 Lingling Dai
Lingling Dai Lizhong Du
Lizhong Du- Department of Neonatology, The Children’s Hospital, National Clinical Research Center for Child Health, Zhejiang University School of Medicine, Hangzhou, China
Pulmonary arterial hypertension (PAH) is a rare but progressive and lethal vascular disease of diverse etiologies, mainly caused by proliferation of endothelial cells, smooth muscle cells in the pulmonary artery, and fibroblasts, which ultimately leads to right-heart hypertrophy and cardiac failure. Recent genetic studies of childhood-onset PAH report that there is a greater genetic burden in children than in adults. Since the first-identified pathogenic gene of PAH, BMPR2, which encodes bone morphogenetic protein receptor 2, a receptor in the transforming growth factor-β superfamily, was discovered, novel causal genes have been identified and substantially sharpened our insights into the molecular genetics of childhood-onset PAH. Currently, some newly identified deleterious genetic variants in additional genes implicated in childhood-onset PAH, such as potassium channels (KCNK3) and transcription factors (TBX4 and SOX17), have been reported and have greatly updated our understanding of the disease mechanism. In this review, we summarized and discussed the advances of genetic variants underlying childhood-onset PAH susceptibility and potential mechanism, and the most promising BMPR2 gene therapy and gene delivery approaches to treat childhood-onset PAH in the future.
Introduction
Pulmonary arterial hypertension (PAH) is a rare but progressive and lethal vascular disease with the clinical definition of mean pulmonary arterial pressure >20 mmHg, normal left atrial pressure, and pulmonary vascular resistance ≥3 Wood units (Simonneau et al., 2019; Ruopp and Cockrill 2022). Histopathology of PAH demonstrates that the occlusion of occluded pulmonary arterioles results from the proliferation of endothelial cells, smooth muscle cells in the pulmonary artery, and fibroblasts, which ultimately results in right-heart hypertrophy and cardiac failure (Tuder et al., 2013). The estimated prevalence of childhood-onset PAH is about 4–16 cases per million, with a severe morbidity and high mortality (Li et al., 2017; Rosenzweig et al., 2019). The childhood-onset PAH has its own unique traits which differ from adult-onset PAH in several important aspects including sex bias, disease etiology, clinical presentation, response to therapy, and survival time, with the prevalence of 15–50 cases per million people for adult-onset PAH. In addition, the higher prevalence of PAH among women than men in adult-onset PAH, which is about three- to fourfold, is not observed in childhood-onset PAH, suggesting less dependence upon sex-specific interacting factors in childhood-onset PAH (Barst et al., 2012; Zhu et al., 2018a; Zhu et al., 2019). Childhood-onset PAH often shows slightly higher mean pulmonary arterial pressure, more decreased cardiac output, and increased pulmonary vascular resistance than adult-onset PAH at diagnosis (Zhu et al., 2018a). If left untreated, patients with PAH often undergo the rapid progression, with a median survival of 2.8 years in adults and 10 months in children (Barst et al., 1999; Runo and Loyd 2003).
Etiologically, according to the Pediatric Pulmonary Hypertension Network (PPHN), nationwide Netherlands PH Service, TOPP (Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension) registry, and REHIPED (REgistro depacientes con HIpertensión Pulmonar PEDiátrica), childhood-onset PAH has a higher proportion of idiopathic PAH (IPAH) and PAH associated with congenital heart disease (PAH-CHD), with PAH-CHD constituting nearly 30.6%–60% of children with PAH, and IPAH and heritable PAH (HPAH) comprising about 23%–53.1% and 5%–18.4%, respectively (van Loon et al., 2011; Barst et al., 2012; Abman et al., 2022; Cruz-Utrilla et al., 2022). In addition, according to the PPHN, the majority of PAH in children were characterized by Eisenmenger syndrome in 10%; systemic-to-pulmonary shunts, excluding Eisenmenger syndrome, in 26%; and post-operative PAH in 25% (Abman et al., 2022).
IPAH is a sporadic form of PAH with unknown etiology, which is usually diagnosed in its later stages due to nonspecific symptoms, and the prevalence rate of IPAH in children is lower than that in adults (about 2.1–4.4 cases per million vs. 5.9–25 cases per million) (Moledina et al., 2010; van Loon et al., 2011; Ivy et al., 2013). In the histopathology, adults with IPAH often have severe intimal fibrosis, plexiform lesions, and irreversible pulmonary vascular changes, while children with IPAH have more pulmonary vascular medial hypertrophy, less intimal fibrosis, and fewer plexiform lesions (Barst et al., 2011). The definition of PAH-CHD in children is various heart defects with specific hemodynamic profiles associated with different temporal evolution patterns of pulmonary vascular disease or with congenital maldevelopment of the pulmonary vasculature (van Loon et al., 2011). In addition, the prevalence rate for pediatric PAH-CHD is higher than that reported in adults (about 15.6–21.9 cases per million vs. 1.7–12 per million) (Humbert et al., 2006; Peacock et al., 2007; van Loon et al., 2011; Li et al., 2017). The mortality rate in PAH-CHD was similar to IPAH and HPAH within 5 years, all of which were about 12% and worse than those of adults (van Loon et al., 2011; Ivy et al., 2013; Abman et al., 2022). It should be noted that the PAH genetics community has found HPAH to comprise familial PAH (FPAH, PAH that occurs in two or more family members) and PAH without a family history in a clear genetic diagnosis. Therefore, patients classified as IPAH at diagnosis should be reclassified as HPAH when a causal genetic variant appears (Welch et al., 2021).
Great genetic burden has been observed in childhood-onset PAH, with at least 35% of PAH in children being attributed to rare deleterious genetic factors (Welch and Chung, 2020). Genetic studies have indicated that mutations of some genes involved in the transforming growth factor-β (TGF-β) pathway, including bone morphogenetic protein receptor 2 (BMPR2), activin receptor-like 1 (ACVRL1), and endoglin (ENG), are associated with childhood-onset PAH (Barst et al., 2011; Rosenzweig et al., 2019). To date, there are at least nine genes associated with childhood-onset PAH. Abnormalities in BMPR2 are the most commonly identified mutations in children with PAH, in approximately 50%–80% of FPAH and 10%–40% of IPAH, with a pattern of autosomal dominant inheritance (Zhu et al., 2018a; Rosenzweig et al., 2019; Zhu et al., 2019; Ivy and Frank, 2021). The innovation of gene detection technology has helped to identify novel genes associated with childhood-onset IPAH, including T-box 4 (TBX4), SRY-related HMG box transcription factor (SOX17), mothers against decapentaplegic 9 (SMAD9), potassium two-pore-domain channel subfamily K member 3 (KCNK3), caveolin-1 (CAV1), and growth differentiation factor 2 (GDF2) (Chida et al., 2012; Garcia-Rivas et al., 2017; Zhu et al., 2018a; Morrell et al., 2019; Rosenzweig et al., 2019; Montani et al., 2021). In addition, pediatric patients are more likely to be affected by IPAH and HPAH, suggesting genetic factors play a significant role in the pathogenesis of pediatric PAH (Berger et al., 2012; Hansmann and Hoeper, 2013). Identification of genetic subtypes of PAH along with known biological functions of the genes would provide important data on the natural progression of disease and is beneficial to help improve risk stratification, treatments, correlations of the phenotype and therapeutic responsiveness, and prognosis. Combined with previous studies and PPHN, we reviewed and summarized the identified risk genes contributing to childhood-onset PAH with large amounts of literature evidence to date: BMPR2, TBX4, SOX17, SMAD9, KCNK3, CAV1, GDF2, ACVRL1, and ENG (Table1). In this review, we also discuss the most promising BMPR2 gene therapy and gene delivery approaches for childhood-onset PAH in the future.
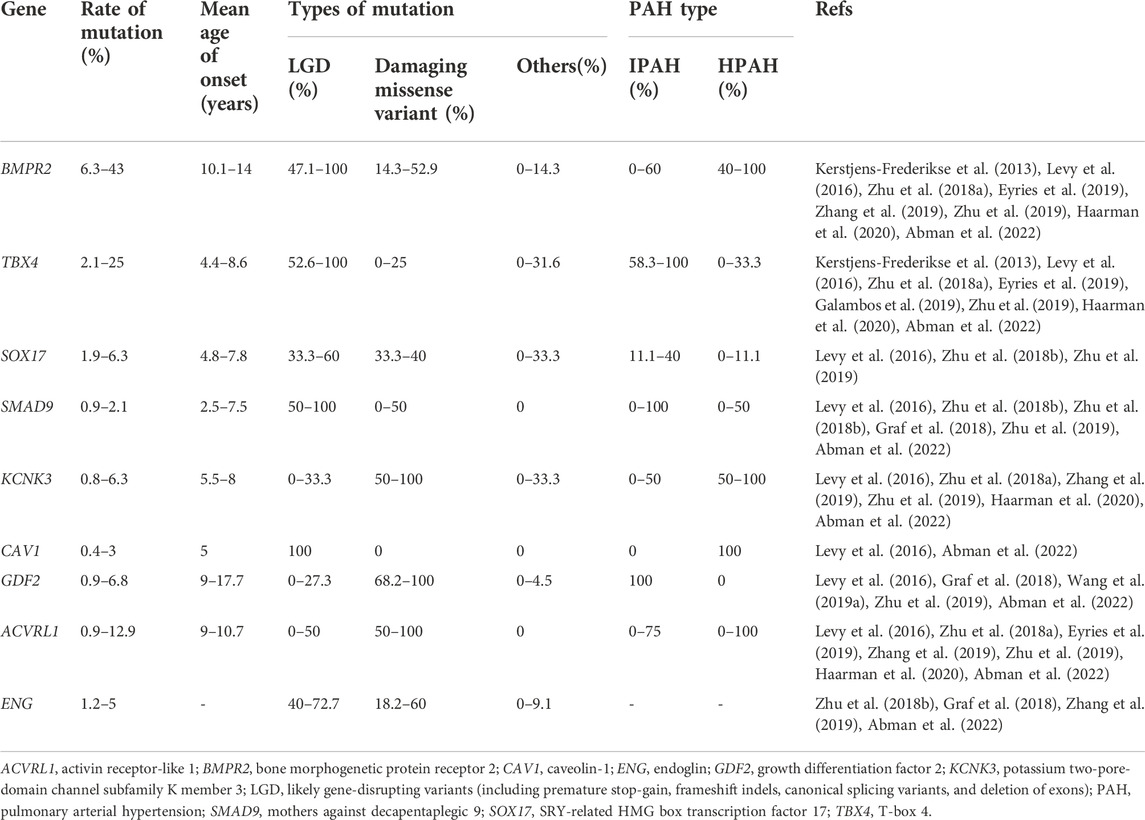
TABLE 1. Summary of genes of childhood-onset PAH.
BMPR2
The detrimental variants in BMPR2 account for nearly 50%–80% of FPAH and 10%–40% of IPAH cases in children (Kerstjens-Frederikse et al., 2013; Levy et al., 2016; Zhu et al., 2018a; Eyries et al., 2019; Zhang et al., 2019; Zhu et al., 2019; Haarman et al., 2020; Abman et al., 2022). The carriers with BMPR2 detrimental variants are always younger, and they have worse hemodynamic characteristics at first consultation. In addition, children with PAH and BMPR2 mutations are unlikely to respond to vasodilators, and it is impossible for them to gain benefit from treatment of calcium channel blockade (Elliott et al., 2006; Rosenzweig et al., 2008).
The mature BMPR2 polypeptide harbors 1,038 amino acids including an extracellular signal peptide, a ligand-binding domain, a single-pass transmembrane domain, an intracellular catalytic kinase domain, and an atypically long cytoplasmic tail, which is a member of the TGFβ superfamily of signaling molecules (Liu et al., 1995; Deng et al., 2000; International et al., 2000). The TGF-β family participates in the control of numerous cellular functions and homeostasis, including proliferation, differentiation, apoptosis, and endothelial–mesenchymal transition (Good et al., 2015). TGF-β is of great importance in the respiratory system, and TGF-β regulates the synthesis of platelet-derived growth factors in endothelial cells, which plays an important role in the process of the vascular smooth muscle cell growth (Roberts and Sporn, 1989; Gore et al., 2014).
The variants of BMPR2 in childhood-onset PAH include missense mutations resulting in the substitutions of amino acids and nonsense mutations, the insertions or deletions of small nucleotides leading to frameshift mutations, gene rearrangements, and splice-site mutations leading to premature protein truncation (Machado et al., 2015; Southgate et al., 2020). It seems that nonsense or frameshift mutations truncate the proteins and result in nonsense-mediated decay, while missense mutations lead to reservation of the mutative proteins in the endoplasmic reticulum or plasma membrane (Frump et al., 2013). Given missense mutations distribute broadly, nearly throughout the BMPR2 exons, and the majority of them are included in key functional domains, especially the ligand-binding domain encoded by exons 2–3 and the highly conservative catalyzed kinase region functionally deciphered by exons 6–9 and 11. In addition, various studies confirming genetic variations in BMPR2 have been limited to the exonic space (Machado et al., 2015; Southgate et al., 2020). Furthermore, BMPR2 haploinsufficiency has been identified as the primary molecular mechanism underlying hereditary PAH.
TBX4
The detrimental variants in TBX4 account for about 2.1%–25% of childhood-onset PAH, and predominantly occur in IPAH cases (Kerstjens-Frederikse et al., 2013; Levy et al., 2016; Zhu et al., 2018a; Eyries et al., 2019; Galambos et al., 2019; Zhu et al., 2019; Haarman et al., 2020; Abman et al., 2022). TBX4 was first suspected as a candidate risk gene of PAH due to the location on chromosome 17q23.1 to 23.2, where microdeletions were associated with severe neurodevelopmental delays and pulmonary hypertension (Ballif et al., 2010; Nimmakayalu et al., 2011). The rare deleterious TBX4 variants have later been reported to have an association with small patella syndrome and PAH (Bongers et al., 2004; Kerstjens-Frederikse et al., 2013). The mean age of onset for the carriers of TBX4 mutations in children with PAH is about 4.4–8.6 years old, which is younger than carriers with BMPR2 mutations (Galambos et al., 2019; Zhu et al., 2019; Southgate et al., 2020). Furthermore, children with PAH who carry TBX4 variants often associate with serious lung, bone, and heart developmental defects (Galambos et al., 2019). However, many PAH cases associated with TBX4 follow a more benign course of disease than BMPR2 carriers (Navas et al., 2016).
