 Xinting Wang1
Xinting Wang1 Hua Zhou1,2*
Hua Zhou1,2* Qian Liu1
Qian Liu1 Peipei Cheng1
Peipei Cheng1 Tingyao Zhao3Tianshu Yang2Yue Zhao4Wanjing Sha1Yanyan Zhao4*Huiyan Qu2*
Tingyao Zhao3Tianshu Yang2Yue Zhao4Wanjing Sha1Yanyan Zhao4*Huiyan Qu2*- 1Institute of Cardiovascular Disease of Integrated Traditional Chinese and Western Medicine, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Department of Cardiovascular Disease, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3Guanghua Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 4Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, China
Cardiovascular diseases (CVDs) are the leading cause of death and disability worldwide. The CVDs are accompanied by inflammatory progression, resulting in innate and adaptive immune responses. Regulatory T cells (Tregs) have an immunosuppressive function and are one of the subsets of CD4+T cells that play a crucial role in inflammatory diseases. Whether using Tregs as a biomarker for CVDs or targeting Tregs to exert cardioprotective functions by regulating immune balance, suppressing inflammation, suppressing cardiac and vascular remodeling, mediating immune tolerance, and promoting cardiac regeneration in the treatment of CVDs has become an emerging research focus. However, Tregs have plasticity, and this plastic Tregs lose immunosuppressive function and produce toxic effects on target organs in some diseases. This review aims to provide an overview of Tregs’ role and related mechanisms in CVDs, and reports on the research of plasticity Tregs in CVDs, to lay a foundation for further studies targeting Tregs in the prevention and treatment of CVDs.
1 Introduction
Regulatory T cells (Tregs), CD4+CD25+Foxp3+Tregs, secrete anti-inflammatory factors such as interleukin (IL)-10 and transforming growth factor-β (TGF-β), which have immunosuppressive effects (1). Tregs account for 5 ~ 10% of all CD4+ T cells. There are two sources of Tregs: derived from the normal thymus (natural Tregs, nTregs); Or derived from peripheral naive CD4+ T cells induced to differentiate into Tregs (inducible Tregs, iTregs). Foxp3 is a specific marker of Tregs, and an essential regulator of Tregs development and function (2). In comparison, the absence of Foxp3 will lead to the loss of Treg function, which is closely associated with severe autoimmune diseases in humans (3) and rodents (4). Tregs play a key role in immune dynamic balance (5) and regulate immunity in Corona Virus Disease 2019 (COVID-19) (6), tumors (7), infectious diseases (8), and transplant rejection (9, 10).
Cardiovascular diseases (CVDs) are the leading cause of death and disability worldwide, with the number of people affected by CVDs increasing from 271 million in 1990 to 523 million in 2019 (11). Inflammation plays a vital role in many CVDs, disrupting the immune balance of the body and causing innate and adaptive immune responses. Tregs can prevent the progression of CVDs by regulating immunity (12). In this review, we summarized the role and related mechanisms of Tregs in the prevention and treatment of CVDs, mainly reflected in the regulation of immune balance, inflammation, cardiac and vascular remodeling, immune tolerance, and cardiac regeneration. CVDs cover heart failure (HF), myocardial infarction (MI), myocarditis, atherosclerosis, hypertension, and atrial fibrillation. The research progress and clinical potential of targeted Tregs therapy for CVDs are further elaborated.
2 Common CVDs and their epidemiology
2.1 Heart failure
2022 AHA/ACC/HFSA defines HF as a complex clinical syndrome with symptoms and signs that result from any structural or functional impairment of ventricular filling or ejection of blood (13). The latest data for 2021 show that the number of HF patients worldwide has increased from 33.5 million in 1990 to 64.3 million in 2017 (14). Up to 25% ~ 40% of patients die of Chronic HF one year after being diagnosed with HF (15, 16). HF is a leading cause of death, affecting more than 24 million people worldwide (17). HF is a significant public health problem in the world with high incidence, re-hospitalization, disability, and mortality (18). The occurrence and development of HF are accompanied by activation and inflammation of the immune system (19), and the immune system regulates inflammation by secreting related factors.
2.2 Myocardial infarction
MI is ischemic necrosis of the myocardium caused by transient or persistent occlusion of the distal coronary artery, associated with high morbidity and mortality, resulting in more than 15 million deaths worldwide every year (20, 21). Due to different medical conditions, MI prevalence varies widely among regions, ranging from 3 to 20% (22–24). Although coronary revascularization treatment strategies can reduce MI mortality (25), MI is associated with inflammation, cardiac remodeling, myocardial fibrosis, and other pathological processes (21, 26), which aggravate clinical cardiovascular malignant events. The immune system plays a critical role in the occurrence and development of post-MI inflammation (27).
2.3 Myocarditis
Myocarditis is an inflammatory disease of myocardium cells with a broad range of clinical and histological manifestations of cardiac pathological immune processes that can lead to acute HF, sudden death, and chronic dilated cardiomyopathy (28, 29). Myocarditis can be attributed to immune responses, viral infections, and bacterial infections. Myocarditis includes periods of acute inflammation, subacute inflammation, and myopathy, resulting in cardiac remodeling, myocardial fibrosis, and cardiac dysfunction (30, 31). Cardiac magnetic resonance imaging (MRI) and molecular detection of viruses by endomyocardial biopsy are effective methods for the clinical diagnosis of myocarditis. However, it is difficult to sample human heart tissue, and it is necessary to explore the pathophysiological mechanisms in experimental animal models (32, 33). Experimental autoimmune myocarditis (EAM) induced by myocardial myosin is a classic model of autoimmune myocarditis (34). Viral myocarditis (VMC) caused by Coxsackievirus B3 (CVB3) infection is the main cause of sudden cardiac death in the young population (30). VMC is characterized by immune-mediated inflammation of the myocardium caused by viral infection (35).
Chronic Chagas disease cardiomyopathy (CCC), progressive inflammation of the heart caused by Trypanosoma cruzi (T.cruzi) infection, manifesting as diffuse myocardial fibrosis, cardiac hypertrophy, myocardial injury, progression to HF, and death, has become a major public health disease in Latin America (36). Chagas’ etiology of HF has become the third most common indication for heart transplantation in South America (37). Parasite-dependent myocardial aggression and immune-mediated tissue damage are key pathological mechanisms of CCC (38, 39). Therefore, targeted modulation of immunity has become a strategy for the treatment of CCC (40).
2.4 Hypertension
The number of hypertensive patients worldwide has grown from 128 million in 1990 to 650 million in 2019, and more than 700 million were unaware of their hypertension status (41). Hypertension is an important risk factor for CVDs, which significantly increases the incidence of coronary heart disease and HF complications. Hypertension is an inflammatory disease, and the inflammatory markers C-reactive protein (CRP), various cytokines, and pathway complement pathway products are increased in patients with hypertension (42).
2.5 Atherosclerosis
In 2020, nearly 2 billion people worldwide suffer from carotid atherosclerosis, which increases the risk of coronary heart disease events (43). In the general middle-aged population, 42.5% had silent coronary atherosclerosis and 5.2% had severe atherosclerosis (coronary significant stenosis ≥50%) (44). Atherosclerosis is a chronic inflammatory disease of the vascular wall which involves cellular immune responses (45). Acute and chronic myocardial ischemia caused by coronary atherosclerosis is the most common cause of HF, and studies have shown that Tregs have atherosclerotic protective effects (46, 47).
3 Tregs-related membrane molecules
3.1 CD4/CD25
T cells specifically recognize antigens presented by antigen-presenting cells (APCs) through T cell receptors (TCRs), and recognize antigens through CD3 molecular transduction, forming TCR-CD3 complexes, generating activation signals, and transmitting them to cells. CD4 recognizes and binds MHC-II molecules, and CD4+T cells specifically recognize exogenous antigens presented by MHC-II molecules. Tregs highly express IL-2 receptor α (IL-2Rα, CD25), It promotes the binding of IL-2 and CD25 without binding with other receptors. It is called CD25-biased IL-2 antibody complexes, which promote the activation and proliferation of Tregs (48). Tregs highly express the high-affinity receptor for IL-2 and competitively prey on IL-2 that is required for the survival of neighboring activated T cells, resulting in suppressed proliferation, followed by apoptosis, of activated T cells.
3.2 CD28
CD28 is a homodimer composed of two identical peptide chains, which is expressed in 90% of CD4+T cells. The costimulatory signal produced by CD28 plays an important role in the activation of T cells. Gain of Tregs function was accomplished by therapeutic administration of superagonistic CD28-specific monoclonal antibodies (CD28-SA) that preferentially activate Tregs over conventional CD4+ T cells in vivo due to a vigorous co-stimulatory signal induced by cross-linking of CD28 molecules (49, 50). CD28 super-agonists, which effectively target Tregs, hold great promise for the treatment of human autoimmune diseases (51).
3.3 CTLA4
Tregs constitutively express the inhibitory receptor cytotoxic T-lymphocyte associated protein 4 (CTLA4, CD152). The cytoplasmic region of CTLA4 has immunoreceptor tyrosine-based inhibitory motifs (ITIMs), which transmit inhibitory signals. Human CTLA-4 haploinsufficiency caused dysregulation of Foxp3+ Tregs, hyperactivation of effector T cells, and lymphocytic infiltration of target organs (52). Deletion of CTLA-4 in mice impairs Tregs’ suppressive function, causing severe autoimmune disease and early lethality, despite normal Foxp3 levels (53, 54).
3.4 PD-1
Programmed cell death protein 1(PD-1, CD279) is a Treg surface costimulatory marker molecule with ligands programmed cell death ligand 1 (PD-L1) and PD-L2. PD-1, when bound to its ligands, can inhibit the proliferation of effector T cells and activated B cells. Furthermore, PD-L1-Ig induced Naïve CD4+ T cells to differentiate, proliferate into CD4+Foxp3+Tregs, and enhanced the immunosuppressive function of Tregs (55, 56).
3.5 CD39/CD73
Ectonucleoside triphosphate diphosphohydrolase-1 (ENTPD1, CD39) and ecto-5 ′-nucleotidase (e5NT, CD73) are expressed on the surface of Tregs. CD39 degrades adenosine triphosphate (ATP) into adenosine diphosphate (ADP)/adenosine monophosphate (AMP), CD73 degrades ADP/AMP into adenosine, and the CD39/CD73 pathway converts pro-inflammatory ATP into adenosine with anti-inflammatory properties, which further exerts immunosuppressive functions and inhibits the activation of T cells and the production of inflammatory mediators (57, 58). CD73 deficiency reduces cardiac chemotaxis of Tregs, impairing the immunosuppressive and protective functions of Tregs during cardiac healing (59). Increased Foxp3 nuclear levels and enhanced CD39 and CD73 transcription in NADPH oxidase 2 (NOX2) KO Tregs effectively inhibit effector T cell proliferation and reverse angiotensin (Ang) II-induced cardiac remodeling (60).
4 Tregs regulate immune balance in CVDs
4.1 Th17/Treg
Naive CD4+ T cells differentiate into different subsets of cells according to different cytokine environments, including 1 helper T (Th1)cells, Th2 cells, Th17 cells (61), and Tregs, which share the exact origin but exhibit opposite effects (62). Th17 cells express the transcription factor retinoid-related orphan receptor-γt (RORγt). Th17 cells, characterized by the production of IL-17, contribute to fibrosis and fibrotic diseases (63), induce autoimmunity, and promote inflammation (64). IL-17 activates the protein kinase C (PKC)β/extracellular signal-regulated kinase 1/2 (ERK1/2)/nuclear factor-κB (NF-κB)-dependent signaling pathway to aggravate the degree of myocardial fibrosis (65). IL-17 activates the MAPK pathway and increases the expression of downstream target genes IL-6, tumor necrosis factor (TNF), C-C Motif Chemokine Ligand (CCL) 20, and C-X-C Motif Chemokine Ligand (CXCL) 1 to worsen cardiac remodeling (66). The microRNA mmu-miR-721, synthesized by Th17 cells, was present in the plasma of mice with acute autoimmune or viral myocarditis, but not in those with AMI. And the human homolog (hsa-miR-Chr8:96) is a novel microRNA that distinguishes myocarditis patients from MI patients (67). Tregs inhibit inflammation and regulate immune balance by secreting IL-10 and TGF-β (1, 68). IL-10 is a key anti-inflammatory mediator. IL-10 treatment significantly improves the left ventricular dilation and ejection fraction of MI mice, promotes the polarization of M2 macrophages to reduce cardiac inflammation, activates fibroblasts to reduce extracellular matrix collagen deposits, and promotes cardiac healing and improves cardiac remodeling (69). However, IL-10 gene deletion enhanced neutrophil infiltration, increased inflammation, enlarged myocardial infarction area, and myocardial necrosis in ischemia-reperfusion mice (70). TGF-β is a crucial enforcer of immune homeostasis and tolerance, and plays an important role in cell development, differentiation, inflammation, and tissue repair (71). However, TGF-β1 gene deletion results in nearly 50% mouse embryonic lethality, with mice born with uncontrolled inflammation and dying at 3-4 weeks (72, 73). Th17/Treg maintains immune dynamic equilibrium when the number and function of Th17 cells and Tregs are balanced.
