DCs and subsequent specific T cells responses.
Specifically, anthracycline chemotherapy drugs can induce ICD, which is a form of apoptosis that induces an effective anti-tumor immune response by activating DCs and subsequent specific T cells responses (150). Recent studies have found that drugs that induce ICD can also regulate anti-tumor CTL immunity through tumor-infiltrating NK and B cells. In human ovarian cancer, platinum-based and taxane-based chemotherapy significantly increased NK cells infiltration and local T cells oligoclonal expansion (116). In human breast cancer, a neoadjuvant regimen of DOX, cyclophosphamide(CTX), and PTX converted infiltrating tumor B cells to a new ICOSL+ phenotype. These newly appearing B cells participated in the formation of TLS and significantly increased the number and cytotoxicity of tumor-specific CD8+ T cells (104).Another topoisomerase II inhibitor, teniposide, can induce ICD, but its mechanism of action is different from that of anthracyclines. Topoisomerase II inhibitors induce proliferation arrest or death of tumor cells by increasing DNA double-strand breaks (130). As mentioned earlier, teniposide activates the endogenous type I interferon (IFN) response in tumor cells and upregulates features of ICD (Figure 3A). In murine colon cancer, teniposide induced potent anti-tumor CD8+ T cells immunity and significant tumor suppression. Administration of teniposide reversed the resistance of KRAS-mutant CT26 colon cancer to PD-1 blockade (151). Although it has positive immunomodulatory effects in mouse tumors, it remains unclear whether teniposide acts as an ICD inducer in human cancers. Considering its ability to activate anti-tumor CTL responses, chemotherapy drugs that induce ICD are thought to be able to enhance the therapeutic effect of ICIs. The combination of DOX and PD-1 or PD-L1 antibodies shows significant anti-tumor effects in various mouse cancers, such as melanoma and breast cancer (152, 153). In human metastatic triple-negative breast cancer (TNBC), short-term treatment with DOX induces sensitivity to PD-1 blockade (154). Similarly, OXA has been shown to enhance the anti-tumor effect of anti-PD-L1 therapy in mouse lung cancer, melanoma, and CRC (155). The combination of PTX and ICIs produce superior tumor suppression in non-immunogenic squamous non-small cell lung cancer (NSCLC) (156).
4.2 Upregulating tumor-specific antigens
Certain chemotherapy drugs can induce tumor cells to express antigens, thereby enhancing T cells recognition and killing of tumor cells.
For example, Irinotecan and Topotecan are derivatives of camptothecin, which can enhance T cells recognition of tumor cells and upregulate tumor-specific antigens (157). An in vitro experiment revealed the upregulation of DAMPs, HMGB1, and heat shock protein 70 (HSP70) after treatment with Irinotecan (131). In melanoma, they can upregulate tumor-specific antigens. Surviving tumor cells upregulate MHC I and Fas expression after treatment with Topotecan (85, 133), making them more susceptible to immune cells killing. PTX can stimulate DC maturation and antigen presentation through various mechanisms, such as the NF-κB and MAPK signaling pathways, TLR4/MyD88 pathways, etc. (158). In addition, PTX can promote an immune response of tumor-specific T cells (123–126) and promote the proliferation of CD8+T cells and T+H1 cells, thereby playing a role in the treatment of tumors. Platinum-based drugs and gemcitabine increase antigen specificity by inducing HLA1 expression (136). OXA can induce upregulation of PD-L1 expression on DCs, while carboplatin upregulates PD-1 mRNA expression. Studies have shown that in patients with head and neck squamous cells carcinoma receiving standard cisplatin treatment, cisplatin can have an immune suppressive effect through upregulation of PD-L1 (121). The expression of PD-L1 may also impede the response of anti-cancer T cells. In vitro, high-dose cisplatin significantly reduced IFN-γ production by T cells (122) and decreased the cytotoxicity of NK cells in ovarian cancer patients (159). Standard doses of 5-FU may produce an immune-stimulatory effect, for example, by promoting antigen uptake by DCs (141). DOX promotes antigen presentation by DCs, promotes the proliferation of CD8+ T cells specific for certain antigens in tumor-draining lymph nodes, and increases IFN-γ production by CD8+ T cells infiltrating the tumor (114, 115). Temozolomide enhances the tumor cells antigen presentation mechanism and enhances T cells recognition (101). Dacarbazine is currently only used in melanoma patients who are not eligible for new therapies or have failed other treatments. Dacarbazine can upregulate NK cells activation and IFN-γ release. Increased levels of IFN-γ lead to upregulation of MHC I expression in tumor cells, which is necessary for T cells recognition (142).
4.3 Inhibiting immune-suppressive cells
Certain chemotherapy drugs can inhibit the activity of immune-suppressive cells, thereby enhancing anti-tumor immune response. For example, platinum-based drugs, CTX, 5-FU, docetaxel, and other chemotherapy drugs can reduce the inhibitory effect of Tregs on the anti-tumor immune response. These chemotherapy drugs can also promote the polarization of tumor-associated macrophages, thereby enhancing their anti-tumor activity. Moreover, the combination of certain ICIs and chemotherapy drugs can further enhance the anti-tumor immune response.
Specifically, Platinum-based drugs reduce the immune-suppressive microenvironment by depleting MDSCs and Tregs (117). PTX can selectively inhibit the number and function of Tregs (114, 123, 124, 160–166). One study found that patients with advanced disease had a significant decrease in the anti-inflammatory cytokine IL-10 levels after receiving PTX treatment (167). PTX can also repolarize TAM2. It has recently been identified as an agonist for TLR4 on TAMs and directly polarizes this anti-inflammatory population towards a pro-inflammatory phenotype (168, 169). Breast cancer patients treated with PTX exhibit peripheral pro-inflammatory features (170). After PTX treatment, ovarian cancer patients have gene enrichment associated with the inflammatory macrophage phenotype (168). Studies have shown that the combination of atezolizumab and nab-PTX prolongs progression-free survival (PFS) in patients with metastatic TNBC (171, 172). Low-dose CTX not only reduces the number of Tregs in tumors but also inhibits Tregs function (143). A recent study found that CTX preferentially targets CCR2+ Tregs in a highly active and proliferative state, namely effector Tregs (173). A clinical trial also showed that repeated low-dose CTX induction of Tregs depletion and enhanced anti-tumor immunity in patients with end-stage metastatic CRC ultimately contributes to prolonged progressive survival (174). CTX can also deplete tumor-infiltrating Tregs and improve the survival rate of mice with neuroblastoma when used in combination with anti-PD-1 therapy (144). Similar to CTX, topoisomerase I inhibitor camptothecin can also inhibit the production and function of Tregs. By removing the suppression of Tregs, irinotecan promotes the initiation and proliferation of CD8+ T cells in draining lymph nodes and inhibits the growth of lung cancer and CRC in a CD8+ T cell-dependent manner (175). Similarly, it has been reported that the FOLFIRI chemotherapy regimen containing irinotecan can reduce the inhibitory activity of peripheral Tregs in CRC patients (132). 5-FU selectively kills MDSCs in vivo while preserving other lymphocyte subtypes (96). Gemcitabine can deplete circulating or tumor-infiltrating MDSCs in various cancers, which benefits the restoration of CTL infiltration and cytotoxic activity (67). The use of standard doses can reduce the number of MDSCs while enhancing the cross-presentation of malignant antigens (136). In pancreatic cancer patients, standard-dose gemcitabine leads to the depletion of Tregs (139). Interestingly, there is no significant decrease in other lymphocyte subtypes after treatment.
4.4 Upregulating immune stimulatory molecules
Certain chemotherapy drugs can upregulate the expression of immune stimulatory molecules, thereby enhancing anti-tumor immune response. For example, PTX and its analogs can upregulate the expression of TAA and MHC I on tumor cells (129).
High-dose methotrexate can cause bone marrow suppression (176), but low-dose methotrexate exhibits immune stimulatory properties (145). In an in vitro experiment, non-cytotoxic low-dose methotrexate concentrations promoted DC maturation by upregulating CD40, CD80, and CD83 (145). In turn, DCs stimulated T cells proliferation, which may lead to a stronger anti-tumor response. This suggests that low-dose methotrexate can be used as an immune stimulant. CTX can induce MHC I expression (136) and deplete Tregs cells (114). Cisplatin also exhibits immune stimulatory properties by upregulating MHC I expression on antigen-presenting cells (118, 119), recruiting effector cells to the tumor site, and stimulating their proliferation (120). A single dose of OXA increased immune cell infiltration in a CRC mouse model (135). In ovarian cancer, a single dose of gemcitabine increased CD8+ T cell tumor infiltration and PD-L1 expression both in vivo and in vitro (139, 177).
5 Clinical application of the combination of chemotherapy and immunotherapy in CRC
As previously discussed, chemotherapy can activate immune regulation through various mechanisms, thereby enhancing patients’ response to immunotherapy. Combination immunotherapy has become an effective strategy for treating certain tumors. Therefore, combining chemotherapy and immunotherapy may be a new treatment strategy. In fact, some studies have already demonstrated the clinical efficacy of this combination strategy. For example, in first-line treatment for NSCLC, Pembrolizumab in combination with chemotherapy has been approved for first-line treatment of advanced non-squamous NSCLC regardless of PD-L1 levels (178). Other promising combinations include Atezolizumab, carboplatin, and etoposide for small cell lung cancer (SCLC), and nab-PTX or PTX in combination with Atezolizumab for advanced/metastatic breast cancer (179). Combination chemotherapy and immunotherapy have been shown to significantly improve patient survival. However, due to tumor heterogeneity and immune escape, a subset of patients with CRC lack response to immunotherapy. Currently, a series of clinical trials are being conducted for the combination of chemotherapy and immunotherapy in MSS/pMMR mCRC, with the aim of finding a breakthrough in treatment for these patients.
FOLFOX plus bevacizumab is the first-line standard of care (SOC) for MSS mCRC. The Checkmate 9X8 study (180) challenged first-line treatment of mCRC with the combination of nivolumab plus mFOLFOX6 and bevacizumab versus mFOLFOX6 and bevacizumab, with 95% of the patients being MSS/pMMR. The phase II results showed that, compared to the control group (current standard treatment regimen), the experimental group had a higher PFS rate starting at 12 months, with significantly improved 15-month PFS rate (45% vs. 21.5%) and 18-month PFS rate (28% vs. 9%), and ORR increased from 46% to 60%.
