 Prakash Sah
Prakash Sah Lauren A. Zenewicz
Lauren A. Zenewicz- Department of Microbiology and Immunology, College of Medicine, The University of Oklahoma Health Sciences Center, Oklahoma City, OK, United States
Innate lymphoid cells (ILCs) are key regulators of tissue homeostasis, inflammation, and immunity to infections. ILCs rapidly respond to environmental cues such as cytokines, microbiota and invading pathogens which regulate their function and phenotype. Even though ILCs are rare cells, they are enriched at barrier surfaces such as the gastrointestinal (GI) tract, and they are often critical to the host’s immune response to eliminate pathogens. On the other side of host-pathogen interactions, pathogenic bacteria also have the means to modulate these immune responses. Manipulation or evasion of the immune cells is often to the pathogen’s benefit and/or to the detriment of competing microbiota. In some instances, specific bacterial virulence factors or toxins have been implicated in how the pathogen modulates immunity. In this review, we discuss the recent progress made towards understanding the role of non-cytotoxic ILCs during enteric bacterial infections, how these pathogens can modulate the immune response, and the implications these have on developing new therapies to combat infection.
Introduction
Innate lymphoid cells (ILCs) are rare lymphocytes that are primarily tissue-resident and often present in mucosal tissues. Lacking specific antigen receptors, ILCs are mainly activated by cytokines and rapidly respond to infections. ILCs represent innate counterparts of T cells as they share several phenotypic and functional features (1). ILCs can be classified into four distinct subsets: NK cells, ILC1s, ILC2s, and ILC3s based on expression of lineage-specifying transcription factors (TF), surface markers and effector functions (2, 3). We will focus on ILCs as NK cells have been well-recognized and studied for decades. ILC1s express T-bet and often protect against intracellular pathogens via production of IFNγ but generally are non-cytotoxic (4). ILC2s express the lineage defining TF GATA3 and produce the type 2 cytokines IL-5 and IL-13, and lesser amounts of IL-4 and IL-9. ILC2s are often key to mediating immune responses to helminths and allergens (5). ILC3 development depends on the TF RORγt and these cells produce IL-22 to maintain tissue barrier function and protect against extracellular pathogens (5, 6). Further, ILCs can have cellular plasticity and can transdifferentiate to other ILC subtypes depending upon the cytokine and/or environmental milieu (3–5).
Mucosal surfaces such as those of the GI tract harbor billions of microorganisms as part of the normal microbiota, which is the first line of defense against bacterial pathogens. Nevertheless, many pathogens enter the host through mucosal surfaces causing infection. The immune system has evolved to provide resistance to pathogens while maintaining homeostasis with the microbiota. ILCs are enriched in mucosal surfaces and orchestrate the early defense against invading pathogens. Although ILC3s are cast to be the ILCs that respond to extracellular bacterial infection, ILC1s and ILC2s also can contribute to the immune responses against these pathogens. For every well-honed immune response to combat a pathogen, a host may have a less than perfect response because of many possible reasons, with a primary one being that the pathogen can fight back. Enteric bacteria produce many toxins and other effector molecules that target host cells and can cause apoptosis or interfere in signaling pathways.
Recent studies have highlighted roles of ILCs during infection and modulation of ILC functions by pathogenic bacteria. Although challenging to test the precise role of virulence factors on ILCs in in vivo models due to the requirement for virulence factors for infectivity and the limitations on the tools to specifically target ILCs and not other immune cells, we have discerned much on how ILCs respond to different gastrointestinal (GI) bacterial infections and how the pathogen may affect this response (Figure 1). Here, we present the current state of the field on modulation of non-cytotoxic ILC functions by enteric bacterial pathogens and how it contributes to pathogenesis or protection against these pathogens.
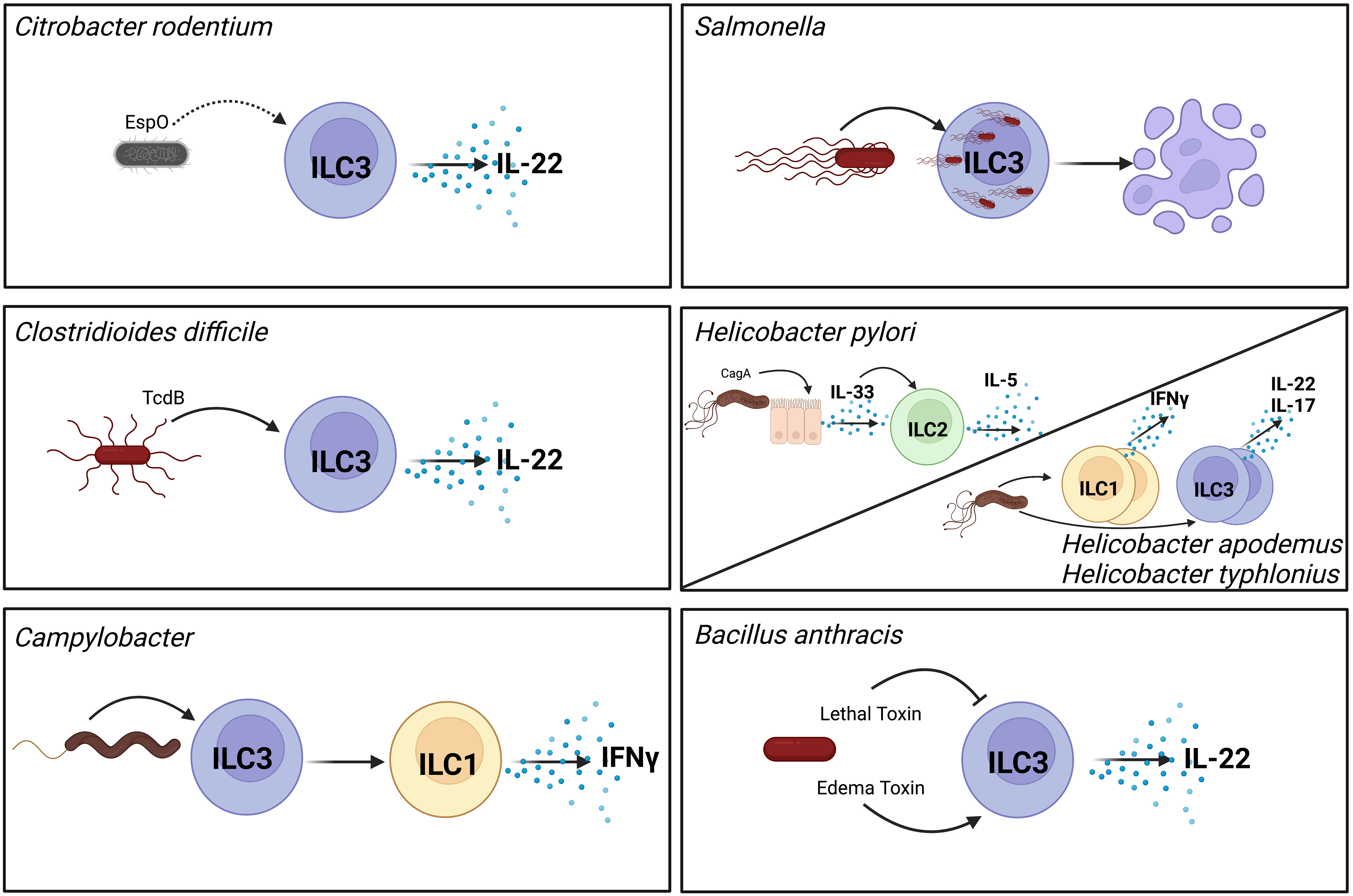
Figure 1 Pathogenic bacteria modulate ILC function through different mechanisms. Citrobacter rodentium produces the effect EspO which indirectly contributes to activation of ILC3s and their production of secreted IL-22 (7). As an intracellular pathogen Salmonella invades ILC3s which induces their pyroptosis and reduces their numbers in the GI tract (8). Toxin B (TcdB) produced by Clostridioides difficile directly activates ILC3s to produce IL-22 (9). Helicobacter pylori activates epithelial cells to produce IL-33 in a CagA-dependent manner. This IL-33 in turns activates ILC2s in the stomach to produce IL-5 (10, 11). Non-gastric Helicobacter species increase frequencies of cytokine producing ILC1s and ILC3s (12). Campylobacter infection induces conversion of ILC3s to ILC1s that can produce IFNγ (13). Bacillus anthracis secretes two toxins with opposing effects on ILC3s. Lethal toxin inhibits MAPK signaling which inhibits ILC3 activation (14). In contrast, edema toxin directly activates ILC3s to produce IL-22 (15). Direct interactions are shown with solid arrows, indirect interactions are shown with dashed arrows. Created with BioRender.com.
