 Anuvinder Kaur1
Anuvinder Kaur1 Muhammad Suleman Riaz1
Muhammad Suleman Riaz1 Valarmathy Murugaiah1
Valarmathy Murugaiah1 Praveen Mathews Varghese1
Praveen Mathews Varghese1 Shiv K. Singh2
Shiv K. Singh2 Uday Kishore1*
Uday Kishore1*
- 1Biosciences, College of Health and Life Sciences, Brunel University London, Uxbridge, United Kingdom
- 2Department of Gastroenterology and Gastrointestinal Oncology, University Medical Center, Goettingen, Germany
Human surfactant protein D (SP-D) is a potent innate immune molecule, which is emerging as a key molecule in the recognition and clearance of altered and non-self targets. Previous studies have shown that a recombinant fragment of human SP-D (rfhSP-D) induced apoptosis via p53-mediated apoptosis pathway in an eosinophilic leukemic cell line, AML14.3D10. Here, we report the ability of rfhSP-D to induce apoptosis via TNF-α/Fas-mediated pathway regardless of the p53 status in human pancreatic adenocarcinoma using Panc-1 (p53mt), MiaPaCa-2 (p53mt), and Capan-2 (p53wt) cell lines. Treatment of these cell lines with rfhSP-D for 24 h caused growth arrest in G1 cell cycle phase and triggered transcriptional upregulation of pro-apoptotic factors such as TNF-α and NF-κB. Translocation of NF-κB from the cytoplasm into the nucleus of pancreatic cancer cell lines was observed via immunofluorescence microscopy following treatment with rfhSP-D as compared to the untreated cells. The rfhSP-D treatment caused upregulation of pro-apoptotic marker Fas, as analyzed via qPCR and western blot, which then triggered caspase cascade, as evident from cleavage of caspase 8 and 3 analyzed via western blot at 48 h. The cell number following the rfhSP-D treatment was reduced in the order of Panc-1 (~67%) > MiaPaCa-2 (~60%) > Capan-2 (~35%). This study appears to suggest that rfhSP-D can potentially be used to therapeutically target pancreatic cancer cells irrespective of their p53 phenotype.
Introduction
Human surfactant protein D (SP-D), a member of soluble C-type lectin family called Collectins, plays a vital role in linking the innate and adaptive immunity to protect against infection, allergy, and inflammation (1). Although its homeostatic role in lungs has been widely studied, its specific functions at extra-pulmonary tissues such as kidney, human trachea, brain, testis, heart, prostate, kidneys, and pancreas are poorly understood (1–3). SP-D deficiency in animal models has been shown to be associated with considerable pathophysiological consequences (4–6). SP-D gene knockout mice showed chronic inflammation and fibrosis due to accumulation of surfactant phospholipids in the lungs, monocytes infiltration, and activation of pro-inflammatory alveolar macrophages (5, 6). The absence of SP-D in children makes them more susceptible to frequent pneumonia as compared to SP-D sufficient children (7). SFTPD (SP-D gene) polymorphisms increase the susceptibility to chronic and infectious lung diseases (8), pneumococcal lung disease (9), emphysema (10), tuberculosis (11, 12), Crohn’s disease, and ulcerative colitis (12).
SP-D has been shown to be a potent innate immune molecule at pulmonary as well as extra-pulmonary mucosal surfaces by virtue of its ability to control inflammatory response and helper T cell polarization (3). The first clue came via a murine model of allergic hypersensitivity, when therapeutic treatment with a recombinant fragment of human SP-D (rfhSP-D) lowered peripheral and pulmonary eosinophilia, in addition to specific IgE levels and Th2 cytokines in the spleen (13, 14). It turned out that rfhSP-D selectively induced apoptosis in sensitized eosinophils derived from allergic patients (15). Using an eosinophilic cell line, AML14.3D10 (a model cell line for leukemia), it was established, via proteomics analysis, that apoptosis induction by rfhSP-D involved upregulation of p53 (16, 17). Another crucial study by Pandit et al. (18) revealed that rfhSP-D was able to induce apoptosis in activated human PBMCs, but not in resting, non-activated PBMCs. These studies, for the first time, raised the possibility that SP-D can have a function of immune surveillance against activated self and perhaps altered self. Recently, human lung adenocarcinoma cells (A549 cell line), when exogenously treated with SP-D, showed suppressed epidermal growth factor (EGF) signaling by reducing the EGF binding to EGFR, which subsequently reduced the cell proliferation, invasion, and migration of cancer cells (19).
Here, we set out to examine a possible pro-apoptotic role of SP-D in pancreatic cancer. Pancreatic cancer is the fourth leading cause of cancer-related mortality in the western world (20, 21) and its 5-year survival rate is ~5% (22). The poor prognosis has been attributed to the silent nature of the tumor in early stages, aggressive phenotype, surgical complications, and lack of targeted efficacious therapies (23). In this study, we show that rfhSP-D, composed of 8 Gly-X-Y repeats, homotrimeric neck and carbohydrate recognition domains (CRDs) (1), induces cell growth arrest in G1 phase and subsequent apoptosis in human pancreatic adenocarcinoma cells using Panc-1, MiaPaCa-2, and Capan-2 cell lines. The apoptosis induction appears to involve TNF-α, NF-κB, and Fas axis, revealing a p53 independent route of apoptosis induction in the p53 mutated Panc-1 and MiaPaCa-2 cell lines and p53-dependent apoptosis in p53 wild type Capan-2 cell line by rfhSP-D.
Materials and Methods
Cell Culture and Treatments
Human pancreatic cancer cells lines, Panc-1 (CRL-1469), MiaPaCa-2 (CRL-1420), and Capan-2 (HTB-80), were obtained from ATCC and used as an in vitro model in this study. All cell lines were cultured at 37°C under 5% v/v CO2 using DMEM-F12 media (Thermo Fisher) containing 10% v/v fetal calf serum with 2 mM l-glutamine, and penicillin (100 U/ml)/streptomycin (100 µg/ml) (Thermo Fisher) until 80–90% confluency was reached.
Expression and Purification of rfhSP-D
Plasmid pUK-D1 (containing cDNA sequences for 8 Gly-X-Y repeats, neck, and CRD region of human SP-D), transformed into Escherichia coli BL21 (λDE3) pLysS (Invitrogen), was used to express rfhSP-D, as described earlier (15, 16). The expression cassette included a short stretch of eight N-terminal Gly–X–Y triplets with substitution of S for P in position 2 (residue 180), followed by the α-helical coiled-coil neck region (residues 203–235) and the globular CRD region (residues 236–355). Endotoxin levels were determined using the QCL-1000 Limulus amebocyte lysate system (Lonza) and the assay was found to be linear over a range of 0.1–1.0 EU/ml (10 EU = 1 ng of endotoxin). The amount of endotoxin levels were <4 pg/μg of the rfhSP-D. Full length native SP-D (FL-SP-D) was purified form lung washings of alveolar proteinosis patients using methods previously described by Strong et al. (24).
Fluorescence Microscopy
All cell lines used in this study (Panc-1, MiaPaCa-2, and Capan-2) were grown on coverslips using 0.5 × 105 cells overnight. Next day, cells were washed three times with PBS before being incubated with rfhSP-D (20 µg/ml) in a serum-free DMEM-F12 medium. For rfhSP-D and FL-SP-D binding analysis, the coverslips were incubated for 1 h with mouse anti-human SP-D (rfhSP-D) and rabbit anti-human SP-D (FL-SP-D) (MRC Immunochemistry Unit, Oxford; 1:200), followed by goat anti-mouse IgG H&L (Cy5) and Goat anti-Rabbit IgG H&L Alexa Fluor 488 (1:500; Abcam), respectively, and Hoechst (1:10,000; Thermo Fisher) for fluorescence microscopy analysis. For apoptosis analysis via fluorescence microscopy using an FITC annexin V apoptosis detection kit with propidium iodide (PI) (BioLegend), the cells were incubated with rfhSP-D (20 µg/ml) for 48 h. After 48 h, the cells were incubated with annexin V binding buffer containing FITC annexin V (1:200), PI (1:200), and Hoechst (1:10,000) for 15 min, and washed twice with PBS before mounting on the slides to visualize under a HF14 Leica DM4000 microscope.
