 Mahdie Jafari1
Mahdie Jafari1 Maryam Kadkhodazadeh2Mina Bahrololoumi Shapourabadi3
Maryam Kadkhodazadeh2Mina Bahrololoumi Shapourabadi3 Nasser Hashemi Goradel4
Nasser Hashemi Goradel4 Mohammad Ali Shokrgozar5
Mohammad Ali Shokrgozar5 Arash Arashkia2Shahriyar Abdoli6*
Arash Arashkia2Shahriyar Abdoli6* Zahra Sharifzadeh1*
Zahra Sharifzadeh1*- 1Department of Immunology, Pasteur Institute of Iran, Tehran, Iran
- 2Department of Molecular Virology, Pasture Institute of Iran, Tehran, Iran
- 3HUM Immune Biotech Company, Tehran, Iran
- 4Department of Medical Biotechnology, School of Advanced Technologies in Medicine, Tehran University of Medical Sciences, Tehran, Iran
- 5National Cell Bank of Iran, Pasteur Institute of Iran, Tehran, Iran
- 6School of Advanced Medical Technologies, Golestan University of Medical Sciences, Gorgan, Iran
Despite the fact that the new drugs and targeted therapies have been approved for cancer therapy during the past 30 years, the majority of cancer types are still remain challenging to be treated. Due to the tumor heterogeneity, immune system evasion and the complex interaction between the tumor microenvironment and immune cells, the great majority of malignancies need multimodal therapy. Unfortunately, tumors frequently develop treatment resistance, so it is important to have a variety of therapeutic choices available for the treatment of neoplastic diseases. Immunotherapy has lately shown clinical responses in malignancies with unfavorable outcomes. Oncolytic virus (OV) immunotherapy is a cancer treatment strategy that employs naturally occurring or genetically-modified viruses that multiply preferentially within cancer cells. OVs have the ability to not only induce oncolysis but also activate cells of the immune system, which in turn activates innate and adaptive anticancer responses. Despite the fact that OVs were translated into clinical trials, with T-VECs receiving FDA approval for melanoma, their use in fighting cancer faced some challenges, including off-target side effects, immune system clearance, non-specific uptake, and intratumoral spread of OVs in solid tumors. Although various strategies have been used to overcome the challenges, these strategies have not provided promising outcomes in monotherapy with OVs. In this situation, it is increasingly common to use rational combinations of immunotherapies to improve patient benefit. With the development of other aspects of cancer immunotherapy strategies, combinational therapy has been proposed to improve the anti-tumor activities of OVs. In this regard, OVs were combined with other biotherapeutic platforms, including various forms of antibodies, nanobodies, chimeric antigen receptor (CAR) T cells, and dendritic cells, to reduce the side effects of OVs and enhance their efficacy. This article reviews the promising outcomes of OVs in cancer therapy, the challenges OVs face and solutions, and their combination with other biotherapeutic agents.
1 Introduction
Cancer is rapidly becoming the leading cause of mortality worldwide. Every year, nineteen million new malignancies are diagnosed, resulting in about ten million deaths (1, 2). Because cancer is a complicated and heterogeneous disease with numerous genetic mutations, current cancer therapies frequently do not achieve the desired outcomes for the majority of malignancies, despite many promises of progress in treatment. Consequently, cancer treatment has become a challenge, so more efficient treatment procedures are required (3, 4). Traditional treatments, such as surgery, chemotherapy, hormone therapy, and radiation therapy, have not only unfavorable adverse effects on individuals in the majority of patients but also yield minimal long-term benefits (5).
Immunotherapy has been a promising approach to cancer treatment over the last two decades. It is target-specific, can be adjusted to the needs of each patient, and has fewer side effects than earlier cancer therapies. Immunotherapy drugs can be more effective against cancer when combined with other therapies, such as radiation therapy, chemotherapy and targeted drugs. As an example, several studies have shown promising results of using a mix of chemotherapy and immunotherapy as a first strike against non-small cell lung cancer (2, 5, 6). To date, various immunotherapeutic approaches have been introduced in cancer treatment, such as pro-inflammatory cytokines, cancer vaccines, adoptive T-cell therapy, antibody-based immunotherapies, and oncolytic viruses (OVs) (7, 8).
Currently, oncolytic virotherapy (OVT) is one of the most popular cancer immunotherapy approaches owing to the flexibility of viral production platforms and providing a multimodal strategy to selectively and efficiently target and destroy tumor cells (9, 10). Furthermore, OV platforms could be applied without depth knowledge of tumor antigens in various malignancies (11). OVs provide multi-mechanistic therapeutic effects against the majority of cancer types, but like with many other current cancer therapies, oncolytic virotherapy still faces challenges and hurdles before becoming an effective anticancer therapy (3). Despite some encouraging outcomes, OVT still is not completely effective in most cases because of some issues such as tumor bulk penetration, anti-viral immune responses, and unfavorable tumor microenvironment (TME) (12, 13). On the other hand, due to off-target infection and sequestrations by non-specific tissues, especially in systemic administration, there are some safety concerns about using OVs as therapeutic agents (14).
Despite this, in clinical trials of monotherapy, OVs with older generations of armings (such as GM-CSF) have elicited a potent and robust response. Newer methods, like combining OVs with immunotherapies to turn “immune-cold” tumors into “immune-hot” ones, can almost certainly make OVs more effective (3, 15, 16). The use of rational combination therapies and targeting have been raised to improve the efficacy of OVs, and these combinations may integrate multiple methodologies and technologies that can increase patient benefit from the treatment (9, 17).
Oncolytic viruses have been used in combination with other cancer treatment modalities of immunotherapy or cell therapy, such as antibodies, nanobodies, bispecific (antibody-based immunotherapies), checkpoint inhibitors, adoptive T-cell therapy, natural killer (NK) cells, and T-cell engagers (BiTE), to improve cancer treatment (10). In this review, the challenges of OVT are discussed in detail, including its low efficiency, safety issues, and delivery methods, and finally, we focus on combining OVs with other biotherapeutic strategies to overcome the challenges.
2 Intro to virotherapy: From concept to bedside
2.1 An overview of virotherapy
The concept of employing viruses to treat cancer cells has existed almost as long as viruses have been discovered (18, 19). For more than a century, viruses have been considered potential cancer-fighting agents (20–22). Since the middle of the 1800s, case reports indicated that spontaneous microbial infections in cancer patients might sometimes temporarily reduce tumor burden and, thus, many therapeutic trials have been conducted employing wild-type non-attenuated viruses in cancer therapy (22–25). In the late 1890s, a finding that a “flu-like” condition accompanied by generalized inflammation corresponded with a reduction in tumor cells in a leukemic patient further confirmed the potential therapeutic significance of viruses, in particular (22, 26). In another case, the measles virus has been shown to be an effective natural anticancer agent in the treatment of Burkitt’s lymphoblastic lymphoma (27).
For a long time, the development of selective and harmless viruses was impeded by a lack of tools for viral genome modification (16, 28, 29). It took a few decades for OVT to reach its full potential when recombinant DNA technology became widely used to increase safety (6, 22, 29). The use of a thymidine kinase (TK)-negative mutant of Herpes Simplex Virus (HSV-1) as a possible treatment for gliomas was the first report of a virus modification to reproduce only in dividing cells. TK-mutated HSV-1 has been demonstrated to reproduce preferentially in cancer cells (30–32). A mutated adenovirus (Ad), dl1520 (also known as ONYX-015), was discovered in 1996 that had the E1B55K gene deleted (33, 34). Since the E1B-55kD gene product can bind to and inactivate p53, it was assumed that the deletion of E1B-55kDa renders the mutant adenovirus unable to inactivate p53 in normal cells and, therefore, the viral replication cycle would not be completed. Moreover, the replication of ONYX-015 might be related to the indirect inactivation of the p53 pathway in tumor cells due to the loss of upstream regulators such as p14ARF (35). Nevertheless, it was shown that the p53 status can not impose a restriction on ONYX-015 replication. Actually, the loss of E1B-55K-mediated late viral RNA export results in inability of ONYX-015 to replicate in normal cells. Since, the tumor cells have a special capacity to efficiently export late viral RNA in the absence of E1B-55K, ONYX-015 would selectively replicate in cancer cells (36). As a result, clinical uses for OVs are increasingly prominent due to technological advances (37, 38).
OVs can selectively reproduce in cancer cells and propagate throughout a tumor without affecting the healthy tissues (39, 40). Despite some viruses’ natural tropism for tumors, the wide range of tumor forms and histologic origins makes it challenging to link OVs to a specific malignancy (41, 42). Additionally, it is crucial to consider the tumor-specificity, possible pathogenicity, immunogenicity, druggability, and the viral stability while choosing a virus.
The administration of OVs, either systemically or locally, in cancer-bearing hosts successfully induces antiviral immunity. As a result, OV treatments activate two separate immune responses: antiviral and anticancer. While antitumor immunity is advantageous, antiviral immune responses, including innate and adaptive, are thought to be harmful to the success of OV-based therapy. Indeed, it is conceivable that antiviral immune responses might impede strong viral replication and spread, reducing direct oncolysis of cancer cells,and therefore the efficiency of OV therapy (43). As a result, the most effective “time window” for most OVs to activate anti-tumoral immunity is within the first 1-2 weeks of administration, before the virus is eliminated. One of the major challenges of OV immunotherapy is to strike a balance between the desirable induction of new anti-tumoral immunity and the competing anti-viral immunity while preventing undesired antiviral effector processes from becoming the dominant response pathway, thereby obstructing the acquisition of acquired anti-tumoral immunity. Because of this, researchers are now looking into a number of ways to treat anti-OV immune responses (44). Many studies are developing strategies to enhance OVs construction, reduce clinical toxicity, design efficient OV delivery systems, and increase efficacy by utilizing contemporary genetic engineering approaches (45). A large number of OVs are being investigated in clinical studies, and an even greater number are being evaluated in preclinical studies. The safety of virotherapy has been shown by clinical researches utilizing various OVs to treat different cancers (42, 46, 47).
2.2 Viruses that have already received regulatory approval for the treatment of cancer
Following 30 years of research and encouraging findings from several clinical trials, the OV has attracted a lot of interest, leading to an OV approved by FDA for cancer treatment (25, 38). Four OVs have been approved for use in the treatment of various malignancies. Despite being licensed in Latvia, the first OV, a picornavirus named Rigvir, was never widely used worldwide (48, 49). In 2005, the Chinese SFDA approved the use of a modified Ad, known as Oncorine (H101), in combination with chemotherapy for the treatment of head and neck cancer (50–52). Talimogene laherparepvec (T-VEC, Imlygic), an attenuated HSV containing granulocyte-macrophage colony-stimulating factor (GM-CSF), was approved by the FDA in October 2015 for the treatment of melanoma in the US (53–56). In Japan, a modified version of the HSV, called Delytact, received time-limited and conditional marketing approval for the treatment of malignant gliomas in 2021 (21, 26, 57–59). A summary of the aforementioned OVs has been presented in Table 1.
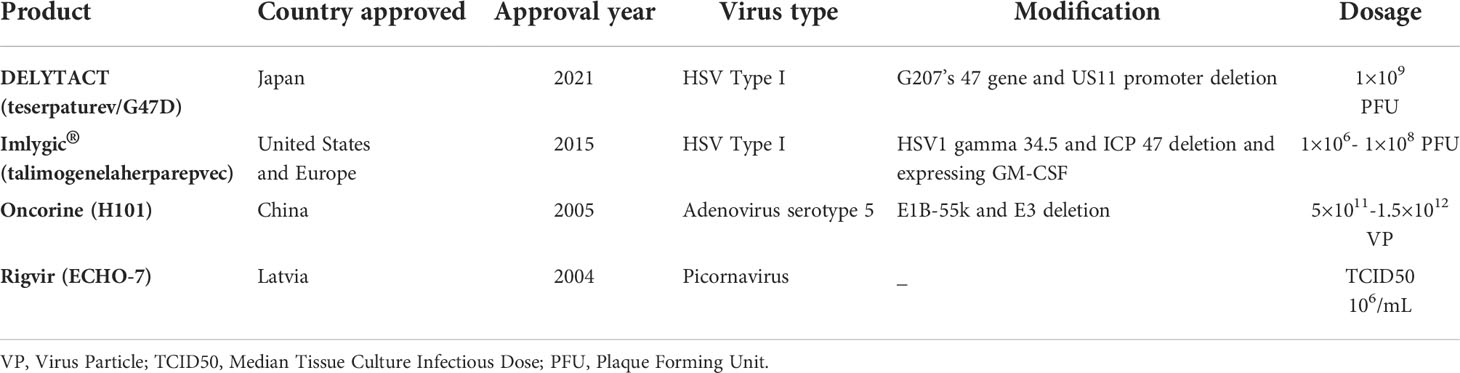
Table 1 Global-approved oncolytic viruses (OVs).
2.3 Action mechanisms of oncolytic viruses: From cytolysis to microenvironment modulation and antitumor immunostimulation
Tumor cells, due to their resistance against apoptosis, appear to be a preferred breeding ground for a wide variety of viruses (22, 60). Viral infection kills tumor cells by several mechanisms including direct cytolytic activity which is thought to be its main oncolytic mechanism; this activity is due to the OVs’ capacity to selectively infect, replicate, and kill cancer cells (Figure 1). It is now generally accepted that virotherapy’s efficacy can be attributed to a number of different processes, including alterations in the tumor’s micro-and macroenvironment and intricate immune control (40, 61, 62). OVs are cytolytic due to viral propagation and host cell bursting (63), and the viral infection may trigger apoptosis in the host cell (20)
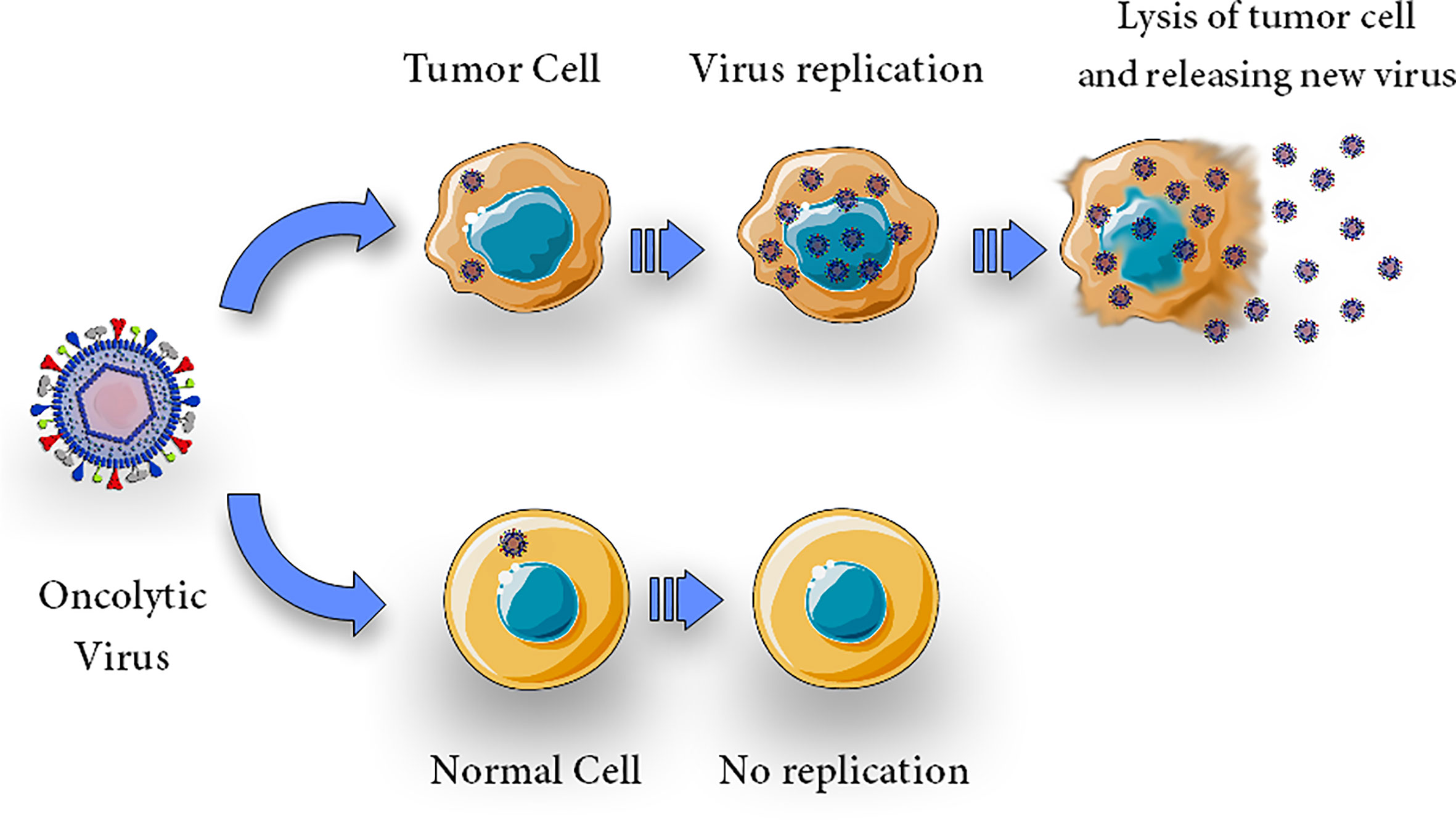
Figure 1 Direct cytolytic activity. Oncolytic virus can selectively infect, replicate, lyse and kill cells. Upon infection with an oncolytic virus, the oncolytic virus replicates in tumor cells and causes oncolysis but does not harm normal cells.
