 Yingnan Liao1
Yingnan Liao1 Liyuan Zhu
Liyuan Zhu- 1Xiamen Key Laboratory of Cardiovascular Disease, Xiamen Cardiovascular Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, China
- 2Institute of Pharmaceutical Science, China Pharmaceutical University, Nanjing, China
Cardiovascular diseases are known as the leading cause of morbidity and mortality worldwide. As an innate immune signaling complex, inflammasomes can be activated by various cardiovascular risk factors and regulate the activation of caspase-1 and the production and secretion of proinflammatory cytokines such as IL-1β and IL-18. Accumulating evidence supports that inflammasomes play a pivotal role in the progression of atherosclerosis, myocardial infarction, and heart failure. The best-known inflammasomes are NLRP1, NLRP3, NLRC4, and AIM2 inflammasomes, among which NLRP3 inflammasome is the most widely studied in the immune response and disease development. This review focuses on the activation and regulation mechanism of inflammasomes, the role of inflammasomes in cardiovascular diseases, and the research progress of targeting NLRP3 inflammasome and IL-1β for related disease intervention.
Highlights
1. Many cardiovascular diseases risk factors, such as obesity and cholesterol crystals (ox-LDL/ox-PLs), can trigger the activation of the inflammasome, thus inducing inflammation.
2. Active inflammasome, especially NLRP3 inflammasome, plays a pivotal role in the development of cardiovascular diseases, such as atherosclerosis, myocardial infarction, and heart failure.
3. As promising targets for cardiovascular diseases, NLRP3 and IL-1 inhibitors have achieved positive clinical effects; it is of great clinical significance to develop more novel and specific inhibitors.
1 Introduction
Although major improvements in treatment and outcomes, cardiovascular diseases (CVDs) remain the leading cause of morbidity and mortality worldwide (1). The primary clinical endpoints of CVDs are heart failure (HF) and CVD death, mainly due to myocardial infarction (MI), which is mainly attributable to atherothrombotic plaques occlusion of blood vessels. Long-term atherosclerotic changes are driven by dyslipidemia and vascular inflammation. Some environmental and genetic factors, including hypertension, diabetes, hyperlipidemia, smoking, and aging, have been linked with CVDs (2). However, the pathological process of CVDs, especially atherosclerosis (AS) and myocardial ischemia/reperfusion (MI/R) injury, is gradually considered to be closely related to the inflammatory response. Therefore, inflammatory pathways may be effective targets in the treatment of CVDs. In fact, several clinical studies have assessed the outcomes of anti-inflammatory therapy in patients with AS (3).
As a multi-protein complex assembled by intracytoplasmic pattern recognition receptors, inflammasome is an important part of the natural immune system. It has been determined that a variety of inflammasomes participate in the host defense response against various pathogens (4). It can recognize pathogen-related molecular patterns or host-derived damage-associated molecular patterns (DAMPs) and recruit and activate the caspase-1, a proinflammatory protease. Activated caspase-1 cleaves the cytokine precursors pro-interleukin-1beta (IL-1β) and pro-IL-18 to produce corresponding mature cytokines in the process of innate immune defense (5). The activation of inflammasome can also induce the caspase-1-dependent programmed cell apoptosis (pyroptosis) and cell death stimulated by inflammation and stress (6). Inflammasome activation results in a very powerful self-amplifying and inflammatory response and thus modulates a variety of inflammatory diseases, such as CVDs. A balanced inflammatory response promotes damage resolution and tissue healing, while excessive inflammasome activation leads to harmful effects such as tissue damage (7). The activation of the inflammasome is considered to be involved in the pathogenesis of various CVDs. The intervention of inflammasome-mediated signaling can reduce inflammation and the severity of diseases (8). Here we review the role and mechanism of NOD, LRR, and PYD domain-containing protein (NLRP) 1, NLRP3, NOD, LRR, and CARD domain-containing protein (NLRC) 4 and absent in melanoma 2 (AIM2) inflammasomes in regulating CVDs, including AS, myocardial ischemic injury, cardiac hypertrophy, and HF; we summarize the therapeutic effect of pharmacological intervention targeting NLRP3 inflammasome and IL-1β in the experimental and clinical studies. In particular, as the core of inflammatory response, a crucial involvement of the NLRP3 inflammasome in CVDs has attracted widespread attention in the academic community in recent years and may provide a new therapeutic possibility for various inflammatory diseases (7). At present, the NLRP3 inflammasome is expected to become a new target for the prevention and treatment of CVDs (9). Issues for the activation and assembly of the NLRP3 inflammasome and the in-depth mechanism of mediating cell pyroptosis, the specific regulatory mechanism of the active ingredients for the NLRP3 inflammasome, and the changing characteristics of NLRP3 inflammasome under CVDs still need to be further studied.
2 Structure and Activation of Inflammasomes
2.1 Structure of Inflammasomes
The basic structure of most inflammasomes is based on the NOD-like receptor or ALR protein family as the receptor protein, apoptosis-associated speck-like protein containing CARD (ASC) as the adaptor protein, and pro-caspase as the effector protein (5).
The NLRP3 inflammasome consists of three components: NLRP3, ASC, and caspase-1. The innate immune receptor NLRP3 protein contains three domains: NACHT, a central nucleotide domain; leucine-rich repeats (LRR); and pyrin domain (PYD). Besides, the adaptor protein ASC contains two interaction domains: PYD and caspase recruitment domain (CARD). In the case of inflammasome assembly, the PYD domain interacts with the PYD domain of ASC, while the CARD structure of ASC can be combined with the CARD domain of the effector protein, during which the ASC acts as a bridge, connecting the receptor protein and the effector protein (4). Through this interaction, once the receptor protein is stimulated, caspase-1 can be finally activated and cleaved. Activated caspase-1 can cut and promote the maturation and release of IL-1β and IL-18 (10), thus causing inflammation. In addition, inflammasome activation can mediate caspase-1-dependent pyroptosis (6) and thus promote the spread of the inflammatory response.
AIM2 contains an N-terminal PYD domain, C-terminal HIN200 domain. AIM2 is a type of DNA sensor that can recognize and bind autologous or foreign DNA through its C-terminal HIN200 domain, which can be completed without relying on sequence (11). DNA sensing by AIM2 results in the assembly of inflammasome, which is a supramolecular multi-protein complex. The HIN200 domain of AIM2 binds to DNA, while the PYD domain (but not that of the other PYHIN family members) establishes interaction with the PYD domain of the adapter molecule ASC, and the CARD domain of ASC associates with the CARD domain of pro-caspase-1 to activate both NF-κB and caspase-1 (11), leading to the assembly of activated AIM2 inflammasome (12).
The special feature of the NLRC4 inflammasome is it does not contain the PYD domain but contains the CARD domain, which means that it does not need the ASC protein as a “bridge” and can directly connect through the CARD domain with the CARD domain of the effector protein (13). The assembly of NLRC4 is accomplished through interaction with NOD-like receptor family apoptosis inhibitory proteins, and the invasion of typhoid fever is an agonist of this process (14). Besides, in contrast to human NLRP1, mouse NLRP1b protein also does not contain the PYD domain but contains the CARD domain, which can directly bind to the effector protein without ASC (15). The structure of the inflammasome is summarized in Figure 1.
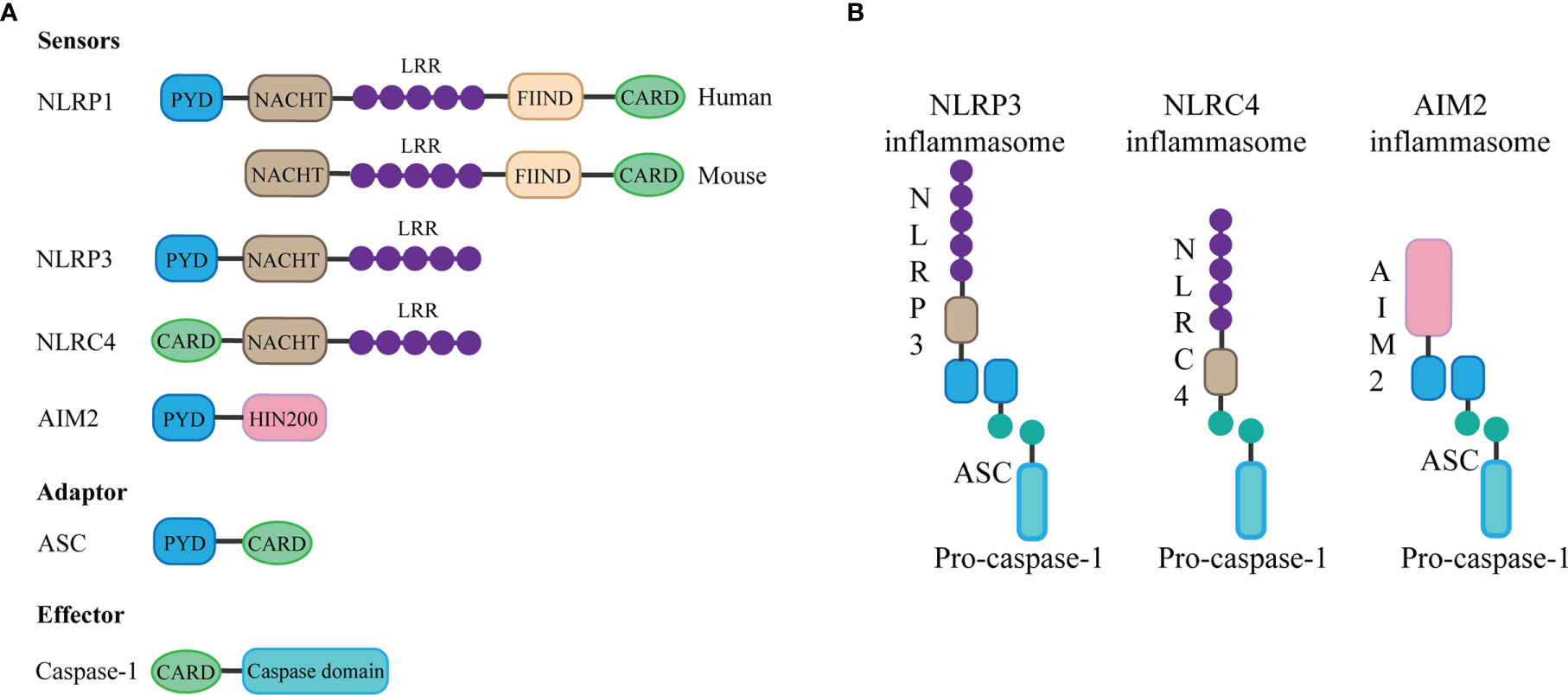
Figure 1 Components and structure of inflammasomes. (A) The basic structure of most inflammasomes is based on the NOD-like receptor or ALR protein family as the sensor protein, ASC as the adaptor protein, and pro-caspase as the effector protein. Different sensor proteins consist of different domains: PYD, NACHT, LRR, function-to-find domains (FIIND), CARD, and HIN domain. (B) The PYD domain of NLRP3 or AIM2 interacts with the PYD domain of ASC, while the CARD domain of ASC can be combined with the CARD domain of the effector protein pro-caspase-1 to form inflammasome. NLRC4 does not contain the PYD domain but contains the CARD domain, which means that it can directly connect through the CARD domain with the CARD domain of the effector protein.
2.2 Expression and Activation of Inflammasomes
Important components of inflammasome such as NLRP3 are not constructively expressed in cardiomyocytes but regulated by NF-κB (16, 17). Proinflammatory stimuli, such as cell debris or microbial products (referred to as DAMPs or pathogen-associated molecular patterns) (16), induce the expressions of NLRP3 and other inflammasome components in cardiomyocytes, leukocytes, fibroblasts, and endothelial cells (ECs) (18, 19). In the initial stages of formation, the inflammasome specks mainly accumulate in the ECs, cardiomyocytes, and fibroblasts. As the leukocytes infiltrate the heart, the majority of specks can be seen within the granulocytes and macrophages. In the post-injury healing phase, the majority of specks are more common in isolated cardiomyocytes or fibroblasts (20, 21).
Although each type of cell in the heart may trigger the NLRP3 inflammasome with appropriate stimulation, the stimulation required and the role of the active inflammasome may differ. Circulating monocytes constitutively express many components of inflammasome and produce large amounts of IL-1β with minimal stimulation (22). Conversely, cardiomyocytes express very low and almost undetectable levels of IL-1β, which is unaffected even in response to high doses of proinflammatory mediators. Besides, cardiomyocytes have low expressions of NLRP3 and caspase-1. However, both monocytes and cardiomyocytes can express and activate IL-18, and cardiomyocytes die of pyroptosis after caspase-1 activation (16, 22). The difference between the secretion of IL-1β and IL-18 secretion may be related to their transcriptional regulation (23).
Activation of NLRP3 inflammasome requires two separate steps: priming and triggering (17). A trigger is required after the activation is completed and the NLRP3 inflammasome components are expressed, which is largely dependent on intracellular K+ concentration. Lysosome instability activates a signaling pathway that indirectly leads to increased membrane permeability for K+ and intracellular efflux (16). Extracellular ATP triggers K+ outflow binding to the purinoreceptor 7 (P2X7). A change in K+ concentration results in a conformational change in NLRP3, thus leading to the recruitment of ASC (16). The use of transgenic mice with constructive NLRP3 activity showed that NLRP3 activation alone was not sufficient to cause cardiac dysfunction (17). Furthermore, patients with cryopyrin-associated periodic symptoms, who have mutations in NLRP3 gene causing continued NLRP3 activity, usually do not appear to have cardiac dysfunction but may be more susceptible to cardiac dysfunction during clinically active phases, proving the indispensability of priming for the activation of NLRP3 inflammasome (24).
The time-dependent activation of the NLRP3 inflammasome in the heart has been studied using animal models. In a model of ischemia–reperfusion injury, the expression of NLRP3 and the activity of the inflammasome in the heart were low within 3 h after acute MI (AMI), significantly increased in the myocardium within 3–24 h, and peaked after 1 and 3 days in mice with reperfused and non-reperfused AMI, respectively, aggravating inflammatory response and the ischemia–reperfusion injury (16, 18, 20, 21, 25). Therefore, delayed treatment with NLRP3 inhibitor (16673-34-0) even by 60 min after reperfusion initiation still exerted its cardioprotective effect, while the protective effect was lost if administered more than 3 h after reperfusion (20).