Modulation of ILC function by bacterial pathogens
Citrobacter rodentium
C. rodentium is a murine Gram-negative pathogen that is widely used to model the human pathogen enteropathogenic Escherichia coli (EPEC) (16). Like EPEC, C. rodentium secretes effectors via a type 3 secretion system (T3SS) into intestinal epithelial cells to modulate host cell processes and establish infection (17). The utility of the C. rodentium infection model extends beyond bacterial pathogenesis studies to reveal fundamental aspects of mucosal immunity (18). C. rodentium infection elicits both innate and adaptive immune responses that are important for control and clearance of the pathogen in the initial and late phases of infection, respectively. ILC3s are critical immune cells in early control of C. rodentium (19–22). Following infection, macrophages and dendritic cells (DCs) are activated by bacterial pathogen-associated molecular patterns (PAMPs) and secrete inflammatory cytokines such as IL-23 and IL-1β which activate ILC3s (23–27). Upon activation, ILC3s rapidly produce IL-22, a critical cytokine in control of C. rodentium infection (5, 19, 28). ILC3s represent a major early source of IL-22 in infection (21, 29), where the cytokine upregulates antimicrobial peptides and mucin production promoting intestinal barrier resistance (19, 28).
C. rodentium T3SS effectors modulate innate immune responses (18). It is not known whether C. rodentium directly interacts with ILC3s. However, a recent study showed that the T3SS effector EspO modulates IL-22 signaling (7). While the study reported infection with C. rodentium lacking EspO reduced IL-22 secretion by colonic explants and a subsequent reduction in antimicrobial peptides, no change in the frequency of IL-22 producing ILC3s or T cells was observed (7). It is not clear why a C. rodentium effector would upregulate IL-22. Modulation of IL-22 by other pathogens is known to promote colonization (30) but this is unlikely in the case of C. rodentium as IL-22 is well-described to be protective in C. rodentium infection (19). C. rodentium is also known to modulate metabolism of intestinal epithelial cells (IECs) (18). T3SS effectors, Map and EspF, cause mitochondrial disruption leading to shift in IEC metabolism to aerobic glycolysis causing increased oxygenation of the intestinal mucosa that supports C. rodentium colonization (31–33). Given the role of hypoxia in regulating ILC3 function (34), the question arises whether this increased oxygenation of the GI tract caused by C. rodentium T3SS effectors modulate ILC3. Lastly, C. rodentium infection can cause metabolic rewiring of ILC3s, enhancing their proliferation and cytokine production (35). These “trained ILC3s” show properties of “innate memory” and are better in controlling re-infection than naive ILC3s in mice (35). In summary, C. rodentium infection has been a valuable model to learn much about pathogen-ILC interactions.
Salmonella
Salmonella enterica encompasses several serovars that cause GI infection (36, 37). In developing countries, typhoidal serovars, S. Typhi and S. Paratyphi, cause enteric fever. Non-typhoidal serovars such as S. enterica serovar Typhimurium (S. Typhimurium) cause food-borne gastroenteritis that are usually self-limiting but can lead to disseminated infection (36). S. Typhimurium is the most widely studied serovar and findings reviewed here pertain to this serovar. Like many other Gram-negative GI pathogens, S. Typhimurium encodes for T3SS, which in Salmonella is two separate T3SS systems that secrete an arsenal of virulence factors (37).
Interferon-γ (IFNγ) is a key cytokine in defense against Salmonella infections (38–41). The main innate source of IFNγ during Salmonella GI infection is NKp46+ T-bet+ ILCs that have decreased RORγt levels (41). The IFNγ production by these ILCs during infection is driven by IL-12 while IL-23 has no significant effect (41). Although ILCs are mainly tissue-resident cells, Kastele et al. showed that Salmonella infection increased the migratory RORγt+ T-bet+ ILC population in the mesenteric lymph nodes and contributed to IFNγ production (42). Further refinement in identifying the IFNγ-producing cells, has shown significant IFNγ production by ILC1s but not ILC3s (8). IFNγ regulates mucin production during Salmonella infection and deficiency of the mucin MUC2 increases susceptibility to infection in a mouse model (39, 43). In line with this, depletion of ILCs or genetic ablation of IFNγ production by ILCs, results in impaired mucus production (41). Additionally, IFNγ-producing ILCs also contribute to intestinal inflammation as interference with IFNγ production by ILCs leads to reduced inflammation (41). These studies suggest that IFNγ production by ILCs induced by Salmonella infection can be both protective as well as pathologic.