Lysed tumor cells produce endogenous danger-associated molecular patterns (DAMPs), tumor-associated antigens (TAAs), virus-derived PAMPs, and immune-stimulatory cytokines, triggering the anti-tumor immune responses (64, 65).
The key and distinguishing characteristic of OVs is their selective amplification and replication in cancer cells, leading to the death of tumor cells without affecting the normal ones. The intensity of their anti-tumor action depends on the modalities of OV-induced cancer cell death (66, 67). Immunovirotherapy, also known as OV immunotherapy or viroimmunotherapy implies an OV infection that causes an inflammatory TME by eliciting anti-tumor immune responses. Furthermore, there is evidence that OVs have the capacity to transform an immunologically “cold” TME into a “hot” one via the production of chemokines and cytokines. It is worth noting that a balance between helpful anti-tumor immunity and harmful anti-virus immune responses is necessary for optimizing immunovirotherapy (68, 69). In order to modify the TME, OVs may also target tumor-associated stroma cells, such as endothelial cells. Immunogenic cell death (ICD) can be caused by OVs, which promote endoplasmic stress, resulting in the release of DAMPs, such as ATP, HMGB1, ectocalreticulin, and pro-inflammatory cytokines (64). STING, TLR1, and TLR3 on immune cells sense PAMPs and DAMPs, establishing a pro-inflammatory microenvironment that stimulates the production of pro-inflammatory cytokines such as type I IFNs, interleukin (IL)-1, IL-6; TNF-a, GM-CSF, and chemokines such as CCL2, CCL3, CCL5, and CXCL10 (70, 71), leading to transformation of immunologically “cold” T-cell into “hot” T-cells (72, 73). Neutrophils and macrophages are attracted to the site of infection by CCL3 and CXCL10 chemokines, which are involved in anti-cancer responses. Aggregation of PAMPs with NK cell virus-recognition receptors causes early NK cell influx (74). Activated cytotoxic NK cells may produce cytolytic components and activate FAS-FASL, killing virus-infected cells. NK cells emit IFNs and TNF-a to excite macrophages, DCs, and T-cells. This activation of NK cells and DCs induces them to produce IFNs, TNF-a, IL-12, IL-6, and chemokines, which work both autocrinely and paracrinely to increase the initial innate response (21, 75, 76).
The tumor-specific T-cell response is the foundation of adaptive immunity against tumor cells during OV infection. Antigens presented in the context of an MHC molecule, co-stimulatory molecules, and cytokines are required for antigen presenting cells (APCs) to activate antigen-specific T-cell responses successfully (77). The released TAAs and neoantigens following tumor cell lysis by OVs are processed by APCs and are presented on their surface with MHC molecules to CD4+ and CD8+ T-cells (78). Also, OV-infected cells or mature APCs release various cytokines and chemokines, which aid in the recruitment and reactivation of T-cells. Both stimulated T-cells and B-cells could promote tumor regression and are capable of eradicating distant or freshly transplanted tumors without relying on an OV (79, 80).
In addition to tumor cells, the tumor’s extracellular matrix (ECM) and vasculature are also affected by OVs. More than 60% of a solid tumor’s mass comes from the ECM, which is a non-cellular compartment made by activated cancer-associated fibroblasts (CAFs). Collagenous matrix, proteoglycans, and hyaluronan build up in the ECM, providing an impenetrable and stiff barrier around cancerous cells. Because of these physical impediments, OVs have a tough time reaching the entire tumor mass (21). Ilkow et al. showed that interaction between CAFs and cancer cells improves vesicular stomatitis virus (VSV)-based therapies (61).
In contrast, tumor cells release transforming growth factor-beta 1 (TGF-1), which promote OV infection in CAFs. Tumor cells produce large quantities of fibroblast growth factor 2, making them vulnerable to viral infection. It has been reported that OAd not only could lyse glioblastoma cells, but also kills glioblastoma-associated stromal cells (81). Figure 2 provides a detailed anti-cacner mechanism of action of OVs.
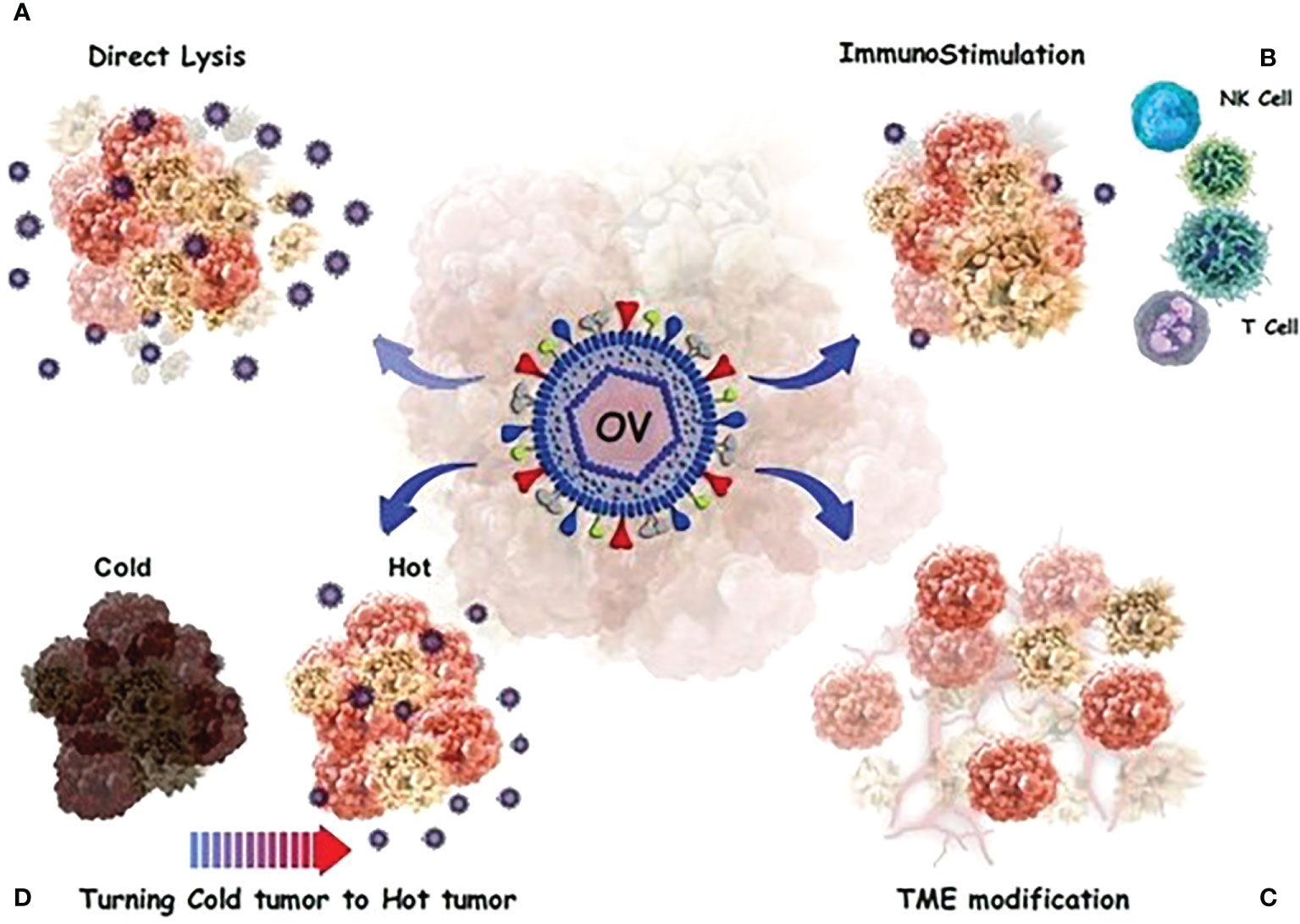
Figure 2 OVs have a direct or indirect toxic effect on tumor cells. (A) Direct oncolysis; The viruses can infect cancer cells and then replicate until the cancer cells rupture. The newborn viruses are then released to infect more cancer cells. (B) Neoantigens and debries from lysed cancer cells activate and recruit dendritic cells (DCs) into the tumor microenvironment and T cells migrate to the site of infection. (C) a number of processes occur, including the alteration of the tumor’s micro-and macro-environment and the control of the immune response in a complex modulation. (D) OVs stimulate innate immunity and turn “cold” tumors into “hot” tumors by stimulating immune cell recruitment and activating systemic anticancer adaptive immunity to reduce tumor growth.
3 Various types of OVs
There are two broad classes of OVs: 1) viruses that have a natural tropism for cancer cells; the naturally cancer-selective OVs utilize the abnormal signaling pathways that support their growth in cancer cells; and 2) those engineered specifically to replicate only in cancer cells (16). The activity of OVs reflects their underlying biology and the host-virus interactions that have evolved in the struggle between pathogenesis and immunity (29). The lack of an anti-viral response in cancer cells is an important mechanism of tumor selectivity for both categories. Interferons (IFNs) are secreted by normal cells in response to viral infection after intracellular pathogen recognition receptors identify viral RNA, DNA, or proteins (PRRs). Hundreds of effector genes, called IFN-stimulated genes (ISGs), are expressed as a result of this signaling cascade and aid in the elimination of the viral infection (16). Myxoma virus (MYXV; poxvirus), Newcastle disease virus (NDV; paramyxovirus), reovirus, Seneca Valley virus (SVV; picornavirus), measles virus (MV; paramyxovirus), poliovirus (PV; picornavirus), vaccinia virus (VV), Ad, HSV, and VSV are some examples of oncolytic viruses (Table 2) (63, 66, 82). Table 2 lists the different types of viruses that have been used for oncolytic purposes.
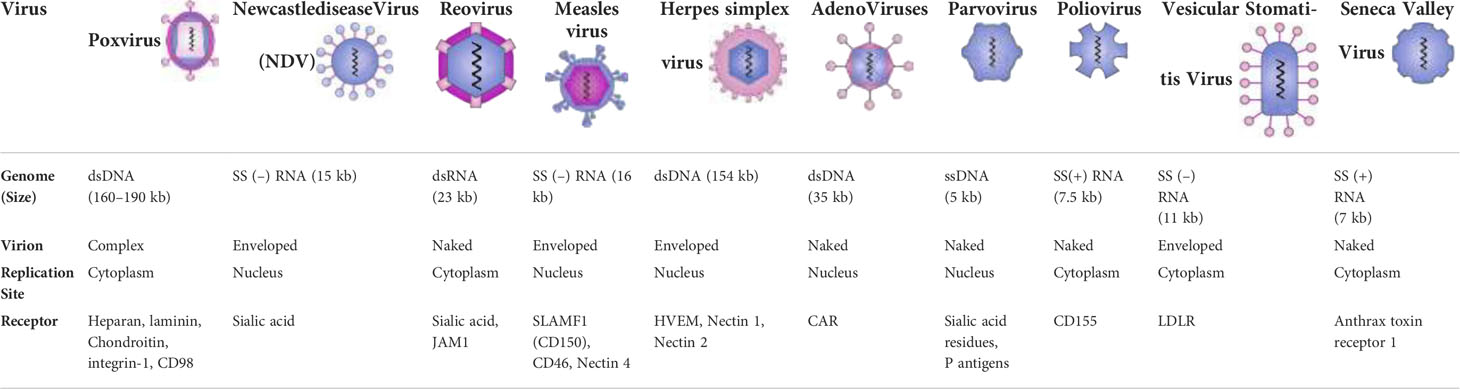
Table 2 Characteristics of major oncolytic viruses.
3.1 Natural tumor-replicating viruses
3.1.1 Poxviruses
One of the most important OV platforms now showing promising outcomes in clinical studies is the vaccinia virus (VV), a member of the Poxviridae family that naturally attacks malignancies (21). Tumors are a recognized target of VV strains owing to the activation of the epidermal growth factor receptor (EGFR) pathway in malignant cells (83). Researchers showed that vascular endothelial growth factor A (VEGF-A) enhances oncolytic VV cytotoxicity by studying the effect of hypoxia on VV infection (84). Tumor-derived VEGF increases VV internalization, leading to enhanced replication and cytotoxicity in both tumor cells and normal respiratory epithelial cells in an AKT-dependent manner (85). Moreover, tumor cells lack the anti-viral cytokines that protect normal cells from viral infection because of their poor interferon (IFN) response. Multiple attenuated VV mutants have been developed to improve tumor-specific targeting and safety in normal tissues (86, 87). The Pexa-Vec (JX-594), an oncolytic VV armed with GM-CSF and disruption of the TK gene, is under investigation in phase I and II clinical trials for treating renal cell carcinoma, advanced breast cancer, and advanced soft-tissue sarcoma (NCT03294083 and NCT02630368). Furthermore, an engineered vaccinia OV, RGV004, encoding a bispecific CD19/CD3 antibody, is in phase I clinical trials for the treatment of refractory/relapsed B-cell lymphoma (NCT04887025). A phase I/II clinical trial showed that Pexa-vec intratumoral injection was safe and effective in treating surgically incurable metastatic melanoma (NCT00429312).
3.1.2 Newcastle disease virus
The Newcastle disease virus (NDV), belonging to the family Paramyxoviridae, is an enclosed virus and contains negative-sense single-stranded RNA (88, 89). The HN protein interacts with sialic acid receptors on the surface of host cells to bind tumor cells, and when the activated F protein joins the viral and host cell membrane, the HN protein fuses with the virus (90). As a result, the virus’s genome penetrates the cytoplasm of the host. NDV can also enter cells by endocytosis and clathrin-mediated endocytosis (91). There is evidence that gene-editing technologies make it simple to introduce foreign genes with anti-tumor activities into the extensive genome of NDV (92). Numerous clinical investigations have shown that NDV has a very excellent safety profile for patients and has considerable anti-cancer activity (93). For instance, because NDV only affects the type I IFN-deficient glioblastoma cells, an inhibitor of IFN signaling eliminates the NDV resistance in type I IFN-positive cells (8, 94). In two clinical studies (NCT03889275 and NCT04613492), the drugs Durvalumab (anti-PD-L1) and attenuated NDV with the GM-CSF and IL-12 genes (MEDI5395 and MEDI9253, respectively) are being used. NDV with durvalumab, is in phase I clinical trial for treating advanced solid tumors (95).
3.1.3 Reovirus
Reovirus (RV), an unenveloped virus containing a double-stranded RNA belongs to the Reoviridae family (96). The oncolytic properties of wild-type reovirus are due to the virus’s preference for replicating in cancer cells (97). Reovirus has the ability to kill cancer cells because of its preferential ability to multiply in cancer cells. Ras overexpression impairs the PKR (protein kinase RNA-activated) pathway, allowing reovirus to infect tumor cells preferentially (98–100). Reolysin (also known as Pelareorep) (101), serotype 3 RV, is the well-known oncolytic RV that as a single agent or in combination with other therapeutic strategies (29), is under investigation in clinical trials (NCT04102618, NCT04445844, and NCT04215146).
3.1.4 Measles virus
The measles virus (MV), which belongs to the genus Morbillivirus in the Paramyxoviridae family, is an enveloped virus containing negative-sense single-stranded RNA (102). Three receptors, CD46, SLAM/CD150, and poliovirus-receptor-like-4, are used by MV to infect host cells (103, 104). However, CD46 is not a tumor-selective receptor because of its expression on normal cells. MV is a hopeful OV candidate due to its good safety profile, which includes the absence of dose-limiting toxicities and spontaneous oncotropism (21, 104, 105).
Heinzerling et al. performed the first Phase I dose-escalation test with a live MV, Edmonston-Zagreb vaccine strain against cutaneous T-cell lymphoma (CTCL) (106). Clinical trials using a measles virus that expresses the human sodium/iodide symporter SLC5A5 are currently being conducted (107). The Mayo Clinic (USA) has launched a number of Phase I/II clinical studies (NCT00390299, NCT02364713, NCT02068794, NCT02700230, NCT01503177, NCT01846091) to examine the clinical safety and usefulness of MV-CEA and MV-NIS (95).