Flow Cytometry
Cell lines were plated in a 6-well plate (0.1 × 107) and incubated with rfhSP-D (20 µg/ml), FL-SP-D (10 and 20 µg/ml), and an untreated control, for 24 and 48 h, followed by cell detachment using 5 mM EDTA, pH 8, and centrifugation at 1,200 × g for 5 min. For cell cycle analysis, the cells were fixed in 70% v/v ethanol for 30 min at 4°C, followed by PBS wash twice at 850 × g. The cells were then treated with ribonuclease (100 µg/ml) to ensure DNA staining without RNA contamination before staining with PI (50 µg/ml). 10,000 cells were then acquired for both treated and untreated samples and the PI histograms were plotted using the set markers within the analysis program of Novocyte Flow Cytometer. For apoptosis analysis via FACS, FITC annexin V apoptosis detection kit with PI (BioLegend) was used, as per manufacturer’s instructions. Compensation parameters were acquired using unstained, untreated FITC stained, and untreated PI stained cells.
MTT Assay
MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) (Thermo Fisher) assay was performed by incubating pancreatic cancer cells (0.1 × 105) in a 96-well microtiter plate with rfhSP-D, FL-SP-D (10 and 20 µg/ml), and an untreated control in serum-free DMEM-F12 medium for 48 h, followed by incubation with 50 µg/µl MTT (5 mg/ml stock) per well for 4 h at 37°C. Majority of the media was removed leaving behind 25 µl per well, which was mixed thoroughly with 50 µl of dimethyl sulfoxide and incubated for another 10 min at 37°C. The absorbance was read at 570 nm using a plate reader.
Western Blot
Cell lines (0.1 × 107 cells) were seeded in a 6-well plate (Nunc) and incubated with rfhSP-D (20 µg/ml), together with an untreated control, in a serum-free DMEM-F12 medium. The cells were lysed within the wells using treatment buffer (50 mM Tris-HCl pH 6.8, 2% v/v β-mercaptoethanol, 2% v/v SDS, 0.1% w/v bromophenol blue, and 10% v/v glycerol) and transferred to pre-cooled microcentrifuge tubes followed by sonication for 15 s. The samples were heated at 100°C for 10 min and subjected to SDS-PAGE (12% w/v) for 90 min at 120 V. The SDS-PAGE separated proteins were then electrophoretically transferred onto a nitrocellulose membrane (Thermo Fisher) using an iBLOT (Thermo Fisher). The membrane was then blocked using 5% w/v dried milk powder (Sigma) in 100 ml PBS for 2 h on a rotatory shaker at room temperature. The membrane was incubated with rabbit anti-human caspase primary antibodies (anti-cleaved caspase 3; anti-cleaved caspase 8; Cell Signaling) at 4°C overnight, followed by incubation with secondary Goat anti-rabbit IgG HRP-conjugate (1:1,000; Promega) for 1 h at room temperature. The membrane was washed with PBST (PBS + 0.05% Tween 20) three times, 10 min each time. The color was developed using 3,3′-diaminobenzidine substrate kit (Thermo Fisher).
Quantitative RT-PCR
Panc-1, MiaPaCa-2, and Capan-2 cells were incubated with and without rfhSP-D (20 µg/ml) for various time points. The cell pellet for each time-point was centrifuged and stored at −80°C. RNA was extracted using GenElute Mammalian Total RNA Purification Kit (Sigma-Aldrich, UK), as per manufacturer’s instructions, followed by treatment with DNase I (Sigma-Aldrich, UK). The absorbance at 260 and 260:280 nm ratio was used to determine the concentration and purity of total RNA, respectively, using NanoDrop 2000/2000c (Thermo-Fisher Scientific). Total RNA (2 µg) was used for cDNA synthesis using High Capacity RNA to cDNA Kit (Applied Biosystems). The forward and reverse primers used in this study were designed using the web based Basic Local Alignment Search Tool and Primer-BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) are given in Table 1.
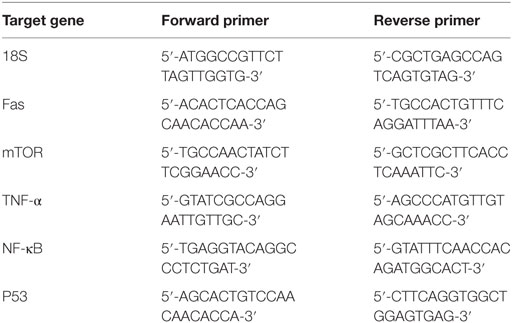
Table 1. Target genes and terminal primers used in the qPCR analysis.
Relative mRNA expression was determined by qPCR reactions performed in triplicates consisting of 10 µl final volume per well [5 µl Power SYBR Green MasterMix (Applied Biosystems), 75 nM of forward and reverse primers, and 500 ng cDNA], using the 7900HT Fast Real-Time PCR System (Applied Biosystems). Samples were initially incubated at 50°C (2 min) and 95°C (10 min), followed by 40 cycles (each cycle for 15 s at 95°C and 1 min at 60°C) for amplification of the template. Human 18S rRNA, an endogenous control, was used to normalize the gene expression. Relative quantification (RQ) value and formula: RQ = 2−ΔΔCt was used to calculate the relative expression of each target.
Statistical Analysis
Graphs were made and statistically analyzed using Graphpad Prism 6.0 by applying an unpaired two-way ANOVA test. Significance of values is based on *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 between treated and untreated samples. Error bars represent the SD or SEM, as indicated in the figure legends.
Results
rfhSP-D Binds to a Range of Pancreatic Cell Lines
The fluorescence microscopy analysis of rfhSP-D and FL-SP-D binding to Panc-1, MiaPaCa-2, and Capan-2 cells revealed its membrane localization following 1 h incubation at 4°C (Figure 1). The rfhSP-D probed with mouse anti-human SP-D-CY5 antibody and FL-SP-D probed with rabbit anti-human SP-D-FITC appeared evenly bound in clusters on the cell membrane, along with nucleus stained positively with Hoechst. All cell lines showed a similar rfhSP-D and FL-SP-D binding pattern. No CY5 or FITC fluorescence was detected in the untreated controls, probed with primary and secondary antibodies, for each cell line, suggesting the rfhSP-D and FL-SP-D binding observed in the treated cell lines was protein-specific.
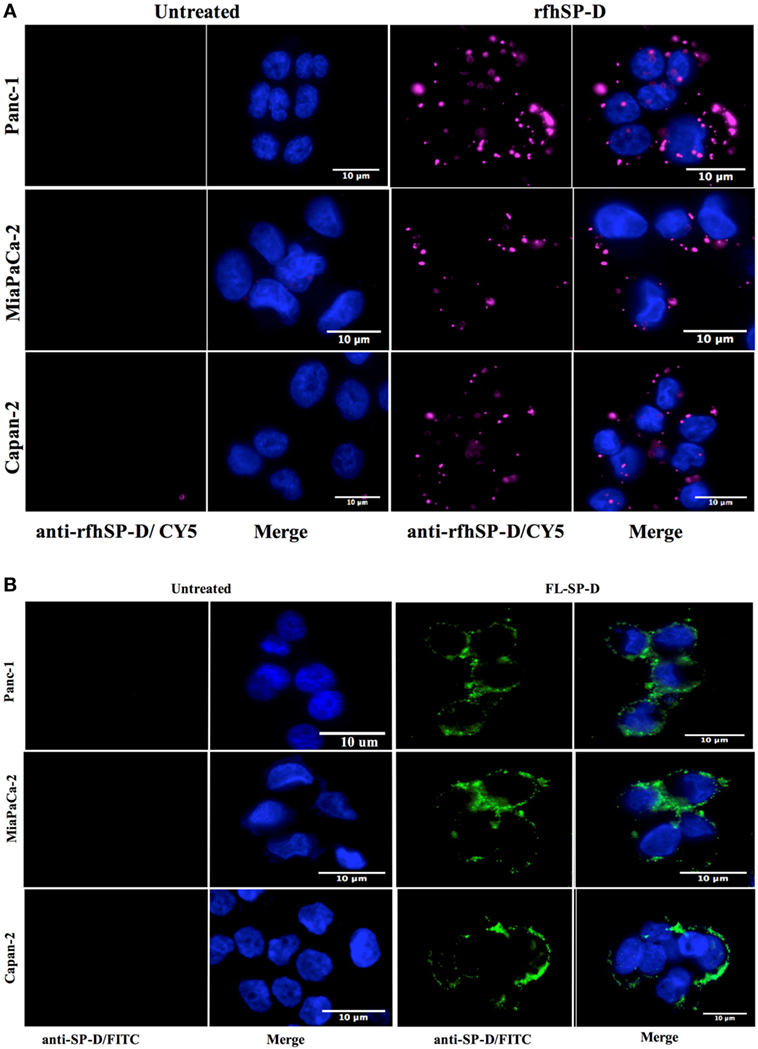
Figure 1. (A) Fluorescence microscopy showing binding of rfhSP-D and (B) FL-SP-D (10 µg/ml; 1 h incubation) to Panc-1, MiaPaCa-2, and Capan-2 cells. The nucleus of the cells was stained with Hoechst. Cells were probed with mouse anti-human SP-D/CY5 (rfhSP-D) and rabbit anti-human/FITC (FL-SP-D); the bound proteins are visible on the cell membrane in the treated cells. No CY5 or FITC fluorescence was detected in the untreated control cells.