Active NLRP3 inflammasome enhances the recruitment and activation of caspase-1. The activated caspase-1 promotes the activation of the precursors of IL-1β and IL-18 to IL-1β and IL-18 as well as the cleavage of gasdermin D (GSDMD) (26). The cleaved GSDMD then forms pores on the cell membrane, triggering proinflammatory cell death, that is, pyroptosis (27). This process is accompanied by the release of IL-1β, IL-18, and alarm elements such as high mobility group proteins 1, which spreads dangerous signals from damaged or dead cells and mobilizes immune cells, especially in the recruitment of neutrophils (28). In addition, oligomeric inflammasome particles can be phagocytosed by surrounding macrophages to amplify the inflammatory response (29).
NLRP3 inflammasome-activating ligands can stimulate the production of reactive oxygen species (ROS), which in turn promote the assembly of inflammasome (30). Consistently, Jurg Tschopp found that mitochondrial-derived ROS are the key signal that regulates the activation of NLRP3 inflammasome (31). At the same time, it was found that autophagy and mitophagy regulate the quality of mitochondria and reduce the number of damaged mitochondria, thus preventing the activation of NLRP3 inflammasome induced by ROS. Autophagy-deficient cells exacerbate the activation of NLRP3 inflammasome. Further, the voltage-dependent anion channels of mitochondria regulate the production of mitochondrial ROS and the activation of NLRP3 inflammasome (31). The mechanism of how ROS triggers the activation of NLRP3 remains to be further investigated (30). The activation process of NLRP3 inflammasome is summarized in Figure 2.
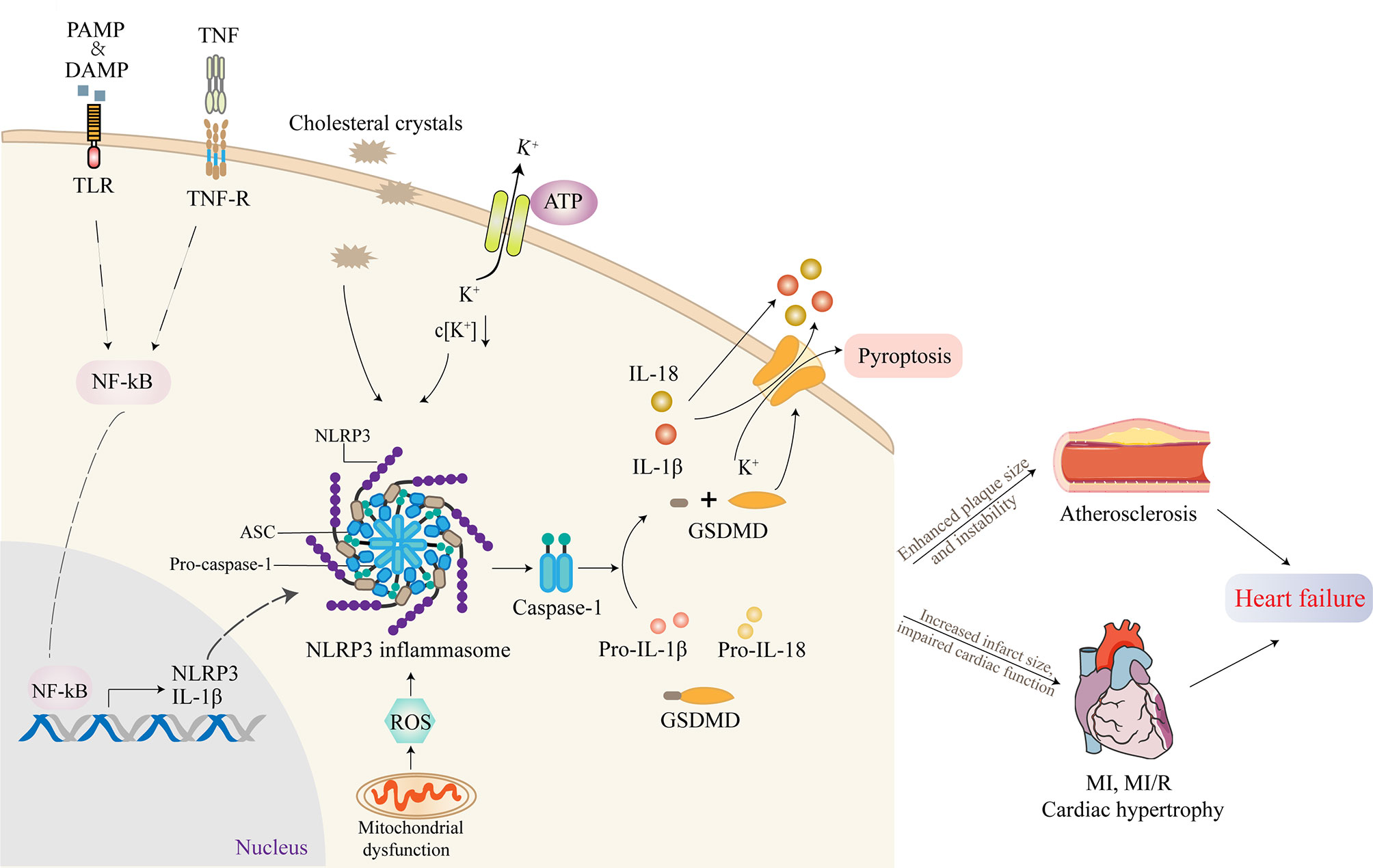
Figure 2 The activation of NLRP3 inflammasome and its role in cardiovascular diseases (CVDs). Inflammasome activation requires two independent steps: priming and triggering. Proinflammatory stimuli, such as PAMP, DAMP, or TNF, induce NF-κB signaling through TLR, TNF-R. The transcription factor NF-κB translocates to the nucleus and induces the expression of the IL-1β precursor pro-IL-1β and NLRP3. Cholesterol crystals or ox-LDL can be phagocytosed through receptor-mediated phagocytosis, leading to the formation of cholesterol crystals in phagolysosomes. Another mechanism of NLRP3 activation involves crystal-induced lysosomal rupture, or ATP-gated P2X7R-mediated decreased intracellular K+ concentration can activate NLRP3 protein. Once activated, NLRP3 recruits adapter molecule ASC, which interacts with pro-caspase-1, resulting in the formation of NLRP3 inflammasome. Active NLRP3 inflammasome promotes the recruitment and activation of caspase-1, which is responsible for the activation of IL-1β and IL-18 as well as the cleavage of GSDMD. The cleaved GSDMD then forms pores on the cell membrane, triggering pyroptosis. Most studies supported that the activation of NLRP3 inflammasome contributed to the development of atherosclerosis (AS), myocardial infarction (MI), myocardial ischemia/reperfusion (MI/R), and cardiac hypertrophy, which ultimately promoted heart failure.
Research has shown that a variety of risk factors for CVDs, obesity, diabetes, and metabolic syndrome can activate NLRP3 inflammasome and promote inflammation (32, 33). Compared with healthy adults, the expressions of the NLRP3 inflammasome, ASC, caspase-1, IL-1β, and IL-18 in the serum of patients with coronary heart disease were significantly increased (34). Elevated levels of NLRP3 mRNA in cardiac hypertrophy, inflammatory response, and ventricular dilatation were observed in mouse models (35). The expression and release of mediators such as IL-1β and IL-18 promote the occurrence and development of AS and affect the stability of AS (36, 37), suggesting their important role in the pathogenesis of CVDs. Intervention in the formation and activation of NLRP3 inflammasome is expected to provide new ideas for the prevention and treatment of CVDs. Inhibition of the NLRP3/IL-1β pathway can alleviate cardiac inflammatory response, improve cardiac contractility, and attenuate cardiomyopathy in sepsis (38). Therefore, it is speculated that the immune pathway mediated by inflammasome is involved in the development of ventricular remodeling in patients with coronary heart disease (39).
So far, there have been conflicting results in research on the potential association between the polymorphism of NLRP3 and CVDs. A study showed that NLRP3 rs10754558 polymorphism is associated with the occurrence and severity of coronary artery disease and the increase in serum concentration of IL-1β (40). On the contrary, another study revealed no association between NLRP3 polymorphism and CVDs (41). However, the cohort sizes of the two studies are relatively small, so larger studies involving more patients will be needed in the future to accurately evaluate the impact of NLRP3 polymorphisms on CVDs.
3 Inflammasomes in Cardiovascular Diseases
3.1 Activation of Inflammasomes in Cardiovascular Diseases
AS is a progressive disease of the arteries, characterized by inflammation of the blood vessel walls and the accumulation of cholesterol, calcium, and cellular debris. Cholesterol crystals (ChCs), a major component of atherosclerotic lesions, are known to directly activate NLRP3 inflammasome by inducing the infiltration of lysosomal protease cathepsin B into the cytoplasm (42), thus inducing a series of inflammation in vivo and in vitro, which links lipid metabolism and inflammation. ChCs were also found to trigger NLRP3 activation in human macrophages and mouse carotid arterial ECs (43). Moreover, ChCs induced severe endothelial dysfunction in mouse coronary arteries, which can be alleviated in Nlrp3−/− mice (44). Besides, it has been found that oxidized low-density lipoprotein (ox-LDL) upregulated the protein levels of NLRP3, caspase-1, and IL-1β in vascular ECs and increased cell pyroptosis, via inhibiting tetmethylcytosine dioxygenase 2, which can be downregulated by blocking the NF-κB pathway and improving mitochondrial functions (45). Oxidized phospholipids (oxPLs), a product of lipid peroxidation, mainly result from tissue injury and lead to proinflammatory or anti-inflammatory responses, depending on the context (46), which have been reported to potentiate oxidative stress, trigger sterile inflammation, and be involved in many infectious and sterile-mediated inflammation in the CVDs, including AS (47). The levels of oxPLs are increased in hyperlipidemic human plasma and atherosclerotic lesions (48). Ox-LDL and oxPLs increase oxidative stress in the endothelium and promote the activation of NLRP3 inflammasome and release of IL-1β, thereby contributing to the atherogenic phenotype of ECs (49). Significantly increased oxPLs were observed in vivo and in vitro following MI/R. Furthermore, oxPLs resulted in rat cardiomyocyte cell death in vitro, and neutralization of oxPL in vivo reduced infarct size after MI/R, which may represent a novel potential therapeutic avenue to attenuate I/R injury and improve cardiac outcomes during AMI (50). Therefore, based on the association between the activation of inflammasome, especially NLRP3, and atherogenic factors, whether the inflammasome might be involved in AS has been extensively studied (51, 52).
Actually, several epidemiological studies have linked aortic NLRP3 expression with CVD prevalence, thus indirectly confirming the importance of the NLRP3 inflammasome in humans. The levels of NLRP3 in aortic (53) and peripheral blood monocytes (54) were elevated in patients with coronary AS and positively correlated with the disease severity and the atherosclerotic risk factors, such as hypertension, smoking, LDL-C, and high-density lipoprotein-C levels, which are possible to be predictors of major adverse cardiac events. Furthermore, NLRP3, ASC, caspase-1, IL-1β, and IL-18 were highly expressed in human carotid atherosclerotic plaques, compared with healthy mesenteric arteries or iliac arteries (55, 56), and the expression levels were even higher in unstable in comparison to stable plaques (55).
The cell debris released during AMI functions as DAMPs, which induces the expressions of key components of the inflammasome through activation of the NF-κB or cell surface receptors, such as IL-1R1 (17). Compared with healthy controls, the NLRP3 and caspase-1 mRNA transcription and protein levels of circulating monocytes in patients with acute coronary syndrome or stable angina pectoris are increased (23).
The expressions of NLRP1 and NLRC4 were increased in patients with primary AS lesions compared with healthy controls, and inflammasome complex was activated by interaction with NLRP1 and NLRC4 receptors (57). Another study has reported that elevated levels of plasma triglycerides and very-low-density lipoprotein cholesterol of patients with AS, manifested as peripheral arterial disease, promoted activation of the NLRP1 inflammasome by NF-κB in human arterial ECs (58). It is suggested that statin can regulate the expression of NLRP1 inflammasome via sterol regulatory-element binding proteins (59). It has been reported that the NLRP1 inflammation pathway is activated after MI, which can be suppressed by the promotion of autophagic flux (60). More and more evidence suggests that NLRP1 may trigger the immune-inflammatory processes in arterial ECs and affect the vessel remodeling; thus, NLRP1 inflammasome inhibition may be a novel therapeutic approach to peripheral arterial disease (61). A genome-wide association study showed that the NLRC4 inflammasome was important for IL-18 production in acute coronary syndrome patients, thus promoting the formation of atherosclerotic plaque (62).
Notably, AIM2 was constitutively expressed in normal intima and media of the human carotid artery as well as the aorta and was upregulated around the necrotic core of human atherosclerotic lesions (63). Abundant AIM2 expression was associated with increased dsDNA deposition in late AS, during which dsDNA released by necrotic cells may activate the AIM2 inflammasome and promote the release of atherosclerotic cytokines, and in return, AIM2-mediated cell death may contribute to the deposition of dsDNA in the necrotic core (64). AIM2 and NLRC4 inflammasomes in cardiac macrophages and cardiomyocytes were found to be increased in the area around left ventricle (LV) infarction, thus resulting in cardiomyocyte death and HF and confirming the previous finding that NLRC4 plays an important role in regulating IL-18 in patients with acute coronary syndromes (62, 65) It has been found that expressions of the inflammasome protein AIM2 and NLRC4 were increased in human HF samples, while the NLRP1/NALP1 and NLRP3 inflammasome showed no change, among which the expression of AIM2 was primarily detected in monocytes/macrophages of failing hearts and has been further verified in animal models (66).
As the important downstream effector of the inflammasome, IL-1 and IL-18 play an important role in AS. IL-1 was shown to be expressed in human atherosclerotic plaques (67) and contribute to the initiation, formation, growth, and rupture of the plaques (68, 69). Animal studies have shown that IL-1β expression levels were elevated in animals with ventricular remodeling (70). The levels of IL-18 were upregulated in mice with either MI/R injury or MI (71, 72). A study reported that plasma levels of IL-18 were increased in patients with congestive HF, which was directly associated with the severity of myocardial damage and dysfunction (73).
In short, many CVD risk factors can lead to the activation of the inflammasome and its downstream molecules, which in turn participate in the pathological process of the disease.