T-box 4 (TBX4) is a transcription factor incorporated in the T-box gene family members. It expresses in the lung and tracheal stroma and in the heart atrium. It plays an important role in limb development, lung growth, and branching. According to the animal models’ studies of the respiratory system development, TBX4 has been proven to interact with fibroblast growth factor (FGF)10 during lung growth and branching (Chapman et al., 1996; Sekine et al., 1999; Sakiyama et al., 2003; Arora et al., 2012).
The variants of TBX4 in pediatric PAH include likely gene-disrupting (LGD) variants (including premature stop-gain, frameshift indels, canonical splicing variants, and deletion of exons), damaging missense variants, heterozygous loss-of-function TBX4 mutations and in-frame deletion, and LGD variants (52.6%–100%) and damaging missense variants (0%–25%) as predominant mutations(Kerstjens-Frederikse et al., 2013; Levy et al., 2016; Vanlerberghe et al., 2017; Zhu et al., 2018a). The mutations in TBX4 related to PAH in children are mainly concentrated in key T-box domains, often leading to impaired amino acid displacements or premature truncation of mutational transcripts (Arora et al., 2012; Kerstjens-Frederikse et al., 2013; Levy et al., 2016). The decreased expression lesions of FGF10 are due to the decrease of TBX4 activity, which is the inducer of bone morphogenetic protein (BMP) 4. The expressions of FGF10 and BMP4 are of great significance to activate the BMP pathway in normal lung development, with reports that TBX4-mutant embryos show failure of endothelial cells to form vessels (Naiche and Papaioannou, 2003; Sountoulidis et al., 2012).
Except for the aforementioned evidence, some reports indicate that the BMP pathway promotes differentiation, while the TGF-β pathway pushes the systems to a higher proliferative state. Decreased activation of the BMP pathway might occur owing to the decreased expression of FGF10 and BMP4 when there are TBX4 mutations which can lead to abnormal lung development and disordered BMP/TGF–β/SMAD signaling pathways. The imbalance between the BMP pathway and TGF-β pathway—toward a higher multiplicative status—perhaps ultimately lead to PAH (Archer et al., 2010).
SOX17
Based on the current literature, rare harmful variants in SOX17 account for about 1.9%–6.3% of children with PAH, in particular with PAH-CHD (Levy et al., 2016; Zhu et al., 2018b; Zhu et al., 2019). The mean age of onset for SOX17 variant carriers is younger than that of BMPR2 variant carriers. Furthermore, variants of SOX17 will increase the risk of other vascular endothelium-related diseases.
SOX17 is a transcription factor that promotes angiogenesis and interacts with mature endothelial molecular mediators to reduce the expression of SOX17 in endothelial cells and to restrict angiogenesis through Notch activation (related to the BMPR2 signaling pathway) (Lee et al., 2014; Kim et al., 2016; Hurst et al., 2017). SOX17 is a highly restricted gene encoding Wnt/β-catenin and Notch signaling transcription factors during development, which is also involved in the development of endoderm, vascular endothelium, hematopoietic cells, and cardiomyocytes (Hudson et al., 1997; Alexander and Stainier, 1999; Sekine et al., 1999; Kanai-Azuma et al., 2002; Zhang et al., 2005; Kim et al., 2007; Liu et al., 2007). The endothelial destiny of CD34 progenitor cells is also determined by SOX17 (Francois et al., 2010; Zhang et al., 2017).
The variants of SOX17 in pediatric PAH include LGD variants, damaging missense variants, and heterozygous SOX17 mutation, and LGD variants (33.3%–60%) and damaging missense variants (33.3%–40%) as the predominant mutations (Zhu et al., 2018b; Zhu et al., 2019; Wang et al., 2021; Welch et al., 2021). Genetic studies in mice showed that SOX17 plays a considerable part in the right development and performance of the pulmonary vascular tree. Inactivation of SOX17 in some specific endothelial cells contributes to arterial damage and embryonic death (Corada et al., 2013). Deletion of SOX17 in mice causes disorders of pulmonary vascular morphogenesis, poor distal lung perfusion, and biventricular hypertrophy which further result in postnatal cardiopulmonary insufficiency and infant mortality (Lange et al., 2014). In addition, a mice endothelial lineage tracing study reports that transcriptional activation of SOX17 through hypoxia-induced factor 1α upregulates cyclin-E1 and contributes to endothelial regeneration after injury of the lungs (Liu et al., 2019). Therefore, there are a variety of mechanisms of SOX17 deficiency or deletion which lead to heart and lung developmental defects or impaired response to hypoxia injury.
SMAD9
Rare deleterious variants in SMAD9 lead to approximately 0.9%–2.1% childhood-onset PAH and predominantly occur in IPAH cases (Levy et al., 2016; Zhu et al., 2018a; Zhu et al., 2018b; Graf et al., 2018; Zhu et al., 2019; Abman et al., 2022). The mean age of onset for SMAD9 variant carriers is younger than that of BMPR2 variant carriers and is about 2.5–7.5 years old (Levy et al., 2016; Zhu et al., 2018a; Zhu et al., 2018b; Graf et al., 2018; Zhu et al., 2019; Abman et al., 2022).
SMAD9 encodes SMAD9, an intracellular signal transducer of the TGF-β pathway (van den Heuvel et al., 2020). As a member of the BMP signal cascade, SMAD9 is located in the downstream in the BMPR2 pathway. Furthermore, SMAD9 is related to the nitric oxide biosynthesis process, the synthesis of various oxidoreductases controlling oxidative stress and homeostasis in the cells (Garcia-Rivas et al., 2017). As functional analyses proved, SMAD9 mutation significantly reduced SMAD transcription activity and heterozygous SMAD9 mutations perturbed microRNA (miRNA) processing (Nasim et al., 2011; Machado et al., 2015). Malfunction of the SMAD signal can affect the proliferation of smooth muscle cells and malfunction of SMAD9 can lead to the high proliferation phenotype, weakened BMP signal, and SMAD-mediated microRNA processing disorder, which further contribute to PAH (Yang et al., 2005; Drake et al., 2011). Interestingly, overexpression of wild-type SMAD9 corrects these defects (Drake et al., 2011; Drake et al., 2015).
KCNK3
Rare deleterious variants in KCNK3, coding for two-pore-domain potassium channels which are expressed in pulmonary arterial smooth muscle cells, contribute to approximately 0.8%–6.3% childhood-onset PAH, and predominantly appear in FPAH. The mean age of onset for KCNK3 carriers is about 5.5–8 years old (Levy et al., 2016; Zhu et al., 2018a; Zhang et al., 2019; Zhu et al., 2019; Haarman et al., 2020; Abman et al., 2022).
It is membrane potential that plays a crucial part in pulmonary artery muscle cell (PASMC) contraction, and dysfunction of potassium channels has been implicated in the pathogenesis of PAH. KCNK3 encodes an outwardly rectifying K+ channel that is sensitive to extracellular pH changes. The high expression of KCNK3 is confirmed in PASMCs of rats, rabbits, and humans. The normal function of this channel is to induce the leakage K+ current, to keep the resting membrane potential, and to contribute to the regulation of the pulmonary vascular tone (Olschewski et al., 2017). The K+ channel is activated to induce K+ outflow, membrane hyperpolarization, and vasodilation (Olschewski et al., 2006; Olschewski, 2010). Genetic and electrophysiological studies suggest that KCNK3 mutation, leading to downregulation of K+ channels, is a rare genetic cause of PAH (Ma et al., 2013). The decrease in activity in the PASMC K+ channel can contribute to cell multiplication, anti-apoptosis, and vasoconstriction, which ultimately leads to the remodeling of vessels (Le Ribeuz et al., 2020).
CAV1
Rare deleterious variants in CAV1, encoding a specific protein which is essential for the integrity of the plasma membrane and the production of nitric oxide, are related to a small number of PAH cases, and contribute to approximately 0.4%–3% childhood-onset PAH. The mean age of onset for CAV1 carriers is about 5 years old (Levy et al., 2016; Zhu et al., 2018a; Zhang et al., 2019; Zhu et al., 2019; Abman et al., 2022).
Caveolae, a specific protein in the membrane, encoded by CAV1, are enriched in mesenchymal cells and lung endothelial cells and are essential for mediating the cascade reaction of TGF-β, G protein, and nitric oxide signal in PAH (Maniatis et al., 2008). Abundant expression of CAV1 is demonstrated in adipocytes, endothelial cells, and fibroblasts (Quest et al., 2004). Truncating mutation, de novo mutation, and frameshift mutation have been described in CAV1-associated PAH (Southgate et al., 2020). Frameshift mutations of CAV1 in PAH cases, revealed by functional analysis, can cause the mutant protein to retain in the endoplasmic reticulum, which is accompanied by the retention of wild-type protein morphology, and furthermore results in significant damage to cell membrane caveolae assembly (Copeland et al., 2017). Furthermore, truncating mutation in CAV1 causes the excessive phosphorylation of SMAD1, SMAD5, and SMAD9, which leads to the decrease in the antiproliferative function of caveolin-1, thus demonstrating that functional gain SMAD may be one of the potential molecular mechanisms of CAV1-associated PAH (Marsboom et al., 2017). In addition, BMPR2 is located in the cell membrane caveolae and interacts directly with the caveolin-1 in different cellular types, consisting of smooth muscle cells of vessels; therefore, the mutations in CAV1 may also cause PAH by interfering with TGF-β signaling (Hartung et al., 2006; Wertz and Bauer, 2008).
GDF2
Rare deleterious variants in GDF2 contribute to approximately 0.9%–6.8% childhood-onset PAH (Levy et al., 2016; Graf et al., 2018; Wang et al., 2019a; Zhu et al., 2019; Abman et al., 2022). Nevertheless, progenitor-specific factors are likely to work in PAH, with GDF2 being a dominant gene in Asian childhood-onset PAH patients compared to European patients (Wang et al., 2019a; Welch and Chung, 2020; Welch et al., 2021). Furthermore, GDF2 has a strong association with IPAH cases and has been identified as one of the leading disease-associated genes in Asian childhood-onset PAH and is secondary to BMPR2 (Wang et al., 2019a).
Expressed principally in the liver, GDF2 is a constituent secreted into circulation. GDF2 plays an important role in regulating vascular biology and angiogenesis by binding to ACVRL1 and BMPR2, and the optimal circulating GDF2 level is crucial for maintaining the resting state of pulmonary vessels. GDF2 is one of the members of the BMP subgroup of the TGF-β superfamily proteins, and its active form in circulation is essential for regulating vascular tension (Du et al., 2003; David et al., 2008). The PAH-specific mutations in GDF2 include frameshift mutations, missense mutations, and nonsense mutations, and these mutations may inhibit biosynthesis and/or secretion of GDF2, leading to a decrease in GDF2 levels in the circulation in PAH patients which may be a cause of PAH (Wang et al., 2019a; Southgate et al., 2020). Furthermore, the results of cell experiments demonstrate pre-treatment of WT GDF2-conditioned medium protects PAECs from apoptosis induced by tumor necrosis factor-α, while these mutants are unable to prevent PAEC apoptosis (Wang et al., 2019a).
ACVRL1
According to the previous studies, rare deleterious variants in ACVRL1 are expressed in approximately 0.9%–12.9% childhood-onset PAH (Levy et al., 2016; Zhu et al., 2018a; Eyries et al., 2019; Zhang et al., 2019; Zhu et al., 2019; Haarman et al., 2020; Abman et al., 2022). The pediatric studies reveal a higher carrier frequency of ACVRL1 variants in Asian childhood-onset PAH than that of predominantly Europeans (6.1%–12.9% vs. 0.9%–2.6%) (Chida et al., 2012; Zhu et al., 2018a; Zhang et al., 2019; Zhu et al., 2019; Haarman et al., 2020). Studies also demonstrate that ACVRL1 has exome-wide extensive mutation enrichment in IPAH cases. Furthermore, BMPR2, GDF2, and ACVRL1 have been identified as the top three disease-related genes in Asian childhood-onset PAH (Wang et al., 2019a). PAH patients harboring ACVRL1 mutations have earlier onset, faster disease progression, and are younger at death than those with BMPR2 mutations (Girerd et al., 2010).
Studies showed that most mutations in ACVRL1 in PAH cases are missense mutations (Machado et al., 2009). Related literature data confirm that ACVRL1 mutation carriers carrying PAH have dominant mutations in exon 10, especially in NANDOR boxes, which are essential for the regulation of TGF-β signaling (Harrison et al., 2005; Girerd et al., 2010). ACVRL1 encodes the BMP receptor, ACVRL1, whose expression is mainly restricted to the vascular and lymphatic endothelia (Seki et al., 2003; Niessen et al., 2010). The disruption of the receptor complex transport was the main cause of the vessel phenotype of PAH deciphered by the functional studies using ACVRL1 mutants (Harrison et al., 2003; Harrison et al., 2005). Further studies show ACVRL1 mutants of PAH are related to the downregulation of SMAD1/5/9 signaling and confirm that the ACVRL1 mutants have negative effects on SMAD1/5 function (Piao et al., 2016). Therefore, ACVRL1 mutations may mainly impact the regulation of the TGF-β signalization pathway. TGF-β signaling pathway dysfunction could promote dysfunction and proliferation of endothelium and/or smooth muscle cells in pulmonary vessels (Trembath et al., 2001).