rfhSP-D Induces Cell Cycle Arrest in G1 Phase in Panc-1 and MiaPaCa-2
Panc-1, MiaPaCa-2, and Capan-2 cell lines were individually treated with rfhSP-D for 24 h to assess whether rfhSP-D induced growth arrest. DNA binding dye, PI, was used to analyze the cell cycle for both treated and untreated cells via DNA quantitation. rfhSP-D induced inhibition of DNA synthesis in treated Panc-1 (68%) and MiaPaCa-2 (50%) in comparison to untreated Panc-1 (3%) and MiaPaCa-2 (2%) cells, respectively, as the cells were arrested in G1 phase (Figure 2). DNA synthesis was unaffected in the untreated cells for both cell lines since Panc-1 (43%) and MiaPaCa-2 (31%) were seen in S phase and Panc-1 (32%) and MiaPaCa-2 (33%) in the G2 phase of cell cycle. The growth arrest was, however, not seen in Capan-2 cell line following the rfhSP-D treatment (data not shown). Growth arrest at 24 h following rfhSP-D treatment prompted the determination of cell fate at a later time point; therefore, all cell lines were analyzed for likely apoptosis at 48 h.
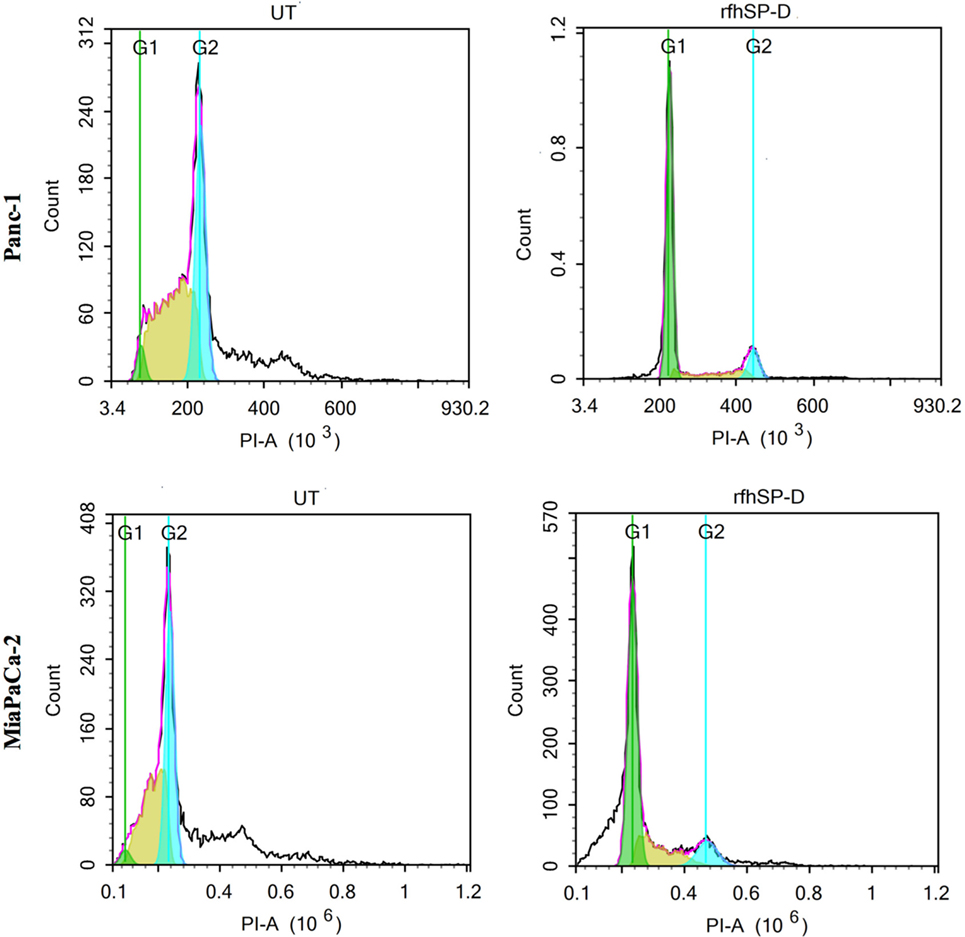
Figure 2. Cell cycle analysis following 24 h treatment of pancreatic cancer cell lines with rfhSP-D. Propidium iodide (PI) was used to stain DNA. PI histograms were plotted using set markers within the analysis program of Novocyte Flow cytometer. The rfhSP-D treated pancreatic cancer cells show arrest in G1 phase in the case of Panc-1 (G1 phase: 68%; S phase: 13%; G2 phase: 11%) and MiaPaCa-2 (G1 phase: ~50%; S phase: 17%; G2 phase: 10%) cell line at 24 h, whereas untreated Panc-1 cells (G1 phase: 3%; S phase: 42%; G2 phase: 32%) and MiaPaCa-2 cells (G1 phase: 2%; S phase: 32%; G2 phase: 33%) progressed to the next cell cycle phases.
rfhSP-D Induces Apoptosis Induction in Pancreatic Cancer Cells by 48 h
The qualitative apoptosis analysis of Panc-1, MiaPaCa-2, and Capan-2 treated with FL-SP-D or rfhSP-D for 48 h using immunofluorescence microscopy (Figure 3A) showed that the cell membrane was disoriented and the PI bound to DNA in the treated cells as compared to untreated cells, where no florescence was detected, indicating that cells were undergoing apoptosis at 48 h.