3.1.5 Picornaviruses
Picornaviruses have promising anti-cancer effects in patients (108). Picornaviruses are tumor-specific due to the overexpression of their entry receptors on cancerous cells, including CD155, integrin a1b2, intercellular adhesion molecule-1 and/or decay-accelerating factor (CVA21), anthrax toxin receptor 1 and sialic acids and anthrax toxin receptor 1 (109). Clinical studies using oncolytic picornaviruses typically go smoothly, and no off-target infections have yet been reported. Intratumoral administration of the oncolytic poliovirus PVSRIPO, the live attenuated, type I poliovirus (Sabin) vaccine harboring an internal ribosome entry site (IRES) of human rhinovirus type 2, has demonstrated initial promise in patients with recurrent glioblastoma multiforme (NCT03712358) (110). In contrast, PVSRIPO infects macrophages and DCs in culture, causing the expression of major histocompatibility complex class II (MHC II) and the generation of IFN-b and IL-12 (9, 87).
3.2 Genetically engineered (modified) oncolytic viruses
3.2.1 Herpes simplex virus
Talimogene laherparepvec (T-VEC; Imlygic), the first OV presently licensed by the FDA, is a member of the Herpesviridae family (56). In addition to T-VEC, other HSV-based OVs have been developed, such as G47δ, oHSV-IL12, G207, and rRp450 (111, 112). The majority of HSV-based vectors carry deletions in ICP34.5, a neurovirulence gene that restricts virus replication to tumor cells overexpressing the Ras gene (113). The inactivation of the ICP6 gene, which encodes a viral homolog of the cellular ribonucleotide reductase (RR), is another mechanism of HSV specificity (114, 115). The mutant virus replication is limited to actively proliferating cancer cells with high levels of RR because this enzyme is necessary for creating deoxyribonucleotides (29). The results of a Phase Ib study using T-VEC in combination with the CTLA4 checkpoint inhibitor, Ipilimumab, in people with advanced melanoma were published by Puzanov et al. (116). Additionally, the combination of OV with nivolumab has showed very promising results (12, 116–118).
3.2.2 Adenoviruses
Adenoviruses (Ads), non-enveloped viruses with icosahedral capsid and double-stranded linear DNA genomes, are members of the Adenoviridae family, specifically the genus Mastadenovirus. The capsid of Ads contains three major proteins, including Hexon, penton-base, and fiber proteins, which give them specific tropism characteristics (119). In clinical research, adenovirus serotype 5 (Ad5) is the most frequently utilized viral vector (120). Ad5 penetrates the targeted cells via interacting its fiber knob protein with coxsackievirus and adenovirus receptors (CARs) (118, 121).
DNX-2401 (Delta-24-RGD; tasadenoturev) is an oncolytic adenovirus, replication -competent adenovirus. A 24-base pair deletion in the E1A gene promotes tumor selectivity by preventing viral replication in normal cells with a functioning Rb pathway. An RGD-motif was added to the fiber H-loop to boost potency, allowing the virus to enter cells through v3 or v5 integrin. On tumor cells, especially glioma stem cells, these integrins are abundant (122). In preclinical models, DNX-2401 kills glioma cells through direct oncolysis and by inducing immunological responses against tumor antigens, resulting in long-term antitumor immunity and tumor regression. DNX-2401 is now being tested in clinical studies for the treatment of recurrent glioblastoma (NCT03896568) (123). Also, additional studies are being conducted to examine its effectiveness against recurrent gliomas when used in conjunction with other treatments, such as checkpoint inhibitors. In one active phase II study, DNX-2401 was injected directly into a recurrent glioblastoma or gliosarcoma followed by pembrolizumab every 3 weeks for up to 2 years or until disease progression (NCT02798406) (38).
H101 is an adenovirus with an E1B deletion that has been approved in China for the treatment of nasopharyngeal carcinoma. H101 was tested in a randomized Phase III clinical study with 160 people who had advanced squamous cell carcinomas of the head and neck or esophagus (124). The patients were randomly assigned to chemotherapy (cisplatin and 5-FU for chemotherapy-naive patients, or adriamycin and 5-FU for patients who had previously received platinum chemotherapy) with or without H101 (5× 1011 to 1.5 ×1012 viral particles per day by intra-tumoral injection) for five consecutive days every three weeks. A total of 123 patients completed treatment, and were able to be evaluated for response. Patients who received cisplatin/5-FU + H101 exhibited a response rate of 78.8%, compared to 39.6% in the cisplatin/5-FU-only cohort. Patients who got the adriamycin/5-FU and H101 virus, as well as the adriamycin/5-FU -only group, both achieved a 50% response rate; however, these groups had a limited number of participants (n = 18). There was a substantial difference in response rate between all patients who got H101 and individuals who only received chemotherapy. The most common adverse events were fever, injection site reactions, and flu-like symptoms. Based on these findings, the Chinese regulatory agencies approved H101 in combination with chemotherapy for the treatment of nasopharyngeal cancer (10). In addition to the aformentioned viruses, several additional viruses, including parvovirus, poliovirus, vesicular stomatitis virus, Seneca valley virus, have been engineered to be used as OVs in combating cancer (21). Table 3 summarizes the types of viruses that are in different phases of clinical trials.
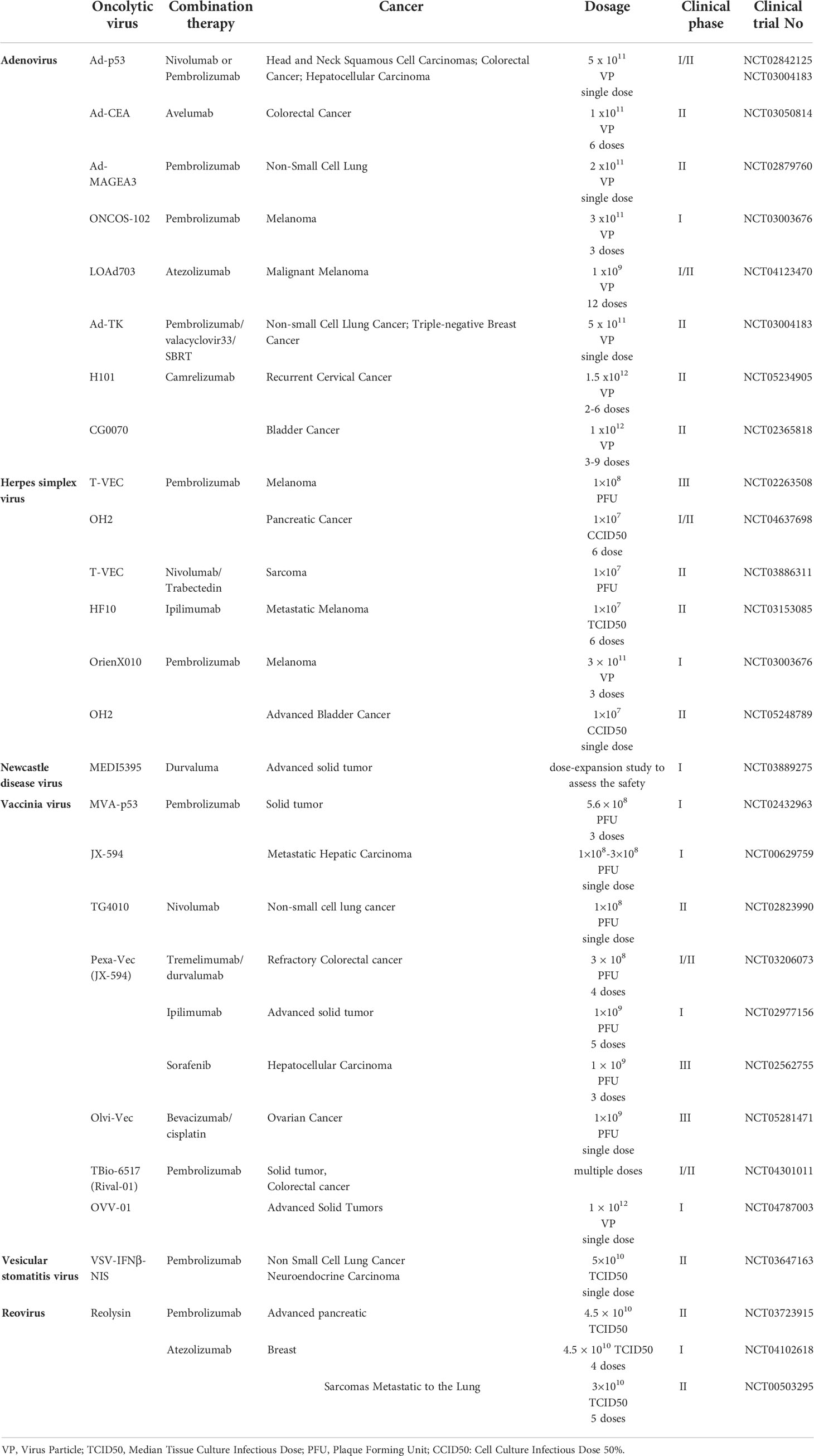
Table 3 Summary of clinical trials of monotherapy and combination therapy of oncolytic viruses.
As a result of genetic engineering, a wide variety of potentially pathogenic viruses have been manipulated for safety and tumor-targeting applications in the past two decades. Genetic modifications including the deletion of viral genes, the use of transcription regulatory elements such as promoters and enhancers, and the alterations of viral surface proteins have been widely used to increase the effectiveness of targeted OVT (82, 125).
4 Tumor targeting strategies of oncolytic viruses
Despite the remarkable preclinical success of OVT, clinical applications remain limited. One of the most significant challenges that must be overcome is viral targeting. Various strategies have been established to achieve targeting OVs toward tumor cells. As previously described, some viruses, such as reovirus and NDV have an intrinsic tropism for tumor cells, whereas the other ones, such as Ad and HSV, should be adapted or engineered to be cancer-specific (126). Virus adaptation to cancer cells is frequently accomplished based on cancer cell modifications, including self-sufficiency in growth signals, resistance to apoptosis, neoantigen expression, and an unlimited replication potential that can be used for OVs selective infection and killing of cancer cells (127). In this regard, different approaches have been used to direct OVs into cancer cells, including modifying the virus’s surface (transductional targeting), introducing specific genes downstream of specific tumor promoters or inserting genetic elements into virus genomes such as miRNA and siRNA to boost OV specificity (transcriptional targeting), and deleting virus genes that are required for replication in normal cells but have little effect on reproduction in cancer cells (33, 128, 129).
4.1 Transductional targeting
Detargeting viruses from their normal cells and retargeting them to a specific cell is a critical step in designing OVs, especially for adenovirus-based oncolytic viruses. As previously stated, Ads as one of the most utilized viruses in cancer treatment, have no innate tropism for cancer cells, whereas they exhibited a broad range of tropism for normal cells due to CAR expression in the majority of normal cells. As a result, an unaltered virus can infiltrate and harm normal cells by systemic injection. Hence, the vector’s inherent tropism should be eliminated to reduce possibly detrimental side effects. Scientists usually use two methods to solve the problem: adding ligands like peptides, antibody fragments, and nanobodies to the structure of the virus, and using bispecific adaptors. Van Erp et al. coupled transcriptional targeting by utilizing a tumor-specific promoter with transductional targeting by using an anti-CEA nanobody incorporated into Ad. CXCR4E1.B2 virus capsids. They showed that using a single specific domain for CEA which was inserted genetically into the Ad fiber could improve the specificity of infection and the ability of Ads to reproduce in cancer cells (130) Another method for modifying the surfaces and tropism of OVs is pseudotyping. This strategy is often done by replacing coat proteins with similar proteins from related serotypes, leading to a new tropism without changing the balance of the genome. In this regard, adenoviral fiber protein pseudotype switching is a reasonable strategy for transductional retargeting. Owing to the upregulation of CD46 on many malignant tumors, researchers replaced Ad5 fiber with the fiber of serotypes 11/35 to target tumor cells (131, 132). Another strategy that has been developed to target viruses toward tumor cells, as previously mentioned, is the use of bispecific adapters. Adaptors are molecules with two ends that bind to the viral proteins and the receptors on the cancer cells. This strategy’s main advantage is the ability to use multiple adaptors to attach to the same vector without affecting the vector`s structure. Due to the overexpression of the high molecular weight melanoma-associated antigen (HMWMAA) on melanoma cells, Curiel et al. designed a bispecific adaptor, scDb MelAd, to target Ad to melanoma cells selectively. They demonstrated significantly reduced infectivity (> 50-fold) of capsid mutant Ads, restored (up to 367-fold increase), CAR-independent and HMWMAA-mediated infectivity of these mutant viruses by scDb MelAd specifically in melanoma cells, compared to a vector with wild-type fibers (133). Additionally, a universal platform for Ad5 detargeting and retargeting using the SpyTag and SpyCatcher system was developed and demonstrated that Ad5 efficiently wasredirected into VEGFR2-expressing cells using an adoptor incorporating SpyCatcher and an anti-VEGFR2 nanobody (under publication data).
4.2 Transcriptional targeting
The effectiveness of transductional techniques has been inadequate for realizing the full promise of virotherapy in the clinic. For instance, early gene therapy experiments used therapeutic genes driven by viral promoters, such as the CMV promoter, which caused non-specific damage in normal cells and tissues as well as cancer cells. However, the use of tumor-specific promoters, which are overexpressed in tumors, stimulates the particular expression of therapeutic genes in a certain tumor, boosting their localized action and reducing mislocalization side effects (134). TTF-1 promoter, glypican-3 protein (GPC3), human secretory leukocyte protease inhibitor (hSLPI), Mucin 1 (MUC1), cyclooxygenase 2 (COX2), epithelial glycoprotein (EPG2), and human telomerase reverse transcriptase (hTERT) are the most common tumor-specific promoters used in transcriptional targeting (7). For instance, combining transcriptional targeting using the tissue-specific SLPI promoter and transductional targeting with the ovarian cancer specific adaptor protein, sCARfC6.5, which contains the coxsackie-adenovirus receptor ectodomain and a single-chain antibody specific for c-erbB-2, increased transgene expression in ovarian tumors while decreasing expression in normal tissues, including the liver, in comparison to single-approach targeting (135). As abovementioned, inserting micro-RNAs (miRNAs) into the virus genome is a new way to improve their specificity and reduce off-target effects. Many teams have used the differential landscape of miRNA expression between normal and malignant cells to hinder OVs proliferation in healthy cells. In one study, it has been demonstrated that adding several copies of a miR-124 recognition sequence into the 3′UTR of the oncolytic HSV-1’s crucial ICP4 gene prevents the virus from infecting normal cells. This phenomenon occurs because of the high expression of miR-124 in healthy neurons but not at all in glioblastoma cells (136, 137). In another study, Luo et al . employed a triple-regulated OAd containing miR143, survivin, and RGD to improve the effects of OAds. They showed that when Ad-RGD-Survivin-ZD55-miR-143 was introduced into cells, it could inhibit cell growth, migration, and invasion, as well as halt cells in the G1 phase and induce cell death (138).
Despite the use of many techniques for targeting viral vectors in cancer virotherapy and boosting the virus’s efficacy in cancer treatment, when the virus must be injected systemically for the treatment of metastatic malignancies, this treatment strategy encounters a number of challenges (125) that must be overcome before the systematic administration of OVs to improve their anti-tumor activities (126). The most common challenges of delivering the virus through the bloodstream are viruses’ identification as foreign agents and elimination from the body before they can reach the tumor site, known as immune-mediated clearance (127), and virus sequesteration by non-specific tissues such as the liver, lungs, and spleen (128). On the other hand, tumors are high-pressure settings with a dense and disorderly collection of cells due to thick stromal tissue and limited lymphatic drainage (42). To address these issues, scientists have designed a variety of approaches, which are described in more detail below.