IL-22 is upregulated during Salmonella infection (44). ILC3s via production of IL-22 and lymphotoxin-α mediate in vitro fucosylation of IECs (45). In vivo ILC3s mediate fucosylation during Salmonella infection and mice deficient in intestinal fucosylation are more susceptible to infection (45). Subsequent studies have reported that IL-22 is not protective in Salmonella infection. In fact, increased IL-22 promotes Salmonella infection by improving its ability to compete with microbiota (30, 46). This is attributed to its resistance to the antimicrobial peptides induced by IL-22 (30, 47, 48). In line with these observations, a recent study found that Salmonella induced IL-22 in ILC3s during infection which promoted infection (8). Flagellin activates antigen-presenting cells to produce IL-23, which in turn activated ILC3s. Further, Salmonella directly invade ILC3s leading to capase-1-mediated pyroptosis of ILC3s in a flagellin-independent manner. ILC3 depletion leads to less mortality and reduced disease severity in Salmonella infected mice (8). Thus, Salmonella-induced ILC3 pyroptosis is a possible host defense mechanism against Salmonella by limiting innate IL-22 production after early induction which benefits the bacteria (8). However, the exact signal(s) that induces ILC3 pyroptosis remain unknown. This example of bacterial modulation of ILC3s by regulating cell death is a bacterial defense mechanism that may prove to be relevant to other GI pathogens.
Clostridioides difficile
C. difficile colitis is the most common nosocomial GI infection occurring in patients with perturbed microbiota owing to use of broad-spectrum antibiotics (49). C. difficile strains can encode for three different toxins: toxin A (TcdA), toxin B (TcdB) and C. difficile transferase (CDT) (50). The combined action of these toxins results in disruption of host cytoskeleton in the GI epithelium and ultimately loss of epithelial integrity. Toxin-mediated inflammation can result in symptoms ranging from mild diarrhea to pseudomembranous colitis (50).
Several studies have described the roles of ILCs during C. difficile infection (CDI) (51–53). Nfil3-/- mice, which due to a lack of the transcriptional regulator nuclear factor, interleukin 3 regulated (NFIL3) have reduced numbers of ILCs owing to a developmental defect in ILC maturation, are highly susceptible to CDI (54). Further, Rag2-/- γc-/- mice lacking both adaptive immune cells and ILCs have increased mortality following CDI compared to RAG-deficient, lacking only adaptative immunity cells or wild-type mice (51). IFNγ is increased in Rag1-/- mice following CDI and selective loss of ILC1s or ILC1-derived IFNγ leads to increased disease severity and mortality (51). The same study also reported an increase in ILC3-associated cytokines such as IL-22 and IL-17 following CDI. However, selective loss of ILC3s or the ILC3-associated cytokine IL-22 exhibited only a modest effect on CDI recovery (51), at least in the absence of an adaptive immune response. In contrast to this, Hasegawa et al. reported increased mortality following CDI in IL-22-deficient mice. Although, lack of IL-22 did not alter C. difficile burden and intestinal damage, IL-22 was important in clearance of pathobionts that translocated following intestinal damage by regulating the complement system (55). Together, these studies show that ILC3s and IL-22 are likely more important during the later stage of infection by preventing translocation of pathobionts whereas ILC1s are more important in the early defense against CDI. The protective role of ILC3s and/or IL-22 in context of CDI has been further confirmed by other studies (34, 53, 56, 57).
ILC2s are important players in immunity against helminth infection and allergic response in the GI tract (5). However, these cells have been recently shown to be important in bacterial infections, including C. difficile (52). IL-33, an activator of ILC2s, was found to be upregulated during CDI in cecal tissues of infected mice. IL-33 protected against CDI by activating ILC2s (52). IL-33-activated ILC2s increased mucin production by goblet cells, improving epithelial barrier function as well as increasing the number of eosinophils in the colon that are protective during CDI (52, 58, 59).
Although, the roles of different ILC subsets have been studied in context of CDI, whether C. difficile or its toxins directly interact with ILCs remains poorly defined. Recently, we reported that TcdB directly activates ILC3s in vitro, inducing IL-22 and other effector molecules (9). The TcdB-mediated activation of ILC3s required the toxin’s enzymatic activity and was in part mediated by inactivation of the small GTPase CDC42 (9). Gene expression analysis revealed that the toxin-mediated activation of ILC3s is distinct from IL-1β-mediated activation (9). Given the protective effects of IL-22 during C. difficile infection, the toxin mediated activation of ILC3s seems surprising. IL-22 may shape the microbiota to favor C. difficile over other species that the pathogen competes for resources. In vivo validation of this observation and function of toxin activated-ILC3s remains to determined.
Helicobacter Spp.
Helicobacter pylori is a highly prevalent Gram-negative pathogen that infects half of the world’s population (60). It causes gastritis and is the strongest risk factor for gastric adenocarcinoma (61). The major virulence factors of H. pylori are type four secretion system (T4SS) effectors, which includes cytotoxin-associated gene A product (CagA) (61). ILC2s are the predominant ILC population in the stomach and increase in number during H. pylori infection (62, 63). IL-33 is increased in gastric mucosa of patients and H. pylori infected mice with strains encoding CagA. In vitro CagA induces IL-33 production by gastric epithelial cells, leading to ILC2 activation and IL-5 production (10, 11). Similarly, increased IL7 mRNA levels are found in the gastric mucosa of H. pylori infected patients (64). In the stomach, ILC2 accumulation and activation is dependent on IL-7 and IL-33, respectively (63). ILC2s coordinate with B cells to produce H. pylori-specific IgA which can coat bacteria in the stomach in a mouse model (63).
Non-gastric Helicobacter species, such as H. apodemus and H. typhlonius, have different effects on ILCs compared to H. pylori (12). In mice lacking T and B cells, colonization of the GI tract with these two Helicobacter species causes activation of ILCs with increased frequencies of IL-22- and/or IL-17-producing ILC3s and IFNγ-producing ILC1s, resulting in GI inflammation. However, infection reduces ILC3 number, particularly T-bet-expressing ILC3s (12). How these Helicobacter species cause reduction in ILC3 numbers remains unknown. The reduction in ILC3s was only observed in mice lacking adaptive immune cells and not wild-type mice suggesting that adaptive immunity can sustain ILC3s during Helicobacter colonization.
Campylobacter
Campylobacter species, mainly C. jejuni and C. coli, are the causative agents of one type of food-borne gastroenteritis (65). Although these Gram-negative infections are mostly self-limiting, long-term immune related intestinal dysfunction has been associated with Campylobacter infections (66, 67). Several studies have examined the role of ILCs during Campylobacter infection, using a mouse model with a predisposition to intestinal inflammation due to a lack of IL-10 signaling (13, 68, 69). IL-23, a primary activator of ILC3s, is key driver of inflammation during Campylobacter infection (69). Jing et al. found that IL-23 regulated production of IFNγ, IL-17 and IL-22 by ILC1s and ILC3s during Campylobacter infection (69). Consistent with previous studies, IFN-γ and IL-17 promoted inflammation while IL-22 was found to be dispensable for inflammation during Campylobacter infection (69). This contrasts with another study in which IL-22 was protective in a mouse model of Campylobacter where IL-10 signaling was intact (70).