ENG
Rare deleterious variants in ENG are seen more rarely and contribute to approximately 1.2%–5% childhood-onset PAH (Zhu et al., 2018b; Graf et al., 2018; Zhang et al., 2019; Abman et al., 2022). The mutations in ACVRL1 or ENG genes primarily lead to PAH associated with hereditary hemorrhagic telangiectasia (HHT), which is a rare autosomal dominantly genetic disease. HHT is caused by telangiectasia enzymes and malformations of arteriovenous, including malformations of pulmonary arteriovenous. Due to different lesion sites, clinical manifestations of malformations of pulmonary arteriovenous are varied, which are primarily brought about by default in type I receptor ACVRL1 and default in type III accessory receptor ENG (Girerd et al., 2010; Ma and Chung, 2017; Southgate et al., 2020).
ENG is one of the members of the TGF-β signaling family and is expressed as the disulfide bond stable homodimer membrane-anchored proteoglycan, which is a marker of the proliferating endothelium. ENG is also an endothelial-specific receptor of BMPs, which lacks the intracellular kinase activity and acts as a co-receptor in complex with ACVRL1 (Saito et al., 2017). ENG shedding releases a soluble form of ENG through proteolytic enzyme cleavage of the extracellular domain of the receptor (Venkatesha et al., 2006; Bernabeu et al., 2009). ENG has been typified as a secondary receptor of the TGF-β–ACVRL1 signal in endothelial cells, which is crucial for regulating endothelial proliferation and migration (Jin et al., 2017; Sugden et al., 2017).
BMP9–BMP10 heterodimer is the high-affinity ligand of a receptor complex including ACVRL1, ENG, and BMPR2 (David et al., 2007; Scharpfenecker et al., 2007; Tillet et al., 2018). When BMP9/10 is activated, the ACVRL1–ENG–BMPR2 receptor complex phosphorylates SMAD1/5/9 and induces the expression of growth factors such as fibroblast growth factor 2 and platelet-derived growth factor B39. Dimers of phospho-SMAD1, phospho-SMAD5, or phospho-SMAD9 associate in a trimeric complex with SMAD4 and translocate into the nucleus of the endothelial cell where they bind to BMP-responsive elements on the promoters of target genes to either enhance or repress their expression (Garcia de Vinuesa et al., 2016; Goumans et al., 2018). The synthesis process of platelet-derived growth factors is regulated by TGF-β through the non-canonical pathway (C-terminal SRC kinase or mitogen-activated protein kinase) in endothelial cells, which can stimulate the growth of vascular smooth muscle cells (Roberts and Sporn, 1989). In addition, HHT is thus now considered by some researchers as a disease of the BMP9/10 pathway rather than a disease of the TGF-β pathway (Tillet and Bailly, 2014).
BMPR2 gene therapy and gene delivery systems
Although important advances in therapeutic management have shown that the quality of life and hemodynamic parameters of PAH have been improved, their benefits to survival and progression of PAH are very limited, as genes influencing PAH are unaffected. They change cell phenotypes and participate in the pathogenesis of PAH by improving the characteristics of hyperproliferation, promoting anti-apoptosis of pulmonary vascular cells, and regulating calcium equilibrium (Leopold and Maron, 2016; Rai et al., 2021). In the past decades, a series of new, mighty technologies enabled precise gene editing to accurately intervene in certain specific steps of the disease. These help to develop safe and effective PAH gene therapies to repair mutations and to recover or downregulate gene expression (Austin et al., 2017). These new regeneration methods have shown great potential in preclinical research studies, but large-scale, well-designed clinical trials are still necessitated for further evaluation of clinical efficacy and safety.
Although at least nine genes are associated with childhood-onset PAH nowadays, BMPR2 has been identified by a body of evidence as the major risk PAH gene, and approximately 50%–80% of HPAH and 10%–40% of IPAH are thought to be driven by BMPR2 mutations; thus, restoring BMPR2 expression to cure PAH seems to be the most reasonable and important approach of genetic therapy (Zhu et al., 2018a; Rosenzweig et al., 2019; Zhu et al., 2019; Ivy and Frank, 2021; Rai et al., 2021; Cruz-Utrilla et al., 2022). In addition, restoration of BMPR2 expression using gene therapy has shown promising results in animal models under research settings. Therefore, we summarized and discussed the advances of BMPR2 gene therapy.
Gene delivery systems
Viral vectors
Retroviral and lentiviral vectors
The retroviruses and lentiviruses are single-stranded RNA viruses characterized by two long-terminal repeated sequences at either end of the genome and a packaging sequence, with the packaging capacity which can be up to 8 kb through a transgenic cassette inserted in a space generated by eliminating the natural viral components (Gag, Pol, and Env) (Szokol et al., 2004; Gandara et al., 2018). The viral RNA is converted into double-stranded DNA by a process of reverse transcription, which can be integrated into the chromosomal DNA of the host in a random way within the viral life cycle through the function of the viral integrase and can prolong and stabilize gene expression (Driskell and Engelhardt, 2003).
The first disadvantage of transfection for retroviral vectors in PAH is that retroviral vectors can only infect the target cells with the actively replicating property; however, the rate of cell proliferation in the lungs is low. In contrast, lentiviruses can transduce quiescent, post-mitotic cells with an efficient and economical transduction rate (Aneja et al., 2009; Lee et al., 2017). However, the requirement of the lentivirus-specific receptor, which is low and localized on the apical surface of the epithelial cells in the lungs, restricts the application of lentiviral vectors for the delivery of lung genes (Thomas et al., 2003; Waehler et al., 2007). A recent strategy to conquer this problem is to utilize pseudotyped lentiviral vectors derived from various origins, for example, Ebola, Sendai virus, influenza, and parainfluenza. Moreover, recent studies have found that G64-pseudotyped and Sendai virus-pseudotyped lentiviral vectors can transduce lung epithelial cells through the apical surface (Ferrari et al., 2007; Mitomo et al., 2010; Sinn et al., 2012). Several preclinical studies have shown that transduction with lentiviral vectors is safe; however, the random integration of viral DNA risks the insertional mutagenesis, the activation of cellular oncogenes, and the inactivation of tumor suppressor genes, which can further lead to tumor formation. Therefore, the long-term safety and efficacy of lentiviral vectors need further clinical studies (Anson, 2004; Mitomo et al., 2010; Alton et al., 2017).
Adenovirus vectors
The recombinant adenovirus (Ad) carries double-stranded DNA characterized by a natural tropism of the respiratory tract, an infection character for both quiescent and proliferating cells, relatively easy to produce and purify at a large scale, making them efficient vectors to transfer the lung gene. The double-stranded DNA of the adenovirus can escape from endolysosomal pathways and remains episomal; therefore, the risk of insertional mutagenesis in retroviral and lentiviral vectors can be avoided in Ad vectors, and the persistence of the therapeutic genes depends on the proliferation of the target cell. As a result, the expression of the transgene will be diluted if the cell proliferates with only one of the daughter cells containing the Ad genome (Driskell and Engelhardt, 2003; St George, 2003). The entry for Ad into the cells is through receptor-mediated endocytosis; however, the receptors are mainly located in the basolateral surface which limits the interactions between vectors and receptors due to the tight junctions of epithelial cells. Moreover, the highly immunogenic nature of Ad vectors can evoke the host immunity to Ad vectors, which limits the gene expression to 2–3 weeks and results in a low level for transfer of the therapeutic gene (Murata et al., 2005; Fausther-Bovendo and Kobinger, 2014). Therefore, the repeated administration on Ad vectors for effectively chronic expression of the therapeutic genes is unsuitable. Except for the aforementioned limitations, there is a body of evidence about the safety use of Ad vectors for lung gene therapy in preclinical rodent models (Chattergoon et al., 2005; Parker et al., 2013; Green et al., 2017).
In order to prolong the expression of transgenes and reduce the immune response of the host, second-generation Ad vectors have been developed by reducing the expression of viral antigens which is caused by deleting the E2 and E4 viral genes (Engelhardt et al., 1994; Chirmule et al., 1998; Cao et al., 2004). In experimental animals, these new vectors could minimize but not delete vector-induced inflammation response, which may be mediated by the antigenic capsid and the low-level expression of late viral genes (Chirmule et al., 1998). Currently, the “gutted/helper-dependent” adenovirus has been engineered in the attempt to further reduce Ad vector immunogenicity by eliminating all viral coding sequences. In experimental animal models, these new vectors showed longer transgene expression with a reduced immunity response of the hosts and the 36-kb packaging capacity (Jozkowicz and Dulak, 2005; Palmer and Ng, 2005; Brunetti-Pierri and Ng, 2008; Vetrini and Ng, 2010).
Adeno-associated virus vectors
Adeno-associated viruses (AAVs) are nonpathogenic, single-stranded DNA parvoviruses with self-replication defects, and AAV vectors are one of the most investigated gene transfer approaches. The recombinant AAV (rAAV) was developed by removing the Rep and Cap genes of the wild-type AAV and inserting the therapeutic transgene cassette, which is engineered as the gene therapy vector with the packaging capacity of up to 5 kb, a better transduction efficiency and long-term expression of the transgene in quiescent and proliferating cells and no risk of insertional mutagenesis because the rAAV persists in the nucleus as an episome (Kearns et al., 1996; Flotte, 2005; Li and Samulski, 2020). In addition, AAV vectors show low cytotoxicity and immunogenicity (Hernandez et al., 1999). Moreover, a body of evidence in experimental animal models has shown AAV can be used to transfer genes to the airway epithelium, alveolar epithelium, pulmonary vascular endothelium, and pleural mesothelioma (Han et al., 2008; Schultz and Chamberlain, 2008; Watanabe et al., 2010; Sondhi et al., 2017).
Nowadays, more than 100 AAV serotypes have been developed since the initially used two AAV serotypes, namely, 2 and 5, in order to improve the tropism of cells and tissues, magnify the therapeutic application fields, and reduce off-target effects (Lisowski et al., 2015). One of the disadvantages of this vector is that AAVs have little room to incorporate large genes because the genome is small. Recently, the strategy of using dual AAV vectors may have provided an approach to solving the limitation of packaging capacity (Halbert et al., 2002; Cooney et al., 2019). The AAV vector packaging capacity increases by developing a parvovirus chimera, with the rAAV genome packaged in the capsid of another parvovirus, such as human bocavirus (HBoV) or gorilla bocavirus (GBoV), and studies have shown the effective transduction efficacy of rAAV/HBoV and rAAV/GBoV in primary human airway epithelial cells, lung organoids, and ferret lungs (Yan et al., 2013; Yan et al., 2017; Fakhiri et al., 2019). Another drawback is that human beings can exhibit neutralizing antibodies to AAVs because they have already been exposed to various AAV serotypes, which further impairs the gene transfer (Schaffer and Maheshri, 2004). The solution strategy that has been put forward to solve this problem is the use of new site-mutated AAVs. Currently, studies have confirmed that these new site-mutated AAVs, such as site-directed mutagenesis (tyrosine to phenylalanine), could evade the immune response due to the pre-existing neutralizing antibodies and improve the AAV tropism and transduction efficiency as well (Wu et al., 2006; Zhong et al., 2008; Lompre et al., 2013).
Other viral vectors
A number of other viruses, such as polyomaviruses, vaccinia virus, baculovirus, and Sendai virus, have been used in lung gene therapy. Polyomaviruses, including simian virus 40 and John Cunningham virus, can deliver long-term expression of transgene in quiescent and proliferating cells by insertion into the host genome with relatively low immunogenicity. In addition, these vectors can be produced and purified in high titers (Vera and Fortes, 2004; Chang et al., 2011). Major concerns about these vectors are the limited packaging capacity of about approximately 2.5–5 kb and the risk of random integration (Grieger and Samulski, 2005).
Vaccinia virus vectors are another potential tool for lung gene transfer, whose packaging capacity can be up to 25 kb, with the characteristics of infection for most cells. Major concerns about vaccinia virus vectors are the induced cytopathic effects, immune responses, and the limited efficacy due to neutralizing antibodies to infections (Guo and Bartlett, 2004; Gregory et al., 2011).
Baculovirus expression vectors, which were initially used for recombinant protein expression, have the potential to serve as the vectors for gene therapy, as they can carry up to 38 kb DNA inserts and transfect various cell types without evoking immune responses in humans and without the risk of insertional mutagenesis. However, the major drawbacks of the vectors are the limited expression duration of the transgene, the requirement of entry through the basolateral membrane, and the rapid virus inactivation by serum complements, which should be further studied to optimize (Hu, 2006; Wang et al., 2020).
Vectors based on the Sendai virus have been developed for the transfer of lung gene with a natural tropism for respiratory epithelial cells and relatively high transduction efficiency (Inoue et al., 2004). However, the transgene expression is transient and the repeated administration can result in diminished gene expression. Currently, the strategy to solve these limitations is pseudotyping Sendai virus envelope proteins to lentiviruses for efficient transfection and sustained gene expression in lung cells (Griesenbach et al., 2012).
Non-viral vectors
Various non-viral vectors have been developed to deliver the therapeutic genetic material as naked DNA or RNA, including mRNA, siRNA, and microRNA, with the aim of enhancing the efficiency of entry into cells. Non-viral vectors can successfully overcome the constraints of the viral vectors, including the packaging capacity of the expression cassette, immunogenicity, biosafety risks, and the risk of insertional mutagenesis. RNA-based gene therapy does not require nuclear localization or transcription, whereas the DNA-based gene therapy requires translocation into the nucleus. An additional benefit is the negligible risk of genomic integration of the delivered sequence. The major limitations of non-viral vectors cannot be neglected, including the transient expression of therapeutic genetic material, the low tropism, and a low transfection efficiency in vivo, while they have significant advantages (Al-Dosari and Gao, 2009; Hill et al., 2016; Durymanov and Reineke, 2018).