3.2 Experimental Evidence for the Role of Inflammasomes in Cardiovascular Diseases
3.2.1 Atherosclerosis
The role and mechanism of inflammasomes in AS have been studied extensively through animal experiments. Latz and colleagues found that in mice lacking Ldlr, inflammasomes contributed to the progression of AS, and mice lacking Nlrp3 or Il-1 were resistant to diet-induced systemic inflammatory cytokine responses and AS (51, 74). Consistently, transplantation with Nlrp3−/−, Asc−/−, or Il-1α/β−/− bone marrow into Ldlr−/− mice markedly decreased early AS in parallel with the reduction in IL-1β and IL-18 levels (51). Moreover, knockdown of Nlrp3 by lentiviral silencing (75) in ApoE−/− mice prevented AS progression and reduced the production of proinflammatory cytokines and the levels of plasma lipids with an increase of macrophage polarization from the M1 to M2 state, which further confirms the importance of NLRP3 inflammasome. It has been found that the NLRP3 inflammasome not only can mediate the early development of early AS but also can promote the formation of vulnerable plaques (76). Thus, the response of inflammasome to ChCs may be a key early trigger of atherosclerotic inflammation.
Cholesterol efflux is mediated by the cholesterol transporters ATP-binding cassette A1 and G1 (ABCA1/G1), transforming cholesterol into high-density lipoprotein, which plays a fundamental role in atherogenesis (77). Bone marrow transplantation from mice with myeloid Abca1/g1−/− along with Nlrp3 or caspase-1/11 double knockout into Ldlr−/− mice fed with a western diet suggested that Nlrp3 and caspase-1/11 deficiency inhibited atherosclerotic lesions in myeloid Abca1/g1-deficient Ldlr−/− mice. The study also showed that inflammasome activation enhanced neutrophil recruitment and neutrophil extracellular trap formation in plaques (78).
Regulating the caspase-1 can help reverse the progression of AS and maintain plaque stability. Two independent studies revealed that deficiency of caspase-1 in ApoE−/− mice showed reduced atherosclerotic lesions fed with a lower cholesterol atherogenic diet (79) or a decreased spontaneous development of lesions with a chow diet for 26 weeks (80). Another study (81) has found that bone marrow transplantation of caspase-1/11−/− mice into Ldlr−/− mice showed a strong reduction in atherosclerotic plaque size, as well as necrotic core content compared with control bone marrow. In a word, these studies indicate that the accumulation of lipids (mainly ox-LDL and ChCs) induces the activation of the NLRP3 inflammasome, which then promotes monocyte adhesion, macrophage transformation into foam cells, and the subsequent development of atherosclerotic lesions (82).
Moreover, in atherosclerotic plaques of ApoE−/− mice, purinergic receptor P2X7 is co-expressed with NLRP3 and plays an important role in AS by promoting phosphorylation of protein kinase R to activate NLRP3 inflammasome (83). Interestingly, microbial pathogens promote AS via the NLRP3 inflammasome. For example, Nlrp3 deficiency inhibited the Chlamydia pneumoniae-accelerated AS (84). Similarly, Porphyromonas gingivalis, a periodontal disease pathogen, activated NLRP3 inflammasome via interacting with pattern recognition receptors and augmented atherogenesis (85).
The activation of the NLRP3 inflammasome is affected by multiple signaling pathways. Therefore, differences in the systemic or local environment may influence the gene dose effect. It is suggested that deletion of Nlrp3 in Ldlr−/− mice inhibited the development of AS due to other pathways participating in the local inflammation, while Nlrp3 deletion alone only had a small effect (78). Besides, another study showed that Nlrp3, Asc, or caspase-1 deletion did not show a reduction in AS development in ApoE-deficient mice fed a high-fat diet (86), while caspase-1 and Cd36 deletion significantly inhibited AS (79, 80, 87), suggesting that the development of atherogenesis may not depend on NLRP3 inflammasome activation (88). These conflicts may be explained by the differences in experimental conditions, including the mouse model, gender, age, the type of atherogenic diet, and high-fat diet feeding time. Actually, ApoE−/− mice used in this study (86) develop diet-induced AS faster and more severely than Ldlr−/− mice used in Latz’s study (51). Moreover, the mice were fed with a stronger atherogenic diet with more than 8-fold higher cholesterol content for 11 weeks, which was 3 weeks longer than in the other study (51). It is possible that excessive dietary cholesterol combined with a prolonged feeding period triggers other inflammatory pathways, potentially weakening the role of NLRP3 inflammasome in AS development. More in-depth studies are needed in the future to clarify the role of the NLRP3 inflammasome.
A recent study (64) has explored the function of the AIM2 inflammasome in AS using ApoE−/− mice and found that Aim2 deletion significantly reduced IL-1β and IL-18 levels within plaques, enhanced atherosclerotic plaque stability, and improved the histopathological features after a high-fat diet. Clonal hematopoiesis, which is very common among the elderly, increases the risk of MI and stroke independent of traditional risk factors, among which the Jak2V617F (Jak2VF) mutations pose the greatest risk of premature coronary heart disease (89, 90). Jak2VF lesions lead to DNA replication stress and activation of the AIM2 inflammasome, thereby aggravating AS, while Aim2 deficiency inhibited AS (91). The mechanism of AIM2 regulating AS involves the following aspects. First, overexpression of Aim2 promoted plaque lesion formation and increased intracellular adhesion molecule-1 (ICAM-1) expression (92), which contributed to monocyte recruitment into arterial endothelium at AS-prone areas through binding lymphocyte function-associated antigen-1. Deletion of Icam-1 in mice reduced aortic lesion size (93). Therefore, increased ICAM-1 level may represent a mechanism for AIM2-induced AS. Second, with the stimulus of ox-LDL, AIM2 had the ability to trigger vascular smooth muscle cell (VSMC) migration and pyroptosis, which are known to be crucial drivers of early AS (94). An in vitro study showed that SMC recruitment and migration were enhanced by Aim2 overexpression and blunted by Aim2 silencing, possibly through regulation of TGF-β/Smad2 signaling and matrix metalloproteinase-2, a key regulator of VSMC migration, providing a mechanism for AIM2 involvement in AS (95). Besides, it has been found that AIM2 mediated VSMC pyroptosis and GSDMD activity through ASC, caspase-1 pathway (92). Third, AIM2 increased the release of proinflammatory cytokines such as IL-1β and IL-18 (64), which were important contributors to the development of lesions, thereby promoting inflammation in atherosclerotic lesions.
Aside from the direct role in AS, AIM2 also has an indirect effect on atherosclerosis. AIM2 induces non-canonical activation of NLRP3 inflammasome via AIM2 inflammasome-mediated pore formation in the cell membrane and subsequent K+ efflux (96). Given the known role of NLRP3 inflammasome in atherosclerosis, it is possible that AIM2 promotes the development of atherosclerosis partially through activating the NLRP3 inflammasome (97).
As downstream effectors of inflammasomes, it has been reported that IL-1α played a key role in the early stage of atherosclerosis, while IL-1β induced inflammation in advanced atherosclerosis in mice (98). Il-1 receptor antagonist (Il-1ra) deficient in ApoE knockout mice developed atheromatous plaques treated with a high-fat diet (68). Consistently, deletion of Il-1r1 in ApoE-deficient mice showed a reduction of atherosclerotic plaques in the aortic root, but not in the brachiocephalic artery (99). Il-1β deficiency reduced atherosclerotic lesions in ApoE−/− mice (100) and Ldlr−/− mice transplanted with bone marrow of Il-1α/Il-1β-deficient mice showed impaired diet-induced atherosclerosis (51). Moreover, IL-18 is expressed in human atheroma cells, such as vascular ECs, SMCs, and macrophages, and also participated in the development of atherosclerosis (101). Il-18 deficiency reduced the development of atherosclerosis in ApoE−/− mice (102). In brief, activated inflammasomes contribute to the development of AS; targeting inflammasomes may be a promising treatment for AS.
3.2.2 Myocardial Infarction
Inflammasome formation is an energy-consuming process that occurs in injured cells and rapidly induces inflammation, which resembles the defense process. However, in the context of sterile inflammation, this inflammation may lead to further injury (16). The evidence that the NLRP3 inflammasome plays an essential role in the development of MI, adverse remodeling, and HF includes the upregulated inflammasome components, the reduced damage caused by inflammasome inhibition, and the involvement of effector molecules (IL-1β and IL-18) in the pathogenesis of MI and HF (8). Endogenous IL-1β and IL-18 also mediated contractile dysfunction following myocardial ischemia (103). Many experimental studies have explored whether the formation of the inflammasome in the injured cardiomyocytes is beneficial or harmful with further injury.
Studies have shown that I/R enhanced NLRP3 inflammasome activation in cardiac microvascular ECs but not cardiomyocytes, and deficiency of Txnip, a contributor to I/R injury, reduced NLRP3 inflammasome activation and infarct size in mouse myocardial tissues (104). Besides, NLRP3 inflammasome is significantly increased in myocardial fibroblasts post-MI, which is essential for myocardial injury. ATP and K+ efflux have been shown to promote NLRP3 inflammasome assembly and IL-1β secretion in cardiac fibroblasts, while a marked reduction of infarct size and improvement of cardiac function were observed in hearts from Nlrp3-deficient mice after ex vivo I/R, but not in Asc-deficient hearts (105), suggesting that NLRP3 inflammasome may be an important contributor to hypoxic damage during ischemia–reperfusion. Consistently, a study has shown that NLRP3 inflammasome-mediated pyroptosis aggravates MI/R injury in diabetic rats (106). Nlrp3 siRNA attenuated macrophage and neutrophil infiltration, cardiomyocyte apoptosis, and infarct size, thus inhibiting MI/R injury (105). Deletion of Asc or caspase-1 reduced myocardial levels of active IL-1β and diminished infarct area and myocardial fibrosis and dysfunction after MI/R injury (19).
However, the role of NLRP3 in AMI is also time-dependent, and Nlrp3 deficiency did not play a protective role during the first few hours of AMI due to low cardiac expression at the onset of ischemia, which was consistent with the 2-step process required for activation (20, 107). Besides, another study showed that deficiency of Nlrp3 increased myocardial infarct size after MI/R injury in vivo, possibly due to dysfunction of the cardioprotective RISK pathway (21). The effect of NLRP3 inflammasome on myocardial injury warrants intensive study.
Taken together, most studies supported that NLRP3 inflammasome can mediate and amplify the inflammatory response and cell pyroptosis, thus aggravating myocardium damage and promoting ventricular remodeling after ischemia with or without reperfused myocardium. Regulating NLRP3 inflammasome can reduce MI/R injury and the area of MI.
Activation of the AIM2 inflammasome is considered to be associated with increased numbers of cardiac M1 macrophages and infarct size, as well as with impaired LV function following MI (65). MicroRNA(miR)-219a attenuated cardiomyocyte apoptosis in a mouse model of MI/R via blocking the AIM2/TLR4 pathway, while overexpression of Aim2 aggravated MI/R injury alleviated by miR-219a (108). So it is assumed that AIM2 inflammasome contributes to MI/R injury probably by promoting the release of IL-18. Collectively, these findings suggest that activation of the AIM2 inflammasome and subsequent release of IL-18 may be a mechanism to exacerbate myocardial damage, LV dysfunction, and LV remodeling after MI.
Besides, NLRP1 inflammasome can be activated by ER stress via the NF-κB signaling pathway and promote MI/R injury. Knockdown of Nlrp1 notably increased cell viability and suppressed hypoxia/reoxygenation-induced cell apoptosis, creatine kinase activity, and lactate dehydrogenase release (109), suggesting the important role of NLRP1 inflammasome in promoting myocardial ischemic injury. Therefore, inhibition of inflammasome activation may be new targets for the prevention and treatment of MI and subsequent HF.
3.2.3 Cardiac Hypertrophy and Heart Failure
HF is the final stage of many cardiac dysfunctions and is the main cause of CVD death. Serum from patients with acute decompensated systolic HF suppressed contractility via an IL-1-dependent and IL-18-dependent mechanism (110). Regulation of NLRP3 inflammasome can decrease cardiac hypertrophy, improve cardiac diastolic and contraction function, and inhibit the development of HF. The study has shown that Nlrp3 knockout or NLRP3 inflammasome inhibition by MCC reduced macrophage accumulation and cardiac fibrosis during AngII infusion, similar to the effect of Ca2+/calmodulin-dependent protein kinase II (CaMKII) δ deletion (111). Furthermore, it has been found that in a mouse model of transverse aortic constriction (TAC), expression of NLRP3 inflammasome was increased in the heart, and cardiomyocyte-specific CaMKIIδ deletion decreased NLRP3 inflammasome levels in cardiomyocytes, assessed by caspase-1 activity and IL-18 activation, inhibited ventricular dilation and contractile dysfunction, and reduced cardiac fibrosis and remodeling, indicating that CaMKIIδ stimulates NLRP3 inflammasome activation in response to pressure overload (112).
However, another study revealed that Nlrp3 deletion impaired cardiac function and that enhanced inflammation, fibrosis, and hypertrophy increased Toll-like receptor 4 expressions in a mouse model of pressure overload interruption, suggesting a negative role of NLRP3 in regulating cardiac remodeling (113). Interestingly, it has been clarified that diabetes further worsened cardiac function and increased HF by promoting the activation of AIM2 and NLRC4 inflammasomes, but not NLRP3 inflammasome (65). Therefore, further investigation is needed to clarify the exact role of the NLRP3 inflammasome.
It has been studied that Nlrp1 deficiency significantly inhibited aortic banding-induced cardiac hypertrophy, inflammation, and fibrosis in vivo and protected against isoproterenol-induced cardiomyocyte hypertrophy in vitro, which was associated with inhibited MAPK, NF-κB, and TGF-β/Smad pathways, indicative of the key role of NLRP1 in the development of HF (114).
Diabetic cardiomyopathy is an important cause of HF. An increased AIM2 expression was observed in the streptozotocin-induced diabetic rat model. Silencing of Aim2 decreased diameter of cardiomyocytes and collagen I and collagen III protein levels, attenuated cardiac dysfunction in vivo, and reduced GSDMD-NT-related pyroptosis in vitro, suggesting that AIM2 promotes diabetic cardiomyopathy by inducing pyroptotic cardiac cell death and ventricular remodeling (115).