4.3 Solutions to the challenges of OVs’ systemic delivery
Complement activation, pre-existing immunity, or the release of inflammatory cytokines (IL-6, IL-12, and TNF) in response to vectors all contribute to OV clearance by the immune system. Some OVs, including vaccinia and HSV-1, produce anti-complement components to evade the immune system (139). HSV-1 secretes glycoprotein E, which functions as an IgG Fc receptor and efficiently inhibits IgG Fc-mediated complement activation as well as antibody-dependent cellular cytotoxicity (ADCC) (140). Pre-existing immunity canoccur due to the virus’s ubiquitous nature (Ad and Reovirus), previous vaccination (vaccinia and MV), or earlier oncolytic viral treatment (141). There are currently various solutions being tested for these issues. Changing the surfaces of viral vectors by shielding with polymers such as (poly ethylene glycol (PEG), poly L-lysine, and N-[2-hydroxypropyl] methacrylamide (HPMA)) and lipidic vesicles often reduces their immunogenicity and increases vector persistence in the bloodstream (142). Cellular carriers, in which cells are taken from a model organism that has been infected and put back in, are another way to deliver OVs. Immune cells, stem cells, and tumor cells have been used to generate experimental OV cell carriers. Among cell carries, stem cells, according to in vitro and in vivo studies, are the most outstanding candidates for systemic delivery of OVs since they allow viruses to infect the target cells and replicate in, conceal them from the immune system, and target tumors (143, 144). In this regard, Mader et al. used mesenchymal stem cells (MSCs) to efficiently deliver oncolytic MV to ovarian cancer and protect the virus from neutralizing anti-viral antibodies. They found that using MSCs as carriers increased their localization and infiltration into tumors and transferred oncolytic MV infection to tumors, leading to enhancing mice survival (143). Immune cells, especially DCs and T-cells, have been used successfully in pre-clinical research to transfer several OVs to tumors. For instance, DCs infected with reovirus have been shown to efficiently transport and deliver their oncolytic payload into melanoma cells, even in the presence of neutralizing antibodies (145). On the other hand, OVs are removed from the bloodstream by mononuclear phagocytic cells, splenic macrophages, and hepatic Kupffer cells in the spleen and liver following systematic administration. This clearance frequently occurs following the decorating of viral particles with antibodies and complement proteins or their interaction with coagulation factors (126). However, some viruses, such as Ads, may bind directly to scavenger receptors on Kupffer cells, resulting in the release of pro-inflammatory cytokines, which may cause severe toxicities (146). The answers to these challenges are fairly similar to the methods outlined for immune response escape. In case of Ads, the hexon protein, as the most frequently structural protein, has a critical role in liver sequestration through interaction with coagulation factor IX and scavenger receptors (147, 148). Different strategies have been developed to avoid this sequestration, such as genetic alteration in the hypervariable region (HVR) of hexon (149), pseudotyping (complete change of HVR) (150), and pharmacological agents (such as warfarin and protein obtained from snake toxin and factor X-binding protein) (151, 152). Surface PEGylation is a popular strategy for reducing non-specific tissue absorption. Kwon et al. detected a substantial 105 increase in tumor to liver ratio when Ad was treated with a PEGylated chitosan specific to the folate receptor compared to naked Ad (153).
4.4 Intratumoral spread of OVs in solid tumors
Tumor physiology is a major issue in cancer treatment since tumors come in a variety of forms and sizes, making it difficult to predict how and where medications, such as OVs, will be absorbed (154). Therefore, viruses transferred within the tumor can only infect and spread cells near blood vessels, leaving the rest of the tumor untreated. Therefore, researchers have focused on establishing mechanically activated transport mechanisms to promote OVs penetration and increase virus anti-cancer activity (155). Most solutions for this barrier rely on virus-encoded matrix-degrading enzymes and anti-fibrotic agents. Diop-Frimpong et al. showed that the penetration and effectiveness of intratumorally injected oncolytic HSV were improved by using Losartan, which is a clinically approved angiotensin II receptor antagonist with anti-fibrotic effects (156). In another experiment, Guedan et al. created a replicating Ad capable of producing soluble sperm hyaluronidase (PH20) (AdwtRGD-PH20). Intratumoral AdwtRGD-PH20 treatment caused hyaluronan (HA) degradation, enhanced viral dispersion, and tumor regression occurred in all of the treated tumors (157). In addition, expressing matrix metalloproteinases-1 and -8 in oncolytic HSV increased viral dispersion and treatment efficiency by breaking down tumor-associated sulfated glycosaminoglycans (158).
5 Oncolytic virotherapy in combination with cancer immunotherapeutics
Although various studies demonstrated viruses potential in eliminating tumor cells, there is currently no report that virotherapy can lead to a complete cure of cancer alone due to the previously mentioned challenges. However, there is ample evidence that OVs can be considered as the basis of combination therapy in different cancers due to their multiple mechanisms of action and their simultaneous effects on tumor cells, immune cells, and the TME (45, 159). Here, we have discussed possible combinations of OVs with different biological products that can overcome monotherapy challenges and limitations in cancer treatment.
These combinations can be classified as 1) Armed recombinant oncolytic viruses that carry the coding sequence of other therapeutic agents which are excellently discussed elsewhere by Kontermann (69), and 2) Combining OVs with other biologic therapeutics separately.
Combining viruses with either the coding sequence or the final protein form of antigen binding biologics)such as antibodies, nanobodies and CAR-T cells(can help viruses overcome some limitations, such as possible off-target side effects and non-specific uptake (Figure 3). Antibodies (Abs) are widely used in targeted therapies and mostly recognize TAAs on tumor cells. Abs are also combined with OVs to improve possibility of attachment of the virus to its target cells. As a proof to this claim, combining OVs with immune checkpoint inhibitor (ICI) antibodies is leading to promising anti-cancer results. Despite this issue, one of the main limitations of antibodies, especially for the treatment of solid tumors, is their poor tumor penetration due to physical barriers. This can be improved by using smaller antibody fragments, such as single-domain antibodies, scFv, and Fab. There are also other functional antigen-binding therapeutic formats with some notable advantages that are used in some other studies in the viro-antibody therapy field that can be translated into the clinical trial. These promising structures can be combined effectively with OVs for better therapeutic efficacy. For improving the combinational therapy outcome, nanobodies can also be used to not only assist OVs in specific targeting, but also helping viruses for more efficient penetration into solid tumors. Table 4 summarizes the various strategies of combination therapy with oncolytic viruses.
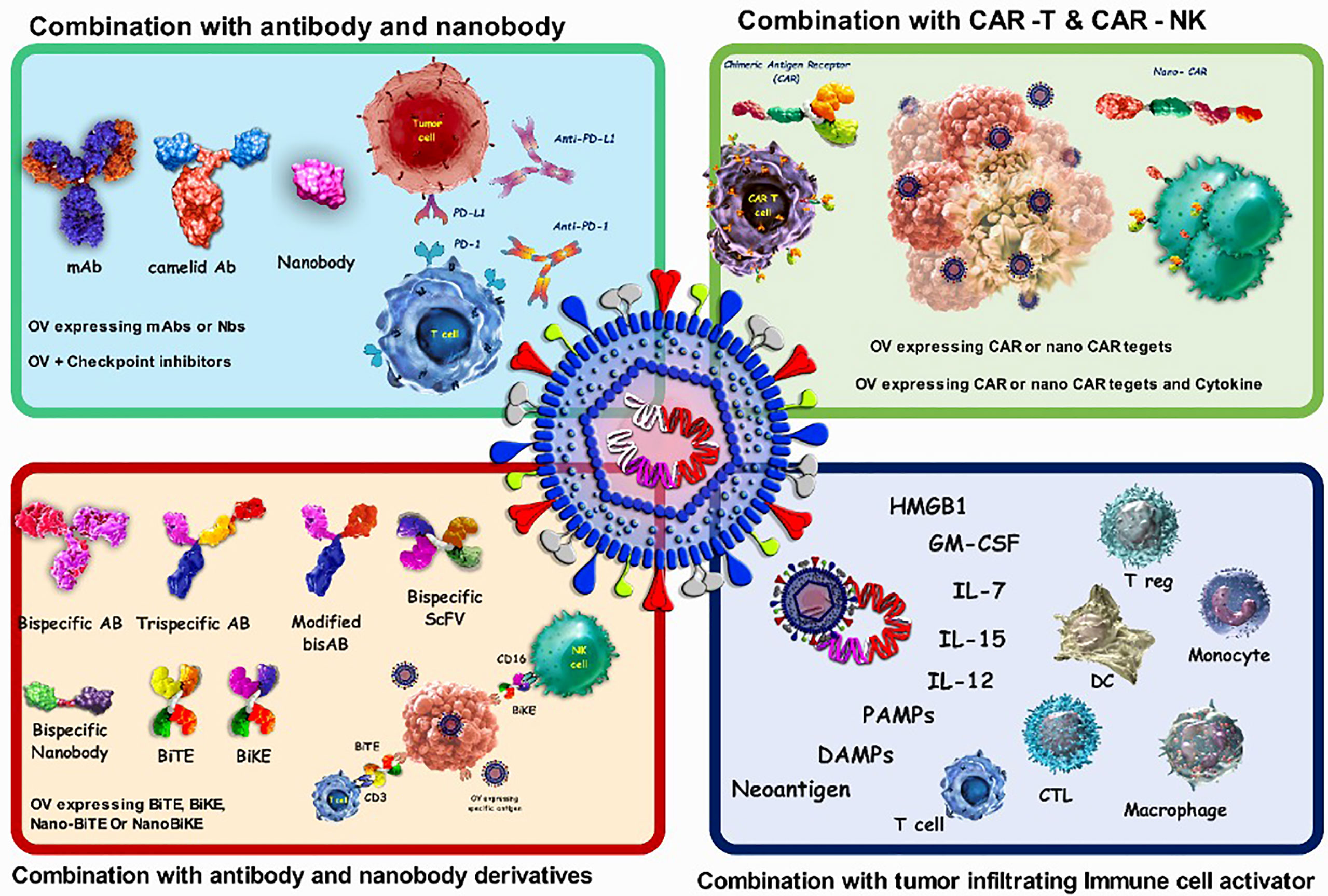
Figure 3 Characteristics of oncolytic virus combination therapy. OVs attack and destroy tumor cells preferentially. Lysis of tumor cells releases neoantigen, PAMPs which trigger PRRs, which then produce inflammatory cytokines and antiviral type I IFNs. Viruses can activate cell death pathways, resulting in immunogenic cell death phenotypes such as necroptosis, pyroptosis, immunogenic apoptosis, and autophagic cell death. Antibodies that target cell surface indicators of immune cells (checkpoint inhibition), cancer cells (targeted therapy), or both (bispecific antibodies) are wellestablished in cancer therapy. combination of oncolytic viruses with antibody and CAR-T cells; CAR-T cells bind to the antigen on the surface of tumor cells and kill them, but they cannot migrate deeper into the dense tumor mass to remove antigennegative tumor cells. also, CAR-NK cells show more anticancer activity than CAR-T cells because they attach to stress ligands on the surface of tumor cells. The oncolytic virus attacks and destroys tumor cells, eliminating the tumor's dense structure.
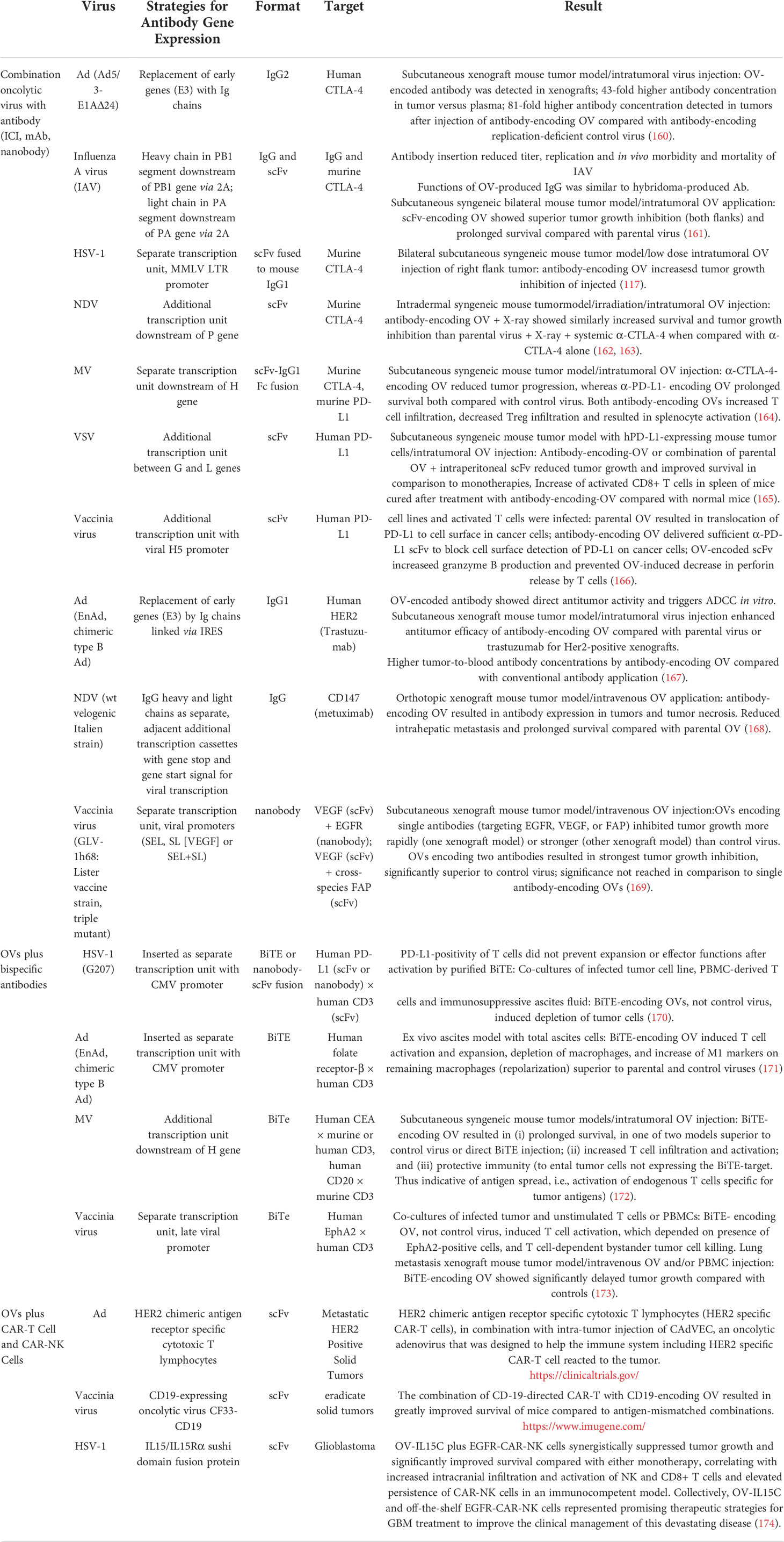
Table 4 Summary of combination therapies with oncolytic viruses and other immunotherapeutic agents.
5.1 Arming oncolytic virus with antibody and its derivatives
5.1.1 OVs plus antibodies
Antibodies with different frameworks have been approved as cancer therapeutics; each has a different mode of action, such as blocking, neutralizing, and activating functions (159, 175, 176). These antibodies have been also coupled to various viruses such as Ad, MV, HSV, NDV, Reovirus, Vaccinia, and VSV in cancer combinational therapy (177–179), and some with promising results have been discussed below.
5.1.1.1 OVs plus immune checkpoint inhibitors
It has been shown that the expression of inhibitory receptors on T-cells, including programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), and their binding to their ligands on the target cells (PDL-1 and B7, respectively) leads to aberrant activation of T-cells. In this regard, blockade of these negative regulators by ICIs can prevent T-cells suppression and improve their optimal activity in combating tumor cells (179). Nevertheless, the clinical response of ICIs correlates with pre-existing anti-tumor immune responses, such as an elevated number of tumor-infiltrating lymphocytes (TILs) and enough expression of immune checkpoints (ligands) on the tumor cells (45, 180). It has been shown that after viral infection, the expression of immune checkpoints upregulates on the surface of tumor cells, and accordingly, one of the most compelling combinational therapies for cancer would be OVs + ICIs (45, 181). In addition, another reason that makes the combination of OVs and ICIs attractive in the treatment of cancer is their different mechanism of action, which is an important parameter from the pharmacological point of view (45).
Zamarin et al. showed that localized OVT by NDV overcomes systemic tumor resistance to ICIs by inflaming the TME. They showed that I.T. administration of NDV not only increased infiltration of the lymphocytes into the injected tumor in the B16 melanoma mice models, but also the anti-tumor effect was observed in a distant tumor without any virus injection. Also, the localized administration of NDV in combinational with systematic administration of CTLA-4 blockade exhibited more efficient anti-cancer outcomes (182). In most studies for assessment of combinational OV-ICI therapy, fully humanized IgG antibody format has been used as ICIs; FDA-approved ICIs, especially those in clinical trials and has been discussed in detail elsewhere (178, 180). For example, although immune checkpoint inhibition is a logical therapeutic candidate against glioblastoma cells due to the increased expression of PD-1 on these cells (along with IL-10 and TGF-b), anti-PD-1 alone could not sufficiently eliminate tumor cells and there was a need for synergistic interactions with OVs. So, Saha et al. showed that a triple combination using anti-CTLA-4, anti-PD-1, and G47D-mIL12 (recombinant HSV virus) cured most mice in two glioma models. This approach not only treated mice, but also protected them against tumor re-challenge. The synergistic activity was associated with increased M1-like macrophages, T effector cells (CD4+ and CD8+), and decreased T regulatory (Treg) cells (183, 184). To emphasize the effect of combination therapy, it should be noted that while none of these agents were effective alone enough, they showed remarkable therapeutic effects in combinational strategy (184). It is worth noting that although Imlygic® has been approved for adult patients with melanoma, there are clinical trials for the assessment of Imlygic® in combination with ICIs for improving the treatment outcomes in melanoma and other cancer types (e.g., Pembrolizumab with Imlygic® or Placebo in Unresected Melanoma, NCT02263508) (181, 185). Other combinations, including this OV and ICIs for triple-negative breast cancer with metastatic liver cancer and colorectal carcinoma are also being evaluated in clinical trials (186).