Campylobacter infection induces production of IFNγ, IL-17 and IL-22 from both ILCs and T cell subsets in mice (68). Similar observations were reported using an ex vivo human gut model of infection (71). Both IFNγ and IL-17 were found to promote intestinal inflammation during Campylobacter infection (68). Two recent studies found that ILC-derived IFNγ contributes to intestinal pathology during infection (13, 69). While ILC1s represent a major source of IFNγ, ILC3s can also convert into IFN-γ-producing ILC1s by expressing the transcription factor T-bet (5, 41). Interestingly, Campylobacter infection induces conversion of ILC3s to ILC1s and these ex-ILC3s in turn produce IFNγ that promotes intestinal inflammation (13). IL-12 and IL-23 have been shown to facilitate in vitro ILC3-ILC1 plasticity (72) and both cytokines are elevated during Campylobacter infection (13, 68). One may speculate that dysregulation of IL-12 and IL-23 levels during Campylobacter infection promotes ILC3 to ILC1 conversion. However, the mechanism by which Campylobacter regulates ILC3 to ILC1 conversion has not yet been elucidated. Campylobacter spp. produce many toxins that modulate host cells, with the best studied being cytolethal distending toxin (CDT) from C. jejuni, that may modulate ILC function. Together, these studies show that ILCs are a major player in inducing intestinal inflammation and promote pathology during Campylobacter infection.
Bacillus anthracis
B. anthracis is the causative agent of anthrax, a rare but deadly infection of the lungs, GI tract or skin (73). The major virulence factors of the Gram-positive B. anthracis are lethal toxin (LT) and edema toxin (ET) (73). LT or ET can both suppress immune function of both innate and adaptive immune cells (74–76). In line with the immunosuppressive action of anthrax toxins, LT can suppress ILC3 activation both in vitro and in vivo in a mouse model (14). LT, a zinc metalloprotease, cleaves mitogen activated protein kinases perturbing cell signaling (77). LT reduced IL-22 production by IL-23-activated ILC3s by inactivating MAPK signaling (14). In contrast, ET as an adenyl cyclase, elevates intracellular cAMP levels in ILC3s leading to their in vitro activation (15). How LT, ET and ILC3s interact in vivo remains to be determined. It is likely that LT-mediated suppression may overcome ET-mediated activation of ILC3s as a study found suppression of the mRNA encoding IL-22-induced antimicrobial peptides, REG3β and REG3γ, in the colons of B. anthracis infected mice (75). ET effects can be dose-dependent, and the toxin can suppress T cell proliferation or promote Th17 differentiation (76, 78), which may have similar implications on related ILC3s. Thus, it is also possible that ET may exert suppressive action on ILC3s in vivo. Although not a focus of our review and has yet to be studied, these toxins likely modulate ILCs in the lungs during respiratory disease. Overall, B. anthracis and its two major toxins modulate ILC3 function.
Crosstalk between ILCs and the adaptive immune system
Although ILCs are mainly tissue-resident cells residing at mucosal surfaces, they are also found in the peripheral blood, bone marrow, and primary and secondary lymphoid organs (79–81). Recently, migratory ILCs have been reported in context of infection (42). Their location in lymphoid organs where the adaptive immune response is initiated suggests a role for ILCs in shaping adaptive immunity. Indeed, several studies have underscored the role of ILCs in regulation of adaptive immunity (reviewed in (81, 82)). ILCs can directly or indirectly influence both T and B cell-mediated responses (82, 83). As one of the first responders to pathogens, ILCs influence the cytokine milieu and thus also influence adaptive immune response (82, 83). ILC2s and ILC3s can also directly interact with T cells by expressing MHCII and acting as antigen presenting cells (APCs) (84–86). ILCs can also express co-stimulatory or auxiliary molecules such as OX40L, ICOS/ICOSL, and PD-L1 (the latter two reported only for ILC2s) which facilitates a direct interaction with T cells thus modulating the adaptive immune response (87–91). Conversely, the adaptive immune system can also regulate ILC function. For example, T cell-derived IFNγ directly limits ILC2 function (92, 93) and regulatory T cells can control ILC2 and ILC3 responses (94–96). T cells can also enhance ILC2 responses via direct interaction with ILC2s mediated by MHCII or other auxiliary molecules listed above (86, 97). Thus, crosstalk between ILCs and adaptive immune cells shape adaptive immunity as well ILC function.
In the healthy GI tract, ILC3s regulate the immune response to the microbiota by regulating T and B cell responses. ILC3s acting as APCs interact with T cells resulting in clonal deletion of microbiota-specific T cells (85). Similarly, ILC3s can act as APCs to interact with T helper follicular cells to regulate the immunoglobulin A (IgA) response to the microbiota (98). ILC3s can also indirectly support IgA production via lymphotoxin signaling in the intestine (99). Ablation of lymphotoxin α signaling in ILC3s results in inhibition of IgA production and composition changes of the GI microbiota (99). Studies on ILC crosstalk during enteric bacterial infection are very limited. As discussed above, ILC3s are important in defense against enteric pathogens such as C. rodentium (21). Several studies have dissected the spatiotemporal interplay of ILCs and the adaptive immune response during C. rodentium infection with ILCs being critical during the early stage of infection and the adaptive response acting later to clear infection (100–102). IL-22, the signature cytokine of ILC3s, regulates the organization and maintenance of colonic lymphoid structures during C. rodentium infection by acting downstream of lymphotoxin signaling (103). Colonic lymphoid structures contain T and B cells and thus are sites of initiation of the adaptive immune response. A recent study showed that Salmonella infection induced migration of intestinal RORγt+ T-bet+ ILCs to mesenteric lymph nodes which contributed to the protective IFNγ response (42). Whether these migratory ILCs contribute to the overall adaptive immune response remains unknown. During colonization by non-gastric Helicobacter species, the adaptive immune system sustains ILC3 number (12). With progress in our understanding of ILC biology, the interplay of ILCs and adaptive immune system has become apparent during healthy, steady-state conditions. However, more studies are needed to uncover the specific nature of ILC and adaptive immune system interactions during enteric infections which will facilitate development of better therapeutics.