Furthermore, as the downstream effector of the inflammasome (including NLRP3 and AIM2), the role of IL-18 in HF has been widely investigated. The study has suggested that IL-18 can cause myocardial action potential prolongation, decrease of K+ current, and increase of calcium activation in diabetic mice and then promote spontaneous contraction of cardiomyocytes through the oxidation and phosphorylation of CaMKII (112). Another clinical study (116) has shown that IL-18 can ultimately lead to HF by promoting myocardial fibrosis and ventricular remodeling. Besides, IL-18 also participated in the cardiomyocyte dysfunction stimulated by IL-1β (110).
Taken together, these findings suggest that inflammasomes play a detrimental role in the progression of CVDs and provide new insights into the molecular mechanisms underlying vascular inflammation (Table 1). It will be an interesting follow-up question clarifying the precise regulatory mechanism of inflammasome activation and downstream genes in specific CVDs.
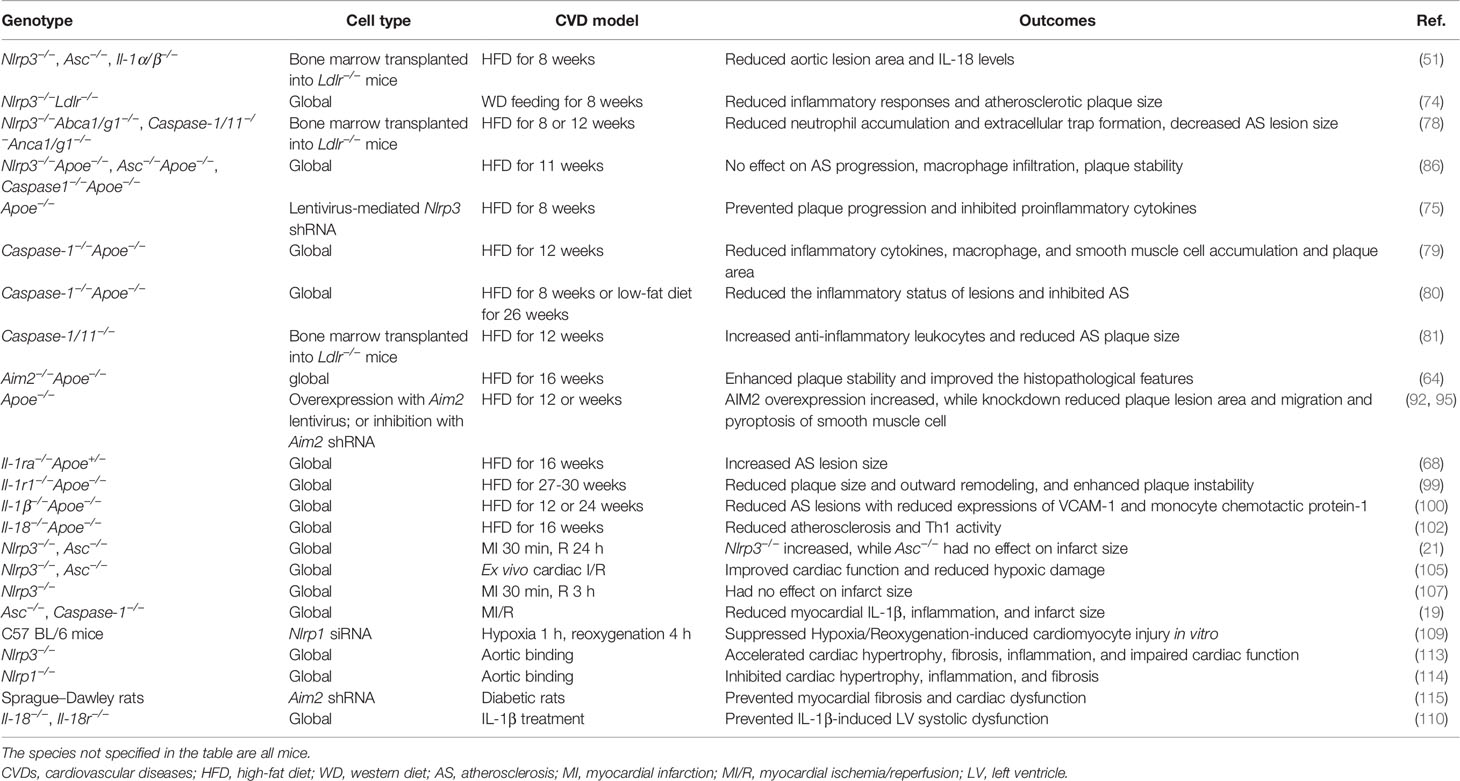
Table 1 Experimental evidence on the role of inflammasomes in animal models of CVDs.
3.3 Pharmacological Intervention of Inflammasomes for Cardiovascular Diseases
Based on the important role of inflammasomes, a variety of studies have explored pharmacological interventions to treat cardiovascular diseases. NLRP3 inhibitor significantly decreased inflammatory cytokines and inhibited the development of atherosclerosis in ApoE−/− mice (117, 118). Marchetti and colleagues designed a novel inhibitor 16673-34-0 derived from glyburide that selectively inhibits NLRP3. This NLRP3 inhibitor significantly reduced infarct size and protected cardiac function after ischemia or reperfusion when given as single or repeated doses, demonstrating the crucial role of NLRP3 inflammasome in MI and I/R models (119, 120). Moreover, MCC950, a known anti-inflammatory small molecule, inhibits the binding of NLRP3 to ASC in mice in vivo, independent of ATPase activity of NLRP3 (121). Treatment with MCC950 decreased infarct size and preserved cardiac function in a pig MI model (20, 122). Consistently, it has been suggested that hematopoietic or myeloid ten-eleven translocation 2 deficiency elevated IL-1β expression and enhanced TAC-induced cardiac remodeling and cardiac dysfunction, thus promoting the development of HF, which was rescued by MCC950, indicating that ten-eleven translocation 2 mediated HF via an IL-1β/NLRP3 inflammasome-dependent mechanism (123). Besides, it was reported that Bay 11-7082, an inhibitor of NLRP3 inflammasome via suppressing the ATPase activity of NLRP3 (124), resulted in a reduction in the formation of the inflammasome in the heart and the infarct size in an I/R model when treated intraperitoneally 10 min before reperfusion (104) or 30 min before ischemia (125). Similarly, treatment with OLT1177, another NLRP3 inhibitor by blocking its ATPase activity, which was originally developed as a topical treatment for degenerative arthritis (126), also reduced the infarct area after MI/R injury (127). Recently, it has been reported that OLT1177 could preserve contractile reserve and diastolic function of the heart in a mouse MI model and may therefore be used to prevent the development of HF in patients with ischemic cardiomyopathy (128). Colchicine, administered at high doses (0.1 mg/kg per day) for 7 days in the AMI mouse model, significantly inhibited NLRP3 inflammasome expression and activation, indicated by reduced NLRP3, ASC, and caspase-1 levels in the MI area; decreased infarct size; alleviated LV remodeling; protected contractile function; and significantly delayed HF development and 7-day survival after MI (129). Acrylamide derivatives were developed, which can covalently bind to NLRP3 to inhibit its ATPase activity. A dose of 50 μmol/L of the compound given 20 min before induction of ischemia in an ex vivo mouse MI model reduced inflammasome activity and infarct size (130).
As a hypoglycemic agent, in addition to lowering blood sugar and reducing the incidence of cardiovascular events in diabetic patients, empagliflozin was also reported to inhibit the activation of the NLRP3 inflammasome, thus reducing cardiac inflammation and preserving cardiac function via a Ca2+-dependent mechanism (131). In consideration of the effect of the inflammasome in cardiac fibrosis, Chinese herbal medicine triptolide and active small molecule pirfenidone have been used to treat fibrosis and were shown to inhibit NLRP3 inflammasome activity, reduce macrophage infiltration and fibrosis, and improve TAC-induced cardiac diastolic and systolic functions (132, 133).
Inhibition of AIM2 with synthetic oligonucleotide A151 reduced the levels of IL-1β and IL-18 and improved plaque stability (64). Administration of probenecid, clinically used as a uricosuric drug, which blocks pannexin-1 channels, significantly reduced AIM2 inflammasome activation in vitro, reduced pressure overload-induced mortality, and restored indices of disease severity in vivo, showing that AIM2 inflammasome activation participates in chronic inflammation in HF (66).
Administration of recombinant IL-1Ra or IL-1β neutralizing antibody to ApoE-deficient mice significantly reduced the diet-induced formation of atheroma (36). In addition, administration of IL-18 by intraperitoneal injections into ApoE−/− mice aggravated atherosclerosis lesions (134). IL-18 neutralizing antibodies have been shown to reduce infarct size following MI/R (71), verifying a pathological role for IL-18 in MI. Daily administration of IL-18 into healthy mice for 7 days led to LV contractile dysfunction and myocardial hypertrophy (135). Correspondingly, it has been reported that treatment with recombinant IL-18 induced mouse interstitial fibrosis, diastolic dysfunction, and myocardial remodeling and increased cardiac osteopontin expression, which are components of the extracellular matrix associated with the fibrotic process during tissue remodeling (116, 136). Conversely, IL-18 neutralizing antibody protected against lipopolysaccharide-induced myocardial dysfunction in mice (137).
To sum up, pharmacological interventions with inflammasome have been shown to be beneficial in the treatment of CVDs in animal studies (Table 2). Further clinical studies are needed to determine whether inhibition of inflammasome can be beneficial for patients with CVDs.
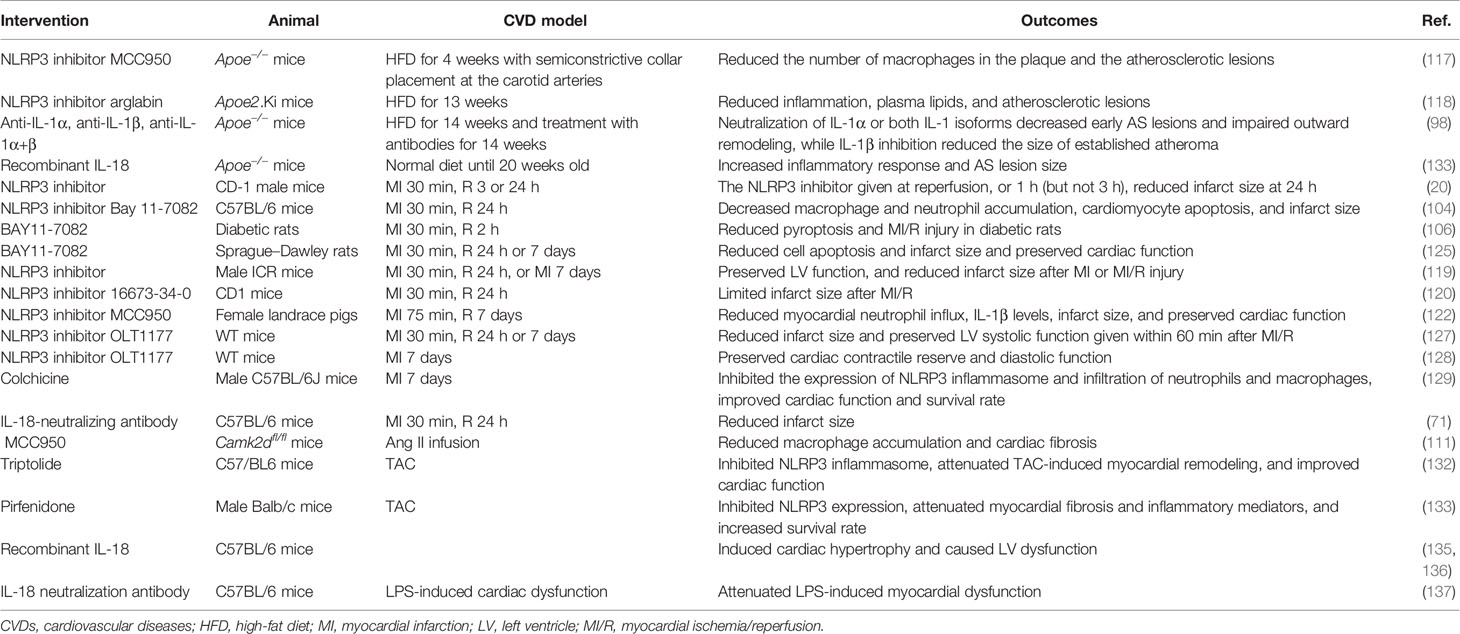
Table 2 Pharmacological interventions of inflammasomes in animal models of CVDs.
4 Clinical Studies on IL-1b and NLRP3 Inflammasome in Cardiovascular Diseases
A variety of studies confirmed that the innate immune responses mediated by inflammasome-dependent cytokines (like IL-1β) promote the development of CVDs such as atherosclerosis and HF (138, 139). Therefore, many clinical studies have been conducted targeting NLRP3 and IL-1β, aiming to discover new prevention and treatment methods for CVDs. The advantage of targeting NLRP3 inflammasome core components is to prevent pyroptosis, which is not affected by IL-1β and IL-18 inhibition. Importantly, the development of NLRP3 inflammasome inhibitors provides the possibility of targeting the harmful effects of NLRP3 (140).
4.1 Canakinumab
Canakinumab, a human IL-1β monoclonal antibody, has been approved by the Food and Drug Administration (FDA) for the treatment of systemic juvenile idiopathic arthritis and some periodic fever syndromes, including cold-heat protein-related periodicity syndromes and familial Mediterranean fever (141, 142). The efficacy of canakinumab in patients with AMI was studied in the multicenter Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) trial (143, 144) involving 10,061 patients with previous AMI (at least 30 days before enrollment) and elevated C-reactive protein (CRP) levels (>2 mg/L), which concluded that canakinumab at a dose of 150 mg significantly reduced the incidence of a recurrent cardiovascular event and hospitalizations for HF in patients with established atherosclerotic disease. CANTOS trial confirms for the first time that targeting an inflammatory mediator can significantly improve the cardiovascular outcome of AMI patients, which verifies the hypothesis of the inflammatory response of coronary heart disease and also provides a basis for the intervention targets of inflammatory response. The limitations of this study are as follows: it included patients with AMI, but it is unknown whether its conclusions can be extended to patients with stable coronary heart disease; the pathogenesis of CVDs is complex, and the intervention of one molecular target in the inflammatory pathway may not be sufficient; besides, as the upstream of the inflammatory response, inhibition of IL-1β by canakinumab-induced severe infection is an adverse effect that cannot be ignored.