Clinically, increased Th17 cells ratio and decreased Tregs ratio lead to pathological manifestations of Th17/Treg immune imbalance, which are widely found in patients with cardiac inflammatory diseases such as acute coronary syndrome (74), congestive HF (75), and rheumatic heart disease (76). Serum IL-17 levels of Th17 characteristic cytokine were significantly increased in HF patients, and IL-10 levels of Tregs characteristic cytokine were significantly decreased (75, 77). The Th17/Treg ratio is an independent predictor for 1-year mortality in patients with MI-related cardiogenic shock (78). Th17/Treg ratio combined with CRP level in serum predicts atrial fibrillation after off-pump coronary artery bypass transplantation (79). Moreover, intensive statin therapy improves Th17/Treg functional imbalance in patients with non-ST elevation acute coronary syndromes undergoing percutaneous coronary intervention, reduces cytokines IL-17, IL-6, and IL-23 secreted by Th17 cells, and increases cytokines IL-10 and TGF-β1 secreted by Tregs (80). The pathological phenomenon of Th17/Treg imbalance is widely found in obese children with systolic hypertension (81), patients with resistant hypertension (82), carotid atherosclerotic hypertension (83), and pulmonary hypertension (84). The Th17/Treg imbalance is a vital contributor to the high incidence of atherosclerosis in systemic lupus erythematosus patients (85). In addition, The Th1/Treg ratio and Th17/Treg ratio were significantly increased in patients with rheumatoid arthritis combined with atrial fibrillation, and the increased Th1/Treg ratio was a risk factor for rheumatoid arthritis combined with atrial fibrillation (86).
Mechanistically, in the studies of the ischemic HF model induced by coronary artery ligation in mice (87), and the HF model induced by abdominal aortic ligation in rats (77), it was found that Th17/Treg ratio was increased in failing myocardium. Th17/Tregs imbalance regulates cardiac fibrosis and heart failure in rats by regulating lysyl oxidase (LOX) expression, Th17 cells aggravate fibrosis-related indicators (matrix metalloproteinase-2/matrix metalloproteinase-9 (MMP-2/9) and collagen I/III) and LOX expression by activating the IL-17/ERK1/2-activating protein-1 (AP-1) pathway, while Tregs inhibit fibrosis-related indicators and LOX expression by activating the IL-10/Janus kinase (JAK) 1-signal transducer and activator of transcription (STAT)3 pathway (77). allogeneic skeletal myoblasts transplantation (allo-SMT) is a potential strategy to treat MI. However, the host immune response to donor skeletal myoblasts is intensified, as evidenced by further Th17/Treg imbalance, which reduces the therapeutic effect of allo-SMT. It was confirmed that transfected vascular endothelial-derived growth factor (VEGF) 165 allo-SMT decreased the expression of RORγt, increased the expression of Foxp3, and promoted the Th17/Treg balance in MI (88). Furthermore, aerobic exercise (89), and catechin (90) interventions can significantly reduce the cardiac Th17/Treg ratio in HF model animals, and improve the cardiac function and immune environment. Targeted inhibition of microRNA-155 significantly reduced cardiac Th17 cell infiltration, and Th17 cells related factor (RORγT, IL-17A, IL-6) expression levels decreased in EAM mice. Targeted inhibition of microRNA-155 resulted in increased expression of Th17 cells related proinflammatory factors (RORγT, IL-17A, IL-21, IL-22) in splenic CD4+ T cells of EAM mice and Treg associated anti-inflammatory factors (Foxp3, TGF-β, IL-10, IL-35) were downregulated without affecting Treg function. Therefore, Targeted inhibition of microRNA-155 attenuated myocardial inflammation, mainly inhibiting Th17 cell immune responses, and then adjusted the immune balance of Th17/Treg (91). Fenofibrate intervention (92) can reduce the severity of EAM disease and cardiac injury by regulating Th17/Treg immune response.
Long-term exposure of parents to particulate matter (PM) 2.5 air pollution may induce increased blood pressure in offspring by mediating an imbalance of the Th17/Treg immune microenvironment (93). Interventions with Lactobacillus fermentum CECT5716 (94), fecal microbiota transplantation (95), and Dieckol (96) attenuate Th17/Treg imbalance in the mesenteric lymph nodes and aorta of spontaneously hypertensive rats (SHR), attenuate endothelial cell dysfunction, and control blood pressure. Electroacupuncture effectively reduces systolic blood pressure by promoting SHR Th17/Treg immune balance (97). Inhibition of serum/glucocorticoid regulated kinase 1 (SGK1) can reduce the translocation of factor forkhead box O1 (FoxO1) from the cytoplasm to the nucleus, ameliorate the Th17/Treg imbalance, and target organ damage to the heart and kidney in Ang II-induced hypertension mice (98).
Th17 cells mediate pro-inflammatory responses to exacerbate atherosclerosis, whereas Tregs exert atheroprotective effects by suppressing inflammation and stabilizing plaques (99). Targeting the Th17/Treg balance has emerged as a strategy for the treatment of atherosclerosis (100). Th17/Treg function is imbalanced during high-fat diet-induced atherosclerosis in age and apolipoprotein E (ApoE)-/- mice (101), and Porphyromonas gingivalis oral infection further exacerbated Th17/Treg imbalance and atherosclerosis plaque deterioration (102). However, pharmacologic interventions by pioglitazone (103), traditional Chinese medicine AnGong Niuhuang Pill (104), and Yangyin Qingre Huoxue Prescription (105) exert anti-atherosclerotic effects by regulating Th17/Treg balance, inhibiting chronic inflammation, reducing plaque collagen fibers, and stabilizing plaques.
4.2 CD4+ T cell subsets and Tregs
The earliest CD4+ T cell subsets to be discovered are Th1 cells and Th2 cells; Secretion of INF- γ, IL-2, and TNF by Th1 cells, and the key transcription factor is T-bet; Th2 cells secrete IL-4, IL-5, and IL-13, and the key transcription factor is GATA-3. Patients with acute coronary syndrome have a decreased proportion of circulating Tregs and an increased proportion of Th1 and Th17 cells. IL-37-treated human dendritic cells acquire a tolerogenic dendritic cells (tDCs) phenotype, with tDCs promoting the expansion of CD4+ T cells into Tregs and reducing Th1 and Th17 populations (106). Blocking angiotensin II (AII) production with angiotensin-converting enzyme (ACE), inhibitors or inhibiting AII signal transduction with angiotensin type 1 receptor (AT1R) blockers inhibited self-reactive Th1 and Th17 cells and promoted CD4+FoxP3+Tregs (107). Cardiac biopsy in patients with dilated cardiomyopathy showed that cardiac T cell infiltration was characterized by differential expression of functional T cell markers, including Th1 markers (IFN-γ, T-bet, Eomesodermin), Tregs (Treg; Foxp3, TGF-β) and cytotoxic T-cells (CTL: Perforin, Granulysin, Granzyme A) increased significantly, while Th17 had no major effect (108). Th1 cells promote inflammation and increase the volume of MI (109). Seven days after MI, the CD4+ T cells in the heart of hyaluronan synthase 3 (HAS3) KO mice were significantly reduced, with CD4+CXCR3+Th1 cells and CD4+CD25+Tregs (110). Progranulin down-regulates the response of Th1 and Th17 cells and the production of inflammatory cytokines by inhibiting the JAK/STAT pathway, and improving CVB3-induced VMC (111). Nicotine activates the cholinergic anti-inflammatory pathway to reduce the inflammatory response of VMC. Nicotine treatment increases the proportion of Th2 cells and Tregs, reduces the proportion of Th1 and Th17 cells in the spleen, and reduces the myocardial injury and inflammatory cell infiltration of VMC (112). VMC mice vagotomy inhibited the activation of JAK2/STAT3 and enhanced NF-κB in spleen CD4+T cells, resulting in an increase in the proportion of Th1 and Th17 cells and a decrease in the proportion of Th2 cells and Tregs in the spleen (113). In atherosclerotic diseases, Th1 plays a pro-inflammatory role while Tregs play an anti-inflammatory role. LCK inhibitor inhibits PP2, inhibits the infiltration of CD4+ T cells in plaque, increases Tregs, and reduces the synthesis of TNF-γ And TNF-α by Th1 cells, Inhibition of PI3K/AKT/mTOR signal activation reduces Th1/Treg ratio and plays an anti-atherosclerotic role (114). Ang II treatment of ApoE-/- mice resulted in plaque enlargement and modulation of CD4 T cell subset activity: increased Th1 and Th17 cells; Decreased Th2 cells and Tregs. Valsartan can reduce the systolic pressure of Ang II treated ApoE-/- mice, promote the differentiation of CD4+T cells into Th2 cells and Tregs, improve the immune balance, and stabilize the atherosclerosis plaque (115). Allergic asthma accelerated atherosclerosis and was accompanied by increased splenic Th2 and Th17 cells and decreased Tregs. Curcumin treatment for 8 weeks attenuates the aggravation of atherosclerotic lesions and stabilizes plaques by decreasing Th2 and Th17 cells and increasing Tregs, which regulate the balance of Th2/Tregs in asthmatic ApoE-/- mice (116). The immune balance involved by Tregs and the differentiation of Naïve CD4+ T cells are shown in Figure 1.
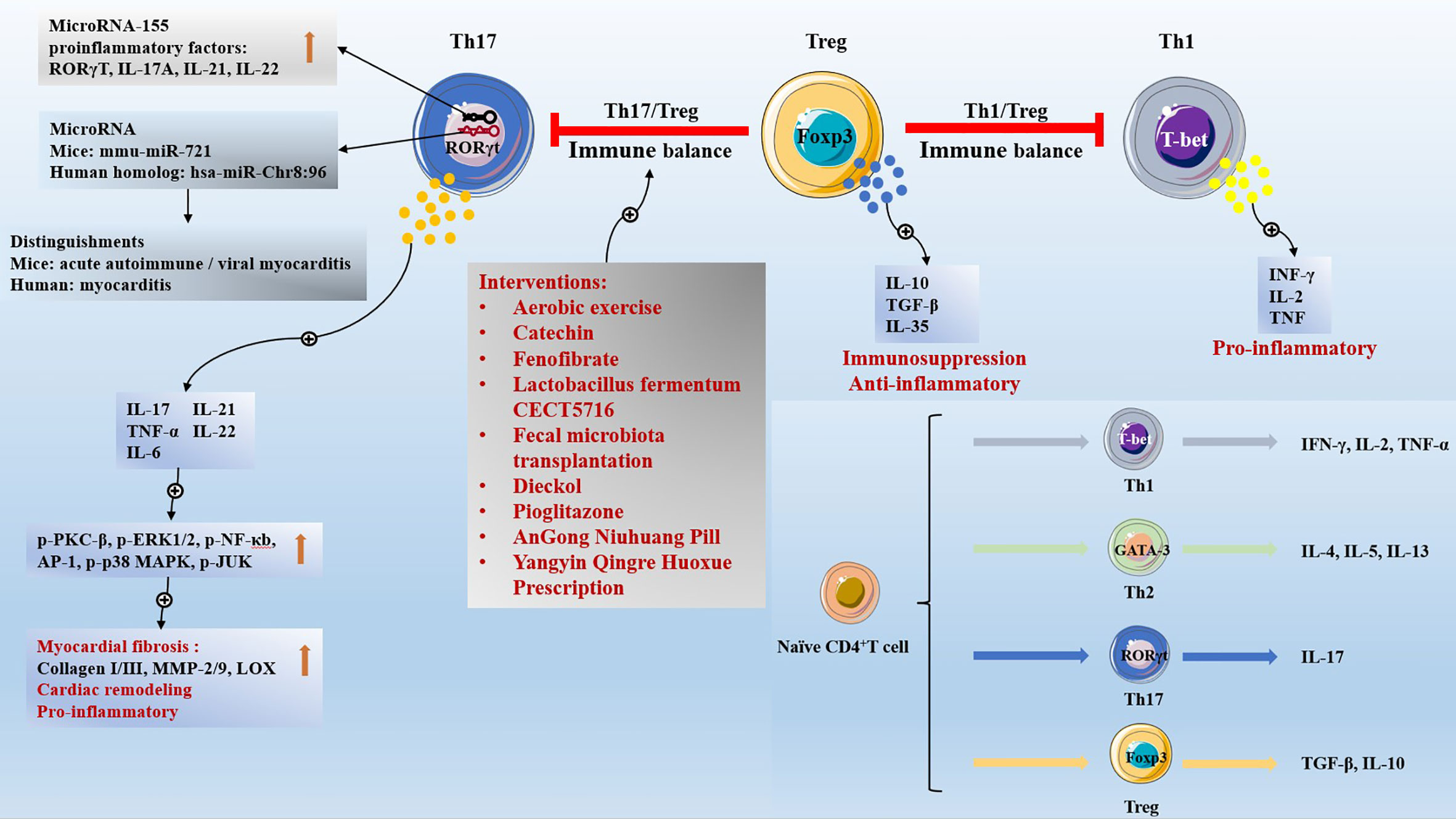
Figure 1 The immune balance involved by Tregs and the differentiation of Naïve CD4+ T cells.
