 Irving Coy Allen
Irving Coy Allen- Department of Biomedical Sciences and Pathobiology, Virginia-Maryland Regional College of Veterinary Medicine, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA
Aberrant inflammation is an enabling characteristic of tumorigenesis. Thus, signaling cascades that alter inflammatory activation and resolution are of specific relevance to disease pathogenesis. Pattern recognition receptors (PRRs) are essential mediators of the host immune response and have emerged as critical elements affecting multiple facets of tumor pathobiology. The nucleotide-binding domain and leucine-rich repeat containing (NLR) proteins are intracellular PRRs that sense microbial and non-microbial products. Members of the NLR family can be divided into functional sub-groups based on their ability to either positively or negatively regulate the host immune response. Recent studies have identified a novel sub-group of non-inflammasome forming NLRs that negatively regulate diverse biological pathways associated with both inflammation and tumorigenesis. Understanding the mechanisms underlying the function of these unique NLRs will assist in the rationale design of future therapeutic strategies targeting a wide spectrum of inflammatory diseases and cancer. Here, we will discuss recent findings associated with this novel NLR sub-group and mechanisms by which these PRRs may function to alter cancer pathogenesis.
Introduction
The intimate association between inflammation and cancer was first noted over 150 years ago by Rudolf Vierchow (1, 2). Indeed today, aberrant inflammation is considered both an emerging hallmark of tumorigenesis and an enabling characteristic of cancer (3). Tumorigenesis is a multistep process and inflammation functions at multiple levels to both antagonize and enhance tumor initiation and progression (3). During the early stages of tumorigenesis, an inflammatory microenvironment serves as an enabling characteristic to activate diverse signaling pathways and drive the progression of pre-malignant and malignant lesions toward cancer (3–5). In later stages, cancer cells typically acquire a diverse repertoire of defense mechanisms that allow the cells to both passively and actively evade immune surveillance and elimination (3, 6, 7). This immune system subversion is an emerging hallmark of cancer and serves to remove the most effective barriers employed by the host to defend against neoplasia, late-stage tumor, and micro-metastasis progression (3).
Pattern recognition receptors (PRRs) are an essential component of the host immune system and significantly contribute to cancer pathobiology. There are 4 major families of PRRs that have been implicated in tumorigenesis, including the toll-like receptors (TLRs), the nucleotide-binding domain and leucine-rich repeat containing (NLR) family of sensors, C-type lectin receptors (CLRs), and RIG-I-like receptors (RLRs) (8). These receptor families function to initiate inflammatory signaling cascades following the direct or indirect recognition of pathogens, damage and stress through sensing highly conserved pathogen-associated molecular patterns (PAMPs), and damage-associated molecular patterns (DAMPs). In addition to their roles in facilitating the immune response, PRRs also play fundamental roles in the regulation of proliferation, cell survival and death, reactive oxygen species generation, angiogenesis, and tissue remodeling and repair (8). In the context of cancer, PRRs drive the immune response following exposure to potentially carcinogenic pathogens, environmental exposures to mutagenic agents and insults, and cancer-associated cellular damage and stress (9–16). In general, increased PRR signaling creates an enriched, pro-inflammatory microenvironment that is favorable for tumor initiation and progression (17). Thus, we find that PRRs are stuck in a “Goldilocks Conundrum.” Robust PRR activation is critical in driving the host immune response following PAMP and DAMP exposure; whereas, an overzealous and persistent immune response driven by PRR activation can cause significant collateral damage to the host tissue that ultimately results in chronic inflammation and cancer.
To date, the majority of studies evaluating PRR signaling in cancer have focused on members of the TLR family. However, new and emerging findings have revealed a significant role for members of the NLR family in contributing either directly or indirectly to a variety of hallmarks associated with cancer, including inflammation, cell death, tumor growth, angiogenesis, invasion, and metastasis (18–26). There are at least 23 distinct NLR and NLR-like proteins that have been identified in humans and 34 family members identified in mice (23, 27–29). The NLR proteins function as cytosolic receptors and sensors to detect intracellular PAMPs and DAMPs. Since their discovery, a variety of names have been used to describe the members of this gene family and their respective proteins. For example, these PRRs have been previously referred to as CATERPILLERs, NOD-like receptors, NACHT-leucine-rich repeats (LRR), and NBD-LRR proteins (28). This resulted in a lack of consistency in the field and resulted in the currently accepted and standardized nomenclature defining the NLRs as the NLR gene family (28). These proteins contain a highly conserved tripartite domain structure (28). The N-terminal domain of the protein is comprised of a variable, but limited number of effector domains that can include combinations of acidic transactivation domains (NLRA proteins), baculoviral inhibitory repeat (BIR)-like domains (NLRB proteins), caspase recruitment domains (NLRC proteins), and pyrin domains (NLRP proteins) (28). These N-terminal domains function to recruit adaptor, intermediary, or effector molecules that drive downstream signaling. The core of the protein is comprised of a conserved NACHT nucleotide-binding domain, which facilitates oligomerization (28). The C-terminal domain of the protein contains multiple LRR elements, which are essential for ligand sensing (28). Each LRR element is typically 28–29 residues in length and each NLR may contain up to 33 individual LRR elements (30, 31).