Targeting ILCs for treatment of enteric bacterial infections
The standard of care for bacterial GI diseases is antibiotics. However, with the ever-growing rise of antibiotic resistance, this treatment may fail for some patients. Targeting the host immune response, or in combination therapy with antibiotics, has the potential to become an effective treatment to combat GI infections. Although ILCs are rare immune cells, they are critical regulators of tissue homeostasis, inflammation, and immunity against GI infections (5). Increased understanding of ILC biology in recent years has resulted in development of new or co-opting T cell-targeted therapies to therapeutically target ILCs as well (104). Existing ILC targeting strategies for therapeutics include administration of cytokines, adoptive transfer of ILCs, antibodies against ILC-related cytokines, ILC depletion, modulation of ILC plasticity, migration and function, and microbiota manipulation (104). These take advantage of pathways that were developed based on T cell biology. Development of treatments that precisely target ILCs and not T cells is a challenge for the field. Furthermore, manipulation of oxygen levels in the GI tract may be an effective means to counteract a pathogen. Thus, targeting ILCs is increasingly considered for clinical therapy as evident by several preclinical or clinical studies on ILC-targeting therapies (104, 105). In fact, several biological therapies targeting ILCs are already approved, and multiple others are in development for Crohn’s disease, a chronic inflammatory disease of GI tract (104, 106).
As discussed, enteric bacterial pathogens often modulate ILC function contributing to a pathologic or defensive outcome. Pathogen-induced cytokine production by ILCs can be to the pathogen’s advantage or contribute to protection of the host. Further, ILC plasticity and migration can also be modulated by enteric bacterial pathogens. Hence, therapeutics targeting ILC biology are an attractive novel option for the treatment of GI infections. Existing ILC-targeting biologic therapeutics could be adapted to treat enteric bacterial infections. Although studies have started to highlight the roles of ILCs in the context of different enteric bacterial infections, more studies on defining the precise roles of ILCs during infection, especially in humans, are needed before existing ILC-targeting strategies can be adapted to treating enteric infections. Such studies will provide a comprehensive understanding of ILC biology in context of infection leading to adaptation of existing or development of new therapeutic strategies targeting ILCs for enteric bacterial infections.
Conclusions
Despite their relatively recent discovery, ILCs have emerged as key players in barrier resistance to prevent breach by pathogens. ILCs are among the first immune cells that these pathogens encounter. ILCs during enteric bacterial infections can have both protective and pathogenic roles depending on the pathogen and context of infection. Enteric pathogens have evolved strategies to evade host immune responses to help establish their infection in the GI tract. In only a few interactions have we identified the specific bacterial virulence or effector molecules that manipulate specific signaling pathways in ILC3s. Future studies need to examine these different pathogens in the shared and novel ways they can modulate ILC responses. Further, many of these findings are established from animal infection models. There is a need for validation in context of human infection to increase the translation of these basic research studies to better patient outcomes. A better understanding of ILC biology in the context of infection has high potential to lead to ILC-targeted therapies for enteric bacterial infections.
Author contributions
PS contributed to the conceptualization and wrote the first draft. LZ contributed to the conceptualization, reviewed and edited the article, and generated the figure. All authors contributed to the article and approved the submitted version.
Funding
Research in the Zenewicz laboratory is supported in part by the National Institute of General Medical Sciences of the National Institutes of Health (P20GM134973) and in part by the National Cancer Institute Cancer Center Support Grant P30CA225520 awarded to the Stephenson Cancer Center.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. innate lymphoid cells: a new paradigm in immunology. Science (2015) 348:aaa6566. doi: 10.1126/science.aaa6566
2. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13:145–9. doi: 10.1038/nri3365
3. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
4. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol (2016) 17:758–64. doi: 10.1038/ni.3482
5. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17:765–74. doi: 10.1038/ni.3489
6. Keir M, Yi Y, Lu T, Ghilardi N. The role of IL-22 in intestinal health and disease. J Exp Med (2020) 217:e20192195. doi: 10.1084/jem.20192195
7. Berger CN, Crepin VF, Roumeliotis TI, Wright JC, Serafini N, Pevsner-Fischer M, et al. The citrobacter rodentium type III secretion system effector EspO affects mucosal damage repair and antimicrobial responses. PloS Pathog (2018) 14:e1007406. doi: 10.1371/journal.ppat.1007406
8. Xiong L, Wang S, Dean JW, Oliff KN, Jobin C, Curtiss,3rd R, et al. Group 3 innate lymphoid cell pyroptosis represents a host defence mechanism against salmonella infection. Nat Microbiol (2022) 7:1087–99. doi: 10.1038/s41564-022-01142-8
9. Pope RL, Chitrakar A, Sah P, Shadid T, Ballard JD, Zenewicz LA. Clostridioides difficile toxin b activates group 3 innate lymphocytes. Infect Immun (2022) 90:e0007322. doi: 10.1128/iai.00073-22
10. Lv YP, Teng YS, Mao FY, Peng LS, Zhang JY, Cheng P, et al. Helicobacter pylori-induced IL-33 modulates mast cell responses, benefits bacterial growth, and contributes to gastritis. Cell Death Dis (2018) 9:457. doi: 10.1038/s41419-018-0493-1
11. Kuo CJ, Chen CY, Lo HR, Feng CL, Wu HY, Huang MZ, et al. Helicobacter pylori induces IL-33 production and recruits ST-2 to lipid rafts to exacerbate inflammation. Cells (2019) 8:1290. doi: 10.3390/cells8101290