5 Tregs regulate inflammation in CVDs
5.1 Myocardial infarction
MI is a sterile inflammatory response disease with exacerbated inflammation in the heart. MI leads to the death of cardiomyocytes exposed to endogenous damage-associated molecular patterns (DAMPs) of the innate immune system, and damps are recognized by pattern recognition receptors (PRRs), which promote the release of chemokines and proinflammatory cytokines that recruit and activate neutrophils, monocytes, and macrophages to the infarct zone, exacerbating cardiac inflammation. Importantly, the monocytes recruited from the circulation are differentiated into macrophages in the infarct zone, which are monocytes/macrophages (Mos/Mps). Mos/Mps are critical immune cells that determine the progression and repair of inflammation after MI. Macrophages have two phenotypes: M1 Macrophages with pro-inflammatory properties, and M2 Macrophages with anti-inflammatory and repair properties (117–119). Treg promoted the polarization of Mos/Mps to M2 type and improved immune homeostasis and cardiac repair after MI. However, in Treg-depleted mice (Foxp3DTR) MI model mice with Treg ablation and MI model mice with Treg depletion, Mos/Mps polarized to the M1 type and intensified the inflammatory response (120). DCs-derived exosomes activated Tregs-mediated M2 type polarization of macrophages, and significantly increased border zone infiltration of Tregs and M2 macrophages in MI model mice, thereby improving cardiac function (121). As a pro-inflammatory mediator of C-C motif chemokine receptor (CCR) 2+ macrophages and DCs, CCL17 inhibits Tregs recruitment by biased activation of CCR4. However, deletion of the CCL17 gene enhanced Tregs recruitment, weakened gene expression of inflammatory macrophages, and improved heart function and cardiac remodeling in MI mice (122). After MI, CXCL12/C-X-C motif chemokine receptor (CXCR) 4 chemotaxis inflammatory cells to the infarct area. CXCR4 antagonist specifically enhanced the recruitment of splenic Treg to the infarct zone by initiating DCs, inhibited the gene expression of pro-inflammatory Mos/Mps, improved cardiac function, and promoted cardiac repair in AMI reperfusion mice (123). Nuclear paraspeckle assembly transcript 1 (NEAT1) is a novel long noncoding RNA (IncRNA) immunomodulator that affects the process of Mos/Mps and T cell differentiation. LncRNA NEAT1 expression is decreased in peripheral blood monocytes of MI patients. Maldifferentiation of Mos/Mps in the bone marrow and blood of NEAT1-/- mice, abnormal differentiation of Tregs in the spleen, increased infiltration of CD68+ macrophages in the aortic wall, and imbalance of the immune system (124). In addition, CD73 derived from CD4+Foxp3+Tregs can bind to CD4+Foxp3-Teffs and reduce IL-1β, TNF-α, IFN-γand IL-17 levels, suppressing inflammatory responses and protecting against MI (59).
5.2 Hypertension
Treg deficiency exacerbates hypertension progression by enhancing innate and adaptive immune responses (125, 126). Depletion of Tregs significantly increased systolic blood pressure (127). Adoptive transfer of Tregs improved hypertension, vasodilatory injury, and immune cell infiltration (128), and inhibited autophagy, oxidative stress, and inflammation to improve hypertensive microvascular function (129). Complement C3a receptor (C3aR) and complement C5a receptor (C5aR) double knockout mediates Tregs function and attenuates Ang II-induced inflammatory cytokine expression, target organ injury, and elevated blood pressure (130). Cystathionine γ lyase-derived hydrogen activates liver kinase B1 (LKB1) and promotes differentiation and proliferation of Tregs, reducing immune inflammation in blood vessels and kidneys, thereby preventing hypertension (128). Doxycycline improves intestinal barrier integrity by reducing Lactobacillus and high plasma L-lactate levels, reducing aortic oxidative stress, increasing Tregs infiltration and IL-10, and improving vascular dysfunction and blood pressure in deoxycorticosterone acetate (DOCA)-salt-induced hypertension model rats (131). Activation of the PD-1/PD-L1 pathway significantly increased Tregs ratio and Foxp3 mRNA expression, and increased the levels of anti-inflammatory factors TGF-β, IL-10, and IL-35 in peripheral blood monocytes (PBMC), improving gestational hypertension (132).
5.3 Atherosclerosis
Atherosclerosis is a vascular inflammatory disease (133). In CVDs, atherosclerotic lesions can cause cardiac ischemia and lead to infarction. Significantly, the adoptive transfer of Tregs dampens inflammatory responses and protects against atherosclerosis (134, 135). Tregs inhibit effector T cells, induce M2-type polarization of macrophages, and accumulate them in plaques, enhancing inflammation dissipating and plaque regression. During lipid-lowering therapy, Tregs in regressing plaques are peripherally induced and characterized by the lack of Neuropilin 1 (Nrp1) and Helios expression (136). Activation of the Tregs/Indoleamine 2,3-dioxygenase axis forms a tolerant immune environment characterized by reducing vascular inflammation and atherosclerotic lesions (137), which has a protective effect on atherosclerotic CVDs. Overexpression of autophagy related 14 (ATG14) can reverse the autophagy dysfunction of macrophages in ApoE-/- mice plaques, inhibit the accumulation of sequestosome 1 (SQSTM1)/P62, promote the differentiation of Tregs and up-regulate the number of Tregs, and reduce the inflammation and lesions of atherosclerosis (138). Recombinant human IL-37 (139) and traditional Chinese medicine Si-Miao-Yong-An decoction (140) can regulate the immune environment and improve atherosclerotic lesions by reducing inflammatory macrophage infiltration and increasing Tregs. Activation of Tregs/Indoleamine 2,3-dioxygenase axis forms a tolerant immune environment characterized by reducing vascular inflammation and atherosclerotic lesions (137), which has a protective effect on atherosclerotic CVDs. However, Inducible T cell costimulatory (ICOS)-/- (141), CD80-/-CD86-/- (135), and hyperhomocysteinaemia (134) can reduce the number of Tregs, suppress the immunosuppressive function, and aggravate the development of atherosclerosis. Tregs depletion exacerbates atherosclerotic lesions, which are associated with hypercholesterolemia caused by abnormal lipoprotein metabolism (142), and exacerbates inflammatory responses by preventing plaque contraction (136).
5.4 Experimental autoimmune myocarditis
Single-cell RNA sequencing analysis of CD45+ cells extracted from the hearts of EAM model mice revealed that Tregs were the predominant T-cell population detected during the subacute inflammatory phase (143). Extracellular vesicles secreted by human-derived heart stromal/Progenitor cells (144), adenovirus vector-mediated gene transfer of CTLA4 Ig fusion protein (145), CD28 superagonists (146), and Oleanolic Acid (147) interventions can protect the heart function and alleviate inflammation of EAM model rodents by increasing the number of Tregs and enhancing the immunosuppressive function of Tregs. Overexpression of Mir-223-3p (148) and Protosappanin A intervention (149) can promote the phenotypic transformation from DCs to tDCs, induce Tregs generation, and inhibit cardiac inflammation and cardiac remodeling in EAM model mice. Of concern, EAM susceptibility differs between strains of mice. Compared to B10.S mice, A.SW mice have a lower ratio of Tregs in vivo, enhanced Th17 cell responses, greater sensitivity to autoimmunity, and more severe disease development in EAM (150).
5.5 Viral myocarditis
Intervention methods such as IL-37 (151) and Valproic acid (152) promote Th17/Treg immune balance and play an anti-inflammatory role, ameliorating CVB3-induced VMC. Cardiac Myosin peptide treatment and OX40 blockade (153), Fasudil (154), and nicotine (112) interventions improved cardiac inflammation and reduced mortality in CVB3-induced VMC mice by enhancing Tregs function. Adoptive transfer of Tregs can regulate TGF-β-Coxsackie-Adenovirus Receptor Pathway (155), promote monocyte differentiation into Ly6ClowCCR2lowCX3CR1high subgroup with anti-inflammatory properties (156), enhance IL-10 secretion (157), and ameliorate cardiac function, inflammatory injury, and myocardial fibrosis in CVB3-induced VMC mice. B-cell deficiency can significantly reduce Tregs, damage Tregs’ immunosuppressive function, and damage myocardial Tregs homeostasis in CVB3-induced VMC mice, whereas adoptive transfer of B cells reverses this phenomenon (158). Latency associated peptide (LAP) is a membrane protein of Tregs. Compared with total Tregs, LAP+Tregs have greater immunomodulatory effects and may serve as a better VMC biomarker (159). In addition, Astragalus Mongholicus (Fisch.) Bge intervention improved cardiac function and peripheral Tregs immune imbalance in children with VMC by reducing miRNA-146b and miRNA-155 levels (160). The release of sex hormones and/or other mediators from the testis inhibits the population of anti-inflammatory cells in the heart, including Tregs, leading to more severe acute myocarditis with CVB3 infection in male mice (161). However, the adoptive transfer of M2 macrophages promoted peripheral Tregs differentiation and reduced cardiac inflammation in CVB3-induced VMC model male mice (162).
5.6 Chronic Chagas disease cardiomyopathy
Tregs are subsets of anti-inflammatory T cells with immunosuppressive functions that help limit tissue damage associated with an immune response triggered by the parasite (163). The mechanism by which immunotherapy with tDCs inhibits the progression of cardiac inflammation and myocardial fibrosis in a mouse model of CCC involves the secretion of IL-10 by tDCs to induce Tregs differentiation and enhance Tregs immunosuppressive function (164). IL-10 is a cytokine that can independently induce Foxp3 expression and Treg differentiation (165), and secretion of IL-10 by tDCS induces Foxp3+Tregs differentiation to regulate immunity (166, 167). Intervention with human recombinant granulocyte colony-stimulating factor (G-CSF) enhances cardiac Tregs recruitment and reduces cardiac inflammation, fibrosis, and parasite load in mice with CCC induced by chronic T.cruzi infection (168). Moreover, during the acute phase of T.cruzi infection, depleting Tregs exacerbated myocardial inflammation and tissue parasite levels, leading to increased mortality in experimental mice (169). In comparison, formyl peptide receptor 2 (FPR2) KO mice had increased Tregs during the acute T.cruzi infection phase, which controlled the protective effect of Th1 cells against T.cruzi infection. However, FPR2-KO mice have reduced Tregs and exacerbated cardiac inflammation and cardiac dysfunction during prolonged chronic T.cruzi infection (170).
5.7 COVID-19-associated myocarditis
A retrospective cohort study of 56963 hospitalized patients with COVID-19 showed that the incidence of acute myocarditis in hospitalized patients with COVID-19 ranged from 0.24 to 0.41%; Chest pain and dyspnea symptoms were the most frequent, accounting for 55.5% and 53.7%, respectively; 38.9% presented with fulminant manifestations; The combined incidence of in-hospital mortality or temporary mechanical circulatory support was 20.4%; At 120 days, the mortality rate was approximately 6.6% (171). Another retrospective cohort study involving 718365 COVID-19 patients showed that the incidence of COVID-19 with myocarditis and 6-month all-cause mortality were 5.0% and 3.9% respectively (172). Although COVID-19-associated myocarditis is very severe, the role of Tregs in it remain understudied.
6 Tregs regulate cardiac remodeling in CVDs
Cardiac remodeling is defined as changes in the size, shape, and function of the heart resulting from pathological conditions (173). Myocardial fibrosis is a qualitative and quantitative change in the myocardial interstitial collagen network characterized by excessive deposition of collagen and other extracellular matrix components. In ischemic heart disease, myocardial fibrosis exacerbates cardiac remodeling, promoting cardiac insufficiency, arrhythmias, and ultimately HF (174, 175). Targeted regulation of myocardial fibrosis and improvement of cardiac remodeling are effective therapeutic strategies for ischemic CVDs (176). T lymphocytes play an essential role in regulating extracellular matrix components and myocardial fibrosis by regulating the expression of myocardial collagen and matrix metalloproteinases, and the role of Tregs in myocardial fibrosis has also received attention (177, 178).
6.1 Myocardial infarction
Studies have shown that the adoptive transfer of Tregs inhibits myocardial fibrosis and cardiac remodeling in MI model animals (179, 180). Tregs can inhibit myocardial fibrosis and improve cardiac remodeling by regulating cardiac fibroblast phenotypes, reducing α-smooth muscle actin (α-SMA) expression, and extracellular matrix collagen deposition (181). Overexpression of Sparc enabled Treg to have a tissue repair phenotype, which helped to improve collagen content and maturity in scars after MI, prevent heart rupture, and improve MI survival rate (182). MI model mice CCR5+ monocytes promote the secretion of anti-inflammatory factor IL-10, mediate Tregs recruitment, inhibit inflammation, and inhibit myocardial fibrosis and cardiac remodeling. However, the expression of cardiac proinflammatory factors in CCR5-/- MI model mice was significantly up-regulated, Tregs recruitment was impaired, and cardiac remodeling continued to worsen (183). IL-2/JES6-1 mAb (JES6-1) complex can improve cardiac function and remodeling by increasing the ratio of Tregs in MI model mouse heart infarct zone, inhibiting inflammation, inducing macrophages to transform from M1 to M2 type (184). Transferred myosin heavy chain α (MYHCA)614–629-specific CD4+T cells selectively accumulated in the myocardium and mediastinal lymph nodes of infarcted mice, acquired Tregs phenotype with a distinct pro-healing gene expression profile, and accelerated the regression of inflammation, promoted proper extracellular matrix deposition in the myocardial scar, and mediated cardioprotection (185).
6.2 Hypertension
Single-cell sequencing analysis of cardiac CD45+ immune cells in transverse aortic constriction-induced non-ischemic, pressure-overload HF model mice revealed that Tregs were activated, and the Tregs-specific molecule PD-1 was upregulated (186). Adoptive transfer of Tregs significantly ameliorated ventricular remodeling and myocardial fibrosis in rats with abdominal aortic constriction-induced HF by suppressing LOX expression via activation of the IL-10/Jak1/STAT3 signaling pathway (77). β-hydroxybutyrate can down-regulate NOX2/glycogen synthase kinase-3β (GSK3β) pathway, increase the number of cardiac Tregs, inhibit inflammation, and improve cardiac function, myocardial fibrosis, and cardiac remodeling in heart failure with preserved ejection fraction (HFpEF) mice (187).