The BACCI phase II trial (181) (NCT0287319) evaluated the efficacy of adding Atezolizumab to Capecitabine and Bevacizumab in refractory mCRC. The addition of Atezolizumab to Capecitabine and Bevacizumab significantly extended progression-free survival (PFS), demonstrating a positive research advancement. This is the first positive study targeting the PD-1/PD-L1 pathway, chemotherapy, and the VEGF pathway, highlighting the need for further analysis and research.
The domestic BBCAPX study (182) is a study of the first-line treatment of MSS/RAS mutation mCRC with sintilimab + CapeOX + bevacizumab. The phase II single-arm trial results showed an ORR of up to 84%, a DCR of 100%, and unexpected conversion to R0 resection in 3 cases (12%). The study results demonstrated that the combination of sintilimab with CapeOX and bevacizumab for the treatment of RAS gene mutations and MSS-type mCRC showed good clinical benefits, with a high objective response rate and unexpected conversion rate, as well as low toxicity and tolerable safety. Based on the results of this phase II study, the ongoing BBCAPX phase III study holds great promise.
The objective of the single-arm phase II MEDITREME trial was to (183) evaluate the efficacy of the combination treatment with pembrolizumab, tremelimumab, and mFOLFOX6 in patients with MSS mCRC. The study results showed that the combination treatment resulted in a 3-month PFS rate of 90.7%, an overall response rate (ORR) of 64.5%, a median PFS (mPFS) of 8.2 months, and overall survival (OS) has not been reached yet.
NIVACOR (NCT04072198) (184) is a single-arm, open-label, multicenter phase II study with a safety assessment phase. Eligible patients with KRAS/BRAF-mutated metastatic CRC can participate and receive first-line treatment. Patients will receive FOLFOXIRI/Bevacizumab in combination with Nivolumab as induction therapy every two weeks, followed by maintenance therapy. Preliminary safety results indicate that this combination regimen is generally well-tolerated with acceptable toxicities. There is a high expectation for positive outcomes.
A study (185) evaluated the efficacy of PD-1/PD-L1 inhibitors in combination with the OXA-fluorouracil-leucovorin (mFOLFOX6) regimen in 30 patients with unresectable mCRC. The results showed a disease stabilization rate of 100% at 8 weeks and an overall response rate of 53% at 24 weeks. OXA and 5-FU led to increased ICD and antigen presentation. The study emphasized the potential benefits of combining chemotherapy with ICIs, as the combination of mFOLFOX6 and anti-PD-1 therapy was within an acceptable toxicity profile. The results showed that ICIs should be given concurrently or early after FOLFOX treatment and demonstrated clinical efficacy in pMMR CRC patients, showing promising results in patients with unresectable CRC.
Chemoradiotherapy also plays an important role in enhancing tumor response to immunotherapy. Current research indicates that radiation therapy can increase the expression of antigens on tumor cells, enhance tumor cell immunogenicity, and promote immune cell infiltration (186, 187). Therefore, combining immunotherapy with radiotherapy may lead to better therapeutic outcomes. For example, Lin et al. (188) used short-course therapy combining radiotherapy, sequential immunotherapy, and chemotherapy to treat CRC patients, which showed a pCR rate of up to 48%. In addition, the ongoing TORCH trial (189) is using toripalimab in combination with chemoradiotherapy or CapeOX in MSS CRC patients, with a proportion of 81.3% achieving cCR or pCR. These results suggest that combining immunotherapy with chemoradiotherapy may be an effective option for the treatment of CRC patients.
Most immunogenic chemotherapy agents have been shown to evoke immune stimulation not only by increasing the immunogenicity of cancer cells, but also by activating effector T cells and suppressing immune suppressor cells. These results suggest that the combination of chemotherapy and ICIs can have a synergistic anticancer effect and indicate that chemotherapy in combination with immunotherapy may be suitable for tumors that respond poorly to ICIs monotherapy. There are also ongoing prospective studies whose safety has been proven feasible, and the results of which are highly anticipated.
6 Preclinical and clinical studies of inducing chemotherapy combined with immunotherapy
Considering the immune-activating effects of chemotherapy drugs, the combination of chemotherapy and ICIs is an appropriate partner to achieve rapid and long-term cancer control. Based on these findings, we propose the concept of inducing chemotherapy, which involves using immunogenic chemotherapy drugs to change the timing before immunotherapy, converting “cold” tumors into “hot” metastases to initiate or restore anti-tumor immune responses, thereby enhancing the efficacy of ICIs (190). Some preclinical studies are being conducted in targeted preclinical models of CRC.
Song et al. (155) investigated the efficacy of OXA and anti-PD-L1 drugs in a microsyngeneic transplantation mouse model based on the CT26 cell line and found that the combination therapy of OXA and anti-PD-L1 drugs significantly slowed tumor growth compared to the use of OXA alone (191).
Dosset et al. conducted an interesting preclinical study using a microsyngeneic mouse model of two MSI-H CRCs (CT26 and MC38) and observed that adding adjuvant ICIs after FOLFOX could induce complete and durable tumor responses, whereas FOLFOX or ICIs alone were ineffective (192). Therefore, adding ICIs enables CD8+ T cells recruited by FOLFOX to induce effective anti-tumor immune responses (140). This is the first description of the PD-1/PD-L1 pathway as part of FOLFOX chemotherapy-induced adaptive immune resistance, indicating that chemotherapy can enhance the efficacy of ICIs (192). This correlation can attract a population of effective T cells which creating a favorable environment for immunotherapy to work. It has been shown to be associated with improved patient survival, especially after the emergence of immunotherapy (193).
The successful outcomes achieved in these preclinical trials provide strong evidence for the future implementation of clinical trials involving inducing chemotherapy (Table 2). Studies have shown that inducing chemotherapy has become one of the standard treatment options for certain tumors. For example, the combination of pembrolizumab with platinum and 5-FU was recently approved for metastatic and recurrent head and neck cancer based on its OS benefit (197).
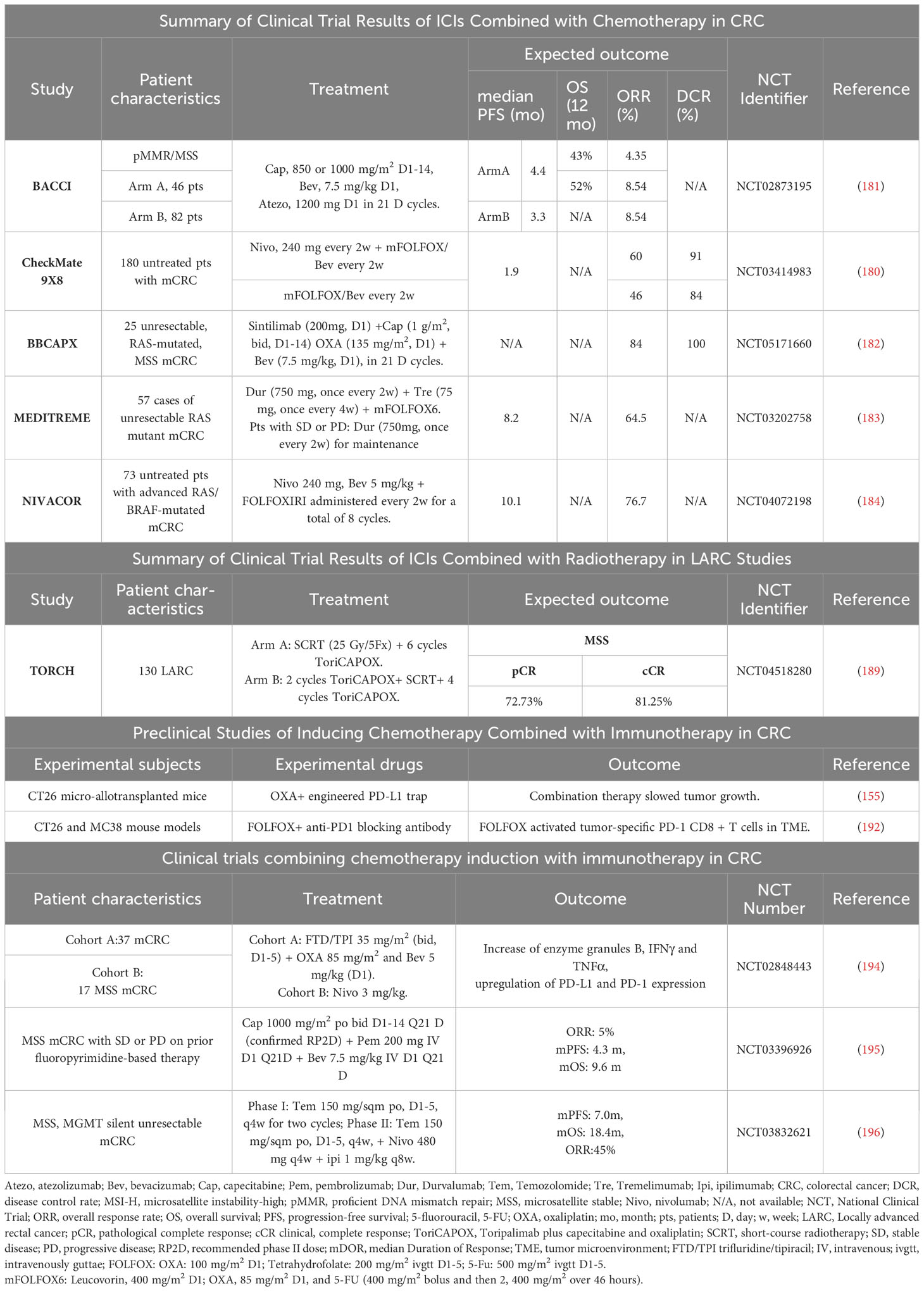
Table 2 Summary of clinical trials and preclinical studies of chemotherapy combined with immunotherapy.