Inflammasome Forming NLRs in Cancer
One of the most fundamental roles of the NLR family is to regulate pro-inflammatory cytokines and chemokines that drive the host innate immune response to pathogens and environmental insults. Key to this response is the proper regulation of IL-1β and IL-18, which are both potent pro-inflammatory cytokines that affect diverse aspects of health and disease (32–37). Both of these cytokines are generated in an immature pro-form that requires post-translational cleavage for activation. A functional sub-group of NLRs has been identified as driving this process through the formation of a multi-protein complex termed the inflammasome (32, 35, 36). Upon activation, the NLR is thought to undergo a conformational change that allows the recruitment and binding of adaptor and effector proteins and inflammasome formation (35). The inflammasome is composed of an NLR that recognizes a specific repertoire of PAMPs and DAMPs, the adaptor protein ASC, and pro-Caspase-1 (32). These sub-units continue to multiplex, ultimately resulting in the maturation and activation of Caspase-1, which subsequently drives the cleavage and activation of IL-1β and IL-18. These inflammasome forming NLRs are by far the best characterized and most highly studied members of the NLR family. To date, at least 6 NLR and NLR-like proteins have been strongly implicated in inflammasome formation, including NLRP1, NLRP3, NLRP6, NLRC4, NLRC5, and the PYHIN family member AIM2 (NLR-like) (32–37). Inflammasome forming NLRs significantly regulate the tumor microenvironment by modulating cytokine production. For example, many of the inflammasome forming NLRs have been shown to significantly attenuate inflammation and tumorigenesis in mouse models of colitis-associated colorectal cancer (CAC) by regulating IL-18 production (18, 19, 21, 22, 38–40). In addition to being a potent pro-inflammatory cytokine, IL-18 is also secreted by epithelial cells to stimulate regeneration and repair and improve barrier function in the colon, thus loss of this cytokine in NLR inflammasome deficient mice enhances tumorigenesis (41). Beyond colon cancer, NLR inflammasome activation may also play important roles in many other types of cancer, including breast cancer, skin cancers, and virus-associated hepatocellular carcinoma (25, 26, 42–47).
Non-Inflammasome Forming NLRs That Negatively Regulate Inflammation
While the inflammasome forming NLRs are the best characterized members of this PRR family, recent studies have identified a functional sub-group of NLRs that negatively regulate inflammation (48–54). This sub-group is currently composed of three NLR family members, NLRP12, NLRX1, and NLRC3 (Figure 1). NLRP12 was one of the first NLR proteins to be described and is the best characterized member of this functional NLR sub-group. NLRP12 was previously known as monarch-1 and PYPAF7 and was originally suggested to form an inflammasome with ASC in overexpression systems (55, 56). In these overexpression studies, transient transfection of NLRP12 and ASC was also shown to induce the transcription of an NF-κB reporter construct (56). Thus, these early in vitro studies initially suggested that NLRP12 was an inflammasome forming NLR and a positive regulator of NF-κB signaling. These findings are also consistent with human data that has identified mutations in NLRP12 linked to a spectrum of hereditary periodic fever syndromes. The disorders associated with NLRP12 mutations are characterized by redox alterations and enhanced secretion of IL-1β, which are similar to the characteristics associated with the family of diseases linked to gain-of-function mutations in the NLRP3 gene (57–59). Interestingly, these diseases are associated with increased caspase-1 activity, are sensitive to therapeutics targeting IL-1β (anakinra), and appear to be independent of NF-κB activation (57–59). However, the ability of NLRP12 to form a functional inflammasome under physiological situations and in the context of human disease appears to occur only under highly specific conditions and is an area of current investigation (60, 61). Indeed, several studies have evaluated NLRP12 inflammasome formation ex vivo and using Nlrp12−/− mice under a variety of conditions and have directly shown that this NLR does not regulate IL-1β/IL-18 maturation (62–69). The prevailing literature associated with NLRP12 indicates that this protein functions as a negative regulator of inflammation by modulating canonical and non-canonical NF-κB signaling (48, 49, 62, 65, 66, 68, 70–73). NLRP12 negatively regulates non-canonical NF-κB signaling through its association with TRAF3 and NF-κB inducing kinase (NIK) (49, 68). This interaction leads to the degradation of NIK and subsequent attenuation of p100 cleavage to p52 (Figure 1). Similarly, NLRP12 attenuates canonical NF-κB signaling through the inhibition of IRAK-1 phosphorylation (48, 66, 71) (Figure 1). In addition to directly mediating the NF-κB cascade, NLRP12 has also been shown to attenuate ERK signaling, though the exact mechanism has yet to be fully resolved (66, 68). Thus, while some conflicting data has been reported, most issues can be resolved by considering the technical limitations of the assays used to define the respective mechanisms and the specific models being evaluated.