Adoptive transfer of Tregs significantly reduced the infiltration of cardiac macrophages in Ang II-infused hypertension mice, improved cardiac inflammation, myocardial hypertrophy, and fibrosis, and inhibited electrical remodeling. The mechanism involved Tregs fixation of connexin 43 (CX43) gap junction protein in intercalated disk regions rather than lateral borders of cardiomyocytes, and reduced the risk of ventricular arrhythmias (188). Tregs with Nox2 deficiency by adoptive transfer significantly inhibited Ang II-induced hypertension and cardiac remodeling, and the effect was better than Tregs (60). In galectin-3 (Gal-3) KO hypertensive model mice, spleen Tregs significantly increased, and cardiac inflammation and myocardial fibrosis were improved (189). Overexpression of developmental endothelial locus-1 (DEL-1) in endothelial cells, combined with recombinant DEL-1 intervention, stabilized the number of αvβ3 integrin-dependent Tregs and Il-10 levels, and improved cardiovascular remodeling and blood pressure levels in Ang II and DOCA-salt-induced hypertension mice (190). Tregs-derived IL-35 had a protective effect on right ventricular systolic pressure and right ventricular dilation in mice with pulmonary hypertension (191). IL-2/JES6-1 complex intervention effectively induced splenic Tregs amplification five times and inhibited Ang II mediated aortic collagen remodeling and atherosclerosis (192).
6.3 Atrial fibrillation
The abundance of Bacteroides Fragilis decreased in elderly patients with atrial fibrillation. Bacteroides Fragilis intervention can reduce the inflammatory response of aging rats by increasing the number of Tregs, inhibiting atrial remodeling, and preventing the occurrence of atrial fibrillation (193). Foxp3 is the direct target gene of miRNA-210. IL-6 promotes the expression of miRNA-210 by regulating HIF-1α, and inhibits Tregs function by targeting Foxp3, promoting myocardial fibrosis and exacerbating atrial fibrillation (194).
7 Tregs regulate plaque regression in atherosclerosis
Traditionally, atherosclerosis is considered to be a cholesterol storage disease caused by the retention of lipoproteins (including low-density lipoprotein, LDL) in the intimal space of arteries. The residual LDL is modified and absorbed by scavenger receptor-mediated phagocytosis, resulting in the continuous growth of fatty infiltration rich in inflammatory white blood cells and the formation of plaque. Plaque regression is an important clinical goal in the treatment of atherosclerosis. The increase of Tregs in plaque is one of the characteristics of plaque regression. The CD45+ cells isolated from aortic arch plaques of atherosclerotic mice were sequenced by single-cell RNA-sequencing, and the expression profiles of Tregs in progressing and regressing plaque were compared. The results showed that the Tregs in progressing plaques had high mRNA levels of thymus-derived or natural Tregs (nTregs) markers Nrp1 and nTregs-activated genes (Itgb1, CTLA4). In contrast, the level of Tregs Nrp1 mRNA in regressing plaque is lower, and the level of mRNA related to the differentiation or maintenance of Tregs is higher (Mif, lgals9, Ly6a), suggesting that Tregs in regressing plaque may come from the peripheral differentiation of naïve T cells (136). Under atherosclerotic pathological conditions, CX3CL1 was selectively recruited to the aortic wall, while CCL4, CXCL11 and CXCL9 mainly increased in lymph nodes. Although CX3CR1 was not significantly expressed in CD4+ T cells, overexpression of CX3CR1 in Tregs showed that the CX3CL1/CX3CR1 axis selectively chemotactic Tregs to the aortic plaque of atherosclerotic mice, reducing lipid deposition, increasing the content of collagen and smooth muscle cells to improve plaque stability, reducing the number of proinflammatory macrophages, and inhibiting the progression of atherosclerosis (195). Anti-CD3 antibody (CD3-Ab) significantly induced the rapid regression of plaque in the treatment of atherosclerosis. The mechanism is that CD3-Ab significantly reduced the infiltration of macrophages and CD4+ T cells in plaque and increased the proportion of Tregs in plaque. However, when the anti-CD25 antibody eliminates the function of Tregs, CD3-Ab cannot induce the regression of atherosclerotic plaque (196).
8 Tregs regulate immune tolerance
Heart transplantation is the only solution for end-stage HF, but it is limited by allogeneic heart rejection. One of the important pathophysiological processes of rejection after transplantation is inflammatory cell infiltration. Tregs mediate immune tolerance and regulate the immune microenvironment after heart transplantation.
Tregs-targeted Nox2 gene deletion (Nox2fl/flFoxp3Cre) mice received allogeneic heart transplantation. Nox2-deficient Treg expressed higher levels of CCR4 and CCR8, driving Tregs to migrate to the transplanted heart and enhancing immunosuppressive function. Reduce the degree of cardiomyocyte necrosis and fibrosis in cardiac grafts (197). IL-34 is an inhibitory Tregs-specific cytokine as well as a tolerance cytokine, which can effectively inhibit allogenic reactive immune response and mediate transplant tolerance (198). The orthogonal IL-2/IL-2R system was used to target Tregs and selectively amplify Tregs to improve cardiac allotransplantation and enhance immune tolerance (199). Low-dose IL-2 can prolong the survival period of chronic cardiac allograft rejection model mice, increase the infiltration of CD4+CD25+Foxp3 Tregs in spleen and graft, increase the percentage of circulating FoxP3+PD-L1+exocrine and FoxP3+CD73+exocrine, and delay the rejection (200). Simvastatin combined with aspirin can activate Tregs to enhance immune tolerance, enhance the protective effect of vascular endothelial cells, and prolong the survival time of cardiac allograft (201). Sirtinol combined with FK506 has a synergistic effect on prolonging cardiac allograft survival, which regulates Th17/Treg balance by down-regulating IL-17A and up-regulating Foxp3 (10). In addition, a clinical study of 91 heart transplant patients showed that a low peripheral Treg/endothelial progenitor cell ratio after heart transplantation was an independent predictor of acute immune rejection (202).
Knockdown of circFSCN1 induced DC transformation into tDC phenotype, which contributed to Tregs amplification, prevented immune rejection of heart transplantation, prolonged allograft survival time, and reduced allograft fibrosis (203). Overexpression of growth differentiation factor 15 (GDF15) in DC enhances effector T cells depletion and promotes Tregs generation through the IDO signaling pathway, thus inhibiting immune rejection in cardiac allograft (204). The combination of marine and tacrolimus inhibited DC maturation through the reactive oxygen species (ROS)/ERK/NF-κB pathway, increased the rate of Tregs, reduced oxidative damage and apoptosis, and alleviated acute rejection of mouse heart allograft (205).
9 Targeted Tregs in the treatment of the neonatal cardiac injury
The neonatal mouse heart was injured from postnatal day (P) 0-7, and Tregs were recruited to directly promote myocardial cell proliferation and cardiac regeneration through paracrine CCL24, growth arrest specific 6 (GAS6), or amphiregulin (AREG). Depleted Tregs aggravate cardiac fibrosis, while adoptive transfer of Tregs reduces fibrosis and enhances the proliferation of injury cardiomyocytes. Single-cell sequencing analysis showed that there was no difference in Tregs transcriptomes whether neonatal hearts were regenerated or not, suggesting that adult Tregs had the same regenerative capacity as long as they were abundant (206). There were significantly more Tregs in the P8 hearts of newborn mice than in the first week after injury (207).
10 Discussion
This review summarized that targeted Tregs effectively treat CVDs and have cardiac protective effects on MI, HF, myocarditis, hypertension, atherosclerosis, atrial fibrillation, heart transplantation, and neonatal heart injury. The specific mechanism involved Tregs regulating immune balance, anti-inflammatory, inhibiting cardiac remodeling and vascular remodeling, mediating immune tolerance, and promoting tissue regeneration and repair (Figure 2). Tregs inhibit the inflammatory response mediated by effector T cells, Th17 being the most significant, and regulate Th17/Treg to promote immune balance. Tregs regulate fibroblast phenotype and inhibit myocardial fibrosis and cardiac remodeling. Tregs promote the M2-type polarization of macrophages, which has anti-inflammatory and repair effects, and inhibit the M1-type polarization of macrophages, which has pro-inflammatory effects, thus enabling the recovery of damaged myocardium. Related intervention methods can promote Tregs amplification, enhance the immunosuppressive function of Tregs and further strengthen immune tolerance by regulating the transformation of DCs into tDCs phenotype. Tregs promote the regeneration of heart muscle cells and the repair of damaged hearts.
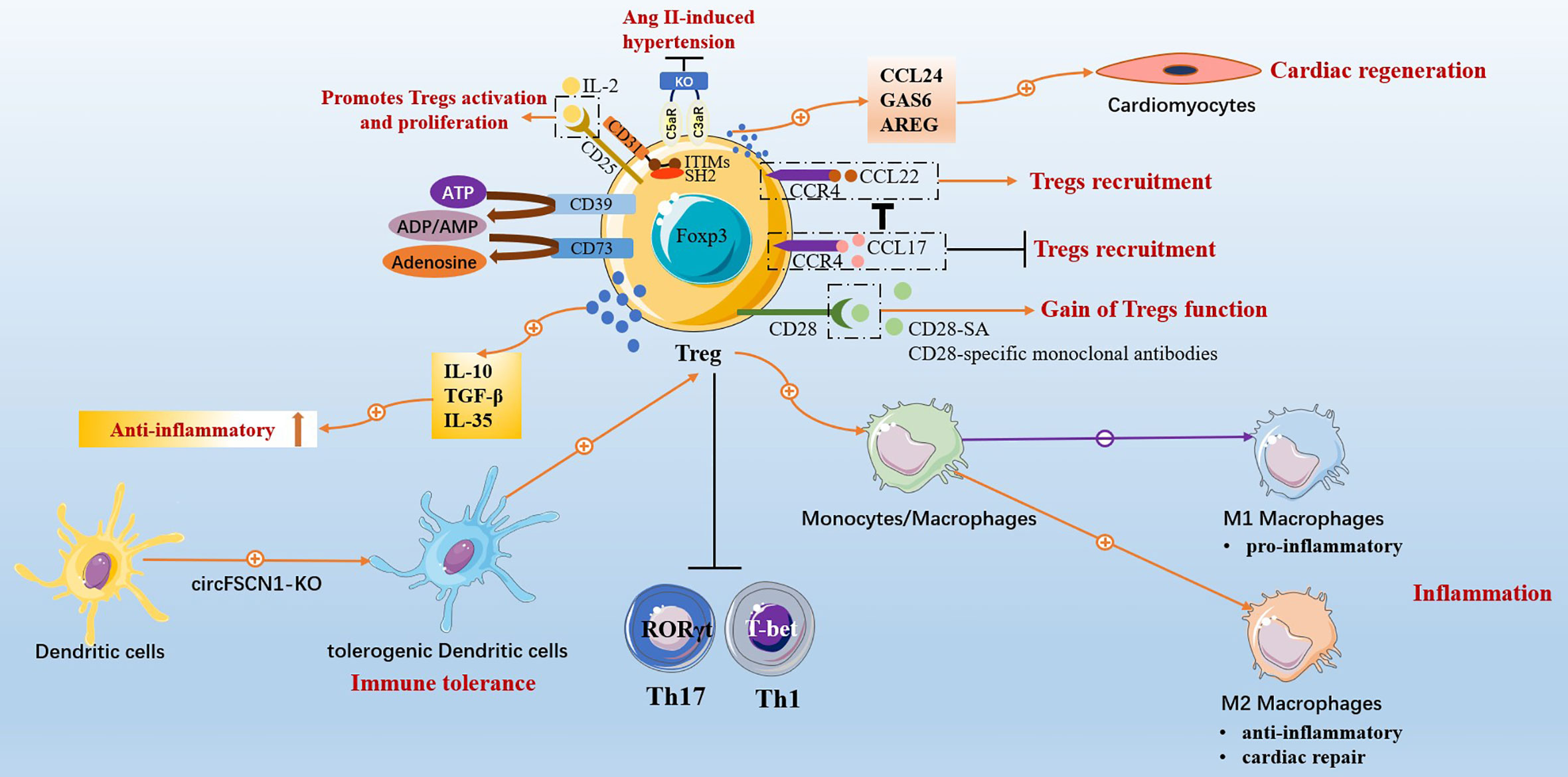
Figure 2 Protective effects of Conventional CD4+CD25+Foxp3+Tregs in cardiovascular diseases.
Conventional CD4+CD25+Foxp3+Tregs have a wide range of benefits in the treatment of CVDs. However, studies have shown the plasticity of Tregs (208), and this plasticity of Tregs has cardiotoxic effects on CVDs. Atherosclerosis can shift Tregs from a protective CXCR3+Treg response to dysfunctional interferon (IFN) γ+Th1/Treg response, driving inflammation and worsening disease progression (209). Tregs in the MI-post HF model mice showed pro-inflammatory Th1 cell characteristics, losing immunomodulatory function, enhancing anti-angiogenesis, and promoting fibrosis. Tregs reconstructed after selective dysfunctional proinflammatory Tregs ablation showed a recovery of immunosuppressive ability (210). The discovery of Tregs’ protean function and phenotypic plasticity in chronic ischemic HF has caused a considerable dispute in the cardiovascular field due to its novelty, challenging the conventional view of the phenotypic stability of Tregs after myocardial injury (211, 212), and generated extensive academic reports (213). HF disease can be divided into ischemic HF and non-ischemic HF. The cardioprotective effect of Tregs in non-ischemic HF and ischemic disease MI has been reviewed previously. While in MI-induced ischemic HF mice model experiments, a dysfunctional pro-inflammatory Tregs phenotype emerged. So, does it mean that Tregs play a typical functional role in a specific animal model? In addition, in the mice model of right lower extremity ischemia induced by right femoral artery ligation, although Tregs had immunosuppressive functions to suppress ongoing inflammation, Tregs had anti-neoangiogenic effects, resulting in foot inadequate perfusion and reduced capillary density (214). Human peripheral blood Tregs have IL-17+/Foxp3+Tregs phenotype and retain immunosuppressive function, while inhibition of Tregs Foxp3 expression in vitro and driven by inflammatory microenvironment show plasticity of IL-17 secretion (215). IL-17+/Foxp3+Tregs exist in the inflammatory intestinal mucosa of patients with Crohn’s disease and exhibit the phenotype of secreting IL-17 (216). The levels of the Th17 plasticity of Tregs are elevated in patients with rheumatoid arthritis (217). In autoimmune arthritis disease, the inflammatory microenvironment induces Foxp3 instability, leading to the trans-differentiation of Tregs into pro-inflammatory Th17 cells phenotypes, accelerating synovial membrane damage (218). So, do IL-17+/Foxp3+Tregs phenotype exist in heart tissue? Or does Foxp3 instability have a similar toxic effect on cardiovascular disease? It is worthy of further exploration. Moreover, Tregs are controversial in the context of myocardial fibrosis. Although much literature has reported that Tregs ameliorate cardiac fibrosis, TGF-β is secreted by Tregs, TGF-β/Smads are key pathways in the induction of fibrosis (219). Of course, the specific mechanism awaits further exploration.