Ma et al. (198) presented the results of a phase III multicenter randomized controlled clinical trial on sequential treatment of locally advanced nasopharyngeal carcinoma with PD-1 inhibitor sintilimab and concurrent chemoradiotherapy after inducing chemotherapy at the 2023 American Society of Clinical Oncology (ASCO) Annual Meeting. Patients were randomly assigned to two groups, one receiving standard GP inducing chemotherapy and concurrent chemoradiotherapy, and the other adding sintilimab to the standard treatment. The primary endpoint was event-free survival (EFS). From December 2018 to March 2020, a total of 425 patients were recruited, and after a median follow-up of 42 months, sintilimab increased the 3-year EFS rate from 76% to 86%, a 10% improvement, and reduced the risk of relapse, metastasis, and death by 41%. The risks of local-regional recurrence and distant metastasis were reduced by 48% and 43%, respectively. This trial is the first to achieve a positive EFS result in all locally advanced head and neck cancers, demonstrating the feasibility of inducing chemotherapy as a promising strategy to optimize anti-tumor treatment. In addition, concurrent chemoradiotherapy after inducing chemotherapy is the standard treatment for locally advanced nasopharyngeal carcinoma. Zhang et al. (199) conducted a multicenter randomized trial, assigning patients to receive concurrent chemoradiotherapy or GP inducing chemotherapy followed by concurrent chemoradiotherapy. The median follow-up time was 69.8 months, and the 5-year OS rate in the inducing chemotherapy group was significantly higher than that in the concurrent chemoradiotherapy group (87.9% vs. 78.8%), with equivalent risks of late toxicities (≥grade 3) (11.3% vs. 11.4%). This study suggests that inducing chemotherapy before concurrent chemoradiotherapy can significantly improve the OS of patients with locally advanced nasopharyngeal carcinoma without increasing the risk of late toxicities.
Based on the effective results obtained from preclinical trials, a series of clinical trials combining chemotherapy induction with immunotherapy have also been conducted in CRC, and significant progress has been achieved.
According to studies, TAM depletion induced by trifluridine/tipiracil (FTD/TPI), OXA, or combination therapy, especially TAM2, results in changes in the TAM1/TAM2 ratio, as well as enhanced infiltration and activation of cytotoxic CD8+ T cells, enhanced production of granzyme B, IFNγ, and TNFα in CD8+ T cells within the tumor, and upregulation of PD-L1 and PD-1 expression (Figure 5) (194). The combination use of FTD/TPI and OXA also induces ICD in vivo, providing a basis for using these drugs to eliminate immune-suppressive cells and improve checkpoint efficacy in patients with metastatic MSS CRC. The combination of FTP/TPI and OXA has been shown to be safe and effective in a phase I human clinical trial (200).
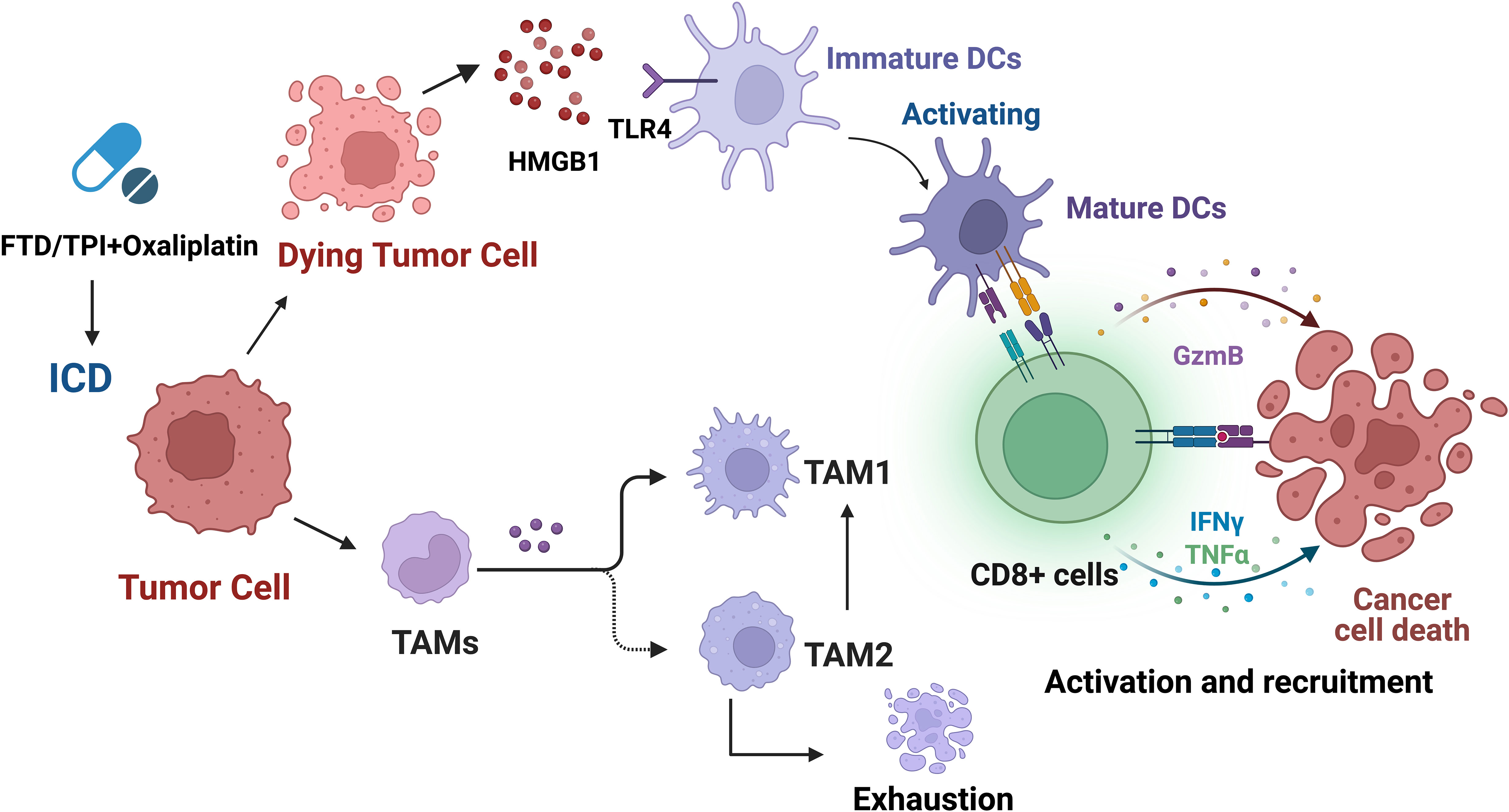
Figure 5 Trifluridine/tipiracil (FTD/TPI), oxaliplatin, or their combination not only induces ICD but also leads to enhanced infiltration and activation of cytotoxic CD8+ T cells, and increases the production of granzyme B, IFNγ, and TNFα in CD8+ T cells within the tumor. In addition, it leads to TAM depletion, especially TAM2, resulting in changes in the TAM1/TAM2 ratio, and upregulation of PD-L1 and PD-1 expression.
A phase II clinical trial (195) is currently ongoing to evaluate the safety, tolerability, and initial efficacy of pembrolizumab in combination with capecitabine and bevacizumab for the treatment of MSS mCRC patients. Bevacizumab, capecitabine, and pembrolizumab are used for treatment in the trial. The study results showed that the ORR among 40 evaluable patients was 5%, with a mPFS of 4.3 months and a mOS of 9.6 months. It is worth noting that MSS mCRC is rarely responsive to monotherapy with pembrolizumab, but capecitabine and bevacizumab may promote immune stimulation. These results suggest that the combination of pembrolizumab with capecitabine and bevacizumab may have some efficacy for MSS mCRC patients. However, it is important to note that the size of this trial is smaller, and further research and large-scale Phase III trials are needed to confirm the effectiveness and safety of this treatment regimen.
The MAYA II phase clinical trial (NCT03832621) (196) studied the combination of Nivolumab, Ipilimumab, and Temozolomide (TMZ) for the treatment of MSS, MGMT silenced unresectable mCRC patients who have not progressed, regardless of RAS mutational status. In the pre-selected 716 patients, 33 patients (24%) achieved disease control, which represents the final study population. The mPFS was 7.0 months, the mOS was 18.4 months, and the ORR was 45%. A series of temozolomide initiation followed by low-dose ipilimumab and nivolumab combination may induce durable clinical benefits in MSS and MGMT silenced mCRC. The initiation of treatment with tremelimumab provides the basic principle for immune sensitization induced by hypermutation in pMMR/MSS (MGMT-silenced) mCRC.
A randomized phase II trial (201) evaluated the safety of immunotherapy in combination with SOC in untreated MSS mCRC patients. Patients were randomized to receive SOC alone or SOC plus immunotherapy, which included mFOLFOX6 + Bevacizumab with or without AdCEA vaccine and Avelumab. In this small, randomized trial, the addition of immunotherapy did not significantly improve mPFS or overall response rate (ORR) compared to SOC alone. However, the SOC + immunotherapy regimen yielded biological activity in the form of substantial increases in multifunctional CD4+ and CD8+ T cells specific for the cascade antigens MUC1 and brachyury. Among them, the MUC1 and Brachyury pathways play important roles in cancer development and immune evasion and have become potential targets for tumor immunotherapy (202, 203).
A study (204) aimed to evaluate the safety, activity, and biomarker patterns of FOLFOX treatment with atezolizumab (anti-PD-L1) and bevacizumab (anti-VEGF-A) in patients with MSS mCRC. As of September 1, 2015, 52% of patients showed RECIST responses, with a mPFS time of 14.1 months and a median response duration of 11.4 months. No unexpected toxicities were observed. Wallin et al. found an increase in the expression of CD8+ T cells and PD-L1 in tumors after FOLFOX treatment alone and after combined FOLFOX, atezolizumab, and bevacizumab treatment. In some patients’ tumors, there was also an increase in cytotoxic T-cell markers (such as IFN-γ, GZMB, EOMES). Patients with increased tumor-infiltrating CD8+ T cells, which were consistent with increases in cytotoxic T-cell markers and PD-L1 expression, showed sustained responses or long-term disease control. These data further confirm that the combination of FOLFOX, atezolizumab, and bevacizumab may promote immune-related activities in CRC, thereby enhancing efficacy.
