 Kishan Kumar Nyati
Kishan Kumar Nyati Tadamitsu Kishimoto
Tadamitsu Kishimoto- Laboratory of Immune Regulation, World Premier International Immunology Frontier Research Center, Osaka University, Osaka, Japan
AT-rich interactive domain 5a (Arid5a) is a nucleic acid binding protein. In this review, we highlight recent advances in the association of Arid5a with inflammation and human diseases. Arid5a is known as a protein that performs dual functions. In in vitro and in vivo studies, it was found that an inflammation-dependent increase in Arid5a expression mediates both transcriptional and post-transcriptional regulatory effects that are implicated in immune regulation and cellular homeostasis. A series of publications demonstrated that inhibiting Arid5a augmented several processes, such as preventing septic shock, experimental autoimmune encephalomyelitis, acute lung injury, invasion and metastasis, immune evasion, epithelial-to-mesenchymal transition, and the M1-like tumor-associated macrophage (TAM) to M2-like TAM transition. In addition, Arid5a controls adipogenesis and obesity in mice to maintain metabolic homeostasis. Taken together, recent progress indicates that Arid5a exhibits multifaceted, both beneficial and detrimental, roles in health and disease and suggest the relevance of Arid5a as a potential therapeutic target.
1 Introduction
Interleukin (IL) 6 is a pleiotropic and multi-functional cytokine (1). Many cells secrete IL6, which is transcriptionally activated by various stimuli, including the inflammatory cytokines, IL1, and tumor necrosis factor α (TNFα) (2). Uncontrolled expression of IL6 is responsible for several autoimmune inflammatory diseases and tumor growth (3). The anti-IL6 receptor antibody, Tocilizumab, has been approved by food and drug administration of the United States for the treatment of autoimmune diseases including rheumatoid arthritis (RA), juvenile idiopathic arthritis, ulcerative colitis, psoriatic arthritis, and giant cell arteritis. This antibody has been further in use to treat Castleman’s disease, polymyalgia rheumatica, and Takayasu’s arteritis. Another anti-IL6 receptor antibody, Sarilumab and an antibody to block IL6, Siltuximab are also being used to treat these diseases. Recently, these drugs (Tocilizumab and Sarilumab) have been recommended by the National Institutes of Health, U.S.A. for the patients who require supplemental oxygen, high-flow oxygen, noninvasive ventilation, or mechanical ventilation. However, blocking IL6 may lead to secondary immune responses. Therefore, IL6 expression must be strictly controlled at the transcriptional and post-transcriptional levels. The transcriptional control of Il6 expression by transcription factors (TFs), such as nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and CCAAT-enhancer-binding proteinβ (C/EBPβ), has been studied extensively (4, 5). However, little is known regarding the manner in which Il6 messenger RNA (mRNA) is post-transcriptionally regulated. The regulation of cytokine mRNA stability by RNA-binding proteins (RBPs) is essential because of their roles in life-threatening human diseases. Previously, we observed that chlorpromazine (CPZ), an anti-histaminic and anti-psychotic drug, inhibits lipopolysaccharide (LPS)-induced IL6 production in macrophages, whereas other inflammatory cytokines were not affected (6). During the study of the molecular mechanism of CPZ-induced inhibition of IL6, we identified a unique molecule, AT-rich interactive domain 5a (Arid5a), which is also inhibited by CPZ. We also determined its unique function in mRNA stability by binding to the 3′ untranslated region (3’ UTR) of Il6 mRNA. However, little is known regarding the manner in which Arid5a regulates mRNAs post-transcriptionally and whether is associated with the development of inflammatory and autoimmune diseases or cancer.
The AT-rich interactive domain (arid) is a nucleic acid binding domain that is evolutionarily conserved in higher eukaryotes. Its consensus sequence consists of approximately 100 amino acid residues and has a helix-turn-helix motif (7–10). Arid has also been identified in other proteins and it represents a family containing 15 members, which are important in development, differentiation, proliferation, and tissue-specific gene expression (11–13). Arid family members are widespread and are found in some protozoa, green algae, higher plants, fungi, and metazoans (14). Of these, Arid5a has gained significant interest in the field of immunology over the past decade and recently in cancer, because of its identification in the regulation of several TFs and inflammatory mRNA transcripts, including Il6, Tbx21, Ox40, Stat3, and Pparγ, through transcriptional and post-transcriptional mechanisms (15–20). Thus, Arid5a performs a dual function depending on its localization. In the resting stage of a cell, Arid5a is localized to the nucleus along with other nuclear proteins and regulates the function of transcription factors. Subsequently, it shuttles to the cytoplasm under inflammatory conditions (21–24), where it is involved in mRNA stabilization (22). During TLR4 signaling, LPS induces Arid5a expression in the early phase of stimulation through an IKK-mediated NFκB signaling mechanism (21) (Figure 1A). Furthermore, overexpression of NFκB subunits (p65 and c-Rel) in LPS-induced HEK293T cells increased the activity of the Arid5a promoter (21). However, another arm of the TLR4 pathway involving TRIF (Toll interleukin-1 receptor homology domain–containing adaptor inducing IFNβ) signaling has recently been shown to promote Arid5a-mediated IL6 expression during the late phase of LPS stimulation (25). Moreover, TLR4 endocytosis resulting from TRIF signaling following LPS stimulation activates non-canonical phosphorylation of STAT1 at Thr749, which promotes the expression of the Arid5a gene (Figure 1A).
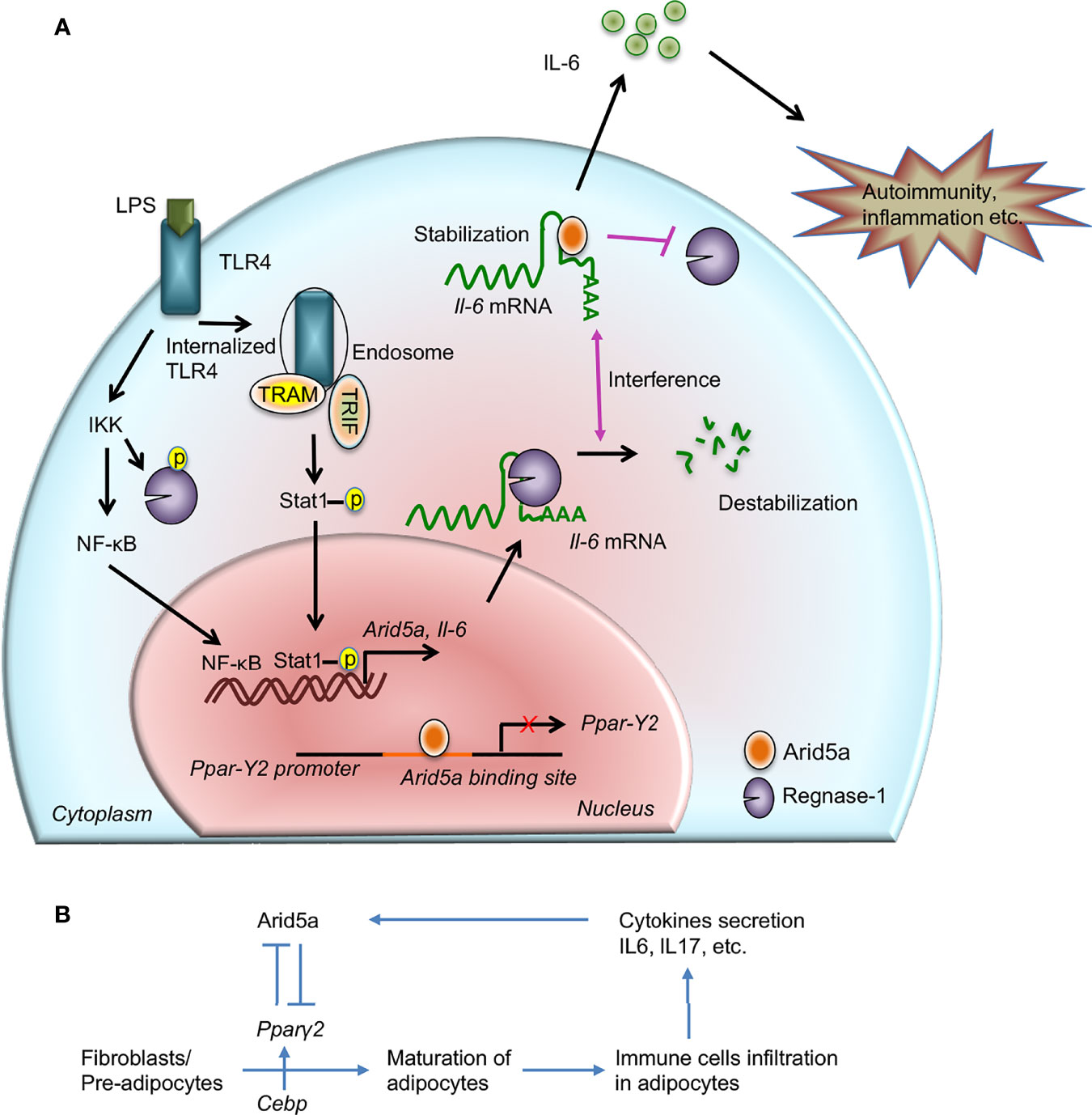
Figure 1 Contribution of AT-rich interactive domain 5A (Arid5a) in interleukin (IL) 6 production and disease. (A) The innate immune response is generated by pathogen-associated molecular patterns, which are recognized by pathogen recognition receptors and lead to expression of proinflammatory mediators. For example, toll-like receptor 4 recognizes lipopolysaccharide and activates the IKK/NFκB signaling pathway. In the early phase of TLR4 signaling, the IKK complex phosphorylates Regnase-1 and also promotes transcription of Il6 and Arid5a. Regnase-1 binds to the Il6 mRNA and degrades it. In the late phase of LPS stimulation, internalization of TLR4 or TLR4 endocytosis under TRIF signaling augments noncanonical phosphorylation of Stat1 which further induces the expression of Arid5a. In the nucleus, Arid5a binds to genes encoding transcription factors, such as Pparγ2, to inhibit their expression. Furthermore, Arid5a is shuttled to the cytoplasm under inflammatory conditions where it interferes with the destabilizing effect of Regnase-1. Arid5a is involved in Il6 mRNA stabilization, and the resulting increase in IL6 production is associated with inflammation and autoimmune diseases. (B) Arid5a and Pparγ2 in adipogenic homeostasis. C/ebp activates Pparγ2, and both are involved in adipogenesis. Immune cells, such as macrophages and T cells, infiltrate adipose tissues and provide a framework to regulate energy homeostasis. In adipose tissue, these cells secrete cytokines, such as IL6 and IL17, which induce Arid5a. Arid5a further represses Pparγ2 transcription in fibroblasts, thereby inhibiting adipocyte development and inhibiting adipogenesis and obesity. “Reprinted from Frontiers in Immunology, 10, Kishan Kumar Nyati, Riddhi Girdhar Agarwal, Praveen Sharma, Tadamitsu Kishimoto, Arid5a regulation and the roles of Arid5a in the inflammatory response and disease, article 2790, copyright (December 2019), under the terms of the creative commons attribution license (CC-BY)”, https://www.frontiersin.org/articles/10.3389/fimmu.2019.02790/full.
Arid5a is indispensable in the development of inflammatory and autoimmune diseases, such as experimental autoimmune encephalomyelitis (EAE) (15), sepsis (17), RA (26), acute lung injury (ALI) (27), as well as breast (20), lung (28), pancreatic (PDAC) (29), colorectal (CRC) (29), and brain (30) cancers. In addition, Arid5a induces chondrocyte differentiation as a transcriptional partner of Sox9, which regulates chondrocyte differentiation through activation of Col2a1, a chondrocyte-specific gene (31). In the nucleus, Arid5a negatively regulates the transcription of the major immediate early (Mie) gene of human cytomegalovirus (HCMV) in a human embryonal carcinoma cell line (32). Furthermore, in the nucleus of fibroblasts, Arid5a suppresses transcription of Pparγ, a master regulator of adipogenesis, thereby inhibiting adipogenesis and obesity in mice (19), whereas increased expression of long noncoding RNA (lncRNA)-AU021063 in breast cancer cells by binding to its promoter region resulted in the regulation of invasion and metastasis (20). By discussing potential mechanisms of Arid5a and its influential roles in immunological disorders and cancer, we aim to encourage discussion on recent advances in Arid5a research. We highlight Arid5a-mediated transcriptional and post-transcriptional regulation of inflammatory molecules. We also describe the role of Arid5a in the development of inflammation and human disease.