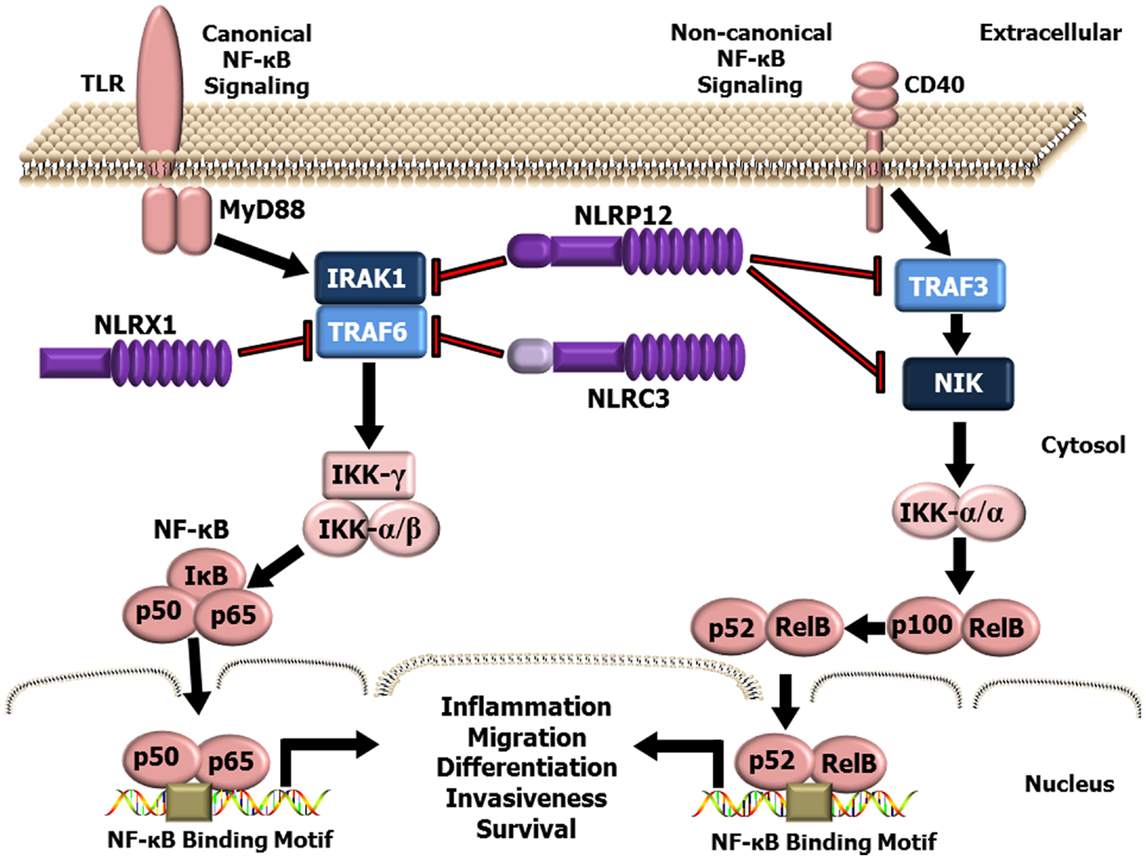
Figure 1. Schematic illustrating NLR attenuation of canonical and non-canonical NF-κB signaling. NF-κB is a master regulator of gene transcription and contributes to several hallmarks of cancer. NLRX1, NLRP12, and NLRC3 negatively regulate NF-κB signaling at multiple levels. NLRX1 interacts with and inhibits TRAF6 and the IKK complex resulting in the attenuation of NF-κB signaling following TLR stimulation. Likewise, NLRC3 was also shown to interact with TRAF6 and attenuate NF-κB signaling through a similar mechanism. NLRP12, has been shown to attenuate both the canonical NF-κB signaling pathway through modulating the phosphorylation of IRAK-1 and the non-canonical NF-κB pathway through interactions with TRAF3 and NIK.
NLRX1 was originally characterized in 2008, and was shown to negatively regulate the host anti-viral immune response (51). NLRX1 is unique among the NLRs due to its mitochondrial localization and its relatively undefined N-terminal domain. Similar to NLRP12, NLRX1 negatively regulates canonical NF-κB signaling (50, 52) (Figure 1). NLRX1 associates with TRAF6 and IκB kinase (IKK) through an activation signal-dependent mechanism (50). Following stimulation, NLRX1 is rapidly ubiquitinated and disassociates from TRAF6 to bind the IKK complex and inhibit subsequent canonical NF-κB activation (50). In addition to attenuating NF-κB signaling, NLRX1 also negatively regulates type-I interferon (IFN-I) signaling through inhibiting the interaction between the PRR Rig-I and the mitochondrial anti-viral signaling (MAVS) protein following virus exposure (50–52, 74, 75) (Figure 2). NLRX1 also functions as a positive regulator of autophagy following virus exposure through interacting with the protein TUFM and the mitochondrial immune signaling complex (MISC), which also includes ATG5, ATG12, and ATG16L1 (74, 75) (Figure 2). Interestingly, autophagy also functions as a negative regulator of IFN-I signaling and provides an additional route for the negative regulatory properties of NLRX1. In addition to regulating NF-κB and IFN-I signaling, subsequent studies have also shown that NLRX1 functions as a positive regulator of ROS production in epithelial cells following Chlamydia trachomatis infection, likely through interactions with the UQCRC2 protein (76, 77) (Figure 2). Thus, it is clear that NLRX1 regulation is quite complex and appears to occur through cell type, temporal and signal-dependent mechanisms.