5.1.1.2 OVs plus monoclonal antibodies other than ICIs
Apart from ICIs, some commercial mAbs with distinct mechanism of actions, have also been combined with OVs to improve the therapy outcome. In one study, the antitumor activity of cetuximab (an epidermal growth factor receptor inhibitor mAb) was assessed in combination with HSV. The result showed that combining cetuximab and HSV could improve distribution of the virus and lead to a synergistic antitumor effect in HT-29 tumor xenograft models (187). In another study, Zhang et al. demonstrated that combination of recombinant oncolytic HSV with Bevacizumab (BEV) (which is an antiangiogenic mAb approved for glioblastoma) in mice-bearing human GBM, was led to improvement of antiangiogenic effect of BEV while decreasing the tumor invasive-like phenotype induced by this drug (188).
5.1.2 OVs plus nanobodies
Nanobodies are the smallest natural antigen-binding constructs with a single variable domain (VHH, ∼15kDa) as the antigen-binding region (189). Nanobodies have unique characteristics, such as easy selection by phage display, ease of manipulation, high stability in harsh conditions, and reaching and recognition of specific hard-to-access epitopes, making them more attractive in combination with other agents in cancer immunotherapy, including OVs (190, 191). For instance, due to the high complexity of glioblastomas and the low accessibility of therapeutic agents to their TME, the combination of viruses and nanobodies is a promising candidate for glioblastoma treatment. In a proof-of-concept study, Gil et al. used an anti-CXCR4 nanobody for retargeting oncolytic HSV toward CXCR4+ GBM cells. CXCR4 is overexpressed in various cancers, including glioblastoma, and usually correlates with a poor prognosis. The results of this study indicated that OVs plus nanobodies were highly encouraging for targeting GBM cells (192).
CD47 acts as a ”don’t eat me signal” to the immune system’s macrophages, making it a potential therapeutic target in some cancers. Different viruses have been engineered to express anti-CD47 antibodies (193) or nanobodies (194) to have a multifaced attack on the tumor cells. In a study, anti-CD47 nanobody-expressing adenovirus reprogramed tumor immune microenvironment and showed excellent anti-tumor immunity (194). This anti-CD47 oncolytic adenovirus could induce durable tumor suppression by changing the TME condition and increasing activated TILs in the tumor site. Systemic anti-tumor effects and memory immune cells were also observed after treatment by this recombinant virus (194).
5.1.3 OVs plus bispecific or trispecific antibodies
Bispecific antibodies (bsAb) are constructs with two different antigen-binding sites with the aim of dual targeting (195). The coding sequence of bsAbs can be inserted into the viral genome to be expressed in the target tissue. Different viruses are engineered to this end, including Ad, HSV, MV and vaccinia (159, 196). Bi/trispecific antibodies can also be used in combination with viruses in various timings and dosages.
5.1.3.1 OVs plus bispecific T-cell engagers
Viruses have been successfully combined with T-cell retargeting bsAb, also called Bispecific T-cell Engager or BiTe (197), to retarget T cells to the targeted tumor cells. In one study, a recombinant adenovirus encoding bsCD3-EpCAM bispecific antibody (bsAb) could effectively activate T-cells in malignant peritoneal and pleural exudates despite the immunosuppressive environment (198). Also, in another study, NDV-BiTe constructs (e.g., antiHN scFv/antiCD3 scFv and antiHN scFv/antiCD28 scFv) were successfully designed and expressed to be evaluated for thier remarkable potentials in tumor immunotherapy especially in breast and colorectal cancers (199, 200).
5.1.3.2 OVs plus natural killer cell engagers
Emerging role of NKs cells in cancer therapy becomes clearer every day and its combination with virotherapy has accelerated the progress of therapeutic processes in cancer. Viruses can also be combined with bispecific NK engagers (201) to retarget NK cells to their targeted tumor cells.
As an examples for this type of combination, Bahrololoumi et al. constructed a bsAb (antiHN scFv/antiCD16 scFv) and a trispecific antibody(antiHN scFv/IL-15/antiCD16 scFv) to bind to the haemagglutinin neuraminidase (HN), a viral protein that is expressed on the surface of the NDV infected tumor cell, and the CD16 activating receptor on the surface of the NK cells for redirecting NK cells toward the tumor cells (201, 202). NDV-Ulster is a non-lytic strain of NDV that was used to inflame the tumoral microenvironment in this study, and also in NDV-based autologous tumor cell vaccines for stimulating the immune response in the patient’s body (93, 178, 203, 204).
5.1.3.3 OVs plus trispecific antibodies
Trispecific antibody (Trike) is a single engineered antibody platform that recognizes and binds to three different targets and is expected to boost immune response significantly (205). There are different studies that combine these trifunctional engagers with OVs with the aim of cancer viro-immunotherpy. In one study, researchers constructed a trispecific immunocytokine (anti-NDV/IL2/anti-CD28) for efficiently targeting tumor cells. This trike could bind to the HN of the NDV infected tumor cells from one side and to the CD28 receptor on the T cells from the other side, while IL-2 promoted T cells function (206, 207). Ravirala et al, showed that combination of oncolytic HSV with bi/tri specific antibodies which could bind to the NKG2D and epidermal growth factor (EGF) from each side, while the trispecific one also contained IL-2 sequence, could significantly enhance infiltration and activation of NK and T cells in the tumor site (208). Various bi/trispecific antibodies that were combined with different viruses have been extensively reviewed elsewhere (209).
5.2 OVs plus CAR-T and CAR-NK cells
Chimeric antigen receptor (CAR)-T cells are T cells that have been genetically engineered to express an artificial receptor to direct them toward a specific target in an MHC-independent manner. The external domain of CAR-T cells consists of an extracellular target antigen binding domain which is usually a single-chain fragment variable (scFv) from a specific monoclonal antibody, attached to the transmembrane and signaling domains of this artificial receptor by a hinge (210). Although some scFv-based CAR-T cell products are currently approved by FDA for B-cell malignancies with encouraging results, this approach still faces some limitations, such as trafficking and tumor infiltration, antigen escape, immunosuppressive microenvironment, and CAR-T cell-associated toxicities (211). As a consequence, CAR-T cells do not exhibit profound anti-tumor effects in solid tumors. The use of VHH-based CAR-T cells (Nanobody CAR-T cells) may could resolve the abovementioned problems (212, 213). There are different antigens that are targeted through Nanobody-based CAR-T cells, such as vascular endothelial growth factor receptor 2 (VEGFR2) (214), human epidermal growth factor receptor 2 (HER2) (215), tumor-associated glycoprotein 72 (TAG‐;72) (216), prostate-specific membrane antigen (PSMA) (217–219), glypican 2 (GPC2), epidermal growth factor receptor (EGFR), B‐;cell maturation antigen (BCMA), PD‐;L1, and EIIIB (212, 220). It has been shown that the combination of CAR-T cells (scFv or VHH-based) with virotherapy can help CAR-T cells overcome their challenges in combat against solid tumors (such as immunosuppressive TME and heterogeneity of the antigens) and increase the immune response dramatically. For instance, Nishio et al. showed that armed oncolytic Ad (with RANTES and IL-15) could increase the efficacy of GD2 targeting CAR-T cell in a neuroblastoma solid tumor model (221). Furthermore, there is evidence that pre-treatment of solid tumors with OVs before the administration of CAR-T cells may lead to better ICD (222, 223). For example, combining recombinant oncolytic Ads containing a coding sequence of different cytokines, such as IL-2, RANTES and TNF-α, could lead to better accumulation and survival of CAR-T cells (223). Some of the combinations, such as Ad/HER-2 targeting CAR-T cell (NCT03740256) and VZV/GD2 targeting CAR-T cell (NCT01953900) therapy, are examples of such combinations being evaluated in clinical trials.
In the case of CAR-NK cells, in combination with HSV and to treat brain cancer metastases, EGFR CAR-NK cells were used intracranially in mice. This combination resulted in significantly longer survival of tumor-bearing mice when compared to monotherapies (224).
OVs attack and destroy tumor cells preferentially. Lysis of tumor cells releases neoantigen, PAMPs which trigger PRRs, which then produce inflammatory cytokines and antiviral type I IFNs. Viruses can activate cell death pathways, resulting in immunogenic cell death phenotypes such as necroptosis, pyroptosis, immunogenic apoptosis, and autophagic cell death. Antibodies that target cell surface indicators of immune cells (checkpoint inhibition), cancer cells (targeted therapy), or both (bispecific antibodies) are well-established in cancer therapy. combination of oncolytic viruses with antibody and CAR-T cells; CAR-T cells bind to the antigen on the surface of tumor cells and kill them, but they cannot migrate deeper into the dense tumor mass to remove antigennegative tumor cells. also, CAR-NK cells show more anticancer activity than CAR-T cells because they attach to stress ligands on the surface of tumor cells. The oncolytic virus attacks and destroys tumor cells, eliminating the tumor’s dense structure.
5.3 Other combinations with OVs
5.3.1 OVs plus autologous DC or T cells
OVs can also be combined with cell therapy to treat different tumors, including solid tumors. For instance, the combination of NDV and dendritic cells (DC (as an exciting platform is being used in the IOZK clinic in Cologne Germany (Immun-Onkologisches Zentrum Köln). Their studies and practices in the IOZK indicated that DCs loaded with the lysate of NDV-infected tumor cells (viral oncolysate, VOL) triggered potent anti-tumor immunity by promoting the secretion of IFN-γ and IL-2 from T-cells. This combinational therapy is now available in the IOZK clinic and patients can benefit from the advantages of this kind of cancer combinational treatment (200, 225). Activation of naïve human T-cells by co-incubating with NDV-infected irradiated autologous tumor cells (ATV-NDV) which can be further modified with bi-specific or tri-specific antibodies can also offer a promising multimodal anti-cancer approach (226).
Altogether, strong evidence confirm that different therapeutic agents can have a measurable therapeutic effect in cancer treatment, but due to the specific and complex biology of cancer and its TME, the therapeutic outcome of these agents lonely, do not contribute to the final treatment of cancer patients. Thus, this is where rational combination therapy of these factors with each other, especially with OVs is much needed.
5.3.2 OVs plus tumor-infiltrating lymphocytes
It has been demonstrated that weak functionality of natural TILs in the tumor site is strongly related to tumor progression (227). OVs can set the scene by inflaming the tumor microenvironment for better functionality of TILs. Feist et al, showed that local injection of poxvirus into a solid tumor in mice, could lead to activation and accumulation of TILs in the tumor site which had a low immunogenicity before virus infection (228). In another study, it was shown that virotherapy with a type of oncolytic adenovirus, could increase the TILs and significantly reduce the tumor size in the immunocompetent mouse model (229). It has also been shown that infecting the tumor with recombinant oncolytic HSV, could unleash the full potential of TILs which led to tumor regression and antitumor immunological memory (230).
6 Conclusions
OVT success, specifically FDA- and regional-approved OVs, has made waves in (pre)clinical areas, attracting both society and the scientific community’s attention. However, some of challenges have limited OVs application as immunotherapy, and their combination with other biotherapeutic platforms has been proposed in cancer therapy. To date, hundreds of combinations of OVs with other biotherapeutic platforms, including antibodies, nanobodies, ICIs, CAR-T cells, and DCs, have been investigated in clinical trials to understand which and how best to provoke anti-cancer immune responses. Some considerations could improve the efficacy of OVs, either as monotherapy or combination therapy. First, the dosage, targeted mechanisms, administration schedule, delivery technologies, and types of OVs could be considered because of their indispensable roles in the outcome of cancer immunotherapy and priming TME in combinational regimens. Second, understanding the interaction between immune cells/system, tumor cells/TME, and OVs and the combinational agents should help make new therapeutic combinations possible. Third, defining reliable biomarkers to distinguish “hot tumors” from “cold ones” can help scientists determine subsequent therapies. Finally, providing beneficial impacts of OVs and their combinational regimens on patients’ life quality requires the contribution of molecular biologists, pharmacologists, immunologists, and clinicians. Indeed, current clinical trials results can help scientists develop new systems of combination therapy and deliver innovative treatments to patients.
Author contributions
ZS conceived the presented idea and SA developed the theory. MJ took the lead in writing the manuscript and she was in charge of overall direction and planning. MJ, MK and MBS wrote the manuscript with input from all authors. NH contributed to the writing of the results and to the editing of the manuscript. MAS and AA contributed to the final version of the manuscript. All authors discussed the results and contributed to the final manuscript
Acknowledgments
We gratefully acknowledge the Immunology Department of Pasteur Institute of Iran for their technical assistance.
Conflict of interest
Author MBS was employed by HUM Immune Biotech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
2. Hemminki O, Manuel J, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol (2020) 13(1):84. doi: 10.1186/s13045-020-00922-1
3. Rahman MM, Mcfadden G. Oncolytic Viruses : Newest frontier for cancer immunotherapy. Cancers (2021) 13(21):5452. doi: 10.3390/cancers13215452
4. Grossman DC, Curry SJ, Owens DK, Barry MJ, Caughey AB, Davidson KW, et al. Behavioral counseling to prevent skin cancer: US preventive services task force recommendation statement. JAMA (2018) 319(11):1134–42. doi: 10.1001/jama.2018.1623
5. Spaas M, Lievens Y. Is the combination of immunotherapy and radiotherapy in non-small cell lung cancer a feasible and effective approach? Front Med (2019) 6(November). doi: 10.3389/fmed.2019.00244
6. Zhang S, Rabkin SD. The discovery and development of oncolytic viruses: Are they the future of cancer immunotherapy? Expert Opin Drug Discov (2021) 16(4):391–410. doi: 10.1080/17460441.2021.1850689
7. Koch MS, Lawler SE, Chiocca EA. HSV-1 oncolytic viruses from bench to Bedside : An overview of current clinical trials. Cancers (2020) 12(12):3514. doi: 10.3390/cancers12123514
8. Malogolovkin A, Gasanov N, Egorov A, Weener M, Ivanov R, Karabelsky A. Combinatorial approaches for cancer treatment using oncolytic Viruses : Projecting the perspectives through clinical trials outcomes. Viruses (2021) 13(7):1271. doi: 10.3390/v13071271
9. Cerqueira OLD, Antunes F, Assis NG, Cardoso EC, Clavijo-Salomón MA, Domingues AC, et al. Perspectives for combining viral oncolysis with additional immunotherapies for the treatment of melanoma. Front Mol Biosci (2022) 9:777775. doi: 10.3389/fmolb.2022.777775
10. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: A new class of immunotherapy drugs. Nat Rev Drug Discovery (2015) 14(9):642–62. doi: 10.1038/nrd4663
11. Jin K-T, Du W-L, Liu Y-Y, Lan H-R, Si J-X, Mou X-Z. Oncolytic virotherapy in solid tumors: The challenges and achievements. Cancers (Basel) (2021) 13(4):588. doi: 10.3390/cancers13040588
12. Jin K, Du W, Liu Y, Lan H, Si J, Mou X. Oncolytic virotherapy in solid Tumors : The challenges and achievements. Cancers (2021) 13(4):588. doi: 10.3390/cancers13040588
13. Vähä-Koskela MJV, Heikkilä JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett (2007) 254(2):178–216. doi: 10.1016/j.canlet.2007.02.002
14. Coelho JF, Ferreira PC, Alves P, Cordeiro R, Fonseca AC, Góis JR, et al. Drug delivery systems: Advanced technologies potentially applicable in personalized treatments. EPMA J (2010) 1(1):164–209. doi: 10.1007/s13167-010-0001-x
15. Oh C-M, Chon HJ, Kim C. Combination immunotherapy using oncolytic virus for the treatment of advanced solid tumors. Int J Mol Sci (2020) 21(20):7743. doi: 10.3390/ijms21207743
16. Malfitano AM, Di Somma S, Iannuzzi CA, Pentimalli F, Portella G. Virotherapy: From single agents to combinatorial treatments. Biochem Pharmacol (2020) 177:113986. doi: 10.1016/j.bcp.2020.113986
17. Finck A, Gill SI, June CH. Cancer immunotherapy comes of age and looks for maturity. Nat Commun (2020) 11(1):3325. doi: 10.1038/s41467-020-17140-5
18. Larson C, Oronsky B, Scicinski J, Fanger GR, Stirn M, Oronsky A, et al. Going viral: a review of replication-selective oncolytic adenoviruses. Oncotarget (2015) 6(24):19976–89. doi: 10.18632/oncotarget.5116
19. Beijerinck MW. Concerning a contagium viwm fluidum as cause of the spot disease of tobacco leaves. Phytopathol Class. (1898) 7(1):33–52.