2 Arid5a-Mediated Regulation of Inflammatory Genes
Gene expression is controlled at multiple checkpoints including at the gene transcription, mRNA translation, and mRNA degradation levels. TFs play a central role in regulating the transcription of a gene, whereas RBPs are known as key players in post-transcriptional regulation. Several RBPs are reported to stabilize or de-stabilize mRNAs by binding to the 3’UTR. The versatility of RNA-binding domains of RBPs and structural flexibility facilitates the control of metabolism of an array of mRNA transcripts (33). The significant role of TFs and RBPs in gene regulation has been established and perturbations in TF-gene and RBP-RNA network activities have been associated with disease development. Accumulating evidence has shown that post-transcriptional regulation controls numerous cellular mechanisms, including proliferation, differentiation, invasion, metastases, apoptosis and angiogenesis, which could lead to a cancerous phenotype (34). It is likely that the transcripts of nearly all known oncogenes and tumor suppressor genes may be controlled by mechanisms that define mRNA stability and, ultimately, competency for translation. As discussed, Arid5a contains a nucleic acid binding domain that enables it to bind to either DNA or RNA. This ability imparts a dual function to Arid5a in the stabilization of mRNAs, including Il6 (15), Tbx21 (encodes Tbet) (17), Stat3 (16), and Ox40 (18), and in the transcription of genes, such as Pparγ (19) and lncRNA-AU020163 (20), some of which are associated with inflammation, autoimmunity, and cancer.
2.1 Transcriptional Regulation
In cardiovascular tissues, Arid5a binds to the N- and C-terminus of estrogen receptor (ER)-alpha and acts as an ER repressor of gene expression in the nucleus (35). It is expressed in cartilage to induce chondrocyte differentiation by interacting with the TF, Sox9, and regulates chondrocyte differentiation through activation of Col2a1, a chondrocyte-specific gene (31). Arid5a actually binds to the promoter of the Col2a1 gene and enhances transcription of Col2a1, which further induces acetylation of histone 3 proteins to regulate chondrocyte differentiation in association with Sox9. Recently, we observed that Arid5a regulates the invasion and metastasis of breast cancer cells (20). Arid5a under IL6 signaling binds to the upstream region of a lncRNA, AU021063, and transcriptionally upregulates the expression of this noncoding RNA, which further leads to the enhanced invasion and metastasis of breast cancer cells in mice by stabilizing the tribbles homolog 3 (Trib3) protein (20). In contrast, Arid5a inhibits transcription of the Mie gene of HCMV by binding to multiple sites in the modulator region (31). We reported that Arid5a is an important negative regulator of energy metabolism in which Arid5a and Pparγ were dynamically counter-regulated by one another. Transcriptome data revealed that Arid5a binds to the promoter region of Pparγ, which is the leading cause of adipogenesis, and represses the transcription of the Pparγ gene (Figure 1A), thereby inhibiting adipogenesis and obesity in mouse fibroblasts (19) (Figure 1B).
2.2 Posttranscriptional Regulation
2.2.1 Macrophages
CPZ was previously demonstrated to exert a specific inhibitory activity on IL6 production in LPS-stimulated mouse macrophages without affecting the production of other cytokines (e.g., TNFα) (6). To determine the underlying mechanism of IL6 inhibition by CPZ, we identified Arid5a, which binds to the Il6 3′ UTR in mouse macrophages in an Il6 3′ UTR RBP assay coupled with mass spectrometry (15). Further in vitro and in vivo experiments established a relationship between Arid5a protein and Il6 mRNA suggesting that the ARID domain of Arid5a binds to the Il6 3′ UTR and requires Il6 mRNA for stability (Figure 1A). Interestingly, Arid5a interrupts the function of Regnase-1 or Zc3h12a (15) in macrophages, which is a novel RNase that harbors a CCCH-type zinc-finger domain and a PIN-like domain. It has the ability to destabilize Il6 mRNA by binding to the stem-loop region in the Il6 3′ UTR (36, 37). This indicates regulation of Il6 mRNA through a balance between Arid5a and Regnase-1 activities (Figure 1A).
2.2.2 Cardiac Fibroblasts
Studies describing a post-transcriptional regulatory role for Arid5a in different cell types have established it as an RBP. Cardiac fibroblasts secrete numerous cytokines and growth factors to regulate cardiac function (38, 39). Angiotensin II-stimulated cardiac fibroblasts release IL6 family proteins that facilitate the growth of cardiomyocytes (39). The β-adrenergic receptor (βAR) agonist also enhances the production of proinflammatory cytokines (40, 41), which cause heart failure. Thus, inhibition of production of proinflammatory cytokines may be a useful strategy for cardiac remodeling. A recent study suggested that β2-adrenergic stimulation induces Arid5a, which further post-transcriptionally upregulates the expression of Il6 mRNA through the cAMP/PKA/CREB pathway in adult cardiac fibroblasts, and indicates that the β2AR/Arid5a/IL6 axis may be a therapeutic target to prevent cardiac inflammation (42).
2.2.3 T Cells Through Regulation of T Cell Differentiation
Reports indicate that Arid5a−/− mice have low numbers of CD4+IL17+ T cells, or Th17 cells, or IFNγ-producing CD4+ T cells (Th1 cells) compared with wild-type mice (16, 17). This indicates a significant involvement of Arid5a in the differentiation of naive Th cells into Th1 and Th17 cells. possibly through the regulation of Tbet and Stat3 mRNA stability, respectively (16) (Figure 2). The differentiating conditions of Th17 with TGFβ1 and IL6 in early stages of CD4+ T cells induces the rapid expression of Arid5a, but not at later times, which indicates a role for Arid5a in the early phase of Th17 cell development (16). Stat3 is induced by IL6 canonical signaling in CD4+ T cells and IL6/Stat3 axis signaling contributes to the development of Th17 cells and is associated with several human diseases, such as multiple sclerosis (MS) and RA (43–46). Arid5a binds to the stem region of the Stat3 3′ UTR (1738–1765) through the R128 residue, where Regnase-1 also binds. Regnase-1 recognizes a unique sequence, UGU, in the loop region of the mouse Stat3 3′ UTR (1738-1765) (47). Furthermore, Regnase-1 reduces the luciferase reporter activity of the Il6 3′ UTR as mentioned previously (37), and impairs the luciferase reporter activity of the Stat3 3’ UTR, which is rescued by overexpression of Arid5a in 293T cells (16). This indicates that both Arid5a and Regnase-1 target Stat3 (16) and indicates that Arid5a prevents Regnase-1 from binding to the Stat3 3′ UTR, leading to interference of Regnase-1 function (16). In addition, the population of Th17 cells was recovered following overexpression of Stat3c in Arid5a−/− CD4+ T cells under Th17-polarizing conditions, which further suggested a central role of Arid5a in Th17 cell differentiation through Stat3 (16).
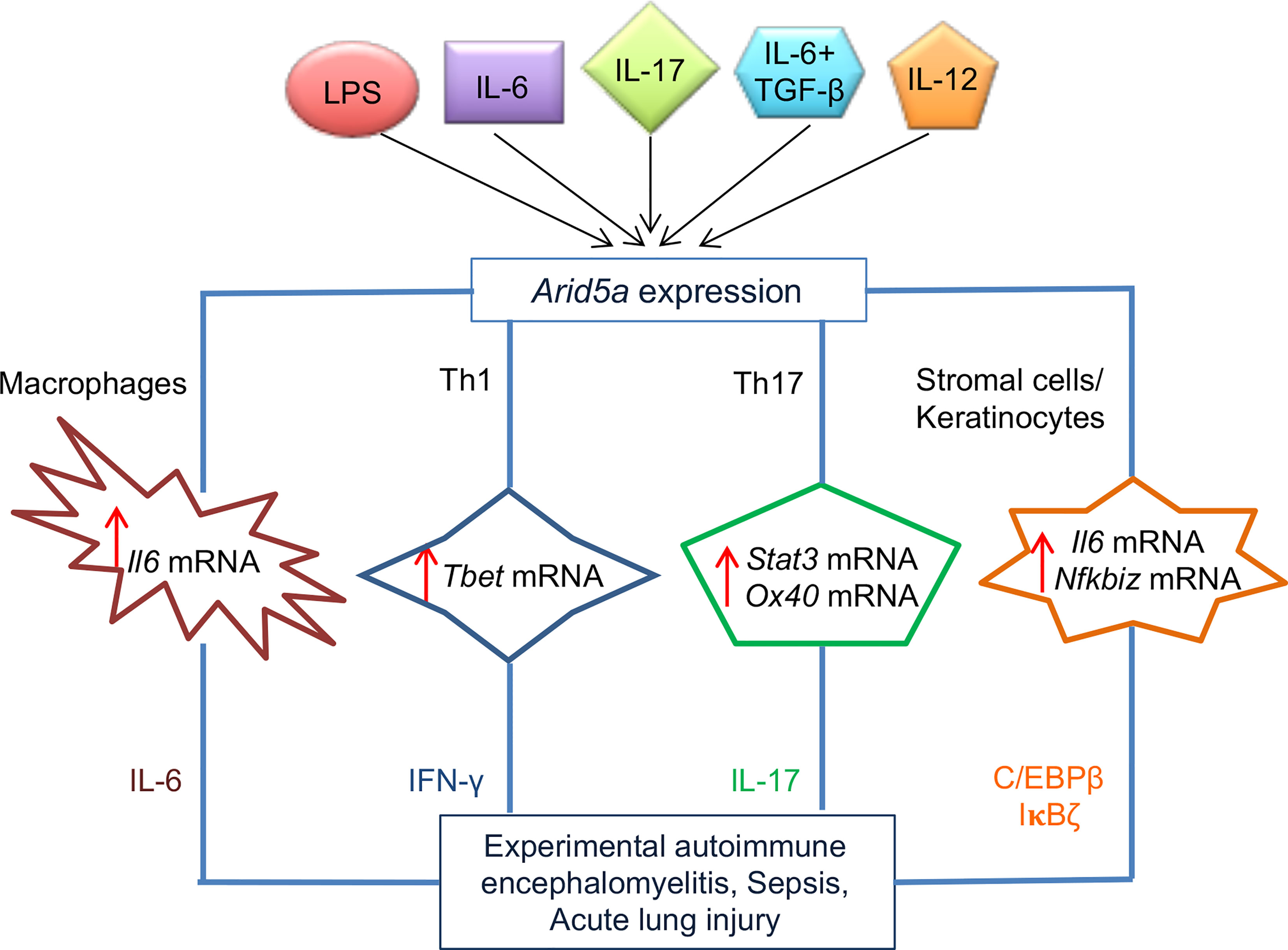
Figure 2 Role of AT-rich interactive domain 5A (Arid5a) in the pathogenesis of sepsis, acute lung injury (ALI), and experimental autoimmune encephalomyelitis (EAE) in mice. Arid5a expression is induced in many immune cells, such as macrophages, T helper (Th)1 cells, Th17 cells, stromal cells, and keratinocytes, after stimulation with lipopolysaccharide (LPS), interleukin (IL)6, IL6 + transforming growth factor (TGF)β, IL17, or IL-2. Arid5a stabilizes a variety of inflammatory mRNAs, including Il6, Tbx21, Stat3, Ox40, and Nfkbiz, in these cells. The stabilization of these target mRNAs results in overproduction of IL6, interferon (IFN)γ, IL17, CCAAT-enhancer-binding proteins (C/EBP)β, and inhibitor of κB (IκB)ζ, which triggers pathogenesis of sepsis, ALI, and EAE in mice. “Reprinted from Trends in Immunology, 41(3), Kishan Kumar Nyati, Mohammad Mahabub-Uz Zaman, Praveen Sharma, Tadamitsu Kishimoto, Arid5a, an RNA-binding protein in immune regulation: RNA stability, inflammation, and autoimmunity, p255–268, copyright (March 2020), with permission from Elsevier”.
Similarly, to regulate Stat3 and Il6 mRNAs, Arid5a regulates the stability of other mRNAs, such as Ox40 in murine CD4+ T cells under Th17-polarizing conditions and Tbx21 in Th1 cells (17, 18). Treatment of CD4+CD62L+ T cells from Arid5a−/− mice with actinomycin D showed a reduction in the half-life of Ox40 mRNA (18), whereas overexpression of Tbx21 3’ UTR and Arid5a constructs in HEK293T Tet-off cells resulted in a significantly higher mRNA stability of Tbx21 following doxycycline treatment (17). In these cells, Arid5a binds to the stem-loop structures of the Ox40 and Tbx21 mRNA transcripts, which further indicates the Arid5a recognition of stem-loop regions of the Stat3 and Il6 3’ UTRs (17, 18) (Figure 2). Similar to Th17 cell differentiation, Th1 cell differentiation is also suppressed in Arid5a−/− mice by decreasing the half-life of Tbx21 mRNA and leading to a reduction in IFNγ production (17) (Figure 2). Taken together, Arid5a controls Th1 and Th17 cell differentiation by regulating the stability of Tbx21, Stat3, and Ox40 mRNAs in T cells.