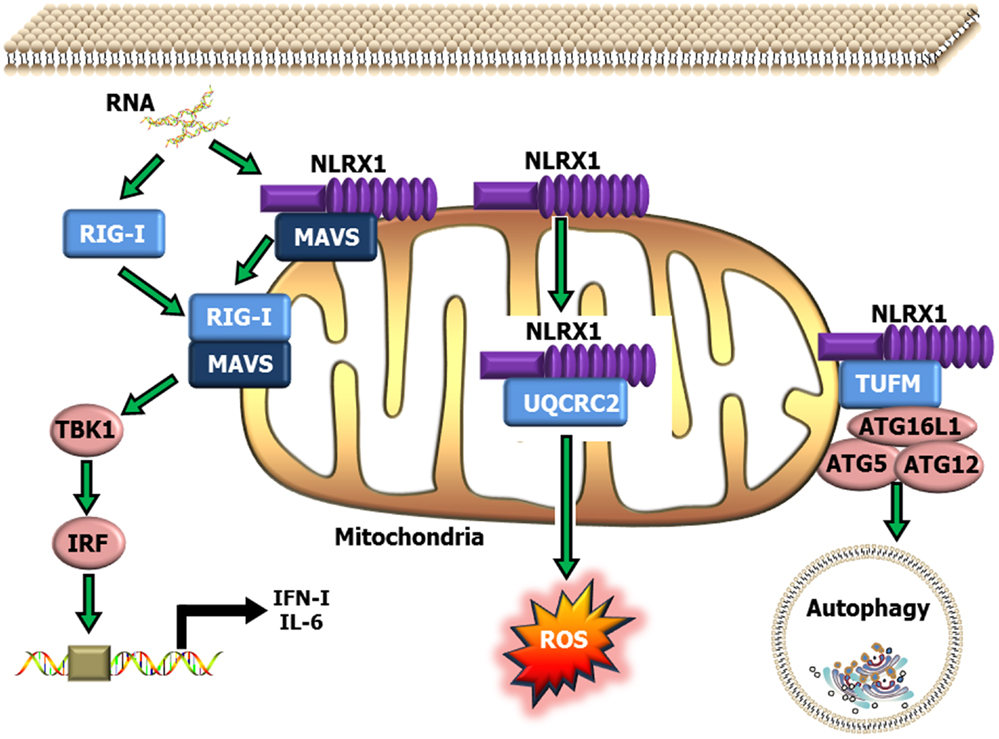
Figure 2. Schematic illustrating NLRX1 regulation of type-I interferon, ROS and autophagy signaling. NLRX1 is localized to the mitochondria, where it has been shown to bind with MAVS and prevent the interaction between MAVS and RIG-I during the host anti-viral response. This interaction significantly attenuates MAVS activation of IRF3 and IRF7 and results in reduced IFN and IL-6 signaling. NLRX1 has also been shown to function as a positive regulator of autophagy through its interactions with the mitochondrial protein TUFM, and the mitochondrial immune signaling complex (MISC), which includes Atg5–Atg12 and ATG16L1. This complex has been shown to be important in promoting virus-induced autophagy and concurrently attenuating IFN signaling. In addition to its role in attenuating host anti-viral signaling, NLRX1 has also been shown to significantly augment ROS generation from the mitochondria through interactions with UQCRC2 following infection with specific species of bacteria.
NLRC3 is the most recently characterized member of this functional sub-group and has been shown to negatively regulate NF-κB and IFN-I signaling (54, 78). NLRC3 was originally identified as a negative regulator of T cell function, in part through delaying the degradation of IκBα (78). Subsequent studies have since revealed that NLRC3 attenuates TLR signaling through interacting with and modulating TRAF6 activity and inhibiting canonical NF-κB signaling (54). NLRC3 has also been recently shown to fine tune the host innate immune response to intracellular DNA, DNA viruses, and c-di-GMP (53). NLRC3 impedes STING-TANK-binding kinase 1 (TBK1) interactions and inhibits STING trafficking, which results in an attenuation of subsequent downstream activation of IFN-I genes (53).
While NLRP12, NLRX1, and NLRC3 each influence a variety of signaling pathways, the convergence on NF-κB signaling appears to be a common strategy among the NLRs in this functional sub-group to attenuate inflammation (Figure 1). Additional mechanistic studies have revealed prevalent NLR–TRAF interactions in these models and support the emerging hypothesis that these NLRs function to inhibit NF-κB signaling through the formation of a multi-protein “TRAFasome” complex (54). Dysregulated NF-κB signaling and the additional pathways modulated by these NLRs are critical features in cancer initiation and progression. Thus, the NLRs that modulate these signaling cascades are highly relevant to cancer pathobiology and additional mechanistic insight will be critical for developing future therapeutic strategies.