12. Bostick JW, Wang Y, Shen Z, Ge Y, Brown J, Chen ZE, et al. Dichotomous regulation of group 3 innate lymphoid cells by nongastric helicobacter species. Proc Natl Acad Sci USA (2019) 116:24760–9. doi: 10.1073/pnas.1908128116
13. Muraoka WT, Korchagina AA, Xia Q, Shein SA, Jing X, Lai Z, et al. Campylobacter infection promotes IFNgamma-dependent intestinal pathology via ILC3 to ILC1 conversion. Mucosal Immunol (2021) 14:703–16. doi: 10.1038/s41385-020-00353-8
14. Seshadri S, Allan DSJ, Carlyle JR, Zenewicz LA. Bacillus anthracis lethal toxin negatively modulates ILC3 function through perturbation of IL-23-mediated MAPK signaling. PloS Pathog (2017) 13:e1006690. doi: 10.1371/journal.ppat.1006690
15. Sah P, Derouen JT, Alexander JL, Zenewicz LA. Group 3 innate lymphocytes (ILC3s) upregulate IL-22 in response to elevated intracellular cAMP levels. Cytokine (2022) 153:155862. doi: 10.1016/j.cyto.2022.155862
16. Borenshtein D, McBee ME, Schauer DB. Utility of the citrobacter rodentium infection model in laboratory mice. Curr Opin Gastroenterol (2008) 24:32–7. doi: 10.1097/MOG.0b013e3282f2b0fb
17. Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, et al. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol (2014) 12:612–23. doi: 10.1038/nrmicro3315
18. Mullineaux-Sanders C, Sanchez-Garrido J, Hopkins EGD, Shenoy AR, Barry R, Frankel G. Citrobacter rodentium-host-microbiota interactions: immunity, bioenergetics and metabolism. Nat Rev Microbiol (2019) 17:701–15. doi: 10.1038/s41579-019-0252-z
19. Jarade A, Di Santo JP, Serafini N. Group 3 innate lymphoid cells mediate host defense against attaching and effacing pathogens. Curr Opin Microbiol (2021) 63:83–91. doi: 10.1016/j.mib.2021.06.005
20. Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity (2008) 29:958–70. doi: 10.1016/j.immuni.2008.11.001
21. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity (2011) 34:122–34. doi: 10.1016/j.immuni.2010.12.009
22. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature (2009) 457:722–5. doi: 10.1038/nature07537
23. Manta C, Heupel E, Radulovic K, Rossini V, Garbi N, Riedel CU, et al. CX(3)CR1(+) macrophages support IL-22 production by innate lymphoid cells during infection with citrobacter rodentium. Mucosal Immunol (2013) 6:177–88. doi: 10.1038/mi.2012.61
24. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med (2014) 211:1571–83. doi: 10.1084/jem.20140678
25. Seo SU, Kuffa P, Kitamoto S, Nagao-Kitamoto H, Rousseau J, Kim YG, et al. Intestinal macrophages arising from CCR2(+) monocytes control pathogen infection by activating innate lymphoid cells. Nat Commun (2015) 6:8010. doi: 10.1038/ncomms9010
26. Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol (2013) 14:937–48. doi: 10.1038/ni.2679
27. Wang B, Lim JH, Kajikawa T, Li X, Vallance BA, Moutsopoulos NM, et al. Macrophage beta2-integrins regulate IL-22 by ILC3s and protect from lethal citrobacter rodentium-induced colitis. Cell Rep (2019) 26:1614–1626 e5. doi: 10.1016/j.celrep.2019.01.054
28. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med (2008) 14:282–9. doi: 10.1038/nm1720
29. Rankin LC, Girard-Madoux MJ, Seillet C, Mielke LA, Kerdiles Y, Fenis A, et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat Immunol (2016) 17:179–86. doi: 10.1038/ni.3332
30. Behnsen J, Jellbauer S, Wong CP, Edwards RA, George MD, Ouyang W, et al. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity (2014) 40:262–73. doi: 10.1016/j.immuni.2014.01.003
31. Ma C, Wickham ME, Guttman JA, Deng W, Walker J, Madsen KL, et al. Citrobacter rodentium infection causes both mitochondrial dysfunction and intestinal epithelial barrier disruption in vivo: role of mitochondrial associated protein (Map). Cell Microbiol (2006) 8:1669–86. doi: 10.1111/j.1462-5822.2006.00741.x
32. Nougayrede JP, Donnenberg MS. Enteropathogenic escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cell Microbiol (2004) 6:1097–111. doi: 10.1111/j.1462-5822.2004.00421.x
33. Berger CN, Crepin VF, Roumeliotis TI, Wright JC, Carson D, Pevsner-Fischer M, et al. Citrobacter rodentium subverts ATP flux and cholesterol homeostasis in intestinal epithelial cells in vivo. Cell Metab (2017) 26:738–752 e6. doi: 10.1016/j.cmet.2017.09.003
34. Fachi JL, Pral LP, Dos Santos JAC, Codo AC, de Oliveira S, Felipe JS, et al. Hypoxia enhances ILC3 responses through HIF-1alpha-dependent mechanism. Mucosal Immunol (2021) 14:828–41. doi: 10.1038/s41385-020-00371-6
35. Serafini N, Jarade A, Surace L, Goncalves P, Sismeiro O, Varet H, et al. Trained ILC3 responses promote intestinal defense. Science (2022) 375:859–63. doi: 10.1126/science.aaz8777
36. Galan JE. Salmonella typhimurium and inflammation: a pathogen-centric affair. Nat Rev Microbiol (2021) 19:716–25. doi: 10.1038/s41579-021-00561-4
37. LaRock DL, Chaudhary A, Miller SI. Salmonellae interactions with host processes. Nat Rev Microbiol (2015) 13:191–205. doi: 10.1038/nrmicro3420
38. Bao S, Beagley KW, France MP, Shen J, Husband AJ. Interferon-gamma plays a critical role in intestinal immunity against salmonella typhimurium infection. Immunology (2000) 99:464–72. doi: 10.1046/j.1365-2567.2000.00955.x
39. Songhet P, Barthel M, Stecher B, Muller AJ, Kremer M, Hansson GC, et al. Stromal IFN-gammaR-signaling modulates goblet cell function during salmonella typhimurium infection. PloS One (2011) 6:e22459. doi: 10.1371/journal.pone.0022459
40. Kupz A, Scott TA, Belz GT, Andrews DM, Greyer M, Lew AM, et al. Contribution of Thy1+ NK cells to protective IFN-gamma production during salmonella typhimurium infections. Proc Natl Acad Sci USA (2013) 110:2252–7. doi: 10.1073/pnas.1222047110
41. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d'Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature (2013) 494:261–5. doi: 10.1038/nature11813
42. Kastele V, Mayer J, Lee ES, Papazian N, Cole JJ, Cerovic V, et al. Intestinal-derived ILCs migrating in lymph increase IFNgamma production in response to salmonella typhimurium infection. Mucosal Immunol (2021) 14:717–27. doi: 10.1038/s41385-020-00366-3
43. Zarepour M, Bhullar K, Montero M, Ma C, Huang T, Velcich A, et al. The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during salmonella enterica serovar typhimurium colitis. Infect Immun (2013) 81:3672–83. doi: 10.1128/IAI.00854-13