Other studies have demonstrated that Arid5a binds directly to the 3′ UTRs of multiple target mRNAs and stabilizes IL17-induced mRNA transcripts, such as Il6 and CXC chemokines (48). Furthermore, Arid5a enhances the translation of IL17-induced TFs, such as C/ebpβ and IκBζ (Figure 2), which attenuates the expression of the Lcn2 gene. Moreover, Arid5a promotes IL17 signaling by stabilizing the expression of several IL17-dependent cytokine mRNA transcripts including Il6, Cxcl1, and Cxcl5. In additional, Arid5a induces translation of IL17-dependent TFs (C/ebpβ and IκBζ) that facilitate transactivation of the Lcn2 promoter. Arid5a failed to activate a mutant of the C/EBP binding element and inhibiting Arid5a further decreased IL17-induced expression of all C/EBP protein isoforms. The expression of Arid5a is increased through a feed-forward loop in IL17-stimulated cells, which further triggers the recruitment of Arid5a to TRAF2 (23, 24, 48). Taken together, Arid5a drives Th17 signaling by regulating IL17-dependent TFs, which may represent an approach to treat IL17-dependent diseases (24).
2.2.4 CRC and PDAC Cells
Although the functions of TFs in cancer are well studied, the role of TFs in RNA-binding activities is less clear. Several RBPs that are required for complete RNA metabolism from production to degradation are mutated in cancer; however, only a few are recognized as major cancer drivers (49). Arid5a, which functions as a TF and RBP, was recently shown to be involved in cancer. Our laboratory recently reported an association of Arid5a with CRC and PDAC (29). We found that the injection of Arid5a−/− tumor cells into immunocompetent and immunodeficient mice resulted in smaller tumors in the immunocompetent mice compared with the immunodeficient mice. This prompted us to consider the possibility of an Arid5a-mediated immunosuppressive effect in the tumor microenvironment (TME). This hypothesis is further supported by the observation of a higher frequency of effector CD8+ T cells, whereas lower numbers of immunosuppressive cells, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), were present in Arid5a−/− tumors compared with wild-type tumors. Subsequently, a series of studied revealed that Arid5a induces the expression of Ido1 and Ccl2 by increasing the half-lives of their mRNA transcripts. IDO1, a rate-limiting metabolic enzyme, degrades tryptophan, which ultimately reduces the activation of effector T cells, but enhances Treg differentiation (50, 51). The inhibition of Ido1 resulted in a T cell-dependent anti-tumor response in mice (52, 53) and CCL2 was involved in the recruitment of neutrophils, MDSCs, and monocytes into the TME, which was associated to tumorigenesis (54, 55). Collectively, Arid5a mediates immune evasion in PDAC and CRC models by inducing an Ido1-dependent local immunosuppressive TME and CCL2-mediated MDSC recruitment (Figure 3A). An in-depth understanding of the role of Arid5a in these cancer models will contribute to the development of prognostic and response biomarkers and may lead to new therapeutic strategies.
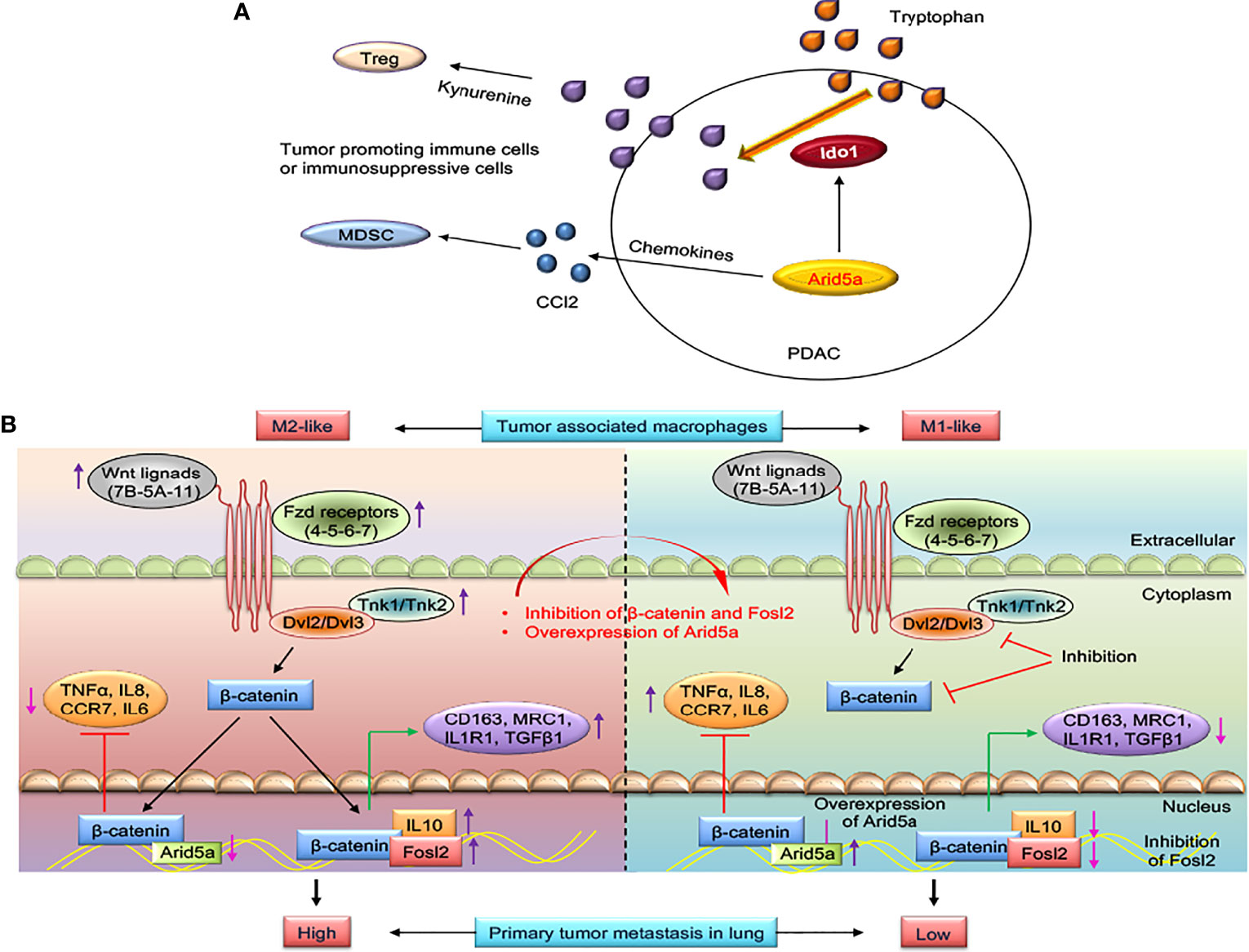
Figure 3 AT-rich interactive domain 5A (Arid5a)-mediated immune evasion in pancreatic cancer and reprogramming of M2-like tumor-associated macrophages (TAMs) to M1-like TAMs in lung cancer. (A) Epithelial-to-mesenchymal transition-transcription factors (EMT-TFs) induces Arid5a expression in mesenchymal subtypes of pancreatic cancer (PDAC) which promotes tumorigenesis by increasing immunosuppressive cells such as Ccl2-mediated recruitment of MDSCs and Ido1-dependent Tregs differentiation. Arid5a stabilizes Ido1 and Ccl2 mRNA transcripts leading to IDO1-mediated degradation of degrade tryptophan which ultimately reduce activation of effector T cells, but enhances Tregs differentiation. Further, CCL2 enhances recruitment of neutrophils, MDSCs, and monocytes into the tumor microenvironment facilitates tumorigenesis. (B) Wnt ligands (Wnt5A-7B-11), frizzled receptors (Fzd4-5-6-8-9), disheveled (Dvl2-3), and Tnk (Tnk1-2) are upregulated in M2-like TAMs, and in this way, transcriptional activation of β-catenin occurs. β-catenin binds to the promoter region of M2 macrophage genes such as IL10 and to TF-activating M2 macrophage genes like Fosl2, CD163, MRC1, IL1R1, and TGFβ1 and thus activates the M2 macrophage program. In contrast, β-catenin suppresses the M1 macrophage program by binding to TF activating M1 macrophage genes such as Arid5a, TNFα, IL8, CCR7, and IL6. With inhibition of β-catenin and Fosl2 and overexpression of Arid5a, M2-like TAMs are reprogrammed into M1-like TAMs; thus, reactivation of antitumor immunity in the tumor microenvironment occurs to restrict primary and metastatic lung tumor growth.
3 Linking Arid5a-Mediated Transcriptional and Post-Transcriptional Regulatory Mechanisms
An earlier study demonstrated the time-dependent subcellular localization of a substantial fraction of Arid5a from the nucleus to the cytoplasm under inflammatory conditions, such as LPS exposure (22). The study has also found that nuclear Il6 mRNA was increased in the early phase of LPS stimulation, whereas cytoplasmic Il6 mRNA was increased in macrophages at later time points. Furthermore, RNA immunoprecipitation assays using anti-Arid5a antibodies coupled with qPCR revealed a 3.9-fold increase in Il6 mRNA bound to cytoplasmic Arid5a and a 2.6-fold increase in nuclear Arid5a following LPS stimulation compared with unstimulated control cells. These data indicate that both nuclear and cytoplasmic Arid5a have the ability to bind to inflammatory mRNAs, such as Il6, even after LPS stimulation. Thus, Arid5a plays different roles in the regulation of mRNAs depending on its subcellular (nuclear or cytoplasmic) localization. It is therefore likely that nuclear and cytoplasmic Arid5a perform dual functions simultaneously which are associated with its transcriptional and post-transcriptional regulatory activities. As mentioned, the dual function of Arid5a depends on time, inflammation, and disease conditions. However, a correlation between these two mechanisms has not been established. These mechanisms warrant further study with respect to the role of Arid5a in reducing inflammation by inhibiting Arid5a-associated transcriptional regulation in the nucleus and post-transcriptional regulation of inflammatory genes in the cytoplasm.
4 Arid5a-Associated Inflammatory and Autoimmune Diseases
4.1 ALI by Regulating IL6 and Reactive Oxygen Species Production
ALI with acute respiratory failure causes acute inflammation in the lungs and is responsible for a high mortality worldwide (56, 57). No treatment exists other than maintaining patients on mechanical ventilation during ALI. Bleomycin-induced pulmonary fibrosis and respiratory inflammation in mice mimic ALI, in which IL6 is abnormally upregulated (58). Il6-deficiency in mice improved recovery from lung fibrosis and ALI following bleomycin administration (59), but the underlying mechanism remains elusive. In preliminary studies, bleomycin-treated Arid5a−/− mice exhibited less reactive oxygen species (ROS) production, lung inflammation, and were found to be resistant to ALI (27). The reduction in ROS affected oxidized 1-palmitoyl-2-arachidonoyl-snglycero-3-phosphocholine (OxPAPC) production. This attenuated IL6 production is because of the absence of post-transcriptional regulation by Arid5a in Arid5a-deficient mice (27) (Figure 2). However, further studies are needed to shed light on Arid5a-mediated regulation of ROS production during ALI.
4.2 Septic Shock by Controlling Tbet-Mediated IFNγ Production and Th1 Cell Differentiation
Endotoxin or LPS present in the Gram-negative bacteria is the primary cause of septic shock, which is defined as a severe systemic inflammatory condition and is one of the bases for “cytokine release syndrome”. Several proinflammatory cytokines, including IFNγ and IL6, play a central role in the development of septic shock and activate T cells and NK cells by binding to specific cell receptors (60). The transcription factor, Tbet, enhances IFNγ production in T cells and directs Th1 cell differentiation (61, 62). Tbet-deficient mice are unable to differentiate naive T cells into Th1 cells and produce low amount of IFNγ (62). Therefore, disruption of Tbet expression results in various Th1 cells-associated diseases and provides evidence of a role for Tbet regulation in the innate immune response (63, 64). The inhibition of Arid5a in LPS-treated mice reduces the IL6 levels in serum (15) and results in the reduced frequency of Th1 and Th17 cells in Arid5a−/− mice (16, 17). In a study of Arid5a in immune regulation, Zaman et al. (17) observed that Arid5a-deficient mice are extremely resistant to LPS-administered endotoxic shock. They also found an association with reduced levels of proinflammatory cytokines, including IFNγ and IL6 compared with wild-type counterparts. Mechanistically, Arid5a targets Tbet mRNA and increases the expression of Tbet mRNA by binding to the 3’ UTR, thus attenuating the production of IFNγ. Moreover, Arid5a deficiency decreased the stability of Tbet mRNA due to a lack of Arid5a-mediated post-transcriptional regulation, which resulted in reduced levels of IFNγ in T cells during Th1 cell polarization (Figure 2). Furthermore, Arid5a−/− mice were resistant to Propionibacterium acnes-primed LPS exposure, indicating that Arid5a regulates IFNγ and IL6 by increasing the stability of Tbet and Il6 mRNA transcripts, respectively. The synergistic amplification of several cytokines likely occurs which eventually results in septic shock. Therefore, inhibiting Arid5a expression may represent a useful approach to limit some of pathologies associated with septic shock.