Negative Regulatory NLRs in Cancer Pathobiology
While several studies have characterized the contribution of the NLRP3, NLRC4, and NLRP6 inflammasomes in tumorigenesis, significantly less is known regarding the role of NLRs that negatively regulate inflammation. Initial studies have focused on NLRP12. In the context of cancer, somatic mutations in human NLRP12 have been detected in several large scale screening studies evaluating a variety of cancer sub-types, including glioblastoma, breast cancer, lung squamous cell carcinoma, melanoma, prostate adenocarcinoma, and colon adenocarcinoma (http://cancergenome.nih.gov/). However, broader linkage with specific populations, causation, and mechanism for each mutation has not yet been established. In mice, NLRP12 has been shown to attenuate colorectal cancer. Using the AOM/DSS model of CAC, Nlrp12−/− mice were shown to develop increased inflammation and tumorigenesis (66, 68). Colon histopathology revealed significant epithelial cell damage and loss of barrier integrity in these animals, which resulted in increased pro-inflammatory cytokine and chemokine production (66, 68). These animals eventually develop extensive pre-cancerous lesions, which result in significantly increased areas of hyperplasia, dysplasia, and adenocarcinoma (66, 68). These studies revealed that NLRP12 attenuates inflammation and tumorigenesis through negatively regulating NF-κB and ERK signaling (66, 68).
While the overall results of each study are quite complementary, it should be noted that a few mechanistic differences were proposed. In one study, the increased tumorigenesis was attributed to an increase in canonical NF-κB signaling (66). NF-κB signaling was evaluated in vivo and in macrophages isolated from wild type and Nlrp12−/− mice following PAMP stimulation and a significant increase in the levels of p-p105, Rel-A, and p65 activity was observed (66). Furthermore, loss of NLRP12 was shown to significantly increase the transcription of a variety of pro-inflammatory mediators associated with canonical NF-κB signaling and colon tumorigenesis, including Il-6, Tnf-α, and Cox2 (66). These findings are consistent with earlier in vitro studies, which demonstrated that NLRP12 functions as an antagonist of TLR and TNFR-induced pro-inflammatory signals, in part through inhibiting IRAK-1 hyper-phosphorylation (48). In the second study, NLRP12 was shown to attenuate colon tumorigenesis through negatively regulating non-canonical NF-κB signaling. While some markers of canonical NF-κB signaling were found to be transiently increased in the absence of NLRP12, this study revealed a significant increase in NIK activation and p100 to p52 cleavage in primary cells and in colon tissues isolated from Nlrp12−/− mice during disease progression (68). These data are highly consistent with previous in vitro studies associating NLRP12 activity with NIK suppression and attenuation of non-canonical NF-κB signaling (49, 79). Loss of NLRP12 resulted in a significant increase in Cxcl12 and Cxcl13 expression in the colons from Nlrp12−/− mice (68). These chemokines are highly associated with non-canonical NF-κB activation and cancer (49, 68, 80–82). CXCL12 (SDF-1) and CXCL13 (BLC) and their respective receptors CXCR4 and CXCR5 have been implicated in tumor growth, metastasis, and are critical for the regulation of the tumor microenvironment in multiple cancer sub-types as a component of the tumor “Immunome” (3, 83–85). Regulation of the NF-κB signaling pathway is highly complex. The apparent discrepancies between these two studies can be reconciled by previous findings, which show that non-canonical NF-κB signaling can influence both the canonical pathway and MAPK signaling (86, 87). It is also highly likely that NLRP12 regulates canonical and non-canonical NF-κB signaling through currently undefined cell type, temporal and/or stimuli-specific mechanisms.
To date, neither NLRX1 nor NLRC3 have been directly evaluated in the context of cancer. As previously stated, both of these NLRs negatively regulate NF-κB signaling and would be expected to attenuate tumorigenesis through mechanisms similar to those described for NLRP12. However, each also regulates pathways other than NF-κB that could dramatically influence cancer pathobiology. For example, NLRX1 has been shown to additionally regulate ROS production and autophagy. The dysregulation of oxidative stress signaling is a well-established and important element of tumor development (88). Similarly, autophagy is thought to have a dual function in cancer, where it can attenuate tumor initiation by suppressing tissue damage and inflammation signaling or it can function as a tumor promoter to sustain metabolism, growth, and survival through metabolite recycling (89, 90). Thus, it is highly likely that NLRX1 will contribute to tumorigenesis; however, it is difficult to speculate which of its many biologic functions will have a greater influence on disease pathogenesis.