44. Godinez I, Raffatellu M, Chu H, Paixao TA, Haneda T, Santos RL, et al. Interleukin-23 orchestrates mucosal responses to salmonella enterica serotype typhimurium in the intestine. Infect Immun (2009) 77:387–98. doi: 10.1128/IAI.00933-08
45. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, A. Lamichhane N, et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science (2014) 345:1254009. doi: 10.1126/science.1254009
46. Grizotte-Lake M, Zhong G, Duncan K, Kirkwood J, Iyer N, Smolenski I, et al. Commensals suppress intestinal epithelial cell retinoic acid synthesis to regulate interleukin-22 activity and prevent microbial dysbiosis. Immunity (2018) 49:1103–1115 e6. doi: 10.1016/j.immuni.2018.11.018
47. Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, et al. Lipocalin-2 resistance confers an advantage to salmonella enterica serotype typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe (2009) 5:476–86. doi: 10.1016/j.chom.2009.03.011
48. Stelter C, Kappeli R, Konig C, Krah A, Hardt WD, Stecher B, et al. Salmonella-induced mucosal lectin RegIIIbeta kills competing gut microbiota. PloS One (2011) 6:e20749. doi: 10.1371/journal.pone.0020749
49. Schnizlein MK, Young VB. Capturing the environment of the clostridioides difficile infection cycle. Nat Rev Gastroenterol Hepatol (2022) 19:508–20. doi: 10.1038/s41575-022-00610-0
50. Abt MC, McKenney PT, Pamer EG. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol (2016) 14:609–20. doi: 10.1038/nrmicro.2016.108
51. Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Susac B, et al. Innate immune defenses mediated by two ILC subsets are critical for protection against acute clostridium difficile infection. Cell Host Microbe (2015) 18:27–37. doi: 10.1016/j.chom.2015.06.011
52. Frisbee AL, Saleh MM, Young MK, Leslie JL, Simpson ME, Abhyankar MM, et al. IL-33 drives group 2 innate lymphoid cell-mediated protection during clostridium difficile infection. Nat Commun (2019) 10:2712. doi: 10.1038/s41467-019-10733-9
53. Fachi JL, Secca C, Rodrigues PB, Mato FCP, Di Luccia B, Felipe JS, et al. Acetate coordinates neutrophil and ILC3 responses against c. difficile through FFAR2. J Exp Med (2020) 217:e20190489. doi: 10.1084/jem.20191520
54. Geiger TL, Abt MC, Gasteiger G, Firth MA, O'Connor MH, Geary CD, et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med (2014) 211:1723–31. doi: 10.1084/jem.20140212
55. Hasegawa M, Yada S, Liu MZ, Kamada N, Munoz-Planillo R, Do N, et al. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity (2014) 41:620–32. doi: 10.1016/j.immuni.2014.09.010
56. Peniche AG, Spinler JK, Boonma P, Savidge TC, Dann SM. Aging impairs protective host defenses against clostridioides (Clostridium) difficile infection in mice by suppressing neutrophil and IL-22 mediated immunity. Anaerobe (2018) 54:83–91. doi: 10.1016/j.anaerobe.2018.07.011
57. Cribas ES, Denny JE, Maslanka JR, Abt MC. Loss of interleukin-10 (IL-10) signaling promotes IL-22-Dependent host defenses against acute clostridioides difficile infection. Infect Immun (2021) 89:e00730-20. doi: 10.1128/IAI.00730-20
58. Cowardin CA, Buonomo EL, Saleh MM, Wilson MG, Burgess SL, Kuehne SA, et al. The binary toxin CDT enhances clostridium difficile virulence by suppressing protective colonic eosinophilia. Nat Microbiol (2016) 1:16108. doi: 10.1038/nmicrobiol.2016.108
59. Buonomo EL, Cowardin CA, Wilson MG, Saleh MM, Pramoonjago P, Petri WA Jr. Microbiota-regulated IL-25 increases eosinophil number to provide protection during clostridium difficile infection. Cell Rep (2016) 16:432–43. doi: 10.1016/j.celrep.2016.06.007
60. Tshibangu-Kabamba E, Yamaoka Y. Helicobacter pylori infection and antibiotic resistance - from biology to clinical implications. Nat Rev Gastroenterol Hepatol (2021) 18:613–29. doi: 10.1038/s41575-021-00449-x
61. Polk DB, Peek RM Jr. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer (2010) 10:403–14. doi: 10.1038/nrc2857
62. Li R, Jiang XX, Zhang LF, Liu XM, Hu TZ, Xia XJ, et al. Group 2 innate lymphoid cells are involved in skewed type 2 immunity of gastric diseases induced by helicobacter pylori infection. Mediators Inflamm (2017) 2017:4927964. doi: 10.1155/2017/4927964
63. Satoh-Takayama N, Kato T, Motomura Y, Kageyama T, Taguchi-Atarashi N, Kinoshita-Daitoku R, et al. Bacteria-induced group 2 innate lymphoid cells in the stomach provide immune protection through induction of IgA. Immunity (2020) 52:635–649 e4. doi: 10.1016/j.immuni.2020.03.002
64. Yamaoka Y, Kita M, Kodama T, Sawai N, Kashima K, Imanishi J. Expression of cytokine mRNA in gastric mucosa with helicobacter pylori infection. Scand J Gastroenterol (1995) 30:1153–9. doi: 10.3109/00365529509101624
65. Kaakoush NO, Castano-Rodriguez N, Mitchell HM, Man SM. Global epidemiology of campylobacter infection. Clin Microbiol Rev (2015) 28:687–720. doi: 10.1128/CMR.00006-15
66. Riddle MS, Gutierrez RL, Verdu EF, Porter CK. The chronic gastrointestinal consequences associated with campylobacter. Curr Gastroenterol Rep (2012) 14:395–405. doi: 10.1007/s11894-012-0278-0
67. O'Brien SJ. The consequences of campylobacter infection. Curr Opin Gastroenterol (2017) 33:14–20. doi: 10.1097/MOG.0000000000000329
68. Malik A, Sharma D, St Charles J, Dybas LA, Mansfield LS. Contrasting immune responses mediate campylobacter jejuni-induced colitis and autoimmunity. Mucosal Immunol (2014) 7:802–17. doi: 10.1038/mi.2013.97
69. Jing X, Korchagina AA, Shein SA, Muraoka WT, Koroleva E, Tumanov AV. IL-23 contributes to campylobacter jejuni-induced intestinal pathology via promoting IL-17 and IFNgamma responses by innate lymphoid cells. Front Immunol (2020) 11:579615. doi: 10.3389/fimmu.2020.579615
70. Heimesaat MM, Grundmann U, Alutis ME, Fischer A, Gobel UB, Bereswill S. The IL-23/IL-22/IL-18 axis in murine campylobacter jejuni infection. Gut Pathog (2016) 8:21. doi: 10.1186/s13099-016-0106-4
71. Edwards LA, Nistala K, Mills DC, Stephenson HN, Zilbauer M, Wren BW, et al. Delineation of the innate and adaptive T-cell immune outcome in the human host in response to campylobacter jejuni infection. PloS One (2010) 5:e15398. doi: 10.1371/journal.pone.0015398
72. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity (2015) 43:146–60. doi: 10.1016/j.immuni.2015.06.019
73. Moayeri M, Leppla SH, Vrentas C, Pomerantsev AP, Liu S. Anthrax pathogenesis. Annu Rev Microbiol (2015) 69:185–208. doi: 10.1146/annurev-micro-091014-104523