Liposome reagents are a class of heterogeneous compounds prepared by combining phospholipids with fatty acid tails and hydrophilic head groups. Liposomes binding to plasmids have been widely used to enhance the internalization of the genetic material into the cytoplasm by endocytosis (Allen and Cullis, 2013; Rezaee et al., 2016). There are two extensively studied classes of liposomal vectors, anionic liposomes, and cationic liposomes. The anionic liposomes cannot directly connect to DNA; thus, DNA must be encapsulated inside the aqueous solution of the vesicle, which limits the packaged size of DNA and must require receptor-mediated endocytosis. The cationic liposomes with a positive charge binding to DNA by electrostatic interaction have larger packaging capacity than anionic liposomes. Currently, strategies to ameliorate liposomes are to permit targeting specific cell types because the liposomes adhere to cells non-specifically and further undergo endocytosis (Gallego et al., 2019).
Nanoparticles comprise the nucleic acid complexed with other materials, such as lipid or polymers including peptides or polysaccharides (Yin et al., 2014). Solid lipid nanoparticles maintain a solid state at physiological temperatures, can be prevented from nucleic acid degradation by the nucleases, and are often used to deliver siRNA (Sondhi et al., 2017). Polymer-based nanoparticles are more stable compared to lipids or liposomes, including natural or synthetic cationic polymers compounded with DNA. Due to higher efficiency of synthetic polymers, such as polyethyleneimine (PEI) or polyethylene glycol (PEG), they are often used to transfer genes despite the risk of cytotoxicity (Yin et al., 2014). Lipid nanoparticles (LNPs) are another fast and highly efficient method of transfecting cells by combining lipid-based components with siRNA or modRNA, and the combination of organic and aqueous components induces hydrophilic–hydrophobic interactions leading to nanoparticle formation (Reichmuth et al., 2016). In addition, the lipidoid components can be optimized by combinatorial chemistry to achieve higher stability of LNPs (Turnbull et al., 2017). However, the expression is transient, and the study showed that expression dropped to almost negligible levels in the second week after administration, which limits this method to the indications for feasible repeated administration or the preferred transient expression (Turnbull et al., 2016) (Table 2).
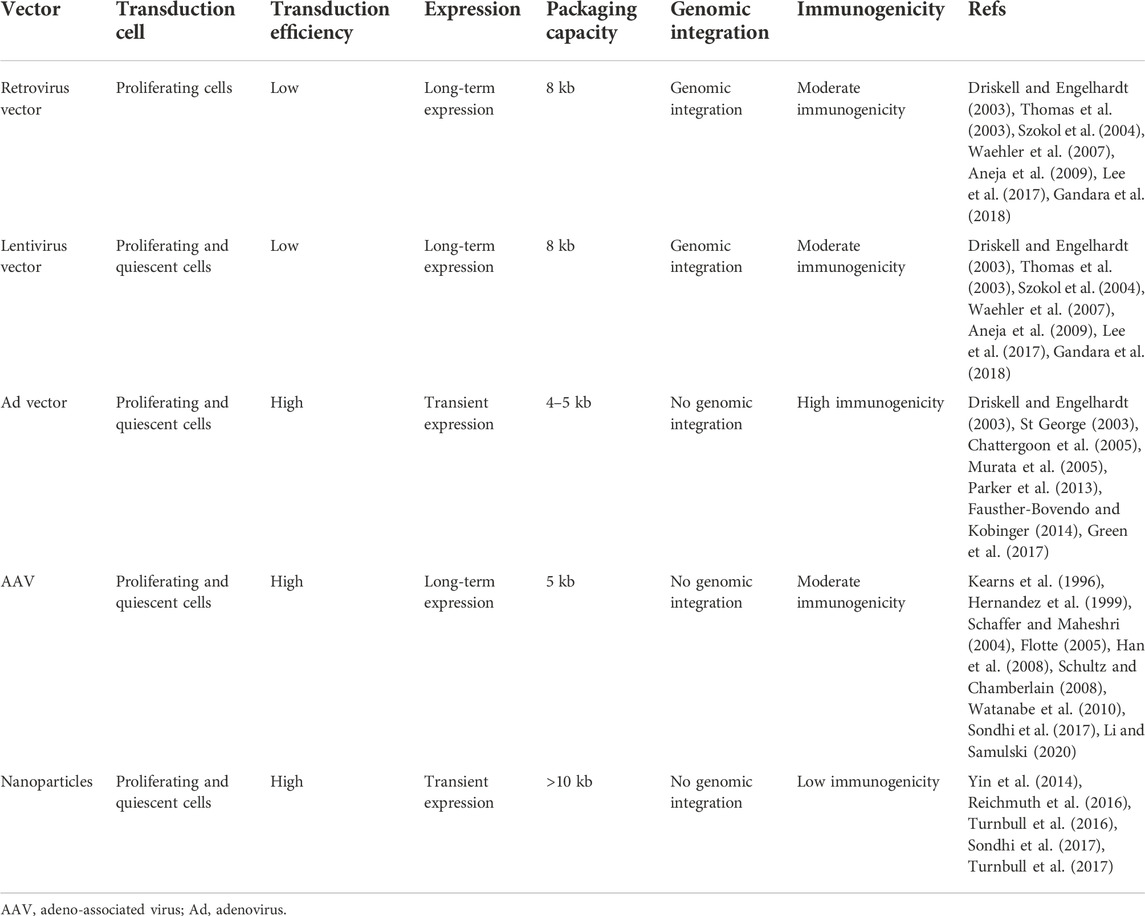
TABLE 2. Summary of vectors for gene transfer.
CRISPR/Cas9 as a gene-editing tool
The focus of gene therapy is to express exogenous DNA to cure diseases caused by gene mutations. CRISPR/Cas9 is a newly emerging, promising gene-editing technology which uses guide RNAs that have complementary sequences to the target site, which attempts to edit and repair mutated genes in vivo, insert new genes, or delete unwanted ones. Gene editing depends on homology-targeted repair or non-homologous DNA end joining, using the cellular mechanism of double-strand breaks in genomic DNA (Sondhi et al., 2017). The formation of the heterologous double-stranded complexes between guide RNA and DNA allows Cas9 nuclease to cleave, making it a simple and effective tool for gene editing (Alapati and Morrisey, 2017). The main drawback of this tool is the need for an adjacent protospacer motif and NGG sequence (Zhang et al., 2014). In addition, CRISPR/Cas9 can be used to regulate the reversible epigenetics of targeted genes. Enzyme-inactivated Cas9 can fuse with activator or repressor domains to activate or silence the target gene expression, respectively. Furthermore, CRISPR/Cas9 can be used for base editing. By fusing the inactivated Cas9 with other deaminases, a nitrogen-containing base can be replaced without producing a double-strand break (Maeder et al., 2013; Qi et al., 2013; Komor et al., 2016).
Preclinical studies have evaluated the therapeutic effects of CRISPR/Cas9 in a number of diseases, including congenital genetic lung diseases, infectious diseases, lung cancer, and immune diseases (Alapati et al., 2019; Ferdosi et al., 2019; Mohammadzadeh et al., 2020; Nair et al., 2020). Moreover, a recent study has reported that CRISPR/Cas9 can edit Bmpr2 in rats (Kabwe et al., 2022). In addition, a clinical trial including 12 patients with metastatic non-small-cell lung cancer reported the clinical safety for the use of CRISPR/Cas9 gene-edited T cells targeting the PD-1 (Cyranoski, 2016). CRISPR/Cas9 holds tremendous potential, but further studies are needed to ensure the specificity of cleavage sites, avoid unexpected off-target effects, improve the efficiency of nuclease delivery, and guide RNAs to target cells.
BMPR2 gene therapy
In 2007, Reynolds and others demonstrated that in rodent experimental models, Bmpr2-containing Ad vector targeting pulmonary vascular endothelial cells could alleviate hypoxic pulmonary hypertension (Reynolds et al., 2007). The investigators surprisingly found that transfer of the Bmpr2 gene can tremendously increase the BMPR2 expression, which increases Smad1/5/8 signaling and reduces the Smad2/3 signaling pathway, and significantly alleviate right ventricle hypertrophy and systolic blood pressure, mean pulmonary arterial pressure, and the hypoxia-induced vascularization (Reynolds et al., 2007). In 2016, other investigators used intravenous injection of Ad-mediated endothelial Bmpr2 gene therapy to reverse PAH in mice carrying a Bmpr2 mutation, as shown by reduced right ventricle systolic pressure and hypertrophy. In addition, they also found that the effects of BMPR2 regulation on non-SMAD signaling increased phosphoinositide-3 kinase, reduced phosphorylated-p38-mitogen-activated protein kinase, and upregulated nitric oxide production in the microvascular endothelial cells, which were related to relief of PAH, such as the decrease in right ventricular, mean pulmonary artery pressure and Fulton Index (Feng et al., 2016; Harper et al., 2016). Meanwhile, two other studies used the same monocrotaline-induced PAH model to explore the effects of Bmpr2 gene therapy but found inconsistent results. One study found the intratracheal delivery of an Ad vector containing the Bmpr2 gene did not cure the disease, despite the good distribution of the gene in the arteriolar network. Another study showed that intravenous injection of Ad-mediated endothelial Bmpr2 gene therapy significantly alleviated right ventricular hypertrophy and pulmonary artery pressure (McMurtry et al., 2007; Harper et al., 2016). The different results may suggest the importance of targeting the endothelium for BMPR2 gene therapy.
However, Ad vectors in these study limit their clinical use due to the risks of insertional mutagenesis reasons and the inflammatory response (Driskell and Engelhardt, 2003; Lee et al., 2017). Recent advances in the gene delivery technology develop new AAV vectors (Daya and Berns, 2008; Naso et al., 2017). At present, there are many AAV variants that show specific tropism characteristics, and moreover, the delivery of AAV-based gene therapy has high, long-term transduction and limited immunogenicity in experimental animals, showing a good application prospect (Mingozzi and High, 2013; Watanabe et al., 2018; Wang et al., 2019b; Bisserier et al., 2020). Currently, this strategy is investigated in preclinical studies of pulmonary hypertension.
Challenges and future directions of pulmonary gene delivery in PAH
PAH is a fatal disease, and there is a growing interest in human gene therapy. The lung is an attractive tissue for gene therapy interventions because various gene delivery strategies, including intranasal, intravenous, intratracheal instillations, or aerosols, have been used for various lung diseases (Weiss, 2002). There is substantial evidence that preclinical PAH gene therapy successfully restores gene expression defects, corrects dysfunctional pathways, improves pulmonary vascular remodeling, inhibits disease progression, and reverses established diseases (Zhao et al., 2006; Reynolds, 2011; Katz et al., 2019). Intratracheal administration via aerosol delivery to the lungs, including intratracheal instillation or inhalation with or without bronchoscopy, showing low endonuclease activity, minimizing systemic side effects, and avoiding liver metabolism and liver absorption, serves as one of the favorable gene transfer pathways (Auricchio et al., 2002; Henning et al., 2010; Katz et al., 2019).
There are some advantages to intravenous administration of non-viral vectors entering the lung, including an almost unlimited packaging capacity, safe application, low immunogenicity, and the ability to simultaneously package both RNA species and DNA. However, various non-viral vectors are less efficient in gene transfer and provide only transient gene expression, and this obstacle can be overcome by repeated injections (Ramamoorth and Narvekar, 2015; van Haasteren et al., 2018). The insertion of tissue-specific DNA nuclear input signals found in some promoter sequences or the combination with the targeting peptide portion showed precise and enhanced cell targeting in the lung (Degiulio et al., 2010; Manunta et al., 2017). Driven by inflammatory response and hypoxia, PAH-lung vascular endothelial dysfunction and increased vascular permeability result in focal rupture of the endothelial basement membrane, and elevated vascular permeability contributes to enhanced nanoparticle accumulation in the diseased lung. Therefore, the nanoparticle-based gene therapy approach serves as a promising strategy for the treatment of PAH (Alton et al., 2015; Ramamoorth and Narvekar, 2015; Manunta et al., 2017).
Although virus vectors have good transduction efficiency, there are still some disadvantages to these vectors, such as the complex production methods, the immunogenicity and cytotoxicity, and the time-consuming and resource-consuming process that requires extensive experience to achieve consistent results for the concentration and titration of Ad, lentivirus, and AVV virus particles. Lentiviruses can randomly insert viral DNA into the host DNA, which leads to the risk of the insertional mutagenesis, the activation of cellular oncogenes, and the inactivation of tumor suppressor genes. Therefore, the extensive detection of these viruses in vector products and in patients is required by many regulatory agencies (Alton et al., 2017; Anson, 2004; Milone and O'Doherty, 2018). Ad vectors are highly effective in transduction of most cell types and tissues, but their capsids mediate potential inflammatory responses. Studies have shown that gene transfer with Ad vectors can lead to the induction of host immune response and apoptosis of vascular endothelial cells, indicating that pulmonary vascular endothelium exhibits specific antiviral immune activity that affects low gene transfer. In this respect, the production of helper-dependent Ad, characterized by more sustained gene expression, reduced immunogenicity, and eliminated capsid-mediated intense inflammatory responses is encouraging for effectively targeting airway basal cells in vivo (Murata et al., 2005; Brunetti-Pierri and Ng, 2006; Cao et al., 2018). AAVs have many benefits over the other vectors, including better transduction efficiency, long-term expression of the transgene, and relatively low immune responses and toxicities in vivo, and gene transfer with the AAV approach is currently among the most frequently used viral vectors systems. Drawbacks of AAV vectors include little room for AAVs to incorporate large genes, neutralizing antibodies to AAVs existing in human beings which further impair the gene transfer, and difficulties in manufacturing of pure AAVs (Schaffer and Maheshri, 2004; Moss et al., 2007; Daya and Berns, 2008; Katz et al., 2017). The discovery of new serotypes, an understanding of the tropism of each, and the subsequent generation of hybrid serotypes or capsid mutants can extensively improve the potential effectiveness of AAVs and will increase the effect of infecting only specific cell types in the lung. There have been many preclinical trials on successful delivery and transduction of AAVs for the safety and long-term efficacy of AAVs (Chamberlain et al., 2016; Verdera et al., 2020). Therefore, we believe that AAV-based therapy may be the most promising tool in the aforementioned approaches that can be safely applied to PAH studies.