4.3 Obesity by Inhibiting Transcription of Pparγ
Obesity can be inhibited by limiting adipogenesis, which is associated with IL6 expression in mice (19, 65). Arid5a knockout mice exhibit low levels of IL6 because of the absence of Arid5a-mediated Il-6 mRNA post-transcriptional upregulation (15). IL6 also induces Arid5a expression (20, 21). This increases the probability of Arid5a involvement in IL6 inhibition of adipogenesis and obesity. We reported that IL6-stimulation in Arid5a−/− 3T3-L1 adipocytes inhibited the differentiation of adipocytes (19) and increased the possibility of a role for Arid5a in inhibiting adipocyte differentiation. Il6-deficiency in mice resulted in higher adipogenesis and obesity, which result from insufficient Arid5a induction. Following stimulation by IL6, Arid5a further represses Pparγ expression, which ameliorates adipogenesis and obesity. Pparγ is a major regulator of adipogenesis and obesity, and its increased expression is responsible for weight gain in animals and humans (66). Pparγ2 was found to be upregulated in Arid5a−/− 3T3-L1 cells and mouse embryonic fibroblasts (MEFs) at early time points of adipocyte differentiation (19). An immunofluorescence assay revealed that in the early phase of adipocyte differentiation, Pparγ2 promoter activity was higher compared with normal conditions in 3T3-L1 cells; however, increased expression of Arid5a, following transfection of an Arid5a expression vector, significantly reduced Pparγ2 promoter activity. The underlying in vitro mechanism may involve the binding of Arid5a to the Pparγ2 promoter, which inhibited Pparγ2 transcription (19). This binding further impaired the interaction of Pparγ2 with C/EBP, which is another regulator of adipogenesis and eventually limits the adipogenesis process in mice (Figure 1B). However, these findings draw further attention to the role of Arid5a in regulating adipogenesis and obesity and provide further insight into the coordinated activity of Arid5a and other targets, such as Sox9, that are involved in these biological processes (67).
4.4 EAE by Coordination of IL6, Stat3, and Ox40 mRNAs Regulation and Th17 Cell Differentiation
EAE is an animal model of MS, which has been associated with a pathogenic role for Th17 cells and abnormal levels of IL6 and Stat3. Since Arid5a regulates Il6 and Stat3 expression in immune and nonimmune cells, Arid5a deficiency protects mice from developing EAE, in which IL17-producing T cells are markedly decreased (15). Furthermore, Stat3 activation is required for the differentiation of Th17 cells (68–70); however, loss of Arid5a in T cells of Arid5a-deficient mice reduces the expression of Stat3 and Stat3-regulated genes, such as Rora, Rorc, and Batf, resulting in a lower number of Th17 cells (16). Inflammatory CD4+ T cells from the spinal cord of EAE mice express Ox40 (71, 72), which further facilitates IL17 expression. CD4+ T cells from Arid5a−/− mice under Th17 polarizing conditions exhibit decreased expression of Ox40 and are unable to induce IL17 expression. In addition, Stat3 deficiency in CD4+CD62L+ T cells had lower expression of Ox40 under Th17-polarizing conditions. Stat3 increases the survival of CD4+ T cells through Ox40, FasL, and Bcl2 expression in wild-type mice compared with Stat3−/− mice (73). Furthermore, Arid5a targets a stem loop in the Ox40 3’ UTR to enhance mRNA stability. Thus, Arid5a and Stat3 are involved in the regulation of Ox40 expression. Furthermore, adoptive transfer of Arid5a-deficient encephalitogenic CD4+ T cells impaired EAE through the Arid5a/Ox40 axis in CD4+ T cells, and is required for IL17 production and EAE development (Figure 2). Collectively, Stat3 and Ox40 are regulated by Arid5a in CD4+ T cells in which IL6 is equally important. The coordinated activities of these molecules may regulate Th17 signaling.
5 Association of Arid5a in Cancer
An association of Arid5a-regulated molecules, such as IL6, Tbet, Stat3, Ox40, and Pparγ, has been widely studied in various cancers and tumor models. Therefore, it is likely that Arid5a contributes to many processes in cancer cells. Interestingly, recent studies have reported a role for Arid5a in many cancers including glioma, breast, pancreatic, lung, and colorectal cancer (Figure 4).
Figure 4 Functions of AT-rich interactive domain 5A (Arid5a) in cancer. Arid5a regulates various cellular processes via transcriptional and post-transcriptional modifications and is associated with the regulation of various cellular activities, as depicted above.
5.1 Arid5a-Mediated Wnt Signaling in Lung Cancer Reprograms M1-Like Tumor-Associated Macrophages (TAMs) Into M2-Like TAMs
Lung cancer is responsible for the majority of cancer-associated deaths in men and women globally (74). The 5-year survival rate for metastatic lung cancer is reported to be approximately 5%, which is considerably lower compared with other cancers (75). The majority of lung cancer cases cannot be treated with surgery. Therefore, chemotherapy and radiotherapy are used to treat these cases; however, side effects of these therapies disrupt normal tissue homeostasis. Even with immune checkpoint blockade therapies targeting cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and PD-1 in are unable to achieve the expected outcomes because of significant toxicity (76–79). Thus, studies are required to identify new treatment options, which may be achieved through an understanding of other tumor-associated cells in the lung.
A central role of β-catenin-mediated Wnt signaling and transcription of fos-like antigen 2 (Fosl2) and Arid5a in the transition of tumor-inhibiting M1-like TAMs into tumor-promoting M2-like TAMs has recently been identified, which suggests a role for TAM-specific signaling in immune evasion in lung cancer (28) (Figure 3B). Polarization of macrophages (M1/M2) upon stimulation by LPS/IFNγ or IL4 modifies the extrinsic and intrinsic properties of macrophages (80) and enables them to act differently in the TME (81, 82). Alternatively, transcriptional activation Fosl2 and Arid5a inhibition by β-catenin in M2-like TAMs and partially regulation of macrophage gene expression by Tnf and Ccr2 results in the suppression of lung tumorigenesis and metastasis in lung cancer models developed using in vitro-trained TAMs and ex vivo-cultured TAMs isolated from mouse and human lung tumors (Figure 3B). Furthermore, high expression of β-catenin and Fosl2 and low expression of Arid5a were found in a transcriptome analysis of lung cancer cases and were correlated with poor prognosis. This study may provide a strategy for developing new treatment options by targeting β-catenin-dependent gene regulation in M2-like TAMs in lung cancer (28).
5.2 Regulation of Invasion and Metastasis by Arid5a in Breast Cancer
Breast cancer-associated deaths in women are estimated a number of 280,000 new cases annually in the United States (83). Despite advances in diagnosis and treatment, the prevention of tumor recurrence and metastasis are the major challenges to achieving complete remission in breast cancer. The clinical efficacy of gene-targeted therapies is also limited; therefore, there is a need to identify new biomarkers and therapeutic targets.
Abnormal IL6 concentrations in breast cancer is associated with the poor survival of patients, cancer progression, invasion, and metastasis, and is increased with tumor grade (84–86). In addition, lncRNAs regulate gene or protein expression at the transcriptional, post-transcriptional, and post-translational levels (87), and were associated with MAPK signaling pathways in breast cancer (88, 89). IL6 signaling, function, and the development of several disorders resulting from IL6 dysregulation have been implicated in the regulation of lncRNAs. The upregulation of lncRNA-UICC and IL6 signaling in cervical cancer promotes tumorigenesis and metastasis in cervical cancer (90). Analysis of transcriptomic and genomic data revealed that the expression of lncRNA-AU021063 was significantly upregulated in IL6-stimulated 4T1 breast cancer cells. Arid5a−/− 4T1 cells exhibited lower expression of lncRNA-AU021063 in a fluorescent in situ hybridization assay, and was restored following ectopic overexpression of Arid5a in these cells under IL6 signaling. Notably, Arid5a regulates genes and mRNAs at the transcriptional and post-transcriptional level, resulting in the development of inflammation and disease (15, 17, 19). ChIP sequencing coupled with qPCR verified the binding of the Arid5a promoter region to lncRNA-AU021063, which coordinates with Arid5a to increase the transcription of lncRNA-AU021063. In addition, Arid5a deficiency in 4T1 cells decreased the invasion and metastasis of breast cancer cells in in vitro and in vivo studies (20). LncRNA-AU021063 contributed to the regulation of Trib3 protein stability. Trib3 is induced by IL6 in breast cancer cells (91), promotes the malignant behavior of ovarian cancer cells through Mek-Erk signaling (92), and induce cell migration, invasion, and metastasis of liver cancer cells (93). Overexpression of lncRNA-AU021063 in breast cancer cells prolonged the activation of the Mek1/2 and Erk1/2 kinases and increased the production of Trib3 protein under IL6 signaling. Furthermore, inhibiting Trib3 expression in 4T1 cells reduced invasion and metastasis of IL6-treated 4T1 cells. This effect could not be restored by overexpressing lncRNA-AU021063, which suggests that Trib3 upregulation mediated by lncRNA-AU021063 contributes to the invasion and metastasis of breast cancer cells during IL6/Arid5a signaling. This study may lead to strategies for using Arid5a and lncRNA for the treatment of breast cancer.
A database mining study recently assessed the expression of Arid family members including Arid5a based on The Cancer Genome Atlas (TCGA) data and also evaluated the prognostic value of each member in breast cancer patients (94). The mRNA expression of Arid3a, Arid3b, Arid4b, Jarid1b, Jarid1c, and Jarid2 was higher, whereas Arid1b, Arid3c, Arid4a, Arid5a, Arid5b, and Jarid1a mRNA expression was lower in tumor tissues compared with controls. Furthermore, the expression of Arid1a, Arid1b, Arid2, Arid3a, Arid4a, and Arid4b in non-luminal subtypes of breast cancer was lower compared with that in luminal subtypes. Conversely, Arid3c, Arid5a, and Jarid2 mRNA expression was higher in non-luminal subtypes of breast cancer. A Kaplan-Meier analysis revealed that lower expression of Arid5a mRNA was associated with poor overall survival in the luminal type and all breast cancer patients. Based on an analysis of online datasets, Arid5a is considered a good candidate for tumor treatment in breast cancer (94); however, it will be necessity to conduct further studies to provide more experimental evidence.
5.3 Immune Evasion by Arid5a in CRC and PDAC
Immunotherapy has become a better option for the treatment of advanced cancers (95). Although, many patients are resistant to immune checkpoint blockade (ICB) (96), it is useful for the treatment of metastatic tumors, and results in sustained tumor regression. The plasticity and heterogenicity of tumors along with the TME contributes to ICB resistance and immune evasion. Immune evasion is a hallmark of cancer activated by an immune checkpoint pathway, such as PD1-PDL1 signaling, or an increase in immunosuppressive factors (97). CRC and PDAC are considered cold tumors and are non-immunogenic because they have low numbers of tumor infiltrating cells (98, 99). The tumor-intrinsic mesenchymal phenotype of CRC and PDAC expedite immune evasion by interacting with stromal immune cells in the TME (96). The epithelial-to-mesenchymal transition (EMT) process enhances invasion and migration, and the immunosuppressive properties of tumor cells; however, a precise molecular mechanism has not been defined.
Recently, we demonstrated an association of Arid5a with the mesenchymal subtypes, CMS4 and QM, and immune evasion in CRC and PDAC (29). In addition, Arid5a mediates immune evasion by recruiting immunosuppressive cells, such as MDSCs and Tregs, and decreasing antitumor effector T cell recruitment and activation in the TME (29) (Figure 3A). Arid5a−/− tumor cells produced smaller tumors in immunocompetent mice compared with immunodeficient mice, which further supports a role for Arid5a in immune evasion and immunosuppression in the TME. During EMT in human and murine pancreatic cancer lines, higher expression of Arid5a was observed, which is due to induction by EMT-TFs, but not by Stat3 in mesenchymal cells of CRC and PDAC. High expression of Arid5a further correlated positively with TGFβ1 and IL6 levels. As mention above, Ido1 and Ccl2 mRNAs are stabilized by Arid5a. Therefore, increased IDO1 activity facilitates immunosuppression by metabolizing tryptophan and results in deceased effector T cell proliferation and promotes Treg differentiation, and CCL2 recruits MDSCs to the tumor more efficiently (29) (Figure 3A). However, the molecular mechanisms underlying the regulation of Arid5a in mesenchymal tumors remain unclear and these mechanisms will be interest in future studies, especially in targeting Arid5a for tumor immunotherapy.