Although inducing chemotherapy combined with immunotherapy has achieved significant results, studies have shown that some chemotherapy drugs exhibit different immunogenic effects depending on their regimen, timing, dose, or administration sequence, even when used in combination with ICIs. One study investigated the impact of drug administration sequence and found that CTX given one day before anti-CTLA-4 therapy resulted in immune-mediated anti-tumor responses. However, when the sequence was reversed, CD8+ T cells underwent massive apoptosis, and the anti-tumor effect of anti-CTLA-4 was weakened (205). Another study tested three different regimens in NSCLC patients: a phase II study evaluating chemotherapy given before ipilimumab, a concurrent regimen, and a control group receiving placebo and chemotherapy (206). The primary endpoint of improved PFS was only achieved in the sequential regimen. A study investigated the effect of various types of chemotherapy on the treatment of metastatic triple-negative breast cancer (TNBC) and found that 2 weeks of low-dose chemotherapy during the induction period was more effective than nivolumab monotherapy (207). In addition, compared to the no-induction period, the number and clonality of T cells in the tumor were higher after chemotherapy-induced treatment (208).
Excitingly, inducing chemotherapy has become one of the standard treatment options for certain tumors, and a series of clinical trials on inducing chemotherapy have been conducted in CRC, achieving promising results. We propose the exploration of personalized treatment plans, gradually reducing the chemotherapy regimen and combining it with immunotherapy after inducing chemotherapy, until only oral chemotherapy is maintained, and eventually achieving maintenance therapy with a single immunotherapy. We look forward to further confirmation and application in the future.
7 Challenges and future
In the field of CRC treatment, the combination of inducing chemotherapy and immunotherapy has emerged as a promising therapeutic strategy. However, there are still challenges and directions that need to be addressed in the future.
The TIME in CRC typically exhibits immunosuppressive features that limit the activity of immune cells. Future research can further explore the underlying mechanisms by which chemotherapy-induced immunotherapy overcomes this immune suppression, such as enhancing the activity of immune cells, modulating the polarization state of tumor-associated macrophages, disrupting tumor vasculature, and so on (87). To improve the success rate of chemotherapy-induced immunotherapy, it is crucial to explore innovative treatment targets/strategies and identify patients who respond better to specific treatment regimens.
scRNA-seq technology provides us with an opportunity to gain in-depth understanding of tumor and immune cell heterogeneity (209, 210). This technology can be used to reveal the roles of different cell subpopulations in chemotherapy-induced immunotherapy, thereby helping optimize treatment strategies and select the most suitable patients (211, 212). Combination therapies of chemotherapy and immunotherapy may yield better treatment outcomes compared to monotherapies. However, determining the optimal combination strategies and dosages remains challenging. Future research should focus on identifying the optimal drug combinations, timing of administration, dosages, and the best concentration-time curves in representative preclinical models (112, 213). Single-cell data can be utilized in future studies to achieve more precise treatment optimization. Other therapies such as photodynamic therapy, photothermal therapy, radiation therapy, and magnetic fluid hyperthermia can further induce ICD in tumor cells, enhance the efficacy of anti-tumor immunotherapy, expand their potential applications, and maximize clinical benefits (214).
Due to the limited predictive ability of current biomarkers such as PD-L1 expression and tumor mutation burden in cancer precision medicine, alternative biomarkers are still being explored (215, 216). A recent study has shown that advanced NSCLC patients with high PD-L1 expression and high immune infiltration can actually respond to PD-1 therapy plus chemotherapy in the first-line setting. For patients lacking PD-L1 expression or immune infiltration, chemotherapy may be a better treatment choice (217). This suggests that in the future, it is also significant to further explore alternative biomarkers in CRC, for guiding precision medicine in the clinical practice of CRC treatment.
Furthermore, due to significant biological differences among CRC patients, personalized immunotherapy approaches become crucial. scRNA-seq may reveal potential mechanisms regulating immune cell exhaustion and identify advanced biomarkers, thereby facilitating the design of novel personalized immunotherapy strategies (11, 218–220). By utilizing scRNA-seq to better understand individual variations, we can design optimal individualized chemotherapy-induced immunotherapy regimens for specific patients. These efforts are expected to provide more effective treatment choices and personalized treatment plans for CRC patients.
8 Conclusion
Immunotherapy has made significant progress in CRC, revolutionizing treatment outcomes. The TIME is closely related to tumor immunotherapy, which is a key obstacle to anti-tumor immunity and may limit the clinical benefits of immunotherapy. Recent studies have shown that some chemotherapy drugs can promote immune activation and enhance the efficacy of immunotherapy. By inducing ICD and exposing new antigens, it can activate CD8+ T cells and enhance the immune response to cancer. A large body of research has shown that most chemotherapy drugs exert immunostimulatory effects by inhibiting immune-suppressive cells or activating effector cells, or by increasing immunogenicity and T cells infiltration. inducing chemotherapy combined with immunotherapy has become a standard part of treatment in some tumors, and clinical trials have demonstrated its feasibility and safety in CRC. These combination therapies typically transform “cold” tumors that are insensitive to immune response into “hot” tumors. We propose a personalized exploration of inducing chemotherapy, gradually reducing chemotherapy regimens after systemic chemotherapy induction, combining with immunotherapy until only oral chemotherapy is maintained, and eventually transitioning to immune monotherapy maintenance treatment. In summary, chemotherapy-induced immunotherapy has enormous potential in the field of CRC. Future research will focus on overcoming the immune-suppressive microenvironment, applying scRNA-seq technology, achieving personalized treatment, researching predictive biomarkers, and optimizing combination therapy, among other challenges, to benefit more cancer patients in the near future.
Author contributions
KJ and HL contributed to the conception and design of the study. SY drafted the initial manuscript and prepared the figures. YH and MY wrote sections of the manuscript. All authors participated in revising the manuscript, read, and approved the final version for submission.
Funding
This work was supported by National Natural Science Foundation of China [grant no. 82104445 to HL], Zhejiang Provincial Science and Technology Projects (grant no. LGF22H160046 to HL), Jinhua Municipal Science and Technology Projects (grants no. 2021-3-040 to KJ, and 2021-3-046 to HL).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin (2023) 73:17–48. doi: 10.3322/caac.21763
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: A review. Jama (2021) 325:669–85. doi: 10.1001/jama.2021.0106
4. Kalyan A, Kircher S, Shah H, Mulcahy M, Benson A. Updates on immunotherapy for colorectal cancer. J Gastrointest Oncol (2018) 9:160–9. doi: 10.21037/jgo.2018.01.17
5. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol (2019) 16:361–75. doi: 10.1038/s41575-019-0126-x
6. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
7. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi: 10.1038/nm1622
8. Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology (2014) 3:e955691. doi: 10.4161/21624011.2014.955691
9. Li Y, Liu X, Zhang X, Pan W, Li N, Tang B. Immune cycle-based strategies for cancer immunotherapy. Adv Funct Mater (2021) 31:2107540. doi: 10.1002/adfm.202107540
10. Grimaldi A, Cammarata I, Martire C, Focaccetti C, Piconese S, Buccilli M, et al. Combination of chemotherapy and PD-1 blockade induces T cell responses to tumor non-mutated neoantigens. Commun Biol (2020) 3:85. doi: 10.1038/s42003-020-0811-x
11. Tieng FYF, Lee LH, Ab Mutalib NS. Deciphering colorectal cancer immune microenvironment transcriptional landscape on single cell resolution - A role for immunotherapy. Front Immunol (2022) 13:959705. doi: 10.3389/fimmu.2022.959705
12. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
13. Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol (2018) 19:1480–92. doi: 10.1016/s1470-2045(18)30700-9
14. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J Clin Oncol (2019) 37:537–46. doi: 10.1200/jco.18.00149
15. Motzer RJ, Rini BI, McDermott DF, Arén Frontera O, Hammers HJ, Carducci MA, et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol (2019) 20:1370–85. doi: 10.1016/s1470-2045(19)30413-9
16. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
17. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol (2018) 36:773–9. doi: 10.1200/jco.2017.76.9901
18. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol (2017) 18:1182–91. doi: 10.1016/s1470-2045(17)30422-9
19. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res (2019) 25:3753–8. doi: 10.1158/1078-0432.Ccr-18-4070
20. André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability–high advanced colorectal cancer. N Engl J Med (2020) 383:2207–18. doi: 10.1056/NEJMoa2017699
21. Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother (2012) 61:1019–31. doi: 10.1007/s00262-011-1172-6
22. Hegde PS, Karanikas V, Evers S. The where, the whenand the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res (2016) 22:1865–74. doi: 10.1158/1078-0432.Ccr-15-1507
23. Higgs B, Robbins PB, Blake-Haskins JA, Morehouse C, Brohawn PZ, Rebelatto M, et al. 15LBA High tumoral IFNγ mRNA, PD-L1 proteinand combined IFNγ mRNA/PD-L1 protein expression associates with response to durvalumab (anti-PD-L1) monotherapy in NSCLC patients. Eur J Cancer (2015) 51:S717. doi: 10.1016/S0959-8049(16)31937-2
24. Le DT, Hubbard-Lucey VM, Morse MA, Heery CR, Dwyer A, Marsilje TH, et al. A blueprint to advance colorectal cancer immunotherapies. Cancer Immunol Res (2017) 5:942–9. doi: 10.1158/2326-6066.Cir-17-0375
25. Ghiringhelli F, Fumet JD. Is there a place for immunotherapy for metastatic microsatellite stable colorectal cancer? Front Immunol (2019) 10:1816. doi: 10.3389/fimmu.2019.01816
26. Kim CW, Chon HJ, Kim C. Combination immunotherapies to overcome intrinsic resistance to checkpoint blockade in microsatellite stable colorectal cancer. Cancers (Basel) (2021) 13. doi: 10.3390/cancers13194906
27. Sahin IH, Ciombor KK, Diaz LA, Yu J, Kim R. Immunotherapy for microsatellite stable colorectal cancers: challenges and novel therapeutic avenues. In: American Society of Clinical Oncology Educational Book (2022) 242–53. doi: 10.1200/edbk_349811
28. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res (2012) 18:1386–94. doi: 10.1158/1078-0432.Ccr-11-2479
29. Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, et al. BRAFV600E co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol Res (2015) 3:602–9. doi: 10.1158/2326-6066.Cir-15-0030
30. Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res (2013) 19:393–403. doi: 10.1158/1078-0432.Ccr-12-1626
31. Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med (2015) 7:279ra41. doi: 10.1126/scitranslmed.aaa4691
32. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res (2013) 19:1225–31. doi: 10.1158/1078-0432.Ccr-12-1630
33. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDOand T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med (2013) 5:200ra116. doi: 10.1126/scitranslmed.3006504
34. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov (2015) 5:43–51. doi: 10.1158/2159-8290.Cd-14-0863
35. Li X, Song W, Shao C, Shi Y, Han W. Emerging predictors of the response to the blockade of immune checkpoints in cancer therapy. Cell Mol Immunol (2019) 16:28–39. doi: 10.1038/s41423-018-0086-z
36. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science (2018) 362. doi: 10.1126/science.aar3593
37. Xiong Y, Wang Y, Tiruthani K. Tumor immune microenvironment and nano-immunotherapeutics in colorectal cancer. Nanomedicine (2019) 21:102034. doi: 10.1016/j.nano.2019.102034
38. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9:34. doi: 10.1186/s13073-017-0424-2
39. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554:538–43. doi: 10.1038/nature25492
40. Gutting T, Burgermeister E, Härtel N, Ebert MP. Checkpoints and beyond - Immunotherapy in colorectal cancer. Semin Cancer Biol (2019) 55:78–89. doi: 10.1016/j.semcancer.2018.04.003
41. Makaremi S, Asadzadeh Z, Hemmat N, Baghbanzadeh A, Sgambato A, Ghorbaninezhad F, et al. Immune checkpoint inhibitors in colorectal cancer: challenges and future prospects. Biomedicines (2021) 9. doi: 10.3390/biomedicines9091075
42. Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat Commun (2018) 9:741. doi: 10.1038/s41467-017-02696-6
43. Lin RL, Zhao LJ. Mechanistic basis and clinical relevance of the role of transforming growth factor-β in cancer. Cancer Biol Med (2015) 12:385–93. doi: 10.7497/j.issn.2095-3941.2015.0015
44. Guo L, Zhang Y, Zhang L, Huang F, Li J, Wang S. MicroRNAs, TGF-β signalingand the inflammatory microenvironment in cancer. Tumour Biol (2016) 37:115–25. doi: 10.1007/s13277-015-4374-2
45. Ischenko I, D'Amico S, Rao M, Li J, Hayman MJ, Powers S, et al. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat Commun (2021) 12:1482. doi: 10.1038/s41467-021-21736-w
46. Klampfer L, Huang J, Corner G, Mariadason J, Arango D, Sasazuki T, et al. Oncogenic Ki-ras inhibits the expression of interferon-responsive genes through inhibition of STAT1 and STAT2 expression. J Biol Chem (2003) 278:46278–87. doi: 10.1074/jbc.M304721200
47. Glodde N, Hölzel M. RAS and PD-L1: A masters' Liaison in cancer immune evasion. Immunity (2017) 47:1007–9. doi: 10.1016/j.immuni.2017.12.001
48. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med (2015) 21:1350–6. doi: 10.1038/nm.3967
49. Owyang SY, Zhang M, Walkup GA, Chen GE, Grasberger H, El-Zaatari M, et al. The effect of CT26 tumor-derived TGF-β on the balance of tumor growth and immunity. Immunol Lett (2017) 191:47–54. doi: 10.1016/j.imlet.2017.08.024
50. Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet (2015) 47:320–9. doi: 10.1038/ng.3225
51. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PloS One (2018) 13:e0191358. doi: 10.1371/journal.pone.0191358
52. Huang XM, Zhang NR, Lin XT, Zhu CY, Zou YF, Wu XJ, et al. Antitumor immunity of low-dose cyclophosphamide: changes in T cells and cytokines TGF-beta and IL-10 in mice with colon-cancer liver metastasis. Gastroenterol Rep (Oxf) (2020) 8:56–65. doi: 10.1093/gastro/goz060
53. Xu Y, Wei Z, Feng M, Zhu D, Mei S, Wu Z, et al. Tumor-infiltrated activated B cells suppress liver metastasis of colorectal cancers. Cell Rep (2022) 40:111295. doi: 10.1016/j.celrep.2022.111295
54. Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res (2019) 25:3074–83. doi: 10.1158/1078-0432.Ccr-18-1942
55. Panarelli NC, Vaughn CP, Samowitz WS, Yantiss RK. Sporadic microsatellite instability-high colon cancers rarely display immunohistochemical evidence of Wnt signaling activation. Am J Surg Pathol (2015) 39:313–7. doi: 10.1097/pas.0000000000000380
56. Sun X, Liu S, Wang D, Zhang Y, Li W, Guo Y, et al. Colorectal cancer cells suppress CD4+ T cells immunity through canonical Wnt signaling. Oncotarget (2017) 8:15168–81. doi: 10.18632/oncotarget.14834
57. Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov (2018) 8:730–49. doi: 10.1158/2159-8290.Cd-17-1327
58. Pai SG, Carneiro BA, Mota JM, Costa R, Leite CA, Barroso-Sousa R, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol (2017) 10:101. doi: 10.1186/s13045-017-0471-6
59. Xue J, Yu X, Xue L, Ge X, Zhao W, Peng W. Intrinsic β-catenin signaling suppresses CD8(+) T-cell infiltration in colorectal cancer. BioMed Pharmacother (2019) 115:108921. doi: 10.1016/j.biopha.2019.108921
60. Feng M, Jin JQ, Xia L, Xiao T, Mei S, Wang X, et al. Pharmacological inhibition of β-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating T(reg) cells. Sci Adv (2019) 5:eaau5240. doi: 10.1126/sciadv.aau5240
61. Ganesh S, Shui X, Craig KP, Park J, Wang W, Brown BD, et al. RNAi-mediated β-catenin inhibition promotes T cell infiltration and antitumor activity in combination with immune checkpoint blockade. Mol Ther (2018) 26:2567–79. doi: 10.1016/j.ymthe.2018.09.005
62. Hafezi S, Rahmani M. Targeting BCL-2 in cancer: advances, challengesand perspectives. Cancers (Basel) (2021) 13. doi: 10.3390/cancers13061292
63. Bortolomeazzi M, Keddar MR, Montorsi L, Acha-Sagredo A, Benedetti L, Temelkovski D, et al. Immunogenomics of colorectal cancer response to checkpoint blockade: analysis of the KEYNOTE 177 trial and validation cohorts. Gastroenterology (2021) 161:1179–93. doi: 10.1053/j.gastro.2021.06.064
64. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunityand cancer immunotherapy. Cancer Cell (2018) 33:570–80. doi: 10.1016/j.ccell.2018.03.015
65. Shaked Y. The pro-tumorigenic host response to cancer therapies. Nat Rev Cancer (2019) 19:667–85. doi: 10.1038/s41568-019-0209-6
66. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
67. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
68. Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med (1994) 179:1317–30. doi: 10.1084/jem.179.4.1317
69. Rawson PM, Molette C, Videtta M, Altieri L, Franceschini D, Donato T, et al. Cross-presentation of caspase-cleaved apoptotic self antigens in HIV infection. Nat Med (2007) 13:1431–9. doi: 10.1038/nm1679
70. Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, et al. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology (2017) 6:e1386829. doi: 10.1080/2162402x.2017.1386829
71. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med (2005) 202:1691–701. doi: 10.1084/jem.20050915
72. Paroli M, Bellati F, Videtta M, Focaccetti C, Mancone C, Donato T, et al. Discovery of chemotherapy-associated ovarian cancer antigens by interrogating memory T cells. Int J Cancer (2014) 134:1823–34. doi: 10.1002/ijc.28515
73. Garg AD, Agostinis P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol Rev (2017) 280:126–48. doi: 10.1111/imr.12574
74. Wang HT, Lee HI, Guo JH, Chen SH, Liao ZK, Huang KW, et al. Calreticulin promotes tumor lymphocyte infiltration and enhances the antitumor effects of immunotherapy by up-regulating the endothelial expression of adhesion molecules. Int J Cancer (2012) 130:2892–902. doi: 10.1002/ijc.26339
75. Saenz R, Futalan D, Leutenez L, Eekhout F, Fecteau JF, Sundelius S, et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med (2014) 12:211. doi: 10.1186/1479-5876-12-211
76. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene (2010) 29:482–91. doi: 10.1038/onc.2009.356
77. Tel J, Hato SV, Torensma R, Buschow SI, Figdor CG, Lesterhuis WJ, et al. The chemotherapeutic drug oxaliplatin differentially affects blood DC function dependent on environmental cues. Cancer Immunol Immunother (2012) 61:1101–11. doi: 10.1007/s00262-011-1189-x
78. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol (2017) 17:97–111. doi: 10.1038/nri.2016.107