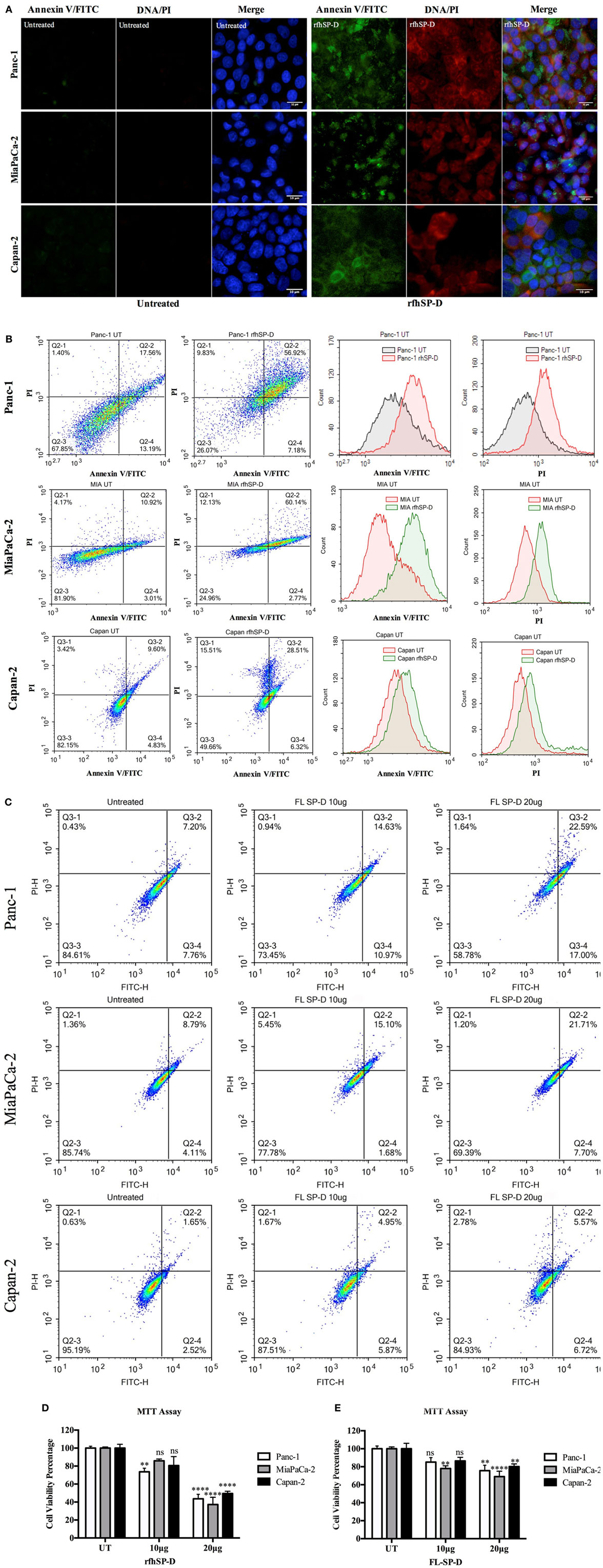
Figure 3. (A) Fluorescence microscopy to analyze apoptosis in pancreatic cancer cell lines following treatment with rfhSP-D. Cells were treated with rfhSP-D for 48 h and apoptosis was analyzed using an annexin V/propidium iodide (PI) staining kit. The cell membrane was positively stained for annexin V and the DNA staining is visible in the treated cells indicating that the cells underwent apoptosis turning the membrane inside out, thus making phosphatidylserine available for annexin V binding; due to the porous membrane, PI was taken in which stained the DNA of apoptotic cells. No such staining was seen in the untreated cells. The nucleus was stained with Hoechst for both treated and untreated cells. (B,C) Quantitative analysis of apoptosis using Flow Cytometer. Cells were treated with rfhSP-D or FL-SP-D for 48 h and apoptosis was analyzed using annexin V with PI kit. 10,000 cells were acquired and plotted for both annexin V/FITC and DNA/PI staining, which showed a shift in the fluorescence intensity of both FITC and PI between treated and untreated cells. Approximately 67% of Panc-1 cells, ~60% MiaPaCa-2 cells, and ~35% Capan-2 cells underwent apoptosis following rfhSP-D treatment and ~25% Panc-1 and MiaPaCa-2 cells following FL-SP-D treatment as compared to untreated cells. No significant difference was seen in Capan-2 cells following FL-SP-D treatment. (D,E) MTT assay to assess cell viability following treatment with rfhSP-D and FL-SP-D (10 and 20 µg/ml) and untreated for 48 h (±SEM, of three independent experiments). Cell numbers were reduced by approximately 70% in the rfhSP-D-treated Panc-1, 60% in MiaPaCa-2, and 45% in Capan-2 cells, as compared to untreated cells. Cell numbers were reduced by approximately 25% in the Panc-1 and MiaPaCa-2 and less than 10% in Capan-2 cells treated with FL-SP-D as compared to untreated cells. Significance was established using the unpaired two-way ANOVA test (**p < 0.01, ****p < 0.0001, ns: non-significant) (n = 3).
The flow cytometry analysis to quantify apoptosis showed significant reduction in the viable cell percentage of Panc-1, MiaPaCa-2, and Capan-2. The rfhSP-D induced apoptosis in ~67% of Panc-1 cells at 48 h, out of which, ~57% Panc-1 cells were both FITC and PI positive and ~7% were FITC alone positive, suggesting annexin V/FITC binding to phosphatidylserine, a cell membrane phospholipid, which is externalized during early apoptotic stage and the passage of PI, a DNA stain, through the porous cell membrane into the nucleus in order to intercalate the DNA. Approximately, 10% cells were PI alone positive, which suggested that these cells were either dead or in late apoptotic stage. The percentage of viable cells, i.e., unstained, in the untreated sample was significantly higher (70%) as compared to treated (26%) (Figure 3B). The rfhSP-D induced apoptosis in MiaPaCa-2 was ~60%. However, rfhSP-D induced apoptosis in Capan-2 (~35%) cell line, which was not as much as in Panc-1 and MiaPaCa-2 cell lines (Figure 3B). The treatment with FL-SP-D (20 µg/ml) for 48 h induced apoptosis in approximately 25% of Panc-1 and MiaPaCa-2 cell lines, and less than 10% in Capan-2 cell line. No significant difference was seen with FL-SP-D (10 µg/ml) treatment for 48 h in all the cell lines investigated in this study (Figure 3C).
The cell viability analysis via MTT assay following rfhSP-D treatment showed ~60% decrease in the cell viability of Panc-1 and MiaPaCa-2 and 45% in Capan-2 as compared to untreated (Figure 3D) and BSA (10 and 20 µg/ml; data not shown) controls. The cell viability analysis via MTT assay following FL-SP-D treatment also showed consistent reduction as seen in flow cytometer analysis (Figure 3E). Apoptosis was further confirmed by analyzing the activation of caspase to determine the pathway involved.
rfhSP-D Activates Cleavage of Caspase 8 and 3
Western blot analysis revealed that caspase 8 and 3 were cleaved in all the cell lines following treatment with rfhSP-D for 48 h (Figure 4). The cleavage of caspase 3, however, was not seen in the untreated cells and faint bands appeared for caspase 8 in the untreated cells (Figure 4), which further confirmed that cell death occurred via apoptosis. Interestingly, although Capan-2 cell line appeared unaffected in terms of cell cycle arrest at 24 h; yet, the cleaved bands for caspase 8 and 3 were seen in Capan-2 treated cells too, which suggested that rfhSP-D can affect the cancer cells via multiple pathways. Caspase 9 was tested as a marker for intrinsic apoptosis pathway; however, no difference was noted between treated and untreated cells (data not shown). Therefore, gene expressions were assessed for pro-apoptotic genes such as Bax, an intrinsic pathway marker, and Fas, an extrinsic pathway marker, to further determine the apoptotic pathway.
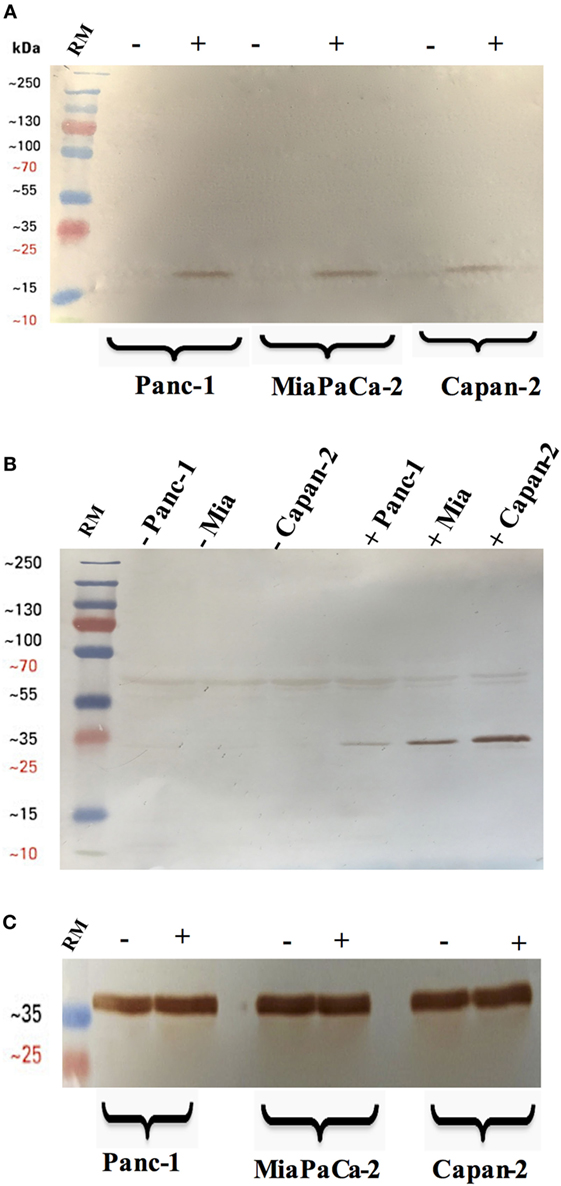
Figure 4. Cleavage of caspase 3 (A) and 8 (B) in pancreatic cancer cell lines following rfhSP-D treatment. Pancreatic cancer cell lines were analyzed for caspase 8 and 3 activation via western blot using anti-rabbit cleaved caspase 3 and 8 (1:1,000) at 4°C overnight, followed by incubation with secondary anti-rabbit IgG HRP-conjugate (1:1,000) for 1 h at room temperature. The membrane was washed with PBST (PBS + 0.05% Tween 20) three times, 10 min each between each step. The bands were developed using 3,3′-diaminobenzidine substrate kit. The cleaved caspase 3 and 8 were detected only in the rfhSP-D treated samples of all cell lines, whereas no bands appeared in the untreated cell samples. Full-length caspase 8 bands are visible around 43 kDa. (C) Anti-GAPDH was used as a loading control.