74. Gottle M, Dove S, Seifert R. Bacillus anthracis edema factor substrate specificity: evidence for new modes of action. Toxins (Basel) (2012) 4:505–35. doi: 10.3390/toxins4070505
75. Lightfoot YL, Yang T, Sahay B, Zadeh M, Cheng SX, Wang GP, et al. Colonic immune suppression, barrier dysfunction, and dysbiosis by gastrointestinal bacillus anthracis infection. PloS One (2014) 9:e100532. doi: 10.1371/journal.pone.0100532
76. Paccani SR, Baldari CT. T Cell targeting by anthrax toxins: two faces of the same coin. Toxins (Basel) (2011) 3:660–71. doi: 10.3390/toxins3060660
77. Friebe S, van der Goot FG, Burgi J. The ins and outs of anthrax toxin. Toxins (Basel) (2016) 8:69. doi: 10.3390/toxins8030069
78. Paccani SR, Benagiano M, Savino MT, Finetti F, Tonello F, D'Elios MM, et al. The adenylate cyclase toxin of bacillus anthracis is a potent promoter of T(H)17 cell development. J Allergy Clin Immunol (2011) 127:1635–7. doi: 10.1016/j.jaci.2010.12.1104
79. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350:981–5. doi: 10.1126/science.aac9593
80. Walker JA, Clark PA, Crisp A, Barlow JL, Szeto A, Ferreira ACF, et al. Polychromic reporter mice reveal unappreciated innate lymphoid cell progenitor heterogeneity and elusive ILC3 progenitors in bone marrow. Immunity (2019) 51:104–118 e7. doi: 10.1016/j.immuni.2019.05.002
81. Kumar V. Innate lymphoid cell and adaptive immune cell cross-talk: a talk meant not to forget. J Leukoc Biol (2020) 108:397–417. doi: 10.1002/JLB.4MIR0420-500RRR
82. Sonnenberg GF, Hepworth MR. Functional interactions between innate lymphoid cells and adaptive immunity. Nat Rev Immunol (2019) 19:599–613. doi: 10.1038/s41577-019-0194-8
83. Withers DR. Innate lymphoid cell regulation of adaptive immunity. Immunology (2016) 149:123–30. doi: 10.1111/imm.12639
84. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature (2013) 498:113–7. doi: 10.1038/nature12240
85. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Immune tolerance. group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science (2015) 348:1031–5. doi: 10.1126/science.aaa4812
86. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
87. Withers DR, Gaspal FM, Bekiaris V, McConnell FM, Kim M, Anderson G, et al. OX40 and CD30 signals in CD4(+) T-cell effector and memory function: a distinct role for lymphoid tissue inducer cells in maintaining CD4(+) T-cell memory but not effector function. Immunol Rev (2011) 244:134–48. doi: 10.1111/j.1600-065X.2011.01057.x
88. Deng T, Suo C, Chang J, Yang R, Li J, Cai T, et al. ILC3-derived OX40L is essential for homeostasis of intestinal tregs in immunodeficient mice. Cell Mol Immunol (2020) 17:163–77. doi: 10.1038/s41423-019-0200-x
89. Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy (2014) 69:1300–7. doi: 10.1111/all.12446
90. Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity (2015) 42:538–51. doi: 10.1016/j.immuni.2015.02.007
91. Schwartz C, Khan AR, Floudas A, Saunders SP, Hams E, Rodewald HR, et al. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J Exp Med (2017) 214:2507–21. doi: 10.1084/jem.20170051
92. Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol (2016) 17:65–75. doi: 10.1038/ni.3308
93. Duster M, Becker M, Gnirck AC, Wunderlich M, Panzer U, Turner JE. T Cell-derived IFN-gamma downregulates protective group 2 innate lymphoid cells in murine lupus erythematosus. Eur J Immunol (2018) 48:1364–75. doi: 10.1002/eji.201747303
94. Mao K, Baptista AP, Tamoutounour S, Zhuang L, Bouladoux N, Martins AJ, et al. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature (2018) 554:255–9. doi: 10.1038/nature25437
95. Bauche D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3(+) regulatory T cells restrain interleukin-23-Producing CX3CR1(+) gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity (2018) 49:342–352 e5. doi: 10.1016/j.immuni.2018.07.007
96. Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I, et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J Allergy Clin Immunol (2017) 139:1468–1477 e2. doi: 10.1016/j.jaci.2016.08.034
97. Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol (2014) 192:2442–8. doi: 10.4049/jimmunol.1300974
98. Melo-Gonzalez F, Kammoun H, Evren E, Dutton EE, Papadopoulou M, Bradford BM, et al. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J Exp Med (2019) 216:728–42. doi: 10.1084/jem.20180871
99. Kruglov AA, Grivennikov SI, Kuprash DV, Winsauer C, Prepens S, Seleznik GM, et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science (2013) 342:1243–6. doi: 10.1126/science.1243364
100. Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity (2012) 37:1061–75. doi: 10.1016/j.immuni.2012.08.024
101. Ahlfors H, Morrison PJ, Duarte JH, Li Y, Biro J, Tolaini M, et al. IL-22 fate reporter reveals origin and control of IL-22 production in homeostasis and infection. J Immunol (2014) 193:4602–13. doi: 10.4049/jimmunol.1401244
102. Silberger DJ, Zindl CL, Weaver CT. Citrobacter rodentium: a model enteropathogen for understanding the interplay of innate and adaptive components of type 3 immunity. Mucosal Immunol (2017) 10:1108–17. doi: 10.1038/mi.2017.47
103. Ota N, Wong K, Valdez PA, Zheng Y, Crellin NK, Diehl L, et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with citrobacter rodentium. Nat Immunol (2011) 12:941–8. doi: 10.1038/ni.2089
104. Cobb LM, Verneris MR. Therapeutic manipulation of innate lymphoid cells. JCI Insight (2021) 6:e146006. doi: 10.1172/jci.insight.146006
105. Bennstein SB, Uhrberg M. Biology and therapeutic potential of human innate lymphoid cells. FEBS J (2022) 289:3967–81. doi: 10.1111/febs.15866
Keywords: GI tract, bacteria, IL-22, ILCs, innate lymphoid cells, modulation of immune response
Citation: Sah P and Zenewicz LA (2023) Modulation of innate lymphoid cells by enteric bacterial pathogens. Front. Immunol. 14:1219072. doi: 10.3389/fimmu.2023.1219072
Received: 08 May 2023; Accepted: 22 June 2023;
Published: 06 July 2023.
Edited by:
Laurel L. Lenz, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Liang Zhou, University of Florida, United StatesAmanda M. Jamieson, Brown University, United States
Copyright © 2023 Sah and Zenewicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lauren A. Zenewicz, lauren-zenewicz@ouhsc.edu