79. Chang CL, Hsu YT, Wu CC, Lai YZ, Wang C, Yang YC, et al. Dose-dense chemotherapy improves mechanisms of antitumor immune response. Cancer Res (2013) 73:119–27. doi: 10.1158/0008-5472.Can-12-2225
80. Lesterhuis WJ, Punt CJ, Hato SV, Eleveld-Trancikova D, Jansen BJ, Nierkens S, et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J Clin Invest (2011) 121:3100–8. doi: 10.1172/jci43656
81. Palombo F, Focaccetti C, Barnaba V. Therapeutic implications of immunogenic cell death in human cancer. Front Immunol (2014) 4:503. doi: 10.3389/fimmu.2013.00503
82. Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle (2009) 8:3723–8. doi: 10.4161/cc.8.22.10026
83. Kwon M, Jung H, Nam GH, Kim IS. The right Timing, right combination, right sequenceand right delivery for Cancer immunotherapy. J Control Release (2021) 331:321–34. doi: 10.1016/j.jconrel.2021.01.009
84. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer (2022) 21:28. doi: 10.1186/s12943-021-01489-2
85. Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PloS One (2012) 7:e32542. doi: 10.1371/journal.pone.0032542
86. Ross ME, Caligiuri MA. Cytokine-induced apoptosis of human natural killer cells identifies a novel mechanism to regulate the innate immune response. Blood (1997) 89:910–8. doi: 10.1182/blood.V89.3.910
87. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
88. Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer (2017) 17:286–301. doi: 10.1038/nrc.2017.17
89. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci (2012) 125:5591–6. doi: 10.1242/jcs.116392
90. Barcellos-Hoff MH, Lyden D, Wang TC. The evolution of the cancer niche during multistage carcinogenesis. Nat Rev Cancer (2013) 13:511–8. doi: 10.1038/nrc3536
91. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19:1423–37. doi: 10.1038/nm.3394
92. Mascaux C, Angelova M, Vasaturo A, Beane J, Hijazi K, Anthoine G, et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature (2019) 571:570–5. doi: 10.1038/s41586-019-1330-0
93. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
94. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature (2011) 480:480–9. doi: 10.1038/nature10673
95. Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangère V, Chevriaux A, et al. Accumulation of MDSC and th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res (2016) 76:5241–52. doi: 10.1158/0008-5472.Can-15-3164
96. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res (2010) 70:3052–61. doi: 10.1158/0008-5472.Can-09-3690
97. Duan Q, Zhang H, Zheng J, Zhang L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer (2020) 6:605–18. doi: 10.1016/j.trecan.2020.02.022
98. Hui Z, Ren Y, Zhang D, Chen Y, Yu W, Cao J, et al. PD-1 blockade potentiates neoadjuvant chemotherapy in NSCLC via increasing CD127(+) and KLRG1(+) CD8 T cells. NPJ Precis Oncol (2023) 7:48. doi: 10.1038/s41698-023-00384-x
99. Corrales L, McWhirter SM, Dubensky TW Jr., Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest (2016) 126:2404–11. doi: 10.1172/jci86892
100. Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol (2014) 35:88–93. doi: 10.1016/j.it.2013.10.010
101. Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst (2017) 109. doi: 10.1093/jnci/djw199
102. Kciuk M, Kołat D, Kałuzińska-Kołat Ż, Gawrysiak M, Drozda R, Celik I, et al. PD-1/PD-L1 and DNA damage response in cancer. Cells (2023) 12. doi: 10.3390/cells12040530
103. Garaude J, Blander JM. ICOStomizing immunotherapies with T(H)17. Sci Transl Med (2010) 2:55ps52. doi: 10.1126/scitranslmed.3001722
104. Lu Y, Zhao Q, Liao JY, Song E, Xia Q, Pan J, et al. Complement signals determine opposite effects of B cells in chemotherapy-induced immunity. Cell (2020) 180:1081–1097.e24. doi: 10.1016/j.cell.2020.02.015
105. Medler TR, Cotechini T, Coussens LM. Immune response to cancer therapy: mounting an effective antitumor response and mechanisms of resistance. Trends Cancer (2015) 1:66–75. doi: 10.1016/j.trecan.2015.07.008
106. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ (2014) 21:15–25. doi: 10.1038/cdd.2013.67
107. Machiels J-PH, Reilly RT, Emens LA, Ercolini AM, Lei RY, Weintraub D, et al. Cyclophosphamide, doxorubicinand paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice1. Cancer Res (2001) 61:3689–97.
108. Foukakis T, Lövrot J, Matikas A, Zerdes I, Lorent J, Tobin N, et al. Immune gene expression and response to chemotherapy in advanced breast cancer. Br J Cancer (2018) 118:480–8. doi: 10.1038/bjc.2017.446
109. Hodge JW, Garnett CT, Farsaci B, Palena C, Tsang K-Y, Ferrone S, et al. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer (2013) 133:624–36. doi: 10.1002/ijc.28070
110. Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov (2012) 11:215–33. doi: 10.1038/nrd3626
111. Tanaka H, Matsushima H, Mizumoto N, Takashima A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res (2009) 69:6978–86. doi: 10.1158/0008-5472.Can-09-1101
112. Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol (2019) 30:219–35. doi: 10.1093/annonc/mdy551
113. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med (2014) 20:1301–9. doi: 10.1038/nm.3708
114. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother (2007) 56:641–8. doi: 10.1007/s00262-006-0225-8
115. Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res (2011) 71:4809–20. doi: 10.1158/0008-5472.Can-11-0753
116. Jiménez-Sánchez A, Cybulska P, Mager KL, Koplev S, Cast O, Couturier DL, et al. Unraveling tumor-immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat Genet (2020) 52:582–93. doi: 10.1038/s41588-020-0630-5
117. Huang X, Cui S, Shu Y. Cisplatin selectively downregulated the frequency and immunoinhibitory function of myeloid-derived suppressor cells in a murine B16 melanoma model. Immunol Res (2016) 64:160–70. doi: 10.1007/s12026-015-8734-1
118. Jackaman C, Majewski D, Fox SA, Nowak AK, Nelson DJ. Chemotherapy broadens the range of tumor antigens seen by cytotoxic CD8(+) T cells in vivo. Cancer Immunol Immunother (2012) 61:2343–56. doi: 10.1007/s00262-012-1307-4
119. Nio Y, Hirahara N, Minari Y, Iguchi C, Yamasawa K, Toga T, et al. Induction of tumor-specific antitumor immunity after chemotherapy with cisplatin in mice bearing MOPC-104E plasmacytoma by modulation of MHC expression on tumor surface. Anticancer Res (2000) 20:3293–9.
120. Hu J, Kinn J, Zirakzadeh AA, Sherif A, Norstedt G, Wikström AC, et al. The effects of chemotherapeutic drugs on human monocyte-derived dendritic cell differentiation and antigen presentation. Clin Exp Immunol (2013) 172:490–9. doi: 10.1111/cei.12060
121. Ock CY, Kim S, Keam B, Kim S, Ahn YO, Chung EJ, et al. Changes in programmed death-ligand 1 expression during cisplatin treatment in patients with head and neck squamous cell carcinoma. Oncotarget (2017) 8:97920–7. doi: 10.18632/oncotarget.18542
122. Tran L, Allen CT, Xiao R, Moore E, Davis R, Park SJ, et al. Cisplatin alters antitumor immunity and synergizes with PD-1/PD-L1 inhibition in head and neck squamous cell carcinoma. Cancer Immunol Res (2017) 5:1141–51. doi: 10.1158/2326-6066.Cir-17-0235
123. Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials (2014) 35:814–24. doi: 10.1016/j.biomaterials.2013.10.003
124. Sevko A, Kremer V, Falk C, Umansky L, Shurin MR, Shurin GV, et al. Application of paclitaxel in low non-cytotoxic doses supports vaccination with melanoma antigens in normal mice. J Immunotoxicol (2012) 9:275–81. doi: 10.3109/1547691X.2012.655343
125. Pfannenstiel LW, Lam SSK, Emens LA, Jaffee EM, Armstrong TD. Paclitaxel enhances early dendritic cell maturation and function through TLR4 signaling in mice. Cell Immunol (2010) 263:79–87. doi: 10.1016/j.cellimm.2010.03.001
126. Yuan L, Wu L, Chen J, Wu Q, Hu S. Paclitaxel acts as an adjuvant to promote both Th1 and Th2 immune responses induced by ovalbumin in mice. Vaccine (2010) 28:4402–10. doi: 10.1016/j.vaccine.2010.04.046
127. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38:729–41. doi: 10.1016/j.immuni.2013.03.003
128. Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol (2012) 189:5147–54. doi: 10.4049/jimmunol.1200274
129. Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol (2015) 6:187. doi: 10.3389/fimmu.2015.00187
130. Bansal S, Bajaj P, Pandey S, Tandon V. Topoisomerases: resistance versus sensitivity, how far we can go? Med Res Rev (2017) 37:404–38. doi: 10.1002/med.21417
131. Frey B, Stache C, Rubner Y, Werthmöller N, Schulz K, Sieber R, et al. Combined treatment of human colorectal tumor cell lines with chemotherapeutic agents and ionizing irradiation can in vitro induce tumor cell death forms with immunogenic potential. J Immunotoxicol (2012) 9:301–13. doi: 10.3109/1547691x.2012.693547
132. Roselli M, Formica V, Cereda V, Jochems C, Richards J, Grenga I, et al. The association of clinical outcome and peripheral T-cell subsets in metastatic colorectal cancer patients receiving first-line FOLFIRI plus bevacizumab therapy. Oncoimmunology (2016) 5:e1188243. doi: 10.1080/2162402x.2016.1188243
133. Alagkiozidis I, Facciabene A, Tsiatas M, Carpenito C, Benencia F, Adams S, et al. Time-dependent cytotoxic drugs selectively cooperate with IL-18 for cancer chemo-immunotherapy. J Transl Med (2011) 9:77. doi: 10.1186/1479-5876-9-77
134. Lin N, Song X, Chen B, Ye H, Wang Y, Cheng X, et al. Leptin is upregulated in epididymitis and promotes apoptosis and IL-1β production in epididymal epithelial cells by activating the NLRP3 inflammasome. Int Immunopharmacol (2020) 88:106901. doi: 10.1016/j.intimp.2020.106901
135. Wang W, Wu L, Zhang J, Wu H, Han E, Guo Q. Chemoimmunotherapy by combining oxaliplatin with immune checkpoint blockades reduced tumor burden in colorectal cancer animal model. Biochem Biophys Res Commun (2017) 487:1–7. doi: 10.1016/j.bbrc.2016.12.180
136. Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer (2010) 102:115–23. doi: 10.1038/sj.bjc.6605465
137. Nowak AK, Robinson BW, Lake RA. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemo-immunotherapy. Cancer Res (2002) 62:2353–8.
138. Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res (2011) 71:5101–10. doi: 10.1158/0008-5472.Can-10-2670
139. Homma Y, Taniguchi K, Nakazawa M, Matsuyama R, Mori R, Takeda K, et al. Changes in the immune cell population and cell proliferation in peripheral blood after gemcitabine-based chemotherapy for pancreatic cancer. Clin Transl Oncol (2014) 16:330–5. doi: 10.1007/s12094-013-1079-0
140. Bruchard M, Mignot G, Derangère V, Chalmin F, Chevriaux A, Végran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med (2013) 19:57–64. doi: 10.1038/nm.2999
141. Galetto A, Buttiglieri S, Forno S, Moro F, Mussa A, Matera L. Drug- and cell-mediated antitumor cytotoxicities modulate cross-presentation of tumor antigens by myeloid dendritic cells. Anticancer Drugs (2003) 14:833–43. doi: 10.1097/00001813-200311000-00010
142. Ugurel S, Paschen A, Becker JC. Dacarbazine in melanoma: from a chemotherapeutic drug to an immunomodulating agent. J Invest Dermatol (2013) 133:289–92. doi: 10.1038/jid.2012.341
143. Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol (2004) 34:336–44. doi: 10.1002/eji.200324181
144. Webb ER, Moreno-Vincente J, Easton A, Lanati S, Taylor M, James S, et al. Cyclophosphamide depletes tumor infiltrating T regulatory cells and combined with anti-PD-1 therapy improves survival in murine neuroblastoma. iScience (2022) 25:104995. doi: 10.1016/j.isci.2022.104995
145. Kaneno R, Shurin GV, Tourkova IL, Shurin MR. Chemomodulation of human dendritic cell function by antineoplastic agents in low noncytotoxic concentrations. J Transl Med (2009) 7:58. doi: 10.1186/1479-5876-7-58
146. Terenzi A, Pirker C, Keppler BK, Berger W. Anticancer metal drugs and immunogenic cell death. J Inorg Biochem (2016) 165:71–9. doi: 10.1016/j.jinorgbio.2016.06.021
147. Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, et al. Trial Watch: Immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology (2015) 4:e1008866. doi: 10.1080/2162402x.2015.1008866
148. Rufo N, Garg AD, Agostinis P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer (2017) 3:643–58. doi: 10.1016/j.trecan.2017.07.002
149. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12:860–75. doi: 10.1038/nrc3380
150. Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res (2011) 71:4821–33. doi: 10.1158/0008-5472.Can-11-0950
151. Wang Z, Chen J, Hu J, Zhang H, Xu F, He W, et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J Clin Invest (2019) 129:4850–62. doi: 10.1172/jci127471
152. Lu J, Liu X, Liao YP, Wang X, Ahmed A, Jiang W, et al. Breast cancer chemo-immunotherapy through liposomal delivery of an immunogenic cell death stimulus plus interference in the IDO-1 pathway. ACS Nano (2018) 12:11041–61. doi: 10.1021/acsnano.8b05189
153. Mei L, Liu Y, Rao J, Tang X, Li M, Zhang Z, et al. Enhanced tumor retention effect by click chemistry for improved cancer immunochemotherapy. ACS Appl Mater Interfaces (2018) 10:17582–93. doi: 10.1021/acsami.8b02954
154. Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med (2019) 25:920–8. doi: 10.1038/s41591-019-0432-4
155. Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun (2018) 9:2237. doi: 10.1038/s41467-018-04605-x
156. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med (2018) 379:2040–51. doi: 10.1056/NEJMoa1810865
157. Maker AV, Ito H, Mo Q, Weisenberg E, Qin LX, Turcotte S, et al. Genetic evidence that intratumoral T-cell proliferation and activation are associated with recurrence and survival in patients with resected colorectal liver metastases. Cancer Immunol Res (2015) 3:380–8. doi: 10.1158/2326-6066.Cir-14-0212
158. Michels T, Shurin GV, Naiditch H, Sevko A, Umansky V, Shurin MR. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J Immunotoxicol (2012) 9:292–300. doi: 10.3109/1547691x.2011.642418
159. Garzetti GG, Ciavattini A, Muzzioli M, Romanini C. Cisplatin-based polychemotherapy reduces the natural cytotoxicity of peripheral blood mononuclear cells in patients with advanced ovarian carcinoma and their in vitro responsiveness to interleukin-12 incubation. Cancer (1999) 85:2226–31. doi: 10.1002/(sici)1097-0142(19990515)85:10<2226::aid-cncr18>3.0.co;2-x
160. Liu N, Zheng Y, Zhu Y, Xiong S, Chu Y. Selective Impairment of CD4+CD25+Foxp3+Regulatory T cells by paclitaxel is explained by Bcl-2/Bax mediated apoptosis. Int Immunopharmacol (2011) 11:212–9. doi: 10.1016/j.intimp.2010.11.021
161. Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, et al. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin Immunol (2008) 129:219–29. doi: 10.1016/j.clim.2008.07.013
162. Vicari AP, Luu R, Zhang N, Patel S, Makinen SR, Hanson DC, et al. Paclitaxel reduces regulatory T cell numbers and inhibitory function and enhances the anti-tumor effects of the TLR9 agonist PF-3512676 in the mouse. Cancer Immunol Immunother (2009) 58:615–28. doi: 10.1007/s00262-008-0586-2
163. Zhu Y, Liu N, Xiong SD, Zheng YJ, Chu YW. CD4+Foxp3+ Regulatory T-cell impairment by paclitaxel is independent of toll-like receptor 4. Scandinavian J Immunol (2011) 73:301–8. doi: 10.1111/j.1365-3083.2011.02514.x
164. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res (2010) 16:4583–94. doi: 10.1158/1078-0432.Ccr-10-0733
165. Li JY, Duan XF, Wang LP, Xu YJ, Huang L, Zhang TF, et al. Selective depletion of regulatory T cell subsets by docetaxel treatment in patients with nonsmall cell lung cancer. J Immunol Res (2014) 2014:286170. doi: 10.1155/2014/286170
166. Roselli M, Cereda V, di Bari MG, Formica V, Spila A, Jochems C, et al. Effects of conventional therapeutic interventions on the number and function of regulatory T cells. Oncoimmunology (2013) 2:e27025. doi: 10.4161/onci.27025
167. Tong AW, Seamour B, Lawson JM, Ordonez G, Vukelja S, Hyman W, et al. Cellular immune profile of patients with advanced cancer before and after taxane treatment. Am J Clin Oncol (2000) 23:463–72. doi: 10.1097/00000421-200010000-00007
168. Wanderley CW, Colón DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res (2018) 78:5891–900. doi: 10.1158/0008-5472.Can-17-3480
169. Cullis J, Siolas D, Avanzi A, Barui S, Maitra A, Bar-Sagi D. Macropinocytosis of nab-paclitaxel drives macrophage activation in pancreatic cancer. Cancer Immunol Res (2017) 5:182–90. doi: 10.1158/2326-6066.Cir-16-0125
170. Pusztai L, Mendoza TR, Reuben JM, Martinez MM, Willey JS, Lara J, et al. Changes in plasma levels of inflammatory cytokines in response to paclitaxel chemotherapy. Cytokine (2004) 25:94–102. doi: 10.1016/j.cyto.2003.10.004
171. Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol (2019) 21:1113–26. doi: 10.1038/s41556-019-0373-7
172. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med (2018) 379:2108–21. doi: 10.1056/NEJMoa1809615
173. Loyher PL, Rochefort J, Baudesson de Chanville C, Hamon P, Lescaille G, Bertolus C, et al. CCR2 influences T regulatory cell migration to tumors and serves as a biomarker of cyclophosphamide sensitivity. Cancer Res (2016) 76:6483–94. doi: 10.1158/0008-5472.Can-16-0984
174. Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart K, et al. Low-dose cyclophosphamide induces antitumor T-cell responses, which associate with survival in metastatic colorectal cancer. Clin Cancer Res (2017) 23:6771–80. doi: 10.1158/1078-0432.Ccr-17-0895
175. Hibino S, Chikuma S, Kondo T, Ito M, Nakatsukasa H, Omata-Mise S, et al. Inhibition of nr4a receptors enhances antitumor immunity by breaking treg-mediated immune tolerance. Cancer Res (2018) 78:3027–40. doi: 10.1158/0008-5472.Can-17-3102
176. Grosflam J, Weinblatt ME. Methotrexate: mechanism of action, pharmacokinetics, clinical indicationsand toxicity. Curr Opin Rheumatol (1991) 3:363–8. doi: 10.1097/00002281-199106000-00006
177. Peng J, Hamanishi J, Matsumura N, Abiko K, Murat K, Baba T, et al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-κB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res (2015) 75:5034–45. doi: 10.1158/0008-5472.Can-14-3098
178. Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med (2018) 379:2220–9. doi: 10.1056/NEJMoa1809064
179. Kang C, Syed YY. Atezolizumab (in combination with nab-paclitaxel): A review in advanced triple-negative breast cancer. Drugs (2020) 80:601–7. doi: 10.1007/s40265-020-01295-y
180. Tabernero J, Yoshino T, Cohn AL, Kochenderfer MD, Holdridge RC, Couture F, et al. Open-label phase II/III study of nivolumab plus standard of care versus standard of care for first-line treatment of metastatic colorectal cancer: Checkmate-9X8. J Clin Oncol (2019) 37:TPS718–8. doi: 10.1200/JCO.2019.37.4_suppl.TPS718
181. Mettu NB, Twohy E, Ou FS, Halfdanarson TR, Lenz HJ, Breakstone R, et al. 533PD - BACCI: A phase II randomized, double-blind, multicenter, placebo-controlled study of capecitabine (C) bevacizumab (B) plus atezolizumab (A) or placebo (P) in refractory metastatic colorectal cancer (mCRC): An ACCRU network study. Ann Oncol (2019) 30:v203. doi: 10.1093/annonc/mdz246.011
182. Fang X, Zhong C, Zhu N, Weng S, Hu H, Wang J, et al. A phase 2 trial of sintilimab (IBI 308) in combination with CAPEOX and bevacizumab (BBCAPX) as first-line treatment in patients with RAS-mutant, microsatellite stable, unresectable metastatic colorectal cancer. J Clin Oncol (2022) 40:3563–3. doi: 10.1200/JCO.2022.40.16_suppl.3563