rfhSP-D Upregulates the Expression of Pro-Apoptotic Marker, Fas
Human pancreatic cancer cells often escape apoptosis by downregulating apoptosis stimulators such as FasL/FasR (25), or pro-apoptotic proteins such as Bax (26). These pro-apoptotic genes, Bax and Fas, for time-points ranging from 2 to 24 h in all the cell lines, were analyzed. Bax was unaffected following the treatment with rfhSP-D in Panc-1 and MiaPaCa-2 cell lines at all time-points (data not shown), which, in addition to unaffected caspase 9, suggested that intrinsic pathway may not have been involved in causing the cell death in these cell lines. Fas expression was unaffected at earlier time-points up to 6 h (data not shown); however, it was upregulated at 12 and 24 h in Panc-1 (log10 ~0.5), MiaPaCa-2 (log10 ~1), and Capan-2 (log10 ~1) cell lines (Figure 5A), which indicated that apoptosis induction by rfhSP-D is likely to take place via the extrinsic pathway. Western blot analysis also showed upregulation of Fas at the protein level in rfhSP-D treated cells as compared to untreated cells (Figure 5B). Since TNF-α and NF-κB are crucial factors in the apoptotic pathway and they can regulate Fas expression (27), the effect of rfhSP-D on the gene expression of TNF-α and NF-κB as well as translocation of NF-κB from the cytoplasm to nucleus was investigated.
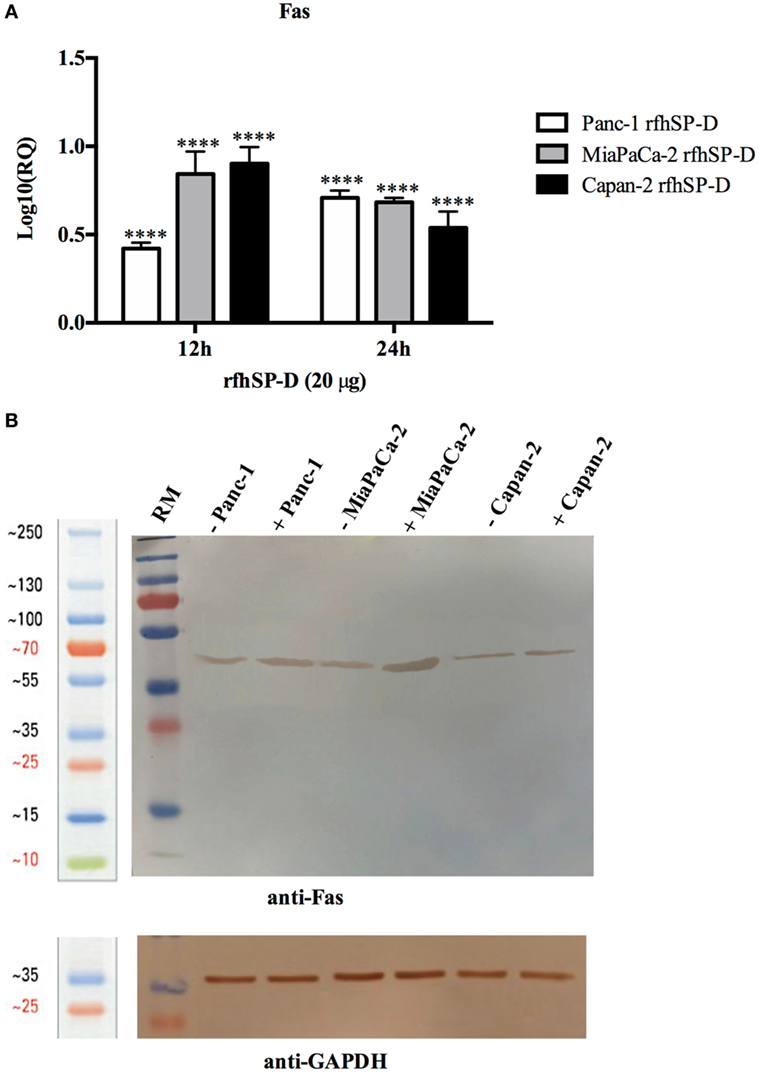
Figure 5. Relative quantification (RQ) of Fas mRNA expression in Panc-1, MiaPaCa-2, and Capan-2 cell lines treated with rfhSP-D (20 µg/ml) for 12 and 24 h. (A) Fas expression was upregulated in the treated samples at 12 and 24 h as compared to untreated cells. Significance was determined using the unpaired two-way ANOVA test (****p < 0.0001) (n = 3). (B) Fas expression via western blot analysis in pancreatic cell lines treated with rfhSP-D for 24 h using rabbit anti-human Fas (1:1,000) at 4°C overnight, followed by incubation with secondary anti-rabbit IgG HRP-conjugate (1:1,000) for 1 h at room temperature. The bands were developed using diaminobenzidine substrate kit. Fas expression at ~50 kDa was upregulated in the treated samples at 24 h for all cells as compared to untreated. Anti-GAPDH used as a loading control.
rfhSP-D Upregulates p53 Expression in Capan-2 Cell Line
The p53 transcript levels were measured by qPCR following the treatment with rfhSP-D at 2, 6, and 12 h in Capan-2 cells and compared with the p53 levels in untreated cells for each time-point. Interestingly, the levels of p53 were upregulated, most significantly at 12 h, which suggested that p53 may also have contributed to the apoptosis in Capan-2 cells (Figure 6).
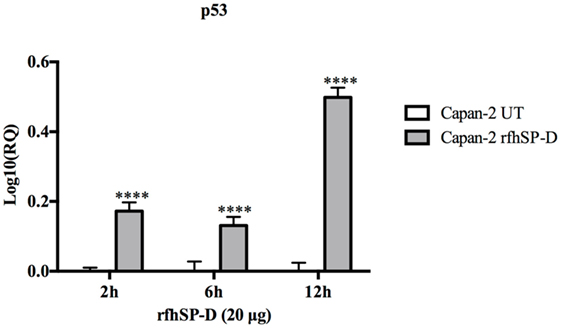
Figure 6. Relative quantification (RQ) of p53 mRNA expression in Capan-2 cell line treated with rfhSP-D (20 µg/ml) for 2, 6, and 12 h. p53 expression was significantly upregulated in the rfhSP-D-treated samples at 2, 6, and 12 h as compared to the untreated. Significance was determined using the unpaired two-way ANOVA test (****p < 0.0001) (n = 3).
rfhSP-D Upregulates the Expression of TNF-α and Causes Nuclear Translocation of NF-κB
Following treatment with rfhSP-D, the analysis of TNF-α mRNA expression levels showed a significant upregulation in Panc-1 (log10 ~0.5), MiaPaCa-2 (log10 ~1), and Capan-2 (log10 ~1) at 12 and 24 h; however, no difference was observed at earlier time-points. Similar transcriptional upregulation was noted for NF-κB for Panc-1 (log10 ~0.4), MiaPaCa-2 (log10 ~0.8), and Capan-2 (log10 ~0.6) at 12 and 24 h (Figure 7A). Immunofluorescence microscopy of Panc-1, MiaPaCa-2, and Capan-2 cell lines showed that NF-κB was translocated to the nucleus at 24 h, which was not seen in the untreated cells (Figure 7B). This further confirmed that NF-κB could play a key role in deciding the apoptotic fate of the pancreatic cancer cells following the rfhSP-D treatment.