20. Xie F, Zheng L. Oncolytic viruses and their application to cancer treatment. Int Arch Clin Pharmacol (2019) 5(1):1–6. doi: 10.23937/2572-3987.1510020
21. Tian Y, Xie D, Yang L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduct Target Ther (2022) 7(1):117. doi: 10.1038/s41392-022-00951-x
22. Kelly E, Russell SJ. History of oncolytic Viruses : Genesis to genetic engineering. Mol Ther (2007) 15(4):651–9. doi: 10.1038/sj.mt.6300108
23. Bouard D, Alazard-Dany D, Cosset F-L. Viral vectors: from virology to transgene expression. Br J Pharmacol (2009) 157(2):153–65. doi: 10.1038/bjp.2008.349
24. Dock G. The influence of complicating diseases upon leukaemia. Am J Med Sci (1904) 127(4):563. doi: 10.1097/00000441-190412740-00001
25. Davola ME, Mossman KL, Davola ME, Mossman KL. Oncolytic viruses : how “ lytic “ must they be for therapeutic efficacy? Oncoimmunology (2019) 8(6):1–7. doi: 10.1080/2162402X.2019.1596006
26. Rahman MM, McFadden G. Oncolytic viruses: Newest frontier for cancer immunotherapy. Cancers (Basel). (2021) 13(21):5452. doi: 10.3390/cancers13215452
27. Bluming AZ, Ziegler JL. Regression of burkitt’s lymphoma in association with measles infection. Lancet (London England). (1971) 2(7715):105–6. doi: 10.1016/S0140-6736(71)92086-1
28. Chhabra N, Kennedy J. A review of cancer immunotherapy toxicity II: adoptive cellular therapies, kinase inhibitors, monoclonal antibodies, and oncolytic viruses. J Med Toxicol (2021) 18(1):1–13. doi: 10.1007/s13181-021-00835-6
29. Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic viruses in cancer treatment a review. JAMA Oncol (2017) 3(6):841–9. doi: 10.1001/jamaoncol.2016.2064
30. Abdoli S, Roohvand F, Teimoori-Toolabi L, Shayan S, Shokrgozar MA. Cytotoxic effect of dual fluorescent-labeled oncolytic herpes simplex virus type 1 on mouse tumorigenic cell lines. Res Pharm Sci (2019) 14(1):27–35. doi: 10.4103/1735-5362.251850
31. Valyi-Nagy T, Gesser RM, Raengsakulrach B, Deshmane SL, Randazzo BP, Dillner AJ, et al. A thymidine kinase-negative HSV-1 strain establishes a persistent infection in SCID mice that features uncontrolled peripheral replication but only marginal nervous system involvement. Virology (1994) 199(2):484–90. doi: 10.1006/viro.1994.1150
32. Russell SJ, Peng K-W. Oncolytic virotherapy: a contest between apples and oranges. Mol Ther (2017) 25(5):1107–16. doi: 10.1016/j.ymthe.2017.03.026
33. Heise C, Sampson-Johannes A, Williams A, Mccormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med (1997) 3(6):639–45. doi: 10.1038/nm0697-639
34. Toth K, Wold WSM. Increasing the efficacy of oncolytic adenovirus vectors. Viruses (2010) 2(9):1844–66. doi: 10.3390/v2091844
35. Ries SJ, Brandts CH, Chung AS, Biederer CH, Hann BC, Lipner EM, et al. Loss of p14ARF in tumor cells facilitates replication of the adenovirus mutant dl1520 (ONYX-015). Nat Med (2000) 6(10):1128–33. doi: 10.1038/80466
36. O’Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell (2004) 6(6):611–23. doi: 10.1016/j.ccr.2004.11.012
37. Boagni DA, Ravirala D, Zhang SX. Current strategies in engaging oncolytic viruses with antitumor immunity. Mol Ther Oncolytics (2021) 22(September):98–113. doi: 10.1016/j.omto.2021.05.002
38. Ripp J, Hentzen S, Saeed A. Oncolytic viruses as an adjunct to immune checkpoint inhibition. Front Biosci (2022) 27(5):151. doi: 10.31083/j.fbl2705151
39. Wong B, Bergeron A, Alluqmani N, Maznyi G, Chen A, Arulanandam R, et al. Dependency of EGFR activation in vanadium-based sensitization to oncolytic virotherapy. Mol Ther Oncolytics (2022) 25:146–59. doi: 10.1016/j.omto.2022.04.004
40. Ricca JM, Oseledchyk A, Walther T, Liu C, Mangarin L, Merghoub T, et al. Pre-existing immunity to oncolytic virus potentiates its immunotherapeutic efficacy. Mol Ther (2018) 26(4):1008–19. doi: 10.1016/j.ymthe.2018.01.019
41. Russell SJ, Peng K-W. Viruses as anticancer drugs. Trends Pharmacol Sci (2007) 28(7):326–33. doi: 10.1016/j.tips.2007.05.005
42. Zheng M, Huang J, Tong A, Yang H. Oncolytic viruses for cancer Therapy : Barriers and recent advances. Mol Ther Oncolytics (2019) 15(37):234–47. doi: 10.1016/j.omto.2019.10.007
43. Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G. Antitumor benefits of antiviral immunity: An underappreciated aspect of oncolytic virotherapies. Trends Immunol (2018) 39(3):209–21. doi: 10.1016/j.it.2017.11.006
44. Lemos de Matos A, Franco LS, McFadden G. Oncolytic viruses and the immune system: The dynamic duo. Mol Ther Methods Clin Dev (2020) 17:349–58. doi: 10.1016/j.omtm.2020.01.001
45. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol (2018) 18(8):498–513. doi: 10.1038/s41577-018-0014-6
46. de Graaf JF, de Vor L, Fouchier RAM, van den Hoogen BG. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev (2018) 41:28–39. doi: 10.1016/j.cytogfr.2018.03.006
47. Allegrezza MJ, Conejo-Garcia JR. Targeted therapy and immunosuppression in the tumor microenvironment. Trends Cancer (2017) 3(1):19–27. doi: 10.1016/j.trecan.2016.11.009
48. Alberts P, Olmane E, Brokāne L, Krastiņa Z, Romanovska M, Kupčs K, et al. Long-term treatment with the oncolytic ECHO-7 virus rigvir of a melanoma stage IV M1c patient, a small cell lung cancer stage IIIA patient, and a histiocytic sarcoma stage IV patient-three case reports. Apmis (2016) 124(10):896–904. doi: 10.1111/apm.12576
49. Alberts P, Tilgase A, Rasa A, Bandere K, Venskus D. The advent of oncolytic virotherapy in oncology: The rigvir® story. Eur J Pharmacol (2018) 837:117–26. doi: 10.1016/j.ejphar.2018.08.042
50. Yu W, Fang H. Clinical trials with oncolytic adenovirus in China. Curr Cancer Drug Targets. (2007) 7(2):141–8. doi: 10.2174/156800907780058817
51. Goradel NH, Alizadeh A, Hosseinzadeh S, Taghipour M, Ghesmati Z, Arashkia A, et al. Oncolytic virotherapy as promising immunotherapy against cancer: mechanisms of resistance to oncolytic viruses. Futur Oncol (2021) 18(2):245–59. doi: 10.2217/fon-2021-0802
52. Goradel NH, Mohajel N, Malekshahi ZV, Jahangiri S, Najafi M, Farhood B, et al. Oncolytic adenovirus: A tool for cancer therapy in combination with other therapeutic approaches. J Cell Physiol (2019) 234(6):8636–46. doi: 10.1002/jcp.27850
53. Harrington KJ, Puzanov I, Hecht JR, Hodi FS, Szabo Z, Murugappan S, et al. Clinical development of talimogene laherparepvec (T-VEC): A modified herpes simplex virus type-1–derived oncolytic immunotherapy. Expert Rev Anticancer Ther (2015) 15(12):1389–403. doi: 10.1586/14737140.2015.1115725
54. Wang Y, Jin J, Wu Z, Hu S, Hu H, Ning Z, et al. Stability and anti-tumor effect of oncolytic herpes simplex virus type 2. Oncotarget (2018) 9(37):24672–83. doi: 10.18632/oncotarget.25122
55. Dharmadhikari N, Mehnert JM, Kaufman HL. Oncolytic virus immunotherapy for melanoma. Curr Treat Options Oncol (2015) 16(3):1–5. doi: 10.1007/s11864-014-0326-0
56. Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol (2017) 18(1):1–15. doi: 10.1007/s40257-016-0238-9
57. Ekeke CN, Russell KL, Joubert K, Bartlett DL, Luketich JD, Soloff AC, et al. Fighting fire with Fire : Oncolytic virotherapy for thoracic malignancies. Ann Surg Oncol (2021) 28(5):2715–27. doi: 10.1245/s10434-020-09477-4
58. Koch MS, Lawler SE, Chiocca EA. HSV-1 oncolytic viruses from bench to bedside: An overview of current clinical trials. Cancers (Basel) (2020) 12(12):3514. doi: 10.3390/cancers12123514
59. Chaurasiya S, Fong Y, Warner SG. Oncolytic virotherapy for Cancer. Clin Exp (2021) 9(4):419. doi: 10.3390/biomedicines9040419
60. Garber K. China Approves world’s first oncolytic virus therapy for cancer treatment. vol. 98. J Natl Cancer Institute. US (2006) 8(5):298–300. doi: 10.1093/jnci/djj111
61. Ilkow CS, Marguerie M, Batenchuk C, Mayer J, Ben Neriah D, Cousineau S, et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat Med (2015) 21(5):530–6. doi: 10.1038/nm.3848
62. Desjardins A, Vlahovic G, Friedman HS. Vaccine therapy, oncolytic viruses, and gliomas. Oncol (willist Park NY). (2016) 30(3):211–8.
63. Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res (2014) 2(4):295–300. doi: 10.1158/2326-6066.CIR-14-0015
64. Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer (2019) 7(1):6. doi: 10.1186/s40425-018-0495-7
65. Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer (2013) 12(1):103. doi: 10.1186/1476-4598-12-103
66. Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol (2008) 6(7):529–40. doi: 10.1038/nrmicro1927
67. Hemminki O, Hemminki A. A century of oncolysis evolves into oncolytic immunotherapy. Oncoimmunol (2016) 5(2):e1074377. doi: 10.1080/2162402X.2015.1074377
68. Russell SJ, Barber GN. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell (2018) 33(4):599–605. doi: 10.1016/j.ccell.2018.03.011
69. Kontermann RE. Viro-antibody therapy : engineering oncolytic viruses for genetic delivery of diverse antibody-based biotherapeutics. InMabs (2021) 13(1):1982447. doi: 10.1080/19420862.2021.1982447
70. Kleijn A, Kloezeman J, Treffers-Westerlaken E, Fulci G, Leenstra S, Dirven C, et al. The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. PloS One (2014) 9(5):e97495. doi: 10.1371/journal.pone.0097495
71. Melchjorsen J. Learning from the messengers: innate sensing of viruses and cytokine regulation of immunity - clues for treatments and vaccines. Viruses (2013) 5(2):470–527. doi: 10.3390/v5020470
72. Kakiuchi Y, Kuroda S, Kanaya N, Kumon K, Tsumura T, Hashimoto M, et al. Local oncolytic adenovirotherapy produces an abscopal effect via tumor-derived extracellular vesicles. Mol Ther (2021) 29(10):2920–30. doi: 10.1016/j.ymthe.2021.05.015
73. Ma J, Ramachandran M, Jin C, Quijano-Rubio C, Martikainen M, Yu D, et al. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis (2020) 11(1):1–5. doi: 10.1038/s41419-020-2236-3
74. Jiang H, Clise-Dwyer K, Ruisaard KE, Fan X, Tian W, Gumin J, et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PloS One (2014) 9(5):e97407. doi: 10.1371/journal.pone.0097407
75. Ramelyte E, Tastanova A, Balázs Z, Ignatova D, Turko P, Menzel U, et al. Oncolytic virotherapy-mediated anti-tumor response: a single-cell perspective. Cancer Cell (2021) 39(3):394–406.e4. doi: 10.1016/j.ccell.2020.12.022
76. Mardi A, Shirokova AV, Mohammed RN, Keshavarz A, Zekiy AO, Thangavelu L, et al. Biological causes of immunogenic cancer cell death ( ICD ) and anti − tumor therapy ; combination of oncolytic virus − based immunotherapy and CAR T − cell therapy for ICD induction. Cancer Cell Int (2022) 22(1):1–21. doi: 10.1186/s12935-022-02585-z
77. Ranki T, Joensuu T, Jäger E, Karbach J, Wahle C, Kairemo K, et al. Local treatment of a pleural mesothelioma tumor with ONCOS-102 induces a systemic antitumor CD8(+) T-cell response, prominent infiltration of CD8(+) lymphocytes and Th1 type polarization. Oncoimmunology (2014) 3(10):e958937. doi: 10.4161/21624011.2014.958937
78. Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci USA (2001) 98(11):6396–401. doi: 10.1073/pnas.101136398
79. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature (2020) 577(7791):549–55. doi: 10.1038/s41586-019-1922-8
80. Gujar SA, Marcato P, Pan D, Lee PWK. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol Cancer Ther (2010) 9(11):2924–33. doi: 10.1158/1535-7163.MCT-10-0590
81. Marchini A, Daeffler L, Pozdeev VI, Angelova A, Rommelaere J. Immune conversion of tumor microenvironment by oncolytic viruses: the protoparvovirus h-1PV case study. Front Immunol (2019) 10:1848. doi: 10.3389/fimmu.2019.01848
82. Tsun A, Miao XN, Wang CM, Yu DC. Oncolytic immunotherapy for treatment of cancer. Prog Cancer Immunother (2016) 241–283. doi: 10.1007/978-94-017-7555-7_5
83. Arulanandam R, Batenchuk C, Angarita FA, Ottolino-Perry K, Cousineau S, Mottashed A, et al. VEGF-mediated induction of PRD1-BF1/Blimp1 expression sensitizes tumor vasculature to oncolytic virus infection. Cancer Cell (2015) 28(2):210–24. doi: 10.1016/j.ccell.2015.06.009
84. Breitbach CJ, De Silva NS, Falls TJ, Aladl U, Evgin L, Paterson J, et al. Targeting tumor vasculature with an oncolytic virus. Mol Ther (2011) 19(5):886–94. doi: 10.1038/mt.2011.26
85. Hiley CT, Chard LS, Gangeswaran R, Tysome JR, Briat A, Lemoine NR, et al. Vascular endothelial growth factor a promotes vaccinia virus entry into host cells via activation of the akt pathway. J Virol (2013) 87(5):2781–90. doi: 10.1128/JVI.00854-12
86. Luker KE, Hutchens M, Schultz T, Pekosz A, Luker GD. Bioluminescence imaging of vaccinia virus : Effects of interferon on viral replication and spread. Virology (2005) 341(2):284–300. doi: 10.1016/j.virol.2005.06.049
87. Naumenko VA, Stepanenko AA, Lipatova AV, Vishnevskiy DA, Chekhonin VP. Infection of non-cancer cells : A barrier or support for oncolytic virotherapy? Mol Ther Oncolytics (2022) 24:663–82. doi: 10.1016/j.omto.2022.02.004
88. Reichard KW, Lorence RM, Cascino CJ, Peeples ME, Walter RJ, Fernando MB, et al. Newcastle Disease virus selectively kills human tumor cells. J Surg Res (1992) 52(5):448–53. doi: 10.1016/0022-4804(92)90310-V
89. Lorence RM, Reichard KW, Katubig BB, Reyes HM, Phuangsab A, Mitchell BR, et al. Complete regression of human neuroblastoma xenografts in athymic mice after local Newcastle disease virus therapy. J Natl Cancer Inst (1994) 86(16):1228–33. doi: 10.1093/jnci/86.16.1228
90. Ganar K, Das M, Sinha S, Kumar S. Newcastle Disease virus: current status and our understanding. Virus Res (2014) 184:71–81. doi: 10.1016/j.virusres.2014.02.016
91. Tan L, Zhang Y, Zhan Y, Yuan Y, Sun Y, Qiu X, et al. Newcastle Disease virus employs macropinocytosis and Rab5a-dependent intracellular trafficking to infect DF-1 cells. Oncotarget (2016) 7(52):86117–33. doi: 10.18632/oncotarget.13345
92. Meng Q, He J, Zhong L, Zhao Y. Advances in the study of antitumour immunotherapy for Newcastle disease virus. Int J Med Sci (2021) 18(11):2294–302. doi: 10.7150/ijms.59185
93. Zamarin D, Palese P. Oncolytic Newcastle disease virus for cancer therapy: old challenges and new directions. Future Microbiol (2012) 7(3):347–67. doi: 10.2217/fmb.12.4
94. Schirrmacher V. Immunobiology of Newcastle disease virus and its use for prophylactic vaccination in poultry and as adjuvant for therapeutic vaccination in cancer patients. Int J Mol Sci (2017) 18(5):1103. doi: 10.3390/ijms18051103
95. Malogolovkin A, Gasanov N, Egorov A, Weener M, Ivanov R, Karabelsky A. Combinatorial approaches for cancer treatment using oncolytic viruses: projecting the perspectives through clinical trials outcomes. Viruses (2021) 13(7):1271. doi: 10.3390/v13071271
96. Millward S, Graham AF. Structural studies on reovirus: discontinuities in the genome. Proc Natl Acad Sci (1970) 65(2):422–9. doi: 10.1073/pnas.65.2.422
97. Norman KL, Lee PW. Reovirus as a novel oncolytic agent. J Clin Invest. (2000) 105(8):1035–8. doi: 10.1172/JCI9871
98. Fernandes J. Oncogenes: The passport for viral oncolysis through PKR inhibition. biomark Cancer (2016) 8:BIC.S33378. doi: 10.4137/BIC.S33378
99. Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the ras signaling pathway by reovirus. EMBO J (1998) 17(12):3351–62. doi: 10.1093/emboj/17.12.3351
100. Gollamudi R, Ghalib MH, Desai KK, Chaudhary I, Wong B, Einstein M, et al. Intravenous administration of reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs (2010) 28(5):641—649. doi: 10.1007/s10637-009-9279-8
101. Chakrabarty R, Tran H, Selvaggi G, Hagerman A, Thompson B, Coffey M. The oncolytic virus, pelareorep, as a novel anticancer agent: a review. Invest New Drugs (2015) 33(3):761–74. doi: 10.1007/s10637-015-0216-8
102. Horikami SM, Moyer SA. Structure, transcription, and replication of measles virus. Curr Top Microbiol Immunol (1995) 191:35–50. doi: 10.1007/978-3-642-78621-1_3
103. Bhattacharjee S, Yadava PK. Measles virus: Background and oncolytic virotherapy. Biochem Biophys Rep (2018) 13:58–62. doi: 10.1016/j.bbrep.2017.12.004
104. Leber MF, Neault S, Jirovec E, Barkley R, Said A, Bell JC, et al. Engineering and combining oncolytic measles virus for cancer therapy. Cytokine Growth Factor Rev (2020) 56:39–48. doi: 10.1016/j.cytogfr.2020.07.005
105. Mühlebach MD. Measles virus in cancer therapy. Curr Opin Virol (2020) 41:85–97. doi: 10.1016/j.coviro.2020.07.016
106. Heinzerling L, Künzi V, Oberholzer PA, Kündig T, Naim H, Dummer R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood (2005) 106(7):2287–94. doi: 10.1182/blood-2004-11-4558
107. Msaouel P, Dispenzieri A, Galanis E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: an overview. Curr Opin Mol Ther (2009) 11(1):43–53.