183. Fumet JD, Chibaudel B, Bennouna J, Borg C, Martin-Babau J, Cohen R, et al. 433P Durvalumab and tremelimumab in combination with FOLFOX in patients with previously untreated RAS-mutated metastatic colorectal cancer: First results of efficacy at one year for phase II MEDITREME trial. Ann Oncol (2021) 32:S551. doi: 10.1016/j.annonc.2021.08.954
184. Damato A, Iachetta F, Normanno N, Bergamo F, Maiello E, Zaniboni A, et al. NIVACOR: Phase II study of nivolumab in combination with FOLFOXIRI/bevacizumab in first-line chemotherapy for advanced colorectal cancer RASm/BRAFm patients. J Clin Oncol (2020) 38:TPS4118–TPS4118. doi: 10.1200/JCO.2020.38.15_suppl.TPS4118
185. Shahda S, Noonan AM, Bekaii-Saab TS, O'Neil BH, Sehdev A, Shaib WL, et al. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. J Clin Oncol (2017) 35:3541–1. doi: 10.1200/JCO.2017.35.15_suppl.3541
186. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst (2013) 105:256–65. doi: 10.1093/jnci/djs629
187. Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol (2015) 16:e498–509. doi: 10.1016/s1470-2045(15)00007-8
188. Lin Z, Cai M, Zhang P, Li X, Cai K, Nie X, et al. Short-course radiotherapy and subsequent CAPOX plus camrelizumab followed by delayed surgery for locally advanced rectal cancer:Short-term results of a phase II trial. J Clin Oncol (2021) 39:63–3. doi: 10.1200/JCO.2021.39.3_suppl.63
189. Wang Y, Xia F, Shen L, Wan J, Zhang H, Wu R, et al. Short-course radiotherapy based total neoadjuvant therapy combined with toripalimab for locally advanced rectal cancer: preliminary findings from a randomized, prospective, multicenter, double-arm, phase II trial (TORCH). Int J Radiat Oncol Biol Phys (2022) 114:e152. doi: 10.1016/j.ijrobp.2022.07.1009
190. Leduc C, Adam J, Louvet E, Sourisseau T, Dorvault N, Bernard M, et al. TPF induction chemotherapy increases PD-L1 expression in tumour cells and immune cells in head and neck squamous cell carcinoma. ESMO Open (2018) 3:e000257. doi: 10.1136/esmoopen-2017-000257
191. Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics (2014) 15:190. doi: 10.1186/1471-2164-15-190
192. Dosset M, Vargas TR, Lagrange A, Boidot R, Végran F, Roussey A, et al. PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology (2018) 7:e1433981. doi: 10.1080/2162402x.2018.1433981
193. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, densityand location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi: 10.1126/science.1129139
194. Limagne E, Thibaudin M, Nuttin L, Spill A, Derangère V, Fumet J-D, et al. Trifluridine/tipiracil plus oxaliplatin improves PD-1 blockade in colorectal cancer by inducing immunogenic cell death and depleting macrophages. Cancer Immunol Res (2019) 7:1958–69. doi: 10.1158/2326-6066.Cir-19-0228
195. Bocobo AG, Wang R, Behr S, Carnevale JC, Cinar P, Collisson EA, et al. Phase II study of pembrolizumab plus capecitabine and bevacizumab in microsatellite stable (MSS) metastatic colorectal cancer (mCRC): Interim analysis. J Clin Oncol (2021) 39:77–7. doi: 10.1200/JCO.2021.39.3_suppl.77
196. Morano F, Raimondi A, Pagani F, Lonardi S, Salvatore L, Cremolini C, et al. Temozolomide followed by combination with low-dose ipilimumab and nivolumab in patients with microsatellite-stable, O(6)-methylguanine-DNA methyltransferase-silenced metastatic colorectal cancer: the MAYA trial. J Clin Oncol (2022) 40:1562–73. doi: 10.1200/jco.21.02583
197. Luo H, Lu J, Bai Y, Mao T, Wang J, Fan Q, et al. Effect of camrelizumab vs placebo added to chemotherapy on survival and progression-free survival in patients with advanced or metastatic esophageal squamous cell carcinoma: the ESCORT-1st randomized clinical trial. Jama (2021) 326:916–25. doi: 10.1001/jama.2021.12836
198. Sun Y, Liu X, Yang K-Y, Zhang N, Jin F, Zou G, et al. PD-1 blockade with sintilimab plus induction chemotherapy and concurrent chemoradiotherapy (IC-CCRT) versus IC-CCRT in locoregionally-advanced nasopharyngeal carcinoma (LANPC): A multicenter, phase 3, randomized controlled trial (CONTINUUM). J Clin Oncol (2023) 41:LBA6002–LBA6002. doi: 10.1200/JCO.2023.41.17_suppl.LBA6002
199. Zhang Y, Chen L, Hu GQ, Zhang N, Zhu XD, Yang KY, et al. Final overall survival analysis of gemcitabine and cisplatin induction chemotherapy in nasopharyngeal carcinoma: A multicenter, randomized phase III trial. J Clin Oncol (2022) 40:2420–5. doi: 10.1200/jco.22.00327
200. Hollebecque A, Calvo A, Andre T, Argiles G, Cervantes A, Leger C, et al. Phase I multicenter, open-label study to establish the maximum tolerated dose (MTD) of trifluridine/tipiracil (TAS-102) and oxaliplatin combination in patients (pts) with metastatic colorectal cancer (mCRC). J Clin Oncol (2018) 36:816–6. doi: 10.1200/JCO.2018.36.4_suppl.816
201. Redman JM, Tsai YT, Weinberg BA, Donahue RN, Gandhy S, Gatti-Mays ME, et al. A randomized phase II trial of mFOLFOX6 + Bevacizumab alone or with adCEA vaccine + Avelumab immunotherapy for untreated metastatic colorectal cancer. Oncologist (2022) 27:198–209. doi: 10.1093/oncolo/oyab046
202. Xu J, Chen M, Wu Y, Zhang H, Zhou J, Wang D, et al. The role of transcriptional factor brachyury on cell cycle regulation in non-small cell lung cancer. Front Oncol (2020) 10:1078. doi: 10.3389/fonc.2020.01078
203. Nath S, Mukherjee P. MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med (2014) 20:332–42. doi: 10.1016/j.molmed.2014.02.007
204. Wallin J, Pishvaian M, Hernandez G, Yadav M, Jhunjhunwala S, Delamarre L, et al. Abstract 2651: Clinical activity and immune correlates from a phase Ib study evaluating atezolizumab (anti-PDL1) in combination with FOLFOX and bevacizumab (anti-VEGF) in metastatic colorectal carcinoma. Cancer Res (2016) 76:2651–1. doi: 10.1158/1538-7445.AM2016-2651
205. Iida Y, Harashima N, Motoshima T, Komohara Y, Eto M, Harada M. Contrasting effects of cyclophosphamide on anti-CTL-associated protein 4 blockade therapy in two mouse tumor models. Cancer Sci (2017) 108:1974–84. doi: 10.1111/cas.13337
206. Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol (2012) 30:2046–54. doi: 10.1200/jco.2011.38.4032
207. Kok M, Voorwerk L, Horlings H, Sikorska K, Vijver K, Slagter M, et al. Adaptive phase II randomized trial of nivolumab after induction treatment in triple negative breast cancer (TONIC trial): Final response data stage I and first translational data. J Clin Oncol (2018) 36:1012–2. doi: 10.1200/JCO.2018.36.15_suppl.1012
208. Gray A, de la Luz Garcia-Hernandez M, van West M, Kanodia S, Hubby B, Kast WM. Prostate cancer immunotherapy yields superior long-term survival in TRAMP mice when administered at an early stage of carcinogenesis prior to the establishment of tumor-associated immunosuppression at later stages. Vaccine (2009) 27 Suppl 6:G52–9. doi: 10.1016/j.vaccine.2009.09.106
209. Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods (2014) 11:41–6. doi: 10.1038/nmeth.2694
210. Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol (2018) 18:35–45. doi: 10.1038/nri.2017.76
211. Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med (2018) 24:1550–8. doi: 10.1038/s41591-018-0136-1
212. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O'Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell (2020) 181:442–459.e29. doi: 10.1016/j.cell.2020.03.048
213. Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res (2015) 3:436–43. doi: 10.1158/2326-6066.Cir-15-0064
214. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
215. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
216. Garassino M, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, Speranza G, et al. OA04.06 evaluation of TMB in KEYNOTE-189: pembrolizumab plus chemotherapy vs placebo plus chemotherapy for nonsquamous NSCLC. J Thorac Oncol (2019) 14:S216–7. doi: 10.1016/j.jtho.2019.08.427
217. Sun D, Liu J, Zhou H, Shi M, Sun J, Zhao S, et al. Classification of tumor immune microenvironment according to programmed death-ligand 1 expression and immune infiltration predicts response to immunotherapy plus chemotherapy in advanced patients with NSCLC. J Thorac Oncol (2023) 18:869–81. doi: 10.1016/j.jtho.2023.03.012
218. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, presentand the future. Cell (2017) 168:613–28. doi: 10.1016/j.cell.2017.01.018
219. Galon J, Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity (2020) 52:55–81. doi: 10.1016/j.immuni.2019.12.018
Keywords: colorectal cancer, immunotherapy, inducing chemotherapy, immunogenic cell death, combination therapy, clinical trial, tumor microenvironment
Citation: Yao S, Han Y, Yang M, Jin K and Lan H (2023) It’s high-time to re-evaluate the value of induced-chemotherapy for reinforcing immunotherapy in colorectal cancer. Front. Immunol. 14:1241208. doi: 10.3389/fimmu.2023.1241208
Received: 16 June 2023; Accepted: 09 October 2023;
Published: 18 October 2023.
Edited by:
Chengwu Zeng, Jinan University, ChinaReviewed by:
Maha Mohamed Saber-Ayad, University of Sharjah, United Arab EmiratesFrancis Yew Fu Tieng, National University of Malaysia, Malaysia
Copyright © 2023 Yao, Han, Yang, Jin and Lan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ketao Jin, jinketao2001@zju.edu.cn; Huanrong Lan, lanhr2018@163.com
†These authors have contributed equally to this work