4.2 Anakinra
The elevated circulating levels of IL-1β as well as IL-1Ra, IL-6, or CRP correlate with worsening HF symptoms and outcomes, clarifying the relevance of inflammation and HF (145, 146). Other clinical trials have also confirmed the importance of the inflammasome and IL-1 in CVDs by administration of anakinra, an IL-1 receptor antagonist. Treatment with anakinra (100 mg daily for 14 days) in patients with ST-segment elevation MI (STEMI) significantly reduced acute systemic inflammatory response and reduced incidence of adverse remodeling and HF at 3 months or long term (147, 148). The present study has several potential drawbacks. First, the number of patients enrolled is small, which is not sufficient to detect effects on clinical outcomes. Second, true baseline acquisition of the imaging studies is lacking, which was performed obtained 24 to 96 h after admission, and therefore, it is hard to account for early changes in size and contractility. A phase II clinical trial involving 182 patients with non-STEMI elevation AMI revealed that anakinra (100 mg daily for 14 days) reduced the acute inflammatory response. The clinical events at 30 days and 3 months were not affected by anakinra treatment, but surprisingly, anakinra significantly increased recurrent ischemic events after 12 months (149). The main limitation of this study is that the treatment lasted only 14 days and that little is known about the nature of late events, which led to the apparent cardiovascular adverse events 1 year later. In a study of 23 rheumatoid arthritis patients with elevated IL-1β levels, anakinra treatment with a dose of 150 mg significantly improved cardiac and vascular function (150). Consistently, it has been revealed that anakinra improved diastolic dysfunction, cardiac function, and quality of life in patients with decompensated systolic HF (151–153). Recently, it has been reported that targeting IL-6 (a downstream mediator of IL-1β) with tocilizumab in STEMI reduced microvascular obstruction, although it has no effect on the final infarct size; it was less beneficial in patients with symptom duration of <3 h (154), suggesting the importance of the timing of the disease course in which the inflammasome is targeted.
4.3 Colchicine
Colchicine was originally used to treat Mediterranean fever and was later approved by the FDA for the treatment of acute gouty arthritis because it was found to inhibit the assembly and activation of NLRP3 inflammasome with the stimulation of monosodium urate crystals (155). Colchicine inhibits the activity of NLRP3 inflammasome via two aspects: inhibition of P2X7 receptor activation and the polymerization of ASC by interfering with the interaction between the pyrin domains (155). Colchicine was found to inhibit mitochondrial transport and subsequent co-localization of ASC to NLRP3, ultimately suppressing the activation of NLRP3 inflammasome (156). Colchicine plays an anti-inflammatory role mainly through inhibiting NLRP3 inflammasome.
In a clinical trial (157), patients with stable coronary artery diseases were randomly treated with a low dose of colchicine (0.5 mg daily) or placebo, for a median time of 2.3 years. Compared with placebo, colchicine significantly reduced the incidence of adverse cardiovascular endpoints, assessed by reduction of the incidence of acute coronary syndrome. But this trial mainly focused on the effects of colchicine on non-fatal cardiovascular events. Despite the trend toward improved mortality, larger studies are needed to confirm whether colchicine may reduce the risk of fatal cardiac events. It would make sense to investigate the value of colchicine in patients recently hospitalized for the acute coronary syndrome, as they remain at high risk of recurrent events due to plaque disruption (158). Consistent with the important role of NLRP3 inflammasome in ischemia–reperfusion injury, in phase II clinical trial, colchicine (initial dose 1.5 mg, 0.5 mg once or twice daily according to body weight) significantly reduced the infarct size of patients with STEMI and inflammatory biomarkers undergoing primary percutaneous coronary intervention (159). However, the limitation of this study is that 26% of the 77 patients in the colchicine group discontinued treatment due to poor tolerance, which is common when colchicine doses exceed 1.0 mg/day. These clinical studies clarify the contributive role of NLRP3 inflammasome in AMI and HF, which may be a priming target. Conversely, the latest clinical trial enrolled in patients with STEMI referred for primary percutaneous coronary intervention suggested that high-dose colchicine at the time of reperfusion and for 5 days (2-mg loading dose followed by 0.5 mg twice a day) did not reduce the infarct size and LV remodeling (160). The inconsistent result may be because in the former study (159), infarct size reduction was assessed by the release of myocardial biomarker, and only a subgroup of patients was assessed on cardiac MRI. Another explanation may be that in the former study, patients’ thrombosis in MI flow was not reported, suggesting potential confounding factors. More reliable evaluation criteria and primary endpoint events in larger studies are needed in the future.
In summary, clinical data suggest that the IL-1 cytokine family contributes to cardiac dysfunction and that blocking IL-1 with anakinra or canakinumab improves cardiac function and prevents hospitalization in patients with HF. Inhibition of NLRP3 inflammasome protects against myocardial injury in AMI and adverse cardiac remodeling in HF. So far, these data are the strongest evidence that the NLRP3 inflammasome and IL-1β play an important role in CVDs (144, 145) (Table 3).
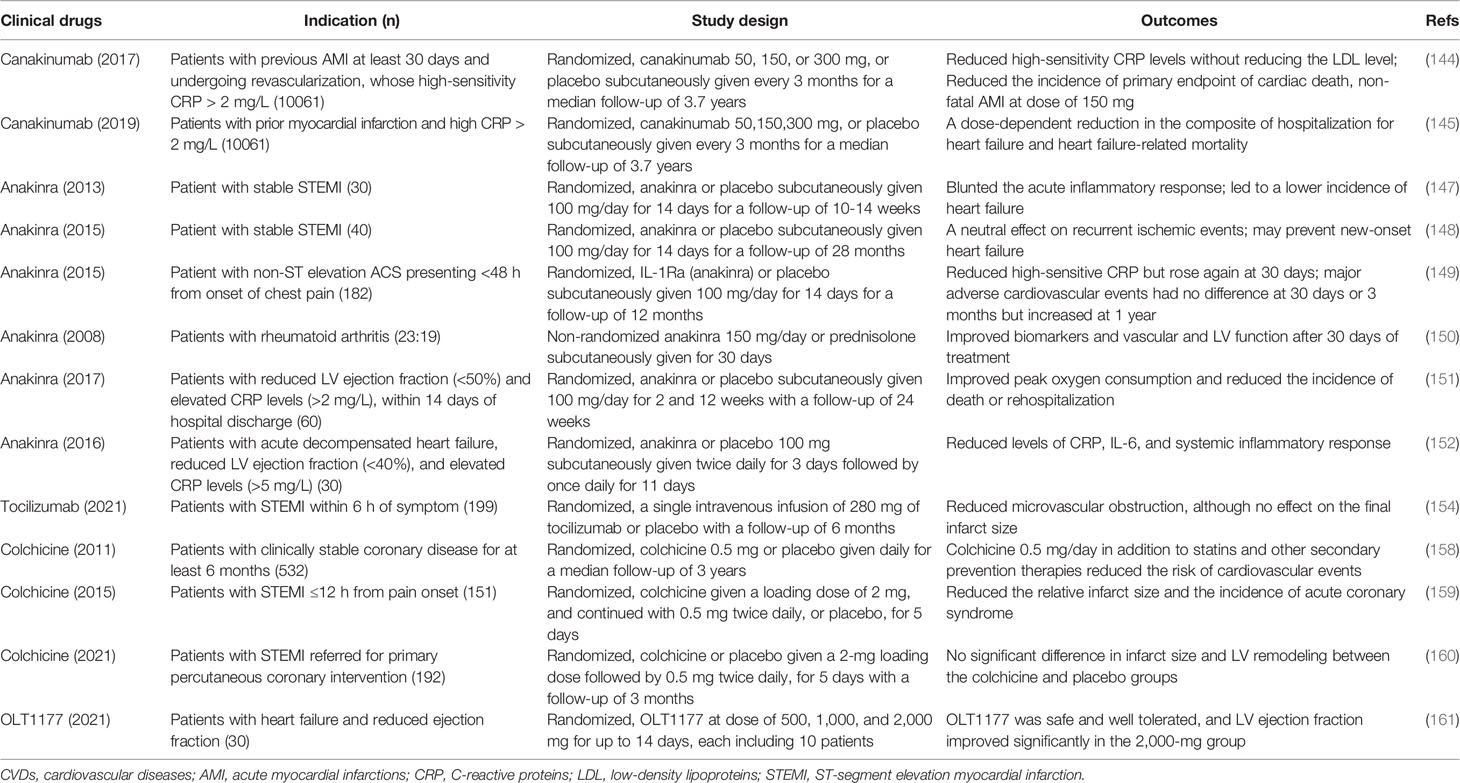
Table 3 Clinical trials of NLRP3 inflammasome and IL-1 or IL-6 blockers in CVDs.
4.4 OLT1177
Recently, the effect and safety of an oral inhibitor of the NLRP3 inflammasome, OLT1177, have been studied in patients with HF and reduced ejection fraction. Subjects were randomized to treatment with OLT1177 for up to 14 days at ascending dose cohorts (500, 1,000, and 2,000 mg), each including 10 patients, and underwent clinical assessment at baseline, day 14, and day 28. Results showed that treatment with OLT1177 for 14 days was safe and well tolerated, and LV ejection fraction improved significantly in the 2,000-mg group (161), suggesting that NLRP3 inhibition can be an important therapeutic target for HF and provide preliminary safety evidence.
CANTOS study is the first successful large-scale anti-inflammatory therapy of CVDs in the clinic and enriches the theoretical system of atherosclerotic CVDs, which is a supplement and improvement to the traditional cholesterol theory. Traditional medicines such as statins and PCSK9 inhibitors reduce cardiovascular risk and play a protective role mainly by lowering blood lipid levels. However, many patients who still have high levels of hypersensitive CRP after statin therapy, indicating a high risk of residual inflammation, often have a poor clinical prognosis. CANTOS study suggests that for these patients, further anti-inflammatory therapy in addition to statin therapy may be considered to maximize cardiovascular benefit.
Most acute coronary events are associated with the rupture of unstable atherosclerotic plaques, and inflammation induced by inflammasome is an important cause of plaque instability and rupture (36, 37). Clinical studies targeting the NLRP3 inflammasome have achieved preliminary clinical benefits in cardiovascular patients, also broadened the targets of inflammatory intervention, and enriched the inflammatory hypothesis. Interestingly, necessary anti-inflammatory therapy with anakinra in rheumatoid arthritis patients also reduced cardiovascular risk and improved cardiac function, further confirming the cardiovascular protective effect of anti-inflammatory therapy.
However, current clinical studies still have some limitations. Clinical trials targeting anakinra and colchicine mainly focused on AMI but few on other types of CVDs; in addition, the scale of most clinical studies is small, and evidence of therapeutic effect in a larger population of CVDs patients is lacking. Whether inhibition of inflammasomes and downstream effectors (such as NLRP3 and IL-1β) can be an effective clinical therapy for the treatment of CVDs needs to be confirmed in larger clinical trials with more inhibitors.
5 Conclusion
In conclusion, a variety of danger signals and pathogenic factors can trigger the priming and activation of the inflammasomes. The activated inflammasomes promote the production and release of the inflammatory cytokines and the systemic inflammatory response, thus playing a key role in the pathological process of CVDs, such as atherosclerosis, myocardial ischemic and non-ischemic injury, and HF. The inflammasome is a promising therapeutic target for CVDs. Therefore, the process of activation and assembly and the precise regulatory mechanism in cell pyroptosis and the progression of CVDs of the inflammasomes are worthwhile to be further studied in the future.
A lot of animal experiments and clinical trials confirm that inflammasomes and the cytokine family, especially NLRP3 inflammasome and IL-1, are central to the pathological response to injury. Therefore, NLRP3 and IL-1 are considered promising targets for CVDs. However, no selective NLRP3 inhibitors are clinically available at present, and the clinical possibilities of IL-1 blockers are being explored. Specific oral NLRP3 inflammasome inhibitors are in clinical development. It will be of great value to the clinic to develop novel inhibitors of inflammasome and proinflammatory cytokines. In the future, there may be inhibitors that target the IL-1 subtype, possibly as well as oral NLRP3 inflammasome inhibitors for a wide range of CVDs (162).
It is worth noting that the optimal timing for administering drugs targeting the inflammasome remains unclear, which depends on the specific drug, type, and severity of CVDs. Future research should also aim to map the oscillations of drug targets such as IL-1β throughout the day so that drugs can be administered at the most effective times (163). Drug target, dose, dosage form, time point, and duration of administration will be the focus of future research to maximize the benefit to the patient.
Author Contributions
YL and LZ are responsible for writing and revising this review. KL participated in the revision of the review. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (82100441).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CVDs, cardiovascular diseases; HF, heart failure; MI, myocardial infarction; AS, atherosclerosis; MI/R, myocardial ischemia/reperfusion; DAMPs, damage-associated molecular patterns; IL-1β, interleukin-1beta; NLRP, NOD, LRR, and PYD domain-containing protein; NLRC, NOD, LRR, and CARD domain-containing protein; AIM2, absent in melanoma 2; ASC, apoptosis-associated speck-like protein containing CARD; PYD, pyrin domain; CARD, caspase recruitment domain; P2X7, purinoreceptor 7; AMI, acute myocardial infarction; GSDMD, gasdermin D; ROS, reactive oxygen species; ChCs, cholesterol crystals; ox-LDL, oxidized low-density lipoprotein; oxPLs, oxidized phospholipids; ABCA1/G1, ATP-binding cassette A1 and G1; ICAM-1, intracellular adhesion molecule-1; IL-1Ra, IL-1 receptor antagonist; LV, left ventricle; TAC, transverse aortic constriction; CANTOS, Canakinumab Anti-Inflammatory Thrombosis Outcome Study; CRP, C-reactive protein; STEMI, ST-segment elevation myocardial infarction.