108. Fallaux FJ, Bout A, van der Velde I, van den Wollenberg DJ, Hehir KM, Keegan J, et al. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum Gene Ther (1998) 9(13):1909–17. doi: 10.1089/hum.1998.9.13-1909
109. Walton RW, Brown MC, Sacco MT, Gromeier M. Engineered oncolytic poliovirus PVSRIPO subverts MDA5-dependent innate immune responses in cancer cells. J Virol (2018) 92(19):e00879–18. doi: 10.1128/JVI.00879-18
110. Desjardins A, Gromeier M, Herndon JE, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med (2018) 379(2):150–61. doi: 10.1056/NEJMoa1716435
111. Aldrak N, Alsaab S, Algethami A, Bhere D, Wakimoto H, Shah K, et al. Oncolytic herpes simplex virus-based therapies for cancer. Cells (2021) 10(6):1541. doi: 10.3390/cells10061541
112. Farassati F, Yang AD, Lee PW. Oncogenes in ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat Cell Biol (2001) 3(8):745–50. doi: 10.1038/35087061
113. Abdoli S, Roohvand F, Teimoori-Toolabi L, Shokrgozar MA, Bahrololoumi M, Azadmanesh K. Construction of various γ34.5 deleted fluorescent-expressing oncolytic herpes simplex type 1 (oHSV) for generation and isolation of HSV-based vectors. Iran BioMed J (2017) 21(4):206–17. doi: 10.18869/acadpub.ibj.21.4.206
114. Jugovic P, Hill AM, Tomazin R, Ploegh H, Johnson DC. Inhibition of major histocompatibility complex class I antigen presentation in pig and primate cells by herpes simplex virus type 1 and 2 ICP47. J Virol (1998) 72(6):5076–84. doi: 10.1128/JVI.72.6.5076-5084.1998
115. Kanai R, Zaupa C, Sgubin D, Antoszczyk SJ, Martuza RL, Wakimoto H, et al. Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol (2012) 86(8):4420–31. doi: 10.1128/JVI.00017-12
116. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34(22):2619–26. doi: 10.1200/JCO.2016.67.1529
117. Thomas S, Kuncheria L, Roulstone V, Kyula JN, Mansfield D, Bommareddy PK, et al. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J Immunother Cancer (2019) 7(1):214. doi: 10.1186/s40425-019-0682-1
118. Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. Journal for immunotherapy of cancer (2020) 8(2):e001486. doi: 10.1136/jitc-2020-001486
119. Vellinga J, van der Heijdt S, Hoeben RC. The adenovirus capsid: major progress in minor proteins. J Gen Virol (2005) 86(Pt 6):1581–8. doi: 10.1099/vir.0.80877-0
120. Arnberg N. Adenovirus receptors: implications for targeting of viral vectors. Trends Pharmacol Sci (2012) 33(8):442–8. doi: 10.1016/j.tips.2012.04.005
121. Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I, et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther (2010) 18(10):1874–84. doi: 10.1038/mt.2010.161
122. Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst (2003) 95(9):652–60. doi: 10.1093/jnci/95.9.652
123. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol Off J Am Soc Clin Oncol (2018) 36(14):1419–27. doi: 10.1200/JCO.2017.75.8219
124. Xia Z-J, Chang J-H, Zhang L, Jiang W-Q, Guan Z-Z, Liu J-W, et al. Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Ai zheng= Aizheng= Chin J Cancer (2004) 23(12):1666–70.
125. Liu T-C, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol (2007) 4(2):101–17. doi: 10.1038/ncponc0736
126. Russell SJ, Peng K, Bell JC. Oncolytic virotherapy. Nat Biotechnol (2012) 30(7):658–70. doi: 10.1038/nbt.2287
127. Kew Y, Levin VA. Advances in gene therapy and immunotherapy for brain tumors. Curr Opin Neurol (2003) 16(6):665–70. doi: 10.1097/00019052-200312000-00004
128. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Sci (80- ). (1991) 252(5007):854–6. doi: 10.1126/science.1851332
129. Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med (2000) 6(7):821–5. doi: 10.1038/77558
130. Van Erp EA, Kaliberova LN, Kaliberov SA, Curiel DT. Retargeted oncolytic adenovirus displaying a single variable domain of camelid heavy-chain-only antibody in a fiber protein. Mol Ther (2015) 2:15001. doi: 10.1038/mto.2015.1
131. Liu Y, Wang H, Yumul R, Gao W, Gambotto A, Morita T, et al. Transduction of liver metastases after intravenous injection of Ad5/35 or Ad35 vectors with and without factor X-binding protein pretreatment. Hum Gene Ther (2009) 20(6):621–9. doi: 10.1089/hum.2008.142
132. Wang H, Liu Y, Li Z, Tuve S, Stone D, Kalyushniy O, et al. In vitro and in vivo properties of adenovirus vectors with increased affinity to CD46. J Virol (2008) 82(21):10567–79. doi: 10.1128/JVI.01308-08
133. Nettelbeck DM, Rivera AA, Kupsch J, Dieckmann D, Douglas JT, Kontermann RE, et al. Retargeting of adenoviral infection to melanoma: combining genetic ablation of native tropism with a recombinant bispecific single-chain diabody (scDb) adapter that binds to fiber knob and HMWMAA. Int J Cancer (2004) 108(1):136–45. doi: 10.1002/ijc.11563
134. Montaño-Samaniego M, Bravo-Estupiñan DM, Méndez-Guerrero O, Alarcón-Hernández E, Ibáñez-Hernández M. Strategies for targeting gene therapy in cancer cells with tumor-specific promoters. Front Oncol (2020) 10:605380. doi: 10.3389/fonc.2020.605380
135. Barker SD, Dmitriev IP, Nettelbeck DM, Liu B, Rivera AA, Alvarez RD, et al. Combined transcriptional and transductional targeting improves the specificity and efficacy of adenoviral gene delivery to ovarian carcinoma. Gene Ther (2003) 10(14):1198–204. doi: 10.1038/sj.gt.3301974
136. Mazzacurati L, Marzulli M, Reinhart B, Miyagawa Y, Uchida H, Goins WF, et al. Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol Ther (2015) 23(1):99–107. doi: 10.1038/mt.2014.177
137. Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, et al. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res (2007) 67(6):2456–68. doi: 10.1158/0008-5472.CAN-06-2698
138. Luo Q, Song H, Deng X, Li J, Jian W, Zhao J, et al. A triple-regulated oncolytic adenovirus carrying microRNA-143 exhibits potent antitumor efficacy in colorectal cancer. Mol Ther (2020) 16:219–29. doi: 10.1016/j.omto.2020.01.005
139. Ferguson MS, Lemoine NR, Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv Virol (2012) 2012:805629. doi: 10.1155/2012/805629
140. Lubinski JM, Lazear HM, Awasthi S, Wang F, Friedman HM. The herpes simplex virus 1 IgG fc receptor blocks antibody-mediated complement activation and antibody-dependent cellular cytotoxicity in vivo. J Virol (2011) 85(7):3239–49. doi: 10.1128/JVI.02509-10
141. Croyle MA, Le HT, Linse KD, Cerullo V, Toietta G, Beaudet A, et al. PEGylated helper-dependent adenoviral vectors: highly efficient vectors with an enhanced safety profile. Gene Ther (2005) 12(7):579–87. doi: 10.1038/sj.gt.3302441
142. Capasso C, Hirvinen M, Cerullo V. Beyond gene delivery: Strategies to engineer the surfaces of viral vectors. Biomedicines (2013) 1(1):3–16. doi: 10.3390/biomedicines1010003
143. Mader EK, Maeyama Y, Lin Y, Butler GW, Russell HM, Galanis E, et al. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin Cancer Res (2009) 15(23):7246–55. doi: 10.1158/1078-0432.CCR-09-1292
144. Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Sci (80- ). (2006) 311(5768):1780–4. doi: 10.1126/science.1121411
145. Ilett EJ, Bárcena M, Errington-Mais F, Griffin S, Harrington KJ, Pandha HS, et al. Internalization of oncolytic reovirus by human dendritic cell carriers protects the virus from neutralization. Clin Cancer Res (2011) 17(9):2767–76. doi: 10.1158/1078-0432.CCR-10-3266
146. Haisma HJ, Boesjes M, Beerens AM, van der Strate BWA, Curiel DT, Pl̈ddemann A, et al. Scavenger receptor A: New route adenovirus 5. Mol Pharm (2009) 6(2):366–74. doi: 10.1021/mp8000974
147. Roberts DM, Nanda A, Havenga MJE, Abbink P, Lynch DM, Ewald BA, et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature (2006) 441(7090):239–43. doi: 10.1038/nature04721
148. Khare R, Reddy VS, Nemerow GR, Barry MA. Identification of adenovirus serotype 5 hexon regions that interact with scavenger receptors. J Virol (2012) 86(4):2293–301. doi: 10.1128/JVI.05760-11
149. Prill J-M, Espenlaub S, Samen U, Engler T, Schmidt E, Vetrini F, et al. Modifications of adenovirus hexon allow for either hepatocyte detargeting or targeting with potential evasion from kupffer cells. Mol Ther (2011) 19(1):83–92. doi: 10.1038/mt.2010.229
150. Zhang Z, Krimmel J, Zhang Z, Hu Z, Seth P. Systemic delivery of a novel liver-detargeted oncolytic adenovirus causes reduced liver toxicity but maintains the antitumor response in a breast cancer bone metastasis model. Hum Gene Ther (2011) 22(9):1137–42. doi: 10.1089/hum.2011.003
151. Parker AL, Waddington SN, Nicol CG, Shayakhmetov DM, Buckley SM, Denby L, et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood (2006) 108(8):2554–61. doi: 10.1182/blood-2006-04-008532
152. Atoda H, Ishikawa M, Mizuno H, Morita T. Coagulation factor X-binding protein from deinagkistrodon acutus venom is a gla domain-binding protein. Biochemistry (1998) 37(50):17361–70. doi: 10.1021/bi981177x
153. Kwon O-J, Kang E, Choi J-W, Kim SW, Yun C-O. Therapeutic targeting of chitosan–PEG–folate-complexed oncolytic adenovirus for active and systemic cancer gene therapy. J Control Release (2013) 169(3):257–65. doi: 10.1016/j.jconrel.2013.03.030
154. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer (2006) 6(8):583–92. doi: 10.1038/nrc1893
155. Hill C, Carlisle R. Achieving systemic delivery of oncolytic viruses. Expert Opin Drug Deliv. (2019) 16(6):607–20. doi: 10.1080/17425247.2019.1617269
156. Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci (2011) 108(7):2909–14. doi: 10.1073/pnas.1018892108
157. Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther (2010) 18(7):1275–83. doi: 10.1038/mt.2010.79
158. Mok W, Boucher Y, Jain RK. Matrix metalloproteinases-1 and-8 improve the distribution and efficacy of an oncolytic virus. Cancer Res (2007) 67(22):10664–8. doi: 10.1158/0008-5472.CAN-07-3107
159. Kontermann RE, Ungerechts G, Nettelbeck DM. Viro-antibody therapy: engineering oncolytic viruses for genetic delivery of diverse antibody-based biotherapeutics. MAbs (2021) 13(1):1982447. doi: 10.1080/19420862.2021.1982447
160. Dias JD, Hemminki O, Diaconu I, Hirvinen M, Bonetti A, Guse K, et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther (2012) 19(10):988–98. doi: 10.1038/gt.2011.176
161. Hamilton JR, Vijayakumar G, Palese P. A recombinant antibody-expressing influenza virus delays tumor growth in a mouse model. Cell Rep (2018) 22(1):1–7. doi: 10.1016/j.celrep.2017.12.025
162. Vijayakumar G, Palese P, Goff PH. Oncolytic Newcastle disease virus expressing a checkpoint inhibitor as a radioenhancing agent for murine melanoma. EBioMedicine (2019) 49:96–105. doi: 10.1016/j.ebiom.2019.10.032
163. Vijayakumar G, McCroskery S, Palese P. Engineering Newcastle disease virus as an oncolytic vector for intratumoral delivery of immune checkpoint inhibitors and immunocytokines. J Virol (2020) 94(3):e01677–19. doi: 10.1128/JVI.01677-19
164. Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther (2014) 22(11):1949–59. doi: 10.1038/mt.2014.160
165. Wu C, Wu M, Liang M, Xiong S, Dong C. A novel oncolytic virus engineered with PD-L1 scFv effectively inhibits tumor growth in a mouse model. Cell Mol Immunol (2019) 16(9):780–2. doi: 10.1038/s41423-019-0264-7
166. Kleinpeter P, Fend L, Thioudellet C, Geist M, Sfrontato N, Koerper V, et al. Vectorization in an oncolytic vaccinia virus of an antibody, a fab and a scFv against programmed cell death -1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology (2016) 5(10):e1220467. doi: 10.1080/2162402X.2016.1220467
167. Liikanen I, Tähtinen S, Guse K, Gutmann T, Savola P, Oksanen M, et al. Oncolytic adenovirus expressing monoclonal antibody trastuzumab for treatment of HER2-positive cancer. Mol Cancer Ther (2016) 15(9):2259–69. doi: 10.1158/1535-7163.MCT-15-0819
168. Wei D, Li Q, Wang X-L, Wang Y, Xu J, Feng F, et al. Oncolytic Newcastle disease virus expressing chimeric antibody enhanced anti-tumor efficacy in orthotopic hepatoma-bearing mice. J Exp Clin Cancer Res (2015) 34:153. doi: 10.1186/s13046-015-0271-1
169. Huang T, Wang H, Chen NG, Frentzen A, Minev B, Szalay AA. Expression of anti-VEGF antibody together with anti-EGFR or anti-FAP enhances tumor regression as a result of vaccinia virotherapy. Mol Ther Oncolytics (2015) 2:15003. doi: 10.1038/mto.2015.3
170. Khalique H, Baugh R, Dyer A, Scott EM, Frost S, Larkin S, et al. Oncolytic herpesvirus expressing PD-L1 BiTE for cancer therapy: exploiting tumor immune suppression as an opportunity for targeted immunotherapy. J Immunother Cancer (2021) 9(4):e001292. doi: 10.1136/jitc-2020-001292
171. Scott EM, Jacobus EJ, Lyons B, Frost S, Freedman JD, Dyer A, et al. Bi-and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J Immunother Cancer (2019) 7(1):1–18. doi: 10.1186/s40425-019-0807-6
172. Schönung M, Meyer J, Nöllke P, Olshen AB, Hartmann M, Murakami N, et al. International consensus definition of DNA methylation subgroups in juvenile myelomonocytic LeukemiaDNA methylation subgroups in JMML. Clin Cancer Res (2021) 27(1):158–68. doi: 10.1158/1078-0432.CCR-20-3184