Author contributions
LLD performed the literature search and edited the manuscript; LD contributed to the design and revision of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81630037).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abman, S. H., Mullen, M. P., Sleeper, L. A., Austin, E. D., Rosenzweig, E. B., Kinsella, J. P., et al. (2022). Characterisation of paediatric pulmonary hypertensive vascular disease from the PPHNet Registry. Eur. Respir. J. 59, 2003337. doi:10.1183/13993003.03337-2020
Al-Dosari, M. S., and Gao, X. (2009). Nonviral gene delivery: Principle, limitations, and recent progress. AAPS J. 11, 671–681. doi:10.1208/s12248-009-9143-y
Alapati, D., and Morrisey, E. E. (2017). Gene editing and genetic lung disease. Basic research meets therapeutic application. Am. J. Respir. Cell Mol. Biol. 56, 283–290. doi:10.1165/rcmb.2016-0301PS
Alapati, D., Zacharias, W. J., Hartman, H. A., Rossidis, A. C., Stratigis, J. D., Ahn, N. J., et al. (2019). In utero gene editing for monogenic lung disease. Sci. Transl. Med. 11, eaav8375. doi:10.1126/scitranslmed.aav8375
Alexander, J., and Stainier, D. Y. (1999). A molecular pathway leading to endoderm formation in zebrafish. Curr. Biol. 9, 1147–1157. doi:10.1016/S0960-9822(00)80016-0
Allen, T. M., and Cullis, P. R. (2013). Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 65, 36–48. doi:10.1016/j.addr.2012.09.037
Alton, E., Armstrong, D. K., Ashby, D., Bayfield, K. J., Bilton, D., Bloomfield, E. V., et al. (2015). Repeated nebulisation of non-viral cftr gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet. Respir. Med. 3, 684–691. doi:10.1016/S2213-2600(15)00245-3
Alton, E. W., Beekman, J. M., Boyd, A. C., Brand, J., Carlon, M. S., Connolly, M. M., et al. (2017). Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 72, 137–147. doi:10.1136/thoraxjnl-2016-208406
Aneja, M. K., Geiger, J. P., Himmel, A., and Rudolph, C. (2009). Targeted gene delivery to the lung. Expert Opin. Drug Deliv. 6, 567–583. doi:10.1517/17425240902927841
Anson, D. S. (2004). The use of retroviral vectors for gene therapy-what are the risks? A review of retroviral pathogenesis and its relevance to retroviral vector-mediated gene delivery. Genet. Vaccines Ther. 2, 9. doi:10.1186/1479-0556-2-9
Archer, S. L., Weir, E. K., and Wilkins, M. R. (2010). Basic science of pulmonary arterial hypertension for clinicians: New concepts and experimental therapies. Circulation 121, 2045–2066. doi:10.1161/CIRCULATIONAHA.108.847707
Arora, R., Metzger, R. J., and Papaioannou, V. E. (2012). Multiple roles and interactions of tbx4 and tbx5 in development of the respiratory system. PLoS Genet. 8, e1002866. doi:10.1371/journal.pgen.1002866
Auricchio, A., O'Connor, E., Weiner, D., Gao, G. P., Hildinger, M., Wang, L., et al. (2002). Noninvasive gene transfer to the lung for systemic delivery of therapeutic proteins. J. Clin. Invest. 110, 499–504. doi:10.1172/JCI15780
Austin, E. D., West, J., Loyd, J. E., and Hemnes, A. R. (2017). Translational advances in the field of pulmonary hypertension molecular medicine of pulmonary arterial hypertension. From population genetics to precision medicine and gene editing. Am. J. Respir. Crit. Care Med. 195, 23–31. doi:10.1164/rccm.201605-0905PP
Ballif, B. C., Theisen, A., Rosenfeld, J. A., Traylor, R. N., Gastier-Foster, J., Thrush, D. L., et al. (2010). Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am. J. Hum. Genet. 86, 454–461. doi:10.1016/j.ajhg.2010.01.038
Barst, R. J., Ertel, S. I., Beghetti, M., and Ivy, D. D. (2011). Pulmonary arterial hypertension: A comparison between children and adults. Eur. Respir. J. 37, 665–677. doi:10.1183/09031936.00056110
Barst, R. J., Maislin, G., and Fishman, A. P. (1999). Vasodilator therapy for primary pulmonary hypertension in children. Circulation 99, 1197–1208. doi:10.1161/01.cir.99.9.1197
Barst, R. J., McGoon, M. D., Elliott, C. G., Foreman, A. J., Miller, D. P., and Ivy, D. D. (2012). Survival in childhood pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 125, 113–122. doi:10.1161/CIRCULATIONAHA.111.026591
Berger, R. M., Beghetti, M., Humpl, T., Raskob, G. E., Ivy, D. D., Jing, Z. C., et al. (2012). Clinical features of paediatric pulmonary hypertension: A registry study. Lancet 379, 537–546. doi:10.1016/S0140-6736(11)61621-8
Bernabeu, C., Lopez-Novoa, J. M., and Quintanilla, M. (2009). The emerging role of tgf-beta superfamily coreceptors in cancer. Biochim. Biophys. Acta 1792, 954–973. doi:10.1016/j.bbadis.2009.07.003
Bisserier, M., Milara, J., Abdeldjebbar, Y., Gubara, S., Jones, C., Bueno-Beti, C., et al. (2020). Aav1.Serca2a gene therapy reverses pulmonary fibrosis by blocking the stat3/foxm1 pathway and promoting the snon/ski axis. Mol. Ther. 28, 394–410. doi:10.1016/j.ymthe.2019.11.027
Bongers, E. M., Duijf, P. H., van Beersum, S. E., Schoots, J., Van Kampen, A., Burckhardt, A., et al. (2004). Mutations in the human tbx4 gene cause small patella syndrome. Am. J. Hum. Genet. 74, 1239–1248. doi:10.1086/421331
Brunetti-Pierri, N., and Ng, P. (2008). Progress and prospects: Gene therapy for genetic diseases with helper-dependent adenoviral vectors. Gene Ther. 15, 553–560. doi:10.1038/gt.2008.14
Brunetti-Pierri, N., and Ng, P. (2006). Progress towards the clinical application of helper-dependent adenoviral vectors for liver and lung gene therapy. Curr. Opin. Mol. Ther. 8, 446–454.
Cao, H., Koehler, D. R., and Hu, J. (2004). Adenoviral vectors for gene replacement therapy. Viral Immunol. 17, 327–333. doi:10.1089/vim.2004.17.327
Cao, H., Ouyang, H., Grasemann, H., Bartlett, C., Du, K., Duan, R., et al. (2018). Transducing airway basal cells with a helper-dependent adenoviral vector for lung gene therapy. Hum. Gene Ther. 29, 643–652. doi:10.1089/hum.2017.201
Chamberlain, K., Riyad, J. M., and Weber, T. (2016). Expressing transgenes that exceed the packaging capacity of adeno-associated virus capsids. Hum. Gene Ther. Methods 27, 1–12. doi:10.1089/hgtb.2015.140
Chang, C. F., Wang, M., Ou, W. C., Chen, P. L., Shen, C. H., Lin, P. Y., et al. (2011). Human jc virus-like particles as a gene delivery vector. Expert Opin. Biol. Ther. 11, 1169–1175. doi:10.1517/14712598.2011.583914
Chapman, D. L., Garvey, N., Hancock, S., Alexiou, M., Agulnik, S. I., Gibson-Brown, J. J., et al. (1996). Expression of the t-box family genes, tbx1-tbx5, during early mouse development. Dev. Dyn. 206, 379–390. doi:10.1002/(SICI)1097-0177(199608)206:4<379::AID-AJA4>3.0.CO;2-F
Chattergoon, N. N., D'Souza, F. M., Deng, W., Chen, H., Hyman, A. L., Kadowitz, P. J., et al. (2005). Antiproliferative effects of calcitonin gene-related peptide in aortic and pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 288, L202–L211. doi:10.1152/ajplung.00064.2004
Chida, A., Shintani, M., Yagi, H., Fujiwara, M., Kojima, Y., Sato, H., et al. (2012). Outcomes of childhood pulmonary arterial hypertension in bmpr2 and alk1 mutation carriers. Am. J. Cardiol. 110, 586–593. doi:10.1016/j.amjcard.2012.04.035
Chirmule, N., Hughes, J. V., Gao, G. P., Raper, S. E., and Wilson, J. M. (1998). Role of e4 in eliciting cd4 t-cell and b-cell responses to adenovirus vectors delivered to murine and nonhuman primate lungs. J. Virol. 72, 6138–6145. doi:10.1128/JVI.72.7.6138-6145.1998
Cooney, A. L., Thornell, I. M., Singh, B. K., Shah, V. S., Stoltz, D. A., McCray, P. B., et al. (2019). A novel aav-mediated gene delivery system corrects cftr function in pigs. Am. J. Respir. Cell Mol. Biol. 61, 747–754. doi:10.1165/rcmb.2019-0006OC
Copeland, C. A., Han, B., Tiwari, A., Austin, E. D., Loyd, J. E., West, J. D., et al. (2017). A disease-associated frameshift mutation in caveolin-1 disrupts caveolae formation and function through introduction of a de novo er retention signal. Mol. Biol. Cell 28, 3095–3111. doi:10.1091/mbc.E17-06-0421
Corada, M., Orsenigo, F., Morini, M. F., Pitulescu, M. E., Bhat, G., Nyqvist, D., et al. (2013). Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat. Commun. 4, 2609. doi:10.1038/ncomms3609
Cruz-Utrilla, A., Gallego-Zazo, N., Tenorio-Castano, J. A., Guillen, I., Torrent-Vernetta, A., Moya-Bonora, A., et al. (2022). Clinical implications of the genetic background in pediatric pulmonary arterial hypertension: Data from the Spanish rehiped registry. Int. J. Mol. Sci. 23, 10433. doi:10.3390/ijms231810433
Cyranoski, D. (2016). Crispr gene-editing tested in a person for the first time. Nature 539, 479. doi:10.1038/nature.2016.20988
David, L., Mallet, C., Keramidas, M., Lamande, N., Gasc, J. M., Dupuis-Girod, S., et al. (2008). Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 102, 914–922. doi:10.1161/CIRCRESAHA.107.165530
David, L., Mallet, C., Mazerbourg, S., Feige, J. J., and Bailly, S. (2007). Identification of bmp9 and bmp10 as functional activators of the orphan activin receptor-like kinase 1 (alk1) in endothelial cells. Blood 109, 1953–1961. doi:10.1182/blood-2006-07-034124
Daya, S., and Berns, K. I. (2008). Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 21, 583–593. doi:10.1128/CMR.00008-08
Degiulio, J. V., Kaufman, C. D., and Dean, D. A. (2010). The sp-c promoter facilitates alveolar type ii epithelial cell-specific plasmid nuclear import and gene expression. Gene Ther. 17, 541–549. doi:10.1038/gt.2009.166
Deng, Z., Morse, J. H., Slager, S. L., Cuervo, N., Moore, K. J., Venetos, G., et al. (2000). Familial primary pulmonary hypertension (gene pph1) is caused by mutations in the bone morphogenetic protein receptor-ii gene. Am. J. Hum. Genet. 67, 737–744. doi:10.1086/303059
Drake, K. M., Comhair, S. A., Erzurum, S. C., Tuder, R. M., and Aldred, M. A. (2015). Endothelial chromosome 13 deletion in congenital heart disease-associated pulmonary arterial hypertension dysregulates smad9 signaling. Am. J. Respir. Crit. Care Med. 191, 850–854. doi:10.1164/rccm.201411-1985LE
Drake, K. M., Zygmunt, D., Mavrakis, L., Harbor, P., Wang, L., Comhair, S. A., et al. (2011). Altered microrna processing in heritable pulmonary arterial hypertension: An important role for smad-8. Am. J. Respir. Crit. Care Med. 184, 1400–1408. doi:10.1164/rccm.201106-1130OC
Driskell, R. A., and Engelhardt, J. F. (2003). Current status of gene therapy for inherited lung diseases. Annu. Rev. Physiol. 65, 585–612. doi:10.1146/annurev.physiol.65.092101.142426
Du, L., Sullivan, C. C., Chu, D., Cho, A. J., Kido, M., Wolf, P. L., et al. (2003). Signaling molecules in nonfamilial pulmonary hypertension. N. Engl. J. Med. 348, 500–509. doi:10.1056/NEJMoa021650
Durymanov, M., and Reineke, J. (2018). Non-viral delivery of nucleic acids: Insight into mechanisms of overcoming intracellular barriers. Front. Pharmacol. 9, 971. doi:10.3389/fphar.2018.00971
Elliott, C. G., Glissmeyer, E. W., Havlena, G. T., Carlquist, J., McKinney, J. T., Rich, S., et al. (2006). Relationship of bmpr2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation 113, 2509–2515. doi:10.1161/CIRCULATIONAHA.105.601930
Engelhardt, J. F., Litzky, L., and Wilson, J. M. (1994). Prolonged transgene expression in cotton rat lung with recombinant adenoviruses defective in e2a. Hum. Gene Ther. 5, 1217–1229. doi:10.1089/hum.1994.5.10-1217