5.4 Arid5a Regulation of Proliferation and Global Protein Synthesis in Prostate Cancer
Prostate cancer (PCa) is a common neoplasm in the urinary tract of men and results in approximately 260,000 deaths annually worldwide (83). PCa is considered a global health issue because of the increasing number of prostate cancer cases in developing countries (100). Serum prostate-specific antigen (PSA) levels and pathological stage are the major diagnostic criteria for PCa. Tumor recurrence and metastasis following surgery is observed in 30% of PCa patients (101). Some cases develop aggressive castration-resistant PCa (102, 103) after androgen deprivation therapy. Therefore, the identification of new molecular markers is necessary to improve the prognosis and diagnosis of PCa patients. IL6 is highly expressed in tumors and modulates progression by affecting proliferation, apoptosis, angiogenesis, and differentiation (104, 105). IL6 levels are positively correlated with the progression of hormone-refractory and metastatic PCa patients (106–108). IL6 induces androgen-independent cell growth in vitro and in vivo (109–112). Interestingly, a recent analysis of microarray datasets from independent PCa cohorts revealed a significant correlation between the expression of Arid5a and Il6 and further identified the transcriptional upregulation of Il6 by Arid5a in human PCa cell lines, suggesting a complementary role of Arid5a in the regulation of Il6 mRNA stability (113).
The inhibition of Arid5a expression in LNCaP PCa cells reduced global protein synthesis and resulted in lowering proliferation despite dihydrotestosterone (DHT)-stimulation (114). Inhibition of Arid5a facilitated cell cycle arrest at the G1 phase in DHT-stimulated cells and further decreased DHT-induced lipid accumulation. Several biological processes, such as cell survival, proliferation, and cell cycle regulatory proteins, such as HIF1a, cyclin D1, and cyclin D3, were attenuated following inhibition of Arid5a expression, but it did not affect mRNA expression which indicates a translational and/or post-translational role for Arid5a in protein regulation. In LNCaP cells, Arid5a deficiency augmented the phosphorylation of eukaryotic translation initiation factor 2a (eIF2a) by activating general control nonderepressible 2 kinase (GCN2) and RNA-dependent protein kinase (PKR)-like endoplasmic reticulum kinase (PERK) (114). These findings indicate a role for Arid5a in the regulation of proliferation and global protein synthesis in PCa; however, further studies are needed to determine the molecular mechanism of Arid5a-mediated regulation of GCN2 and PERK kinases, tumorigenesis, and tumor progression in PCa.
5.5 Predicting the Diagnostic and Prognostic Significance of Arid5a in Glioma
Glioma is an intracranial malignant primary tumor, which accounts for nearly 27% of central nervous system (CNS) tumors (115). Morbidity, mortality, and recurrence rates are higher in gliomas compared with other CNS tumors (116, 117). Grade 2 and 3 gliomas are considered low-grade gliomas (LGGs) and have characteristics of slow growth and low malignancy, whereas grade 4 glioma, known as glioblastoma multiform (GBM), is an invasive tumor with high recurrence and mortality (118, 119). Because of this, the successful treatment of gliomas is difficult (116) despite advanced treatment options such as surgical resection, postoperative adjuvant radiotherapy, and temozolomide-based chemotherapy. The survival of treated-glioma patients is limited because of the poor prognosis and high recurrence rate (120, 121). Next-generation gene sequencing has identified several molecular markers in gliomas as alternative options for diagnosis and improved prognosis. Two most of these biomarkers include isocitrate dehydrogenase (IDH) mutation and 1p/19q combined deletion, which have been widely used in clinical diagnosis and therapy (122). Therefore, the identification of biomarkers has attracted the interest of researchers and clinicians for determining cancer risk, improving prognosis, and predicting tumor recurrence.
A recent study determined the diagnostic and prognostic significance of Arid5a in gliomas and discovered a possible biological function for Arid5a (30). Low grade gliomas and GBM datasets were analyzed using online database, such as TCGA, and inversely correlated high expression of Arid5a with the prognosis of all stages of gliomas. Moreover, high expression of Arid5a is likely a marker of poor prognosis in glioma. In contrast, low expression of Arid5a was associated with poor prognosis of lung cancer in a previous study (28). Based on datasets from the Chinese Glioma Genome Atlas (CGGA) and TCGA RNA sequencing, as well as clinical and molecular characterization, the expression of Arid5a was increased with higher glioma tumor grade. In addition, Arid5a expression was higher in IDH-wild-type gliomas compared with IDH-mutant gliomas and in gliomas without 1p/19q co-deletion compared with 1p/19q co-deletion in gliomas. Earlier studies reported that wild-type IDH lacks 1p/19q co-deletion and higher tumor grade was associated with poor prognosis in glioma (123, 124). Another study reported high expression of Arid5a expression is associated with complex tumor malignancy (30), suggesting that Arid5a may be a useful marker for the degree of tumor malignancy.
The analysis of glioma samples in the TCGA datasets revealed enrichment of apoptosis, cytokine-cytokine receptor interactions, JAK-STAT signaling, leukocyte transendothelial migration, and toll-like receptor signaling in the high Arid5a expression group. In addition, cell adhesion molecules, ECM receptor interactions, JAK-STAT signaling, leukocyte transendothelial migration, and p53 signaling were also enriched in the high Arid5a expression group of gliomas from CGGA datasets by gene set enrichment analysis. Arid5a regulates Il6 and Stat3 mRNAs (15, 16, 23), which are further associated with the modulation of p53 gene expression and protein degradation (125, 126). Thus, Arid5a may affect the occurrence, development, and clinical prognosis of glioma by regulating the p53 and JAK-STAT signaling pathways in glioma patients, as well as cell apoptosis. Also, high levels of Arid5a expression may regulate the immune response in glioma. A gene ontology analysis suggested a correlation of inflammatory response, immune response, and IFNγ-mediated signaling pathway with higher Arid5a expression in glioma, which may further reveal mechanisms that affect the growth, and proliferation of cancer cells. The dual nature of IFNγ in cancer may increase tumor growth by facilitating an immunosuppressive TME (127). In contrast, it involves the suppression of angiogenesis and enhanced immunogenicity in tumors (128). Furthermore, Arid5a increases Tbet-meditated IFNγ production in CD4+ T cells (17). Therefore, it is likely that Arid5a modulates the IFNγ-mediated signaling pathway to support TME. However, the role of Arid5a in glioma has been primarily based on database mining; therefore, comprehensive studies utilizing cell culture, animal models, and/or patient samples is needed to confirm a regulatory function of Arid5a in glioma.
6 Arid5a Implies Non-Canonical IL6 Signaling in Cancer
Previous studies of Arid5a have indicated an association of Arid5a with canonical IL6 signaling. IL6 induces Arid5a mRNA expression in macrophages and MEFs (15, 21). Furthermore, Arid5a expression is inhibited in Il6-knockdown MEFs activated by LPS exposure (21), although Arid5a mRNA expression cannot be induced in Stat3-knockdown MEFs, despite LPS stimulation. These findings suggest that Stat3 under conditions of canonical IL6 signaling plays an important role in Arid5a expression. Further studies revealed that phosphorylated Stat3 binds to the promoter region of Arid5a, thus increasing its expression. Taken together, IL6-Stat3 signaling has been considered to act synergistically with LPS signaling to induce Arid5a expression in MEFs, suggesting that IL6 regulates Arid5a expression through a positive feedback loop that is regulated by TLR4 signaling (21). Interestingly, Arid5a was subsequently found to post-transcriptionally regulate Stat3 mRNA in Th17 cells (16). Arid5a binds to the stem structure of the Stat3 3’ UTR to stabilize the transcript. Stat3 plays a key role in regulating the fate of naive CD4+ T cells (129, 130), and in Arid5a-deficient mice, the rate at which naive CD4+ T cells differentiate into Th17 cells is reduced because of the lower level of IL17-producing T cells and hence the relatively low expression of IL17A. Accordingly, the level of Stat3 in Arid5a−/− T cells contributes to the impairment of Th17 cell differentiation. In contrast, as discussed earlier, we demonstrated a role for Arid5a in immune evasion by augmenting tryptophan metabolism and chemokine expression in cancer (29). Moreover, Arid5a expression was associated with the mesenchymal phenotypes of CRC and PDAC, and Arid5a is involved in immune evasion by promoting tumor infiltration by MDSCs and Tregs, and by suppressing the recruitment and activation of antitumor effector T cells (Figure 3A). The activation of the IL6/Jak/Stat3 signaling pathway upregulates EMT-TFs, such as Zeb1, Zeb2, Snai1, Snai2, Twist1, Twist2, and Stat3, through which metastasis is enhanced through the induction of EMT (131). Our data also suggest the involvement of Arid5a in inducing the mesenchymal properties of EMT cell lines, such as KPC. However, analysis of PDAC RNAseq data from TCGA and KPC RNA seq revealed that Arid5a expression is significantly correlated with the expression of cytokines associated with EMT induction, such as TGFβ1 and IL6, and of representative EMT-TFs, but not Stat3 (29). This indicates a diversity signaling pathways in cancer models, which are more likely to be different from signaling in MEFs, and other immune cells, such as Th17 and macrophages.
7 Therapeutic Approaches Targeting Arid5a
The inflammatory cytokines such as TNFα and IL1β are produced by the immune cells in response to pathogen attacks on humans. This generated immune response sometimes exacerbates the expression of cytokines, such as IL6, which was observed during inflammation and other disease conditions, including sepsis and influenza (132, 133). The excessive production of IL6 could be dangerous. Blocking or inhibiting IL6 may be a useful treatment in the prevention of IL6-regulated diseases (134). However, an in vivo study suggested that knocking out IL6 in mice was not shown recovery from sepsis (135) owing to the role of IL6 in host defense mechanism from pathogens and tissue damage (136). Therefore, it is likely that the complete elimination of IL6 can be lethal, and it calls for a substitute approach to control IL-6. Arid5a as discussed increases the expression of inflammatory mRNAs including IL6 by increasing their half-life. Targeting Arid5a may limit the production of these inflammatory mediators. No drug designs have been established so far those target endogenous proteins. Cytosolic antibodies against Trim21 that linked to an Fc receptor were observed in degradation of their analogous endogenous target proteins via ubiquitin-proteasome system raising hope to exploit a similar approach to target and degrade endogenous proteins such as Arid5a with antibodies. It could pose to be a potent therapeutic option to balance the action of not only IL6 but also controls inflammation.
Earlier studies have shown Arid5a is able to enhance the production of IFNγ by stabilizing T-bet mRNA (17, 23). Also, Zhou et al. have recently reported upregulation of Arid5a in glioma is positively correlated with inflammation, immune response, IFNγ-mediated signaling, and apoptosis which affect the proliferation and growth of cancer cells. IFNγ has a dual function in cancer immunology. It suppresses proliferation and induces the expression of class I MHC on cancer cells that results in enhancement of immunogenicity, and suppression of angiogenesis in tumors (128). In contrast, IFNγ stimulation can increase tumor growth and accelerate the formation of 5tumor immunosuppressive microenvironment (127). Therefore, controlling IFNγ signaling further aids in the regulation of TME via Arid5a.
Reported that JAK-STAT signaling is critically involved in the regulation of glioma cell survival, growth, and invasion and is known as one of the potential targets for gene therapy (137, 138). As mentioned, Arid5a regulates Il6 and Stat3 mRNAs (15, 16, 23), which are further associated with the modulation of p53 gene expression and protein degradation (125, 126). This suggests that Arid5a may be associated with the JAK-STAT and p53 signaling in tumorigenesis. Moreover, Arid5a may affect the occurrence, development, and clinical prognosis of glioma by regulating the p53 and JAK-STAT signaling pathways in glioma patients, as well as cell apoptosis. In addition, IL-6 promotes Arid5a expression through Stat3 activation (15). Furthermore, Arid5a stabilizes Il6 and Stat3 mRNAs after stimulation by IL6 via a feedback loop, which involves in the pathogenesis of inflammatory autoimmune diseases (15, 16). It is also reported that immature dendritic cells accelerate tumor immune tolerance and promote tumor growth (139). There are other reports showing activation of Stat3 by IL6 can inhibit the maturation and activation of dendritic cells (140), while Arid5a functions are associated with IL6/STAT3 signaling (24). Therefore, it is likely that Arid5a may be implicated with the regulation of the function of dendritic cells that can also be disrupted by inhibiting one or more of its downstream targets. For example, Stat3 inhibitor was shown reductions in inflammation and tumor development (141), which may prompt us to the application of a similar strategy against inflammatory diseases where IL6 is overproduced, such as sepsis and EAE. Several stimuli IL1β, IFNγ, LPS, and OxPAPC (15, 16, 21, 27) have been shown to induce expression of Arid5a which results in IL6 overproduction; therefore, there may be an alternative strategy to suppress Arid5a expression by inhibiting these stimuli would ultimately reduce the IL6 expression and associated diseases. Finally, in-depth research of the expression and function of Arid5a not only in immune-related diseases but also in cancers can provide accurate diagnostic values and reliable prognostic pieces of evidence, and may thus improve the clinical therapeutics of these diseases.