References
1. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation (2017) 135:e146–603. doi: 10.1161/CIR.0000000000000491
2. Heianza Y, Qi L. Impact of Genes and Environment on Obesity and Cardiovascular Disease. Endocrinology (2019) 160:81–100. doi: 10.1210/en.2018-00591
3. Welsh P, Grassia G, Botha S, Sattar N, Maffia P. Targeting Inflammation to Reduce Cardiovascular Disease Risk: A Realistic Clinical Prospect? Br J Pharmacol (2017) 174:3898–913. doi: 10.1111/bph.13818
4. Broz P, Dixit VM. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
5. Latz E, Xiao TS, Stutz A. Activation and Regulation of the Inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi: 10.1038/nri3452
6. Miao EA, Rajan JV, Aderem A. Caspase-1-Induced Pyroptotic Cell Death. Immunol Rev (2011) 243:206–14. doi: 10.1111/j.1600-065X.2011.01044.x
7. Toldo S, Mezzaroma E, Buckley LF, Potere N, Di Nisio M, Biondi-Zoccai G, et al. Targeting the NLRP3 Inflammasome in Cardiovascular Diseases. Pharmacol Ther (2021) 236:108053. doi: 10.1016/j.pharmthera.2021.108053
8. van Hout GPJ, Bosch L. The Inflammasomes in Cardiovascular Disease. Exp Suppl (2012) (2018) 108:9–40. doi: 10.1007/978-3-319-89390-7_2
9. Li PL. Cardiovascular Pathobiology of Inflammasomes: Inflammatory Machinery and Beyond. Antioxid Redox Signal (2015) 22:1079–83. doi: 10.1089/ars.2015.6319
10. Dinarello CA. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu Rev Immunol (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
11. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 Recognizes Cytosolic dsDNA and Forms a Caspase-1-Activating Inflammasome With ASC. Nature (2009) 458:514–8. doi: 10.1038/nature07725
12. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An Orthogonal Proteomic-Genomic Screen Identifies AIM2 as a Cytoplasmic DNA Sensor for the Inflammasome. Nat Immunol (2009) 10:266–72. doi: 10.1038/ni.1702
13. Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a Human Caspase-1-Activating Protein Related to Apaf-1. J Biol Chem (2001) 276:28309–13. doi: 10.1074/jbc.C100250200
14. Qu Y, Misaghi S, Newton K, Maltzman A, Izrael-Tomasevic A, Arnott D and Dixit VM. NLRP3 Recruitment by NLRC4 During Salmonella Infection. J Exp Med (2016) 213:877–85. doi: 10.1084/jem.20132234
15. Guey B, Bodnar M, Manié SN, Tardivel A and Petrilli V. Caspase-1 Autoproteolysis is Differentially Required for NLRP1b and NLRP3 Inflammasome Function. Proc Natl Acad Sci U S A (2014) 111:17254–9. doi: 10.1073/pnas.1415756111
16. Toldo S, Mezzaroma E, Mauro AG, Salloum F, Van Tassell BW, Abbate A. The Inflammasome in Myocardial Injury and Cardiac Remodeling. Antioxid Redox Signal (2015) 22:1146–61. doi: 10.1089/ars.2014.5989
17. Toldo S, Mezzaroma E, McGeough MD, Peña CA, Marchetti C, Sonnino C, et al. Independent Roles of the Priming and the Triggering of the NLRP3 Inflammasome in the Heart. Cardiovasc Res (2015) 105:203–12. doi: 10.1093/cvr/cvu259
18. Rees Smith B, Furmaniak J. Structural Analysis of the TSH Receptor. Horm Metab Res Suppl Ser (1990) 23:28–32.
19. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, et al. Inflammasome Activation of Cardiac Fibroblasts is Essential for Myocardial Ischemia/Reperfusion Injury. Circulation (2011) 123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777
20. Toldo S, Marchetti C, Mauro AG, Chojnacki J, Mezzaroma E, Carbone S, et al. Inhibition of the NLRP3 Inflammasome Limits the Inflammatory Injury Following Myocardial Ischemia-Reperfusion in the Mouse. Int J Cardiol (2016) 209:215–20. doi: 10.1016/j.ijcard.2016.02.043
21. Sandanger Ø, Gao E, Ranheim T, Bliksøen M, Kaasbøll OJ, Alfsnes K, et al. NLRP3 Inflammasome Activation During Myocardial Ischemia Reperfusion is Cardioprotective. Biochem Biophys Res Commun (2016) 469:1012–20. doi: 10.1016/j.bbrc.2015.12.051
22. Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential Requirement for the Activation of the Inflammasome for Processing and Release of IL-1beta in Monocytes and Macrophages. Blood (2009) 113:2324–35. doi: 10.1182/blood-2008-03-146720
23. Toldo S, Abbate A. The NLRP3 Inflammasome in Acute Myocardial Infarction. Nat Rev Cardiol (2018) 15:203–14. doi: 10.1038/nrcardio.2017.161
24. Pörksen G, Lohse P, Rösen-Wolff A, Heyden S, Förster T, Wendisch J, et al. Periodic Fever, Mild Arthralgias, and Reversible Moderate and Severe Organ Inflammation Associated With the V198M Mutation in the CIAS1 Gene in Three German Patients–Expanding Phenotype of CIAS1 Related Autoinflammatory Syndrome. Eur J Haematol (2004) 73:123–7. doi: 10.1111/j.1600-0609.2004.00270.x
25. Mastrocola R, Penna C, Tullio F, Femminò S, Nigro D, Chiazza F, et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxid Med Cell Longev (2016) 2016:5271251. doi: 10.1155/2016/5271251
26. McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, et al. Caspase-1 Inhibition Prevents Glial Inflammasome Activation and Pyroptosis in Models of Multiple Sclerosis. Proc Natl Acad Sci U S A (2018) 115:E6065–74. doi: 10.1073/pnas.1722041115
27. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
28. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in Pyroptosis. Acta Pharm Sin B (2021) 11:2768–82. doi: 10.1016/j.apsb.2021.02.006
29. Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 Inflammasome is Released as a Particulate Danger Signal That Amplifies the Inflammatory Response. Nat Immunol (2014) 15:738–48. doi: 10.1038/ni.2919
31. Zhou R, Yazdi AS, Menu P and Tschopp J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
32. Pavillard LE, Marín-Aguilar F, Bullon P, Cordero MD. Cardiovascular Diseases, NLRP3 Inflammasome, and Western Dietary Patterns. Pharmacol Res (2018) 131:44–50. doi: 10.1016/j.phrs.2018.03.018
33. Sokolova M, Ranheim T, Louwe MC, Halvorsen B, Yndestad A, Aukrust P. NLRP3 Inflammasome: A Novel Player in Metabolically Induced Inflammation-Potential Influence on the Myocardium. J Cardiovasc Pharmacol (2019) 74:276–84. doi: 10.1097/FJC.0000000000000704
34. Rong J, Xu J, Liu Q, Xu J, Mou T, Zhang X, et al. Anti-Inflammatory Effect of Up-Regulated microRNA-221-3p on Coronary Heart Disease via Suppressing NLRP3/ASC/pro-Caspase-1 Inflammasome Pathway Activation. Cell Cycle (Georgetown Tex) (2020) 19:1478–91. doi: 10.1080/15384101.2020.1754562
35. Yue R, Zheng Z, Luo Y, Wang X, Lv M, Qin D, et al. NLRP3-Mediated Pyroptosis Aggravates Pressure Overload-Induced Cardiac Hypertrophy, Fibrosis, and Dysfunction in Mice: Cardioprotective Role of Irisin. Cell Death Discov (2021) 7:50. doi: 10.1038/s41420-021-00434-y
36. Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H, et al. Monoclonal Antibodies Targeting IL-1 Beta Reduce Biomarkers of Atherosclerosis In Vitro and Inhibit Atherosclerotic Plaque Formation in Apolipoprotein E-Deficient Mice. Atherosclerosis (2011) 216:313–20. doi: 10.1016/j.atherosclerosis.2011.02.026
37. Mallat Z, Corbaz A, Scoazec A, Graber P, Alouani S, Esposito B, et al. Interleukin-18/Interleukin-18 Binding Protein Signaling Modulates Atherosclerotic Lesion Development and Stability. Circ Res (2001) 89:E41–5. doi: 10.1161/hh1901.098735
38. Busch K, Kny M, Huang N, Klassert TE, Stock M, Hahn A, et al. Inhibition of the NLRP3/IL-1β Axis Protects Against Sepsis-Induced Cardiomyopathy. J Cachexia Sarcopenia Muscle (2021) 12:1653–68. doi: 10.1002/jcsm.12763
39. Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, et al. The Inflammasome Promotes Adverse Cardiac Remodeling Following Acute Myocardial Infarction in the Mouse. Proc Natl Acad Sci U S A (2011) 108:19725–30. doi: 10.1073/pnas.1108586108
40. Zhou D, Wang X, Chen T, Wen W, Liu Y, Wu Y, et al. The NLRP3 Rs10754558 Polymorphism Is Associated With the Occurrence and Prognosis of Coronary Artery Disease in the Chinese Han Population. BioMed Res Int (2016) 2016:3185397. doi: 10.1155/2016/3185397
41. Zhao X, Gu C, Yan C, Zhang X, Li Y, Wang L, et al. NALP3-Inflammasome-Related Gene Polymorphisms in Patients With Prehypertension and Coronary Atherosclerosis. BioMed Res Int (2016) 2016:7395627. doi: 10.1155/2016/7395627
42. Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S, Kovanen PT, et al. Cholesterol Crystals Activate the NLRP3 Inflammasome in Human Macrophages: A Novel Link Between Cholesterol Metabolism and Inflammation. PloS One (2010) 5:e11765. doi: 10.1371/journal.pone.0011765
43. Koka S, Xia M, Chen Y, Bhat OM, Yuan X, Boini KM, et al. Endothelial NLRP3 Inflammasome Activation and Arterial Neointima Formation Associated With Acid Sphingomyelinase During Hypercholesterolemia. Redox Biol (2017) 13:336–44. doi: 10.1016/j.redox.2017.06.004
44. Zhang Y, Li X, Pitzer AL, Chen Y, Wang L, Li PL. Coronary Endothelial Dysfunction Induced by Nucleotide Oligomerization Domain-Like Receptor Protein With Pyrin Domain Containing 3 Inflammasome Activation During Hypercholesterolemia: Beyond Inflammation. Antioxid Redox Signal (2015) 22:1084–96. doi: 10.1089/ars.2014.5978
45. Zhaolin Z, Jiaojiao C, Peng W, Yami L, Tingting Z, Jun T, et al. OxLDL Induces Vascular Endothelial Cell Pyroptosis Through miR-125a-5p/TET2 Pathway. J Cell Physiol (2019) 234:7475–91. doi: 10.1002/jcp.27509
46. Zhivaki D, Kagan JC. Innate Immune Detection of Lipid Oxidation as a Threat Assessment Strategy. Nat Rev Immunol (2021), 1–9. doi: 10.1038/s41577-021-00618-8
47. Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell (2001) 104:503–16. doi: 10.1016/S0092-8674(01)00238-0
48. Capoulade R, Chan KL, Yeang C, Mathieu P, Bossé Y, Dumesnil JG, et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J Am Coll Cardiol (2015) 66:1236–46. doi: 10.1016/j.jacc.2015.07.020
49. Miller YI, Shyy JY. Context-Dependent Role of Oxidized Lipids and Lipoproteins in Inflammation. Trends Endocrinol Metab (2017) 28:143–52. doi: 10.1016/j.tem.2016.11.002
50. Yeang C, Hasanally D, Que X, Hung MY, Stamenkovic A, Chan D, et al. Reduction of Myocardial Ischaemia-Reperfusion Injury by Inactivating Oxidized Phospholipids. Cardiovasc Res (2019) 115:179–89. doi: 10.1093/cvr/cvy136
51. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 Inflammasomes are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature (2010) 464:1357–61. doi: 10.1038/nature08938
52. Lukens JR, Dixit VD, Kanneganti TD. Inflammasome Activation in Obesity-Related Inflammatory Diseases and Autoimmunity. Discov Med (2011) 12:65–74.