Eyries, M., Montani, D., Nadaud, S., Girerd, B., Levy, M., Bourdin, A., et al. (2019). Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur. Respir. J. 53, 1801371. doi:10.1183/13993003.01371-2018
Fakhiri, J., Schneider, M. A., Puschhof, J., Stanifer, M., Schildgen, V., Holderbach, S., et al. (2019). Novel chimeric gene therapy vectors based on adeno-associated virus and four different mammalian bocaviruses. Mol. Ther. Methods Clin. Dev. 12, 202–222. doi:10.1016/j.omtm.2019.01.003
Fausther-Bovendo, H., and Kobinger, G. P. (2014). Pre-existing immunity against ad vectors: Humoral, cellular, and innate response, what's important? Hum. Vaccin. Immunother. 10, 2875–2884. doi:10.4161/hv.29594
Feng, F., Harper, R. L., and Reynolds, P. N. (2016). Bmpr2 gene delivery reduces mutation-related pah and counteracts tgf-beta-mediated pulmonary cell signalling. Respirology 21, 526–532. doi:10.1111/resp.12712
Ferdosi, S. R., Ewaisha, R., Moghadam, F., Krishna, S., Park, J. G., Ebrahimkhani, M. R., et al. (2019). Multifunctional crispr-cas9 with engineered immunosilenced human t cell epitopes. Nat. Commun. 10, 1842. doi:10.1038/s41467-019-09693-x
Ferrari, S., Griesenbach, U., Iida, A., Farley, R., Wright, A. M., Zhu, J., et al. (2007). Sendai virus-mediated cftr gene transfer to the airway epithelium. Gene Ther. 14, 1371–1379. doi:10.1038/sj.gt.3302991
Flotte, T. R. (2005). Adeno-associated virus-mediated gene transfer for lung diseases. Hum. Gene Ther. 16, 643–648. doi:10.1089/hum.2005.16.643
Francois, M., Koopman, P., and Beltrame, M. (2010). Soxf genes: Key players in the development of the cardio-vascular system. Int. J. Biochem. Cell Biol. 42, 445–448. doi:10.1016/j.biocel.2009.08.017
Frump, A. L., Lowery, J. W., Hamid, R., Austin, E. D., and de Caestecker, M. (2013). Abnormal trafficking of endogenously expressed bmpr2 mutant allelic products in patients with heritable pulmonary arterial hypertension. PLoS One 8, e80319. doi:10.1371/journal.pone.0080319
Galambos, C., Mullen, M. P., Shieh, J. T., Schwerk, N., Kielt, M. J., Ullmann, N., et al. (2019). Phenotype characterisation of tbx4 mutation and deletion carriers with neonatal and paediatric pulmonary hypertension. Eur. Respir. J. 54, 1801965. doi:10.1183/13993003.01965-2018
Gallego, I., Villate-Beitia, I., Martinez-Navarrete, G., Menendez, M., Lopez-Mendez, T., Soto-Sanchez, C., et al. (2019). Non-viral vectors based on cationic niosomes and minicircle DNA technology enhance gene delivery efficiency for biomedical applications in retinal disorders. Nanomedicine 17, 308–318. doi:10.1016/j.nano.2018.12.018
Gandara, C., Affleck, V., and Stoll, E. A. (2018). Manufacture of third-generation lentivirus for preclinical use, with process development considerations for translation to good manufacturing practice. Hum. Gene Ther. Methods 29, 1–15. doi:10.1089/hgtb.2017.098
Garcia de Vinuesa, A., Abdelilah-Seyfried, S., Knaus, P., Zwijsen, A., and Bailly, S. (2016). Bmp signaling in vascular biology and dysfunction. Cytokine Growth Factor Rev. 27, 65–79. doi:10.1016/j.cytogfr.2015.12.005
Garcia-Rivas, G., Jerjes-Sanchez, C., Rodriguez, D., Garcia-Pelaez, J., and Trevino, V. (2017). A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Med. Genet. 18, 82. doi:10.1186/s12881-017-0440-5
Girerd, B., Montani, D., Coulet, F., Sztrymf, B., Yaici, A., Jais, X., et al. (2010). Clinical outcomes of pulmonary arterial hypertension in patients carrying an acvrl1 (alk1) mutation. Am. J. Respir. Crit. Care Med. 181, 851–861. doi:10.1164/rccm.200908-1284OC
Good, R. B., Gilbane, A. J., Trinder, S. L., Denton, C. P., Coghlan, G., Abraham, D. J., et al. (2015). Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am. J. Pathol. 185, 1850–1858. doi:10.1016/j.ajpath.2015.03.019
Gore, B., Izikki, M., Mercier, O., Dewachter, L., Fadel, E., Humbert, M., et al. (2014). Key role of the endothelial TGF-β/ALK1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PLoS One 9, e100310. doi:10.1371/journal.pone.0100310
Goumans, M. J., Zwijsen, A., Ten Dijke, P., and Bailly, S. (2018). Bone morphogenetic proteins in vascular homeostasis and disease. Cold Spring Harb. Perspect. Biol. 10, a031989. doi:10.1101/cshperspect.a031989
Graf, S., Haimel, M., Bleda, M., Hadinnapola, C., Southgate, L., Li, W., et al. (2018). Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 9, 1416. doi:10.1038/s41467-018-03672-4
Green, D. E., Murphy, T. C., Kang, B. Y., Bedi, B., Yuan, Z., Sadikot, R. T., et al. (2017). Peroxisome proliferator-activated receptor-gamma enhances human pulmonary artery smooth muscle cell apoptosis through microrna-21 and programmed cell death 4. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L371–L383. doi:10.1152/ajplung.00532.2016
Gregory, S. M., Nazir, S. A., and Metcalf, J. P. (2011). Implications of the innate immune response to adenovirus and adenoviral vectors. Future Virol. 6, 357–374. doi:10.2217/fvl.11.6
Grieger, J. C., and Samulski, R. J. (2005). Packaging capacity of adeno-associated virus serotypes: Impact of larger genomes on infectivity and postentry steps. J. Virol. 79, 9933–9944. doi:10.1128/JVI.79.15.9933-9944.2005
Griesenbach, U., Inoue, M., Meng, C., Farley, R., Chan, M., Newman, N. K., et al. (2012). Assessment of f/hn-pseudotyped lentivirus as a clinically relevant vector for lung gene therapy. Am. J. Respir. Crit. Care Med. 186, 846–856. doi:10.1164/rccm.201206-1056OC
Guo, Z. S., and Bartlett, D. L. (2004). Vaccinia as a vector for gene delivery. Expert Opin. Biol. Ther. 4, 901–917. doi:10.1517/14712598.4.6.901
Haarman, M. G., Kerstjens-Frederikse, W. S., Vissia-Kazemier, T. R., Breeman, K. T. N., Timens, W., Vos, Y. J., et al. (2020). The genetic epidemiology of pediatric pulmonary arterial hypertension. J. Pediatr. 225, 65–73. doi:10.1016/j.jpeds.2020.05.051
Halbert, C. L., Allen, J. M., and Miller, A. D. (2002). Efficient mouse airway transduction following recombination between aav vectors carrying parts of a larger gene. Nat. Biotechnol. 20, 697–701. doi:10.1038/nbt0702-697
Han, Z., Zhong, L., Maina, N., Hu, Z., Li, X., Chouthai, N. S., et al. (2008). Stable integration of recombinant adeno-associated virus vector genomes after transduction of murine hematopoietic stem cells. Hum. Gene Ther. 19, 267–278. doi:10.1089/hum.2007.161
Hansmann, G., and Hoeper, M. M. (2013). Registries for paediatric pulmonary hypertension. Eur. Respir. J. 42, 580–583. doi:10.1183/09031936.00065713
Harper, R. L., Reynolds, A. M., Bonder, C. S., and Reynolds, P. N. (2016). Bmpr2 gene therapy for pah acts via smad and non-smad signalling. Respirology 21, 727–733. doi:10.1111/resp.12729
Harrison, R. E., Berger, R., Haworth, S. G., Tulloh, R., Mache, C. J., Morrell, N. W., et al. (2005). Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 111, 435–441. doi:10.1161/01.CIR.0000153798.78540.87
Harrison, R. E., Flanagan, J. A., Sankelo, M., Abdalla, S. A., Rowell, J., Machado, R. D., et al. (2003). Molecular and functional analysis identifies alk-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J. Med. Genet. 40, 865–871. doi:10.1136/jmg.40.12.865
Hartung, A., Bitton-Worms, K., Rechtman, M. M., Wenzel, V., Boergermann, J. H., Hassel, S., et al. (2006). Different routes of bone morphogenic protein (bmp) receptor endocytosis influence bmp signaling. Mol. Cell. Biol. 26, 7791–7805. doi:10.1128/MCB.00022-06
Henning, A., Hein, S., Schneider, M., Bur, M., and Lehr, C. M. (2010). Pulmonary drug delivery: Medicines for inhalation. Handb. Exp. Pharmacol. 197, 171–192. doi:10.1007/978-3-642-00477-3_6
Hernandez, Y. J., Wang, J., Kearns, W. G., Loiler, S., Poirier, A., and Flotte, T. R. (1999). Latent adeno-associated virus infection elicits humoral but not cell-mediated immune responses in a nonhuman primate model. J. Virol. 73, 8549–8558. doi:10.1128/JVI.73.10.8549-8558.1999
Hill, A. B., Chen, M., Chen, C. K., Pfeifer, B. A., and Jones, C. H. (2016). Overcoming gene-delivery hurdles: Physiological considerations for nonviral vectors. Trends Biotechnol. 34, 91–105. doi:10.1016/j.tibtech.2015.11.004
Hu, Y. C. (2006). Baculovirus vectors for gene therapy. Adv. Virus Res. 68, 287–320. doi:10.1016/S0065-3527(06)68008-1
Hudson, C., Clements, D., Friday, R. V., Stott, D., and Woodland, H. R. (1997). Xsox17alpha and -beta mediate endoderm formation in xenopus. Cell 91, 397–405. doi:10.1016/s0092-8674(00)80423-7
Humbert, M., Sitbon, O., Chaouat, A., Bertocchi, M., Habib, G., Gressin, V., et al. (2006). Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 173, 1023–1030. doi:10.1164/rccm.200510-1668OC
Hurst, L. A., Dunmore, B. J., Long, L., Crosby, A., Al-Lamki, R., Deighton, J., et al. (2017). TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 8, 14079. doi:10.1038/ncomms14079
Inoue, M., Tokusumi, Y., Ban, H., Shirakura, M., Kanaya, T., Yoshizaki, M., et al. (2004). Recombinant sendai virus vectors deleted in both the matrix and the fusion genes: Efficient gene transfer with preferable properties. J. Gene Med. 6, 1069–1081. doi:10.1002/jgm.597
International, P. P. H. C., Lane, K. B., Machado, R. D., Pauciulo, M. W., Thomson, J. R., Phillips, J. A., et al. (2000). Heterozygous germline mutations in bmpr2, encoding a tgf-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 26, 81–84. doi:10.1038/79226
Ivy, D., and Frank, B. S. (2021). Update on pediatric pulmonary arterial hypertension. Curr. Opin. Cardiol. 36, 67–79. doi:10.1097/HCO.0000000000000822
Ivy, D. D., Abman, S. H., Barst, R. J., Berger, R. M., Bonnet, D., Fleming, T. R., et al. (2013). Pediatric pulmonary hypertension. J. Am. Coll. Cardiol. 62, D117–D126. doi:10.1016/j.jacc.2013.10.028
Jin, Y., Muhl, L., Burmakin, M., Wang, Y. X., Duchez, A. C., Betsholtz, C., et al. (2017). Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through vegfr2 signalling. Nat. Cell Biol. 19:639–652. doi:10.1038/ncb3534
Jozkowicz, A., and Dulak, J. (2005). Helper-dependent adenoviral vectors in experimental gene therapy. Acta Biochim. Pol. 52, 589–599. doi:10.18388/abp.2005_3419
Kabwe, J. C., Sawada, H., Mitani, Y., Oshita, H., Tsuboya, N., Zhang, E., et al. (2022). Crispr-mediated bmpr2 point mutation exacerbates late pulmonary vasculopathy and reduces survival in rats with experimental pulmonary hypertension. Respir. Res. 23, 87. doi:10.1186/s12931-022-02005-w
Kanai-Azuma, M., Kanai, Y., Gad, J. M., Tajima, Y., Taya, C., Kurohmaru, M., et al. (2002). Depletion of definitive gut endoderm in sox17-null mutant mice. Development 129, 2367–2379. doi:10.1242/dev.129.10.2367
Katz, M. G., Fargnoli, A. S., Gubara, S. M., Fish, K., Weber, T., Bridges, C. R., et al. (2019). Targeted gene delivery through the respiratory system: Rationale for intratracheal gene transfer. J. Cardiovasc. Dev. Dis. 6, E8. doi:10.3390/jcdd6010008
Katz, M. G., Fargnoli, A. S., Weber, T., Hajjar, R. J., and Bridges, C. R. (2017). Use of adeno-associated virus vector for cardiac gene delivery in large-animal surgical models of heart failure. Hum. Gene Ther. Clin. Dev. 28, 157–164. doi:10.1089/humc.2017.070
Kearns, W. G., Afione, S. A., Fulmer, S. B., Pang, M. C., Erikson, D., Egan, M., et al. (1996). Recombinant adeno-associated virus (aav-cftr) vectors do not integrate in a site-specific fashion in an immortalized epithelial cell line. Gene Ther. 3, 748–755.