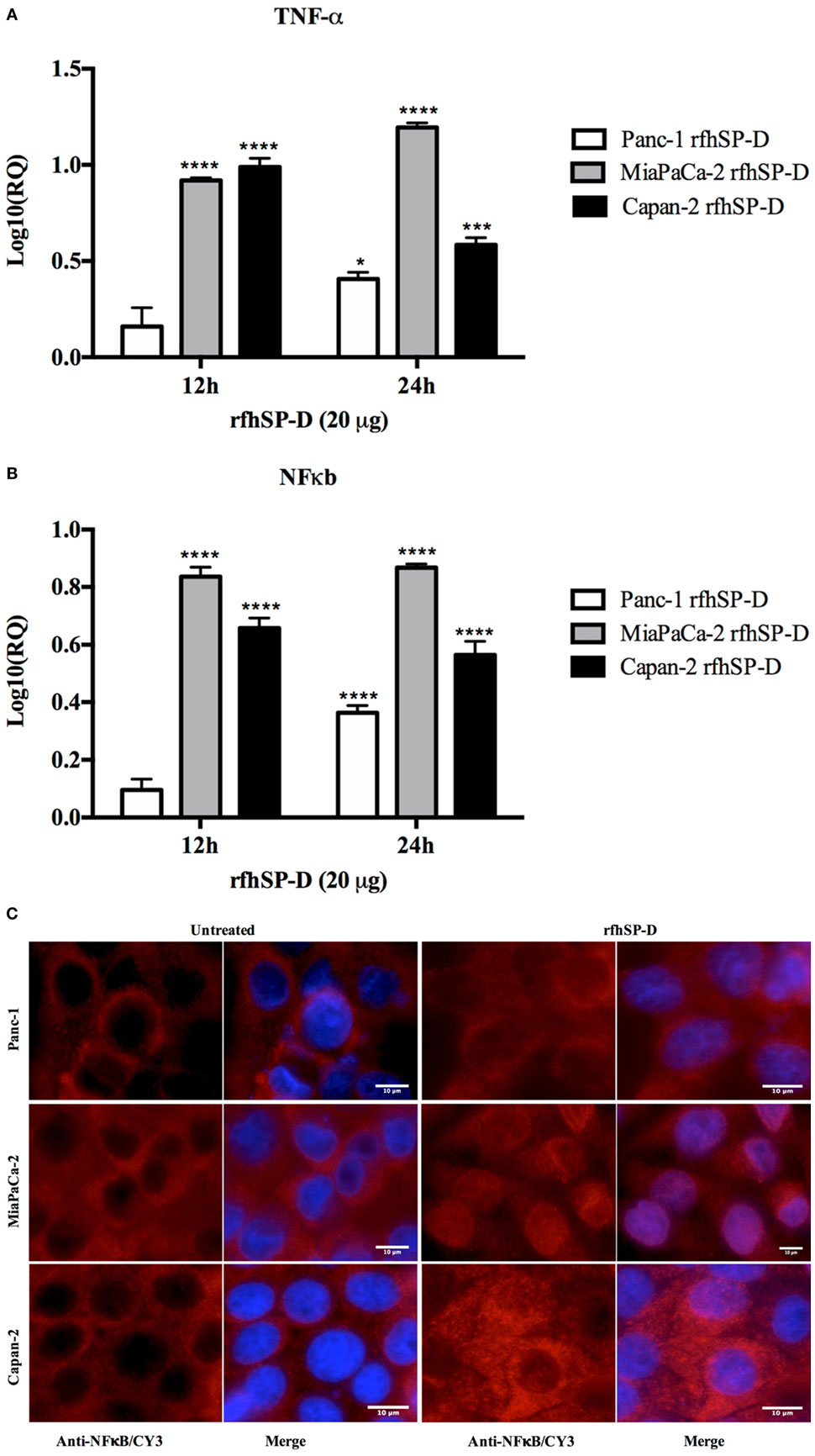
Figure 7. Relative quantification (RQ) comparisons of TNF-α (A) and NF-κB (B) mRNA expression in Panc-1, MiaPaCa-2, and Capan-2 cell lines treated with rfhSP-D (20 µg/ml) for 12 and 24 h. The transcriptional expressions of both TNF-α and NF-κB were upregulated in the treated samples at 12 and 24 h as compared to untreated. Significance was determined using the unpaired two-way ANOVA test (*p < 0.05, ***p < 0.001, ****p < 0.0001) (n = 3). (C) Immunofluorescence microscopy to determine the translocation of NF-κB into nucleus following rfhSP-D treatment. Anti-NF-κB stained positively in the nucleus of treated cells as compared to untreated in all cell lines at 24 h.
rfhSP-D Downregulates the Survival Pathway, mTOR
The mTOR is often deregulated in the pancreatic cancer (28) and its activation is associated with poor prognosis (29). Upon treatment with rfhSP-D, mRNA expression of mTOR was downregulated in Panc-1 and MiaPaCa-2 cell line at 12 h (Figure 8A), however, no difference was seen in Capan-2 (data not shown). In addition, immunofluorescence analysis revealed that in comparison to the untreated cells, a significant decrease in the cytoplasmic levels and an increased accumulation of mTOR in the nucleus of MiaPaCa-2 cells was evident (Figure 8B), where it has been shown to be present in its inactive form (30).
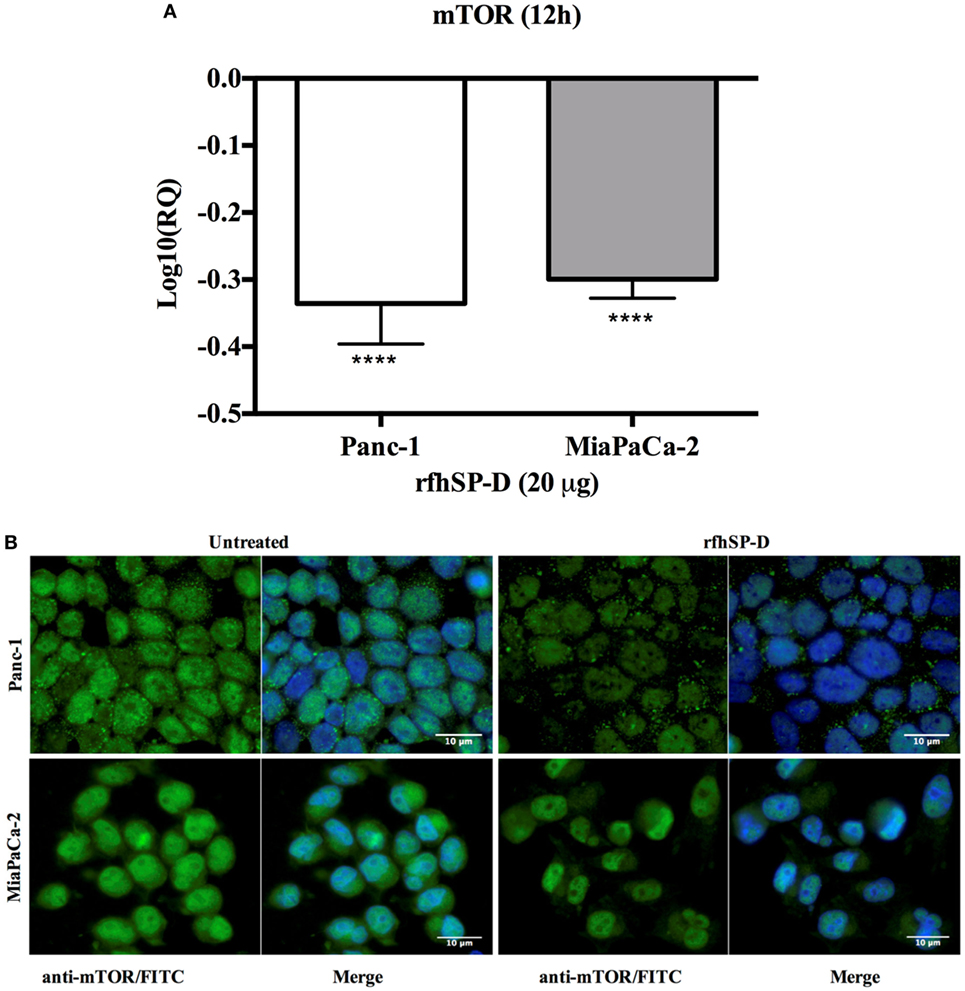
Figure 8. rfhSP-D downregulated the survival pathway, mTOR. (A) rfhSP-D treatment of Panc-1 and MiaPaCa-2 cells downregulated the mRNA expression of mTOR (****p < 0.0001). (B) Immunofluorescence microscopy showed reduced cytoplasmic levels of mTOR following treatment as compared to the untreated. Nuclear accumulation is clearly visible in the rfhSP-D treated cells.
Discussion
In this study, we show that a recombinant fragment of human surfactant protein D (rfhSP-D) induces apoptosis in a range of pancreatic cancer cell lines. We show that rfhSP-D induces apoptosis regardless of p53 status using two p53 mutated, aggressive cell lines, Panc-1 (derived from head of the pancreas), MiaPaCa-2 (derived from the body and tail of the pancreas), and a p53 wild type, non-aggressive cell line, Capan-2 (derived from head of the pancreas) (31).