8 Concluding Remarks and Future Directions
The association of Arid5a with inflammation and autoimmune diseases and a variety of cancers has been revealed. The mechanisms underlying these diseases may occur through Arid5a-mediated transcriptional and post-transcriptional regulation of various genes and mRNA transcripts. For example, Arid5a binds to the promoter of Pparγ to suppress transcription, which leads to inhibition of adipogenesis and obesity and to lncRNA-AU021063 to augment invasion and metastasis in breast cancer. It also binds to the 3ʹ UTR of the Il6, Stat3, and Tbet transcripts to enhance stability, which further exacerbates the pathogenesis of EAE and septic shock. This demonstrates the dual function of the Arid5a protein to bind both DNA and RNA to facilitate the pathogenesis of inflammatory and autoimmune diseases and cancer.
Whereas the Il6 gene is transcriptionally upregulated by Arid5a in prostate cancer cells, Arid5a increases the half-life of Il6 mRNA at the post-transcriptional level in immune cells, such as macrophages, which further suggests a cell-specific function of Arid5a. It is likely that IL6 and the diseases in which IL6 is overproduced may be regulated, in part, by Arid5a, and deserves further study. Nonetheless, many of the Arid5a targets identified function as TFs, such as Tbet and Stat3, Ox40 as a co-stimulatory receptor in T cell activation, or IL6 as a cytokine signal. This increases the possibility of the existence of secondary targets that have yet to be identified, although their significance is poorly understood. Certainly, many other such targets will likely be discovered in the future.
Arid5a exhibits multifactorial functions, both beneficial and detrimental, in different disease environments. Some of these involve regulating the innate and adaptive immune response, modulating cell proliferation, Th1 and Th17 cells differentiation, stabilizing mRNAs, regulating TFs, and maintaining cellular homeostasis. Thus, further studies of the Arid5a protein and its interaction with its target molecules, and revealing a relationship between transcriptional and post-transcriptional mechanism mediated by Arid5a, will provide insight into the role of Arid5a in diseases and improve therapeutic and diagnostic methods. Nonetheless, Arid5a controls several biological processes in cancer such as invasion, metastasis, reprogramming M2-like TAMs to M1-like TAMs, immune suppression/immune evasion in the TME, and regulation of lncRNAs. This suggests that Arid5a represents a therapeutic target in cancer. However, an in-depth understanding of Arid5a in the biological processes of cancer and associated TME requires further study. Although Arid5a has emerged as a new target for tumors, inflammation, and immunological diseases, further studies are needed to elucidate the precise functions and underlying molecular mechanisms in patients and in vivo models.
Author Contributions
KN and TK conceptualized and conceived the manuscript. KN drafted the manuscript including literature search, reading and writing. KN and TK edited and critically evaluated the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Kishimoto Foundation and the Advanced Postdoc Program at the Immunology Frontier Research Center, Osaka University, Japan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge Ms. Mari Okawa for her assistance.
Glossary

References
1. Kishimoto T. Interleukin-6: From Basic Science to Medicine-40 Years in Immunology. Annu Rev Immunol (2005) 23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806
2. Legrand-Poels S, Schoonbroodt S, Piette J. in a Colon Cancer Cell Line. Biochem J (2000) 349:765–73. doi: 10.1042/bj3490765
3. Neurath MF, Finotto S. IL-6 Signaling in Autoimmunity, Chronic Inflammation and Inflammation-Associated Cancer. Cytokine Growth Factor Rev (2011) 22:83–9. doi: 10.1016/j.cytogfr.2011.02.003
4. Libermann TA, Baltimore D. Activation of Interleukin-6 Gene Expression Through the NF-Kappa B Transcription Factor. Mol Cell Biol (1990) 10:2327–34. doi: 10.1128/mcb.10.5.2327-2334.1990
5. Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, et al. Transcription Factors NF-IL6 and NF-Kappa B Synergistically Activate Transcription of the Inflammatory Cytokines, Interleukin 6 and Interleukin 8. Proc Natl Acad Sci USA (1993) 90:10193–7. doi: 10.1073/pnas.90.21.10193
6. Masuda K, Kimura A, Hanieh H, Nguyen NT, Nakahama T, Chinen I, et al. Aryl Hydrocarbon Receptor Negatively Regulates LPS-Induced IL-6 Production Through Suppression of Histamine Production in Macrophages. Int Immunol (2011) 23:637–45. doi: 10.1093/intimm/dxr072
7. Yuan YC, Whitson RH, Liu Q, Itakura K, Chen Y. A Novel DNA-Binding Motif Shares Structural Homology to DNA Replication and Repair Nucleases and Polymerases. Nat Struct Biol (1998) 5:959–64. doi: 10.1038/2934
8. Iwahara J, Clubb RT. Solution Structure of the DNA Binding Domain From Dead Ringer, a Sequence-Specific AT-Rich Interaction Domain (ARID). EMBO J (1999) 18:6084–94. doi: 10.1093/emboj/18.21.6084
9. Tu X, Wu J, Xu Y, Shi Y. 1H, 13C and 15N Resonance Assignments and Secondary Structure of ADR6 DNA-Binding Domain. J Biomol NMR (2001) 21:187–8. doi: 10.1023/a:1012434510376
10. Kim S, Zhang Z, Upchurch S, Isern N, Chen Y. Structure and DNA-Binding Sites of the SWI1 AT-Rich Interaction Domain (ARID) Suggest Determinants for Sequence-Specific DNA Recognition. J Biol Chem (2004) 279:16670–6. doi: 10.1074/jbc.M312115200
11. Wilsker D, Patsialou A, Dallas PB, Moran E. ARID Proteins: A Diverse Family of DNA Binding Proteins Implicated in the Control of Cell Growth, Differentiation, and Development. Cell Growth Differ (2002) 13:95–106.
12. Kortschak RD, Tucker PW, Saint R. ARID Proteins Come in From the Desert. Trends Biochem Sci (2000) 25:294–9. doi: 10.1016/s0968-0004(00)01597-8
13. Hurlstone AF, Olave IA, Barker N, van Noort M, Clevers H. Cloning and Characterization of hELD/OSA1, a Novel BRG1 Interacting Protein. Biochem J (2002) 364:255–64. doi: 10.1042/bj3640255
14. Wilsker D, Probst L, Wain HM, Maltais L, Tucker PW, Moran E. Nomenclature of the ARID Family of DNA-Binding Proteins. Genomics (2005) 86:242–51. doi: 10.1016/j.ygeno.2005.03.013
15. Masuda K, Ripley B, Nishimura R, Mino T, Takeuchi O, Shioi G, et al. Arid5a Controls IL-6 mRNA Stability, Which Contributes to Elevation of IL-6 Level In Vivo. Proc Natl Acad Sci USA (2013) 110:9409–14. doi: 10.1073/pnas.1307419110
16. Masuda K, Ripley B, Nyati KK, Dubey PK, Zaman MM, Hanieh H, et al. Arid5a Regulates Naive CD4+ T Cell Fate Through Selective Stabilization of Stat3 mRNA. J Exp Med (2016) 213:605–19. doi: 10.1084/jem.20151289
17. Zaman MM, Masuda K, Nyati KK, Dubey PK, Ripley B, Wang K, et al. Arid5a Exacerbates IFN-γ-Mediated Septic Shock by Stabilizing T-Bet mRNA. Proc Natl Acad Sci USA (2016) 113:11543–8. doi: 10.1073/pnas.1613307113
18. Hanieh H, Masuda K, Metwally H, Chalise JP, Mohamed M, Nyati KK, et al. Arid5a Stabilizes OX40 mRNA in Murine CD4+ T Cells by Recognizing a Stem-Loop Structure in its 3′UTR. Eur J Immunol (2018) 48:593–604. doi: 10.1002/eji.201747109
19. Chalise JP, Hashimoto S, Parajuli G, Kang S, Singh SK, Gemechu Y, et al. Feedback Regulation of Arid5a and Ppar-γ2 Maintains Adipose Tissue Homeostasis. Proc Natl Acad Sci USA (2019) 116:15128–33. doi: 10.1073/pnas.1906712116
20. Nyati KK, Hashimoto S, Singh SK, Tekguc M, Metwally H, Liu YC, et al. The Novel Long Noncoding RNA AU021063, Induced by IL-6/Arid5a Signaling, Exacerbates Breast Cancer Invasion and Metastasis by Stabilizing Trib3 and Activating the Mek/Erk Pathway. Cancer Lett (2021) 520:295–306. doi: 10.1016/j.canlet.2021.08.004
21. Nyati KK, Masuda K, Zaman MM, Dubey PK, Millrine D, Chalise JP, et al. TLR4-Induced NF-κb and MAPK Signaling Regulate the IL-6 mRNA Stabilizing Protein Arid5a. Nucleic Acids Res (2017) 45:2687–703. doi: 10.1093/nar/gkx064
22. Higa M, Oka M, Fujihara Y, Masuda K, Yoneda Y, Kishimoto T. Regulation of Inflammatory Responses by Dynamic Subcellular Localization of RNA-Binding Protein Arid5a. Proc Natl Acad Sci USA (2018) 115:E1214–20. doi: 10.1073/pnas.1719921115
23. Nyati KK, Agarwal RG, Sharma P, Kishimoto T. Arid5a Regulation and the Roles of Arid5a in the Inflammatory Response and Disease. Front Immunol (2019) 10:2790. doi: 10.3389/fimmu.2019.02790
24. Nyati KK, Zaman MM, Sharma P, Kishimoto T. Arid5a, an RNA-Binding Protein in Immune Regulation: RNA Stability, Inflammation, and Autoimmunity. Trends Immunol (2020) 41:255–68. doi: 10.1016/j.it.2020.01.004
25. Metwally H, Tanaka T, Li S, Parajuli G, Kang S, Hanieh H, et al. Noncanonical STAT1 Phosphorylation Expands Its Transcriptional Activity Into Promoting LPS-Induced IL-6 and IL-12p40 Production. Sci Signal (2020) 13:eaay0574. doi: 10.1126/scisignal.aay0574
26. Saito Y, Kagami S, Sanayama Y, Ikeda K, Suto A, Kashiwakuma D, et al. AT-Rich Interactive Domain-Containing Protein 5a Functions as a Negative Regulator of Retinoic Acid Receptor-Related Orphan Nuclear Receptor γt-Induced Th17 Cell Differentiation. Arthritis Rheumatol (2014) 66:1185–94. doi: 10.1002/art.38324
27. Dubey PK, Masuda K, Nyati KK, -Uz Zaman MM, Chalise JP, Millrine D, et al. Arid5a-Deficient Mice are Highly Resistant to Bleomycin-Induced Lung Injury. Int Immunol (2017) 29:79–85. doi: 10.1093/intimm/dxx004
28. Sarode P, Zheng X, Giotopoulou GA, Weigert A, Kuenne C, Günther S, et al. Reprogramming of Tumor-Associated Macrophages by Targeting β-Catenin/Fosl2/Arid5a Signaling: A Potential Treatment of Lung Cancer. Sci Adv (2020) 6:eaaz6105. doi: 10.1126/sciadv.aaz6105
29. Parajuli G, Tekguc M, Wing JB, Hashimoto A, Okuzaki D, Hirata T, et al. Arid5a Promotes Immune Evasion by Augmenting Tryptophan Metabolism and Chemokine Expression. Cancer Immunol Res (2021) 9:862–76. doi: 10.1158/2326-6066.CIR-21-0014
30. Zhou Q, Zhou J, Fan J. Expression and Prognostic Value of Arid5a and its Correlation With Tumor-Infiltrating Immune Cells in Glioma. Front Oncol (2021) 11:638803. doi: 10.3389/fonc.2021.638803
31. Amano K, Hata K, Muramatsu S, Wakabayashi M, Takigawa Y, Ono K, et al. Arid5a Cooperates With Sox9 to Stimulate Chondrocyte-Specific Transcription. Mol Biol Cell (2011) 22:1300–11. doi: 10.1091/mbc.E10-07-0566
32. Huang TH, Oka T, Asai T, Okada T, Merrills BW, Gertson PN, et al. Repression by a Differentiation-Specific Factor of the Human Cytomegalovirus Enhancer. Nucleic Acids Res (1996) 24:1695–701. doi: 10.1093/nar/24.9.1695
33. Pereira B, Billaud M, Almeida R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer (2017) 3:506–28. doi: 10.1016/j.trecan.2017.05.003