The global burden of CVDs is still increasing. Although Tregs are a crucial target for the treatment of CVDs, there is still a lack of evidence from a large number of clinical randomized controlled trials. A few clinical trials in patients with CVDs have focused on measuring Tregs’ number, ratio, and function as a biomarker of disease severity. However, in terms of improving CVDs, whether endogenous Tregs are added, or exogenous Tregs are injected to enhance Tregs function, many basic experimental studies and rigorous efficacy and safety assessments are still needed before they can be used in clinical trials. This review summarized the clinical trials and basic experimental studies of targeted Tregs for CVDs, laying a foundation for further research on Tregs.
Author contributions
XW: writing, editing and review of the manuscript. QL, PC, and TZ: revised the article. YZ, TY, and WS: Assisted with literature search. HZ, HQ, and YYZ: designed the conception and figures. All authors contributed to the article and approved the submitted version.
Funding
The work was supported by the Three-year Action Plan of Shanghai Shenkang Medical Development Center (Grant/Award Number: SHDC2020CR1053B); the National Natural Science Foundation of China (Grant/Award Number: 82274306, 82204859, 81973656).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol (2005) 6:345–52. doi: 10.1038/ni1178
2. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi: 10.1126/science.1079490
3. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi: 10.1038/83713
4. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
5. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
6. Harb H, Benamar M, Lai PS, Contini P, Griffith JW, Crestani E, et al. Notch4 signaling limits regulatory T-cell-mediated tissue repair and promotes severe lung inflammation in viral infections. Immunity (2021) 54:1186–99. doi: 10.1016/j.immuni.2021.04.002
7. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol (2017) 17:703–17. doi: 10.1038/nri.2017.75
8. Adibzadeh SM, Abdollahpour-Alitappeh M, Mahdavi M, Ranjbar R, Ahmadi K, Taheri RA, et al. Immunologic balance of regulatory T cell/T helper 17 responses in gastrointestinal infectious diseases: Role of miRNAs. Microb Pathog (2019) 131:135–43. doi: 10.1016/j.micpath.2019.03.029
9. Walsh PT, Taylor DK, Turka LA. Tregs and transplantation tolerance. J Clin Invest (2004) 114:1398–403. doi: 10.1172/JCI200423238
10. Ye Q, Zhang M, Wang Y, Fu S, Han S, Wang L, et al. Sirtinol regulates the balance of Th17/Treg to prevent allograft rejection. Cell Biosci (2017) 7:55. doi: 10.1186/s13578-017-0182-2
11. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: Update from the GBD 2019 study. J Am Coll Cardiol (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
12. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol (2016) 13:167–79. doi: 10.1038/nrcardio.2015.169
13. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation (2022) 145(18):e876–94. doi: 10.1161/CIR.0000000000001062
14. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics–2014 update: a report from the American heart association. Circulation (2014) 129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80
15. Nabeebaccus A, Zheng S, Shah AM. Heart failure-potential new targets for therapy. Br Med Bull (2016) 119:99–110. doi: 10.1093/bmb/ldw025
16. Tamargo J, Caballero R, Delpon E. New drugs in preclinical and early stage clinical development in the treatment of heart failure. Expert Opin Investig Drugs (2019) 28:51–71. doi: 10.1080/13543784.2019.1551357
17. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2020 update: A report from the American heart association. Circulation (2020) 141:e139–596. doi: 10.1161/CIR.0000000000000757
18. Ponikowski P, Anker SD, AlHabib KF, Cowie MR, Force TL, Hu S, et al. Heart failure: preventing disease and death worldwide. ESC Heart Fail (2014) 1:4–25. doi: 10.1002/ehf2.12005
19. Zhang Y, Bauersachs J, Langer HF. Immune mechanisms in heart failure. Eur J Heart Fail (2017) 19:1379–89. doi: 10.1002/ejhf.942
20. Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation (2000) 101:2981–8. doi: 10.1161/01.CIR.101.25.2981
21. Seropian IM, Toldo S, Van Tassell BW, Abbate A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol (2014) 63:1593–603. doi: 10.1016/j.jacc.2014.01.014
22. Chapman AR, Lee KK, McAllister DA, Cullen L, Greenslade JH, Parsonage W, et al. Association of high-sensitivity cardiac troponin I concentration with cardiac outcomes in patients with suspected acute coronary syndrome. Jama (2017) 318:1913–24. doi: 10.1001/jama.2017.17488
23. Alaour B, Liew F, Kaier TE. Cardiac troponin - diagnostic problems and impact on cardiovascular disease. Ann Med (2018) 50:655–65. doi: 10.1080/07853890.2018.1530450
24. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics-2018 update: A report from the American heart association. Circulation (2018) 137:e67–e492. doi: 10.1161/CIR.0000000000000558
25. Lawton JS, Tamis-Holland JE, Bangalore S, Bates ER, Beckie TM, Bischoff JM, et al. 2021 ACC/AHA/SCAI guideline for coronary artery revascularization: A report of the American college of Cardiology/American heart association joint committee on clinical practice guidelines. Circulation (2022) 145:e18–e114. doi: 10.1161/CIR.0000000000001038
26. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res (2016) 365:563–81. doi: 10.1007/s00441-016-2431-9
27. Saparov A, Ogay V, Nurgozhin T, Chen W, Mansurov N, Issabekova A, et al. Role of the immune system in cardiac tissue damage and repair following myocardial infarction. Inflammation Res (2017) 66:739–51. doi: 10.1007/s00011-017-1060-4
28. Fung G, Luo H, Qiu Y, Yang D, McManus B. Myocarditis. Circ Res (2016) 118:496–514. doi: 10.1161/CIRCRESAHA.115.306573
29. Sagar S, Liu PP, Cooper LJ. Myocarditis. Lancet (2012) 379:738–47. doi: 10.1016/S0140-6736(11)60648-X
30. Pollack A, Kontorovich AR, Fuster V, Dec GW. Viral myocarditis–diagnosis, treatment options, and current controversies. Nat Rev Cardiol (2015) 12:670–80. doi: 10.1038/nrcardio.2015.108
31. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European society of cardiology working group on myocardial and pericardial diseases. Eur Heart J (2013) 34:2636–2648, 2648a. doi: 10.1093/eurheartj/eht210
32. Blyszczuk P. Myocarditis in humans and in experimental animal models. Front Cardiovasc Med (2019) 6:64. doi: 10.3389/fcvm.2019.00064
33. Protonotarios A, Marelli-Berg F. Towards precision disease-modelling in experimental myocarditis. Cardiovasc Res (2020) 116:1656–7. doi: 10.1093/cvr/cvaa057
34. Neu N, Klieber R, Fruhwirth M, Berger P. Cardiac myosin-induced myocarditis as a model of postinfectious autoimmunity. Eur Heart J (1991) 12 Suppl D:117–20. doi: 10.1093/eurheartj/12.suppl_D.117
35. Marchant DJ, Boyd JH, Lin DC, Granville DJ, Garmaroudi FS, McManus BM. Inflammation in myocardial diseases. Circ Res (2012) 110:126–44. doi: 10.1161/CIRCRESAHA.111.243170
36. Bocchi EA, Bestetti RB, Scanavacca MI, Cunha NE, Issa VS. Chronic chagas heart disease management: From etiology to cardiomyopathy treatment. J Am Coll Cardiol (2017) 70:1510–24. doi: 10.1016/j.jacc.2017.08.004
37. Benatti RD, Oliveira GH, Bacal F. Heart transplantation for chagas cardiomyopathy. J Heart Lung Transplant (2017) 36:597–603. doi: 10.1016/j.healun.2017.02.006
38. Caldas IS, Santos EG, Novaes RD. An evaluation of benznidazole as a chagas disease therapeutic. Expert Opin Pharmacother (2019) 20:1797–807. doi: 10.1080/14656566.2019.1650915
39. Marin-Neto JA, Cunha-Neto E, Maciel BC, Simoes MV. Pathogenesis of chronic chagas heart disease. Circulation (2007) 115:1109–23. doi: 10.1161/CIRCULATIONAHA.106.624296
40. Santos ES, Silva D, Dos RB, Barreto BC, Cardoso C, Ribeiro DSR, et al. Immunomodulation for the treatment of chronic chagas disease cardiomyopathy: A new approach to an old enemy. Front Cell Infect Microbiol (2021) 11:765879. doi: 10.3389/fcimb.2021.765879
41. Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet (2021) 398:957–80. doi: 10.1016/S0140-6736(21)01330-1
42. Xiao L, Harrison DG. Inflammation in hypertension. Can J Cardiol (2020) 36:635–47. doi: 10.1016/j.cjca.2020.01.013
43. Song P, Fang Z, Wang H, Cai Y, Rahimi K, Zhu Y, et al. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: A systematic review, meta-analysis, and modelling study. Lancet Glob Health (2020) 8:e721–9. doi: 10.1016/S2214-109X(20)30117-0
44. Bergstrom G, Persson M, Adiels M, Bjornson E, Bonander C, Ahlstrom H, et al. Prevalence of subclinical coronary artery atherosclerosis in the general population. Circulation (2021) 144:916–29. doi: 10.1161/CIRCULATIONAHA.121.055340
45. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
46. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol (2015) 35:280–7. doi: 10.1161/ATVBAHA.114.303568
47. Spitz C, Winkels H, Burger C, Weber C, Lutgens E, Hansson GK, et al. Regulatory T cells in atherosclerosis: Critical immune regulatory function and therapeutic potential. Cell Mol Life Sci (2016) 73:901–22. doi: 10.1007/s00018-015-2080-2
48. Karakus U, Sahin D, Mittl P, Mooij P, Koopman G, Boyman O. Receptor-gated IL-2 delivery by an anti-human IL-2 antibody activates regulatory T cells in three different species. Sci Transl Med (2020) 12(574):eabb9283. doi: 10.1126/scitranslmed.abb9283
49. Beyersdorf N, Hanke T, Kerkau T, Hunig T. Superagonistic anti-CD28 antibodies: potent activators of regulatory T cells for the therapy of autoimmune diseases. Ann Rheum Dis (2005) 64 Suppl 4:v91–5. doi: 10.1136/ard.2005.042564
50. Lin CH, Hunig T. Efficient expansion of regulatory T cells in vitro and in vivo with a CD28 superagonist. Eur J Immunol (2003) 33:626–38. doi: 10.1002/eji.200323570
51. Beyersdorf N, Hanke T, Kerkau T, Hunig T. CD28 superagonists put a break on autoimmunity by preferentially activating CD4+CD25+ regulatory T cells. Autoimmun Rev (2006) 5:40–5. doi: 10.1016/j.autrev.2005.06.001
52. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345:1623–7. doi: 10.1126/science.1255904
53. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
54. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in ctla-4. Science (1995) 270:985–8. doi: 10.1126/science.270.5238.985
55. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi: 10.1084/jem.20090847
56. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
57. Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood (2004) 104:3986–92. doi: 10.1182/blood-2004-06-2066
58. Takenaka MC, Robson S, Quintana FJ. Regulation of the T cell response by CD39. Trends Immunol (2016) 37:427–39. doi: 10.1016/j.it.2016.04.009
59. Zhuang R, Meng Q, Ma X, Shi S, Gong S, Liu J, et al. CD4(+)FoxP3(+)CD73(+) regulatory T cell promotes cardiac healing post-myocardial infarction. Theranostics (2022) 12:2707–21. doi: 10.7150/thno.68437
60. Emmerson A, Trevelin SC, Mongue-Din H, Becker PD, Ortiz C, Smyth LA, et al. Nox2 in regulatory T cells promotes angiotensin II-induced cardiovascular remodeling. J Clin Invest (2018) 128:3088–101. doi: 10.1172/JCI97490
61. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discovery (2012) 11:763–76. doi: 10.1038/nrd3794
62. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi: 10.1038/nature04753
63. Zhang M, Zhang S. T Cells in fibrosis and fibrotic diseases. Front Immunol (2020) 11:1142. doi: 10.3389/fimmu.2020.01142
64. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6:1133–41. doi: 10.1038/ni1261
65. Liu Y, Zhu H, Su Z, Sun C, Yin J, Yuan H, et al. IL-17 contributes to cardiac fibrosis following experimental autoimmune myocarditis by a PKCbeta/Erk1/2/NF-kappaB-dependent signaling pathway. Int Immunol (2012) 24:605–12. doi: 10.1093/intimm/dxs056