Following the treatment with rfhSP-D, Panc-1 and MiaPaCa-2 cells were arrested in G1 phase at 24 h, whereas untreated cells progressed to S and G2 phase. In addition, upregulation of Fas, an apoptosis stimulator, and pro-apoptotic TNF-α (and associated transcription factor, NF-κB) at 24 h was consistent with the cleavage of caspase 8 and 3 at 48 h. These findings indicated that cell death is likely to occur via TNF-α/Fas-mediated apoptosis pathway (32–34). The cell viability after 48 h of rfhSP-D treatment was reduced in the order of Panc-1 > MiaPaCa-2 > Capan-2, which coincided with the approximate growth arrested percentage of Panc-1 and MiaPaCa-2 at 24 h. Although Capan-2 cells were not arrested in the cell cycle, yet they underwent apoptosis at 48 h, which may be attributed to their increased sensitivity to Fas-mediated apoptosis as compared to other two cell lines (25) and upregulation of p53 transcripts following the treatment with rfhSP-D, as reported previously (16). Treatment with FL-SP-D induced apoptosis in approximately 25% of Panc-1 and MiaPaCa-2, compared to Panc-1 (~67%) > MiaPaCa-2 (~60%) > Capan-2 (~35) by rfhSP-D. This quantitative difference is likely to be due to difference in the molar ration of the two proteins at the same concentration.
Fas is a type I membrane protein that belongs to TNF superfamily (35, 36) that undergoes trimerization upon binding to its physiological ligand, FasL, to form a Fas-associated death domain protein (FADD) via its cytoplasmic domain (37, 38). It then activates downstream caspase cascade, which subsequently causes cleavage of caspase 3 as the terminal molecular event during apoptosis (39, 40). When the Panc-1, MiaPaCa-2, and Capan-2 cell lines were treated with rfhSP-D, Fas remained unaffected up to 12 h. Upregulation of Fas transcripts as well as protein was seen at 24 h, indicating that TNF-α (41) and NF-κB (33) might also be affected since they are well known to tightly regulate the Fas-mediated apoptosis pathway. TNF-α, another member of TNF superfamily, acts via TNFR2 to increase the susceptibility of the target cells to Fas-mediated death; in addition, it stimulates the downstream NF-κB signaling (42) by recruitment and activation of inhibitor of IκB kinases (IKK), which in turn enables its translocation to the nucleus where transcription of NF-κB-dependent genes such as Fas occurs (33, 43).
In this study, transcriptional levels of both NF-κB and TNF-α were upregulated at the same time-point as Fas, which was largely anticipated (33). In addition, the immunofluorescence microscopy revealed NF-κB translocation to nucleus at 24 h in the rfhSP-D-treated cells as compared to the untreated counterpart, which suggested that TNF-α induced canonical NF-κB pathway (44). NF-κB can regulate both pro- as well as anti-apoptotic genes, depending upon its canonical or non-canonical signaling (43, 44). Interestingly, canonical NF-κB has been shown to bind directly to the Fas promoter to facilitate cell death via Fas-mediated pathway (33). NF-κB plays an important role in deciding the cell fate as its canonical activation acts a transcription factor of Fas, which upon stimulation induces apoptosis signaling (32, 33). However, SP-D has been shown to regulate steady-state NF-κB activation in alveolar macrophages of SP-D deficient mice (45). Interestingly, SP-D has also been shown to trigger TNF-α production in human CCR2+ inflammatory monocytes (46). These studies present an interesting central role of SP-D and their interdependent regulation, which could be important in deciding the cell viability/apoptosis. Moreover, cleaved caspase 8 and 3 were seen at 48 h, whereas intrinsic apoptosis markers such as caspase 9 and Bax remained unaffected (27), in all rfhSP-D-treated pancreatic cancer cell lines as compared to untreated cells, which further confirmed the cell death via Fas-mediated pathway alone. In addition, mTOR pathway was downregulated following the treatment with rfhSP-D, which is crucial for cell survival and proliferation, and thus, to protect the cancer cells from apoptosis (47). These findings are also supported by studies such as targeting mTOR pathway using rapamycin (48), or its regulating component RICTOR knockdown (49), significantly reduces the pancreatic cancer cell growth. Interestingly, immunofluorescence microscopy showed that rfhSP-D causes nuclear accumulation of mTOR in the treated cells, which may have a transcriptional role. However, the nuclear versions do not form an intact mTORC1 required for regulatory signaling pathways (30).
rfhSP-D bound all the pancreatic cell lines tested in this study: Panc-1, MiaPaCa-2, and Capan-2 (Figure 1A). However, the putative SP-D receptor or the ligand on the pancreatic cancer cell surface is not yet known. Recently, an interaction between the CRD region of human SP-D and N-glycans of EGFR has been reported which led to downregulated EGF signaling in human lung adenocarcinoma, A549 cell line cells (19).
In conclusion, rfhSP-D upregulates pro-apoptotic factors such as TNF-α, NF-κB, and Fas to activate caspase cascade to induce apoptosis in pancreatic cancer cell lines, which needs further exploration in orthotropic murine models. Majority of the conventional anti-cancer therapies only target the rapidly proliferating cancer cells, therefore, new strategies involving immune molecules such as rfhSP-D that target the signaling pathways to reduce the cell growth merit further investigation as these would not only help eliminate the tumor but could also influence recurrence or migratory capacity of the tumor cells.
Author Contributions
AK carried out most crucial experiments and was supported by MR, VM, and PV. SS provided ideas for crucial experiments and offered important reagents. AK wrote the first draft. UK led the study and helped with the manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
SS is supported by a Max-Elder Research Grant.
References
1. Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, et al. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol (2006) 43:1293–315. doi:10.1016/j.molimm.2005.08.004
2. Ujma S, Horsnell WG, Katz AA, Clark HW, Schafer G. Non-pulmonary immune functions of surfactant proteins A and D. J Innate Immun (2017) 9:3–11. doi:10.1159/000451026
3. Nayak A, Dodagatta-Marri E, Tsolaki AG, Kishore U. An insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Front Immunol (2012) 3:131. doi:10.3389/fimmu.2012.00131
4. Schaub B, Westlake RM, He H, Arestides R, Haley KJ, Campo M, et al. Surfactant protein D deficiency influences allergic immune responses. Clin Exp Allergy (2004) 34:1819–26. doi:10.1111/j.1365-2222.2004.02068.x
5. Wert SE, Yoshida M, LeVine AM, Ikegami M, Jones T, Ross GF, et al. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc Natl Acad Sci U S A (2000) 97:5972–7. doi:10.1073/pnas.100448997
6. Ikegami M, Na CL, Korfhagen TR, Whitsett JA. Surfactant protein D influences surfactant ultrastructure and uptake by alveolar type II cells. Am J Physiol Lung Cell Mol Physiol (2005) 288:L552–61. doi:10.1152/ajplung.00142.2004
7. Griese M, Steinecker M, Schumacher S, Braun A, Lohse P, Heinrich S. Children with absent surfactant protein D in bronchoalveolar lavage have more frequently pneumonia. Pediatr Allergy Immunol (2008) 19:639–47. doi:10.1111/j.1399-3038.2007.00695.x
8. Foreman MG, Kong X, DeMeo DL, Pillai SG, Hersh CP, Bakke P, et al. Polymorphisms in surfactant protein-D are associated with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol (2011) 44:316–22. doi:10.1165/rcmb.2009-0360OC
9. Lingappa JR, Dumitrescu L, Zimmer SM, Lynfield R, McNicholl JM, Messonnier NE, et al. Identifying host genetic risk factors in the context of public health surveillance for invasive pneumococcal disease. PLoS One (2011) 6:e23413. doi:10.1371/journal.pone.0023413
10. Ishii T, Hagiwara K, Ikeda S, Arai T, Mieno MN, Kumasaka T, et al. Association between genetic variations in surfactant protein D and emphysema, interstitial pneumonia, and lung cancer in a Japanese population. COPD (2012) 9:409–16. doi:10.3109/15412555.2012.676110