34. Jewer M, Findlay SD, Postovit LM. Post-Transcriptional Regulation in Cancer Progression: Microenvironmental Control of Alternative Splicing and Translation. J Cell Commun Signal (2012) 6:233–48. doi: 10.1007/s12079-012-0179-x
35. Georgescu SP, Li JH, Lu Q, Karas RH, Brown M, Mendelsohn ME. Modulator Recognition Factor 1, an AT-Rich Interaction Domain Family Member, is a Novel Corepressor for Estrogen Receptor α. Mol Endocrinol (2005) 19:2491–501. doi: 10.1210/me.2004-0311
36. Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase Essential for Controlling Immune Responses by Regulating mRNA Decay. Nature (2009) 458:1185–90. doi: 10.1038/nature07924
37. Iwasaki H, Takeuchi O, Teraguchi S, Matsushita K, Uehata T, Kuniyoshi K, et al. The Iκb Kinase Complex Regulates the Stability of Cytokine-Encoding mRNA Induced by TLR-IL-1R by Controlling Degradation of REGNASE-1. Nat Immunol (2011) 12:1167–75. doi: 10.1038/ni.2137
38. Porter KE, Turner NA. Cardiac Fibroblasts: At the Heart of Myocardial Remodeling. Pharmacol Ther (2009) 123:255–78. doi: 10.1016/j.pharmthera.2009.05.002
39. Sano M, Fukuda K, Kodama H, Pan J, Saito M, Matsuzaki J, et al. Interleukin-6 Family of Cytokines Mediate Angiotensin II-Induced Cardiac Hypertrophy in Rodent Cardiomyocytes. J Biol Chem (2000) 275:29717–23. doi: 10.1074/jbc.M003128200
40. de Lucia C, Eguchi A, Koch WJ. New Insights in Cardiac Beta-Adrenergic Signaling During Heart Failure and Aging. Front Pharmacol (2018) 9:904. doi: 10.3389/fphar.2018.00904
41. Murray DR, Prabhu SD, Chandrasekar B. Chronic Beta-Adrenergic Stimulation Induces Myocardial Proinflammatory Cytokine Expression. Circulation (2000) 101:2338–41. doi: 10.1161/01.cir.101.20.2338
42. Tanaka S, Imaeda A, Matsumoto K, Maeda M, Obana M, Fujio Y. β2-Adrenergic Stimulation Induces Interleukin-6 by Increasing Arid5a, a Stabilizer of mRNA, Through cAMP/PKA/CREB Pathway in Cardiac Fibroblasts. Pharmacol Res Perspect (2020) 8:e00590. doi: 10.1002/prp2.590
43. Muranski P, Restifo NP. Essentials of Th17 Cell Commitment and Plasticity. Blood (2013) 121:2402–14. doi: 10.1182/blood-2012-09-378653
44. O’Reilly S, Cant R, Ciechomska M, van Laar JM. Interleukin-6: A New Therapeutic Target in Systemic Sclerosis? Clin Transl Immunol (2013) 2:e4. doi: 10.1038/cti.2013.2
45. Dong C. Targeting Th17 Cells in Immune Diseases. Cell Res (2014) 24:901–3. doi: 10.1038/cr.2014.92
46. Masuda K, Kishimoto T. CD5: A New Partner for IL-6. Immunity (2016) 44:720–2. doi: 10.1016/j.immuni.2016.03.011
47. Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels HH, Ori D, et al. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell (2015) 161:1058–73. doi: 10.1016/j.cell.2015.04.029
48. Amatya N, Childs EE, Cruz JA, Aggor FEY, Garg AV, Berman AJ, et al. IL-17 Integrates Multiple Self-Reinforcing, Feed-Forward Mechanisms Through the RNA Binding Protein Arid5a. Sci Signal (2018) 11:eaat4617. doi: 10.1126/scisignal.aat4617
49. Choi PS, Thomas-Tikhonenko A. RNA-Binding Proteins of COSMIC Importance in Cancer. J Clin Invest (2021) 131:e151627. doi: 10.1172/JCI151627
50. van Baren N, Van den Eynde BJ. Tumoral Immune Resistance Mediated by Enzymes That Degrade Tryptophan. Cancer Immunol Res (2015) 3:978–85. doi: 10.1158/2326-6066.CIR-15-0095
51. van den Eynde BJ. Arid5a: A Missing Link Between EMT and Tumoral Immune Resistance. Cancer Immunol Res (2021) 9:854. doi: 10.1158/2326-6066.CIR-21-0432
52. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-Dioxygenase Expression in Human Cancers: Clinical and Immunologic Perspectives. Clin Cancer Res (2011) 17:6985–91. doi: 10.1158/1078-0432.CCR-11-1331
53. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-Dioxygenase is a Critical Resistance Mechanism in Antitumor T Cell Immunotherapy Targeting CTLA-4. J Exp Med (2013) 210:1389–402. doi: 10.1084/jem.20130066
54. Li BH, Garstka MA, Li ZF. Chemokines and Their Receptors Promoting the Recruitment of Myeloid-Derived Suppressor Cells Into the Tumor. Mol Immunol (2020) 117:201–15. doi: 10.1016/j.molimm.2019.11.014
55. Nagarsheth N, Wicha MS, Zou W. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat Rev Immunol (2017) 17:559–72. doi: 10.1038/nri.2017.49
56. Johnson ER, Matthay MA. Acute Lung Injury: Epidemiology, Pathogenesis, and Treatment. J Aerosol Med Pulm Drug Deliv (2010) 23:243–52. doi: 10.1089/jamp.2009.0775
57. Ragaller M, Richter T. Acute Lung Injury and Acute Respiratory Distress Syndrome. J Emerg Trauma Shock (2010) 3:43–51. doi: 10.4103/0974-2700.58663
58. Goldman JL, Sammani S, Kempf C, Saadat L, Letsiou E, Wang T, et al. Pleiotropic Effects of Interleukin-6 in a “Two-Hit” Murine Model of Acute Respiratory Distress Syndrome. Pulm Circ (2014) 4:280–8. doi: 10.1086/675991
59. Saito F, Tasaka S, Inoue K, Miyamoto K, Nakano Y, Ogawa Y, et al. Role of Interleukin-6 in Bleomycin-Induced Lung Inflammatory Changes in Mice. Am J Respir Cell Mol Biol (2008) 38:566–71. doi: 10.1165/rcmb.2007-0299OC
60. Farrar MA, Schreiber RD. The Molecular Cell Biology of Interferon-Gamma and its Receptor. Annu Rev Immunol (1993) 11:571–611. doi: 10.1146/annurev.iy.11.040193.003035
61. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell (2000) 100:655–69. doi: 10.1016/s0092-8674(00)80702-3
62. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct Effects of T-Bet in TH1 Lineage Commitment and IFN-Gamma Production in CD4 and CD8 T Cells. Science (2002) 295:338–42. doi: 10.1126/science.1065543
64. Lazarevic V, Glimcher LH, Lord GM. T-Bet: A Bridge Between Innate and Adaptive Immunity. Nat Rev Immunol (2013) 13:777–89. doi: 10.1038/nri3536
65. Wedell-Neergaard AS, Lehrskov LL, Christensen RH, Legaard GE, Dorph E, Larsen MK, et al. Exercise-Induced Changes in Visceral Adipose Tissue Mass are Regulated by IL-6 Signaling: A Randomized Controlled Trial. Cell Metab (2019) 29:844–55. doi: 10.1016/j.cmet.2018.12.007
66. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. Pparγ Signaling and Metabolism: The Good, the Bad and the Future. Nat Med (2013) 19:557–66. doi: 10.1038/nm.3159
67. Gulyaeva O, Nguyen H, Sambeat A, Heydari K, Sul HS. Sox9-Meis1 Inactivation is Required for Adipogenesis, Advancing Pref-1(+) to PDGFRalpha(+) Cells. Cell Rep (2018) 25:1002–17.e4. doi: 10.1016/j.celrep.2018.09.086
68. Crotty S. T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
69. Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, et al. Cutting Edge: An In Vivo Requirement for STAT3 Signaling in Th17 Development and Th17-Dependent Autoimmunity. J Immunol (2007) 179:4313–7. doi: 10.4049/jimmunol.179.7.4313
70. Ma CS, Deenick EK. Human T Follicular Helper (Tfh) Cells and Disease. Immunol Cell Biol (2014) 92:64–71. doi: 10.1038/icb.2013.55
71. Weinberg AD, Bourdette DN, Sullivan TJ, Lemon M, Wallin JJ, Maziarz R, et al. Selective Depletion of Myelin Reactive T Cells With the Anti-OX40 Antibody Ameliorates Autoimmune Encephalomyelitis. Nat Med (1996) 2:183–9. doi: 10.1038/nm0296-183
72. Weinberg AD. OX40: Targeted Immunotherapy Implications for Tempering Autoimmunity and Enhancing Vaccines. Trends Immunol (2002) 23:102–9. doi: 10.1016/s1471-4906(01)02127-5
73. Oh HM, Yu CR, Golestaneh N, Amadi-Obi A, Lee YS, Eseonu A, et al. STAT3 Protein Promotes T-Cell Survival and Inhibits Interleukin-2 Production Through Up-Regulation of Class O Forkhead Transcription Factors. J Biol Chem (2011) 286:30888–97. doi: 10.1074/jbc.M111.253500
74. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
75. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2019. CA Cancer J Clin (2019) 69:7–34. doi: 10.3322/caac.21551
76. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic Activity of Cytotoxic T Lymphocyte-Associated Antigen 4 Antibody Blockade in Previously Vaccinated Metastatic Melanoma and Ovarian Carcinoma Patients. Proc Natl Acad Sci USA (2003) 100:4712–7. doi: 10.1073/pnas.0830997100
77. Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, et al. Cancer Regression and Autoimmunity Induced by Cytotoxic T Lymphocyte-Associated Antigen 4 Blockade in Patients With Metastatic Melanoma. Proc Natl Acad Sci USA (2003) 100:8372–7. doi: 10.1073/pnas.1533209100
78. Chae YK, Arya A, Iams W, Cruz MR, Chandra S, Choi J, et al. Current Landscape and Future of Dual Anti-CTLA4 and PD-1/PD-L1 Blockade Immunotherapy in Cancer; Lessons Learned From Clinical Trials With Melanoma and Non-Small Cell Lung Cancer (NSCLC). J Immunother Cancer (2018) 6:39. doi: 10.1186/s40425-018-0349-3
79. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol (2018) 8:86. doi: 10.3389/fonc.2018.00086
80. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
81. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages Sequentially Change Their Functional Phenotype in Response to Changes in Microenvironmental Influences. J Immunol (2005) 175:342–9. doi: 10.4049/jimmunol.175.1.342
82. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations Across Individuals and Species. Immunity (2019) 50:1317–34. doi: 10.1016/j.immuni.2019.03.009
83. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
84. Sui X, Tsuji K, Tajima S, Tanaka R, Muraoka K, Ebihara Y, et al. Erythropoietin-Independent Erythrocyte Production: Signals Through Gp130 and C-Kit Dramatically Promote Erythropoiesis From Human CD34+ Cells. J Exp Med (1996) 183:837–45. doi: 10.1084/jem.183.3.837
85. Murakami-Mori K, Taga T, Kishimoto T, Nakamura S. The Soluble Form of the IL-6 Receptor (sIL-6R Alpha) is a Potent Growth Factor for AIDS-Associated Kaposi’s Sarcoma (KS) Cells; the Soluble Form of Gp130 Iis Antagonistic for sIL-6R Alpha-Induced AIDS-KS Cell Growth. Int Immunol (1996) 8:595–602. doi: 10.1093/intimm/8.4.595
86. Mihara M, Moriya Y, Kishimoto T, Ohsugi Y. Interleukin-6 (IL-6) Induces the Proliferation of Synovial Fibroblastic Cells in the Presence of Soluble IL-6 Receptor. Br J Rheumatol (1995) 34:321–5. doi: 10.1093/rheumatology/34.4.321
87. Dykes IM, Emanueli C. Transcriptional and Post-Transcriptional Gene Regulation by Long non-Coding RNA. Genomics Proteomics Bioinf (2017) 15:177–86. doi: 10.1016/j.gpb.2016.12.005
88. Dahariya S, Paddibhatla I, Kumar S, Raghuwanshi S, Pallepati A, Gutti RK. Long non-Coding RNA: Classification, Biogenesis and Functions in Blood Cells. Mol Immunol (2019) 112:82–92. doi: 10.1016/j.molimm.2019.04.011
89. Dashti S, Taherian-Esfahani Z, Kholghi-Oskooei V, Noroozi R, Arsang-Jang S, Ghafouri-Fard S, et al. In Silico Identification of MAPK14-Related lncRNAs and Assessment of Their Expression in Breast Cancer Samples. Sci Rep (2020) 10:8316. doi: 10.1038/s41598-020-65421-2
90. Su K, Zhao Q, Bian A, Wang C, Cai Y, Zhang Y. A Novel Positive Feedback Regulation Between Long Noncoding RNA UICC and IL-6/STAT3 Signaling Promotes Cervical Cancer Progression. Am J Cancer Res (2018) 8:1176–89.