66. Chang SL, Hsiao YW, Tsai YN, Lin SF, Liu SH, Lin YJ, et al. Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure. J Mol Cell Cardiol (2018) 122:69–79. doi: 10.1016/j.yjmcc.2018.08.005
67. Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell (2020) 183:94–109. doi: 10.1016/j.cell.2020.08.031
68. Schmetterer KG, Neunkirchner A, Pickl WF. Naturally occurring regulatory T cells: markers, mechanisms, and manipulation. FASEB J (2012) 26:2253–76. doi: 10.1096/fj.11-193672
69. Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, et al. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol (2017) 112:33. doi: 10.1007/s00395-017-0622-5
70. Yang Z, Zingarelli B, Szabo C. Crucial role of endogenous interleukin-10 production in myocardial ischemia/reperfusion injury. Circulation (2000) 101:1019–26. doi: 10.1161/01.CIR.101.9.1019
71. Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol (2014) 32:51–82. doi: 10.1146/annurev-immunol-032713-120257
72. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature (1992) 359:693–9. doi: 10.1038/359693a0
73. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U.S.A. (1993) 90:770–4. doi: 10.1073/pnas.90.2.770
74. Cheng X, Yu X, Ding YJ, Fu QQ, Xie JJ, Tang TT, et al. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol (2008) 127:89–97. doi: 10.1016/j.clim.2008.01.009
75. Li N, Bian H, Zhang J, Li X, Ji X, Zhang Y. The Th17/Treg imbalance exists in patients with heart failure with normal ejection fraction and heart failure with reduced ejection fraction. Clin Chim Acta (2010) 411:1963–8. doi: 10.1016/j.cca.2010.08.013
76. Bas HD, Baser K, Yavuz E, Bolayir HA, Yaman B, Unlu S, et al. A shift in the balance of regulatory T and T helper 17 cells in rheumatic heart disease. J Investig Med (2014) 62:78–83. doi: 10.2310/JIM.0000000000000023
77. Lu M, Qin X, Yao J, Yang Y, Zhao M, Sun L. Th17/Treg imbalance modulates rat myocardial fibrosis and heart failure by regulating LOX expression. Acta Physiol (Oxf) (2020) 230:e13537. doi: 10.1111/apha.13537
78. Del REMM, Bohm M, Link A. The Th17/Treg imbalance in patients with cardiogenic shock. Clin Res Cardiol (2014) 103:301–13. doi: 10.1007/s00392-013-0656-0
79. He Y, Chen X, Guo X, Yin H, Ma N, Tang M, et al. Th17/Treg ratio in serum predicts onset of postoperative atrial fibrillation after off-pump coronary artery bypass graft surgery. Heart Lung Circ (2018) 27:1467–75. doi: 10.1016/j.hlc.2017.08.021
80. Ma X, Liu S, Li T, Yuan H. Intensive statin treatment ameliorate the Th17/Treg functional imbalance in patients with non-ST elevation acute coronary syndrome underwent percutaneous coronary intervention. Clin Cardiol (2020) 43:379–85. doi: 10.1002/clc.23326
81. Calcaterra V, Croce S, Vinci F, De Silvestri A, Cordaro E, Regalbuto C, et al. Th17 and treg balance in children with obesity and metabolically altered status. Front Pediatr (2020) 8:591012. doi: 10.3389/fped.2020.591012
82. Imiela AM, Mikolajczyk TP, Siedlinski M, Dobrowolski P, Konior-Rozlachowska A, Wrobel A, et al. Th17/Treg imbalance in patients with primary hyperaldosteronism and resistant hypertension. Pol Arch Intern Med (2022) 132(3):132. doi: 10.20452/pamw.16171
83. Liu Z, Zhao Y, Wei F, Ye L, Lu F, Zhang H, et al. Treatment with telmisartan/rosuvastatin combination has a beneficial synergistic effect on ameliorating Th17/Treg functional imbalance in hypertensive patients with carotid atherosclerosis. Atherosclerosis (2014) 233:291–9. doi: 10.1016/j.atherosclerosis.2013.12.004
84. Gaowa S, Zhou W, Yu L, Zhou X, Liao K, Yang K, et al. Effect of Th17 and treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediators Inflammation (2014) 2014:247372. doi: 10.1155/2014/247372
85. Zhu M, Mo H, Li D, Luo X, Zhang L. Th17/Treg imbalance induced by increased incidence of atherosclerosis in patients with systemic lupus erythematosus (SLE). Clin Rheumatol (2013) 32:1045–52. doi: 10.1007/s10067-013-2237-z
86. Wang X, Fan H, Wang Y, Yin X, Liu G, Gao C, et al. Elevated peripheral T helper cells are associated with atrial fibrillation in patients with rheumatoid arthritis. Front Immunol (2021) 12:744254. doi: 10.3389/fimmu.2021.744254
87. Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, et al. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Heart Fail (2017) 10:e3688. doi: 10.1161/CIRCHEARTFAILURE.116.003688
88. Liu R, Guo C, Yang C, Xu D, Wang C. VEGF165 attenuates the Th17/Treg imbalance that exists when transplanting allogeneic skeletal myoblasts to treat acute myocardial infarction. Inflammation Res (2013) 62:69–79. doi: 10.1007/s00011-012-0553-4
89. Chen Z, Yan W, Mao Y, Ni Y, Zhou L, Song H, et al. Effect of aerobic exercise on treg and Th17 of rats with ischemic cardiomyopathy. J Cardiovasc Transl Res (2018) 11:230–5. doi: 10.1007/s12265-018-9794-0
90. Zhang Q, Hu LQ, Yin CS, Chen P, Li HQ, Sun X, et al. Catechin ameliorates cardiac dysfunction in rats with chronic heart failure by regulating the balance between Th17 and treg cells. Inflammation Res (2014) 63:619–28. doi: 10.1007/s00011-014-0734-4
91. Yan L, Hu F, Yan X, Wei Y, Ma W, Wang Y, et al. Inhibition of microRNA-155 ameliorates experimental autoimmune myocarditis by modulating Th17/Treg immune response. J Mol Med (Berl) (2016) 94:1063–79. doi: 10.1007/s00109-016-1414-3
92. Cheng H, Xi Y, Chi X, Wu Y, Liu G. Fenofibrate treatment of rats with experimental autoimmune myocarditis by alleviating Treg/Th17 disorder. Cent Eur J Immunol (2016) 41:64–70. doi: 10.5114/ceji.2016.58817
93. Pan K, Jiang S, Du X, Zeng X, Zhang J, Song L, et al. Parental PM2.5 exposure changes Th17/Treg cells in offspring, is associated with the elevation of blood pressure. Environ Toxicol (2021) 36:1152–61. doi: 10.1002/tox.23114
94. Robles-Vera I, Toral M, de la Visitacion N, Sanchez M, Romero M, Olivares M, et al. The probiotic lactobacillus fermentum prevents dysbiosis and vascular oxidative stress in rats with hypertension induced by chronic nitric oxide blockade. Mol Nutr Food Res (2018) 62:e1800298. doi: 10.1002/mnfr.201800298
95. Toral M, Robles-Vera I, de la Visitacion N, Romero M, Sanchez M, Gomez-Guzman M, et al. Role of the immune system in vascular function and blood pressure control induced by faecal microbiota transplantation in rats. Acta Physiol (Oxf) (2019) 227:e13285. doi: 10.1111/apha.13285
96. Oh S, Shim M, Son M, Jang JT, Son KH, Byun K. Attenuating effects of dieckol on endothelial cell dysfunction via modulation of Th17/Treg balance in the intestine and aorta of spontaneously hypertensive rats. Antioxidants (Basel) (2021) 10(2):298. doi: 10.3390/antiox10020298
97. Wang Y, Zhang L, Li L, Hu H, Pan P, Zhang B, et al. Electroacupuncture improves blood pressure in SHRs by regulating the immune balance between Th17 and treg. Evid Based Complement Alternat Med (2020) 2020:5375981. doi: 10.1155/2020/5375981
98. Du YN, Tang XF, Xu L, Chen WD, Gao PJ, Han WQ. SGK1-FoxO1 signaling pathway mediates Th17/Treg imbalance and target organ inflammation in angiotensin II-induced hypertension. Front Physiol (2018) 9:1581. doi: 10.3389/fphys.2018.01581
99. Huang L, Zheng Y, Yuan X, Ma Y, Xie G, Wang W, et al. Decreased frequencies and impaired functions of the CD31(+) subpopulation in treg cells associated with decreased FoxP3 expression and enhanced treg cell defects in patients with coronary heart disease. Clin Exp Immunol (2017) 187:441–54. doi: 10.1111/cei.12897
100. He X, Liang B, Gu N. Th17/Treg imbalance and atherosclerosis. Dis Markers (2020) 2020:8821029. doi: 10.1155/2020/8821029
101. Xie JJ, Wang J, Tang TT, Chen J, Gao XL, Yuan J, et al. The Th17/Treg functional imbalance during atherogenesis in ApoE(-/-) mice. Cytokine (2010) 49:185–93. doi: 10.1016/j.cyto.2009.09.007
102. Yang J, Wu J, Zhang R, Yao M, Liu Y, Miao L, et al. Porphyromonas gingivalis oral infection promote T helper 17/Treg imbalance in the development of atherosclerosis. J Dent Sci (2017) 12:60–9. doi: 10.1016/j.jds.2016.10.003
103. Tian Y, Chen T, Wu Y, Yang L, Wang L, Fan X, et al. Pioglitazone stabilizes atherosclerotic plaque by regulating the Th17/Treg balance in AMPK-dependent mechanisms. Cardiovasc Diabetol (2017) 16:140. doi: 10.1186/s12933-017-0623-6
104. Fan Q, Liu Y, Rao J, Zhang Z, Xiao W, Zhu T, et al. Anti-atherosclerosis effect of angong niuhuang pill via regulating Th17/Treg immune balance and inhibiting chronic inflammatory on ApoE(-/-) mice model of early and mid-term atherosclerosis. Front Pharmacol (2019) 10:1584. doi: 10.3389/fphar.2019.01584
105. Qiu R, Long J, Zhou L, Ma Y, Zhao L, Liu F, et al. Yangyin qingre huoxue method in traditional Chinese medicine ameliorates atherosclerosis in ApoE(-/-) mice suffering from high-fat diet and HSP65 aggression. Evid Based Complement Alternat Med (2019) 2019:2531979. doi: 10.1155/2019/2531979
106. Mao X, Zhu R, Zhang F, Zhong Y, Yu K, Wei Y, et al. IL-37 plays a beneficial role in patients with acute coronary syndrome. Mediators Inflammation (2019) 2019:9515346. doi: 10.1155/2019/9515346
107. Platten M, Youssef S, Hur EM, Ho PP, Han MH, Lanz TV, et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci U.S.A. (2009) 106:14948–53. doi: 10.1073/pnas.0903958106
108. Noutsias M, Rohde M, Goldner K, Block A, Blunert K, Hemaidan L, et al. Expression of functional T-cell markers and T-cell receptor vbeta repertoire in endomyocardial biopsies from patients presenting with acute myocarditis and dilated cardiomyopathy. Eur J Heart Fail (2011) 13:611–8. doi: 10.1093/eurjhf/hfr014
109. Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation (2006) 114:2056–64. doi: 10.1161/CIRCULATIONAHA.106.649244
110. Piroth M, Gorski DJ, Hundhausen C, Petz A, Gorressen S, Semmler D, et al. Hyaluronan synthase 3 is protective after cardiac ischemia-reperfusion by preserving the T cell response. Matrix Biol (2022) 112:116–31. doi: 10.1016/j.matbio.2022.08.008
111. Li L, Li L, Xiao L, Shangguan J. Progranulin ameliorates coxsackievirus-B3-induced viral myocarditis by downregulating Th1 and Th17 cells. Exp Cell Res (2018) 367:241–50. doi: 10.1016/j.yexcr.2018.04.001
112. De-Pu Z, Li-Sha G, Guang-Yi C, Xiaohong G, Chao X, Cheng Z, et al. The cholinergic anti-inflammatory pathway ameliorates acute viral myocarditis in mice by regulating CD4(+) T cell differentiation. Virulence (2018) 9:1364–76. doi: 10.1080/21505594.2018.1482179
113. Yue-Chun L, Gu XH, Li-Sha G, Zhou DP, Xing C, Guo XL, et al. Vagus nerve plays a pivotal role in CD4+ T cell differentiation during CVB3-induced murine acute myocarditis. Virulence (2021) 12:360–76. doi: 10.1080/21505594.2020.1869384
114. Liu J, Guo Z, Zhang Y, Wu T, Ma Y, Lai W, et al. LCK inhibitor attenuates atherosclerosis in ApoE(-/-) mice via regulating T cell differentiation and reverse cholesterol transport. J Mol Cell Cardiol (2020) 139:87–97. doi: 10.1016/j.yjmcc.2020.01.003
115. Meng K, Zeng Q, Lu Q, Lin Y, Wu B, Yu K, et al. Valsartan attenuates atherosclerosis via upregulating the Th2 immune response in prolonged angiotensin II-treated ApoE(-/-) mice. Mol Med (2015) 21:143–53. doi: 10.2119/molmed.2014.00195
116. Gao S, Zhang W, Zhao Q, Zhou J, Wu Y, Liu Y, et al. Curcumin ameliorates atherosclerosis in apolipoprotein e deficient asthmatic mice by regulating the balance of Th2/Treg cells. Phytomedicine (2019) 52:129–35. doi: 10.1016/j.phymed.2018.09.194
117. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res (2020) 116:1101–12. doi: 10.1093/cvr/cvz336
118. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204:3037–47. doi: 10.1084/jem.20070885
119. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol (2013) 62:24–35. doi: 10.1016/j.yjmcc.2013.04.023
120. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res (2014) 115:55–67. doi: 10.1161/CIRCRESAHA.115.303895
121. Zhang Y, Cai Z, Shen Y, Lu Q, Gao W, Zhong X, et al. Hydrogel-load exosomes derived from dendritic cells improve cardiac function via treg cells and the polarization of macrophages following myocardial infarction. J Nanobiotechnol (2021) 19:271. doi: 10.1186/s12951-021-01016-x
122. Feng G, Bajpai G, Ma P, Koenig A, Bredemeyer A, Lokshina I, et al. CCL17 aggravates myocardial injury by suppressing recruitment of regulatory T-cells. Circulation (2022) 145(10):765–82. doi: 10.1161/CIRCULATIONAHA.121.055888
123. Wang Y, Dembowsky K, Chevalier E, Stuve P, Korf-Klingebiel M, Lochner M, et al. C-X-C motif chemokine receptor 4 blockade promotes tissue repair after myocardial infarction by enhancing regulatory T cell mobilization and immune-regulatory function. Circulation (2019) 139:1798–812. doi: 10.1161/CIRCULATIONAHA.118.036053
124. Gast M, Rauch BH, Haghikia A, Nakagawa S, Haas J, Stroux A, et al. Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc Res (2019) 115:1886–906. doi: 10.1093/cvr/cvz085
125. Katsuki M, Hirooka Y, Kishi T, Sunagawa K. Decreased proportion of Foxp3+ CD4+ regulatory T cells contributes to the development of hypertension in genetically hypertensive rats. J Hypertens (2015) 33:773–83. doi: 10.1097/HJH.0000000000000469
126. Mian MO, Barhoumi T, Briet M, Paradis P, Schiffrin EL. Deficiency of T-regulatory cells exaggerates angiotensin II-induced microvascular injury by enhancing immune responses. J Hypertens (2016) 34:97–108. doi: 10.1097/HJH.0000000000000761
127. Pollow DJ, Uhlorn JA, Sylvester MA, Romero-Aleshire MJ, Uhrlaub JL, Lindsey ML, et al. Menopause and FOXP3(+) treg cell depletion eliminate female protection against T cell-mediated angiotensin II hypertension. Am J Physiol Heart Circ Physiol (2019) 317:H415–23. doi: 10.1152/ajpheart.00792.2018
128. Cui C, Fan J, Zeng Q, Cai J, Chen Y, Chen Z, et al. CD4(+) T-cell endogenous cystathionine gamma lyase-hydrogen sulfide attenuates hypertension by sulfhydrating liver kinase B1 to promote T regulatory cell differentiation and proliferation. Circulation (2020) 142:1752–69. doi: 10.1161/CIRCULATIONAHA.119.045344
129. Radwan E, Mali V, Haddox S, El-Noweihi A, Mandour M, Ren J, et al. Treg cells depletion is a mechanism that drives microvascular dysfunction in mice with established hypertension. Biochim Biophys Acta Mol Basis Dis (2019) 1865:403–12. doi: 10.1016/j.bbadis.2018.10.031
130. Chen XH, Ruan CC, Ge Q, Ma Y, Xu JZ, Zhang ZB, et al. Deficiency of complement C3a and C5a receptors prevents angiotensin II-induced hypertension via regulatory T cells. Circ Res (2018) 122:970–83. doi: 10.1161/CIRCRESAHA.117.312153
131. Robles-Vera I, de la Visitacion N, Toral M, Sanchez M, Romero M, Gomez-Guzman M, et al. Changes in gut microbiota induced by doxycycline influence in vascular function and development of hypertension in DOCA-salt rats. Nutrients (2021) 13(9):2971. doi: 10.3390/nu13092971
132. Jiang L, Tang C, Gong Y, Liu Y, Rao J, Chen S, et al. PD-1/PD-L1 regulates treg differentiation in pregnancy-induced hypertension. Braz J Med Biol Res (2018) 51:e7334. doi: 10.1590/1414-431x20187334
133. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
134. Feng J, Zhang Z, Kong W, Liu B, Xu Q, Wang X. Regulatory T cells ameliorate hyperhomocysteinaemia-accelerated atherosclerosis in apoE-/- mice. Cardiovasc Res (2009) 84:155–63. doi: 10.1093/cvr/cvp182
135. Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med (2006) 12:178–80. doi: 10.1038/nm1343
136. Sharma M, Schlegel MP, Afonso MS, Brown EJ, Rahman K, Weinstock A, et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res (2020) 127:335–53. doi: 10.1161/CIRCRESAHA.119.316461
137. Forteza MJ, Polyzos KA, Baumgartner R, Suur BE, Mussbacher M, Johansson DK, et al. Activation of the regulatory T-Cell/Indoleamine 2,3-dioxygenase axis reduces vascular inflammation and atherosclerosis in hyperlipidemic mice. Front Immunol (2018) 9:950. doi: 10.3389/fimmu.2018.00950
138. Zhang H, Ge S, Ni B, He K, Zhu P, Wu X, et al. Augmenting ATG14 alleviates atherosclerosis and inhibits inflammation via promotion of autophagosome-lysosome fusion in macrophages. Autophagy (2021) 17:4218–30. doi: 10.1080/15548627.2021.1909833
139. Ji Q, Meng K, Yu K, Huang S, Huang Y, Min X, et al. Exogenous interleukin 37 ameliorates atherosclerosis via inducing the treg response in ApoE-deficient mice. Sci Rep (2017) 7:3310. doi: 10.1038/s41598-017-02987-4
140. Chen XN, Ge QH, Zhao YX, Guo XC, Zhang JP. Effect of Si-Miao-Yong-An decoction on the differentiation of monocytes, macrophages, and regulatory T cells in ApoE(-/-) mice. J Ethnopharmacol (2021) 276:114178. doi: 10.1016/j.jep.2021.114178
141. Gotsman I, Grabie N, Gupta R, Dacosta R, MacConmara M, Lederer J, et al. Impaired regulatory T-cell response and enhanced atherosclerosis in the absence of inducible costimulatory molecule. Circulation (2006) 114:2047–55. doi: 10.1161/CIRCULATIONAHA.106.633263
142. Klingenberg R, Gerdes N, Badeau RM, Gistera A, Strodthoff D, Ketelhuth DF, et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J Clin Invest (2013) 123:1323–34. doi: 10.1172/JCI63891
143. Hua X, Hu G, Hu Q, Chang Y, Hu Y, Gao L, et al. Single-cell RNA sequencing to dissect the immunological network of autoimmune myocarditis. Circulation (2020) 142:384–400. doi: 10.1161/CIRCULATIONAHA.119.043545
144. Akhmerov A, Rogers R, de Couto G, Valle J, Li L, Ibrahim A, et al. Regulatory T cell activation, proliferation, and reprogramming induced by extracellular vesicles. J Heart Lung Transplant (2021) 40:1387–95. doi: 10.1016/j.healun.2021.06.005
145. Wei L, Wei-Min L, Cheng G, Bao-Guo Z. Upregulation of CD4+CD25+ T lymphocyte by adenovirus-mediated gene transfer of CTLA4Ig fusion protein in experimental autoimmune myocarditis. Autoimmunity (2006) 39:289–98. doi: 10.1080/08916930600758035
146. Wang S, Liu J, Wang M, Zhang J, Wang Z. Treatment and prevention of experimental autoimmune myocarditis with CD28 superagonists. Cardiology (2010) 115:107–13. doi: 10.1159/000256660
147. Martin R, Cordova C, San RJ, Gutierrez B, Cachofeiro V, Nieto ML. Oleanolic acid modulates the immune-inflammatory response in mice with experimental autoimmune myocarditis and protects from cardiac injury. therapeutic implications for the human disease. J Mol Cell Cardiol (2014) 72:250–62. doi: 10.1016/j.yjmcc.2014.04.002
148. Chen L, Hou X, Zhang M, Zheng Y, Zheng X, Yang Q, et al. MicroRNA-223-3p modulates dendritic cell function and ameliorates experimental autoimmune myocarditis by targeting the NLRP3 inflammasome. Mol Immunol (2020) 117:73–83. doi: 10.1016/j.molimm.2019.10.027
149. Wu J, Liu M, Mang G, Yu S, Chen Q, Li T, et al. Protosappanin a protects against experimental autoimmune myocarditis, and induces metabolically reprogrammed tolerogenic DCs. Pharmacol Res (2019) 146:104269. doi: 10.1016/j.phrs.2019.104269
150. Chen P, Baldeviano GC, Ligons DL, Talor MV, Barin JG, Rose NR, et al. Susceptibility to autoimmune myocarditis is associated with intrinsic differences in CD4(+) T cells. Clin Exp Immunol (2012) 169:79–88. doi: 10.1111/j.1365-2249.2012.04598.x
151. An B, Liu X, Li G, Yuan H. Interleukin-37 ameliorates coxsackievirus B3-induced viral myocarditis by modulating the Th17/Regulatory T cell immune response. J Cardiovasc Pharmacol (2017) 69:305–13. doi: 10.1097/FJC.0000000000000476
152. Jin H, Guo X. Valproic acid ameliorates coxsackievirus-B3-induced viral myocarditis by modulating Th17/Treg imbalance. Virol J (2016) 13:168. doi: 10.1186/s12985-016-0626-z
153. Fousteri G, Dave A, Morin B, Omid S, Croft M, von Herrath MG. Nasal cardiac myosin peptide treatment and OX40 blockade protect mice from acute and chronic virally-induced myocarditis. J Autoimmun (2011) 36:210–20. doi: 10.1016/j.jaut.2011.01.006
154. Dai K, Wang Y, Tai S, Ni H, Lian H, Yu Y, et al. Fasudil exerts a cardio-protective effect on mice with coxsackievirus B3-induced acute viral myocarditis. Cardiovasc Ther (2018) 36:e12477. doi: 10.1111/1755-5922.12477
155. Shi Y, Fukuoka M, Li G, Liu Y, Chen M, Konviser M, et al. Regulatory T cells protect mice against coxsackievirus-induced myocarditis through the transforming growth factor beta-coxsackie-adenovirus receptor pathway. Circulation (2010) 121:2624–34. doi: 10.1161/CIRCULATIONAHA.109.893248
156. Pappritz K, Savvatis K, Miteva K, Kerim B, Dong F, Fechner H, et al. Immunomodulation by adoptive regulatory T-cell transfer improves coxsackievirus B3-induced myocarditis. FASEB J (2018), j201701408R. doi: 10.1096/fj.201701408R
157. Cao Y, Xu W, Xiong S. Adoptive transfer of regulatory T cells protects against coxsackievirus B3-induced cardiac fibrosis. PloS One (2013) 8:e74955. doi: 10.1371/journal.pone.0074955
158. Lu J, Cen Z, Tang Q, Dong J, Qin L, Wu W. The absence of b cells disrupts splenic and myocardial treg homeostasis in coxsackievirus B3-induced myocarditis. Clin Exp Immunol (2022) 208:1–11. doi: 10.1093/cei/uxac015
159. Wang X, Ge J, Chen R. LAP(+) treg is a better biomarker than total treg in viral myocarditis. J Med Virol (2019) 91:886–9. doi: 10.1002/jmv.25378
160. Zhang Z, Dai X, Qi J, Ao Y, Yang C, Li Y. Astragalus mongholicus (Fisch.) bge improves peripheral treg cell immunity imbalance in the children with viral myocarditis by reducing the levels of miR-146b and miR-155. Front Pediatr (2018) 6:139. doi: 10.3389/fped.2018.00139
161. Frisancho-Kiss S, Coronado MJ, Frisancho JA, Lau VM, Rose NR, Klein SL, et al. Gonadectomy of male BALB/c mice increases Tim-3(+) alternatively activated M2 macrophages, Tim-3(+) T cells, Th2 cells and treg in the heart during acute coxsackievirus-induced myocarditis. Brain Behav Immun (2009) 23:649–57. doi: 10.1016/j.bbi.2008.12.002
162. Li K, Xu W, Guo Q, Jiang Z, Wang P, Yue Y, et al. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ Res (2009) 105:353–64. doi: 10.1161/CIRCRESAHA.109.195230
163. Belkaid Y, Blank RB, Suffia I. Natural regulatory T cells and parasites: a common quest for host homeostasis. Immunol Rev (2006) 212:287–300. doi: 10.1111/j.0105-2896.2006.00409.x
164. Santos ES, de Aragao-Franca LS, Meira CS, Cerqueira JV, Vasconcelos JF, Nonaka C, et al. Tolerogenic dendritic cells reduce cardiac inflammation and fibrosis in chronic chagas disease. Front Immunol (2020) 11:488. doi: 10.3389/fimmu.2020.00488
165. Boks MA, Kager-Groenland JR, Haasjes MS, Zwaginga JJ, van Ham SM, Ten BA. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction–a comparative study of human clinical-applicable DC. Clin Immunol (2012) 142:332–42. doi: 10.1016/j.clim.2011.11.011
166. Choo EH, Lee JH, Park EH, Park HE, Jung NC, Kim TH, et al. Infarcted myocardium-primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the regulatory T cell and macrophage polarization. Circulation (2017) 135:1444–57. doi: 10.1161/CIRCULATIONAHA.116.023106
167. Lan YY, Wang Z, Raimondi G, Wu W, Colvin BL, de Creus A, et al. “Alternatively activated” dendritic cells preferentially secrete IL-10, expand Foxp3+CD4+ T cells, and induce long-term organ allograft survival in combination with CTLA4-ig. J Immunol (2006) 177:5868–77. doi: 10.4049/jimmunol.177.9.5868
168. Vasconcelos JF, Souza BS, Lins TF, Garcia LM, Kaneto CM, Sampaio GP, et al. Administration of granulocyte colony-stimulating factor induces immunomodulation, recruitment of T regulatory cells, reduction of myocarditis and decrease of parasite load in a mouse model of chronic chagas disease cardiomyopathy. FASEB J (2013) 27:4691–702. doi: 10.1096/fj.13-229351