53. Zheng F, Xing S, Gong Z, Xing Q. NLRP3 Inflammasomes Show High Expression in Aorta of Patients With Atherosclerosis. Heart Lung Circ (2013) 22:746–50. doi: 10.1016/j.hlc.2013.01.012
54. Afrasyab A, Qu P, Zhao Y, Peng K, Wang H, Lou D, et al. Correlation of NLRP3 With Severity and Prognosis of Coronary Atherosclerosis in Acute Coronary Syndrome Patients. Heart Vessels (2016) 31:1218–29. doi: 10.1007/s00380-015-0723-8
55. Shi X, Xie WL, Kong WW, Chen D, Qu P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J Stroke Cerebrovasc Dis (2015) 24:2455–66. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024
56. Paramel Varghese G, Folkersen L, Strawbridge RJ, Halvorsen B, Yndestad A, Ranheim T, et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J Am Heart Assoc (2016) 5:e003031. doi: 10.1161/JAHA.115.003031
57. Borborema MEA, Crovella S, Oliveira D, de Azevêdo Silva J. Inflammasome Activation by NLRP1 and NLRC4 in Patients With Coronary Stenosis. Immunobiology (2020) 225:151940. doi: 10.1016/j.imbio.2020.151940
58. Bleda S, de Haro J, Varela C, Ferruelo A, Acin F. Elevated Levels of Triglycerides and Vldl-Cholesterol Provoke Activation of Nlrp1 Inflammasome in Endothelial Cells. Int J Cardiol (2016) 220:52–5. doi: 10.1016/j.ijcard.2016.06.193
59. Yu SY, Dong B, Tang L, Zhou SH. Statin Regulates NLRP1 Inflammasome Expression Through SREBP1: A Novel Anti-Atherosclerotic Mechanism. Int J Cardiol (2017) 247:11. doi: 10.1016/j.ijcard.2017.05.016
60. Yang Y, Li J, Rao T, Fang Z, Zhang J. The Role and Mechanism of Hyperoside Against Myocardial Infarction in Mice by Regulating Autophagy via NLRP1 Inflammation Pathway. J Ethnopharmacol (2021) 276:114187. doi: 10.1016/j.jep.2021.114187
61. Wang W, Wang C, Gong Y, Zhang X. Inhibition of NLRP1 Inflammasome Might be a Novel Therapeutic Target in the Treatment of Peripheral Arterial Disease. Int J Cardiol (2018) 256:29. doi: 10.1016/j.ijcard.2017.10.125
62. Johansson Å, Eriksson N, Becker RC, Storey RF, Himmelmann A, Hagström E, et al. NLRC4 Inflammasome Is an Important Regulator of Interleukin-18 Levels in Patients With Acute Coronary Syndromes: Genome-Wide Association Study in the PLATelet Inhibition and Patient Outcomes Trial (PLATO). Circ Cardiovasc Genet (2015) 8:498–506. doi: 10.1161/CIRCGENETICS.114.000724
63. Hakimi M, Peters A, Becker A, Böckler D, Dihlmann S. Inflammation-Related Induction of Absent in Melanoma 2 (AIM2) in Vascular Cells and Atherosclerotic Lesions Suggests a Role in Vascular Pathogenesis. J Vasc Surg (2014) 59:794–803. doi: 10.1016/j.jvs.2013.03.048
64. Paulin N, Viola JR, Maas SL, de Jong R, Fernandes-Alnemri T, Weber C, et al. Double-Strand DNA Sensing Aim2 Inflammasome Regulates Atherosclerotic Plaque Vulnerability. Circulation (2018) 138:321–3. doi: 10.1161/CIRCULATIONAHA.117.033098
65. Durga Devi T, Babu M, Mäkinen P, Kaikkonen MU, Heinaniemi M, Laakso H, et al. Aggravated Postinfarct Heart Failure in Type 2 Diabetes Is Associated With Impaired Mitophagy and Exaggerated Inflammasome Activation. Am J Pathol (2017) 187:2659–73. doi: 10.1016/j.ajpath.2017.08.023
66. Onódi Z, Ruppert M, Kucsera D, Sayour AA, Tóth VE, Koncsos G, et al. AIM2-Driven Inflammasome Activation in Heart Failure. Cardiovasc Res (2021) 117:2639–51. doi: 10.1093/ehjci/ehaa946.0847
67. Dewberry R, Holden H, Crossman D, Francis S. Interleukin-1 Receptor Antagonist Expression in Human Endothelial Cells and Atherosclerosis. Arterioscler Thromb Vasc Biol (2000) 20:2394–400. doi: 10.1161/01.ATV.20.11.2394
68. Isoda K, Sawada S, Ishigami N, Matsuki T, Miyazaki K, Kusuhara M, et al. Lack of Interleukin-1 Receptor Antagonist Modulates Plaque Composition in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol (2004) 24:1068–73. doi: 10.1161/01.ATV.0000127025.48140.a3
69. Chamberlain J, Evans D, King A, Dewberry R, Dower S, Crossman D, et al. Interleukin-1beta and Signaling of Interleukin-1 in Vascular Wall and Circulating Cells Modulates the Extent of Neointima Formation in Mice. Am J Pathol (2006) 168:1396–403. doi: 10.2353/ajpath.2006.051054
70. Ono K, Matsumori A, Shioi T, Furukawa Y and Sasayama S. Cytokine Gene Expression After Myocardial Infarction in Rat Hearts: Possible Implication in Left Ventricular Remodeling. Circulation (1998) 98:149–56. doi: 10.1161/01.CIR.98.2.149
71. Venkatachalam K, Prabhu SD, Reddy VS, Boylston WH, Valente AJ, Chandrasekar B. Neutralization of Interleukin-18 Ameliorates Ischemia/Reperfusion-Induced Myocardial Injury. J Biol Chem (2009) 284:7853–65. doi: 10.1074/jbc.M808824200
72. Woldbaek PR, Tønnessen T, Henriksen UL, Florholmen G, Lunde PK, Lyberg T, et al. Increased Cardiac IL-18 mRNA, Pro-IL-18 and Plasma IL-18 After Myocardial Infarction in the Mouse; a Potential Role in Cardiac Dysfunction. Cardiovasc Res (2003) 59:122–31. doi: 10.1016/S0008-6363(03)00339-0
73. Seta Y, Kanda T, Tanaka T, Arai M, Sekiguchi K, Yokoyama T, et al. Interleukin-18 in Patients With Congestive Heart Failure: Induction of Atrial Natriuretic Peptide Gene Expression. Res Commun Mol Pathol Pharmacol (2000) 108:87–95.
74. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell (2018) 172:162–175.e14. doi: 10.1016/j.cell.2017.12.013
75. Zheng F, Xing S, Gong Z, Mu W, Xing Q. Silence of NLRP3 Suppresses Atherosclerosis and Stabilizes Plaques in Apolipoprotein E-Deficient Mice. Mediators Inflamm (2014) 2014:507208. doi: 10.1155/2014/507208
76. Li Q, Leng K, Liu Y, Sun H, Gao J, Ren Q, et al. The Impact of Hyperglycaemia on PKM2-Mediated NLRP3 Inflammasome/Stress Granule Signalling in Macrophages and its Correlation With Plaque Vulnerability: An In Vivo and In Vitro Study. Metab: Clin Exp (2020) 107:154231. doi: 10.1016/j.metabol.2020.154231
77. Ou X, Gao JH, He LH, Yu XH, Wang G, Zou J, et al. Angiopoietin-1 Aggravates Atherosclerosis by Inhibiting Cholesterol Efflux and Promoting Inflammatory Response. Biochim Biophys Acta Mol Cell Biol Lipids (2020) 1865:158535. doi: 10.1016/j.bbalip.2019.158535
78. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation (2018) 138:898–912. doi: 10.1161/CIRCULATIONAHA.117.032636
79. Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, et al. Critical Role of Caspase-1 in Vascular Inflammation and Development of Atherosclerosis in Western Diet-Fed Apolipoprotein E-Deficient Mice. Biochem Biophys Res Commun (2012) 425:162–8. doi: 10.1016/j.bbrc.2012.07.058
80. Gage J, Hasu M, Thabet M and Whitman SC. Caspase-1 Deficiency Decreases Atherosclerosis in Apolipoprotein E-Null Mice. Can J Cardiol (2012) 28:222–9. doi: 10.1016/j.cjca.2011.10.013
81. Hendrikx T, Jeurissen ML, van Gorp PJ, Gijbels MJ, Walenbergh SM, Houben T, et al. Bone Marrow-Specific Caspase-1/11 Deficiency Inhibits Atherosclerosis Development in Ldlr(-/-) Mice. FEBS J (2015) 282:2327–38. doi: 10.1111/febs.13279
82. Wang Y, Liu X, Shi H, Yu Y, Yu Y, Li M and Chen R. NLRP3 Inflammasome, an Immune-Inflammatory Target in Pathogenesis and Treatment of Cardiovascular Diseases. Clin Trans Med (2020) 10:91–106. doi: 10.1002/ctm2.13
83. Peng K, Liu L, Wei D, Lv Y, Wang G, Xiong W, et al. P2X7R is Involved in the Progression of Atherosclerosis by Promoting NLRP3 Inflammasome Activation. Int J Mol Med (2015) 35:1179–88. doi: 10.3892/ijmm.2015.2129
84. Tumurkhuu G, Dagvadorj J, Porritt RA, Crother TR, Shimada K, Tarling EJ, et al. Chlamydia Pneumoniae Hijacks a Host Autoregulatory IL-1β Loop to Drive Foam Cell Formation and Accelerate Atherosclerosis. Cell Metab (2018) 28:432–448.e4. doi: 10.1016/j.cmet.2018.05.027
85. Brown PM, Kennedy DJ, Morton RE, Febbraio M. CD36/SR-B2-TLR2 Dependent Pathways Enhance Porphyromonas Gingivalis Mediated Atherosclerosis in the Ldlr KO Mouse Model. PloS One (2015) 10:e0125126. doi: 10.1371/journal.pone.0125126
86. Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, et al. Atherosclerosis in ApoE-Deficient Mice Progresses Independently of the NLRP3 Inflammasome. Cell Death Dis (2011) 2:e137. doi: 10.1038/cddis.2011.18
87. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 Coordinates NLRP3 Inflammasome Activation by Facilitating Intracellular Nucleation of Soluble Ligands Into Particulate Ligands in Sterile Inflammation. Nat Immunol (2013) 14:812–20. doi: 10.1038/ni.2639
88. de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol (2014) 6::a016287. doi: 10.1101/cshperspect.a016287
89. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
90. Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited Causes of Clonal Haematopoiesis in 97,691 Whole Genomes. Nature (2020) 586:763–8. doi: 10.1038/s41586-020-2819-2
91. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 Inflammasome Exacerbates Atherosclerosis in Clonal Haematopoiesis. Nature (2021) 592:296–301. doi: 10.1038/s41586-021-03341-5
92. Pan J, Han L, Guo J, Wang X, Liu D, Tian J, et al. AIM2 Accelerates the Atherosclerotic Plaque Progressions in ApoE-/- Mice. Biochem Biophys Res Commun (2018) 498:487–94. doi: 10.1016/j.bbrc.2018.03.005
93. Nageh MF, Sandberg ET, Marotti KR, Lin AH, Melchior EP, Bullard DC, et al. Deficiency of Inflammatory Cell Adhesion Molecules Protects Against Atherosclerosis in Mice. Arterioscler Thromb Vasc Biol (1997) 17:1517–20. doi: 10.1161/01.ATV.17.8.1517
94. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular Smooth Muscle Cells in Atherosclerosis. Nat Rev Cardiol (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
95. Pan J, Lu L, Wang X, Liu D, Tian J, Liu H, et al. AIM2 Regulates Vascular Smooth Muscle Cell Migration in Atherosclerosis. Biochem Biophys Res Commun (2018) 497:401–9. doi: 10.1016/j.bbrc.2018.02.094
96. Cunha LD, Silva ALN, Ribeiro JM, Mascarenhas DPA, Quirino GFS, Santos LL, et al. AIM2 Engages Active But Unprocessed Caspase-1 to Induce Noncanonical Activation of the NLRP3 Inflammasome. Cell Rep (2017) 20:794–805. doi: 10.1016/j.celrep.2017.06.086
97. Zhao ZZ, Zheng XL, Jiang ZS. Emerging Roles of Absent in Melanoma 2 in Cardiovascular Diseases. Clin Chim Acta Int J Clin Chem (2020) 511:14–23. doi: 10.1016/j.cca.2020.08.031
98. Vromman A, Ruvkun V, Shvartz E, Wojtkiewicz G, Santos Masson G, Tesmenitsky Y, et al. Stage-Dependent Differential Effects of Interleukin-1 Isoforms on Experimental Atherosclerosis. Eur Heart J (2019) 40:2482–91. doi: 10.1093/eurheartj/ehz008
99. Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL, et al. Genetic Inactivation of IL-1 Signaling Enhances Atherosclerotic Plaque Instability and Reduces Outward Vessel Remodeling in Advanced Atherosclerosis in Mice. J Clin Invest (2012) 122:70–9. doi: 10.1172/JCI43713
100. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of Interleukin-1beta Decreases the Severity of Atherosclerosis in ApoE-Deficient Mice. Arterioscler Thromb Vasc Biol (2003) 23:656–60. doi: 10.1161/01.ATV.0000064374.15232.C3
101. Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schönbeck U. Expression of Interleukin (IL)-18 and Functional IL-18 Receptor on Human Vascular Endothelial Cells, Smooth Muscle Cells, and Macrophages: Implications for Atherogenesis. J Exp Med (2002) 195:245–57. doi: 10.1084/jem.20011022
102. Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, et al. Reduced Atherosclerosis in Interleukin-18 Deficient Apolipoprotein E-Knockout Mice. Cardiovasc Res (2003) 59:234–40. doi: 10.1016/S0008-6363(03)00343-2
103. Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of Caspase 1 Reduces Human Myocardial Ischemic Dysfunction via Inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci U S A (2001) 98:2871–6. doi: 10.1073/pnas.041611398
104. Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao C, et al. TXNIP Mediates NLRP3 Inflammasome Activation in Cardiac Microvascular Endothelial Cells as a Novel Mechanism in Myocardial Ischemia/Reperfusion Injury. Basic Res Cardiol (2014) 109:415. doi: 10.1007/s00395-014-0415-z
105. Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, et al. The NLRP3 Inflammasome is Up-Regulated in Cardiac Fibroblasts and Mediates Myocardial Ischaemia-Reperfusion Injury. Cardiovasc Res (2013) 99:164–74. doi: 10.1093/cvr/cvt091
106. Qiu Z, Lei S, Zhao B, Wu Y, Su W, Liu M, et al. NLRP3 Inflammasome Activation-Mediated Pyroptosis Aggravates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats. Oxid Med Cell Longev (2017) 2017:9743280. doi: 10.1155/2017/9743280
107. Jong WM, Leemans JC, Weber NC, Juffermans NP, Schultz MJ, Hollmann MW, et al. Nlrp3 Plays No Role in Acute Cardiac Infarction Due to Low Cardiac Expression. Int J Cardiol (2014) 177:41–3. doi: 10.1016/j.ijcard.2014.09.148
108. Li Y, Xing N, Yuan J, Yang J. Sevoflurane Attenuates Cardiomyocyte Apoptosis by Mediating the miR-219a/AIM2/TLR4/MyD88 Axis in Myocardial Ischemia/Reperfusion Injury in Mice. Cell Cycle (Georgetown Tex) (2020) 19:1665–76. doi: 10.1080/15384101.2020.1765512
109. Cao L, Chen Y, Zhang Z, Li Y, Zhao P. Endoplasmic Reticulum Stress-Induced NLRP1 Inflammasome Activation Contributes to Myocardial Ischemia/Reperfusion Injury. Shock (Augusta Ga) (2019) 51:511–8. doi: 10.1097/SHK.0000000000001175
110. Toldo S, Mezzaroma E, O’Brien L, Marchetti C, Seropian IM, Voelkel NF, et al. Interleukin-18 Mediates Interleukin-1-Induced Cardiac Dysfunction. Am J Physiol Heart Circ Physiol (2014) 306:H1025–31. doi: 10.1152/ajpheart.00795.2013