Conclusion
The recent characterization of this unique sub-group of NLRs that function to attenuate inflammation emphasizes the point that a significant number of the identified NLR proteins in humans have yet to be adequately characterized. Identifying the unique regulatory and signaling pathways modulated by these NLRs is an essential step toward ultimately developing effective therapeutics targeting these proteins and the pathways they modulate. Characterizing unidentified ligands, cell type and temporal regulatory mechanisms, and redundant functions of these NLR family members will significantly improve our understanding of the contribution of these proteins in maintaining immune system homeostasis. It is also clear that NLRs significantly impact cancer pathobiology, beyond colorectal cancer. Additional studies are necessary to better define the contribution of both inflammasome forming NLRs and non-inflammasome forming NLRs in modulating the hallmarks of cancer.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grant K01 DK092355 and a Virginia-Maryland Regional College of Veterinary Medicine Internal Research Grant. The software application ScienceSlides Suite 2011 (VisiScience) was used to generate Figures 1 and 2.
References
1. Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol (2012) 30:677–706. doi:10.1146/annurev-immunol-020711-075008
2. Heidland A, Klassen A, Rutkowski P, Bahner U. The contribution of Rudolf Virchow to the concept of inflammation: what is still of importance? J Nephrol (2006) 19(Suppl 10):S102–9.
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
4. Cook J, Hagemann T. Tumour-associated macrophages and cancer. Curr Opin Pharmacol (2013) 13:595–601. doi:10.1016/j.coph.2013.05.017
5. Dutsch-Wicherek M, Kazmierczak W. Creation of a suppressive microenvironment by macrophages and cancer-associated fibroblasts. Front Biosci (Landmark Ed) (2013) 18:1003–16. doi:10.2741/4159
6. Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, et al. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol (2014) 122:91–128. doi:10.1016/B978-0-12-800267-4.00003-1
7. Biragyn A, Longo DL. Neoplastic “Black Ops”: cancer’s subversive tactics in overcoming host defenses. Semin Cancer Biol (2012) 22:50–9. doi:10.1016/j.semcancer.2012.01.005
8. Kutikhin AG, Yuzhalin AE. Inherited variation in pattern recognition receptors and cancer: dangerous liaisons? Cancer Manag Res (2012) 4:31–8. doi:10.2147/CMAR.S28688
9. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (2008) 320:674–7. doi:10.1126/science.1156995
10. Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A (2008) 105:9035–40. doi:10.1073/pnas.0803933105
11. Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol (2007) 17:1140–5. doi:10.1016/j.cub.2007.05.074
12. Hong S, Hwang I, Lee YS, Park S, Lee WK, Fernandes-Alnemri T, et al. Restoration of ASC expression sensitizes colorectal cancer cells to genotoxic stress-induced caspase-independent cell death. Cancer Lett (2013) 331:183–91. doi:10.1016/j.canlet.2012.12.020
13. Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, et al. Discovery of a viral NLR homolog that inhibits the inflammasome. Science (2011) 331:330–4. doi:10.1126/science.1199478
14. Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol (2004) 5:1166–74. doi:10.1038/ni1131
15. Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem (1999) 274:17406–9. doi:10.1074/jbc.274.25.17406
16. Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via toll-like receptor 2. J Immunol (1999) 163:1–5.
17. Kipanyula MJ, Seke Etet PF, Vecchio L, Farahna M, Nukenine EN, Nwabo Kamdje AH. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell Signal (2013) 25:403–16. doi:10.1016/j.cellsig.2012.10.014
18. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32:379–91. doi:10.1016/j.immuni.2010.03.003
19. Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med (2010) 207:1045–56. doi:10.1084/jem.20100050
20. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut (2010) 59:1192–9. doi:10.1136/gut.2009.197822
21. Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A (2010) 107:21635–40. doi:10.1073/pnas.1016814108
22. Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci U S A (2013) 110:9862–7. doi:10.1073/pnas.1307575110
23. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi:10.1016/j.cell.2010.01.040
24. Liu W, Luo Y, Dunn JH, Norris DA, Dinarello CA, Fujita M. Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma. J Invest Dermatol (2013) 133:518–27. doi:10.1038/jid.2012.317
25. Drexler SK, Bonsignore L, Masin M, Tardivel A, Jackstadt R, Hermeking H, et al. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc Natl Acad Sci U S A (2012) 109:18384–9. doi:10.1073/pnas.1209171109
26. Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J Biol Chem (2010) 285:6477–88. doi:10.1074/jbc.M109.064907
27. Ting JP, Davis BK. CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu Rev Immunol (2005) 23:387–414. doi:10.1146/annurev.immunol.23.021704.115616
28. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR gene family: a standard nomenclature. Immunity (2008) 28:285–7. doi:10.1016/j.immuni.2008.02.005
29. Chen GY. Role of Nlrp6 and Nlrp12 in the maintenance of intestinal homeostasis. Eur J Immunol (2014) 44:321–7. doi:10.1002/eji.201344135
30. Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol (2001) 11:725–32. doi:10.1016/S0959-440X(01)00266-4
31. Motyan JA, Bagossi P, Benko S, Tozser J. A molecular model of the full-length human NOD-like receptor family CARD domain containing 5 (NLRC5) protein. BMC Bioinformatics (2013) 14:275. doi:10.1186/1471-2105-14-275
32. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity (2004) 20:319–25. doi:10.1016/S1074-7613(04)00046-9
33. Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell (2014) 156:1045–59. doi:10.1016/j.cell.2014.01.026
34. Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol (2011) 186:1333–7. doi:10.4049/jimmunol.1003111
35. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi:10.1016/S1097-2765(02)00599-3
36. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430:213–8. doi:10.1038/nature02664
37. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi:10.1038/nature07725
38. Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol (2010) 185:4912–20. doi:10.4049/jimmunol.1002046
39. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A (2011) 108:9601–6. doi:10.1073/pnas.1100981108
40. Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol (2011) 186:7187–94. doi:10.4049/jimmunol.1100412
41. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi:10.1016/j.cell.2011.04.022
42. Burdette D, Haskett A, Presser L, McRae S, Iqbal J, Waris G. Hepatitis C virus activates interleukin-1beta via caspase-1-inflammasome complex. J Gen Virol (2012) 93:235–46. doi:10.1099/vir.0.034033-0
43. Dunn JH, Ellis LZ, Fujita M. Inflammasomes as molecular mediators of inflammation and cancer: potential role in melanoma. Cancer Lett (2012) 314:24–33. doi:10.1016/j.canlet.2011.10.001
44. Jin L, Yuan RQ, Fuchs A, Yao Y, Joseph A, Schwall R, et al. Expression of interleukin-1beta in human breast carcinoma. Cancer (1997) 80:421–34. doi:10.1002/(SICI)1097-0142(19970801)80:3<421::AID-CNCR10>3.0.CO;2-Z
45. Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog (2013) 9:e1003330. doi:10.1371/journal.ppat.1003330
46. Pantschenko AG, Pushkar I, Anderson KH, Wang Y, Miller LJ, Kurtzman SH, et al. The interleukin-1 family of cytokines and receptors in human breast cancer: implications for tumor progression. Int J Oncol (2003) 23:269–84. doi:10.3892/ijo.23.2.269
47. Snoussi K, Strosberg AD, Bouaouina N, Ben Ahmed S, Chouchane L. Genetic variation in pro-inflammatory cytokines (interleukin-1beta, interleukin-1alpha and interleukin-6) associated with the aggressive forms, survival, and relapse prediction of breast carcinoma. Eur Cytokine Netw (2005) 16:253–60.
48. Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem (2005) 280:39914–24. doi:10.1074/jbc.M502820200
49. Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ, et al. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J Immunol (2007) 178:1256–60.
50. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity (2011) 34:843–53. doi:10.1016/j.immuni.2011.02.022
51. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature (2008) 451:573–7. doi:10.1038/nature06501
52. Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity (2011) 34:854–65. doi:10.1016/j.immuni.2011.03.026
53. Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity (2014) 40(3):329–41. doi:10.1016/j.immuni.2014.01.010
54. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol (2012) 13:823–31. doi:10.1038/ni.2378
55. Williams KL, Taxman DJ, Linhoff MW, Reed W, Ting JP. Cutting edge: monarch-1: a pyrin/nucleotide-binding domain/leucine-rich repeat protein that controls classical and nonclassical MHC class I genes. J Immunol (2003) 170:5354–8.
56. Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem (2002) 277:29874–80. doi:10.1074/jbc.M203915200
57. Jeru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A (2008) 105:1614–9. doi:10.1073/pnas.0708616105
58. Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum (2011) 63:830–9. doi:10.1002/art.30170
59. Jeru I, Hentgen V, Normand S, Duquesnoy P, Cochet E, Delwail A, et al. Role of interleukin-1beta in NLRP12-associated autoinflammatory disorders and resistance to anti-interleukin-1 therapy. Arthritis Rheum (2011) 63:2142–8. doi:10.1002/art.30378
60. Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza Mdo C, Franklin BS, et al. Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog (2014) 10:e1003885. doi:10.1371/journal.ppat.1003885
61. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity (2012) 37:96–107. doi:10.1016/j.immuni.2012.07.006
62. Arthur JC, Lich JD, Ye Z, Allen IC, Gris D, Wilson JE, et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J Immunol (2010) 185:4515–9. doi:10.4049/jimmunol.1002227
63. Meixenberger K, Pache F, Eitel J, Schmeck B, Hippenstiel S, Slevogt H, et al. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol (2010) 184:922–30. doi:10.4049/jimmunol.0901346
64. Tsuchiya K, Hara H, Kawamura I, Nomura T, Yamamoto T, Daim S, et al. Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J Immunol (2010) 185:1186–95. doi:10.4049/jimmunol.1001058
65. Pinheiro AS, Eibl C, Ekman-Vural Z, Schwarzenbacher R, Peti W. The NLRP12 pyrin domain: structure, dynamics, and functional insights. J Mol Biol (2011) 413:790–803. doi:10.1016/j.jmb.2011.09.024
66. Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell (2011) 20:649–60. doi:10.1016/j.ccr.2011.10.022
67. Allen IC, Lich JD, Arthur JC, Jania CM, Roberts RA, Callaway JB, et al. Characterization of NLRP12 during the development of allergic airway disease in mice. PLoS One (2012) 7:e30612. doi:10.1371/journal.pone.0030612
68. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity (2012) 36:742–54. doi:10.1016/j.immuni.2012.03.012
69. Allen IC, McElvania-TeKippe E, Wilson JE, Lich JD, Arthur JC, Sullivan JT, et al. Characterization of NLRP12 during the in vivo host immune response to Klebsiella pneumoniae and Mycobacterium tuberculosis. PLoS One (2013) 8:e60842. doi:10.1371/journal.pone.0060842
70. Arthur JC, Lich JD, Aziz RK, Kotb M, Ting JP. Heat shock protein 90 associates with monarch-1 and regulates its ability to promote degradation of NF-kappaB-inducing kinase. J Immunol (2007) 179:6291–6.