169. Mariano FS, Gutierrez FR, Pavanelli WR, Milanezi CM, Cavassani KA, Moreira AP, et al. The involvement of CD4+CD25+ T cells in the acute phase of trypanosoma cruzi infection. Microbes Infect (2008) 10:825–33. doi: 10.1016/j.micinf.2008.04.009
170. Das DPR, Rabelo R, Leite PG, Cramer A, Botelho A, Cruz JS, et al. Role of formyl peptide receptor 2 (FPR2) in modulating immune response and heart inflammation in an experimental model of acute and chronic chagas disease. Cell Immunol (2021) 369:104427. doi: 10.1016/j.cellimm.2021.104427
171. Ammirati E, Lupi L, Palazzini M, Hendren NS, Grodin JL, Cannistraci CV, et al. Prevalence, characteristics, and outcomes of COVID-19-Associated acute myocarditis. Circulation (2022) 145:1123–39. doi: 10.1161/CIRCULATIONAHA.121.056817
172. Buckley B, Harrison SL, Fazio-Eynullayeva E, Underhill P, Lane DA, Lip G. Prevalence and clinical outcomes of myocarditis and pericarditis in 718,365 COVID-19 patients. Eur J Clin Invest (2021) 51:e13679. doi: 10.1111/eci.13679
173. Hashimoto H, Olson EN, Bassel-Duby R. Therapeutic approaches for cardiac regeneration and repair. Nat Rev Cardiol (2018) 15:585–600. doi: 10.1038/s41569-018-0036-6
174. Gyongyosi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur J Heart Fail (2017) 19:177–91. doi: 10.1002/ejhf.696
175. Gonzalez A, Schelbert EB, Diez J, Butler J. Myocardial interstitial fibrosis in heart failure: Biological and translational perspectives. J Am Coll Cardiol (2018) 71:1696–706. doi: 10.1016/j.jacc.2018.02.021
176. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther (2009) 123:255–78. doi: 10.1016/j.pharmthera.2009.05.002
177. Wei L. Immunological aspect of cardiac remodeling: T lymphocyte subsets in inflammation-mediated cardiac fibrosis. Exp Mol Pathol (2011) 90:74–8. doi: 10.1016/j.yexmp.2010.10.004
178. Yu Q, Horak K, Larson DF. Role of T lymphocytes in hypertension-induced cardiac extracellular matrix remodeling. Hypertension (2006) 48:98–104. doi: 10.1161/01.HYP.0000227247.27111.b2
179. Sharir R, Semo J, Shimoni S, Ben-Mordechai T, Landa-Rouben N, Maysel-Auslender S, et al. Experimental myocardial infarction induces altered regulatory T cell hemostasis, and adoptive transfer attenuates subsequent remodeling. PloS One (2014) 9:e113653. doi: 10.1371/journal.pone.0113653
180. Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N, et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol (2012) 107:232. doi: 10.1007/s00395-011-0232-6
181. Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol (2014) 307:H1233–42. doi: 10.1152/ajpheart.00328.2014
182. Xia N, Lu Y, Gu M, Li N, Liu M, Jiao J, et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation (2020) 142:1956–73. doi: 10.1161/CIRCULATIONAHA.120.046789
183. Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol (2010) 176:2177–87. doi: 10.2353/ajpath.2010.090759
184. Zeng Z, Yu K, Chen L, Li W, Xiao H, Huang Z. Interleukin-2/Anti-Interleukin-2 immune complex attenuates cardiac remodeling after myocardial infarction through expansion of regulatory T cells. J Immunol Res (2016) 2016:8493767. doi: 10.1155/2016/8493767
185. Rieckmann M, Delgobo M, Gaal C, Buchner L, Steinau P, Reshef D, et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest (2019) 129:4922–36. doi: 10.1172/JCI123859
186. Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T, et al. Single-cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation (2019) 140:2089–107. doi: 10.1161/CIRCULATIONAHA.119.041694
187. Liao S, Tang Y, Yue X, Gao R, Yao W, Zhou Y, et al. Beta-hydroxybutyrate mitigated heart failure with preserved ejection fraction by increasing treg cells via Nox2/GSK-3beta. J Inflammation Res (2021) 14:4697–706. doi: 10.2147/JIR.S331320
188. Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, et al. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation (2009) 119:2904–12. doi: 10.1161/CIRCULATIONAHA.108.832782
189. Gonzalez GE, Rhaleb NE, D’Ambrosio MA, Nakagawa P, Liao TD, Peterson EL, et al. Cardiac-deleterious role of galectin-3 in chronic angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol (2016) 311:H1287–96. doi: 10.1152/ajpheart.00096.2016
190. Failer T, Amponsah-Offeh M, Neuwirth A, Kourtzelis I, Subramanian P, Mirtschink P, et al. Developmental endothelial locus-1 protects from hypertension-induced cardiovascular remodeling via immunomodulation. J Clin Invest (2022) 132(6):e126155. doi: 10.1172/JCI126155
191. Wan N, Rong W, Zhu W, Jia D, Bai P, Liu G, et al. Tregs-derived interleukin 35 attenuates endothelial proliferation through STAT1 in pulmonary hypertension. Ann Transl Med (2021) 9:926. doi: 10.21037/atm-21-1952
192. Majeed B, Tawinwung S, Eberson LS, Secomb TW, Larmonier N, Larson DF. Interleukin-2/Anti-Interleukin-2 immune complex expands regulatory T cells and reduces angiotensin II-induced aortic stiffening. Int J Hypertens (2014) 2014:126365. doi: 10.1155/2014/126365
193. Zhang Y, Sun D, Zhao X, Luo Y, Yu H, Zhou Y, et al. Bacteroides fragilis prevents aging-related atrial fibrillation in rats via regulatory T cells-mediated regulation of inflammation. Pharmacol Res (2022) 177:106141. doi: 10.1016/j.phrs.2022.106141
194. Chen Y, Chang G, Chen X, Li Y, Li H, Cheng D, et al. IL-6-miR-210 suppresses regulatory T cell function and promotes atrial fibrosis by targeting Foxp3. Mol Cells (2020) 43:438–47. doi: 10.1016/j.phrs.2022.106141
195. Bonacina F, Martini E, Svecla M, Nour J, Cremonesi M, Beretta G, et al. Adoptive transfer of CX3CR1 transduced-T regulatory cells improves homing to the atherosclerotic plaques and dampens atherosclerosis progression. Cardiovasc Res (2021) 117:2069–82. doi: 10.1093/cvr/cvaa264
196. Kita T, Yamashita T, Sasaki N, Kasahara K, Sasaki Y, Yodoi K, et al. Regression of atherosclerosis with anti-CD3 antibody via augmenting a regulatory T-cell response in mice. Cardiovasc Res (2014) 102:107–17. doi: 10.1093/cvr/cvu002
197. Trevelin SC, Zampetaki A, Sawyer G, Ivetic A, Brewer AC, Smyth LA, et al. Nox2-deficient tregs improve heart transplant outcomes via their increased graft recruitment and enhanced potency. JCI Insight (2021) 6(18):e149301. doi: 10.1172/jci.insight.149301
198. Bezie S, Picarda E, Ossart J, Tesson L, Usal C, Renaudin K, et al. IL-34 is a treg-specific cytokine and mediates transplant tolerance. J Clin Invest (2015) 125:3952–64. doi: 10.1172/JCI81227
199. Hirai T, Ramos TL, Lin PY, Simonetta F, Su LL, Picton LK, et al. Selective expansion of regulatory T cells using an orthogonal IL-2/IL-2 receptor system facilitates transplantation tolerance. J Clin Invest (2021) 131(8):e139991. doi: 10.1172/JCI139991
200. Ravichandran R, Itabashi Y, Fleming T, Bansal S, Bowen S, Poulson C, et al. Low-dose IL-2 prevents murine chronic cardiac allograft rejection: Role for IL-2-induced T regulatory cells and exosomes with PD-L1 and CD73. Am J Transplant (2022) 22:2180–94. doi: 10.1111/ajt.17101
201. Zhu J, Gao B. Simvastatin combined with aspirin increases the survival time of heart allograft by activating CD4(+)CD25(+) treg cells and enhancing vascular endothelial cell protection. Cardiovasc Pathol (2015) 24:173–8. doi: 10.1016/j.carpath.2014.09.001
202. Choi DH, Chmura SA, Ramachandran V, Dionis-Petersen KY, Kobayashi Y, Nishi T, et al. The ratio of circulating regulatory cluster of differentiation 4 T cells to endothelial progenitor cells predicts clinically significant acute rejection after heart transplantation. J Heart Lung Transplant (2018) 37:496–502. doi: 10.1016/j.healun.2017.10.012
203. Wang B, Zhou Q, Li A, Li S, Greasley A, Skaro A, et al. Preventing alloimmune rejection using circular RNA FSCN1-silenced dendritic cells in heart transplantation. J Heart Lung Transplant (2021) 40:584–94. doi: 10.1016/j.healun.2021.03.025
204. Zhang Y, Zhang G, Liu Y, Chen R, Zhao D, McAlister V, et al. GDF15 regulates malat-1 circular RNA and inactivates NFkappaB signaling leading to immune tolerogenic DCs for preventing alloimmune rejection in heart transplantation. Front Immunol (2018) 9:2407. doi: 10.3389/fimmu.2018.02407
205. Zheng S, Chen Y, Wang Z, Che Y, Wu Q, Yuan S, et al. Combination of matrine and tacrolimus alleviates acute rejection in murine heart transplantation by inhibiting DCs maturation through ROS/ERK/NF-kappaB pathway. Int Immunopharmacol (2021) 101:108218. doi: 10.1016/j.intimp.2021.108218
206. Li J, Yang KY, Tam R, Chan VW, Lan HY, Hori S, et al. Regulatory T-cells regulate neonatal heart regeneration by potentiating cardiomyocyte proliferation in a paracrine manner. Theranostics (2019) 9:4324–41. doi: 10.7150/thno.32734
207. Li J, Liang C, Yang KY, Huang X, Han MY, Li X, et al. Specific ablation of CD4(+) T-cells promotes heart regeneration in juvenile mice. Theranostics (2020) 10:8018–35. doi: 10.7150/thno.42943
208. Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol (2009) 21:281–5. doi: 10.1016/j.coi.2009.05.007
209. Butcher MJ, Filipowicz AR, Waseem TC, McGary CM, Crow KJ, Magilnick N, et al. Atherosclerosis-driven treg plasticity results in formation of a dysfunctional subset of plastic IFNgamma+ Th1/Tregs. Circ Res (2016) 119:1190–203. doi: 10.1161/CIRCRESAHA.116.309764
210. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation (2019) 139:206–21. doi: 10.1161/CIRCULATIONAHA.118.036065
211. Wang D, Yang X, Zhong H. Letter by Wang et al. regarding article, “Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy”. Circulation (2019) 139:e1033–4.
212. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, et al. Response by bansal et al. to letter regarding article, “Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy”. Circulation (2019) 139:e1035–6. doi: 10.1161/CIRCULATIONAHA.119.040737
213. Frangogiannis NG. Protean functions and phenotypic plasticity of regulatory T cells in chronic ischemic heart failure. Circulation (2019) 139:222–5. doi: 10.1161/CIRCULATIONAHA.118.036524
214. Zouggari Y, Ait-Oufella H, Waeckel L, Vilar J, Loinard C, Cochain C, et al. Regulatory T cells modulate postischemic neovascularization. Circulation (2009) 120:1415–25. doi: 10.1161/CIRCULATIONAHA.109.875583
215. Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood (2009) 113:4240–9. doi: 10.1182/blood-2008-10-183251
216. Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology (2011) 140:957–65. doi: 10.1053/j.gastro.2010.12.002
217. Wang T, Sun X, Zhao J, Zhang J, Zhu H, Li C, et al. Regulatory T cells in rheumatoid arthritis showed increased plasticity toward Th17 but retained suppressive function in peripheral blood. Ann Rheum Dis (2015) 74:1293–301. doi: 10.1136/annrheumdis-2013-204228
218. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20:62–8. doi: 10.1038/nm.3432
Keywords: regulatory T cells, cardiovascular diseases, immune balance, inflammation, cardiac remodeling, immune tolerance, cardiac regeneration
Citation: Wang XT, Zhou H, Liu Q, Cheng PP, Zhao TY, Yang TS, Zhao Y, Sha WJ, Zhao YY and Qu HY (2023) Targeting regulatory T cells for cardiovascular diseases. Front. Immunol. 14:1126761. doi: 10.3389/fimmu.2023.1126761
Received: 18 December 2022; Accepted: 13 February 2023;
Published: 23 February 2023.
Edited by:
Yuekang Xu, Anhui Normal University, ChinaReviewed by:
Bin Li, Shanghai Jiao Tong University, ChinaThomas Kerkau, University of Würzburg, Germany
Copyright © 2023 Wang, Zhou, Liu, Cheng, Zhao, Yang, Zhao, Sha, Zhao and Qu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Zhou, zhouhua@shutcm.edu.cn; Yanyan Zhao, fille_@126.com; Huiyan Qu, 1520043084@qq.com