111. Willeford A, Suetomi T, Nickle A, Hoffman HM. Miyamoto S and Heller Brown J. Camkiiδ-Mediated Inflammatory Gene Expression and Inflammasome Activation in Cardiomyocytes Initiate Inflammation and Induce Fibrosis. JCI Insight (2018) 3:e97054. doi: 10.1172/jci.insight.97054
112. Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, et al. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca(2+)/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation (2018) 138:2530–44. doi: 10.1161/CIRCULATIONAHA.118.034621
113. Li F, Zhang H, Yang L, Yong H, Qin Q, Tan M, et al. NLRP3 Deficiency Accelerates Pressure Overload-Induced Cardiac Remodeling via Increased TLR4 Expression. J Mol Med (Berlin Germany) (2018) 96:1189–202. doi: 10.1007/s00109-018-1691-0
114. Zong J, Li FF, Liang K, Dai R, Zhang H, Yan L, et al. Nuclear Localization Leucine-Rich-Repeat Protein 1 Deficiency Protects Against Cardiac Hypertrophy by Pressure Overload. Cell Physiol Biochem (2018) 48:75–86. doi: 10.1159/000491664
115. Wang X, Pan J, Liu H, Zhang M, Liu D, Lu L, et al. AIM2 Gene Silencing Attenuates Diabetic Cardiomyopathy in Type 2 Diabetic Rat Model. Life Sci (2019) 221:249–58. doi: 10.1016/j.lfs.2019.02.035
116. Yu Q, Vazquez R, Khojeini EV, Patel C, Venkataramani R, Larson DF. IL-18 Induction of Osteopontin Mediates Cardiac Fibrosis and Diastolic Dysfunction in Mice. Am J Physiol Heart Circ Physiol (2009) 297:H76–85. doi: 10.1152/ajpheart.01285.2008
117. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slütter B, et al. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler Thromb Vasc Biol (2017) 37:1457–61. doi: 10.1161/ATVBAHA.117.309575
118. Abderrazak A, Couchie D, Mahmood DF, Elhage R, Vindis C, Laffargue M, et al. Anti-Inflammatory and Antiatherogenic Effects of the NLRP3 Inflammasome Inhibitor Arglabin in ApoE2.Ki Mice Fed a High-Fat Diet. Circulation (2015) 131:1061–70. doi: 10.1161/CIRCULATIONAHA.114.013730
119. Marchetti C, Toldo S, Chojnacki J, Mezzaroma E, Liu K, Salloum FN, et al. Pharmacologic Inhibition of the NLRP3 Inflammasome Preserves Cardiac Function After Ischemic and Nonischemic Injury in the Mouse. J Cardiovasc Pharmacol (2015) 66:1–8. doi: 10.1097/FJC.0000000000000247
120. Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, et al. A Novel Pharmacologic Inhibitor of the NLRP3 Inflammasome Limits Myocardial Injury After Ischemia-Reperfusion in the Mouse. J Cardiovasc Pharmacol (2014) 63:316–22. doi: 10.1097/FJC.0000000000000053
121. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat Med (2015) 21:248–55. doi: 10.1038/nm.3806
122. van Hout GP, Bosch L, Ellenbroek GH, de Haan JJ, van Solinge WW, Cooper MA, et al. The Selective NLRP3-Inflammasome Inhibitor MCC950 Reduces Infarct Size and Preserves Cardiac Function in a Pig Model of Myocardial Infarction. Eur Heart J (2017) 38:828–36. doi: 10.1093/eurheartj/ehw247
123. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J Am Coll Cardiol (2018) 71:875–86. doi: 10.1016/j.jacc.2017.12.037
124. Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, et al. Anti-Inflammatory Compounds Parthenolide and Bay 11-7082 are Direct Inhibitors of the Inflammasome. J Biol Chem (2010) 285:9792–802. doi: 10.1074/jbc.M109.082305
125. Kim YS, Kim JS, Kwon JS, Jeong MH, Cho JG, Park JC, et al. BAY 11-7082, a Nuclear Factor-κb Inhibitor, Reduces Inflammation and Apoptosis in a Rat Cardiac Ischemia-Reperfusion Injury Model. Int Heart J (2010) 51:348–53. doi: 10.1536/ihj.51.348
126. Marchetti C, Swartzwelter B, Koenders MI, Azam T, Tengesdal IW, Powers N, et al. NLRP3 Inflammasome Inhibitor OLT1177 Suppresses Joint Inflammation in Murine Models of Acute Arthritis. Arthritis Res Ther (2018) 20:169. doi: 10.1186/s13075-018-1664-2
127. Toldo S, Mauro AG, Cutter Z, Van Tassell BW, Mezzaroma E, Del Buono MG, et al. The NLRP3 Inflammasome Inhibitor, OLT1177 (Dapansutrile), Reduces Infarct Size and Preserves Contractile Function After Ischemia Reperfusion Injury in the Mouse. J Cardiovasc Pharmacol (2019) 73:215–22. doi: 10.1097/FJC.0000000000000658
128. Aliaga J, Bonaventura A, Mezzaroma E, Dhakal Y, Mauro AG, Abbate A, et al. Preservation of Contractile Reserve and Diastolic Function by Inhibiting the NLRP3 Inflammasome With OLT1177(®) (Dapansutrile) in a Mouse Model of Severe Ischemic Cardiomyopathy Due to Non-Reperfused Anterior Wall Myocardial Infarction. Molecules (Basel Switzerland) (2021) 26:3534. doi: 10.3390/molecules26123534
129. Fujisue K, Sugamura K, Kurokawa H, Matsubara J, Ishii M, Izumiya Y, et al. Colchicine Improves Survival, Left Ventricular Remodeling, and Chronic Cardiac Function After Acute Myocardial Infarction. Circ J (2017) 81:1174–82. doi: 10.1253/circj.CJ-16-0949
130. Cocco M, Miglio G, Giorgis M, Garella D, Marini E, Costale A, et al. Design, Synthesis, and Evaluation of Acrylamide Derivatives as Direct NLRP3 Inflammasome Inhibitors. ChemMedChem (2016) 11:1790–803. doi: 10.1002/cmdc.201600055
131. Byrne NJ, Matsumura N, Maayah ZH, Ferdaoussi M, Takahara S, Darwesh AM, et al. Empagliflozin Blunts Worsening Cardiac Dysfunction Associated With Reduced NLRP3 (Nucleotide-Binding Domain-Like Receptor Protein 3) Inflammasome Activation in Heart Failure. Circ Heart Fail (2020) 13:e006277. doi: 10.1161/CIRCHEARTFAILURE.119.006277
132. Li R, Lu K, Wang Y, Chen M, Zhang F, Shen H, et al. Triptolide Attenuates Pressure Overload-Induced Myocardial Remodeling in Mice via the Inhibition of NLRP3 Inflammasome Expression. Biochem Biophys Res Commun (2017) 485:69–75. doi: 10.1016/j.bbrc.2017.02.021
133. Wang Y, Wu Y, Chen J, Zhao S and Li H. Pirfenidone Attenuates Cardiac Fibrosis in a Mouse Model of TAC-Induced Left Ventricular Remodeling by Suppressing NLRP3 Inflammasome Formation. Cardiology (2013) 126:1–11. doi: 10.1159/000351179
134. Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 Enhances Atherosclerosis in Apolipoprotein E(-/-) Mice Through Release of Interferon-Gamma. Circ Res (2002) 90:E34–8. doi: 10.1161/hh0202.105292
135. Woldbaek PR, Sande JB, Strømme TA, Lunde PK, Djurovic S, Lyberg T, et al. Daily Administration of Interleukin-18 Causes Myocardial Dysfunction in Healthy Mice. Am J Physiol Heart Circ Physiol (2005) 289:H708–14. doi: 10.1152/ajpheart.01179.2004
136. Platis A, Yu Q, Moore D, Khojeini E, Tsau P, Larson D. The Effect of Daily Administration of IL-18 on Cardiac Structure and Function. Perfusion (2008) 23:237–42. doi: 10.1177/0267659108101511
137. Raeburn CD, Dinarello CA, Zimmerman MA, Calkins CM, Pomerantz BJ, McIntyre RC Jr, et al. Neutralization of IL-18 Attenuates Lipopolysaccharide-Induced Myocardial Dysfunction. Am J Physiol Heart Circ Physiol (2002) 283:H650–7. doi: 10.1152/ajpheart.00043.2002
138. Ramji DP, Davies TS. Cytokines in Atherosclerosis: Key Players in All Stages of Disease and Promising Therapeutic Targets. Cytokine Growth Factor Rev (2015) 26:673–85. doi: 10.1016/j.cytogfr.2015.04.003
139. Dihlmann S, Erhart P, Mehrabi A, Nickkholgh A, Lasitschka F, Böckler D, et al. Increased Expression and Activation of Absent in Melanoma 2 Inflammasome Components in Lymphocytic Infiltrates of Abdominal Aortic Aneurysms. Mol Med (Cambridge Mass) (2014) 20:230–7. doi: 10.2119/molmed.2013.00162
140. Mezzaroma E, Abbate A, Toldo S. NLRP3 Inflammasome Inhibitors in Cardiovascular Diseases. Molecules (Basel Switzerland) (2021) 26:976. doi: 10.3390/molecules26040976
141. Maggio MC, Ragusa SS, Corsello G. Early Treatment of Systemic Juvenile Idiopathic Arthritis With Canakinumab and Complete Remission After 2 Years of Treatment Suspension: Case Report of an Adolescent Girl. Clin Drug Invest (2019) 39:491–4. doi: 10.1007/s40261-019-00766-9
142. Çakan M, Karadağ ŞG, Ayaz NA. Canakinumab in Colchicine Resistant Familial Mediterranean Fever and Other Pediatric Rheumatic Diseases. Turk J Pediatr (2020) 62:167–74. doi: 10.24953/turkjped.2020.02.001
143. Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β Inhibition and the Prevention of Recurrent Cardiovascular Events: Rationale and Design of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J (2011) 162:597–605. doi: 10.1016/j.ahj.2011.06.012
144. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy With Canakinumab for Atherosclerotic Disease. N Engl J Med (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
145. Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation (2019) 139:1289–99. doi: 10.1161/CIRCULATIONAHA.118.038010
146. van Wezenbeek J, Canada JM, Ravindra K, Carbone S, Kadariya D, Trankle CR, et al. Determinants of Cardiorespiratory Fitness in Patients With Heart Failure Across a Wide Range of Ejection Fractions. Am J Cardiol (2020) 125:76–81. doi: 10.1016/j.amjcard.2019.09.036
147. Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, et al. Effects of Interleukin-1 Blockade With Anakinra on Adverse Cardiac Remodeling and Heart Failure After Acute Myocardial Infarction [From the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) Pilot Study]. Am J Cardiol (2013) 111:1394–400. doi: 10.1016/j.amjcard.2013.01.287
148. Abbate A, Kontos MC, Abouzaki NA, Melchior RD, Thomas C, Van Tassell BW, et al. Comparative Safety of Interleukin-1 Blockade With Anakinra in Patients With ST-Segment Elevation Acute Myocardial Infarction (From the VCU-ART and VCU-ART2 Pilot Studies). Am J Cardiol (2015) 115:288–92. doi: 10.1016/j.amjcard.2014.11.003
149. Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B, et al. The Effect of Interleukin-1 Receptor Antagonist Therapy on Markers of Inflammation in non-ST Elevation Acute Coronary Syndromes: The MRC-ILA Heart Study. Eur Heart J (2015) 36:377–84. doi: 10.1093/eurheartj/ehu272
150. Ikonomidis I, Lekakis JP, Nikolaou M, Paraskevaidis I, Andreadou I, Kaplanoglou T, et al. Inhibition of Interleukin-1 by Anakinra Improves Vascular and Left Ventricular Function in Patients With Rheumatoid Arthritis. Circulation (2008) 117:2662–9. doi: 10.1161/CIRCULATIONAHA.107.731877
151. Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ Heart Fail (2017) 10:e004373. doi: 10.1161/CIRCHEARTFAILURE.117.004373
152. Van Tassell BW, Abouzaki NA, Oddi Erdle C, Carbone S, Trankle CR, Melchior RD, et al. Interleukin-1 Blockade in Acute Decompensated Heart Failure: A Randomized, Double-Blinded, Placebo-Controlled Pilot Study. J Cardiovasc Pharmacol (2016) 67:544–51. doi: 10.1097/FJC.0000000000000378
153. Van Tassell BW, Buckley LF, Carbone S, Trankle CR, Canada JM, Dixon DL, et al. Interleukin-1 Blockade in Heart Failure With Preserved Ejection Fraction: Rationale and Design of the Diastolic Heart Failure Anakinra Response Trial 2 (D-Hart2). Clin Cardiol (2017) 40:626–32. doi: 10.1002/clc.22719
154. Broch K, Anstensrud AK, Woxholt S, Sharma K, Tøllefsen IM, Bendz B, et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients With Acute ST-Segment Elevation Myocardial Infarction. J Am Coll Cardiol (2021) 77:1845–55. doi: 10.1016/j.jacc.2021.02.049
155. Leung YY, Yao Hui LL, Kraus VB. Colchicine–Update on Mechanisms of Action and Therapeutic Uses. Semin Arthritis Rheum (2015) 45:341–50. doi: 10.1016/j.semarthrit.2015.06.013
156. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-Driven Spatial Arrangement of Mitochondria Promotes Activation of the NLRP3 Inflammasome. Nat Immunol (2013) 14:454–60. doi: 10.1038/ni.2550
157. Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J Am Coll Cardiol (2013) 61:404–10. doi: 10.1016/j.jacc.2012.10.027
158. Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS, et al. A Prospective Natural-History Study of Coronary Atherosclerosis. N Engl J Med (2011) 364:226–35. doi: 10.1056/NEJMoa1002358
159. Deftereos S, Giannopoulos G, Angelidis C, Alexopoulos N, Filippatos G, Papoutsidakis N, et al. Anti-Inflammatory Treatment With Colchicine in Acute Myocardial Infarction: A Pilot Study. Circulation (2015) 132:1395–403. doi: 10.1161/CIRCULATIONAHA.115.017611
160. Mewton N, Roubille F, Bresson D, Prieur C, Bouleti C, Bochaton T, et al. Effect of Colchicine on Myocardial Injury in Acute Myocardial Infarction. Circulation (2021) 144:859–69. doi: 10.1161/CIRCULATIONAHA.121.056177
161. Wohlford GF, Van Tassell BW, Billingsley HE, Kadariya D, Canada JM, Carbone S, et al. Phase 1b, Randomized, Double-Blinded, Dose Escalation, Single-Center, Repeat Dose Safety and Pharmacodynamics Study of the Oral NLRP3 Inhibitor Dapansutrile in Subjects With NYHA II-III Systolic Heart Failure. J Cardiovasc Pharmacol (2020) 77:49–60. doi: 10.1097/FJC.0000000000000931
162. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ Res (2020) 126:1260–80. doi: 10.1161/CIRCRESAHA.120.315937
Keywords: inflammasome, atherosclerosis, myocardial infarction, heart failure, cardiac hypertrophy
Citation: Liao Y, Liu K and Zhu L (2022) Emerging Roles of Inflammasomes in Cardiovascular Diseases. Front. Immunol. 13:834289. doi: 10.3389/fimmu.2022.834289
Received: 13 December 2021; Accepted: 07 March 2022;
Published: 07 April 2022.
Edited by:
Guo-Chang Fan, University of Cincinnati, United StatesReviewed by:
Andrea Baragetti, University of Milan, ItalyXuchu Que, University of California, San Diego, United States
Hui-hui Yang, Central South University, China
Copyright © 2022 Liao, Liu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liyuan Zhu, zhuliyuan1218@foxmail.com; zly931218@163.com