71. Ye Z, Lich JD, Moore CB, Duncan JA, Williams KL, Ting JP. ATP binding by monarch-1/NLRP12 is critical for its inhibitory function. Mol Cell Biol (2008) 28:1841–50. doi:10.1128/MCB.01468-07
72. Wagner RN, Proell M, Kufer TA, Schwarzenbacher R. Evaluation of Nod-like receptor (NLR) effector domain interactions. PLoS One (2009) 4:e4931. doi:10.1371/journal.pone.0004931
73. Zaki MH, Man SM, Vogel P, Lamkanfi M, Kanneganti TD. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc Natl Acad Sci U S A (2014) 111:385–90. doi:10.1073/pnas.1317643111
74. Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity (2012) 36:933–46. doi:10.1016/j.immuni.2012.03.025
75. Lei Y, Wen H, Ting JP. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy (2013) 9:432–3. doi:10.4161/auto.23026
76. Abdul-Sater AA, Said-Sadier N, Lam VM, Singh B, Pettengill MA, Soares F, et al. Enhancement of reactive oxygen species production and chlamydial infection by the mitochondrial Nod-like family member NLRX1. J Biol Chem (2010) 285:41637–45. doi:10.1074/jbc.M110.137885
77. Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci (2009) 122:3161–8. doi:10.1242/jcs.051193
78. Conti BJ, Davis BK, Zhang J, O’Connor W Jr, Williams KL, Ting JP. CATERPILLER 16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. J Biol Chem (2005) 280:18375–85. doi:10.1074/jbc.M413169200
79. Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, et al. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J (2004) 23:4202–10. doi:10.1038/sj.emboj.7600391
80. Madge LA, May MJ. Classical NF-kappaB activation negatively regulates noncanonical NF-kappaB-dependent CXCL12 expression. J Biol Chem (2010) 285:38069–77. doi:10.1074/jbc.M110.147207
81. Kew RR, Penzo M, Habiel DM, Marcu KB. The IKKalpha-dependent NF-kappaB p52/RelB noncanonical pathway is essential to sustain a CXCL12 autocrine loop in cells migrating in response to HMGB1. J Immunol (2012) 188:2380–6. doi:10.4049/jimmunol.1102454
82. Tando T, Ishizaka A, Watanabe H, Ito T, Iida S, Haraguchi T, et al. Requiem protein links RelB/p52 and the Brm-type SWI/SNF complex in a noncanonical NF-kappaB pathway. J Biol Chem (2010) 285:21951–60. doi:10.1074/jbc.M109.087783
83. Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG, et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer (2013) 49:219–30. doi:10.1016/j.ejca.2012.05.005
84. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity (2013) 39:782–95. doi:10.1016/j.immuni.2013.10.003
85. Restifo NP. A “big data” view of the tumor “immunome”. Immunity (2013) 39:631–2. doi:10.1016/j.immuni.2013.10.002
86. Dhawan P, Richmond A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J Biol Chem (2002) 277:7920–8. doi:10.1074/jbc.M112210200
87. Zarnegar B, Yamazaki S, He JQ, Cheng G. Control of canonical NF-kappaB activation through the NIK-IKK complex pathway. Proc Natl Acad Sci U S A (2008) 105:3503–8. doi:10.1073/pnas.0707959105
88. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov (2013) 12:931–47. doi:10.1038/nrd4002
89. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer (2012) 12:401–10. doi:10.1038/nrc3262
Keywords: Nod-like receptors, NLRP12, NLRX1, NLRC3, NF-κB, TRAF, cancer, pattern recognition receptors
Citation: Allen IC (2014) Non-inflammasome forming NLRs in inflammation and tumorigenesis. Front. Immunol. 5:169. doi: 10.3389/fimmu.2014.00169
Received: 22 March 2014; Paper pending published: 24 March 2014;
Accepted: 29 March 2014; Published online: 22 April 2014.
Edited by:
Anton G. Kutikhin, Research Institute for Complex Issues of Cardiovascular Diseases, RussiaReviewed by:
Anton G. Kutikhin, Research Institute for Complex Issues of Cardiovascular Diseases, RussiaArseniy E. Yuzhalin, University of Oxford, UK
Copyright: © 2014 Allen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irving Coy Allen, Department of Biomedical Sciences and Pathobiology, Virginia-Maryland Regional College of Veterinary Medicine, Virginia Polytechnic Institute and State University, IDRF 140, 295 Duck Pond Drive, Blacksburg, VA 24061, USA e-mail: icallen@vt.edu