91. Yu JM, Sun W, Wang ZH, Liang X, Hua F, Li K, et al. TRIB3 Supports Breast Cancer Stemness by Suppressing FOXO1 Degradation and Enhancing SOX2 Transcription. Nat Commun (2019) 10:5720. doi: 10.1038/s41467-019-13700-6
92. Wang S, Wang C, Li X, Hu Y, Gou R, Guo Q, et al. Down-Regulation of TRIB3 Inhibits the Progression of Ovarian Cancer via MEK/ERK Signaling Pathway. Canc Cell Int (2020) 20:418. doi: 10.1186/s12935-020-01509-z
93. Hua F, Mu R, Liu J, Xue J, Wang Z, Lin H, et al. TRB3 Interacts With SMAD3 Promoting Tumor Cell Migration and Invasion. J Cell Sci (2011) 124:3235–46. doi: 10.1242/jcs.082875
94. Zhang J, Hou S, You Z, Li G, Xu S, Li X, et al. Expression and Prognostic Values of ARID Family Members in Breast Cancer. Aging (Albany NY) (2021) 13:5621–37. doi: 10.18632/aging.202489
95. Galon J, Bruni D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity (2020) 52:55–81. doi: 10.1016/j.immuni.2019.12.018
96. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
97. Spranger S, Gajewski TF. Mechanisms of Tumor Cell–Intrinsic Immune Evasion. Annu Rev Cancer Biol (2018) 2:213–28. doi: 10.1146/annurev-cancerbio-030617-050606
98. Vonderheide RH. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell (2018) 33:563–9. doi: 10.1016/j.ccell.2018.03.008
99. Galon J, Bruni D. Approaches to Treat Immune Hot, Altered and Cold Tumours With Combination Immunotherapies. Nat Rev Drug Discov (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
100. Zhu Y, Wang HK, Qu YY, Ye DW. Prostate Cancer in East Asia: Evolving Trend Over the Last Decade. Asian J Androl (2015) 17:48–57. doi: 10.4103/1008-682X.132780
101. Tomita N, Uchiyama K, Mizuno T, Imai M, Sugie C, Ayakawa S, et al. Impact of Advanced Radiotherapy Techniques and Dose Intensification on Toxicity of Salvage Radiotherapy After Radical Prostatectomy. Sci Rep (2020) 10:114. doi: 10.1038/s41598-019-57056-9
102. Graham J, Baker M, Macbeth F, Titshall V, Guideline Development Group. Diagnosis and Treatment of Prostate Cancer: Summary of NICE Guidance. BMJ (2008) 336:610–2. doi: 10.1136/bmj.39498.525706.AD
103. Wong YNS, Ferraldeschi R, Attard G, de Bono J. Evolution of Androgen Receptor Targeted Therapy for Advanced Prostate Cancer. Nat Rev Clin Oncol (2014) 11:365–76. doi: 10.1038/nrclinonc.2014.72
104. Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of Interleukin-6 in Cancer Progression and Therapeutic Resistance. Tumour Biol (2016) 37:11553–72. doi: 10.1007/s13277-016-5098-7
105. Bharti R, Dey G, Mandal M. Cancer Development, Chemoresistance, Epithelial to Mesenchymal Transition and Stem Cells: A Snapshot of IL-6 Mediated Involvement. Cancer Lett (2016) 375:51–61. doi: 10.1016/j.canlet.2016.02.048
106. Nguyen DP, Li J, Tewari AK. Inflammation and Prostate Cancer: The Role of Interleukin 6 (IL-6). BJU Int (2014) 113:986–92. doi: 10.1111/bju.12452
107. Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma Levels of Interleukin-6 and its Soluble Receptor are Associated With Prostate Cancer Progression and Metastasis. Urology (2001) 58:1008–15. doi: 10.1016/s0090-4295(01)01405-4
108. George DJ, Halabi S, Shepard TF, Sanford B, Vogelzang NJ, Small EJ, et al. The Prognostic Significance of Plasma Interleukin-6 Levels in Patients With Metastatic Hormone-Refractory Prostate Cancer: Results From Cancer and Leukemia Group B 9480. Clin Cancer Res (2005) 11:1815–20. doi: 10.1158/1078-0432.CCR-04-1560
109. Lee SO, Lou W, Hou M, de Miguel F, Gerber L, Gao AC. Interleukin-6 Promotes Androgen-Independent Growth in LNCaP Human Prostate Cancer Cells. Clin Cancer Res (2003) 9:370–6.
110. Gu L, Talati P, Vogiatzi P, Romero-Weaver AL, Abdulghani J, Liao Z, et al. Pharmacologic Suppression of JAK1/2 by JAK1/2 Inhibitor AZD1480 Potently Inhibits IL-6-Induced Experimental Prostate Cancer Metastases Formation. Mol Cancer Ther (2014) 13:1246–58. doi: 10.1158/1535-7163.MCT-13-0605
111. Duscharla D, Reddy Kami Reddy KRK, Dasari C, Bhukya S, Ummanni R. Interleukin-6 Induced Overexpression of Valosin-Containing Protein (VCP)/p97 is Associated With Androgen-Independent Prostate Cancer (AIPC) Progression. J Cell Physiol (2018) 233:7148–64. doi: 10.1002/jcp.26639
112. Handle F, Puhr M, Schaefer G, Lorito N, Hoefer J, Gruber M, et al. The STAT3 Inhibitor Galiellalactone Reduces IL6-Mediated AR Activity in Benign and Malignant Prostate Models. Mol Cancer Ther (2018) 17:2722–31. doi: 10.1158/1535-7163.MCT-18-0508
113. Ikeuchi W, Wakita Y, Zhang G, Li C, Itakura K, Yamakawa T. AT-Rich Interaction Domain 5A Regulates the Transcription of Interleukin-6 Gene in Prostate Cancer Cells. Prostate (2021) 82:97–106. doi: 10.1002/pros.24251
114. Sun C, Chesnokov V, Itakura K. Knockdown Arid5a Suppresses Proliferation of LNCaP Prostate Cancer Cell Through the Inhibition of Global Protein Synthesis. Proc Am Assoc Cancer Res Annu Meet; Cancer Res (2017) 77(13 Suppl). doi: 10.1158/1538-7445.AM2017-4489
115. Miller JJ, Shih HA, Andronesi OC, Cahill DP. Isocitrate Dehydrogenase-Mutant Glioma: Evolving Clinical and Therapeutic Implications. Cancer (2017) 123:4535–46. doi: 10.1002/cncr.31039
116. Picca A, Berzero G, Di Stefano AL, Sanson M. The Clinical Use of Idh1 and Idh2 Mutations in Gliomas. Expert Rev Mol Diagn (2018) 18:1041–51. doi: 10.1080/14737159.2018.1548935
117. Siegelin MD, Schneider E, Westhoff MA, Wirtz CR, Karpel-Massler G. Current State and Future Perspective of Drug Repurposing in Malignant Glioma. Semin Cancer Biol (2021) 68:92–104. doi: 10.1016/j.semcancer.2019.10.018
118. Huang SP, Chan YC, Huang SY, Lin YF. Overexpression of Psat1 Gene is a Favorable Prognostic Marker in Lower-Grade Gliomas and Predicts a Favorable Outcome in Patients With Idh1 Mutations and Chromosome 1p19q Codeletion. Cancers (Basel) (2019) 12:13. doi: 10.3390/cancers12010013
119. Wang H, Wang X, Xu L, Zhang J, Cao H. Prognostic Significance of Age-Related Genes in Patients With Lower Grade Glioma. J Cancer (2020) 11:3986–99. doi: 10.7150/jca.41123
120. Liang T, Wang X, Wang F, Feng E, You G. Galectin-9: A Predictive Biomarker Negatively Regulating Immune Response in Glioma Patients. World Neurosurg (2019) 132:e455–62. doi: 10.1016/j.wneu.2019.08.117
121. Ludwig K, Kornblum HI. Molecular Markers in Glioma. J Neurooncol (2017) 134:505–12. doi: 10.1007/s11060-017-2379-y
122. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
123. Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM, et al. Glioma. Nat Rev Dis Primers (2015) 1:15017. doi: 10.1038/nrdp.2015.17
124. Di C, Mladkova N, Lin J, Fee B, Rivas M, Chunsheng K, et al. AJAP1 Expression Modulates Glioma Cell Motility and Correlates With Tumor Growth and Survival. Int J Oncol (2018) 52:47–54. doi: 10.3892/ijo.2017.4184
125. Brighenti E, Calabrese C, Liguori G, Giannone FA, Trerè D, Montanaro L, et al. Interleukin 6 Downregulates P53 Expression and Activity by Stimulating Ribosome Biogenesis: A New Pathway Connecting Inflammation to Cancer. Oncogene (2014) 33:4396–406. doi: 10.1038/onc.2014.1
126. Niu G, Wright KL, Ma Y, Wright GM, Huang M, Irby R, et al. Role of Stat3 in Regulating P53 Expression and Function. Mol Cell Biol (2005) 25:7432–40. doi: 10.1128/MCB.25.17.7432-7440.2005
127. Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual Faces of Ifnγ in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin Cancer Res (2016) 22:2329–34. doi: 10.1158/1078-0432.CCR-16-0224
128. Farhood B, Najafi M, Mortezaee K. CD8+ Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J Cell Physiol (2019) 234:8509–21. doi: 10.1002/jcp.27782
129. O’Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic Views of STAT Function in CD4+ T Helper Cell Differentiation. Nat Rev Immunol (2011) 11:239–50. doi: 10.1038/nri2958
130. Hunter CA, Jones SA. IL-6 as a Keystone Cytokine in Health and Disease. Nat Immunol (2015) 16:448–57. doi: 10.1038/ni.3153
131. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell (2017) 168:670–91. doi: 10.1016/j.cell.2016.11.037
132. Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, et al. Endothelial Cells are Central Orchestrators of Cytokine Amplification During Influenza Virus Infection. Cell (2011) 146:980–91. doi: 10.1016/j.cell.2011.08.015
133. Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, et al. The TLR4 Antagonist Eritoran Protects Mice From Lethal Influenza Infection. Nature (2013) 497:498–502. doi: 10.1038/nature12118
134. Riedemann NC, Neff TA, Guo RF, Bernacki KD, Laudes IJ, Sarma JV, et al. Protective Effects of IL-6 Blockade in Sepsis are Linked to Reduced C5a Receptor Expression. J Immunol (2003) 170:503–7. doi: 10.4049/jimmunol.170.1.503
135. Remick DG, Bolgos G, Copeland S, Siddiqui J. Role of Interleukin-6 in Mortality From and Physiologic Response to Sepsis. Infect Immun (2005) 73:2751–7. doi: 10.1128/IAI.73.5.2751-2757.2005
136. Narazaki M, Kishimoto T. The Two-Faced Cytokine IL-6 in Host Defense and Diseases. Int J Mol Sci (2018) 19:E3528. doi: 10.3390/ijms19113528
137. Gu J, Li G, Sun T, Su Y, Zhang X, Shen J, et al. Blockage of the STAT3 Signaling Pathway With a Decoy Oligonucleotide Suppresses Growth of Human Malignant Glioma Cells. J Neurooncol (2008) 89:9–17. doi: 10.1007/s11060-008-9590-9
138. Stechishin OD, Luchman HA, Ruan Y, Blough MD, Nguyen SA, Kelly JJ, et al. On-Target JAK2/STAT3 Inhibition Slows Disease Progression in Orthotopic Xenografts of Human Glioblastoma Brain Tumor Stem Cells. Neuro Oncol (2013) 15:198–207. doi: 10.1093/neuonc/nos302
139. Ma J, Usui Y, Takeuchi M, Okunuki Y, Kezuka T, Zhang L, et al. Human Uveal Melanoma Cells Inhibit the Immunostimulatory Function of Dendritic Cells. Exp Eye Res (2010) 91:491–9. doi: 10.1016/j.exer.2010.06.025
140. de Haas N, de Koning C, di Blasio S, Flórez-Grau G, de Vries IJM, Hato SV. STAT Family Protein Expression and Phosphorylation State During moDC Development Is Altered by Platinum-Based Chemotherapeutics. J Immunol Res (2019) 2019:7458238. doi: 10.1155/2019/7458238
Keywords: Arid5a, cancer, inflammation, IL6, immune regulation, mRNA stability
Citation: Nyati KK and Kishimoto T (2022) Recent Advances in the Role of Arid5a in Immune Diseases and Cancer. Front. Immunol. 12:827611. doi: 10.3389/fimmu.2021.827611
Received: 02 December 2021; Accepted: 31 December 2021;
Published: 19 January 2022.
Edited by:
Vijay Kumar, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Moutih Rafei, Université de Montréal, CanadaPhilip Cohen, Temple University, United States
Copyright © 2022 Nyati and Kishimoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kishan Kumar Nyati, nyati15@ifrec.osaka-u.ac.jp; nyati15@gmail.com; Tadamitsu Kishimoto, kishimoto@ifrec.osaka-u.ac.jp
†Present address: Kishan Kumar Nyati, Arthritis and Clinical Immunology Program, Oklahoma Medical Research Foundation, Oklahoma City, OK, United States