11. Silveyra P, Floros J. Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury. Front Biosci (Landmark Ed) (2012) 17:407–29. doi:10.2741/3935
12. Tanaka M, Arimura Y, Goto A, Hosokawa M, Nagaishi K, Yamashita K, et al. Genetic variants in surfactant pulmonary-associated protein D (SFTPD) and Japanese susceptibility to ulcerative colitis. Inflamm Bowel Dis (2009) 15:918–25. doi:10.1002/ibd.20936
13. Madan T, Kishore U, Singh M, Strong P, Clark H, Hussain EM, et al. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest (2001) 107:467–75. doi:10.1172/JCI10124
14. Singh M, Madan T, Waters P, Parida SK, Sarma PU, Kishore U. Protective effects of a recombinant fragment of human surfactant protein D in a murine model of pulmonary hypersensitivity induced by dust mite allergens. Immunol Lett (2003) 86:299–307. doi:10.1016/S0165-2478(03)00033-6
15. Mahajan L, Madan T, Kamal N, Singh VK, Sim RB, Telang SD, et al. Recombinant surfactant protein-D selectively increases apoptosis in eosinophils of allergic asthmatics and enhances uptake of apoptotic eosinophils by macrophages. Int Immunol (2008) 20:993–1007. doi:10.1093/intimm/dxn058
16. Mahajan L, Pandit H, Madan T, Gautam P, Yadav AK, Warke H, et al. Human surfactant protein D alters oxidative stress and HMGA1 expression to induce p53 apoptotic pathway in eosinophil leukemic cell line. PLoS One (2013) 8:e85046. doi:10.1371/journal.pone.0085046
17. Mahajan L, Gautam P, Dodagatta-Marri E, Madan T, Kishore U. Surfactant protein SP-D modulates activity of immune cells: proteomic profiling of its interaction with eosinophilic cells. Expert Rev Proteomics (2014) 11:355–69. doi:10.1586/14789450.2014.897612
18. Pandit H, Thakur G, Koippallil Gopalakrishnan AR, Dodagatta-Marri E, Patil A, Kishore U, et al. Surfactant protein D induces immune quiescence and apoptosis of mitogen-activated peripheral blood mononuclear cells. Immunobiology (2016) 221(2):310–22. doi:10.1016/j.imbio.2015.10.004
19. Hasegawa Y, Takahashi M, Ariki S, Asakawa D, Tajiri M, Wada Y, et al. Surfactant protein D suppresses lung cancer progression by downregulation of epidermal growth factor signaling. Oncogene (2015) 34:4285–6. doi:10.1038/onc.2014.20
20. Malvezzi M, Bertuccio P, Rosso T, Rota M, Levi F, La Vecchia C, et al. European cancer mortality predictions for the year 2015: does lung cancer have the highest death rate in EU women? Ann Oncol (2015) 26:779–86. doi:10.1093/annonc/mdv001
21. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin (2016) 66:7–30. doi:10.3322/caac.21332
22. Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, et al. Recent progress in pancreatic cancer. CA Cancer J Clin (2013) 63:318–48. doi:10.3322/caac.21190
23. Ansari D, Tingstedt B, Andersson B, Holmquist F, Sturesson C, Williamsson C, et al. Pancreatic cancer: yesterday, today and tomorrow. Future Oncol (2016) 12:1929–46. doi:10.2217/fon-2016-0010
24. Strong P, Kishore U, Morgan C, Lopez Bernal A, Singh M, Reid KB. A novel method of purifying lung surfactant proteins A and D from the lung lavage of alveolar proteinosis patients and from pooled amniotic fluid. J Immunol Methods (1998) 220:139–49. doi:10.1016/S0022-1759(98)00160-4
25. von Bernstorff W, Spanjaard RA, Chan AK, Lockhart DC, Sadanaga N, Wood I, et al. Pancreatic cancer cells can evade immune surveillance via nonfunctional Fas (APO-1/CD95) receptors and aberrant expression of functional Fas ligand. Surgery (1999) 125:73–84. doi:10.1016/S0039-6060(99)70291-6
26. Friess H, Lu Z, Graber HU, Zimmermann A, Adler G, Korc M, et al. Bax, but not Bcl-2, influences the prognosis of human pancreatic cancer. Gut (1998) 43:414–21. doi:10.1136/gut.43.3.414
27. Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anti-cancer chemotherapy. Oncogene (2006) 25:4798–811. doi:10.1038/sj.onc.1209608
28. Semba S, Moriya T, Kimura W, Yamakawa M. Phosphorylated Akt/PKB controls cell growth and apoptosis in intraductal papillary-mucinous tumor and invasive ductal adenocarcinoma of the pancreas. Pancreas (2003) 26:250–7. doi:10.1097/00006676-200304000-00008
29. Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell (2011) 42:36–49. doi:10.1016/j.molcel.2011.02.020
30. Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol (2013) 203:563–74. doi:10.1083/jcb.201306041
31. Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas (2010) 39:425–35. doi:10.1097/MPA.0b013e3181c15963
32. Kaur A, Sultan SH, Murugaiah V, Pathan AA, Alhamlan FS, Karteris E, et al. Human C1q induces apoptosis in an ovarian cancer cell line via tumor necrosis factor pathway. Front Immunol (2016) 7:599. doi:10.3389/fimmu.2016.00599
33. Liu F, Bardhan K, Yang D, Thangaraju M, Ganapathy V, Waller JL, et al. NF-κB directly regulates Fas transcription to modulate Fas-mediated apoptosis and tumor suppression. J Biol Chem (2012) 287:25530–40. doi:10.1074/jbc.M112.356279
34. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science (1998) 281:1305–8. doi:10.1126/science.281.5381.1305
35. Armitage RJ. Tumor necrosis factor receptor superfamily members and their ligands. Curr Opin Immunol (1994) 6:407–13. doi:10.1016/0952-7915(94)90119-8
36. Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem (1998) 254:439–59. doi:10.1046/j.1432-1327.1998.2540439.x
37. Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem (1995) 270:7795–8. doi:10.1074/jbc.270.14.7795
38. Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell (1995) 81:505–12. doi:10.1016/0092-8674(95)90071-3
39. Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem (1998) 273:9357–60. doi:10.1074/jbc.273.16.9357
40. Zheng TS, Schlosser SF, Dao T, Hingorani R, Crispe IN, Boyer JL, et al. Caspase-3 controls both cytoplasmic and nuclear events associated with Fas-mediated apoptosis in vivo. Proc Natl Acad Sci U S A (1998) 95:13618–23. doi:10.1073/pnas.95.23.13618
41. Elzey BD, Griffith TS, Herndon JM, Barreiro R, Tschopp J, Ferguson TA. Regulation of Fas ligand-induced apoptosis by TNF. J Immunol (2001) 167:3049–56. doi:10.4049/jimmunol.167.6.3049
42. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell (2003) 114:181–90. doi:10.1016/S0092-8674(03)00521-X
43. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol (2009) 1:a000034. doi:10.1101/cshperspect.a000034
44. Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol (2007) 8:49–62. doi:10.1038/nrm2083
45. Yoshida M, Korfhagen TR, Whitsett JA. Surfactant protein D regulates NF-κB and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol (2001) 166:7514–9. doi:10.4049/jimmunol.166.12.7514
46. Barrow AD, Palarasah Y, Bugatti M, Holehouse AS, Byers DE, Holtzman MJ, et al. OSCAR is a receptor for surfactant protein D that activates TNF-alpha release from human CCR2+ inflammatory monocytes. J Immunol (2015) 194:3317–26. doi:10.4049/jimmunol.1402289
47. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci (2009) 122:3589–94. doi:10.1242/jcs.051011
48. Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci Rep (2013) 3:3230. doi:10.1038/srep03230
Keywords: pancreatic cancer, innate immunity, surfactant protein D, apoptosis, immune surveillance
Citation: Kaur A, Riaz MS, Murugaiah V, Varghese PM, Singh SK and Kishore U (2018) A Recombinant Fragment of Human Surfactant Protein D induces Apoptosis in Pancreatic Cancer Cell Lines via Fas-Mediated Pathway. Front. Immunol. 9:1126. doi: 10.3389/fimmu.2018.01126
Received: 13 October 2017; Accepted: 03 May 2018;
Published: 04 June 2018
Edited by:
Janos G. Filep, Université de Montréal, CanadaReviewed by:
Taruna Madan, National Institute for Research in Reproductive Health (ICMR), IndiaSoren Werner Karlskov Hansen, University of Southern Denmark Odense, Denmark
Kenneth Reid, University of Oxford, United Kingdom
Copyright: © 2018 Kaur, Riaz, Murugaiah, Varghese, Singh and Kishore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uday Kishore, uday.kishore@brunel.ac.uk, ukishore@hotmail.com