Kerstjens-Frederikse, W. S., Bongers, E. M., Roofthooft, M. T., Leter, E. M., Douwes, J. M., Van Dijk, A., et al. (2013). Tbx4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 50, 500–506. doi:10.1136/jmedgenet-2012-101152
Kim, I., Saunders, T. L., and Morrison, S. J. (2007). Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell 130, 470–483. doi:10.1016/j.cell.2007.06.011
Kim, K., Kim, I. K., Yang, J. M., Lee, E., Koh, B. I., Song, S., et al. (2016). Soxf transcription factors are positive feedback regulators of vegf signaling. Circ. Res. 119, 839–852. doi:10.1161/CIRCRESAHA.116.308483
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi:10.1038/nature17946
Lange, A. W., Haitchi, H. M., LeCras, T. D., Sridharan, A., Xu, Y., Wert, S. E., et al. (2014). Sox17 is required for normal pulmonary vascular morphogenesis. Dev. Biol. 387, 109–120. doi:10.1016/j.ydbio.2013.11.018
Le Ribeuz, H., Capuano, V., Girerd, B., Humbert, M., Montani, D., and Antigny, F. (2020). Implication of potassium channels in the pathophysiology of pulmonary arterial hypertension. Biomolecules 10, E1261. doi:10.3390/biom10091261
Lee, C. S., Bishop, E. S., Zhang, R., Yu, X., Farina, E. M., Yan, S., et al. (2017). Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 4, 43–63. doi:10.1016/j.gendis.2017.04.001
Lee, S. H., Lee, S., Yang, H., Song, S., Kim, K., Saunders, T. L., et al. (2014). Notch pathway targets proangiogenic regulator sox17 to restrict angiogenesis. Circ. Res. 115, 215–226. doi:10.1161/CIRCRESAHA.115.303142
Leopold, J. A., and Maron, B. A. (2016). Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int. J. Mol. Sci. 17, 761. doi:10.3390/ijms17050761
Levy, M., Eyries, M., Szezepanski, I., Ladouceur, M., Nadaud, S., Bonnet, D., et al. (2016). Genetic analyses in a cohort of children with pulmonary hypertension. Eur. Respir. J. 48, 1118–1126. doi:10.1183/13993003.00211-2016
Li, C., and Samulski, R. J. (2020). Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 21, 255–272. doi:10.1038/s41576-019-0205-4
Li, L., Jick, S., Breitenstein, S., Hernandez, G., Michel, A., and Vizcaya, D. (2017). Pulmonary arterial hypertension in the USA: An epidemiological study in a large insured pediatric population. Pulm. Circ. 7, 126–136. doi:10.1086/690007
Lisowski, L., Tay, S. S., and Alexander, I. E. (2015). Adeno-associated virus serotypes for gene therapeutics. Curr. Opin. Pharmacol. 24, 59–67. doi:10.1016/j.coph.2015.07.006
Liu, F., Ventura, F., Doody, J., and Massague, J. (1995). Human type ii receptor for bone morphogenic proteins (bmps): Extension of the two-kinase receptor model to the bmps. Mol. Cell. Biol. 15, 3479–3486. doi:10.1128/mcb.15.7.3479
Liu, M., Zhang, L., Marsboom, G., Jambusaria, A., Xiong, S., Toth, P. T., et al. (2019). Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nat. Commun. 10, 2126. doi:10.1038/s41467-019-10134-y
Liu, Y., Asakura, M., Inoue, H., Nakamura, T., Sano, M., Niu, Z., et al. (2007). Sox17 is essential for the specification of cardiac mesoderm in embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A. 104, 3859–3864. doi:10.1073/pnas.0609100104
Lompre, A. M., Hadri, L., Merlet, E., Keuylian, Z., Mougenot, N., Karakikes, I., et al. (2013). Efficient transduction of vascular smooth muscle cells with a translational aav2.5 vector: A new perspective for in-stent restenosis gene therapy. Gene Ther. 20, 901–912. doi:10.1038/gt.2013.13
Ma, L., and Chung, W. K. (2017). The role of genetics in pulmonary arterial hypertension. J. Pathol. 241, 273–280. doi:10.1002/path.4833
Ma, L., Roman-Campos, D., Austin, E. D., Eyries, M., Sampson, K. S., Soubrier, F., et al. (2013). A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 369, 351–361. doi:10.1056/NEJMoa1211097
Machado, R. D., Eickelberg, O., Elliott, C. G., Geraci, M. W., Hanaoka, M., Loyd, J. E., et al. (2009). Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 54, S32–S42. doi:10.1016/j.jacc.2009.04.015
Machado, R. D., Southgate, L., Eichstaedt, C. A., Aldred, M. A., Austin, E. D., Best, D. H., et al. (2015). Pulmonary arterial hypertension: A current perspective on established and emerging molecular genetic defects. Hum. Mutat. 36, 1113–1127. doi:10.1002/humu.22904
Maeder, M. L., Linder, S. J., Cascio, V. M., Fu, Y., Ho, Q. H., and Joung, J. K. (2013). Crispr rna-guided activation of endogenous human genes. Nat. Methods 10, 977–979. doi:10.1038/nmeth.2598
Maniatis, N. A., Shinin, V., Schraufnagel, D. E., Okada, S., Vogel, S. M., Malik, A. B., et al. (2008). Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1-/- mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L865–L873. doi:10.1152/ajplung.00079.2007
Manunta, M. D. I., Tagalakis, A. D., Attwood, M., Aldossary, A. M., Barnes, J. L., Munye, M. M., et al. (2017). Delivery of enac sirna to epithelial cells mediated by a targeted nanocomplex: A therapeutic strategy for cystic fibrosis. Sci. Rep. 7, 700. doi:10.1038/s41598-017-00662-2
Marsboom, G., Chen, Z., Yuan, Y., Zhang, Y., Tiruppathi, C., Loyd, J. E., et al. (2017). Aberrant caveolin-1-mediated smad signaling and proliferation identified by analysis of adenine 474 deletion mutation (c.474dela) in patient fibroblasts: A new perspective on the mechanism of pulmonary hypertension. Mol. Biol. Cell 28, 1177–1185. doi:10.1091/mbc.E16-11-0790
McMurtry, M. S., Moudgil, R., Hashimoto, K., Bonnet, S., Michelakis, E. D., and Archer, S. L. (2007). Overexpression of human bone morphogenetic protein receptor 2 does not ameliorate monocrotaline pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L872–L878. doi:10.1152/ajplung.00309.2006
Milone, M. C., and O'Doherty, U. (2018). Clinical use of lentiviral vectors. Leukemia 32, 1529–1541. doi:10.1038/s41375-018-0106-0
Mingozzi, F., and High, K. A. (2013). Immune responses to aav vectors: Overcoming barriers to successful gene therapy. Blood 122, 23–36. doi:10.1182/blood-2013-01-306647
Mitomo, K., Griesenbach, U., Inoue, M., Somerton, L., Meng, C., Akiba, E., et al. (2010). Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with sendai virus envelopes. Mol. Ther. 18, 1173–1182. doi:10.1038/mt.2010.13
Mohammadzadeh, I., Qujeq, D., Yousefi, T., Ferns, G. A., Maniati, M., and Vaghari-Tabari, M. (2020). Crispr/cas9 gene editing: A new therapeutic approach in the treatment of infection and autoimmunity. IUBMB Life 72, 1603–1621. doi:10.1002/iub.2296
Moledina, S., Hislop, A. A., Foster, H., Schulze-Neick, I., and Haworth, S. G. (2010). Childhood idiopathic pulmonary arterial hypertension: A national cohort study. Heart 96, 1401–1406. doi:10.1136/hrt.2009.182378
Montani, D., Girerd, B., Jais, X., Laveneziana, P., Lau, E. M. T., Bouchachi, A., et al. (2021). Genetic counselling in a national referral centre for pulmonary hypertension. Eur. Respir. J. 58, 541–552. doi:10.1183/13993003.00717-2015
Morrell, N. W., Aldred, M. A., Chung, W. K., Elliott, C. G., Nichols, W. C., Soubrier, F., et al. (2019). Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 53, 1801899. doi:10.1183/13993003.01899-2018
Moss, R. B., Milla, C., Colombo, J., Accurso, F., Zeitlin, P. L., Clancy, J. P., et al. (2007). Repeated aerosolized aav-cftr for treatment of cystic fibrosis: A randomized placebo-controlled phase 2b trial. Hum. Gene Ther. 18, 726–732. doi:10.1089/hum.2007.022
Murata, T., Hori, M., Lee, S., Nakamura, A., Kohama, K., Karaki, H., et al. (2005). Vascular endothelium has a local anti-adenovirus vector system and glucocorticoid optimizes its gene transduction. Arterioscler. Thromb. Vasc. Biol. 25, 1796–1803. doi:10.1161/01.ATV.0000174130.75958.b7
Naiche, L. A., and Papaioannou, V. E. (2003). Loss of tbx4 blocks hindlimb development and affects vascularization and fusion of the allantois. Development 130, 2681–2693. doi:10.1242/dev.00504
Nair, J., Nair, A., Veerappan, S., and Sen, D. (2020). Translatable gene therapy for lung cancer using crispr cas9-an exploratory review. Cancer Gene Ther. 27, 116–124. doi:10.1038/s41417-019-0116-8
Nasim, M. T., Ogo, T., Ahmed, M., Randall, R., Chowdhury, H. M., Snape, K. M., et al. (2011). Molecular genetic characterization of smad signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 32, 1385–1389. doi:10.1002/humu.21605
Naso, M. F., Tomkowicz, B., Perry, W. L., and Strohl, W. R. (2017). Adeno-associated virus (aav) as a vector for gene therapy. BioDrugs 31, 317–334. doi:10.1007/s40259-017-0234-5
Navas, P., Tenorio, J., Quezada, C. A., Barrios, E., Gordo, G., Arias, P., et al. (2016). Molecular analysis of bmpr2, tbx4, and kcnk3 and genotype-phenotype correlations in Spanish patients and families with idiopathic and hereditary pulmonary arterial hypertension. Rev. Esp. Cardiol. 69, 1011–1019. doi:10.1016/j.rec.2016.03.029
Niessen, K., Zhang, G., Ridgway, J. B., Chen, H., and Yan, M. (2010). Alk1 signaling regulates early postnatal lymphatic vessel development. Blood 115, 1654–1661. doi:10.1182/blood-2009-07-235655
Nimmakayalu, M., Major, H., Sheffield, V., Solomon, D. H., Smith, R. J., Patil, S. R., et al. (2011). Microdeletion of 17q22q23.2 encompassing tbx2 and tbx4 in a patient with congenital microcephaly, thyroid duct cyst, sensorineural hearing loss, and pulmonary hypertension. Am. J. Med. Genet. A 155A, 418–423. doi:10.1002/ajmg.a.33827
Olschewski, A., Li, Y., Tang, B., Hanze, J., Eul, B., Bohle, R. M., et al. (2006). Impact of task-1 in human pulmonary artery smooth muscle cells. Circ. Res. 98, 1072–1080. doi:10.1161/01.RES.0000219677.12988.e9
Olschewski, A. (2010). Targeting task-1 channels as a therapeutic approach. Adv. Exp. Med. Biol. 661, 459–473. doi:10.1007/978-1-60761-500-2_30
Olschewski, A., Veale, E. L., Nagy, B. M., Nagaraj, C., Kwapiszewska, G., Antigny, F., et al. (2017). Task-1 (kcnk3) channels in the lung: From cell biology to clinical implications. Eur. Respir. J. 50, 1700754. doi:10.1183/13993003.00754-2017
Palmer, D. J., and Ng, P. (2005). Helper-dependent adenoviral vectors for gene therapy. Hum. Gene Ther. 16, 1–16. doi:10.1089/hum.2005.16.1
Parker, A. L., White, K. M., Lavery, C. A., Custers, J., Waddington, S. N., and Baker, A. H. (2013). Pseudotyping the adenovirus serotype 5 capsid with both the fibre and penton of serotype 35 enhances vascular smooth muscle cell transduction. Gene Ther. 20, 1158–1164. doi:10.1038/gt.2013.44
Peacock, A. J., Murphy, N. F., McMurray, J. J., Caballero, L., and Stewart, S. (2007). An epidemiological study of pulmonary arterial hypertension. Eur. Respir. J. 30, 104–109. doi:10.1183/09031936.00092306
Piao, C., Zhu, Y., Zhang, C., Xi, X., Liu, X., Zheng, S., et al. (2016). Identification of multiple acvrl1 mutations in patients with pulmonary arterial hypertension by targeted exome capture. Clin. Sci. 130, 1559–1569. doi:10.1042/CS20160247
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., et al. (2013). Repurposing crispr as an rna-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. doi:10.1016/j.cell.2013.02.022
Quest, A. F., Leyton, L., and Parraga, M. (2004). Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem. Cell Biol. 82, 129–144. doi:10.1139/o03-071
Rai, N., Shihan, M., Seeger, W., Schermuly, R. T., and Novoyatleva, T. (2021). Genetic delivery and gene therapy in pulmonary hypertension. Int. J. Mol. Sci., 22.