173. Yu F, Hong B, Song X-T. A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Transl Med (2017) 3(4):122. doi: 10.4103/ctm.ctm_13_17
174. Ma R, Lu T, Li Z, Teng K-Y, Mansour AG, Yu M, et al. An oncolytic virus expressing IL15/IL15Rα combined with off-the-Shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res (2021) 81(13):3635–48. doi: 10.1158/0008-5472.CAN-21-0035
175. Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, et al. Development of therapeutic antibodies for the treatment of diseases. J BioMed Sci (2020) 27(1):1–30. doi: 10.1186/s12929-019-0592-z
176. Goulet DR, Atkins WM. Considerations for the design of antibody-based therapeutics. J Pharm Sci (2020) 109(1):74–103. doi: 10.1016/j.xphs.2019.05.031
177. Briolay T, Petithomme T, Fouet M, Nguyen-Pham N, Blanquart C, Boisgerault N. Delivery of cancer therapies by synthetic and bio-inspired nanovectors. Mol Cancer (2021) 20(1):1–24. doi: 10.1186/s12943-021-01346-2
178. Russell L, Peng KW, Russell SJ, Diaz RM. Oncolytic viruses: Priming time for cancer immunotherapy. BioDrugs (2019) 33(5):485–501. doi: 10.1007/s40259-019-00367-0
179. Sivanandam V, LaRocca CJ, Chen NG, Fong Y, Warner SG. Oncolytic viruses and immune checkpoint inhibition: The best of both worlds. Mol Ther - Oncolytics (2019) 13:93–106. doi: 10.1016/j.omto.2019.04.003
180. Chiu M, Armstrong EJL, Jennings V, Foo S, Crespo-Rodriguez E, Bozhanova G, et al. Combination therapy with oncolytic viruses and immune checkpoint inhibitors. Expert Opin Biol Ther (2020) 20(6):635–52. doi: 10.1080/14712598.2020.1729351
181. Senior M. Checkpoint inhibitors go viral. Nat Biotechnol (2019) 37(1):12–7. doi: 10.1038/nbt.4327
182. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med (2014) 6(226):226ra32. doi: 10.1126/scitranslmed.3008095
183. Saha D, Martuza RL, Rabkin SD. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy (2018) 10(9):779–86. doi: 10.2217/imt-2018-0009
184. Saha D, Martuza RL, Rabkin SD. Curing glioblastoma : Oncolytic HSV-IL12 and checkpoint blockade. Oncoscience (2017) 4(July):67–9. doi: 10.18632/oncoscience.359
185. Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discovery (2019) 18(9):689–706. doi: 10.1038/s41573-019-0029-0
186. Nettelbeck DM, Leber MF, Altomonte J, Angelova A, Beil J, Berchtold S, et al. Virotherapy in germany–recent activities in virus engineering, preclinical development, and clinical studies. Viruses (2021) 13(8):1–29. doi: 10.3390/v13081420
187. Wu Z, Ichinose T, Naoe Y, Matsumura S, Villalobos IB, Eissa IR, et al. Combination of cetuximab and oncolytic virus canerpaturev synergistically inhibits human colorectal cancer growth. Mol Ther - Oncolytics (2019) 13(June):107–15. doi: 10.1016/j.omto.2019.04.004
188. Zhang W, Fulci G, Buhrman JS, Stemmer-Rachamimov AO, Chen JW, Wojtkiewicz GR, et al. Bevacizumab with angiostatin-armed oHSV increases antiangiogenesis and decreases bevacizumab-induced invasion in U87 glioma. Mol Ther (2012) 20(1):37–45. doi: 10.1038/mt.2011.187
189. Yang EY, Shah K. Nanobodies: Next generation of cancer diagnostics and therapeutics. Front Oncol (2020) 10(July). doi: 10.3389/fonc.2020.01182
190. Rahbarizadeh F, Ahmadvand D, Sharifzadeh Z. Nanobody; an old concept and new vehicle for immunotargeting. Immunol Invest. (2011) 40(3):299–338. doi: 10.3109/08820139.2010.542228
191. Najmeddin A, Bahrololoumi M, Behdani M, Dorkoosh F. Nanobodies as powerful pulmonary targeted biotherapeutics against SARS-CoV-2, pharmaceutical point of view. BBA-General Subj. (2021) 1865(11):129974. doi: 10.1016/j.bbagen.2021.129974
192. Gil JS, Dubois M, Neirinckx V, Lombard A, Coppieters N, D’Arrigo P, et al. Nanobody-based retargeting of an oncolytic herpesvirus for eliminating CXCR4+ GBM cells: a proof-of-principle. Mol Ther - Oncolytics (2022) 26:35–48. doi: 10.1016/j.omto.2022.06.002
193. Xu B, Tian L, Chen J, Wang J, Ma R, Dong W, et al. An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nat Commun (2021) 12(1):1–7. doi: 10.1038/s41467-021-26003-6
194. Zhang B, Shu Y, Hu S, Qi Z, Chen Y, Innovation C. An oncolytic adenovirus armed with a nanobody against CD47 reprograms tumor immune microenvironment and drives durabl antitumor immunity. Available at SSRN 3914649.
195. Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery (2019) 18(8):585–608. doi: 10.1038/s41573-019-0028-1
196. Heidbuechel JPW, Engeland CE. Oncolytic viruses encoding bispecific T cell engagers: a blueprint for emerging immunovirotherapies. J Hematol Oncol (2021) 14(1):1–24. doi: 10.1186/s13045-021-01075-5
197. Haas C, Lulei M, Fournier P, Arnold A, Schirrmacher V. A tumor vaccine containing anti-CD3 and anti-CD28 bispecific antibodies triggers strong and durable antitumor activity in human lymphocytes. Int J Cancer (2006) 118(3):658–67. doi: 10.1002/ijc.21390
198. Freedman JD, Hagel J, Scott EM, Psallidas I, Gupta A, Spiers L, et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol Med (2017) 9(8):1067–87. doi: 10.15252/emmm.201707567
199. Schirrmacher V. Oncolytic Newcastle disease virus as a prospective anti-cancer therapy. a biologic agent with potential to break therapy resistance. Expert Opin Biol Ther (2015) 15(12):1757–71. doi: 10.1517/14712598.2015.1088000
200. Schirrmacher V, van Gool S, Stuecker W. Breaking therapy resistance: An update on oncolytic Newcastle disease virus for improvements of cancer therapy. Biomedicines (2019) 7(3):66. doi: 10.3390/biomedicines7030066
201. Bahrololoumi M, Momburg F, Roohvand F, Jarahian M. International immunopharmacology bi / tri-specific antibodies ( HN-Fc-CD16 and HN-Fc-IL-15-CD16 ) cross-linking natural killer ( NK ) -CD16 and Newcastle disease virus ( NDV ) -HN , enhanced NK activation for cancer immunotherapy. Int Immunopharmacol (2021) 96:107762. doi: 10.1016/j.intimp.2021.107762
202. Bahrololoumi Shapourabadi M, Roohvand F, Arashkia A, Mohajel N, Abdoli S, Shahosseini Z, et al. Expression and purification of a bispecific antibody against CD16 and hemagglutinin neuraminidase (HN) in e. coli for cancer immunotherapy. Rep Biochem Mol Biol (2020) 9(1):50–7. doi: 10.29252/rbmb.9.1.50
203. Schirrmacher V, Haas C, Bonifer R, Ahlert T, Gerhards R, Ertel C. Human tumor cell modification by virus infection: An efficient and safe way to produce cancer vaccine with pleiotropic immune stimulatory properties when using Newcastle disease virus. Gene Ther (1999) 6(1):63–73. doi: 10.1038/sj.gt.3300787
204. Schirrmacher V, Fournier P. Newcastle Disease virus: A promising vector for viral therapy, immune therapy, and gene therapy of cancer. Gene Therapy of Cancer (2009) 2009:565–605.
205. Garfall AL, June CH. Three is a charm for an antibody to fight cancer microbial clues to a liver disease. Nature (2019) 575:450–1. doi: 10.1038/d41586-019-03495-3
206. Aigner M, Janke M, Lulei M, Beckhove P, Fournier P, Schirrmacher V. An effective tumor vaccine optimized for costimulation via bispecific and trispecific fusion proteins. Int J Oncol (2008) 32(4):777–89. doi: 10.3892/ijo.32.4.777
207. Fournier P, Schirrmacher V. Bispecific antibodies and trispecific immunocytokines for targeting the immune system against cancer: Preparing for the future. BioDrugs (2013) 27(1):35–53. doi: 10.1007/s40259-012-0008-z
208. Ravirala D, Mistretta B, Gunaratne PH, Pei G, Zhao Z, Zhang X. Co-Delivery of novel bispecific and trispecific engagers by an amplicon vector augments the therapeutic effect of an HSV-based oncolytic virotherapy. J Immunother Cancer (2021) 9(7):1–14. doi: 10.1136/jitc-2021-002454
209. Guo ZS, Lotze MT, Zhu Z, Storkus WJ, Song XT. Bi- and tri-specific T cell engager-armed oncolytic viruses: Next-generation cancer immunotherapy. Biomedicines (2020) 8(8):1–16. doi: 10.3390/biomedicines8070204
210. Miliotou AN, Papadopoulou LC. CAR T-cell therapy: A new era in cancer immunotherapy. Curr Pharm Biotechnol (2018) 19(1):5–18. doi: 10.2174/1389201019666180418095526
211. Sterner RC, Sterner RM. CAR-T cell therapy : current limitations and potential strategies. Blood Cancer J (2021) 11(4):69. doi: 10.1038/s41408-021-00459-7
212. Safarzadeh Kozani P, Naseri A, Mirarefin SMJ, Salem F, Nikbakht M, Evazi Bakhshi S, et al. Nanobody-based CAR-T cells for cancer immunotherapy. biomark Res (2022) 10(1):1–18. doi: 10.1186/s40364-022-00371-7
213. Gamboa L, Zamat AH, Vanover D, Thiveaud CA, Peck HE, Phuengkham H, et al. Sensitizing solid tumors to CAR-mediated cytotoxicity using synthetic antigens. bioRxiv (2021). doi: 10.1101/2021.12.11.472238
214. Hajari Taheri F, Hassani M, Sharifzadeh Z, Behdani M, Arashkia A, Abolhassani M. T Cell engineered with a novel nanobody-based chimeric antigen receptor against VEGFR2 as a candidate for tumor immunotherapy. IUBMB Life (2019) 71(9):1259–67. doi: 10.1002/iub.2019
215. Jamnani FR, Rahbarizadeh F, Shokrgozar MA, Ahmadvand D, Mahboudi F, Sharifzadeh Z. Targeting high affinity and epitope-distinct oligoclonal nanobodies to HER2 over-expressing tumor cells. Exp Cell Res (2012) 318(10):1112–24. doi: 10.1016/j.yexcr.2012.03.004
216. Sharifzadeh Z, Rahbarizadeh F, Shokrgozar MA, Ahmadvand D, Mahboudi F, Jamnani FR, et al. Genetically engineered T cells bearing chimeric nanoconstructed receptors harboring TAG-72-specific camelid single domain antibodies as targeting agents. Cancer Lett (2013) 334(2):237–44. doi: 10.1016/j.canlet.2012.08.010
217. Hassani M, Hajari Taheri F, Sharifzadeh Z, Arashkia A, Hadjati J, van Weerden WM, et al. Construction of a chimeric antigen receptor bearing a nanobody against prostate a specific membrane antigen in prostate cancer. J Cell Biochem (2019) 120(6):10787–95. doi: 10.1002/jcb.28370
218. Evazalipour M, D’Huyvetter M, Tehrani BS, Abolhassani M, Omidfar K, Abdoli S, et al. Generation and characterization of nanobodies targeting PSMA for molecular imaging of prostate cancer. Contrast Media Mol Imaging. (2014) 9(3):211–20. doi: 10.1002/cmmi.1558
219. Hassani M, Hajari Taheri F, Sharifzadeh Z, Arashkia A, Hadjati J, van Weerden WM, et al. Engineered jurkat cells for targeting prostate-specific membrane antigen on prostate cancer cells by nanobody-based chimeric antigen receptor. Iran BioMed J (2020) 24(2):81–8. doi: 10.29252/ibj.24.2.81
220. Bao C, Gao Q, Li LL, Han L, Zhang B, Ding Y, et al. The application of nanobody in CAR-T therapy. Biomolecules (2021) 11(2):1–18. doi: 10.3390/biom11020238
221. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res (2014) 74(18):5195–205. doi: 10.1158/0008-5472.CAN-14-0697
222. Rezaei R, Esmaeili H, Ghaleh G, Farzanehpour M, Dorostkar R, Ranjbar R, et al. Combination therapy with CAR T cells and oncolytic viruses : a new era in cancer immunotherapy. Cancer Gene Ther (2021) 1:1–14. doi: 10.1038/s41417-021-00359-9
223. Guedan S, Alemany R. CAR-T cells and oncolytic viruses: Joining forces to overcome the solid tumor challenge. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02460
224. Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget (2016) 7(19):27764–77. doi: 10.18632/oncotarget.8526
225. Schirrmacher V, Stücker W, Lulei M, Bihari A-S, Sprenger T. Long-term survival of a breast cancer patient with extensive liver metastases upon immune and virotherapy: A case report. Immunotherapy (2015) 7(8):855–60. doi: 10.2217/imt.15.48
226. Schirrmacher V, Fournier P. Multimodal cancer therapy involving oncolytic Newcastle disease virus, autologous immune cells and bispecific antibodies. Front Oncol (2014) 4:1–5. doi: 10.3389/fonc.2014.00224
227. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA (2005) 102(51):18538–43. doi: 10.1073/pnas.0509182102
228. Feist M, Zhu Z, Dai E, Ma C, Liu Z, Giehl E, et al. Oncolytic virus promotes tumor-reactive infiltrating lymphocytes for adoptive cell therapy. Cancer Gene Ther (2021) 28(1–2):98–111. doi: 10.1038/s41417-020-0189-4
229. Morales-Molina A, Rodríguez-Milla MÁ, Gimenez-Sanchez A, Perisé-Barrios AJ, García-Castro J. Cellular virotherapy increases tumor-infiltrating lymphocytes (Til) and decreases their pd-1+ subsets in mouse immunocompetent models. Cancers (Basel). (2020) 12(7):1–17. doi: 10.3390/cancers12071920
Keywords: oncolytic virotherapy, cancer immunotherapy, nanobody, antibody, combination therapy, immunovirotherapy, T cells, Nk cells
Citation: Jafari M, Kadkhodazadeh M, Shapourabadi MB, Goradel NH, Shokrgozar MA, Arashkia A, Abdoli S and Sharifzadeh Z (2022) Immunovirotherapy: The role of antibody based therapeutics combination with oncolytic viruses. Front. Immunol. 13:1012806. doi: 10.3389/fimmu.2022.1012806
Received: 05 August 2022; Accepted: 27 September 2022;
Published: 13 October 2022.
Edited by:
Zong Sheng Guo, Roswell Park Comprehensive Cancer Center, United StatesReviewed by:
Rajesh Kumar, National Institutes of Health (NIH), United StatesZuqiang Liu, Allegheny Health Network, United States
Copyright © 2022 Jafari, Kadkhodazadeh, Shapourabadi, Goradel, Shokrgozar, Arashkia, Abdoli and Sharifzadeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zahra Sharifzadeh, zsharifzadeh@gmail.com; Shahriyar Abdoli, shahriyaran@gmail.com