Ramamoorth, M., and Narvekar, A. (2015). Non viral vectors in gene therapy- an overview. J. Clin. Diagn. Res. 9, GE01–06. doi:10.7860/JCDR/2015/10443.5394
Reichmuth, A. M., Oberli, M. A., Jaklenec, A., Langer, R., and Blankschtein, D. (2016). Mrna vaccine delivery using lipid nanoparticles. Ther. Deliv. 7, 319–334. doi:10.4155/tde-2016-0006
Reynolds, A. M., Xia, W., Holmes, M. D., Hodge, S. J., Danilov, S., Curiel, D. T., et al. (2007). Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L1182–L1192. doi:10.1152/ajplung.00020.2006
Reynolds, P. N. (2011). Gene therapy for pulmonary hypertension: Prospects and challenges. Expert Opin. Biol. Ther. 11, 133–143. doi:10.1517/14712598.2011.542139
Rezaee, M., Oskuee, R. K., Nassirli, H., and Malaekeh-Nikouei, B. (2016). Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J. Control. Release 236, 1–14. doi:10.1016/j.jconrel.2016.06.023
Roberts, A. B., and Sporn, M. B. (1989). Regulation of endothelial cell growth, architecture, and matrix synthesis by tgf-beta. Am. Rev. Respir. Dis. 140, 1126–1128. doi:10.1164/ajrccm/140.4.1126
Rosenzweig, E. B., Abman, S. H., Adatia, I., Beghetti, M., Bonnet, D., Haworth, S., et al. (2019). Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur. Respir. J. 53, 1801916. doi:10.1183/13993003.01916-2018
Rosenzweig, E. B., Morse, J. H., Knowles, J. A., Chada, K. K., Khan, A. M., Roberts, K. E., et al. (2008). Clinical implications of determining bmpr2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J. Heart Lung Transpl. 27, 668–674. doi:10.1016/j.healun.2008.02.009
Runo, J. R., and Loyd, J. E. (2003). Primary pulmonary hypertension. Lancet 361, 1533–1544. doi:10.1016/S0140-6736(03)13167-4
Ruopp, N. F., and Cockrill, B. A. (2022). Diagnosis and treatment of pulmonary arterial hypertension: A review. JAMA 327, 1379–1391. doi:10.1001/jama.2022.4402
Saito, T., Bokhove, M., Croci, R., Zamora-Caballero, S., Han, L., Letarte, M., et al. (2017). Structural basis of the human endoglin-bmp9 interaction: Insights into bmp signaling and hht1. Cell Rep. 19, 1917–1928. doi:10.1016/j.celrep.2017.05.011
Sakiyama, J., Yamagishi, A., and Kuroiwa, A. (2003). Tbx4-fgf10 system controls lung bud formation during chicken embryonic development. Development 130, 1225–1234. doi:10.1242/dev.00345
Schaffer, D. V., and Maheshri, N. (2004). Directed evolution of aav mutants for enhanced gene delivery. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2004, 3520–3523. doi:10.1109/IEMBS.2004.1403990
Scharpfenecker, M., van Dinther, M., Liu, Z., van Bezooijen, R. L., Zhao, Q., Pukac, L., et al. (2007). Bmp-9 signals via alk1 and inhibits bfgf-induced endothelial cell proliferation and vegf-stimulated angiogenesis. J. Cell Sci. 120, 964–972. doi:10.1242/jcs.002949
Schultz, B. R., and Chamberlain, J. S. (2008). Recombinant adeno-associated virus transduction and integration. Mol. Ther. 16, 1189–1199. doi:10.1038/mt.2008.103
Seki, T., Yun, J., and Oh, S. P. (2003). Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ. Res. 93, 682–689. doi:10.1161/01.RES.0000095246.40391.3B
Sekine, K., Ohuchi, H., Fujiwara, M., Yamasaki, M., Yoshizawa, T., Sato, T., et al. (1999). Fgf10 is essential for limb and lung formation. Nat. Genet. 21, 138–141. doi:10.1038/5096
Simonneau, G., Montani, D., Celermajer, D. S., Denton, C. P., Gatzoulis, M. A., Krowka, M., et al. (2019). Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 53, 1801913. doi:10.1183/13993003.01913-2018
Sinn, P. L., Cooney, A. L., Oakland, M., Dylla, D. E., Wallen, T. J., Pezzulo, A. A., et al. (2012). Lentiviral vector gene transfer to porcine airways. Mol. Ther. Nucleic Acids 1, e56. doi:10.1038/mtna.2012.47
Sondhi, D., Stiles, K. M., De, B. P., and Crystal, R. G. (2017). Genetic modification of the lung directed toward treatment of human disease. Hum. Gene Ther. 28, 3–84. doi:10.1089/hum.2016.152
Sountoulidis, A., Stavropoulos, A., Giaglis, S., Apostolou, E., Monteiro, R., Chuva de Sousa Lopes, S. M., et al. (2012). Activation of the canonical bone morphogenetic protein (bmp) pathway during lung morphogenesis and adult lung tissue repair. PLoS One 7, e41460. doi:10.1371/journal.pone.0041460
Southgate, L., Machado, R. D., Graf, S., and Morrell, N. W. (2020). Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 17, 85–95. doi:10.1038/s41569-019-0242-x
St George, J. A. (2003). Gene therapy progress and prospects: Adenoviral vectors. Gene Ther. 10, 1135–1141. doi:10.1038/sj.gt.3302071
Sugden, W. W., Meissner, R., Aegerter-Wilmsen, T., Tsaryk, R., Leonard, E. V., Bussmann, J., et al. (2017) Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat. Cell Biol. 19:653–665. doi:10.1038/ncb3528
Szokol, J. W., Murphy, G. S., Vender, J. S., and Nitsun, M. (2004). Gene therapy in heart and lung disease. Curr. Opin. Anaesthesiol. 17, 13–20. doi:10.1097/00001503-200402000-00004
Thomas, C. E., Ehrhardt, A., and Kay, M. A. (2003). Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 4, 346–358. doi:10.1038/nrg1066
Tillet, E., and Bailly, S. (2014). Emerging roles of bmp9 and bmp10 in hereditary hemorrhagic telangiectasia. Front. Genet. 5, 456. doi:10.3389/fgene.2014.00456
Tillet, E., Ouarne, M., Desroches-Castan, A., Mallet, C., Subileau, M., Didier, R., et al. (2018). A heterodimer formed by bone morphogenetic protein 9 (bmp9) and bmp10 provides most bmp biological activity in plasma. J. Biol. Chem. 293, 10963–10974. doi:10.1074/jbc.RA118.002968
Trembath, R. C., Thomson, J. R., Machado, R. D., Morgan, N. V., Atkinson, C., Winship, I., et al. (2001). Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 345, 325–334. doi:10.1056/NEJM200108023450503
Tuder, R. M., Archer, S. L., Dorfmuller, P., Erzurum, S. C., Guignabert, C., Michelakis, E., et al. (2013). Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 62, D4–D12. doi:10.1016/j.jacc.2013.10.025
Turnbull, I. C., Eltoukhy, A. A., Anderson, D. G., and Costa, K. D. (2017). Lipidoid mrna nanoparticles for myocardial delivery in rodents. Methods Mol. Biol. 1521, 153–166. doi:10.1007/978-1-4939-6588-5_10
Turnbull, I. C., Eltoukhy, A. A., Fish, K. M., Nonnenmacher, M., Ishikawa, K., Chen, J., et al. (2016). Myocardial delivery of lipidoid nanoparticle carrying modrna induces rapid and transient expression. Mol. Ther. 24, 66–75. doi:10.1038/mt.2015.193
van den Heuvel, L. M., Jansen, S. M. A., Alsters, S. I. M., Post, M. C., van der Smagt, J. J., Handoko-De Man, F. S., et al. (2020). Genetic evaluation in a cohort of 126 Dutch pulmonary arterial hypertension patients. Genes 11, E1191. doi:10.3390/genes11101191
van Haasteren, J., Hyde, S. C., and Gill, D. R. (2018). Lessons learned from lung and liver in-vivo gene therapy: Implications for the future. Expert Opin. Biol. Ther. 18, 959–972. doi:10.1080/14712598.2018.1506761
van Loon, R. L., Roofthooft, M. T., Hillege, H. L., ten Harkel, A. D., van Osch-Gevers, M., Delhaas, T., et al. (2011). Pediatric pulmonary hypertension in The Netherlands: Epidemiology and characterization during the period 1991 to 2005. Circulation 124, 1755–1764. doi:10.1161/CIRCULATIONAHA.110.969584
Vanlerberghe, C., Jourdain, A. S., Dieux, A., Toutain, A., Callewaert, B., Dupuis-Girod, S., et al. (2017). Small patella syndrome: New clinical and molecular insights into a consistent phenotype. Clin. Genet. 92, 676–678. doi:10.1111/cge.13103
Venkatesha, S., Toporsian, M., Lam, C., Hanai, J., Mammoto, T., Kim, Y. M., et al. (2006). Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 12, 642–649. doi:10.1038/nm1429
Vera, M., and Fortes, P. (2004). Simian virus-40 as a gene therapy vector. DNA Cell Biol. 23, 271–282. doi:10.1089/104454904323090903
Verdera, H. C., Kuranda, K., and Mingozzi, F. (2020). Aav vector immunogenicity in humans: A long journey to successful gene transfer. Mol. Ther. 28, 723–746. doi:10.1016/j.ymthe.2019.12.010
Vetrini, F., and Ng, P. (2010). Gene therapy with helper-dependent adenoviral vectors: Current advances and future perspectives. Viruses 2, 1886–1917. doi:10.3390/v2091886
Waehler, R., Russell, S. J., and Curiel, D. T. (2007). Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 8, 573–587. doi:10.1038/nrg2141
Wang, D., Tai, P. W. L., and Gao, G. (2019). Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378. doi:10.1038/s41573-019-0012-9
Wang, T. M., Wang, S. S., Xu, Y. J., Zhao, C. M., Qiao, X. H., Yang, C. X., et al. (2021). Sox17 loss-of-function mutation underlying familial pulmonary arterial hypertension. Int. Heart J. 62, 566–574. doi:10.1536/ihj.20-711
Wang, X. J., Lian, T. Y., Jiang, X., Liu, S. F., Li, S. Q., Jiang, R., et al. (2019). Germline bmp9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 53, 1801609. doi:10.1183/13993003.01609-2018
Wang, Z., Li, M., Ji, Y., Yang, M., Yang, W., Wang, J., et al. (2020). Development of a novel bivalent baculovirus vectors for complement resistance and sustained transgene expression and its application in anti-angiogenesis gene therapy. Biomed. Pharmacother. 123, 109765. doi:10.1016/j.biopha.2019.109765
Watanabe, M., Boyer, J. L., and Crystal, R. G. (2010). Aavrh.10-mediated genetic delivery of bevacizumab to the pleura to provide local anti-vegf to suppress growth of metastatic lung tumors. Gene Ther. 17, 1042–1051. doi:10.1038/gt.2010.87
Watanabe, S., Ishikawa, K., Plataki, M., Bikou, O., Kohlbrenner, E., Aguero, J., et al. (2018). Safety and long-term efficacy of aav1.Serca2a using nebulizer delivery in a pig model of pulmonary hypertension. Pulm. Circ. 8, 2045894018799738. doi:10.1177/2045894018799738
Weiss, D. J. (2002). Delivery of gene transfer vectors to lung: Obstacles and the role of adjunct techniques for airway administration. Mol. Ther. 6, 148–152. doi:10.1006/mthe.2002.0662
Welch, C. L., Austin, E. D., and Chung, W. K. (2021). Genes that drive the pathobiology of pediatric pulmonary arterial hypertension. Pediatr. Pulmonol. 56, 614–620. doi:10.1002/ppul.24637
Welch, C. L., and Chung, W. K. (2020). Genetics and other omics in pediatric pulmonary arterial hypertension. Chest 157, 1287–1295. doi:10.1016/j.chest.2020.01.013
Wertz, J. W., and Bauer, P. M. (2008). Caveolin-1 regulates bmprii localization and signaling in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 375, 557–561. doi:10.1016/j.bbrc.2008.08.066
Wu, Z., Asokan, A., and Samulski, R. J. (2006). Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. 14, 316–327. doi:10.1016/j.ymthe.2006.05.009
Yan, Z., Feng, Z., Sun, X., Zhang, Y., Zou, W., Wang, Z., et al. (2017). Human bocavirus type-1 capsid facilitates the transduction of ferret airways by adeno-associated virus genomes. Hum. Gene Ther. 28, 612–625. doi:10.1089/hum.2017.060
Yan, Z., Keiser, N. W., Song, Y., Deng, X., Cheng, F., Qiu, J., et al. (2013). A novel chimeric adenoassociated virus 2/human bocavirus 1 parvovirus vector efficiently transduces human airway epithelia. Mol. Ther. 21, 2181–2194. doi:10.1038/mt.2013.92
Yang, X., Long, L., Southwood, M., Rudarakanchana, N., Upton, P. D., Jeffery, T. K., et al. (2005). Dysfunctional smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ. Res. 96, 1053–1063. doi:10.1161/01.RES.0000166926.54293.68
Yin, H., Kanasty, R. L., Eltoukhy, A. A., Vegas, A. J., Dorkin, J. R., and Anderson, D. G. (2014). Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 15, 541–555. doi:10.1038/nrg3763
Zhang, C., Basta, T., and Klymkowsky, M. W. (2005). Sox7 and sox18 are essential for cardiogenesis in xenopus. Dev. Dyn. 234, 878–891. doi:10.1002/dvdy.20565
Zhang, F., Wen, Y., and Guo, X. (2014). Crispr/cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 23, R40–R46. doi:10.1093/hmg/ddu125
Zhang, H. S., Liu, Q., Piao, C. M., Zhu, Y., Li, Q. Q., Du, J., et al. (2019). Genotypes and phenotypes of Chinese pediatric patients with idiopathic and heritable pulmonary arterial hypertension-a single-center study. Can. J. Cardiol. 35, 1851–1856. doi:10.1016/j.cjca.2019.07.628
Zhang, L., Jambusaria, A., Hong, Z., Marsboom, G., Toth, P. T., Herbert, B. S., et al. (2017). Sox17 regulates conversion of human fibroblasts into endothelial cells and erythroblasts by dedifferentiation into cd34(+) progenitor cells. Circulation 135, 2505–2523. doi:10.1161/CIRCULATIONAHA.116.025722
Zhao, Y. D., Courtman, D. W., Ng, D. S., Robb, M. J., Deng, Y. P., Trogadis, J., et al. (2006). Microvascular regeneration in established pulmonary hypertension by angiogenic gene transfer. Am. J. Respir. Cell Mol. Biol. 35, 182–189. doi:10.1165/rcmb.2005-0115OC
Zhong, L., Li, B., Mah, C. S., Govindasamy, L., Agbandje-McKenna, M., Cooper, M., et al. (2008). Next generation of adeno-associated virus 2 vectors: Point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. U. S. A. 105, 7827–7832. doi:10.1073/pnas.0802866105
Zhu, N., Gonzaga-Jauregui, C., Welch, C. L., Ma, L., Qi, H., King, A. K., et al. (2018). Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ. Genom. Precis. Med. 11, e001887. doi:10.1161/CIRCGEN.117.001887
Zhu, N., Pauciulo, M. W., Welch, C. L., Lutz, K. A., Coleman, A. W., Gonzaga-Jauregui, C., et al. (2019). Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 11, 69. doi:10.1186/s13073-019-0685-z
Keywords: genetics, gene therapy, PAH, BMPR2, childhood-onset
Citation: Dai L and Du L (2022) Genes in pediatric pulmonary arterial hypertension and the most promising BMPR2 gene therapy. Front. Genet. 13:961848. doi: 10.3389/fgene.2022.961848
Received: 05 June 2022; Accepted: 03 November 2022;
Published: 24 November 2022.
Edited by:
Zhihong Liu, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Sudhiranjan Gupta, VISN 17 Center of Excellence for Research on Returning War Veterans, United StatesPeiran Yang, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Copyright © 2022 Dai and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lizhong Du, dulizhong@zju.edu.cn